


Keywords: albuminuria, angiotensin II, dipeptidyl peptidase-4 inhibition, immune cells, kidney
Abstract
Recent evidence suggests that dipeptidyl peptidase-4 (DPP4) inhibition with saxagliptin (Saxa) is renoprotective under comorbid conditions associated with activation of the renin-angiotensin-aldosterone system (RAAS), such as diabetes, obesity, and hypertension, which confer a high cardiovascular risk. Immune system activation is now recognized as a contributor to RAAS-mediated tissue injury, and, importantly, immunomodulatory effects of DPP4 have been reported. Accordingly, we examined the hypothesis that DPP4 inhibition with Saxa attenuates angiotensin II (ANG II)-induced kidney injury and albuminuria via attenuation of immune activation in the kidney. To this end, male mice were infused with either vehicle or ANG II (1,000 ng/kg/min, s.c.) for 3 wk and received either placebo or Saxa (10 mg/kg/day, p.o.) during the final 2 wk. ANG II infusion increased kidney, but not plasma, DPP4 activity in vivo as well as DPP4 activity in cultured proximal tubule cells. The latter was prevented by angiotensin receptor blockade with olmesartan. Further, ANG II induced hypertension and kidney injury characterized by mesangial expansion, mitochondrial damage, reduced brush border megalin expression, and albuminuria. Saxa inhibited DPP4 activity ∼50% in vivo and attenuated ANG II-mediated kidney injury, independent of blood pressure. Further mechanistic experiments revealed mitigation by Saxa of proinflammatory and profibrotic mediators activated by ANG II in the kidney, including CD8+ T cells, resident macrophages (CD11bhiF4/80loLy6C−), and neutrophils. In addition, Saxa improved ANG II suppressed anti-inflammatory regulatory T cell and T helper 2 lymphocyte activity. Taken together, these results demonstrate, for the first time, blood pressure-independent involvement of renal DPP4 activation contributing to RAAS-dependent kidney injury and immune activation.
NEW & NOTEWORTHY This work highlights the role of dipeptidyl peptidase-4 (DPP4) in promoting ANG II-mediated kidney inflammation and injury. Specifically, ANG II infusion in mice led to increases in blood pressure and kidney DPP4 activity, which then led to activation of CD8+ T cells, Ly6C− macrophages, and neutrophils and suppression of anti-inflammatory T helper 2 lymphocytes and regulatory T cells. Collectively, this led to kidney injury, characterized by mesangial expansion, mitochondrial damage, and albuminuria, which were mitigated by DPP4 inhibition independent of blood pressure reduction.
INTRODUCTION
Kidney injury is common under comorbid conditions associated with inappropriate activation of the renin-angiotensin-aldosterone system (RAAS), including hypertension, diabetes, and obesity, which confer a high cardiovascular disease (CVD) risk (1–3). Indeed, these conditions play a major role in the progression of chronic kidney disease (CKD) to end-stage renal disease and dialysis (4, 5). Importantly, although RAAS blockade is recommended for renoprotection, it is often insufficient to completely prevent the progression of kidney injury or is not well tolerated (6, 7). Recently, the Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus-Thrombolysis in Myocardial Infarction 53 (SAVOR TIMI 53) trial demonstrated that inhibition of dipeptidyl peptidase-4 (DPP4) with saxagliptin (Saxa) reduced albuminuria (marker for kidney injury) in high-risk CVD patients with diabetes and hypertension (8, 9). This renoprotective effect of Saxa occurred independent of CKD stage and glycemic control, suggesting nonmetabolic protective mechanisms of this commonly prescribed diabetes medication warranting further examination (9). Other smaller clinical trials involving linagliptin (Tradjenta, Bohringer Ingelheim) and sitagliptin (Januvia, Merck and Co., Inc.) have shown similar benefits of DPP4 inhibition (10–12).
Several animal models of diabetes and/or obesity and hypertension treated with DPP4 inhibitors have shown improvement in kidney injury (glomerular and proximal tubular) and proteinuria/albuminuria (13). Although these are conditions of RAAS activation, none of the studies have examined whether DPP4 inhibitors directly attenuate ANG II-mediated kidney injury in rodent models. In this regard, a recent study showed that ANG II can directly stimulate DPP4 activity in the kidney and in cultured proximal tubule cells; however, this study did not address whether DPP4 inhibition can prevent ANG II-mediated kidney injury (14). The salutary effects of DPP4 inhibition on the kidney have been most frequently reported to occur without reduction in blood pressure (BP) (15–18). In a couple of studies, BP reduction has been reported with DPP4 inhibition. One study demonstrated attenuation of hypertension in an N-nitro-l-arginine methyl ester-induced model, whereas the other in a spontaneously hypertensive rat model (19, 20). This is similar to those few human studies where small decreases in BP following DPP4 inhibition have been reported, compared with many studies with no change in BP (21–24). Inflammation and immune system activation, which are common features in diabetes and/or obesity and hypertension, are reduced with DPP4 inhibition, suggesting that these mechanisms may play a major role in DPP4-mediated kidney injury (13, 16, 17, 25–27). However, the interactions between immune cell populations, relevant cytokines, and gene expression have not been examined in detail.
DPP4 or CD26, as it is known in the immunology field, is an exopeptidase that cleaves after NH2-terminal proline or alanine at the second position and either alters action of [neuropeptide Y (NPY), peptide YY] or inactivates [glucagon-like peptide 1 (GLP1), chemokine (C-X-C motif) ligand 12 (CXCL12), granulocyte maturation-colony stimulating factor (GM-CSF), monocyte chemoattractant protein-1 (MCP1)] its substrates (28). In addition, CD26 functions as a costimulator in T cell activation, proliferation, B cell activation [T helper (Th) function] and transendothelial migration (29). DPP4 has been shown to be involved in inflammation in the kidney and other organs (13, 30). Its levels have been shown to be higher in the kidney in conditions of obesity, diabetes and hypertension (13), and immune system activation plays a significant role in kidney injury in these conditions (31–33). The effects on ANG II-mediated infiltration of the kidney with immune cells is also well documented (32, 34, 35). Although DPP4 levels have been demonstrated to be increased along with activity in these conditions, the direct inhibitory effects on ANG II-mediated injury have not been examined.
Therefore, we hypothesized that DPP4 inhibition with Saxa would ameliorate the kidney injury and proteinuria caused by ANG II and that suppression of inflammation and/or immunomodulation will play a significant role in this process. To address this, we infused ANG II (1,000 ng/kg/min) into mice to induce hypertension, kidney injury, and immune system activation. We treated these mice with Saxa or vehicle after 1 wk of established hypertension and performed a comprehensive examination of the immune system to determine the effects of DPP4 inhibition.
METHODS
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Missouri. Male C57BL/6J mice were randomly divided into four groups: vehicle controls (Con), vehicle plus Saxa (Con + Saxa), ANG II, and ANG II plus Saxa (ANG II + Saxa). Saxa (10 mg/kg) was provided in peanut butter to achieve a plasma level of ∼300 nM (36). Groups not treated with Saxa received peanut butter alone. All mice had ad libitum access to standard chow and water in a climate controlled facility with a 12:12-h light:dark cycle.
ANG II Kidney Injury Model
ANG II (Sigma-Aldrich; 1,000 ng/kg/min in sterile saline) was infused for 3 wk via osmotic minipumps (Alzet model 1004; Alzet Corp., Mountain View, CA) beginning at 11 wk of age and animals were euthanized at 14 wk of age. In animals treated with Saxa, Saxa or control peanut butter treatment was initiated at 12 wk of age (1 wk after beginning of ANG II infusion) for 2 wk. Previous studies have demonstrated established hypertension in mice using this dose of ANG II after 2–4 days of infusion (37). Thus, in this treatment paradigm, Saxa treatment is initiated following establishment of ANG II-dependent hypertension. The treatment protocol is provided in Supplemental Fig. S1 (all Supplemental Material is available at https://doi.org/10.6084/m9.figshare.13138454). Plasma aldosterone was assessed at the end of the study as an index of ANG II pump effectiveness and was measured by radioimmunoassay at the Vanderbilt Hormone Assay & Analytical Services Core.
Radiotelemetry BP Measurement
C57BL/6J mice at 10 wk of age were anesthetized (2–4% isoflurane in air containing 40% O2) and instrumented via the carotid artery with an abdominal aorta catheter attached to a TA11PA-C10 radiotransmitter (Data Sciences International, St. Paul, MN), as previously described (38). The mice recovered for 7 days after surgery to regain normal circadian rhythms before treatments were initiated. During BP measurements, mice were individually housed in a quiet room where no other activities were permitted. Conscious mice were monitored in 300-s bins every 15 min for three 12-12-h light-dark cycles (sampling rate = 1,000 Hz) before initiating experimental treatments and weekly during treatments. Data were collected and stored for analysis by Dataquest A.R.T. software (Transoma Medical, St. Paul, MN). Parameters evaluated include systolic BP (SBP), diastolic BP (DBP), mean arterial pressure (MAP), heart rate (HR), and spontaneous cage activity [counts of lateral movement per minute (cpm)].
24-h Urinary Albumin Excretion
The 24-h urine collections were obtained on individually housed mice during the final week of treatment, using the MMC100 metabolic cages (Hatteras Instruments) with free access to water and food. After collection, urine was vortexed at max speed for 30 s and immediately centrifuged for 10 min (500 g) to remove heavy debris, and the supernatant was stored at −80°C before analysis. Albuminuria was determined by Albuwell-M ELISA (Exocell Inc., Philadelphia, PA), and creatinine was measured via enzymatic assay by Comparative Clinical Pathology Service, LLC (Columbia, MO).
DPP4 Activity Assay
Blood was collected in EDTA tubes, and plasma was stored at −80°C. Frozen plasma and kidney tissue extracts prepared as for OK cells were thawed on ice (17). For the activity assay, 20 µL plasma-EDTA or 100 µg protein were diluted in DPP4 assay buffer [Tris·HCl pH 8.0, NaCl 150 mM, and protease inhibitor cocktail (Roche Diagnostics)] in a black 96-well plate for a final volume of 50 µL. A total of 50 µL of the DPP4 substrate 200 mM H-Ala-Pro-AFC (200 mM; I-1680, Bachem, Torrance, CA) was added to each well and incubated for 10 min at room temperature in the dark. Fluorescence was measured with a Synergy Microplate Reader (excitation, 405 nm; emission, 535 nm). Results are calculated as relative light units (RLUs) and presented relative to appropriate control group.
Immunohistochemistry and Transmission Electron Microscopy
Immunohistochemistry and transmission electron microscopy (TEM) was performed as previously described (17, 27). Briefly, for immunohistochemistry, kidney sections were stained with anti-mouse/rat megalin antibody and periodic acid-Schiff (PAS) stain. Semiquantitation of mesangial expansion was done based on number of nuclei and staining intensity. More than three nuclei together and/or increased “pink” staining intensity was classified as having mesangial expansion.
Oxidative Stress
Kidney lipid peroxide content was assessed in whole cell lysates using a thiobarbituric acid-reactive substance (TBARS) assay, according to the manufacturer's instructions (Zeptometrix Corp., Buffalo, NY).
Isolation of Kidney Immune Cells
Mice were euthanized on 6 separate days in groups of four to contain a representative from each group (Con, Con + Saxa, ANG II, and ANG II + Saxa) on each day. Kidney immune cells were isolated according to our previously published method (39). Briefly, after mice were perfused via the heart with ice cold saline, kidneys were removed, the capsule was removed, and the kidneys were placed in plastic bags and minced in a Stomacher80 Biomaster. The kidney suspension was then passed through a 70-µm filter at 50 g after which the cells obtained were incubated on ice for 5 min in red blood cell (RBC) lysis buffer (Ebioscience, San Diego, CA). After pelleting the cells and resuspending in 36% Percoll, the cells were gently layered onto 72% Percoll and spun for 20 min at room temperature. The “buffy coat” was carefully suctioned and washed in FACS buffer (Ebioscience, San Diego, CA). Cells were counted on a hemocytometer, and equal number for each sample were used for labeling. We used separate kidney immune cell aliquots for the lymphoid and myeloid cell populations per the gating strategy described previously (39) and adapted to this work (Supplemental Figs. S2 and S3).
Flow Cytometry
Kidney immune cells were blocked with Fc block for 15 min on ice and stained with the following antibodies for 30 min: T cell panel—CD45-BV421 (30-F11), CD4-APC Cy7 (GK1.5), CD8α-BV785 (53-6.7), CD44-PerCP Cy5.5 (IM7), CD62L-BV605 (MEL-14), CD127-PE Cy7 (A7R34), CX3CR1-PE Cy7 (SA011F11), Ly6C-BV605 (HK1.4) and CD26-PE (H194-112) (Biolegend), FVD-BV510 (EBioscience); macrophage panel—lineage (Ly6G, CD3, NK1.1, B19)-FITC, CD45-BV421 (30-F11), F4/80-APC (BM8), CD11b-PerCP Cy5.5 (M1/70), CD11c-BV785 (N418), CD26-PE (H194-112), CX3CR1-PE Cy7 (SA011F11), CCR2-APC Cy7 (QA18A56) and Ly6C-BV605 (HK1.4) (Biolegend), FVD-BV510 (EBioscience). Full minus one (FMO) gating strategy was used to ensure specificity of the populations. The labeled cells were sorted on BD LSRFortessa X20 (BD Biosciences) and data were analyzed with FlowJo software and presented as percent changes relative to the corresponding control sample processed on the same day to account for any day-to-day variation.
mRNA Transcript Quantitation
Real-time quantitative PCR was done for 18 s rRNA, Arg, CCR7, CD68, Col1A, FoxP3, GAPDH, ICAM-1, IL-17, IL-23, MCP1/CCL2, osteoponin (OPN), and RORγT using SYBR green reagent and Bio-Rad iCycler. Fold changes in expression were quantitated by the 2ΔCt method. Semiquantitative PCR was done using real-time PCR primers and standard PCR reagents. Primer sequences are provided in Supplemental Table S1.
Kidney Cytokine Assay
Bioplex cytokine assay (Bio-Plex Pro Mouse Cytokine Assay No. M60009RDPD) was performed on kidney lysates. Samples were assayed in triplicate on a Bio-Rad Bioplex system in 96-well format, according to manufacturer instructions.
Cell Culture
T35OK-ANG II type 1 A receptor (AT1AR) (OK) cells stably expressing rat AT1ARs were grown in DMEM-F12 medium supplemented with 10% FBS, 100 µg/mL penicillin/streptomycin, 200 µg/mL G418, insulin, transferrin, dexamethasone, and epidermal growth factor (40). These cells are opossum-derived proximal tubule cells characterized by robust expression of transporters and channels and are widely utilized for physiological studies of tubular function. OK cells grown in starvation media (all of the above, except 0.1% FBS was substituted for 10% FBS) O/N were treated with either olmesartan (1 µM) or Saxa (1 µM) or vehicle 60 min before treatment with ANG II (10−8 M). OK cells were lysed after 1 h of ANG II stimulation in DPP4 assay buffer containing 0.1% Triton X-100 and protease inhibitors but no SDS for measurement of DPP4 activity.
Statistical Analysis
All data are presented as means ± SE. Two-way ANOVA and Fisher’s LSD post hoc t tests were performed to examine differences in outcomes among the various treatment groups. Between-group differences were considered significant at P values of ≤0.05, and significant trends were considered at P values between 0.051 and 0.1 and are explicitly indicated for clarity.
RESULTS
ANG II Increases and Saxa Reduces Kidney DPP4 Activity
As expected, ANG II (1,000 ng/kg/min) elicited an increase in plasma aldosterone levels along with heart hypertrophy and a reduction in body weight, the latter primarily due to reduced fat depot weights (Table 1). None of these ANG II-mediated effects were impacted by concomitant Saxa treatment, and Saxa reduced plasma insulin only in the setting of ANG II infusion (Table 1).
Table 1.
Phenotypic characteristics of study animals by group
| Phenotypic Characteristics | Control | Control + Saxagliptin | ANG II | ANG II + Saxagliptin |
|---|---|---|---|---|
| Body weight, g | 27.5 ± 0.8 | 27.2 ± 0.5 | 24.0 ± 0.5* | 25.2 ± 0.9* |
| Heart weight, mg | 118 ± 8 | 116 ± 3 | 148 ± 8* | 142 ± 9* |
| HW/TL | 67 ± 5 | 68 ± 1 | 87 ± 4* | 83 ± 6* |
| Kidney weight, mg | 334 ± 9 | 332 ± 9 | 291 ± 9* | 303 ± 15* |
| Epididymal fat weight, mg | 332 ± 18 | 294 ± 18 | 165 ± 27* | 206 ± 31* |
| Retroperitoneal fat weight, mg | 52 ± 7 | 50 ± 5 | 29 ± 5* | 31 ± 9* |
| Gastrocnemius muscle complex weight, mg | 318 ± 6 | 331 ± 17 | 317 ± 11 | 344 ± 41 |
| Plasma aldosterone, pg·mL−1 | 246 ± 18 | 230 ± 36 | 998 ± 180* | 1,426 ± 273* |
| Plasma insulin, ng·mL−1 | 0.24 ± 0.06 | 0.39 ± 0.13 | 0.18 ± 0.04 | 0.14 ± 0.01† |
Values are means ± SE; n = 6–10 mice per group. Statistical significance was determined using two-way ANOVA followed by Fisher’s LSD post hoc analysis. *P < 0.05 vs. control and control + saxagliptin, †P < 0.05 vs. control + saxagliptin. ANG II, angiotensin II; HW/TL, heart weight normalized to tibia length.
Consistent with our previous observations in ANG II-infused (200 ng/kg/min) mice (14), ANG II infusion increased kidney, but not plasma, DPP4 activity (Fig. 1, A and B). Saxa treatment suppressed DPP4 activity in both the vehicle and ANG II-treated groups by ∼50% (Fig. 1, A and B). Furthermore, we have previously demonstrated direct effects of ANG II to increase proximal tubule DPP4 activity in T35OK-AT1R cells (14). Consistent with that study, we observed increased DPP4 activity in T35OK-AT1R cells in response to 24-h ANG II treatment that was prevented by AT1R blockade with olmesartan, demonstrating an ANG II-AT1R-DPP4 signaling axis in proximal tubule cells (Fig. 1C). Saxa suppressed DPP4 activity in T35OK-AT1R cells regardless of treatment group (Fig. 1C).
Figure 1.

ANG II increased DPP4 activity in kidney and proximal tubule cells but not in plasma. DPP4 activity, assessed by gly-pro-p nitroanilide breakdown, was not changed in plasma by ANG II (A) but was increased in the kidney (B) and in cultured T35OK-AT1R proximal tubule cells (C) by ANG II. Saxa treatment suppressed plasma and kidney DPP4 activity ∼50% in vivo. n = 8–11 mice per group for plasma, 5–8 mice per group for kidney, and 4–5 mice per group for T35OK-AT1R cells. Statistical significance was determined using two-way ANOVA followed by Fisher’s LSD post hoc analysis. *P < 0.05 vs. all other groups, †P < 0.05 for Con + Saxa vs. Con, ‡P < 0.05 for ANG II + Saxa vs. ANG II. ANG II, angiotensin II; AT1AR, ANG II type 1 A receptor; Con, control; DPP4, dipeptidyl peptidase-4; Olme, olmesartan; Saxa, saxagliptin.
Saxa Suppresses ANG II-Induced Kidney Injury and Albuminuria but Not Hypertension
ANG II infusion is known to cause kidney injury and increase albuminuria in mice. Kidney injury, quantified by measurement of plasma blood urea nitrogen (BUN) and creatinine, revealed that ANG II increased creatinine and Saxa treatment significantly decreased creatinine (Fig. 2A). BUN was not changed by either treatment (Fig. 2B). Concomitant with a reduction in DPP4 activity, Saxa reduced ANG II-induced albuminuria (Fig. 2C). As expected (37, 41, 42), ANG II infusion increased SBP (Fig. 2D); however, Saxa treatment for 2 wk in the setting of established ANG II-hypertension did not reduce SBP (Fig. 2D), MAP [Con: 114 ± 4 mmHg, Con + Saxa: 114 ± 4 mmHg, ANG II: 138 ± 8 mmHg (P < 0.05 vs. Con/Con + Saxa), ANG II + Saxa: 140 ± 6 mmHg (P < 0.05 vs. Con/Con + Saxa)], or DBP [Con: 99 ± 6 mmHg, Con + Saxa: 97 ± 5 mmHg, ANG II: 126 ± 8 mmHg (P < 0.05 vs. Con/Con + Saxa), and ANG II + Saxa: 123 ± 8 mmHg (P < 0.05 vs. Con/Con + Saxa)] at the final time point.
Figure 2.

Saxa mitigates kidney injury and albuminuria but not established ANG II hypertension. ANG II-mediated kidney injury was indicated by increased serum creatinine levels (A), while BUN levels did not change (B). Saxa treatment significantly improved creatinine levels while exerting no effect on BUN. ANG II-induced albuminuria was reduced by Saxa (C) independent of changes in systolic BP, assessed by radiotelemetry prior to ANG II/vehicle treatment, after 1 wk of ANG II/vehicle treatment, and after 2 wk of ANG II/vehicle plus Saxa/placebo (D). n = 6–11 mice per group. Statistical significance was determined using two-way ANOVA followed by Fisher’s LSD post hoc analysis. *P < 0.05 vs. Con/Con + Saxa (A) and vs. all other groups (C), ‡P < 0.05 vs. ANG II, ¥P = 0.057 vs. ANG II. ANG II, angiotensin II; BP, blood pressure; Con, control; Saxa, saxagliptin; SBP, systolic blood pressure.
DPP4 Inhibition Mitigates ANG II-Mediated Kidney Ultrastructural Injury
Analysis of kidney ultrastructure revealed partial reversal of ANG II-mediated mesangial expansion, assessed with TEM and PAS staining, by Saxa (Fig. 3A, top). Podocyte architecture (effacement, slit pore diaphragm integrity) was unchanged although ANG II-mediated kidney injury involved increased expression of the podocyte slit pore diaphragm protein nephrin (Fig. 3A and Supplemental Fig. S4A), similar to previous work (43). Saxa treatment, interestingly, further increased nephrin protein expression, the significance of which is unknown (Supplemental Fig. S4A). In addition, Saxa treatment reduced kidney TBARs, an index of oxidative stress, but only in the presence of ANG II infusion (Supplemental Fig. S4B). Lastly, ANG II had deleterious effects on proximal tubule mitochondrial ultrastructure (44) that was attenuated by Saxa (Fig. 3B, top, and Supplemental Fig. S5), and ANG II-mediated reduction of megalin protein abundance in the brush border membranes was abrogated by Saxa (Fig. 3B, bottom).
Figure 3.
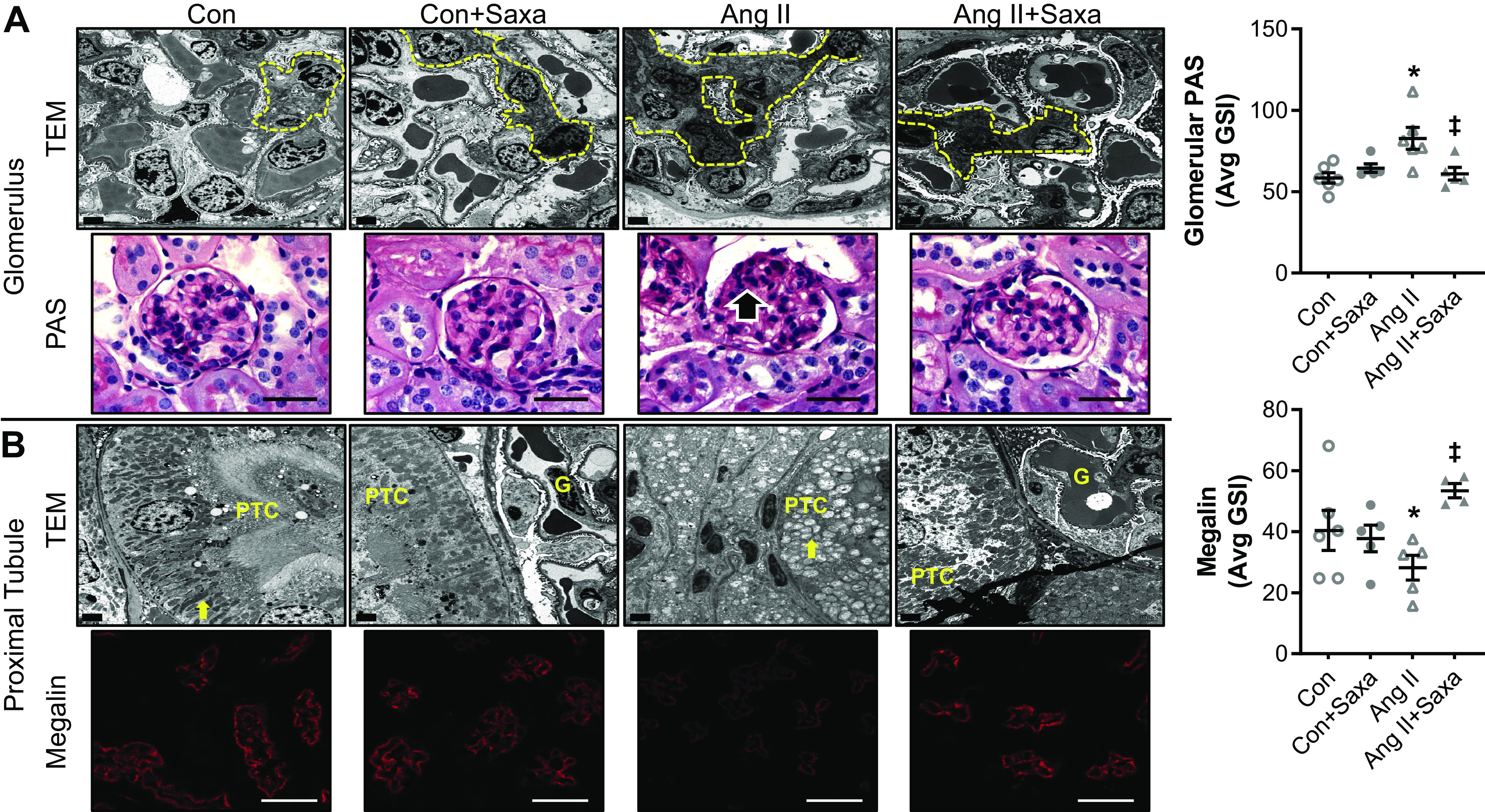
ANG II-induced kidney cortical injury is attenuated by Saxa. Glomeruli of ANG II-infused mice demonstrated focal and segmental mesangial expansion (dashed yellow lines, black arrow), assessed by TEM and PAS staining (600×), that was attenuated by Saxa (A). Proximal tubules of ANG II-infused mice exhibited loss of microvillar architecture and basilar canaliculi and emergence of mitochondrial rounding, assessed by TEM, as well as reduced brush border megalin expression that were both attenuated by Saxa (B). Glomerular PAS staining and brush border megalin expression quantitation are shown in panels on the right. n = 4–6 mice per group. Statistical significance was determined using two-way ANOVA followed by Fisher’s LSD post hoc analysis. *P < 0.05 vs. all other groups, ‡P < 0.05 vs. ANG II. ANG II, angiotensin II; Con, control; G, glomerulus; PAS, periodic acid-Schiff; PTC, proximal tubule cell; Saxa, saxagliptin; TEM, transmission electron microscopy.
DPP4 Inhibition Modulates ANG II-Mediated Activation of the Adaptive Immune System in the Kidney
In light of evidence of immune modulation by both ANG II and DPP4 signaling in the kidney and peripheral tissues (17, 45–52), we examined the adaptive immune response to ANG II and Saxa by flow cytometry (39). First, ANG II infusion induced an overall increase in CD45+ cells in the kidney that was unaffected by concomitant Saxa treatment (Table 2). There was no change in the proportion of total CD8+ or CD8+CD26+ cells with either treatment; however, the mean fluorescence intensity (MFI) of CD8+CD26hi T cells in the ANG II group was increased versus Con, and this increase was abrogated in the ANG II + Saxa group (Table 2 and Fig. 4A). There was tendency for an increased proportion of CD26 expression (P = 0.09) on the effector memory (EM) population (CD8+CD62L−CD44hi) by ANG II that was not suppressed significantly by Saxa (Table 2). There was no significant change in proportions of CD127+ or CD127− subsets of the EM population nor the central memory (CM) population (CD8+CD62L+CD44hi) with ANG II or Saxa. Among CD4+ T cells, ANG II significantly reduced the proportion of CD4+CD45+ cells, and this was not affected by Saxa (Table 2). ANG II did not change the proportion of CD4+ EM cells; however, CD127+ EM cells tended to be increased by ANG II (P = 0.052) and CD127− EM cells tended to decrease (P = 0.061). Saxa did not change either of these ANG II effects on CD4+ EM subpopulations. In contrast, there was a tendency for Saxa to increase CM population when compared with Con group (P = 0.041 by pairwise comparison). Lastly, with regard to kidney B cells (B&NK+CX3CR1−), Saxa tended to increase B cells in the setting of ANG II infusion (P = 0.072). There were no significant changes in CD26 expression on B cells (Supplemental Fig. S6).
Table 2.
Kidney lymphoid immune cell populations by group
| Cell Type | Cell Gates | Control | Control + Saxagliptin | ANG II | ANG II + Saxagliptin |
|---|---|---|---|---|---|
| Leukocytes | CD45+ | 1.90 ± 0.21 | 2.17 ± 0.33 | 3.60 ± 0.49* | 3.36 ± 0.62* |
| CD4+ T cells | CD4+ | 1.00 ± 0.00 | 0.99 ± 0.08 | 0.81 ± 0.06* | 0.81 ± 0.06* |
| CD4+ CD26Lo | 50.80 ± 1.84 | 50.14 ± 1.69 | 54.62 ± 2.73 | 48.82 ± 2.52 | |
| CD4+ Memory T cells | CD4+ CD62L− CD44Hi | 1.00 ± 0.00 | 0.86 ± 0.05 | 1.06 ± 0.08 | 1.01 ± 0.10 |
| CD4+ CD62L− CD44Hi CD127+ | 1.00 ± 0.00 | 1.40 ± 0.29 | 1.65 ± 0.19* | 1.62 ± 0.15* | |
| CD4+ CD62L− CD44Hi CD127− | 1.00 ± 0.00 | 0.92 ± 0.05 | 0.93 ± 0.02* | 0.89 ± 0.02* | |
| CD4+ CD62L+ CD44+ | 1.00 ± 0.00 | 1.20 ± 0.23 | 1.27 ± 0.36 | 1.91 ± 0.41* | |
| CD8+ T cells | CD8+ | 1.00 ± 0.00 | 0.84 ± 0.05 | 0.98 ± 0.16 | 0.82 ± 0.12 |
| CD8+ CD26Hi | Median (Comp-PE-A) | 1.00 ± 0.00 | 1.06 ± 0.05 | 1.91 ± 0.45* | 0.95 ± 0.06‡ | |
| CD8+ Memory T cells | CD8+ CD62L− CD44Hi | 1.00 ± 0.00 | 0.84 ± 0.09 | 1.00 ± 0.18 | 0.73 ± 0.09 |
| CD8+ CD62L− CD44+ CD127+ | 1.00 ± 0.00 | 1.23 ± 0.18 | 1.14 ± 0.13 | 1.11 ± 0.08 | |
| CD8+ CD62L− CD44+ CD127− | 1.00 ± 0.00 | 0.90 ± 0.08 | 0.91 ± 0.05 | 0.94 ± 0.04 | |
| CD8+ CD62L+ CD44Hi CM | 1.00 ± 0.00 | 0.70 ± 0.15 | 1.30 ± 0.09 | 1.30 ± 0.32 | |
| CD8+ CD62L− CD44Hi CD26+ | 1.00 ± 0.00 | 0.98 ± 0.11 | 1.26 ± 0.15¥ | 1.02 ± 0.09 | |
| B cells | B&NK cells/CX3CR1− | 1.00 ± 0.00 | 1.04 ± 0.05 | 1.13 ± 0.08 | 1.21 ± 0.08* |
Values are means ± SE (absolute or relative to control); n = 4–6 mice per group. Statistical significance was determined using two-way ANOVA followed by Fisher’s LSD post hoc analysis. *P < 0.05 vs. control; ‡P < 0.05 vs. ANG II; ¥P = 0.05–0.09 vs. control. ANG II, angiotensin II.
Figure 4.

Saxa suppresses ANG II-mediated activation of kidney immune cells. Surface expression of CD26 was measured by flow cytometry (A and B). ANG II-induced increases of CD26Bright CD8+ T lymphocytes (A) and CD26+ profibrotic macrophages (CD11b+F4/80−Ly6C−; B) in the kidney were attenuated by Saxa. Representative histogram of the macrophage data is shown on the right. C: dot plot representation of ANG II-induced increase of neutrophils in the kidney that was attenuated by Saxa. n = 6 mice per group. Statistical significance was determined using two-way ANOVA followed by Fisher’s LSD post hoc analysis. *P < 0.05 vs. all other groups, ‡P < 0.05 vs. ANG II, §P = 0.08 vs. Con. ANG II, angiotensin II; Con, control; Saxa, saxagliptin.
T cell populations were also analyzed in the blood and spleen to assess whether the findings noted above were unique to the kidney (Supplemental Table S2). ANG II and ANG II + Saxa groups had decreased proportion of CD4+ in the PBMCs (P < 0.05 by Fisher’s multiple pairwise comparison, P = 0.074 by ANOVA) while, similar to the kidney, there was no effect on the CD8+ population. There was no change in either the CD26+ CD4+ or CD8+ T cell populations, and there was no significant treatment effect of either ANG II or Saxa in the PBMCs, which contrasted with the kidney. In the spleen, neither ANG II nor Saxa had any effect on proportion of CD4+ or CD8+ T cells (Supplemental Table S2). Both Saxa alone and ANG II groups had a tendency to increase CD4+CD26Bright cells in the spleen (P < 0.05 by Fisher’s multiple pairwise comparison, P = 0.051 by ANOVA). The CM CD4+CD44+CD62L+ cells tended to be increased with all treatments (P < 0.05 with Saxa and ANG II + Saxa, P = 0.050 with ANG II, P = 0.118 by ANOVA). There was no treatment effect of either Saxa or ANG II on the CD8+ memory cell population. Taken together, the effects of ANG II and Saxa on T cell populations varied by tissue and, in the kidney, some of the ANG II-induced proinflammatory populations were attenuated by Saxa treatment (Table 2 and Supplemental Table S2).
DPP4 Inhibition Modulates ANG II-Mediated Activation of the Innate Immune System in the Kidney
With respect to mononuclear cells, we examined the proinflammatory and profibrotic subtypes of kidney mononuclear cells (2, 30, 39, 53). Among macrophages, ANG II treatment did not change CD11b+F4/80lo or CD11b+F4/80− subpopulations while Saxa tended to increase CD11b+F4/80lo macrophages (P = 0.071; Table 3). Further separation based on the migration marker Ly6C revealed no change in Ly6Chi, Ly6Clo, and Ly6C−CD11b+F4/80lo macrophages in any group. Since CD11b+F4/80loLy6C− macrophages represent a profibrotic macrophage subset, we examined the proportion of CD26+ CD11b+F4/80loLy6C− cells and found a trend to be increased by ANG II (P = 0.08) that was prevented by Saxa (Table 3 and Fig. 4B). Use of CCR2 as marker of macrophages revealed that a very low proportion of CD11b+F4/80loLy6C− macrophages were CCR2+ further confirming that CD11b+F4/80loLy6C− macrophages were likely resident macrophages. Furthermore, there was a tendency (P < 0.05 by pairwise comparison with the Con group) for Saxa to suppress CD11b+F4/80loLy6Clo macrophages (Table 3). Next, we examined CD11b+F4/80− macrophages based on Ly6C expression and observed that Saxa reduced the proportion of Ly6C−, similar to CD11b+F4/80lo macrophages above, while increasing the Ly6Clo subset when compared with ANG II. Furthermore, ANG II increased the proportion of CD26hi CD11b+F4/80− macrophages, and this was suppressed by Saxa (Table 3), and there was a trend for increase in CD26lo CD11b+F4/80− macrophages with ANG II (P < 0.05, pairwise comparison with Con). Together, these data suggest that DPP4 inhibition shifts the balance toward recruitment of more repair-type macrophages and suppression of inflammatory and profibrotic phenotypes (Table 3). Next, we examined the contribution of dendritic cells, and there was no change in the cell proportions of CD11bneg-loF4/80hi dendritic cells in either the ANG II or ANG II + Saxa groups (Table 3). These cells were mostly CXCR1+, CD11c+, and CCR2−, confirming that the CD11bneg-loF4/80hi cells are indeed residential dendritic cells of the kidney (Table 3). The only exception to this result was the tendency to increase the CX3CR1+CD11c− subset of CD11bneg-loF4/80hi dendritic cells by ANG II. In addition, we observed an ANG II-induced reduction in kidney natural killer (NK) cells that was not affected by Saxa (B&NK/CX3CRhi, Table 3). Lastly, ANG II increased the proportion of neutrophils among CD45+ cells, and this was mitigated by Saxa (Fig. 4C and Table 3), further demonstrating amelioration of ANG II-induced inflammation in the kidney by Saxa (P < 0.05).
Table 3.
Kidney myeloid immune cell populations by group
| Cell Type | Cell Gates | Control | Control + Saxagliptin | ANG II | Ang II +Saxagliptin |
|---|---|---|---|---|---|
| Neutrophils and natural killer cells | CD11b+ F4/80Lo Lineage+ Ly6C+ CD26+ | 7.29 ± 1.63 | 8.11 ± 1.75 | 19.56 ± 8.04¥ | 8.47 ± 2.89 |
| CD11b+ F4/80Lo macrophages | CD11b+ Lineage− CD11b+ F4/80Lo | 1.00 ± 0.00 | 1.03 ± 0.15 | 1.49 ± 0.37 | 1.65 ± 0.27§ |
| CD11b+ Lineage− CD11b+ F4/80Lo Ly6C− | 1.00 ± 0.00 | 0.94 ± 0.08 | 0.96 ± 0.06 | 0.88 ± 0.10 | |
| CD11b+ F4/80Lo Lineage− Ly6C− CD26+ | 1.00 ± 0.00 | 1.09 ± 0.08 | 1.19 ± 0.07* | 0.89 ± 0.09‡ | |
| CD11b+ Lineage− CD11b+ F4/80Lo Ly6C− CCR2+ | 1.00 ± 0.00 | 0.93 ± 0.16 | 0.99 ± 0.13 | 0.74 ± 0.05 | |
| CD11b+ Lineage− CD11b+ F4/80Lo Ly6CHi | 1.00 ± 0.00 | 1.05 ± 0.11 | 1.03 ± 0.06 | 1.12 ± 0.11 | |
| CD11b+ Lineage− CD11b+ F4/80Lo Ly6CHi CCR2+ | 1.00 ± 0.00 | 1.12 ± 0.12 | 1.05 ± 0.14 | 1.05 ± 0.16 | |
| CD11b+ Lineage− CD11b+ F4/80Lo Ly6CLo | 1.00 ± 0.00 | 1.19 ± 0.06 | 1.10 ± 0.13 | 1.20 ± 0.13 | |
| CD11b+ Lineage− CD11b+ F4/80Lo Ly6CLo CCR2+ | 1.00 ± 0.00 | 0.88 ± 0.06¥ | 1.04 ± 0.05 | 0.90 ± 0.01‡ | |
| CD11b+ F4/80− macrophages | CD11b+ F4/80− Lineage− | 1.00 ± 0.00 | 0.89 ± 0.14 | 0.92 ± 0.21 | 0.82 ± 0.08 |
| CD11b+ Lineage− CD11b+ F4/80− Ly6C− | 1.00 ± 0.00 | 0.92 ± 0.04 | 1.07 ± 0.07 | 0.84 ± 0.04*‡ | |
| CD11b+ Lineage− CD11b+ F4/80− Ly6CLo | 1.00 ± 0.00 | 1.27 ± 0.02 | 0.86 ± 0.14 | 1.22 ± 0.16‡ | |
| CD11b+ Lineage− CD11b+ F4/80− Ly6CHi | 1.00 ± 0.00 | 0.95 ± 0.12 | 0.93 ± 0.16 | 1.21 ± 0.19 | |
| CD11b+ Lineage− CD11b+ F4/80− CD26Hi | 1.00 ± 0.00 | 1.28 ± 0.10* | 1.32 ± 0.02* | 1.06 ± 0.11‡ | |
| CD11b+ Lineage− CD11b+ F4/80− CD26Lo | 1.00 ± 0.00 | 1.14 ± 0.17 | 1.52 ± 0.22* | 1.39 ± 0.19 | |
| Dendritic cells | CD11bNeg-Lo F4/80Hi Lineage− | 1.00 ± 0.00 | 1.19 ± 0.10 | 1.08 ± 0.07 | 1.03 ± 0.06 |
| CD11bNeg-Lo F4/80Hi Lineage− CCR2− | 90.62 ± 4.48 | 88.85 ± 5.54 | 92.20 ± 3.11 | 90.68 ± 4.38 | |
| CD11bNeg-Lo F4/80Hi Lineage− CX3CR1+ | 99.88 ± 0.08 | 99.95 ± 0.03 | 99.70 ± 0.15 | 99.60 ± 0.22 | |
| CD11bNeg-Lo F4/80Hi Lineage− CX3CR1+ CD11c+ | 96.53 ± 0.36 | 95.72 ± 0.62 | 95.15 ± 0.54 | 95.67 ± 0.70 | |
| CD11bNeg-Lo F4/80Hi Lineage− CX3CR1+ CD11c− | 1.00 ± 0.00 | 1.27 ± 0.18 | 1.50 ± 0.22 | 1.24 ± 0.15 | |
| Natural killer cells | B&NK Cells/CX3CR1Hi | 1.00 ± 0.00 | 0.96 ± 0.19 | 0.70 ± 0.10¥ | 0.66 ± 0.04¥ |
| B&NK Cells/CX3CR1Lo Ly6C− | 78.62 ± 2.97 | 78.80 ± 2.34 | 81.95 ± 1.43 | 80.88 ± 2.12 | |
| B&NK Cells/CX3CR1Lo Ly6CHi | 9.01 ± 0.68 | 11.06 ± 1.44 | 8.75 ± 1.02 | 7.47 ± 0.41 | |
| B&NK Cells/CX3CR1Lo Ly6CLo | 10.40 ± 1.35 | 10.14 ± 1.22 | 9.32 ± 0.95 | 8.92 ± 0.62 | |
| Neutrophils | CD45+ T-B-NK1.1−Ly6G+ | 5.43 ± 0.28 | 6.23 ± 0.45 | 8.81 ± 1.37* | 5.76 ± 0.47‡ |
Values are means ± SE (absolute or relative to control); n = 4–6 mice per group. Statistical significance was determined using two-way ANOVA followed by Fisher’s LSD post hoc analysis. *P < 0.05 vs. control; ‡P < 0.05 vs. ANG II; ¥P = 0.05–0.09 vs. control; §P = 0.07 vs. ANG II. ANG II, angiotensin II.
Saxa Modulates Anti-Inflammatory Genes and Cytokines in the Kidney
To better understand the modulation of the lymphoid and myeloid cell populations by ANG II and Saxa, we explored gene expression and quantitated levels of cytokines/chemokines in the kidney (Fig. 5, A and B). Both ANG II and Saxa increased OPN gene expression (Fig. 5A). MCP1/CCL2 was not changed by any treatment (Supplemental Fig. S7A). RORγT, a transcription factor from Th17 cells, was not changed by ANG II but was increased in the ANG II + Saxa group (Fig. 5A). IL23p19 (Th17 activation cytokine) and IL17A expression were both downregulated by ANG II infusion but not impacted by Saxa. The anti-inflammatory M2 macrophage marker Arg was increased in the ANG II + Saxa group. Further, FoxP3 gene expression, as a marker for regulatory T cells (Tregs), was reduced by ANG II and tended to be restored by Saxa in the kidney (P = 0.079). CCR7 expression, a marker of CM cells, was not changed in CD4+CD44+CD62L+ cells by ANG II but was increased in the ANG II + Saxa group (Supplemental Fig. S7B) (54). In the Con + Saxa group, the expression of OPN was increased while IL-23, FoxP3, CD68, and ICAM-1 expression were all reduced, indicating differential impact of Saxa on kidney gene expression under control versus ANG II conditions (Fig. 5A). To further assess regulation of kidney inflammation specifically in the setting of ANG II infusion, we measured pro- and anti-inflammatory cytokine levels in the kidney by Bioplex cytokine assay and limited it to three groups (Con, ANG II, and ANG II + Saxa). This analysis revealed increased granulocyte colony-stimulating factor (G-CSF) with ANG II infusion that was not affected by Saxa (Fig. 5B, right). Further, the neutrophil chemoattractant protein keratinocytes-derived chemokine (KC) and TNF-α were not affected by either ANG II or Saxa (Supplemental Fig. S8). ANG II infusion suppressed kidney IL-2 and IL-10 but did not significantly suppress IL-9 and IL-13 expression (Fig. 5B, left). Lastly, in ANG II-treated animals, Saxa treatment restored IL-2 expression and increased IL-9 expression but did not significantly restore IL-10 and IL-13 levels (Fig. 5B, left).
Figure 5.

Saxa differentially impacts ANG II-mediated cytokine and gene expression changes in the kidney. ANG II-induced changes in kidney cytokines were assessed at the transcript (A) and protein level (B) in the absence or presence of Saxa. n = 5–6 mice per group (A) and n = 3–4 mice per group (B). Statistical significance was determined using two-way ANOVA followed by Fisher's LSD post hoc analysis. *P < 0.05 vs. Con, †P < 0.05 vs. Con + Saxa, ‡P < 0.05 vs. ANG II, §P = 0.067 vs. Con + Saxa, ¥P = 0.051–0.08 vs. ANG II, **P = 0.068 vs Con. ANG II, angiotensin II; Con, control; Saxa, saxagliptin.
DISCUSSION
The recent SAVOR-TIMI 53 trial demonstrated that DPP4 inhibition with Saxa reduced albuminuria in high-risk CVD patients with comorbid conditions independent of estimated glomerular filtration rate and glycemic control, suggesting renoprotection afforded by Saxa. Activation of the RAAS is widely recognized as a primary mediator of hypertension and kidney injury in comorbid conditions such as type 2 diabetes mellitus and obesity. Therefore, to address potential mechanisms, we examined whether Saxa demonstrated salutary effects in the setting of established ANG II-mediated hypertension and kidney injury. To that end, we demonstated a kidney ANG II-AT1R-DPP4 axis and that Saxa mitigates ANG II-mediated glomerular and tubular injury and albuminuria. These salutary effects involved attenuation of ANG II-mediated activation of kidney T cells, macrophages, and neutrophils, independent of BP. Taken together, these data reveal RAAS-DPP4-dependent contributions to kidney injury in comorbid conditions and highlight attenuation of RAAS-dependent kidney immune activation as a renoprotective action of Saxa.
The nature of the kidney injury induced by ANG II in this study is consistent with previous published work (2, 48, 55, 56). Specifically, ANG II induced modest albuminuria (∼6-fold increase) compared with control mice, associated with patchy (i.e., focal and segmental) glomerulosclerosis (43, 55, 56), with mesangial expansion and tubulointerstitial injury (57). Further, we observed ANG II-induced mitochondrial fragmentation and loss of basilar canaliculi, suggesting increased oxidative stress and tubular injury, in conjunction with reduced brush border megalin expression, suggesting decreased uptake of protein and/or albumin (58). Interestingly, Saxa treatment significantly attenuated ANG II-induced kidney injury, oxidative stress, and inflammation but not hypertension. The lack of effect of Saxa on BP is consistent with available evidence demonstrating either no change or modest BP-lowering effects of DPP4 inhibitors (59). Some preclinical and clinical studies have reported BP-lowering effects of Saxa, whereas others such as SAVOR-TIMI53 did not observe BP changes, suggesting possible context-specific effects of DPP4 inhibition on BP warranting further exploration (8, 60–62). Moreover, Saxa not being a complete angiotensin receptor blocker likely suppresses a downstream arm of angiotensin receptor signaling leading to differential effects on hypertension and kidney injury. Taken together, Saxa treatment attenuated ANG II-mediated injury and albuminuria, independent of BP, highlighting critical local mechanisms of protection in the kidney.
DPP4 activity is increased in comorbid conditions of obesity, type 2 diabetes mellitus, and under conditions of RAAS activation, specifically that induced by ANG II (13, 14, 17, 27, 30, 63–65). Previously, we tested the hypothesis that the RAAS can activate DPP4 and reported that ANG II stimulation of OK-AT1AR cells increased DPP4 activity that was suppressed by both AT1R and DPP4 inhibition (14). In this study, Saxa suppressed ANG II-mediated DPP4 activation in OK-AT1AR cells and in kidney lysates, consistent with the notion that ANG II directly activates DPP4. In addition, we measured insulin and TNF-α, two known stimulators of DPP4, and reported no increase in either of the ANG II treatment groups. In further support of this notion, Saxa is highly selective for DPP4 and does not inhibit DPP8/9, FAP, etc. at the concentration used in this study (36). Furthermore, there was no increase in plasma DPP4 activity with ANG II, suggesting that increased kidney DPP4 activity alone is likely responsible for ANG II-mediated effects. In related studies, we have observed that DPP4 inhibition partially mitigates classical ANG II-mediated activation of ERK, mTOR (unpublished data), and EGFR pathways (14), suggesting downstream interaction between these systems contributing to the renoprotection afforded by Saxa. Taken together, our results strongly support the hypothesis that DPP4 inhibition in the kidney directly mitigated ANG II-mediated deleterious effects in our model.
Since both ANG II (34, 46, 54, 66, 67) and DPP4 (28, 29, 68) are known to exert their effects via immune system activation, we examined this interaction via comprehensive flow cytometric analysis. Indeed, CD26 contributes to the regulation of development, maturation, and migration of CD4+/CD8+ T and NK cells, cytokine secretion, T cell-dependent antibody production, and immunoglobulin isotype switching of B cells (28, 29). In addition to an increase in whole kidney lysate DPP4 activity, we observed an ANG II-induced increase in CD26 expression on CD8+ and CD8+CD44hiCD62L− EM T lymphocytes. Of note, CD26 expression is regulated in response to differentiation and activation status of immune cells and, importantly, activation of lymphocyte CD26 leads to cell proliferation (28, 29, 69). For example, among resting peripheral blood mononuclear cells, only a small subset of T cells expresses CD26 at high density on the surface (CD26bright T cells) (29, 69). In contrast, CD26bright T cells proliferate strongly in response to soluble antigens and allogeneic cells, secrete Th1 type cytokines, and acquire capability for transendothelial migration thus contributing to tissue inflammation as suggested by our data in the setting of ANG II infusion (29, 69). CD8+ cells have been implicated in mediating hypertension in response to ANG II (70, 71), and our data suggest a novel role for DPP4-dependent activation of these cells contributing to ANG II-induced kidney injury. In this study, ANG II increased CD26hi expression (MFI) on CD8+ cells as well as CD26hi expression on proinflammatory CD8+CD44hiCD62L− EM cells, which was reversed by Saxa. Our findings on CD8+ T cells should be taken in context of minor CD8α antigen presence on NK1.1 and dendritic cells since we gated CD8+ cells off of CD45+ cells and not CD45+CD3+ cells. In our study, NK1.1 and dendritic cell contribution was negligible, thereby strongly supporting our conclusion on the role of CD8+ T cells. ANG II also tended to increase CD4+CD44+CD62L−CD127+ EM T cells in the kidney, which were not affected by Saxa. EM T cells are thought to be proinflammatory, have been shown to accumulate in the kidney during repeated stimuli with ANG II, and produce proinflammatory Th1 and Th17 cytokines (32). In addition, mice lacking accumulation of EM T cells were protected against kidney injury (32). Overall, in our study, Saxa did not have much effect on the CD4+ population in the kidney. To determine whether ANG II-mediated kidney injury was via local activation of the immune system versus systemic, we compared our kidney data with that in the peripheral blood and spleen. There was a reduction in CD4+ T cells in blood similar to the kidney with ANG II and ANG II + Saxa, and there was an increase in CD4+CD26bright cells with ANG II in the spleen. Taken together, these data suggest that the predominant way DPP4 inhibition affects protection from ANG II-induced kidney injury is via suppression of CD8+ T cell activation in the kidney.
Beyond kidney T cells, our data suggest further modulation of kidney macrophage populations by DPP4 as possible mechanisms of renoprotection during ANG II infusion. Indeed, DPP4 has been shown to activate adipose tissue macrophages and dendritic cells in obese humans and leads to visceral fat inflammation (30). Further, CD11b+F4/80lo and CD11b+F4/80− macrophages/monocytes that are Ly6C− have been reported as profibrotic in the kidney (72). Accordingly, ANG II increased CD26hi expression on these cells that was suppressed by Saxa. In addition, Saxa decreased the proportion of CD11b+F4/80−Ly6C−, while increasing CD11b+F4/80−Ly6Clo together suggesting attenuation of these profibrotic macrophages and increase in repair-type macrophages underlies Saxa-mediated reduction of ANG II-induced kidney injury (72). In addition to T cells and monocytes, we observed a tendency for decreased numbers of NK cells migrating to the kidney in the ANG II group that was unchanged with Saxa treatment in addition to no change in NK cell CD26 expression. This suggests little role of these cells in Saxa-mediated effects, as CD26 is induced upon stimulation of NK cells (73). Kidney neutrophils (CD11b+T, B&NK−Ly6G+), however, were increased by ANG II infusion consistent with increased oxidative stress and inflammation, while Saxa significantly reduced ANG II-induced renal infiltration of neutrophils. ANG II is known to increase neutrophil migration and accumulation predominantly via G-CSF and KC (74–76). In our study, we observed an increase in G-CSF and no change in KC levels in the kidney with ANG II that were not changed by Saxa, suggesting alternate regulation of kidney neutrophil recruitment by DPP4 in RAAS activation that warrants further study. Further, kidney TBARS were significantly suppressed by Saxa in the setting of ANG II infusion. Taken together, these data highlight local ANG II-DPP4-dependent modulation of kidney immunity, both innate and acquired, and inflammation as primary mechanisms underlying kidney injury and albuminuria associated with RAAS activation that are attenuated by DPP4 inhibition.
Local tissue immune modulation is heavily influenced by local cytokine production to influence recruitment or differentiation of resident/infiltrating leukocytes (47, 77, 78). Overall, our data suggest that ANG II induces a proinflammatory shift in the local cytokine/chemokine profile that is countered by Saxa treatment. Specifically, ANG II infusion reduced anti-inflammatory IL-10 expression (35), secreted by both Tregs and Th2 lymphocytes, and reduced gene expression of the Treg marker FoxP3. Interestingly, Saxa did not prevent the ANG II-induced reduction of IL-10 but did increase IL-9 and tended to increase FoxP3, all Treg markers, in the setting of ANG II infusion. In addition, Saxa increased the M2 macrophage marker Arg, suggesting a shift toward a less inflammatory M2 macrophage profile (77). These data suggest that activation of Tregs (IL-9, FoxP3) and M2 macrophages by Saxa may counter the adverse effects of ANG II on the kidney. Critical to this process may be IL-2, as evidence suggests IL-2 may be a master switch that promotes T lymphocyte differentiation, particularly FoxP3+ Treg cells (79). Indeed, the ANG II-mediated reduction of kidney IL-2 was prevented by Saxa in concert with the noted upregulation of Treg marker genes/cytokines. In contrast to anti-inflammatory genes and cytokines, ANG II did not increase proinflammatory genes and cytokines except for G-CSF and osteopontin. Increased G-CSF may have both profibrotic and antifibrotic properties and is a known chemoattractant for neutrophils. Osteopontin can affect macrophage differentiation and monocyte/lymphocyte migration, promote wound healing, and reduce vascular calcification. In our study, osteopontin expression was increased in all treatment groups compared with the Con group, which suggests either a compensatory increase (ANG II) or a treatment effect (Saxa). Together, our data suggest that the renoprotective impact of Saxa in the setting of RAAS activation involves novel increases in the differentiation and activity of anti-inflammatory Tregs, Th2 lymphocytes, and M2 macrophages to counter the deleterious effects of ANG II signaling on kidney function.
Lastly, it should be noted that this study examined the impact of DPP4 inhibition on ANG II-induced kidney injury only in male mice. Females have greater incidence of CKD and higher hospitalization rates with end-stage renal disease. However, young female mice are protected from ANG II-induced hypertension and kidney injury due to AT2R-mediated and other mechanisms, when compared with male mice (80, 81) complicating investigations into sex differences. The SAVOR TIMI 53 trial that demonstrated improved kidney outcomes in high-risk CVD patients included both male and female patients; thus, further preclinical work in females is warranted.
In summary, our data demonstrate that DPP4 inhibition with Saxa mitigates ANG II-dependent activation of the immune system and associated kidney injury and albuminuria independent of a reduction in BP. Importantly, these changes were associated with suppression of proinflammatory and profibrotic subset of macrophages, neutrophils, CD8+ T cells concomitant with activation of Tregs, Th2 T lymphocytes, and M2 macrophages by Saxa in our model. These findings support a role for DPP4 inhibition in ameliorating kidney injury in comorbid conditions of RAAS activation including diabetes, hypertension, and obesity.
GRANTS
R.N. was supported by National Institutes of Health (NIH) Grant K08DK115886, S.B.B. was supported by Department of Veterans Affairs Grant CDA2 BX002030 and NIH Grant R01HL136386, A.W.-C. was supported by Department of Veteran Affairs Merit Grant BX003391, J.R.S. was supported by NIH Grant R01HL107910, and S.C.M. was supported by NIH Grant R01ES022966.
DISCLOSURES
R.N. and S.B.B. received an Investigator-Initiated Grant from Astra Zeneca. R.N. was supported by a grant from Dialysis Clinics Inc. The rest of the authors had nothing to disclose.
AUTHOR CONTRIBUTIONS
R.N., J.R.S., S.C.M., S.B.B., A.I.M., C.S., and A.W-C. conceived and designed research; R.N., S.C.M., A.I.M., C.S., J.A., J.H., M.R.H., M.J., and A.A. performed experiments; R.N., S.C.M., S.B.B., A.I.M., C.S., J.H., M.R.H., M.J., and A.A. analyzed data; R.N., S.C.M., S.B.B., A.I.M., C.S., M.R.H., M.J., and A.A. interpreted results of experiments; R.N., S.B.B., J.H., and M.R.H. prepared figures; R.N. and S.B.B. drafted manuscript; R.N., J.R.S., S.B.B., and M.R.H. edited and revised manuscript; R.N., J.R.S., S.C.M., S.B.B., A.I.M., C.S., J.A., J.H., M.R.H., M.J., A.A., and A.W-C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Terry L. Carmack and Lisa D. Watkinson of the Truman Memorial Veterans’ Hospital Radiopharmaceutical Sciences Institute for surgical assistance and Dongqing Chen for assisting with telemetry measurements. We thank RADIL/IDEXX Bioanalytics for their assistance with the megalin immunostaining and the University of Missouri Veterinary Medical Diagnostic Laboratory and Mona Garro for assisting with PAS staining. We thank Yujiang Fang and Qian Bai for assisting with the PCR and Charles Wiedmeyer at Comparative Clinical Pathology Services for assisting with plasma and urine chemistry.
REFERENCES
- 1.Hall JE. The kidney, hypertension, and obesity. Hypertension 41: 625–633, 2003. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]
- 2.Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Pritzl P, Floege J, Schwartz SM. Renal injury from angiotensin II-mediated hypertension. Hypertension 19: 464–474, 1992. doi: 10.1161/01.hyp.19.5.464. [DOI] [PubMed] [Google Scholar]
- 3.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol 6: 319–330, 2010. doi: 10.1038/nrneph.2010.58. [DOI] [PubMed] [Google Scholar]
- 4.KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis 49: S12–S154, 2007. doi: 10.1053/j.ajkd.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Inker LA, Astor BC, Fox CH, Isakova T, Lash JP, Peralta CA, Kurella Tamura M, Feldman HI. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am J Kidney Dis 63: 713–735, 2014. doi: 10.1053/j.ajkd.2014.01.416. [DOI] [PubMed] [Google Scholar]
- 6.Georgianos PI, Agarwal R. Revisiting RAAS blockade in CKD with newer potassium-binding drugs. Kidney Int 93: 325–334, 2018. doi: 10.1016/j.kint.2017.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345: 851–860, 2001. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 8.Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I.; SAVOR-TIM1 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 369: 1317–1326, 2013. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 9.Udell JA, Bhatt DL, Braunwald E, Cavender MA, Mosenzon O, Steg PG, Davidson JA, Nicolau JC, Corbalan R, Hirshberg B, Frederich R, Im K, Umez-Eronini AA, He P, McGuire DK, Leiter LA, Raz I, Scirica BM.; SAVOR-TIM1 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes and moderate or severe renal impairment: observations from the SAVOR-TIMI 53 Trial. Diabetes Care 38: 696–705, 2015. doi: 10.2337/dc14-1850. [DOI] [PubMed] [Google Scholar]
- 10.Groop P-H, Cooper ME, Perkovic V, Emser A, Woerle H-J, von Eynatten M. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes Care 36: 3460–3468, 2013. doi: 10.2337/dc13-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hattori S. Sitagliptin reduces albuminuria in patients with type 2 diabetes. Endocr J 58: 69–73, 2011. doi: 10.1507/endocrj.K10E-382. [DOI] [PubMed] [Google Scholar]
- 12.Mori H, Okada Y, Arao T, Tanaka Y. Sitagliptin improves albuminuria in patients with type 2 diabetes mellitus. J Diabetes Invest 5: 313–319, 2014. doi: 10.1111/jdi.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nistala R, Savin V. Diabetes, hypertension and chronic kidney disease progression: role of DPP4. Am J Physiol Renal Physiol 312: F661–F670, 2017. doi: 10.1152/ajprenal.00316.2016. [DOI] [PubMed] [Google Scholar]
- 14.Aroor A, Zuberek M, Duta C, Meuth A, Sowers JR, Whaley-Connell A, Nistala R. Angiotensin II stimulation of DPP4 activity regulates megalin in the proximal tubules. Int J Mol Sci 17: 780, 2016. doi: 10.3390/ijms17050780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alter ML, Ott IM, von Websky K, Tsuprykov O, Sharkovska Y, Krause-Relle K, Raila J, Henze A, Klein T, Hocher B. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney Blood Press Res 36: 119–130, 2012. doi: 10.1159/000341487. [DOI] [PubMed] [Google Scholar]
- 16.Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, Ishigaki Y, Kitada M, Srivastava SP, Koya D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 63: 2120–2131, 2014. doi: 10.2337/db13-1029. [DOI] [PubMed] [Google Scholar]
- 17.Nistala R, Habibi J, Lastra G, Manrique C, Aroor AR, Hayden MR, Garro M, Meuth A, Johnson M, Whaley-Connell A, Sowers JR. Prevention of obesity-induced renal injury in male mice by DPP4 inhibition. Endocrinology 155: 2266–2276, 2014. doi: 10.1210/en.2013-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharkovska Y, Reichetzeder C, Alter M, Tsuprykov O, Bachmann S, Secher T, Klein T, Hocher B. Blood pressure and glucose independent renoprotective effects of dipeptidyl peptidase-4 inhibition in a mouse model of type-2 diabetic nephropathy. J Hypertens 32: 2211–2223, 2014. doi: 10.1097/hjh.0000000000000328. [DOI] [PubMed] [Google Scholar]
- 19.Abd El Motteleb DM, Elshazly SM. Renoprotective effect of sitagliptin against hypertensive nephropathy induced by chronic administration of L-NAME in rats: role of GLP-1 and GLP-1 receptor. Eur J Pharmacol 720: 158–165, 2013. doi: 10.1016/j.ejphar.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 20.Pacheco BP, Crajoinas RO, Couto GK, Davel AP, Lessa LM, Rossoni LV, Girardi AC. Dipeptidyl peptidase IV inhibition attenuates blood pressure rising in young spontaneously hypertensive rats. J Hypertens 29: 520–528, 2011. doi: 10.1097/hjh.0b013e328341939d. [DOI] [PubMed] [Google Scholar]
- 21.Kishimoto S, Kinoshita Y, Matsumoto T, Maruhashi T, Kajikawa M, Matsui S, Hashimoto H, Takaeko Y, Kihara Y, Chayama K, Goto C, Mohamad Yusoff F, Nakashima A, Noma K, Higashi Y. Effects of the dipeptidyl peptidase 4 inhibitor alogliptin on blood pressure in hypertensive patients with type 2 diabetes mellitus. Am J Hypertens 32: 695–702, 2019. doi: 10.1093/ajh/hpz065. [DOI] [PubMed] [Google Scholar]
- 22.Mistry GC, Maes AL, Lasseter KC, Davies MJ, Gottesdiener KM, Wagner JA, Herman GA. Effect of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on blood pressure in nondiabetic patients with mild to moderate hypertension. J Clin Pharmacol 48: 592–598, 2008. doi: 10.1177/0091270008316885. [DOI] [PubMed] [Google Scholar]
- 23.Nilsson PM, Diez J. DPP-4 inhibition and blood pressure lowering in perspective. J Hypertens 34: 184–187, 2016. doi: 10.1097/HJH.0000000000000814. [DOI] [PubMed] [Google Scholar]
- 24.Takamiya Y, Okamura K, Shirai K, Okuda T, Kobayashi K, Urata H. Multicenter prospective observational study of teneligliptin, a selective dipeptidyl peptidase-4 inhibitor, in patients with poorly controlled type 2 diabetes: focus on glycemic control, hypotensive effect, and safety Chikushi Anti-Diabetes Mellitus Trial-Teneligliptin (CHAT-T). Clin Exp Hypertens 42: 197–204, 2020. doi: 10.1080/10641963.2019.1601207. [DOI] [PubMed] [Google Scholar]
- 25.Lee JE, Kim JE, Lee MH, Song HK, Ghee JY, Kang YS, Min HS, Kim HW, Cha JJ, Han JY, Han SY, Cha DR. DA-1229, a dipeptidyl peptidase IV inhibitor, protects against renal injury by preventing podocyte damage in an animal model of progressive renal injury. Lab Invest 96: 547–560, 2016. doi: 10.1038/labinvest.2016.34. [DOI] [PubMed] [Google Scholar]
- 26.Liu WJ, Xie SH, Liu YN, Kim W, Jin HY, Park SK, Shao YM, Park TS. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in streptozotocin-induced diabetic rats. J Pharmacol Exp Ther 340: 248–255, 2012. doi: 10.1124/jpet.111.186866. [DOI] [PubMed] [Google Scholar]
- 27.Nistala R, Habibi J, Aroor A, Sowers JR, Hayden MR, Meuth A, Knight W, Hancock T, Klein T, Demarco VG, Whaley-Connell A. DPP4 Inhibition attenuates filtration barrier injury and oxidant stress in the zucker obese rat. Obesity (Silver Spring) 22: 2172–2179, 2014. doi: 10.1002/oby.20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De MI, Korom S, Van Damme J, Scharpe S. CD26, let it cut or cut it down. Immunol Today 20: 367–375, 1999. doi: 10.1016/s0167-5699(99)01486-3. [DOI] [PubMed] [Google Scholar]
- 29.Ohnuma K, Dang NH, Morimoto C. Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol 29: 295–301, 2008. doi: 10.1016/j.it.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Zhong J, Rao X, Deiuliis J, Braunstein Z, Narula V, Hazey J, Mikami D, Needleman B, Satoskar AR, Rajagopalan S. A potential role for dendritic cell/macrophage-expressing DPP4 in obesity-induced visceral inflammation. Diabetes 62: 149–157, 2013. doi: 10.2337/db12-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, Wolf D, Patsch W, Rosenkranz AR, Eller P. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 60: 2954–2962, 2011. doi: 10.2337/db11-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res 118: 1233–1243, 2016. doi: 10.1161/CIRCRESAHA.115.308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 121: 2111–2117, 2011. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luft FC, Dechend R, Muller DN. Immune mechanisms in angiotensin II-induced target-organ damage. Ann Med 44 Suppl 1: S49–S54, 2012. doi: 10.3109/07853890.2011.653396. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev 97: 1127–1164, 2017. doi: 10.1152/physrev.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang A, Dorso C, Kopcho L, Locke G, Langish R, Harstad E, Shipkova P, Marcinkeviciene J, Hamann L, Kirby MS. Potency, selectivity and prolonged binding of saxagliptin to DPP4: maintenance of DPP4 inhibition by saxagliptin in vitro and ex vivo when compared to a rapidly-dissociating DPP4 inhibitor. BMC Pharmacol 12: 2, 2012. doi: 10.1186/1471-2210-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim H-S, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 103: 17985–17990, 2006. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson MS, Demarco VG, Heesch CM, Whaley-Connell AT, Schneider RI, Rehmer NT, Tilmon RD, Ferrario CM, Sowers JR. Sex differences in baroreflex sensitivity, heart rate variability, and end organ damage in the TGR(mRen2)27 rat. Am J Physiol Heart Circ Physiol 301: H1540–H1550, 2011. doi: 10.1152/ajpheart.00593.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nistala R, Meuth A, Smith C, Annayya A. Reliable and high efficiency extraction of kidney immune cells. J Vis Exp 114: 54368, 2016. doi: 10.3791/54368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nistala R, Andresen BT, Pulakat L, Meuth A, Sinak C, Mandavia C, Thekkumkara T, Speth RC, Whaley-Connell A, Sowers JR. Angiotensin type 1 receptor resistance to blockade in the opossum proximal tubule cell due to variations in the binding pocket. Am J Physiol Renal Physiol 304: F1105–F1113, 2013. doi: 10.1152/ajprenal.00127.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol 13: 2860–2868, 2002. doi: 10.1097/01.asn.0000035087.11758.ed. [DOI] [PubMed] [Google Scholar]
- 42.Sugino H, Ozono R, Kurisu S, Matsuura H, Ishida M, Oshima T, Kambe M, Teranishi Y, Masaki H, Matsubara H. Apoptosis is not increased in myocardium overexpressing type 2 angiotensin II receptor in transgenic mice. Hypertension 37: 1394–1398, 2001. doi: 10.1161/01.hyp.37.6.1394. [DOI] [PubMed] [Google Scholar]
- 43.Jia J, Ding G, Zhu J, Chen C, Liang W, Franki N, Singhal PC. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol 28: 500–507, 2008. doi: 10.1159/000113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamogashira T, Hayashi K, Fujimoto C, Iwasaki S, Yamasoba T. Functionally and morphologically damaged mitochondria observed in auditory cells under senescence-inducing stress. NPJ Aging Mech Dis 3: 2, 2017. doi: 10.1038/s41514-017-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 57: 469–476, 2011. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 46.Crowley SD, Rudemiller NP. Immunologic effects of the renin-angiotensin system. J Am Soc Nephrol 28: 1350–1361, 2017. doi: 10.1681/ASN.2016101066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirchhoff F, Krebs C, Abdulhag UN, Meyer-Schwesinger C, Maas R, Helmchen U, Hilgers KF, Wolf G, Stahl RAK, Wenzel U. Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int 73: 643–650, 2008. doi: 10.1038/sj.ki.5002689. [DOI] [PubMed] [Google Scholar]
- 49.Mervaala E, Muller DN, Park JK, Dechend R, Schmidt F, Fiebeler A, Bieringer M, Breu V, Ganten D, Haller H, Luft FC. Cyclosporin A protects against angiotensin II-induced end-organ damage in double transgenic rats harboring human renin and angiotensinogen genes. Hypertension 35: 360–366, 2000. doi: 10.1161/01.hyp.35.1.360. [DOI] [PubMed] [Google Scholar]
- 50.Muller DN, Shagdarsuren E, Park J-K, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol 161: 1679–1693, 2002. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest 125: 1189–1202, 2015. doi: 10.1172/JCI76327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan S, Marguet D, Dobers J, Reutter W, Fan H. Deficiency of CD26 results in a change of cytokine and immunoglobulin secretion after stimulation by pokeweed mitogen. Eur J Immunol 33: 1519–1527, 2003. doi: 10.1002/eji.200323469. [DOI] [PubMed] [Google Scholar]
- 53.Lara LS, McCormack M, Semprum-Prieto LC, Shenouda S, Majid DSA, Kobori H, Navar LG, Prieto MC. AT1 receptor-mediated augmentation of angiotensinogen, oxidative stress, and inflammation in ANG II-salt hypertension. Am J Physiol Renal Physiol 302: F85–F94, 2012. doi: 10.1152/ajprenal.00351.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itani HA, Harrison DG. Memories that last in hypertension. Am J Physiol Renal Physiol 308: F1197–F1199, 2015. doi: 10.1152/ajprenal.00633.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liao T-D, Yang X-P, Liu Y-H, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension 52: 256–263, 2008. doi: 10.1161/HYPERTENSIONAHA.108.112706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Z, Huang XR, Lan HY. Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am J Physiol Renal Physiol 302: F986–F997, 2012. doi: 10.1152/ajprenal.00595.2011. [DOI] [PubMed] [Google Scholar]
- 57.Lombardi D, Gordon KL, Polinsky P, Suga S, Schwartz SM, Johnson RJ. Salt-sensitive hypertension develops after short-term exposure to angiotensin II. Hypertension 33: 1013–1019, 1999. doi: 10.1161/01.hyp.33.4.1013. [DOI] [PubMed] [Google Scholar]
- 58.Hosojima M, Sato H, Yamamoto K, Kaseda R, Soma T, Kobayashi A, Suzuki A, Kabasawa H, Takeyama A, Ikuyama K, Iino N, Nishiyama A, Thekkumkara TJ, Takeda T, Suzuki Y, Gejyo F, Saito A. Regulation of megalin expression in cultured proximal tubule cells by angiotensin II type 1A receptor- and insulin-mediated signaling cross talk. Endocrinology 150: 871–878, 2009. doi: 10.1210/en.2008-0886. [DOI] [PubMed] [Google Scholar]
- 59.Scheen AJ. Cardiovascular effects of gliptins. Nat Rev Cardiol 10: 73–84, 2013. doi: 10.1038/nrcardio.2012.183. [DOI] [PubMed] [Google Scholar]
- 60.Mason RP, Jacob RF, Kubant R, Ciszewski A, Corbalan JJ, Malinski T. Dipeptidyl peptidase-4 inhibition with saxagliptin enhanced nitric oxide release and reduced blood pressure and sICAM-1 levels in hypertensive rats. J Cardiovasc Pharmacol 60: 467–473, 2012. doi: 10.1097/fjc.0b013e31826be204. [DOI] [PubMed] [Google Scholar]
- 61.Ott C, Raff U, Schmidt S, Kistner I, Friedrich S, Bramlage P, Harazny JM, Schmieder RE. Effects of saxagliptin on early microvascular changes in patients with type 2 diabetes. Cardiovasc Diabetol 13: 19, 2014. doi: 10.1186/1475-2840-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sakai M, Uchii M, Myojo K, Kitayama T, Kunori S. Critical role of renal dipeptidyl peptidase-4 in ameliorating kidney injury induced by saxagliptin in Dahl salt-sensitive hypertensive rats. Eur J Pharmacol 761: 109–115, 2015. doi: 10.1016/j.ejphar.2015.04.023. [DOI] [PubMed] [Google Scholar]
- 63.Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, Bluher M, Czech MP, Tabas I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 555: 673–677, 2018. doi: 10.1038/nature26138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee SA, Kim YR, Yang EJ, Kwon E-J, Kim SH, Kang SH, Park DB, Oh B-C, Kim J, Heo ST, Koh G, Lee DH. CD26/DPP4 levels in peripheral blood and T cells in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 98: 2553–2561, 2013. doi: 10.1210/jc.2012-4288. [DOI] [PubMed] [Google Scholar]
- 65.Yang J, Campitelli J, Hu G, Lin Y, Luo J, Xue C. Increase in DPP-IV in the intestine, liver and kidney of the rat treated with high fat diet and streptozotocin. Life Sci 81: 272–279, 2007. doi: 10.1016/j.lfs.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 66.Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol 3: 128, 2012. doi: 10.3389/fphys.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leibowitz A, Schiffrin EL. Immune mechanisms in hypertension. Curr Hypertens Rep 13: 465–472, 2011. doi: 10.1007/s11906-011-0224-9. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka T, Kameoka J, Yaron A, Schlossman SF, Morimoto C. The costimulatory activity of the CD26 antigen requires dipeptidyl peptidase IV enzymatic activity. Proc Natl Acad Sci USA 90: 4586–4590, 1993. doi: 10.1073/pnas.90.10.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klemann C, Wagner L, Stephan M, von Horsten S. Cut to the chase: a review of CD26/dipeptidyl peptidase-4's (DPP4) entanglement in the immune system. Clin Exp Immunol 185: 1–21, 2016. doi: 10.1111/cei.12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med 215: 21–33, 2018. [Erratum in J Exp Med 215: 719, 2018]doi: 10.1084/jem.20171773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li C-I, Shyr Y, Harrison DG. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 64: 1108–1115, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin SL, Castano AP, Nowlin BT, Lupher ML Jr, Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol 183: 6733–6743, 2009. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- 73.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 91: 521–530, 1997. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 74.Elferink JG, de Koster BM. The stimulation of human neutrophil migration by angiotensin IL: its dependence on Ca2+ and the involvement of cyclic GMP. Br J Pharmacol 121: 643–648, 1997. doi: 10.1038/sj.bjp.0701167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang H-M, Wang H-X, Yang H, Zeng X-J, Tang C-S, Du J, Li H-H. Role for granulocyte colony stimulating factor in angiotensin II-induced neutrophil recruitment and cardiac fibrosis in mice. Am J Hypertens 26: 1224–1233, 2013. doi: 10.1093/ajh/hpt095. [DOI] [PubMed] [Google Scholar]
- 76.Nabah YN, Mateo T, Estelles R, Mata M, Zagorski J, Sarau H, Cortijo J, Morcillo EJ, Jose PJ, Sanz M-J. Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation 110: 3581–3586, 2004. doi: 10.1161/01.CIR.0000148824.93600.F3. [DOI] [PubMed] [Google Scholar]
- 77.Ma L-J, Corsa BA, Zhou J, Yang H, Li H, Tang Y-W, Babaev VR, Major AS, Linton MF, Fazio S, Hunley TE, Kon V, Fogo AB. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am J Physiol Renal Physiol 300: F1203–F1213, 2011. doi: 10.1152/ajprenal.00468.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rudemiller NP, Crowley SD. Interactions between the immune and the renin-angiotensin systems in hypertension. Hypertension 68: 289–296, 2016. doi: 10.1161/HYPERTENSIONAHA.116.06591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Majeed B, Tawinwung S, Eberson LS, Secomb TW, Larmonier N, Larson DF. Interleukin-2/anti-interleukin-2 immune complex expands regulatory T cells and reduces angiotensin II-induced aortic stiffening. Int J Hypertens 2014: 126365, 2014. doi: 10.1155/2014/126365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mirabito KM, Hilliard LM, Head GA, Widdop RE, Denton KM. Pressor responsiveness to angiotensin II in female mice is enhanced with age: role of the angiotensin type 2 receptor. Biol Sex Differ 5: 13, 2014. doi: 10.1186/s13293-014-0013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xue B, Pamidimukkala J, Lubahn DB, Hay M. Estrogen receptor-alpha mediates estrogen protection from angiotensin II-induced hypertension in conscious female mice. Am J Physiol Heart Circ Physiol 292: H1770–H1776, 2007. doi: 10.1152/ajpheart.01011.2005. [DOI] [PubMed] [Google Scholar]