

Abstract
Recent advancements in detection methods have made protein condensates, also called granules, a major area of study, but tools to characterize these assemblies need continued development to keep up with evolving paradigms. We have optimized a protocol to separate condensates from cells using chemical crosslinking followed by size-exclusion chromatography (SEC). After SEC fractionation, the samples can be characterized by a variety of approaches including enzyme-linked immunosorbent assay (ELISA), dynamic light scattering, electron microscopy, and mass spectrometry. The protocol described here has been optimized for cultured mammalian cells and E. coli expressing recombinant proteins. Since the lysates are fractionated by size, this protocol can be modified to study other large protein assemblies, including the nuclear pore complex, and for other tissues or organisms.
Keywords: granule, liquid-liquid phase separation, condensates, size-exclusion chromatography, formaldehyde crosslinking, protein assemblies, non-membrane bound organelles
Introduction
The formation of phase separated protein condensates has become a high-profile topic of research and is considered to give rise to several non-membrane bound organelles within cells (Banani, Lee, Hyman, & Rosen, 2017; Mitrea & Kriwacki, 2016; Zbinden, Perez-Berlanga, De Rossi, & Polymenidou, 2020) (Figure 1A). Condensates are formed by weak interactions that result in separation of proteins and nucleic acids out of solution (Figure 1B) (Kato & McKnight, 2017; Peran & Mittag, 2020; H. Wu & Fuxreiter, 2016). Some condensates appear very dynamic and fluid-like and have thus been described as liquid-liquid phase separation (A & Weber, 2019; McSwiggen, Mir, Darzacq, & Tjian, 2019) (Figure 1C). Studies have driven condensate formation using low temperatures, salt concentration, pH, and RNA concentration (Adame-Arana, Weber, Zaburdaev, Prost, & Julicher, 2020; Kato et al., 2012; Lin, Protter, Rosen, & Parker, 2015; Riback et al., 2020; Sanders et al., 2020; Schwartz, Wang, Podell, & Cech, 2013). Molecular crowding can also be an important driver of phase separation and high molecular weight sugar polymers like polyethylene glycol and Ficoll can mimic the dense molecular environment within cells (Banani et al., 2017; Kato & McKnight, 2017; Molliex et al., 2015).
Figure 1.
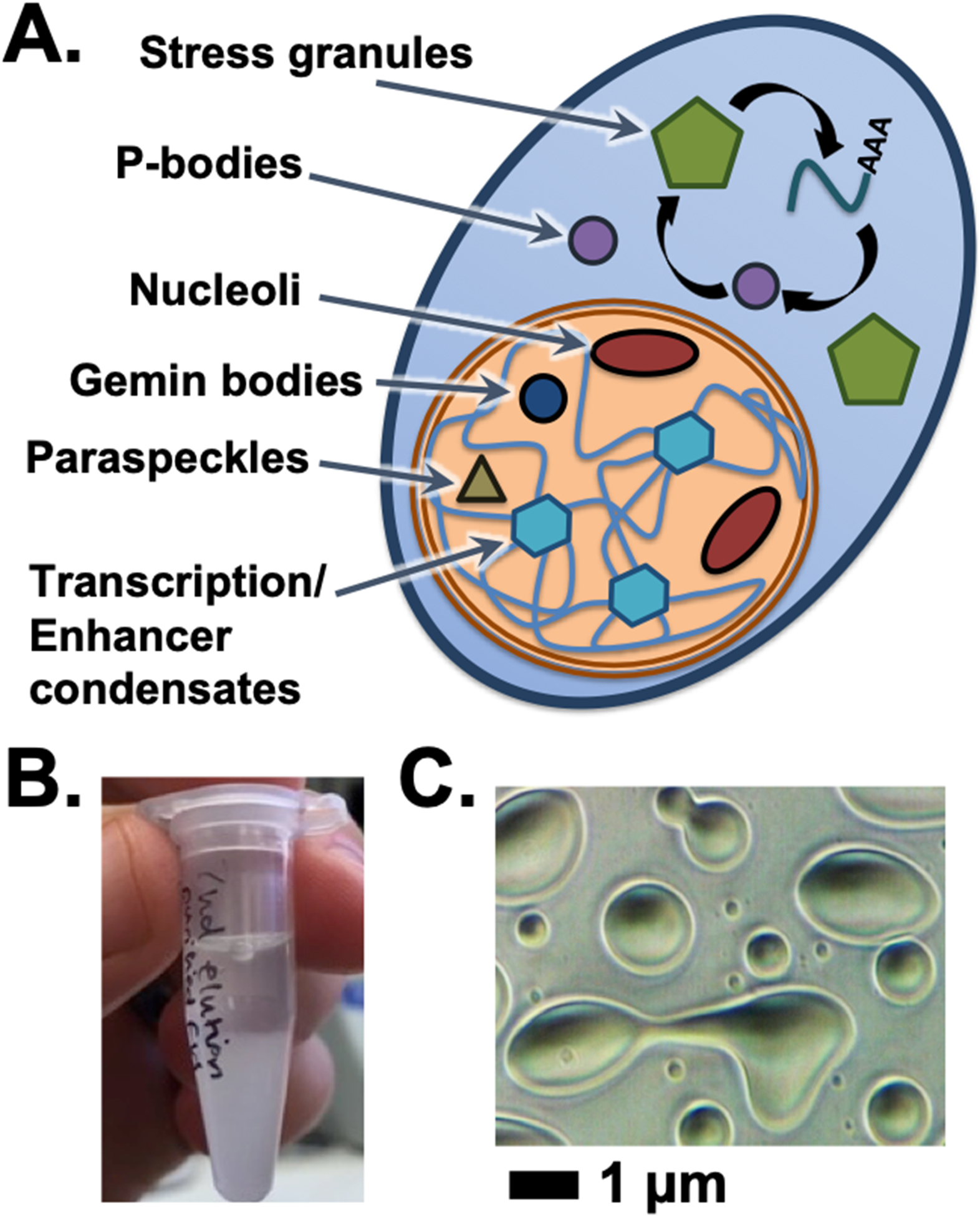
Condensates in cells and in vitro. (A) The cell has a number of granule bodies and non-membrane bound organelles, including those listed here in the cytoplasm or nucleus. (B) Phase separation can be visually observed by the accumulation of turbidity and eventually separation of protein condensate by gravity at sufficiently high protein concentrations. (C) Microscopic scale condensates allow the fluid and dynamic properties of protein phase separation to appear as condensates assume droplet shapes, fuse together, and wet surfaces on which they settle.
Prominent proteins in the growing field of phase separation readily form homogenous condensates in vitro, or simple heterogenous condensates of a few purified proteins (Boija et al., 2018; Lin et al., 2015; Springhower, Rosen, & Chook, 2020; Wang et al., 2018). The physical properties of condensates have been investigated with a range of approaches, including visual observation (Ceballos, McDonald, & Elbaum-Garfinkle, 2018; Kato et al., 2012), fluorescence-based techniques (Ladouceur et al., 2020; Taylor, Wei, Stone, & Brangwynne, 2019), turbidity (Qamar et al., 2018; Schwartz et al., 2013), small molecule interactions (Han et al., 2012; Kroschwald et al., 2015; Wheeler, Matheny, Jain, Abrisch, & Parker, 2016), NMR (Murthy & Fawzi, 2020; Wong, Kim, Muhandiram, Forman-Kay, & Kay, 2020), and electron microscopy (Thompson et al., 2018; G. Wu et al., 2017). The study of phase separation in the cell is complicated by several factors (Milkovic & Mittag, 2020). First, the molecular crowding critical to maintaining phase separation is lost when cells are lysed, causing condensates to break into small complexes or monomers (Molliex et al., 2015). Second, condensates studied in vitro are much larger than those formed in cells and therefore condensates in cells are less amenable to study by microscopy or techniques that require large amounts of protein. Finally, condensates formed in vitro are often studied under very non-physiological conditions, such as low temperatures, high protein concentrations, homogenous solutions, or at non-biological stoichiometries, which may interfere with extrapolation of observed properties to the cell (A & Weber, 2019; Case, Zhang, Ditlev, & Rosen, 2019; Hofweber et al., 2018; Qamar et al., 2018).
Here we describe methods to extract large protein assemblies comprised of weak protein-protein interactions, such as granules and condensates, from cells. In this method, cells are crosslinked with formaldehyde, lysed with sonication, and separated by size-exclusion chromatography (SEC) (Thompson et al., 2018) (Figure 2A). Condensates, and other large protein assemblies, elute from the column early, while smaller protein complexes and monomers are found later in the elution profile (Figure 2B). Our protocol was created and optimized for the study of granules associated with transcription machinery. Other large assemblies in the cell can be monitored as well, including nuclear pore complexes, which we have observed in our early SEC fractions with transmission electron microscopy (Figure 3). Extension of this protocol for targets such as ribosomes or solubilized amyloids may require optimization and established protocols to study these specific targets may be incorporated to augment the methods described here (G. Wu et al., 2017).
Figure 2.
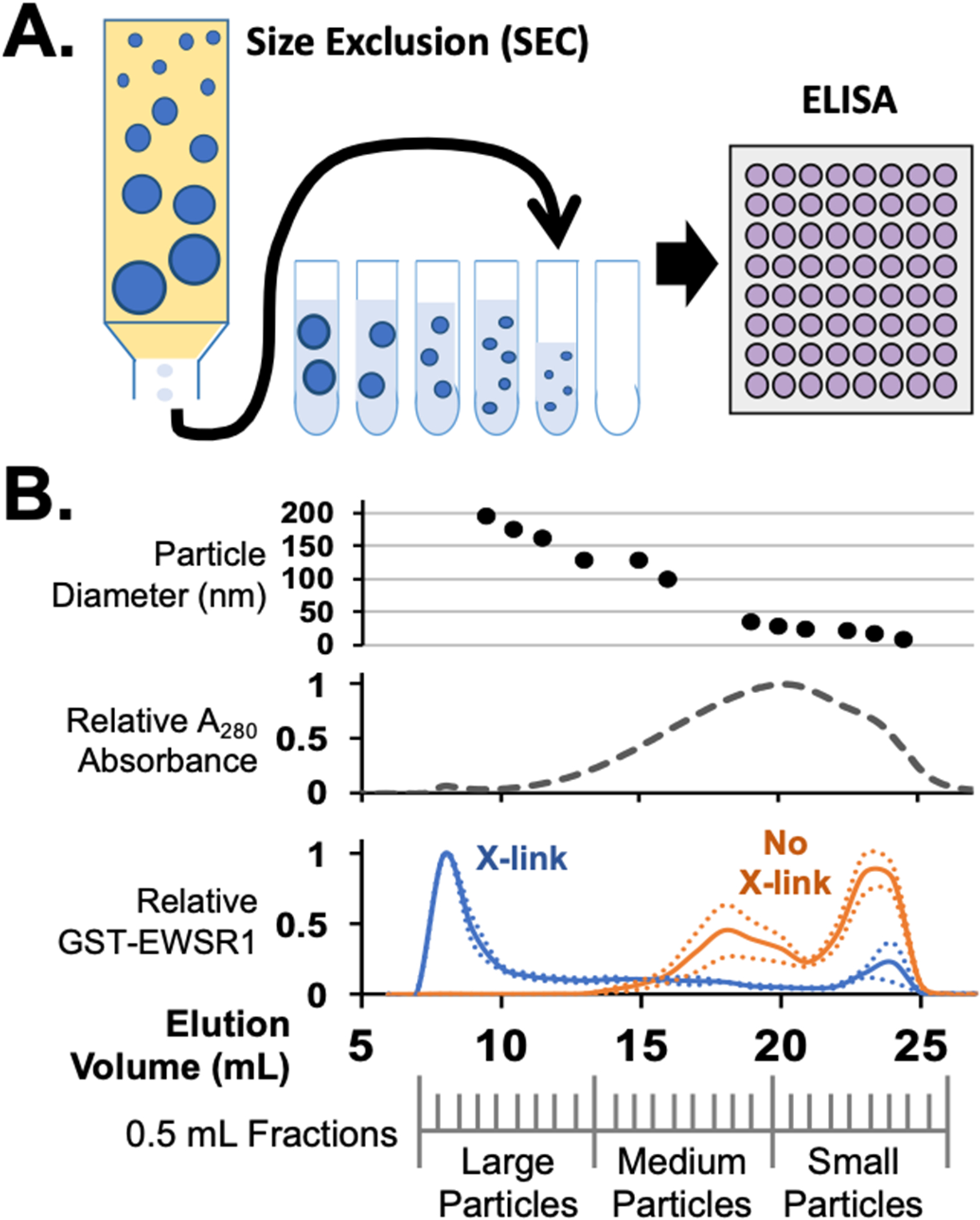
Separation of cell condensates by size exclusion chromatography. (A) Cell assemblies and condensates stabilized by formaldehyde crosslinking can be separated by SEC. ELISA assays of fractions collected can reveal the elution profile for specific crosslinked proteins that elute. (B) Example of SEC data from E. coli lysates expressing the protein GST-EWSR1. Particle sizes eluted can be measured by either DLS or TEM. Shown here is DLS data indicating particles between 200 and 10 nm measured between early and late fractions respectively, top. By monitoring UV absorption, most crosslinked proteins in a cell lysate elute in late fractions as small complexes or monomers. Protein eluting from SEC of a no-crosslink cell lysate produce the same UV absorption profile. In an ELISA, GST-EWSR1 was found to elute primarily in early fractions as large particles, blue. Without crosslinking, GST-EWSR1 assemblies break apart and ELISA signals are primarily in the late fractions, orange, consistent with medium to small particles.
Figure 3.
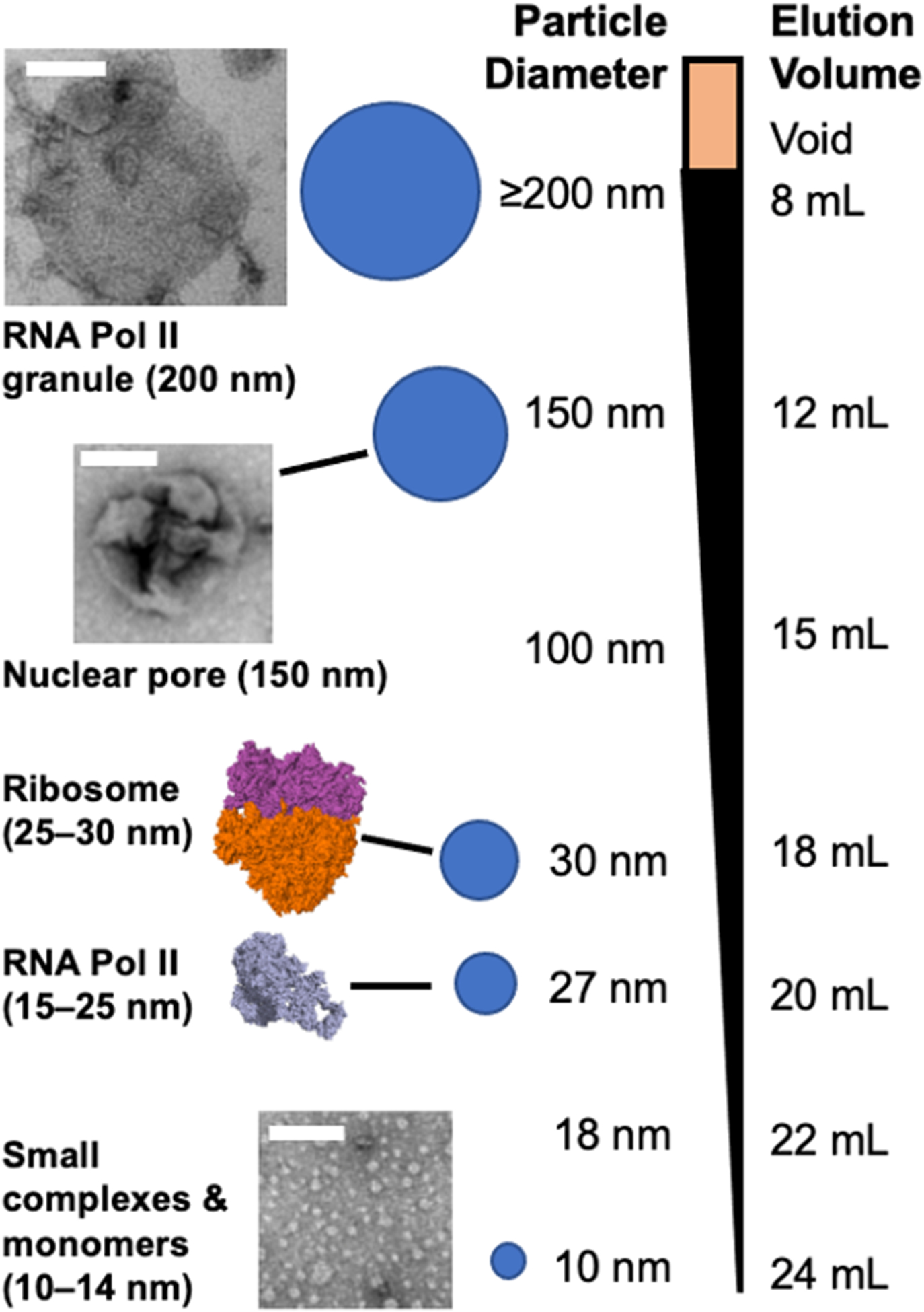
A variety of granules, protein assemblies, and large or small protein complexes can be separated in SEC. The range of particles separable using the CL2B SEC matrix can resolve large condensates, such as nuclear pore complexes and protein granules that associate with RNA Pol II during transcription. In the middle to late fractions elute large to small complexes of ordinary size, including ribosomes, RNA Pol II, and monomers.
Our work developing this protocol has focused on its application to cultured mammalian cells and E. coli cells expressing a recombinant protein of interest that forms condensates (Thompson et al., 2018). Both contexts can yield comparable results, and each provides some unique advantages. In immortalized human cell lines, the proteins studied are endogenously expressed which provides native post-translational modifications and protein interactors. Recombinant protein expression in E. coli is fast, inexpensive, provides a simplified cellular environment to observe condensate formation, and is easy to genetically manipulate. After SEC, many techniques that more directly compare to ex vivo studies of phase separation, can examine the cellular condensates, including dynamic light scattering, mass spectrometry, and electron microscopy (Kawaguchi et al., 2020; Thompson et al., 2018).
STRATEGIC PLANNING
Formaldehyde Crosslinking Condensates before Cell Lysis
The interactions between protein and RNA components of large protein assemblies and condensates are often weak and sensitive to changes in concentrations. Cell lysis perturbs this careful balance and leads to disassembly. However, protein-protein interactions within condensates, assemblies, or granules can be chemically crosslinked while in the cell to stabilize them throughout subsequent procedures.
Sepharose CL2B as a matrix for size exclusion chromatography
This protocol uses a Sepharose CL2B column matrix for SEC because it offers a large fractionation range that separates very large particles from smaller, ordinary-sized protein complexes and monomers (Figure 3). Sepharose CL2B is available from Cytiva as a customized chromatography column. Alternatively, the CL2B resin can be commercially purchased for packing a column yourself, according to the manufacturer instructions.
Note: if you are packing your column, additional equipment may be necessary.
Sepharose CL2B offers separation for particles of globular protein masses between 7 × 105 and 4 × 107 (Boing et al., 2014; Lobb, Lima, & Moller, 2017). Monomeric proteins will elute from the column late and large assemblies elute in early fractions. Assemblies larger than the resolution of the column will elute in the void volume, and there will be greater heterogeneity in particle sizes near these early eluting volumes. High-resolution characterization, such as transmission electron microscopy (TEM) can provide a more accurate assessment of particle sizes in these fractions.
This technique is limited in its application to exceptionally large assemblies, such as nucleoli and large stress granules, that are too large to pass through pre-column filters (typically 0.45 μm) on fast protein liquid chromatography (FPLC) systems (Mao, Zhang, & Spector, 2011; Souquere et al., 2009). Procedures to monitor protein loss during sample preparation are included below to help determine whether this factor is limiting your results. Other methods have been developed for isolation of nucleoli (Z. F. Li & Lam, 2015; Muramatsu, Hayashi, Onishi, Sakai, & Takai, 1974) and stress granules (Wheeler, Jain, Khong, & Parker, 2017).
Basic Protocol 1: SEC separation of crosslinked mammalian cell lysates
Chemical crosslinking of protein interactions provides additional flexibility in sample preparation relative to standard protocols. In this procedure, we leverage the enhanced stability between protein interactors in several ways. For example, the addition of urea to buffers reduces aggregation and interactions that might form after cell lysis. Protein particles, assemblies, or monomers bound to nucleic acids artificially appear larger on SEC elution profiles due to the large size of any bound nucleic acids. Nucleic acids can also act to tether non-interacting proteins and artificially increase their size in this manner. For this reason, sonication and nuclease digestion steps are added to limit interactions tethered by nucleic acids, not protein-protein interactions. Crosslinking allows for aggressive steps, like sonication, to minimize or remove nucleic acids in samples. On the other hand, crosslinking can stabilize protein-protein interactions that form in a nucleic acid dependent manner. These considerations must be kept in mind when interpreting data from crosslinked samples.
Materials
Mammalian cells: e.g. HeLa (Cell Bio Labs, cat. no. AKR-213) or HEK293T/17 (ATCC, cat. no. CRL-11268)
Cell growth media: e.g. DMEM (Corning, cat. no. 10–017-CV) containing 10% (v/v) Fetal Bovine Serum (Peak Serum, Inc., cat. no PS-FB1)
Dulbeccos’s Phosphate Buffered Saline (Sigma, cat. no. 59331C)
Formaldehyde solution (Sigma, cat. no. F8775)
Phosphate Buffered Saline (PBS)
1.25 M glycine in PBS
Sonication Buffer (see recipe in Reagents and Solutions)
Benzonase Nuclease (Millipore, cat. no. E1014)
SEC Running Buffer (see recipe in Reagents and Solutions)
Serological pipets
15-cm dish (CELLTREAT, cat. no. 229695)
Microscope
Cell scraper (CELLTREAT, cat. no. 229315)
50 mL conical tube (VWR, cat. no. 89039–656)
Micropipettes
Pipette Tips
Pipet Aid
Refrigerated benchtop centrifuge
15 mL conical tube (VWR, cat. no. 89039–664)
Bioruptor Pico Sonication Device (Diagenode, cat. no. B01060010)
15 mL Bioruptor Pico tubes and sonication beads (Diagenode, cat. no. C01020031)
1.5 mL microcentrifuge tubes
0.45 μM centrifugal filter (EMD Millipore, cat. no. UFC30HV00)
Spectrophotometer
Sepharose CL2B 10 × 300 chromatography column
Äkta Pure FPLC (Cytiva, cat. no. 29018224)
Test Tubes (Labcon, cat. no. 3411-800-000)
Fraction collector (Cytiva, cat. no. 29011362)
Gastight Syringe (VWR, cat. no. 60376-274)
Mammalian cell growth
-
1
Grow cells to confluency in 3 × 15 cm dishes. Assess confluency by microscopy.
Three confluent 15 cm dishes are sufficient for HeLa and HEK293 cells. Exceptionally large cell types, such as human fibroblasts, can lower cell numbers and protein yield per dish/flask. Adjust for these cell types, as well as those growing in suspension.
Crosslinking of mammalian cells
-
2
Remove growth media.
For cells that are weakly adhesive care must be taken to not disturb the cell monolayer.
-
3
Wash the cells with 10 mL of PBS at room temperature.
-
4
Aspirate PBS.
To verify treatments made to cells (e.g. protein expression or knockdown), after aspirating scrape a 1-inch square of cells and transfer to a 1.5 mL microcentrifuge tube in 600 μL PBS. Cells can be pelleted by centrifugation (500–1000 × g), PBS removed, and frozen dry at −80 °C for subsequent analysis by western blotting or quantitative real-time PCR.
-
5
Dilute formaldehyde to 1% (v/v) in PBS.
-
6
Add 10 mL of 1% formaldehyde in PBS to each dish to crosslink the cells. Incubate for 15 minutes at room temperature.
-
7
Quench the remaining formaldehyde by adding 1 mL of 1.25 M glycine in PBS to each plate and incubate for 5 minutes.
-
8
Transfer all but 2 mL of the buffer from each plate into a single 50 mL conical tube.
-
9
Scrape cells from each plate and transfer to the 50 mL conical tube.
-
10
Centrifuge the cells at 500 × g for 5 minutes at 4 °C.
-
11
Remove the supernatant and resuspend the cell pellet in 10 mL of fresh PBS.
-
12
Transfer the cells to a clean 15 mL conical tube.
It is helpful for the tube to have gradation marks below 1 mL.
-
13
Centrifuge the cells at 500 × g for 5 minutes at 4 °C.
-
14
Remove the PBS.
Cell pellets can often be stored in −80 °C for later use, however this may lead to aggregation during freezing or thawing. Reduced levels of a protein of interest or inconsistent elution profiles observed in SEC may indicate freshly harvested cells should be used. In these cases, proceed to SEC immediately.
Cell lysis and preparation for SEC
-
15
Resuspend the cell pellet in 5 packed cell volumes of Sonication Buffer.
Gradation lines on the tube can be used to estimate the volume of the cells. Alternatively, the pellet size can be visually compared to a volume of water added to another tube. Use 5 times this volume of buffer.
-
16
Add 10 μL Benzonase Nuclease to the resuspended cells and incubate for 10 minutes at room temperature.
-
17
Cool a benchtop centrifuge and the Bioruptor Pico to 4 °C.
-
18
Fill a Bioruptor 15 mL conical tube with sonication beads up to the second gradation line.
This amount is recommended by the manufacturer for a sample volume of 2 mL.
-
19
Equilibrate Bioruptor beads in 2 mL of Sonication Buffer.
-
20
Remove buffer from the beads.
-
21
Transfer the cells to the equilibrated beads in the Bioruptor 15 mL conical tube.
-
22
Place the tube in the Bioruptor Pico and sonicate for 30 cycles of 30 seconds on followed by 30 seconds off.
-
23
Transfer the sonicated lysate to a clean 2 mL microcentrifuge tube without disturbing the sonication beads.
Sonication beads typically settle immediately to the tube bottom. If beads remain dispersed, briefly centrifuge before transferring the lysate.
-
24
Remove cell debris by centrifugation for 30 minutes at maximum speed, >18,000 × g, and 4 °C.
Size Exclusion Chromatography
-
25
Filter the lysate using a 0.45 μm centrifugal filter.
-
26
Estimate the apparent protein concentration in the lysate by UV absorption at 280 nm, A280. Assume 1 Abs = 1 mg/mL.
It should be noted that free nucleotides or small nucleic acid fragments are likely to contribute significantly to the absorbance and that the A260/A280 will likely remain >1.2. However, the absorbance value can ensure consistent loading of SEC and assist comparisons of data from different cell lines.
-
27
Equilibrate Sepharose CL2B column with 2 column volumes of SEC Running Buffer.
-
28
Fill fraction collector with collection tubes.
-
29
Inject 1 ± 0.3 mg of total protein. Run the column at a flow rate of 0.2 mL/min for 2 column volumes.
We typically set fraction collection to be 0.5 mL fractions. For sample size injections described here, 0.5 mL is adequate for at least 3 downstream ELISAs.
-
30
Fractions can be stored at −80 °C.
Alternate Protocol 1: Preparation of non-crosslinked mammalian cells
Condensates formed by weak interactions are not expected to maintain their integrity following cell lysis without the assistance of crosslinking. Nevertheless, this can be tested by gently lysing non-crosslinked cells to run SEC. Since the interactions and protein complexes are not stabilized by chemical crosslinks, buffer components like urea and detergents are not recommended. This protocol digests nucleic acids with benzonase nuclease instead of the sonication step in Basic Protocol 1. It is expected that, in contrast to Basic Protocol 1, large assemblies like granules will break up during cell lysis, therefore the protein components will now elute later.
Materials
Trypan Blue
Hypotonic Lysis Buffer (see recipe in Reagents and Solutions)
Low Salt Buffer (see recipe in Reagents and Solutions)
Cytoplasmic Extract Buffer (see recipe in Reagents and Solutions)
Hemocytometer
27-gauge needle
MultiTherm Shaker (Benchmark Scientific, cat. no. H5000-HC)
Additional reagents and materials listed in Basic Protocol 1
Growth of mammalian cells
-
1
Grow cells to confluency in 3 × 15 cm dishes.
Preparation of nuclear and cytosolic extracts
-
2
Remove media from the 15 cm dishes.
For cells that are weakly adhesive, care must be taken to not disturb the cell monolayer.
-
3
Wash the cells one time with 10 mL PBS. Remove the PBS.
-
4
Add 3 mL of cold PBS to each dish and harvest the cells by scraping.
-
5
Transfer the cells to a 15 mL conical tube.
It is helpful for the tube to have graduation marks below 1 mL.
-
6
Centrifuge the cells at 500 × g for 5 minutes at 4 °C.
-
7
Remove the supernatant.
-
8
Resuspend in 10 mL cold PBS.
-
9
Transfer a 50 μL aliquot to a microcentrifuge tube containing 100 μL PBS and 50 μL Trypan Blue.
-
10
Count the cells.
-
11
Centrifuge the remaining cells in the 15 mL conical at 500 × g for 5 minutes at 4 °C.
-
12
Discard the supernatant and measure the packed cell volume.
Nuclei purification by hypotonic lysis
-
13
Resuspend the cells in 5 packed cell volumes of cold Hypotonic Lysis Buffer.
-
14
Incubate the cells on ice for 15 minutes.
-
15
Lyse the cells by passing the sample through a 27-gauge needle 5 times.
-
16
Transfer lysate to a microcentrifuge tube.
-
17
Repeat steps 9–10 and calculate the percentage of nuclei versus whole cells.
-
18
Centrifuge the remaining lysate at 10,000 × g for 5 minutes at 4 °C. Remove the supernatant.
If interested in the cytoplasmic fraction, save the supernatant at this time by adding 10X Cytoplasmic Extract buffer to a concentration of 1X. Resume the protocol at step 25.
-
19
Wash the pellet by resuspending in 5 packed cell volumes of Low Salt Buffer.
-
20
Centrifuge at 10,000 × g for 5 minutes at 4 °C. Remove the supernatant.
-
21
Measure the packed nuclei volume.
Nucleic acid digestion
-
22
Resuspend in 0.5 packed nuclei volume of cold Low Salt Buffer. Add 100 U Benzonase. Incubate 10 minutes at room temperature.
-
23
Add 0.5 packed nuclei volume of cold High Salt Buffer slowly and without stirring or shaking.
-
24
Incubate at 4 °C for 30 minutes while shaking.
-
25
Centrifuge at maximum speed (> 18,000 × g) for 30 minutes at 4 °C.
-
26
Proceed with Basic Protocol 1 step 25.
Basic Protocol 2: SEC separation of E. coli lysate
This adaptation of Basic Protocol 1 is for E. coli cells. The molecular crowding in E. coli cells is comparable to that of eukaryotic cells. Recombinant proteins expressed in E. coli can form condensates in a model environment isolated from interactors or modifiers found in their native cell type. Some additional considerations apply when expressing recombinant proteins. Overexpression can overwhelm the cell and lead to protein sequestration in inclusion bodies. Moreover, proteins that form condensates can be particularly prone to aggregation. A solubility tag such as MBP can buffer against aggregation and improve consistency in results. Protein expression conditions for a specific protein of interest can also be optimized for induction time, temperature, and IPTG concentration. This protocol crosslinks E. coli cells after induction and prepares lysates for SEC using a Sepharose CL2B column matrix.
Materials
Chemically competent E. coli cells for expression, such as BL21 (NEB, cat. no. C2530H)
Plasmid DNA
SOC media
LB agar plates with appropriate antibiotics
LB growth medium (Miller’s)
Isopropyl β-D-1-thiogalactopyranoside (IPTG)
Liquid nitrogen
E. coli Lysis Buffer (see recipe in Reagents and Solutions)
Programmable heating block
Shaking incubator
37 °C incubator
15 mL culture tubes
250 mL baffle flask(s)
Cuvettes
Weighing scale
50 mL conical tubes
Additional reagents and materials for crosslinking, lysis, and SEC from Basic Protocol 1
Transformation of chemically competent E. coli cells
-
1
Set heat block to 42 °C and shaking incubator to 37 °C to preheat.
-
2
Thaw 50 μL chemically competent cells on ice for 30 minutes.
-
3
Add 20 ng of plasmid DNA to the cells. Incubate on ice 30 minutes.
Keep the volume of the DNA solution added <10% of the volume of the cells.
-
4
Heat shock cells by incubating in a heat block at 42 °C for 30 seconds. Return cells to ice for 2 minutes.
-
5
Add 1 mL SOC media to the cells.
-
6
Incubate cells in a falcon tube with shaking at 37 °C and 200 rpm for 1 hour.
-
7
Spread 50, 100, and 200 μL of the cell solution on 3 different LB agar plates (with antibiotics).
-
8
Incubate plates inverted at 37 °C overnight.
-
9
Store plates at 4 °C.
Growth of transformed E. coli and protein expression
-
10
Inoculate a single, isolated colony into 4 mL LB media with antibiotics.
-
11
Grow overnight at 37 °C and 200 rpm. Do not grow longer than 16 hours.
-
12
Transfer 1 mL of overnight growth to 65 mL LB with antibiotics in a 250 mL baffle flask.
-
13
Grow at 37 °C shaking at 200 rpm until the cells reach an OD600 of 0.4–0.6.
This step typically takes 2–4 hours.
-
14
Transfer 100 μL of cells in suspension to a clean 1.5 mL tube for a “pre-induction” sample. Store at −20 °C.
-
15
Induce protein expression by adding IPTG to a 1 mM final concentration and allow cells to grow 2–4 more hours at 37 °C.
Induction conditions may require optimization (Francis & Page, 2010). Optimization parameters include OD600, time, temperature, and amount of IPTG. Inclusion bodies may be visible as dark banding in cell pellets after centrifugation. Inclusion body formation may be reduced or avoided by using a lower induction temperature. For a low temperature induction, after reaching optimal OD600 at 37 °C, cool the incubator and cells to 17 °C. Once cool, add IPTG and incubate for 16 hours at 17 °C.
Formaldehyde crosslinking of E. coli
-
16
Weigh two 50 mL conical tubes.
-
17
Transfer 100 μL of cells in suspension to a clean 1.5 mL tube for a “post-induction” sample. Store at −20 °C.
The “pre-induction” and “post-induction” samples can be lysed in 1X SDS loading buffer and compared in SDS-PAGE with Coomassie staining. A band at the expected molecular weight for your expressed protein should appear prominently in the “post-induction” sample only. If the expected molecular weight of the exogenously expressed protein is obscured by highly expressed E. coli proteins, additional steps, such as western blotting, may be required to check induction efficiency.
-
18
Transfer 25 mL of the cell growths to each 50 mL conical tube and centrifuge for 10 minutes at 4000 × g and 4 °C.
Traditional protocols may allow freezing of cell pellets for later use. However, we find it important that cells are crosslinked before freezing.
-
19
Remove supernatant and wash the cells once by gently resuspending both pellets in PBS. Centrifuge again for 10 minutes at 4000 × g and 4 °C. Discard supernatant.
-
20
Dilute formaldehyde to 1% (v/v) in PBS.
-
21
Resuspend one cell pellet with 45 mL PBS+ 1% formaldehyde.
-
22
Resuspend the second tube in 45 mL of PBS.
This will act as a no crosslinking control sample.
-
23
Incubate resuspended cells for 20 minutes at room temperature.
-
24
Quench the formaldehyde by adding 2.25 mL of 2.5 M glycine then incubate an additional 5 minutes at room temperature.
-
25
Pellet cells by centrifugation for 10 minutes at 4000 × g and 4 °C. Discard supernatants.
-
26
Wash cells once by gently resuspending pellets in PBS followed by centrifugation for 10 minutes at 4000 × g and 4 °C.
-
27
Decant or aspirate off all supernatant without disturbing the cell pellet.
-
28
Proceed to step 29 or freeze the cell pellets in liquid nitrogen to be stored at −80 °C.
Preparation of E. coli lysate for size-exclusion chromatography
-
29
Cool a benchtop centrifuge and Bioruptor Pico to 4 °C.
-
30
Fill a Bioruptor 15 mL conical tube with 2 lines of beads.
-
31
Equilibrate Bioruptor beads in 2 mL of E. coli lysis buffer.
-
32
Weigh the tube containing the cell pellet to determine mass. Subtract the weight of the tube.
We typically obtain 0.4–0.8 g of cell pellet at this step.
-
33
Resuspend crosslinked E. coli cell pellets in PBS and transfer enough volume to contain 0.2 g of cells to a fresh tube.
-
34
Harvest the cells by centrifugation at 4000 × g for 10 minutes at 4 °C.
-
35
Resuspend the cells in 2 mL of E. coli lysis buffer.
-
36
Remove buffer from the equilibrated beads and transfer resuspended cells to the 15 mL Bioruptor tube containing 2 lines of beads and incubate for 10 minutes at room temperature.
-
37
Sonicate for 30 cycles of 30 seconds on and 30 seconds off at 4 °C.
-
38
Transfer lysate to a 2 mL microcentrifuge tube.
-
39
Centrifuge for 30 minutes at 4 °C at maximum speed (> 18,000 × g).
-
40
Transfer the supernatant to a clean microcentrifuge tube.
If the protein of interest is potentially insoluble, consider saving the pellet and a portion of this supernatant for comparison by SDS-PAGE or Western blotting. If a large portion of the protein is found in the pellet sample, the protein induction or lysis conditions may require optimization.
-
41
Filter 700 μL of lysate (supernatant) using a 0.45 μM PVDF centrifugal filter.
If the filter becomes clogged, increasing the volume of lysis buffer in step 35 may reduce non-specific aggregation resulting from a lysate that is too concentrated.
SEC purification of granules from cell lysate
-
42
Estimate the apparent protein concentration in the lysate by UV absorption at 280 nm, A280. Assume 1 Abs = 1 mg/mL.
It should be noted that free nucleotides or small nucleic acid fragments are likely to contribute significantly to the absorbance and that the A260/A280 will likely remain >1.2. However, the absorbance value can ensure consistent loading of SEC. We typically obtain lysate concentrations around 20 mg/mL from the above protocol. Instances where the lysate concentration is much lower than that, usually leads to poor results downstream due to low signal.
-
43
Equilibrate Sepharose CL2B column with SEC Running Buffer.
-
44
Inject 1 ± 0.3 mg of total protein on a 10 × 300 Sepharose CL2B size exclusion chromatography column and collect 0.5 mL fractions. The column flow rate should be 0.2 mL/min with SEC Running Buffer.
We find injecting ≥1.5 mg protein can result in non-specific sticking to the column, resulting in protein eluting at >1 column volume or large particles in late fractions. Overloading may damage the column. Begin with 1 mg total protein and decrease if overloading is suspected.
-
45
Store fractions at −80 °C.
Support Protocol 1: Detecting protein of interest by ELISA
Crosslinking interferes with SDS-PAGE during the analysis of proteins by western blotting. Protocols to reverse formaldehyde crosslinks cannot guarantee complete removal of crosslinks or recovery of samples. The ELISA is a useful alternative to identify and quantify a protein of interest in crosslinked samples. For this assay, antibody specificity should be validated by western blot analysis of non-crosslinked lysates since the ELISA does not separate signals by molecular weight. Antibody specificity in the ELISA can be further validated using lysates with reduced protein expression, knockdown, or knockout.
Materials
SEC Running Buffer (see recipe in Reagents and Solutions)
Tris buffered saline with 0.1% (v/v) Tween (TBS-T)
5% non-fat dry milk (NFDM) in TBS-T
2.5% NFDM in TBS-T
Primary antibody
HRP conjugated secondary antibody (e.g. Fisher Scientific cat. no. PI31462 or PI31432)
ELISA Substrate Solution (Thermo Scientific cat. no. 34028 or 37074)
Micropipettes
High-bind 96-well plate (Grenier Bio-One, cat. no. 655061)
Orbital Shaker
Plate reader (Biotek Synergy Neo2)
ELISA
-
Load 10–150 μL of each fraction into a 96-well High-Bind plate.
Variability in antibody quality and protein expression levels may necessitate optimizing the amount of fraction used in the ELISA. 25 μL is an appropriate starting point for the E. coli fractions. 150 μL is appropriate for mammalian cell-based fractions. Anywhere within 10–150 μL may yield the best signal to noise ratio. The choice of plate type is determined by the substrate used for detection. Opaque white plates are needed for chemiluminescent detection while a clear plate is used for a colorimetric detection reagent.
-
Add SEC Running buffer to a final volume of 150 μL in each well. This will improve consistent binding across wells.
In order to maximize the number of ELISAs per SEC run, loading every other fraction typically provides a sufficient resolution for elution profiles. Fractions remaining may verify unclear or potential outlier signals and allow more ELISAs to detect additional proteins.
Incubate plate overnight at 4 °C for protein to bind.
Aspirate each well and wash with 200 μL of TBS-T for 5 minutes. Repeat this for a total of 4 washes.
Add 200 μL of 5% NFDM in TBS-T to each well. Incubate for 2 hours at room temperature.
Wash each well with 200 μL of TBS-T for 5 minutes and gentle shaking. Repeat for a total of 3 washes.
-
Dilute primary antibody in 2.5% NFDM in TBS-T.
Appropriate antibody dilutions vary and can be optimized beforehand by measuring the linearity of signal change over a serial dilution of antibody. Optimized dilutions typically range from 1:500–1:4000.
Add 175 μL of the primary antibody solution to each well. Incubate for 2 hours at room temperature or overnight at 4 °C with gentle shaking.
Wash each well with 200 μL of TBS-T for 5 minutes with gentle shaking. Repeat for a total of 4 washes.
-
Dilute secondary antibody in 2.5% NFDM in TBS-T.
As with the primary antibody, appropriate dilutions may need to be optimized. Optimal dilutions found in our assays range from 1:20000–1:40000.
Add 175 μL of secondary antibody solution to each well. Incubate for 1 hour at room temperature with gentle shaking.
Wash each well with 200 μL of TBS-T for 5 minutes with gentle shaking. Repeat for a total of 4 washes.
-
Incubate ELISA substrate per manufacturer instructions.
Mammalian cells typically require more sensitive detection solutions and recommend a maximum sensitivity substrate such as SuperSignal ELISA Femto (Thermo Scientific cat. no. 37074). Recombinant protein expression in E. coli yields high signals and an intermediate sensitivity substrate is sufficient, such as Ultra TMB-ELISA substrate (Thermo Scientific cat. no. 34028).
-
Measure substrate signals as directed by manufacturer instruction.
High levels of protein expression in E. coli can result in high background signals that alter the apparent elution profile for the protein. In these cases, it is important to optimize beforehand the amounts of sample added to the ELISA through serial dilutions of SEC fractions.
Support Protocol 2: TCA Precipitation of SEC Fractions
TCA precipitation of the proteins in SEC fractions may be useful to exchange buffer or remove urea from samples. This protocol also offers a quick approach to combine fractions collected during SEC. Use care in handling TCA, as it is extremely hazardous.
Materials
SEC fractions from Basic Protocol 1 or 2
TCA
Ice
Acetone
Micropipettes
1.5 mL tubes
Benchtop centrifuge
95 °C Heat block
TCA Precipitation of SEC Fractions
Add 1 volume of TCA to every 4 parts sample (e.g. 125 μL of TCA to 500 μL SEC fraction) in a microcentrifuge tube.
Incubate the mixture for 10 minutes on ice.
-
Centrifuge the sample at 18,500 × g for 5 minutes.
Note the outside of the tube where the pellet is expected after centrifugation, to avoid disturbing pellets that may not be visible.
-
Carefully remove the supernatant.
For large amounts of protein, a fluffy white pellet may be seen. Low amounts of protein may not yield a visible pellet.
Wash the pellet with 200 μL of cold acetone.
Centrifuge the sample at 18,500 × g for 5 minutes.
Remove the supernatant and repeat steps 5–6.
Carefully remove the supernatant.
Heat the open tube in a 95 °C heat block for 5–10 minutes to drive off any residual acetone.
Resuspend protein in desired buffer or store dry at 4 °C.
REAGENTS AND SOLUTIONS
Sterilization of the following solutions is not necessary.
10X Cytoplasmic Extract Buffer
0.333 M HEPES, pH 7.9
0.936 M NaCl
15.65 mM EDTA
50.5 mM Dithiothreitol (DTT)*
Store at 4 °C without DTT for up to 2 months. DTT must be added just before use.
E. coli Lysis Buffer
1 M Urea
1 M KCl
50 mM Tris-HCl, pH 8
Cool to 4 °C before use. Make fresh.
High Salt Buffer
0.8 M NaCl
20 mM HEPES, pH 7.9
5% glycerol
0.2 mM EDTA
6 M urea
0.5 mM DTT
1× protease inhibitors
Cool to 4 °C before use. Make fresh.
Hypotonic Lysis Buffer
10 mM HEPES, pH 7.9
1.5 mM MgCl2
10 mM NaCl
0.5 mM DTT
1× protease inhibitors
Store at 4 °C without DTT and protease inhibitors for up to 2 months. DTT and protease inhibitors must be added just before use.
Low Salt Buffer
20 mM HEPES, pH 7.9
5% glycerol
1.5 mM MgCl2
6 M urea
0.5 mM DTT*
1× protease inhibitors
Cool to 4 °C before use. Make fresh.
SEC Running Buffer
20 mM HEPES, pH 7.9
100 mM NaCl
0.2 mM EDTA
5% glycerol
6 M urea
0.5 mM DTT
Filter through a 0.45 μm filter and cool to 4 °C before use. Make fresh.
Sonication Buffer
0.4 M NaCl
20 mM HEPES, pH 7.9
5% glycerol
0.75 mM MgCl2
6 M urea
0.5 mM DTT
1× protease inhibitors
Cool to 4 °C before use. Make fresh.
COMMENTARY
Background Information
Condensates are dynamic structures within cells. They concentrate a heterogeneous subset of proteins, nucleic acids, and even small molecules away from the surrounding cell environment (Banani et al., 2017; Klein et al., 2020; Mitrea & Kriwacki, 2016; Springhower et al., 2020). Other molecular species freely diffuse around or within them. Interactions between the condensate’s constituents are weak. It remains to be seen if a definition for condensates will encompass all weakly interacting non-membrane bound organelles (A & Weber, 2019; Chong et al., 2018; McSwiggen et al., 2019). Alternatively, the field may grow to distinguish subsets of cellular assemblies that resemble liquid-liquid phase separation in appearance but form through another chemical mechanism. Continued innovation and adaptation of experimental approaches will improve interpretations of and relationships between in vitro and in vivo studies of phase separation processes.
Here, we describe a crosslinking approach to stabilize weak interactions in protein assemblies or condensates to allow separation or isolation from cells. Formaldehyde is cell permeable and crosslinks primary and secondary amines separated by <8 Å (Coscia et al., 2020; Tayri-Wilk et al., 2020). Formaldehyde crosslinking is employed to analyze protein interactomes and structure in many molecular biology protocols, including chromatin immunoprecipitation, immunocytochemistry, and mass spectrometry (Hoffman, Frey, Smith, & Auble, 2015; Y. Li et al., 2016; Sinz, 2006; Sutherland, Toews, & Kast, 2008; Toews, Rogalski, Clark, & Kast, 2008). Comparison between crosslinked and non-crosslinked samples can help distinguish crosslinked condensates from protein aggregates and rigid assemblies like hydrogels (Thompson et al., 2018).
Protocols described here use mammalian cell lines or E. coli expressing recombinant proteins. Study of a protein in its native cell environment can reveal its behavior when influenced or modified by cellular interactors. However, in complex granules containing many proteins, nucleic acids, and metabolites, the contribution of one specific protein may be difficult to deconvolute or compare to observations of simple condensates in vitro. Recombinant expression in a model cell, such as E. coli, can offer an intermediate environment to isolate the individual activity of a protein in forming assemblies or condensates. It is expected that this approach can be adapted to other biological systems or model organisms.
We have found that 6 M urea can minimize non-specific aggregation in crosslinked samples. Ionic or non-ionic detergents could also serve this purpose, but we found urea preferable, as detergents can interfere with some downstream applications. Guanidinium hydrochloride is one common substitute for urea. When adapting this protocol, some factors may influence the choice to use urea or detergents. For example, if samples are heated, urea can promote modifications to lysine residues that can affect analysis by mass spectrometry. Urea may also promote some proteins to aggregate. Moreover, some proteins or aggregates may remain insoluble in 6 M urea and require the use of detergents. Recent published protocols may offer guidance to accommodate the use of detergents in the place of urea to improve solubility or prevent aggregation (Lu, Wisniewski, & Mann, 2009; Nagaraj, Lu, Mann, & Wisniewski, 2008; Wierer & Mann, 2016).
After crosslinking and lysing cells, ELISAs can effectively detect crosslinked proteins in large particles, small complexes, as well as monomers in SEC fractions or immunoprecipitations. An alternative approach to protein detection in crosslinked samples is the expression of a fluorescently tagged protein. Direct detection of fluorescence intensity in samples is rapid and avoids expensive antibodies. To monitor particle size in SEC fractions, dynamic light scattering or TEM can be used (Thompson et al., 2018). It may also be useful to perform immunoprecipitation to enrich for a protein of interest and determine binding partners by mass spectrometry or visualize large assemblies in electron microscopy (Kawaguchi et al., 2020; Thompson et al., 2018).
Protocols have also been published to apply ultracentrifugation followed by immunoprecipitation to enrich for whole or semi-intact stress granules (Jain et al., 2016; Wheeler et al., 2017). This alternative protocol may prove more suitable depending on experimental aims. Either protocol may augment or validate results of the other approach and deepen our understanding of the defining characteristics of large protein assemblies, condensates, and membrane-less organelles.
Critical Parameters and Troubleshooting
Optimization of protein expression in E. coli
Overexpression of proteins can lead to aggregation and inclusion body formation (Francis & Page, 2010). Strategies may be explored to minimize this aggregation, but their effectiveness can vary depending on the protein expressed. Proteins that form condensates through liquid-liquid phase separation are particularly prone to such aggregation. Protein tags, such as MBP and GST, can increase solubility and prevent or slow aggregation. For IPTG inducible promoters, expression at a low temperature, such as 17 °C, or using IPTG concentrations as low as 0.1 mM can prevent formation of inclusion bodies. Alternative bacterial strains can also reduce aggregation. One example would be ArcticExpress cells (Agilent 230191), which provide additional chaperones to help protein folding and reduce aggregation.
Monitoring loss of protein
We have not found large amounts of protein loss to be a problem, but it should be closely monitored. A large loss in protein after lysis can indicate non-specific aggregation, which is addressed by altering expression, lysis, or buffer conditions. Samples of a lysate taken just after sonication, centrifugation, and filtration can be used to monitor protein loss at each step. Total protein concentration can be estimated by standard methods or a protein of interest by ELISA. While small percentages of loss are expected, large losses can bias results to reflect only the most soluble fraction of the protein. Reducing the number of cells lysed can improve protein solubility. Another source of protein loss is that the centrifugal filter may clog, or protein may adhere to the filter. Diluting lysates or an alternate membrane material may prevent these losses.
Column Maintenance
Proper column maintenance is essential to achieving good separation (Cytiva, www.cytivalifesciences.com). Cell lysates can oversaturate or clog the column matrix in SEC. Optimizing sample amount to be injected can avoid protein sticking to or clogging of the column matrix. Filters at the top of the column bed and in the FPLC system should be replaced regularly. Inconsistent protein elution profiles or visible column compression can indicate improper care of the column, which may require repacking.
High salt concentrations in SEC running buffer can be hard on the FPLC system. It is important to regularly clean the column and FPLC system lines with 0.5 M sodium hydroxide followed by water until a neutral pH is achieved. The amount of time that salty buffers are in the system should be minimized. Store the system in water if used every day and in 20% ethanol if used infrequently. Consult your FPLC system manual to ensure proper care.
We have monitored column performance by dynamic light scattering (DLS). DLS analysis of SEC fractions measures particle size across SEC elution volumes. Changes to this elution profile can also indicate poor column performance, which can be addressed by cleaning, sample dilution, or repacking the column.
ELISA optimization
We have found ELISAs to be sensitive to protein overexpression. If overloaded, results will be inconsistent with proteins appearing to elute differently in repeated experiments. Oversaturation may be observed as broad, flat elution profiles or high baseline signal in fractions after the end of the column volume. Diluting SEC fractions before adding to ELISAs may reveal signals that are unchanged with decreased sample amounts, which can indicate the signal is at or above the limit of detection for the assay. Optimization at each step of the ELISA is important (Lin et al., 2015).
Antibody specificity is a critical factor in a successful and interpretable ELISA. Antibody performance in other protocols cannot ensure performance in the ELISA. Antibody specificity must be verified carefully using a whole cell lysate not containing the protein of interest. For mammalian cells, this can be achieved through an siRNA knockdown of the endogenous protein. In E. coli experiments, an empty vector control can be used. Many antibodies to mammalian proteins may not be tested for specificity in bacterial lysates and antibody performance may vary when recombinant proteins are expressed in another organism.
Data Analysis
SEC is a technique that separates particles by size and the limit of resolution for SEC is determined by the column matrix. In our experience, the CL2B matrix can resolve from 14 nm to 200 nm (Figure 2B). One of the easiest structures to identify in SEC fractions from cell lysates is the 150 nm nuclear pore complex. Other large protein particles observed in our studies have ranged from 100 to 300 nm in diameter. The volume of granules is mostly water; thus, granules dehydrate and collapse under the vacuum when imaged by TEM. Small proteins in the latest SEC fractions are observed by TEM to be 8–14 nm in diameter (Figure 3). SEC does not discriminate between condensates formed by liquid-liquid phase separation and other types of large protein assemblies. A no-crosslink lysate may help to determine if non-specific aggregation has occurred.
Recombinant expression of proteins in E. coli may allow unnatural condensates, assemblies, or aggregates to form. Overexpression can result in the formation of inclusion bodies which can decrease protein yield and alter protein fold/assembly state. Additionally, high protein concentrations can result in condensate formation that might not naturally occur in native cell environments. Eukaryotic proteins may also be post-translationally modified or fold differently in their native cell which can alter assembly activity in E. coli. Thus, parallel characterizations of a protein in its native cell type can offer an important comparison to use while interpreting key findings.
Understanding Results
After lysate is separated by SEC, many downstream applications can be applied to check for data quality as well as further study large particle assemblies. These approaches can include DLS, electron microscopy, ELISA, mass spectrometry, and immunoprecipitation.
When performing DLS to monitor column performance, several parameters must be monitored. It is important to prevent dust particles from entering samples as these can overwhelm the DLS signal and artificially increase the size readings obtained for your particles. Another key factor is buffer composition. The high amount of urea in SEC Running buffer or highly concentrated samples can cause some interference with DLS readings. Thus, sample dilutions or buffer exchange may be necessary to ensure data quality.
Electron microscopy is another way that particle size can be measured (Kato et al., 2012; Schwartz et al., 2013; Sun et al., 2011; Thompson et al., 2018). This technique is more time intensive but offers the additional benefit that particle morphology can be observed (Figure 3). We have negative stained samples using uranyl acetate for TEM (Thompson et al., 2018). Viewing particles in SEC fractions can verify the size of particles and can reveal if they are the result of non-specific aggregation.
ELISAs are useful for monitoring the elution profile of specific proteins. This targeted approach can confirm if a protein is present in large assemblies and quantify the relative amounts of the protein in large assemblies or smaller complexes and monomers.
Finally, mass spectrometry can be applied to SEC fractions to identify other protein interactors enriched in large versus small protein particles (Lu et al., 2009; Nagaraj et al., 2008). We have successfully performed mass spectrometry to identify proteins in SEC fractions. Mass spectrometry can also identify the interactome of a specific protein immunoprecipitated from pooled SEC fractions. The addition of an immunoprecipitation step following SEC enriches for particles containing a protein of interest from the pool of similarly sized protein assemblies. Interactomes for these large assemblies cannot indicate direct binding between the two proteins, as interactions can be through shared binding partners (Jain et al., 2016; Kawaguchi et al., 2020; Wheeler et al., 2017).
Time Considerations
Preparing cell lysate for column injection takes 2 hours and can be done while the column is equilibrating. Running an SEC column of the size described here generally takes 3 hours due to the low-pressure limit for the Sepharose CL2B matrix. ELISAs typically require a minimum of 1.5 days due to incubation steps. TCA precipitation can be performed in 1 hour.
Acknowledgements
This work was supported by funding from the National Institutes of Health (R21CA238499) and the American Cancer Society (RSG-18-237-01-DMC) to J.C.S. R.A.V. was additionally supported by the Initiative to Maximize Student Diversity/NIH program R25 GM062584 and the Sloan Scholar, Sloan Foundation’s Indigenous Graduate Partnership (SIGP) Program.
Literature Cited
- A P, & Weber SC (2019). Evidence for and against Liquid-Liquid Phase Separation in the Nucleus. Noncoding RNA, 5(4). doi: 10.3390/ncrna5040050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adame-Arana O, Weber CA, Zaburdaev V, Prost J, & Julicher F (2020). Liquid Phase Separation Controlled by pH. Biophys J, 119(8), 1590–1605. doi: 10.1016/j.bpj.2020.07.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, & Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol, 18(5), 285–298. doi: 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, … Young RA (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell, 175(7), 1842–1855 e1816. doi: 10.1016/j.cell.2018.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boing AN, van der Pol E, Grootemaat AE, Coumans FA, Sturk A, & Nieuwland R (2014). Single-step isolation of extracellular vesicles by size-exclusion chromatography. J Extracell Vesicles, 3. doi: 10.3402/jev.v3.23430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case LB, Zhang X, Ditlev JA, & Rosen MK (2019). Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science, 363(6431), 1093–1097. doi: 10.1126/science.aau6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos AV, McDonald CJ, & Elbaum-Garfinkle S (2018). Methods and Strategies to Quantify Phase Separation of Disordered Proteins. Methods Enzymol, 611, 31–50. doi: 10.1016/bs.mie.2018.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, … Tjian R (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. doi: 10.1126/science.aar2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F, Doll S, Bech JM, Schweizer L, Mund A, Lengyel E, … Mann M (2020). A streamlined mass spectrometry-based proteomics workflow for large-scale FFPE tissue analysis. J Pathol, 251(1), 100–112. doi: 10.1002/path.5420 [DOI] [PubMed] [Google Scholar]
- Francis DM, & Page R (2010). Strategies to optimize protein expression in E. coli. Curr Protoc Protein Sci, Chapter 5, Unit 5 24 21–29. doi: 10.1002/0471140864.ps0524s61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, … McKnight SL (2012). Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell, 149(4), 768–779. doi: 10.1016/j.cell.2012.04.016 [DOI] [PubMed] [Google Scholar]
- Hoffman EA, Frey BL, Smith LM, & Auble DT (2015). Formaldehyde crosslinking: a tool for the study of chromatin complexes. J Biol Chem, 290(44), 26404–26411. doi: 10.1074/jbc.R115.651679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, … Dormann D (2018). Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell, 173(3), 706–719 e713. doi: 10.1016/j.cell.2018.03.004 [DOI] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, & Parker R (2016). ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell, 164(3), 487–498. doi: 10.1016/j.cell.2015.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, … McKnight SL (2012). Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell, 149(4), 753–767. doi: 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, & McKnight SL (2017). A Solid-State Conceptualization of Information Transfer from Gene to Message to Protein. Annu Rev Biochem. doi: 10.1146/annurev-biochem-061516-044700 [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Rollins MG, Moinpour M, Morera AA, Ebmeier CC, Old WM, & Schwartz JC (2020). Changes to the TDP-43 and FUS Interactomes Induced by DNA Damage. J Proteome Res, 19(1), 360–370. doi: 10.1021/acs.jproteome.9b00575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Boija A, Afeyan LK, Hawken SW, Fan M, Dall’Agnese A, … Young RA (2020). Partitioning of cancer therapeutics in nuclear condensates. Science, 368(6497), 1386–1392. doi: 10.1126/science.aaz4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, Mateju D, Malinovska L, Nuske E, Poser I, … Alberti S (2015). Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife, 4, e06807. doi: 10.7554/eLife.06807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladouceur AM, Parmar BS, Biedzinski S, Wall J, Tope SG, Cohn D, … Weber SC (2020). Clusters of bacterial RNA polymerase are biomolecular condensates that assemble through liquid-liquid phase separation. Proc Natl Acad Sci U S A, 117(31), 18540–18549. doi: 10.1073/pnas.2005019117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Collins M, An J, Geiser R, Tegeler T, Tsantilas K, … Bowser R (2016). Immunoprecipitation and mass spectrometry defines an extensive RBM45 protein-protein interaction network. Brain Res, 1647, 79–93. doi: 10.1016/j.brainres.2016.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZF, & Lam YW (2015). A new rapid method for isolating nucleoli. Methods Mol Biol, 1228, 35–42. doi: 10.1007/978-1-4939-1680-1_4 [DOI] [PubMed] [Google Scholar]
- Lin Y, Protter DS, Rosen MK, & Parker R (2015). Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell, 60(2), 208–219. doi: 10.1016/j.molcel.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb RJ, Lima LG, & Moller A (2017). Exosomes: Key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev Biol, 67, 3–10. doi: 10.1016/j.semcdb.2017.01.004 [DOI] [PubMed] [Google Scholar]
- Lu A, Wisniewski JR, & Mann M (2009). Comparative proteomic profiling of membrane proteins in rat cerebellum, spinal cord, and sciatic nerve. J Proteome Res, 8(5), 2418–2425. doi: 10.1021/pr8010364 [DOI] [PubMed] [Google Scholar]
- Mao YS, Zhang B, & Spector DL (2011). Biogenesis and function of nuclear bodies. Trends Genet, 27(8), 295–306. doi:S0168–9525(11)00076-X [pii] 10.1016/j.tig.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwiggen DT, Mir M, Darzacq X, & Tjian R (2019). Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev, 33(23–24), 1619–1634. doi: 10.1101/gad.331520.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milkovic NM, & Mittag T (2020). Determination of Protein Phase Diagrams by Centrifugation. Methods Mol Biol, 2141, 685–702. doi: 10.1007/978-1-0716-0524-0_35 [DOI] [PubMed] [Google Scholar]
- Mitrea DM, & Kriwacki RW (2016). Phase separation in biology; functional organization of a higher order. Cell Commun Signal, 14, 1. doi: 10.1186/s12964-015-0125-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, … Taylor JP (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell, 163(1), 123–133. doi: 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Hayashi Y, Onishi T, Sakai M, & Takai K (1974). Rapid isolation of nucleoli from detergent purified nuclei of various tumor and tissue culture cells. Exp Cell Res, 88(2), 245–251. doi: 10.1016/0014-4827(74)90250-x [DOI] [PubMed] [Google Scholar]
- Murthy AC, & Fawzi NL (2020). The (un)structural biology of biomolecular liquid-liquid phase separation using NMR spectroscopy. J Biol Chem, 295(8), 2375–2384. doi: 10.1074/jbc.REV119.009847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N, Lu A, Mann M, & Wisniewski JR (2008). Detergent-based but gel-free method allows identification of several hundred membrane proteins in single LC-MS runs. J Proteome Res, 7(11), 5028–5032. doi: 10.1021/pr800412j [DOI] [PubMed] [Google Scholar]
- Peran I, & Mittag T (2020). Molecular structure in biomolecular condensates. Curr Opin Struct Biol, 60, 17–26. doi: 10.1016/j.sbi.2019.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, … St George-Hyslop P (2018). FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell, 173(3), 720–734 e715. doi: 10.1016/j.cell.2018.03.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback JA, Zhu L, Ferrolino MC, Tolbert M, Mitrea DM, Sanders DW, … Brangwynne CP (2020). Composition-dependent thermodynamics of intracellular phase separation. Nature, 581(7807), 209–214. doi: 10.1038/s41586-020-2256-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kedersha N, Lee DSW, Strom AR, Drake V, Riback JA, … Brangwynne CP (2020). Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell, 181(2), 306–324 e328. doi: 10.1016/j.cell.2020.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, & Cech TR (2013). RNA seeds higher-order assembly of FUS protein. Cell Rep, 5(4), 918–925. doi: 10.1016/j.celrep.2013.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A (2006). Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev, 25(4), 663–682. doi: 10.1002/mas.20082 [DOI] [PubMed] [Google Scholar]
- Souquere S, Mollet S, Kress M, Dautry F, Pierron G, & Weil D (2009). Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J Cell Sci, 122(Pt 20), 3619–3626. doi: 10.1242/jcs.054437 [DOI] [PubMed] [Google Scholar]
- Springhower CE, Rosen MK, & Chook YM (2020). Karyopherins and condensates. Curr Opin Cell Biol, 64, 112–123. doi: 10.1016/j.ceb.2020.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, & Gitler AD (2011). Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol, 9(4), e1000614. doi: 10.1371/journal.pbio.1000614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland BW, Toews J, & Kast J (2008). Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J Mass Spectrom, 43(6), 699–715. doi: 10.1002/jms.1415 [DOI] [PubMed] [Google Scholar]
- Taylor NO, Wei MT, Stone HA, & Brangwynne CP (2019). Quantifying Dynamics in Phase-Separated Condensates Using Fluorescence Recovery after Photobleaching. Biophys J, 117(7), 1285–1300. doi: 10.1016/j.bpj.2019.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayri-Wilk T, Slavin M, Zamel J, Blass A, Cohen S, Motzik A, … Kalisman N (2020). Mass spectrometry reveals the chemistry of formaldehyde cross-linking in structured proteins. Nat Commun, 11(1), 3128. doi: 10.1038/s41467-020-16935-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson VF, Victor RA, Morera AA, Moinpour M, Liu MN, Kisiel CC, … Schwartz JC (2018). Transcription-Dependent Formation of Nuclear Granules Containing FUS and RNA Pol II. Biochemistry, 57(51), 7021–7032. doi: 10.1021/acs.biochem.8b01097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toews J, Rogalski JC, Clark TJ, & Kast J (2008). Mass spectrometric identification of formaldehyde-induced peptide modifications under in vivo protein cross-linking conditions. Anal Chim Acta, 618(2), 168–183. doi: 10.1016/j.aca.2008.04.049 [DOI] [PubMed] [Google Scholar]
- Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, … Hyman AA (2018). A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell, 174(3), 688–699 e616. doi: 10.1016/j.cell.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JR, Jain S, Khong A, & Parker R (2017). Isolation of yeast and mammalian stress granule cores. Methods. doi: 10.1016/j.ymeth.2017.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JR, Matheny T, Jain S, Abrisch R, & Parker R (2016). Distinct stages in stress granule assembly and disassembly. Elife, 5. doi: 10.7554/eLife.18413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierer M, & Mann M (2016). Proteomics to study DNA-bound and chromatin-associated gene regulatory complexes. Hum Mol Genet, 25(R2), R106–R114. doi: 10.1093/hmg/ddw208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LE, Kim TH, Muhandiram DR, Forman-Kay JD, & Kay LE (2020). NMR Experiments for Studies of Dilute and Condensed Protein Phases: Application to the Phase-Separating Protein CAPRIN1. J Am Chem Soc, 142(5), 2471–2489. doi: 10.1021/jacs.9b12208 [DOI] [PubMed] [Google Scholar]
- Wu G, Wang S, Tian Z, Zhang N, Sheng H, Dai W, & Qian F (2017). Elucidating the weak protein-protein interaction mechanisms behind the liquid-liquid phase separation of a mAb solution by different types of additives. Eur J Pharm Biopharm, 120, 1–8. doi: 10.1016/j.ejpb.2017.07.012 [DOI] [PubMed] [Google Scholar]
- Wu H, & Fuxreiter M (2016). The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell, 165(5), 1055–1066. doi: 10.1016/j.cell.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbinden A, Perez-Berlanga M, De Rossi P, & Polymenidou M (2020). Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev Cell, 55(1), 45–68. doi: 10.1016/j.devcel.2020.09.014 [DOI] [PubMed] [Google Scholar]