

Keywords: channel-activating protease 1, epithelial Na channel, Fisher rat thyroid cells, furin, PNGaseF
Abstract
Extracellular proteases can activate the epithelial Na channel (ENaC) by cleavage of the γ subunit. Here, we investigated the cleavage state of the channel in the kidneys of mice and rats on a low-salt diet. We identified the cleaved species of channels expressed in Fisher rat thyroid cells by coexpressing the apical membrane-bound protease channel-activating protease 1 (CAP1; prostasin). To compare the peptides produced in the heterologous system with those in the mouse kidney, we treated both lysates with PNGaseF to remove N-linked glycosylation. The apparent molecular mass of the smallest COOH-terminal fragment of γENaC (52 kDa) was indistinguishable from that of the CAP1-induced species in Fisher rat thyroid cells. Similar cleaved peptides were observed in total and cell surface fractions of the rat kidney. This outcome suggests that most of the subunits at the surface have been processed by extracellular proteases. This was confirmed using nonreducing gels, in which the NH2- and COOH-terminal fragments of γENaC are linked by a disulfide bond. Under these conditions, the major cleaved form in the rat kidney had an apparent molecular mass of 56 kDa, ∼4 kDa lower than that of the full-length form, consistent with excision of a short peptide by two proteolytic events. We conclude that the most abundant γENaC species in the apical membrane of rat and mouse kidneys on a low-Na diet is the twice-cleaved, presumably activated form.
NEW & NOTEWORTHY We have identified the major aldosterone-dependent cleaved form of the epithelial Na channel (ENaC) γ subunit in the kidney as a twice-cleaved peptide. This form appears to be identical in size with a subunit cleaved in vitro by the extracellular protease channel-activating protease 1 (prostasin). In the absence of reducing agents, it has an overall molecular mass less than that of the intact subunit, consistent with the excision of an inhibitory domain.
INTRODUCTION
Currents through the epithelial Na channel (ENaC) can be strongly enhanced by extracellular proteases, particularly when measured in heterologous expression systems (1–3). This activation arises from proteolysis of the γENaC subunit that has already been cleaved once by intracellular proteases such as furin, leading to excision of an inhibitory peptide from the subunit (1). The identities of the proteases that mediate the second cleavage in vivo are uncertain. Channel-activating protease 1 (CAP1) or prostasin is one candidate. This protein was identified as an ENaC activator using expression cloning (4). It is a glycosylphosphatidylinositol-anchored membrane protein that is expressed in the kidney and is excreted in the urine in an aldosterone-dependent fashion (5). In the kidney itself, both the αENaC and γENaC subunits exist in cleaved states, and the amounts of the cleaved forms correlate with the physiological activation of the channels by aldosterone (6, 7). However, the precise cleaved form within the cells, particularly of γENaC, is less clear. The difference in apparent molecular mass of cleaved and intact forms estimated from Western blots is ∼15 kDa (6, 7). This is close to the predicted change in size for a single cleavage by furin (15–16 kDa), but it may also be compatible with the twice-cleaved form given the uncertainties in the determination of mass from Western blots and complications due to glycosylation. The question has clinical implications. During nephrotic syndrome, serum proteases such as plasmin may be present in the urine at high concentrations and could activate incompletely cleaved channels, contributing to the fluid retention characteristic of this disease state (8–10).
Here, we address this problem by comparing the apparent molecular mass of ENaC in the kidney with that cleaved by the membrane-bound extracellular protease CAP1 when expressed in cultured cells. To circumvent possible differences in glycosylation, we treated samples with PNGaseF to remove N-linked sugars. We found that the major γ-subunit species in kidneys from mice and rats on a low-salt diet appear to be identical, within the sensitivity of our detection methods, to the CAP1-cleaved species in Fisher rat thyroid (FRT) cells.
METHODS
Animals
All procedures using animals were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. We used adult C57BL6 mice (20–30 g) and Sprague–Dawley rats (150–200 g) of either sex (Charles River). Animals were maintained either on normal laboratory chow (0.3% Na by weight) or on a low-Na diet (0.004% Na, Cat. No. 902902, MP Biomedicals, Solon, OH).
Cell Culture
FRT cells were cultured in DMEM/F-12 medium supplemented with 8% FBS at 37°C in a 5% CO2 incubator. To study ENaC expression, FRT cells were transfected with plasmids encoding wild-type (WT) or mutant mouse ENaC subunits (0.5 µg per construct) directly on 24-mm Transwell filters using Lipofectamine 3000 (L3000008, Invitrogen), according to the manufacturer’s instructions. In some cases, the γ subunit had an NH2-terminal HA and a COOH-terminal V5 tag. In some experiments, an equal amount of mouse CAP1 plasmid was cotransfected with mouse ENaC. When FRT cells reached confluency on filters (normally ∼3–4 days), cells were scraped off the filters and lysed in Goldstein solution (100 mM NaCl, 40 mM KCl, 20 mM HEPES, 1 mM EDTA, 10% glycerol, 1% Nonidet P-40, and 0.4% deoxycholate, pH 7.4) supplemented with a protease inhibitor cocktail. Insoluble material was removed by centrifugation at 20,000 g, and the supernatant was collected for later analysis.
Western Blot Analysis
Whole-kidney microsomes from rat or mouse kidneys were prepared for Western blot analysis by mincing one kidney and disrupting the tissue with a Dounce homogenizer in sucrose lysis buffer with protease inhibitor cocktail (Sigma). The homogenate was filtered through a nylon strainer (70-μm mesh) to remove intact tissue and centrifuged at 100,000 g for 90 min to obtain a microsomal pellet. This was suspended in 3 mL of lysis buffer and frozen for later analysis. After measurement of protein concentration, samples were prepared for electrophoresis as described previously (11). Lanes were loaded with equal amounts of protein. FRT cell lysates were prepared for Western blots in the same way, except that each lane was loaded with equal volumes of lysate.
To remove N-linked sugar residues, proteins were treated with PNGaseF (New England Biolabs), according to the manufacturer’s instructions. For preparation of samples for nonreducing gels, DTT was omitted from the reaction mixture.
Biotinylation of rat kidney membrane proteins followed a protocol described previously (12–15). Briefly, kidneys were perfused with an impermeant biotinylating reagent ([sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithioproprionate (sulfo-NHS-SS-biotin), Campbell Science, Rockville, IL] in situ. After quenching the biotinylation reaction with Tris buffer, the kidney was removed and microsomes were prepared from it. Microsomes were solubilized with 3% Triton X-100, and surface proteins from equal amounts of microsomal protein were isolated using neutravidin beads and eluted as described below. For preparation of biotinylated proteins for PNGase treatment, proteins were eluted without SDS in buffer containing DTT (final concentration 170 mM) in 10 mM Tris, pH 7.4, for 40 min at 80°C.
For isolation of ENaC at the apical cell membrane of FRT cells, filters were washed four times with 1 mL ice-cold PBS. A sodium borate buffer (137 mM NaCl, 15 mM sodium borate, pH 9.0) was added to both sides of the filters, whereas only the apical surface was labeled with 1 mg/mL sulfo-NHS biotin at 4°C for 12 min (2×). Excess biotin was quenched with 1 mL culture medium with 10% FBS (2×) and then rinsed with PBS (2×) before cells were lysed in 0.5 mL Goldstein solution. This was added to 150 µL of a 50% suspension of Ultra Link neutravidin beads (Pierce) and kept overnight at 4°C with gentle rocking. The beads were washed three times with wash buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris·HCl pH 7.4, and 1% Triton X-100), twice with high-salt wash buffer (500 mM NaCl, 5 mM EDTA, 50 mM Tris·HCl pH 7.4, and 0.1% Triton X-100), and twice with no-salt wash buffer (10 mM Tris·HCl, pH 7.4). After transferring the beads to a clean tube, the supernatant was decanted and protein bound to the beads was eluted in 35 µL of 5× Laemmli sample buffer plus 25 µL of 0.5 M DTT for 15 min at 80°C.
Samples were electrophoresed on 4–12% Bis-TRIS gels (Invitrogen), and the proteins were transferred electrophoretically to PVDF membranes. After being blocked, membranes were incubated overnight at 4°C in a 1:500 dilution of an antibody directed against the COOH-terminal peptide of γENaC (6). Anti-rabbit IgG conjugated with alkaline phosphatase was used as a secondary antibody. Bound antibody was visualized with a Syngene PXi6 Gel and Blot Imaging System using a chemiluminescence substrate (Western Breeze, Invitrogen). Band densities were quantified using Photoshop.
Statistics
Where relevant, data are reported as means ± SE and were compared using two-tailed t tests. P < 0.05 was considered to indicate statistically significant differences.
RESULTS
To identify the species of γENaC produced by cleavage by extracellular protease, we transfected mouse ENaC into FRT cells with and without CAP1. Cell extracts were separated using SDS-PAGE and probed using an antibody raised against a COOH-terminal peptide of the γ subunit. Figure 1 shows the results. Two main bands are seen without CAP1, namely, full-length γENaC (∼80 kDa) and a more diffuse band at 70–75 kDa. The main effect of inclusion of CAP1 was the appearance of a new band that migrated at ∼65 kDa. Furthermore, both the full-length band and the lower diffuse band at 70–75 kDa were decreased in intensity. The latter may represent the furin-cleaved γ subunit, although this is uncertain. The difference in molecular mass compared with the full-length subunit is less than predicted (∼15 kDa) for furin cleavage, but it is possible that differences in glycan maturation account for this discrepancy. Full-length subunits that remain in the presence of CAP1 presumably represent channels that have not undergone trafficking to the apical membrane through the Golgi apparatus and retain immature glycans (13). Probing untransfected cells with this antibody revealed a single, faint, nonspecific band migrating at 55 kDa (Fig. 1, right).
Figure 1.
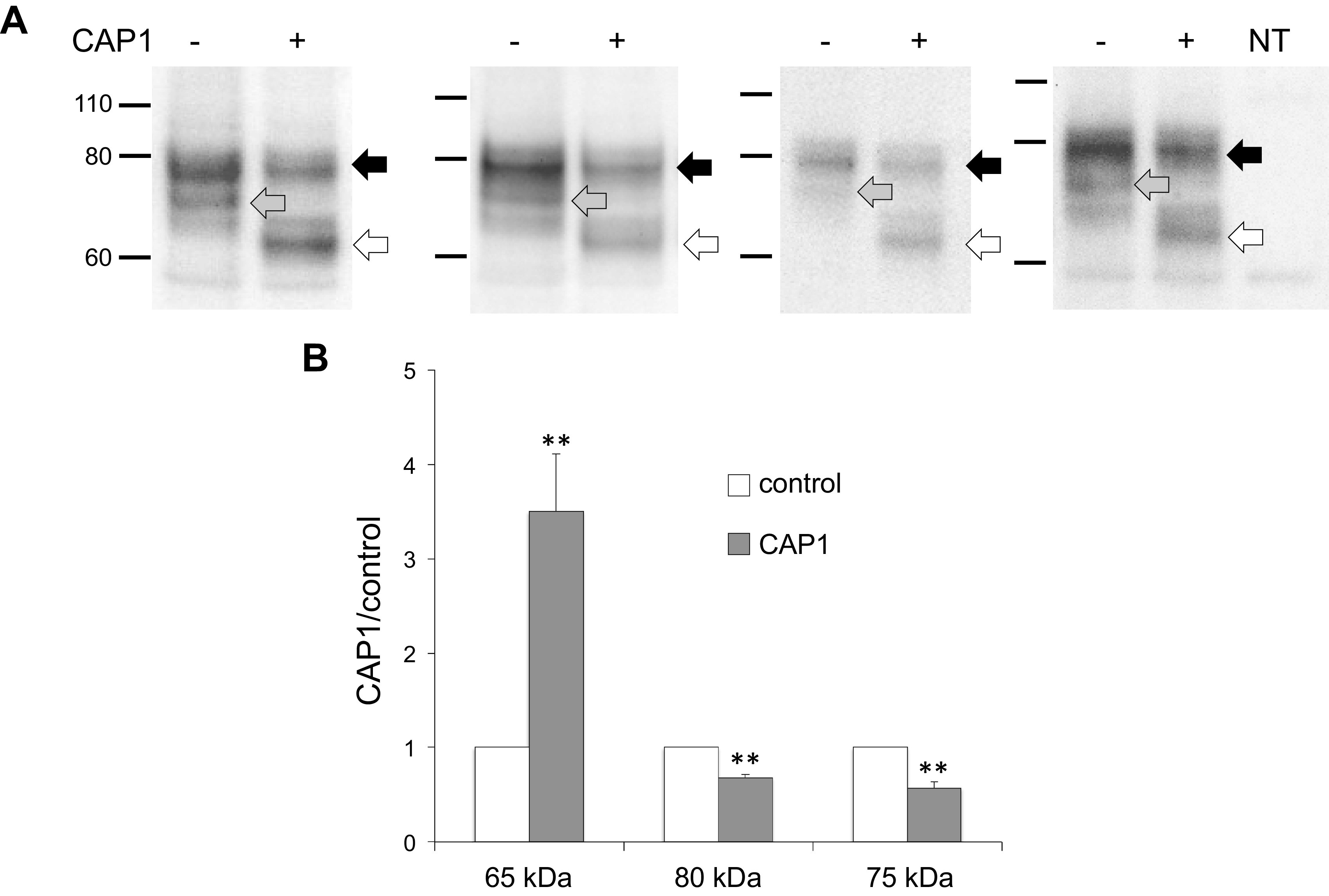
Western blots of γENaC expressed in FRT cells. A: cells were transfected with WT γENaC together with αENaC and βENaC, with or without CAP1. Each pair of lanes represents a different transfection. Each lane was loaded with 10 µL of lysate from a different filter. CAP1 increased the abundance of a cleaved form of the subunit migrating at ∼65 kDa (white arrows) and decreased the intensities of the full-length species (black arrows) and of a diffuse band migrating around 75 kDa (gray arrows). The narrow band below 60 kDa is nonspecific, as it was also observed in nontransfected cells (right lane). B: densitometry of 80 kDa, 75 kDA, and 65 kDa bands from the blots in (A). Data were normalized in the absence of CAP1 and represent means ± SE; n = 4. **P < 0.01 vs. control by a paired two-tailed t test. CAP1, channel activating protease 1; ENaC, epithelial Na channel; FRT, Fisher rat thyroid; WT, wild-type.
To further characterize these cleaved species, we used a mutant γENaC in which four basic residues (RKRK) in the prostasin consensus cleavage site were replaced by glutamine (Q4), preventing processing at the cell surface by the exogenous protease (16). CAP1 coexpression failed to produce the 65 kDa band in this mutant (Fig. 2A), although a faint band migrating between the 70–75 kDa and 65 kDa bands appeared. We also used a mutant CAP1 with an amino acid substitution in the active site (S283A) that lacks proteolytic activity (17). Coexpression of this protein with WT ENaC also failed to induce the 65 kDa cleaved species (Fig. 2B). The apparent molecular masses of the bands in this blot are slightly lower than in the bands shown in Fig. 2A because the γENaC construct lacked epitope tags.
Figure 2.

Characterization of the fully cleaved form of γENaC expressed in FRT cells. A: effect of mutations at the CAP1 cleavage site. Cells were transfected with αENaC and βENaC plus either WT γENaC or with a mutant in which four critical arginine residues at the presumed CAP1 cleavage site were mutated to glutamine (Q4). Each lane was loaded with 9 µL of lysate from a different filter. The Q4 mutations prevented the formation of the fully cleaved species. A faint band at ∼70 kDa appeared with only the Q4 mutant. We have not identified this species. The apparent mass of the subunits is slightly larger than that in Fig. 1 or Fig. 2B due to the presence of NH2- and COOH-terminal epitope tags. The blot is representative of three independent experiments. B: effect of a mutation in the CAP1 active site. Cells were transfected with WT ENaC together with either WT CAP1 or the S283A mutant of the protease. Each lane was loaded with 9 µL of lysate. The CAP1 mutation eliminated the appearance of the fully cleaved γENaC species. The blot is representative of three independent experiments. CAP1, channel activating protease 1; ENaC, epithelial Na channel; FRT, Fisher rat thyroid; WT, wild-type.
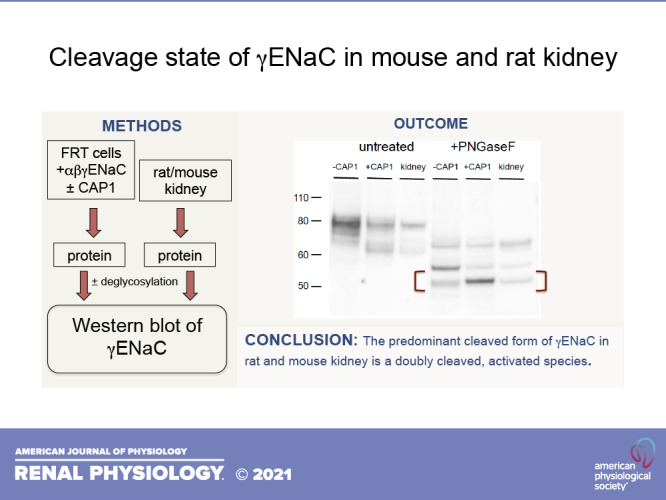
Figure 3 shows a comparison of the cleaved γENaC species in FRT cells with those in the mouse kidney. Again, we used nonepitope-tagged constructs for better comparison with the native species. The kidneys were from mice on normal chow. In addition, we treated all samples with PNGaseF, removing N-linked sugars, to avoid possible differences in glycosylation between the kidney and the cell line. Under these conditions, we resolved three different peptides, which we identify with full-length, furin-cleaved, and doubly cleaved subunits. The migration of the native and heterologously expressed peptides was nearly identical. The shortest peptide from the kidney corresponds to the band that is increased in abundance in FRT cells expressing CAP1. In this blot, the band induced by CAP1 ran slightly slower than that from the mouse kidney, but this was not a consistent finding. In four experiments, the estimated difference in molecular mass of the two peptides was 0.02 ± 0.06 kDa.
Figure 3.

Western blot of γENaC expressed in FRT cells and in mouse kidney before and after treatment with PNGaseF. Cells were transfected with untagged WT γENaC together with αENaC and βENaC, with or without CAP1. Kidney microsomes (30 µg protein) were obtained from a mouse on a control diet. FRT cell lanes were loaded with samples from 7 µL (untreated) or 10 µL (PNGaseF-treated) of cell lysate. As in Fig. 1., CAP1 increased the abundance of a cleaved form of the subunit migrating at ∼65 kDa before PNGaseF treatment. Kidney microsomes showed a presumed full-length subunit migrating at 80 kDa and a major cleaved form at 65 kDa. After PNGaseF treatment three major bands were observed in both FRT cells and kidney samples. These corresponded to a full-length species at 65 kDa, a furin-cleaved form at 55 kDa, and a fully cleaved form at 50 kDa. CAP1 increased the amount of the 50 kDa species and decreased that of the 55 kDa form. The blot is representative of three separate experiments. CAP1, channel activating protease 1; ENaC, epithelial Na channel; FRT, Fisher rat thyroid; WT, wild-type.
As the fully cleaved species in the kidney was identified in total microsomes, we next addressed the possibility that a different peptide might be produced in the apical membrane by comparing γENaC from microsomes and surface fractions after treatment with PNGaseF. For this experiment, we used kidneys from rats on a low-Na diet for 1 wk. This increases yields of material from the neutravidin eluate, facilitating the additional processing with deglycosylating enzyme. The surface fractions were obtained by biotinylating kidneys in situ and separating biotinylated proteins using neutravidin affinity beads (12, 13). Figure 4 shows that surface fractions contain predominantly cleaved forms of γENaC, as previously reported (12, 13). The cleaved forms from total microsomes and surface fractions migrated at the same position, both before and after PNGaseF treatment. In this experiment, we resolved a doublet in the deglycosylated samples in both microsomes and neutravidin eluates. We do not know the nature of the doublet, which we did not observe in all experiments. We conclude that the major cleaved γENaC species at the cell surface are the same as those observed in the cell as a whole and represent subunits cleaved at or near the site of CAP1-dependent cleavage.
Figure 4.

Comparison of γENaC species in total microsomes and surface fractions of rat kidney. Rats were fed a low-Na diet for 1 wk. Microsomes and neutravidin eluates were obtained from two different biotinylated rat kidneys, before and after treatment with PNGaseF. Microsome lanes were loaded with 30 µg of protein. Eluate lanes were loaded with material corresponding to 600 µg of starting material. Before treatment, whole microsomes had a full-length band migrating at 80 kDa and a broader cleaved band migrating at ∼65 kDa. The 65 kDa band predominated in the eluates. After treatment with PNGaseF, three species were apparent in whole microsome fractions migrating at 65 kDa, 55 kDa, and a doublet at 50 kDa. In the eluate, the 50 kDa doublet predominated. Results are typical of three blots, although the doublet was not always resolved. ENaC, epithelial Na channel.
We also compared the cleaved species in total cell lysates and surface fractions in FRT cells. As shown in Fig. 5, the main species on the surface were indistinguishable from those in the whole-cell extract. In these experiments, most of the γENaC in the cell and on the surface was in the full-length form. This was likely due to the presence of Na+ and absence of amiloride in the apical culture medium. Previous studies showed that ENaC-dependent Na+ entry into FRT cells inhibits proteolytic processing of channels (18, 19). We were unable to obtain enough material from the neutravidin eluates to carry out deglycosylation protocols.
Figure 5.

Comparison of γENaC species in total cell extracts and surface fractions of FRT cells expressing ENaC. Cells were transfected with WT αENaC, βENaC, and γENaC with or without CAP1. Total lysate lanes were loaded with 7 µL of lysates from a single filter. Eluate lanes were loaded with eluates from Neutravidin beads loaded with 180 µL of the lysate. The main CAP1-dependent cleaved form was identical in total and surface fractions. Similar results were obtained in four independent experiments. CAP1, channel activating protease 1; ENaC, epithelial Na channel; FRT, Fisher rat thyroid; WT, wild-type.
We confirmed these conclusions by investigating the behavior of γENaC in nonreducing gels. Here, we made use of the high likelihood that after cleavage, the NH2- and COOH-terminal fragments are held together through a highly conserved disulfide bond between C100 and C289 in the mouse sequence (20–22). In this case, if the peptide chain is cut just once, the molecular mass of the connected fragments of the subunit would not change, and the cleaved and intact forms would migrate similarly in the gel. However, after a second cleavage, the molecular mass would decrease and the mobility of the linked fragments would increase. Rat kidneys were used for this experiment because under most conditions only a single cleaved form is detected, simplifying the interpretation. Figure 6 shows a blot of kidney microsomes from rats on control and low-Na diets under nonreducing conditions. Previous studies showed that in reducing gels, the full-length form predominates in control rats, whereas the cleaved form is more abundant in animals on a low-Na diet (12, 15). In the absence of glycosidases, the cleaved form (Fig. 6, lane d) migrated slightly below that of the full-length (Fig. 6, lane a). The difference was much less than that observed with reducing gels, indicating that the NH2- and COOH-terminal fragments indeed remain joined, as previously reported (23). The bands are more easily resolved after treatment with PNGaseF (Fig. 6, lanes a and c). The difference between the apparent molecular masses was 3–4 kDa, slightly less than that predicted, assuming the removal of amino acids between the furin and CAP1 sites (4.7 kDa). Given the uncertainties of nonlinearity of migration rates with molecular mass, we do not consider the discrepancy to be significant.
Figure 6.

Western blot of γENaC in microsomes of rat kidney before and after treatment with PNGaseF under nonreducing conditions. Rats were fed either control or low-Na diets. All lanes were loaded with material corresponding to 20 µg of microsomal protein. In rats on a control diet, the full-length form predominates. In these gels, this form migrates at 75 kDa before (lane a) and 60 kDa after (lane b) PNGaseF treatment. In rats on a low-Na diet, the doubly cleaved forms predominate (lanes c and d). These migrate as a broad band at 70 kDa before and at ∼56 kDa after PNGaseF. The white arrows show the locations expected of the fully cleaved species under reducing conditions. The positions of the cleaved forms are consistent with excision of 3 to 4 kDa from the protein. The blot is representative of three different experiments. ENaC, epithelial Na channel.
DISCUSSION
The role of proteolysis of ENaC subunits in controlling channel function emerged from two converging lines of study. Vallet et al. (4) found that currents through ENaC expressed in Xenopus oocytes were enhanced by coexpression of cRNA encoding a serine protease termed CAP1, later identified as prostasin. Shortly after that, Masilamani et al. (7) reported that γENaC subunits in the kidney existed in both full-length and cleaved states, with the abundance of the latter increasing in states of elevated aldosterone, when channel activity is high. Later studies identified the cleaved state of the αENaC subunit, which is also increased with elevated aldosterone (6).
Subsequent studies revealed that full activation of the channel requires that both α- and γENaC be cleaved twice (17, 24). In the case of αENaC, both proteolytic events occur when the channel is intracellular, likely mediated by furin, a serine protease expressed mainly in the Golgi apparatus. γENaC is cleaved once in the Golgi, and a second time when the channel is in the plasma membrane, presumably the event catalyzed by CAP1 and other extracellular proteases. The second cleavage event excises inhibitory peptides from the subunits (17, 24). This raises the issue of whether the cleaved species of γENaC observed in the kidney corresponds to once- or twice-cleaved subunits.
In most studies, the difference in apparent molecular mass between full-length and cleaved γ subunits is ∼15 kDa (6, 7). This is consistent with the predicted change (15–16 kDa) due to a single cleavage at the furin consensus site at E138 (rat) or E144 (mouse). The further decrease in mass expected from the second cleavage is 4–5 kDa. However, it is difficult to infer exact molecular masses from Western blots, especially in proteins that have extensive glycosylation or other posttranslational modifications.
We addressed this issue by directly comparing γENaC from kidneys with that from heterologous expression systems in which the cleavage can be manipulated. We further used deglycosylated preparations to eliminate differences due to the amount of sugar added to the proteins. The main result was that the band from the mouse kidney with the lowest apparent molecular mass has the same electrophoretic mobility as does the species induced by CAP1 coexpression in the heterologous system. We also identified an intermediate or partially cleaved band from the kidney that comigrates with a species whose abundance is decreased by CAP1. This is presumably the furin-cleaved subunit.
Measurements using nonreducing gels supported these conclusions. Under these conditions, the NH2- and COOH-terminal fragments are expected to stay together due to a disulfide bond (21, 22). After deglycosylation, the cleaved protein from rat kidney has an apparent molecular mass 3–4 kDa lower than that of the intact species, consistent with the excision of an inhibitory peptide. The estimated size of the excised fragment was ∼1 kDa smaller than that predicted from the furin and CAP1 sites; this is within the uncertainty of the estimates. A previous study reached a similar conclusion based on the inability of an antibody raised against the inhibitory peptide to immunoprecipitate the cleaved form of the γ subunit (25).
CAP1 presumably acts on ENaC at the cell surface of the FRT cells. That does not mean that all of the twice-cleaved γENaC is at the surface, however, as the channels will be internalized from the apical membrane into endosomes and much of the pool of cleaved γENaC could reside in that compartment.
Although the size of the cleaved form in the kidney is compatible with proteolysis by CAP1, this does not exclude the possibility of other proteases acting on the channels in vivo. Other candidates for the second cleavage include kallikrein (26–28) and plasmin (29), both of which can cleave and activate ENaC in vitro. The identification of the key extracellular proteases in vivo remains uncertain.
The results have implications for the mechanisms of fluid retention in nephrotic syndrome. Several groups have shown that in this condition, plasma proteases such as plasmin are filtered by the kidney and have suggested that these proteases, either directly or indirectly, may hyperactivate ENaC through excessive cleavage (8–10). This would lead to reabsorption of excess Na and ultimately to fluid retention and edema. The process may be complex, as in a mouse model of nephrotic syndrome, neither urinary plasminogen activator nor plasminogen was found to be necessary for Na retention (30, 31). However, aldosterone could have enhanced ENaC cleavage in these animals, as levels of the steroid were not suppressed. Our Western blots suggest that, when rats or mice are fed a low-Na diet, most of the γENaC that reaches the cell surface is already cleaved at or near the CAP1 site. In contrast, when rats are fed a control Na diet, most of the γ subunit in whole-kidney lysates is uncleaved (12, 15). The expression of cleaved γENaC in the biotinylated fractions is reduced, whereas the amount of the full-length subunit is actually increased (12, 15). The extent of proteolytic processing of the γ subunit in rodent models of nephrotic syndrome is unclear, and the methods developed here to distinguish noncleaved, furin-cleaved, and doubly cleaved γENaC may be helpful in addressing this issue.
A clear prediction of this model of nephrotic syndrome is that ENaC currents in native kidney tissues should be activatable by exogenous proteases. Measurements in rat cortical collecting ducts (CCDs) showed no detectable activation by trypsin when basal activity was increased by aldosterone (12). Nesterov et al. (32) measured somewhat larger increases in current in mouse CCDs treated with trypsin. These were variable and averaged around 30–40% of baseline values. Overall, these results indicate that, at least in rodent kidneys under the conditions associated with elevated levels of aldosterone, most ENaC at the surface exists in a doubly cleaved form and that further activation by excess protease activity in urine will be modest. The status of the channels at the surface under basal conditions is less clear. In CCDs from control animals, a trypsin-induced current was detected that was small in absolute magnitude although large in terms of fractional increase (12).
In summary, we have shown that the most abundant and the most completely proteolytically cleaved species of γENaC in the rodent kidney, and particularly the main species at the cell surface under conditions of elevated aldosterone, is identical or nearly identical to that cleaved by CAP1/prostasin in vitro.
GRANTS
This work was supported by National Institutes of Health Grants RO1DK111380 (to L.G.P.), KO1DK103834 (to S.S.), RO3DK119752 (to S.S.), RO1HL147818 (to T.R.K.), and P30DK079307 (to T.R.K.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.F., S.S., T.R.K., and L.G.P. conceived and designed research; G.F. and S.S. performed experiments; G.F., S.S., and L.G.P. analyzed data; G.F., S.S., T.R.K., and L.G.P. interpreted results of experiments; L.G.P. prepared figures; L.G.P. drafted manuscript; G.F., S.S., T.R.K., and L.G.P. edited and revised manuscript; G.F., S.S., T.R.K., and L.G.P. approved final version of manuscript.
REFERENCES
- 1.Kleyman TR, Carattino MD, Hughey RP. ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J Biol Chem 284: 20447–20451, 2009. doi: 10.1074/jbc.r800083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleyman TR, Eaton DC. Regulating ENaC's gate. Am J Physiol Cell Physiol 318: C150–C162, 2020. doi: 10.1152/ajpcell.00418.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol 71: 361–379, 2008. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- 4.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature 389: 607–610, 1997. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 5.Narikiyo T, Kitamura K, Adachi M, Miyoshi T, Iwashita K, Shiraishi N, Nonoguchi H, Chen L-M, Chai KX, Chao J, Tomita K. Regulation of prostasin by aldosterone in the kidney. J Clin Invest 109: 401–408, 2002. doi: 10.1172/JCI13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ergonul Z, Frindt G, Palmer LG. Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol 291: F683–F693, 2006. doi: 10.1152/ajprenal.00422.2005. [DOI] [PubMed] [Google Scholar]
- 7.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Artunc F, Wörn N, Schork A, Bohnert BN. Proteasuria - the impact of active urinary proteases on sodium retentin in nephrotic syndrome. Acta Physiol (Oxf) 225: e13249, 2019. doi: 10.1111/apha.13249. [DOI] [PubMed] [Google Scholar]
- 9.Hinrichs GR, Jensen BL, Svenningsen P. Mechanisms of sodium retention in nephrotic syndrome. Curr Opin Nephrol Hypertens 29: 207–212, 2020. doi: 10.1097/MNH.0000000000000578. [DOI] [PubMed] [Google Scholar]
- 10.Passero CJ, Hughey RP, Kleyman TR. New role for plasmin in sodium homeostasis. Curr Opin Nephrol Hypertens 19: 13–19, 2010. doi: 10.1097/MNH.0b013e3283330fb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frindt G, Yang L, Uchida S, Weinstein AM, Palmer LG. Am J Physiol Renal Physiol 313: F62–F73, 2017. doi: 10.1152/ajprenal.00668.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frindt G, Ergonul Z, Palmer LG. Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131: 617–627, 2008. doi: 10.1085/jgp.200809989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frindt G, Gravotta D, Palmer LG. Regulation of ENaC trafficking in rat kidney. J Gen Physiol 147: 217–227, 2016. doi: 10.1085/jgp.201511533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890–F897, 2010. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol 297: F1249–F1255, 2009. doi: 10.1152/ajprenal.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 17.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin and prostasin-dependent release of an inhibitory peptide from the gamma subunit. J Biol Chem 282: 6153–6160, 2007. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- 18.Heidrich E, Carattino MD, Hughey RP, Pilewski JM, Kleyman TR, Myerburg MM. Intracellular Na+ regulates epithelial Na+ channel maturation. J Biol Chem 290: 11569–11577, 2015. doi: 10.1074/jbc.M115.640763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knight KK, Wentzlaff DM, Snyder PM. Intracellular sodium regulates proteolytic activation of the epithelial sodium channel. J Biol Chem 283: 27477–27482, 2008. doi: 10.1074/jbc.M804176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Firsov D, Robert-Nicoud M, Gruender S, Schild L, Rossier BC. Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC). Identification of cysteines essential for channel expression at the cell surface. J Biol Chem 274: 2743–2749, 1999. doi: 10.1074/jbc.274.5.2743. [DOI] [PubMed] [Google Scholar]
- 21.Noreng S, Bharadwaj A, Posert R, Yoshioka C, Baconguis I. Structure of the human epithelial sodium channel by cryo-electron microscopy. eLife 7: e39340, 2018. doi: 10.7554/eLife.39340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheng S, Maarouf AB, Bruns JB, Hughey RP, Kleyman TR. Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J Biol Chem 282: 20180–20190, 2007. doi: 10.1074/jbc.M611761200. [DOI] [PubMed] [Google Scholar]
- 23.Frindt G, Bertog M, Korbmacher C, Palmer LG. Ubiquitination of renal ENaC subunits in vivo. Am J Physiol Renal Physiol 318: F1113–F1121, 2020. doi: 10.1152/ajprenal.00609.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carattino MD, Sheng S, Bruns JB, Pilewski JM, Hughey RP, Kleyman TR. The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its alpha subunit. J Biol Chem 281: 18901–18907, 2006. doi: 10.1074/jbc.M604109200. [DOI] [PubMed] [Google Scholar]
- 25.Carattino MD, Mueller GM, Palmer LG, Frindt G, Rued AC, Hughey RP, Kleyman TR. Prostasin interacts with the epithelial Na+ channel and facilitates cleavage of the γ-subunit by a second protease. Am J Physiol Renal Physiol 307: F1080–F1087, 2014. doi: 10.1152/ajprenal.00157.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haerteis S, Schork A, Dorffel T, Bohnert BN, Nacken R, Worn M, Xiao M, Essigke D, Janessa A, Schmaier AH, Feener EP, Haring H-U, Bertog M, Korbmacher C, Artunc F. Plasma kallikrein activates the epithelial sodium channel in vitro but is not essential for volume retention in nephrotic mice. Acta Physiol (Oxf) 224: e13060, 2018. doi: 10.1111/apha.13060. [DOI] [PubMed] [Google Scholar]
- 27.Patel AB, Chao J, Palmer LG. Tissue kallikrein activation of the epithelial Na channel. Am J Physiol Renal Physiol 303: F540–F550, 2012. doi: 10.1152/ajprenal.00133.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Picard N, Eladari D, El Moghrabi S, Planes C, Bourgeois S, Houillier P, Wang Q, Burnier M, Deschenes G, Knepper MA, Meneton P, Chambrey R. Defective ENaC processing and function in tissue kallikrein-deficient mice. J Biol Chem 283: 4602–4611, 2008. doi: 10.1074/jbc.M705664200. [DOI] [PubMed] [Google Scholar]
- 29.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR. Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem 283: 36586–36591, 2008. doi: 10.1074/jbc.M805676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohnert BN, Daiminger S, Worn M, Sure F, Staudner T, Ilyaskin AV, Batbouta F, Janessa A, Schneider JC, Essigke D, Kanse S, Haerteis S, Korbmacher C, Artunc F. Urokinase-type plasminogen activator (uPA) is not essential for epithelial sodium channel (ENaC)-mediated sodium retention in experimental nephrotic syndrome. Acta Physiol (Oxf) 227: e13286, 2019. doi: 10.1111/apha.13286. [DOI] [PubMed] [Google Scholar]
- 31.Xiao M, Bohnert BN, Aypek H, Kretz O, Grahammer F, Aukschun U, Worn M, Janessa A, Essigke D, Daniel C, Amann K, Huber TB, Plow EF, Birkenfeld AL, Artunc F. Plasminogen deficiency does not prevent sodium retention in a genetic mouse model of experimental nephrotic syndrome. Acta Physiol (Oxf) 231: e13512, 2021. doi: 10.1111/apha.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nesterov V, Dahlmann A, Bertog M, Korbmacher C. Trypsin can activate the epithelial sodium channel (ENaC) in microdissected mouse distal nephron. Am J Physiol Renal Physiol 295: F1052–F1062, 2008. doi: 10.1152/ajprenal.00031.2008. [DOI] [PubMed] [Google Scholar]