

Abstract
Redirection of T cell cytotoxicity by the CAR structure may not be sufficient for optimal anti-tumor function in the patient tumor microenvironment. Co-modifying CAR T cells to secrete different classes of proteins can be used to optimize CAR T cell function, overcome suppressive signals and/or alter the tumor microenvironment milieu. These modifications aim to improve initial responses to therapy and enhance the durability of response. Furthermore, CAR T cells can deliver these molecules locally to the tumor microenvironment, avoiding systemic distribution. This approach has been tested in preclinical models using a variety of different classes of agonistic and antagonistic proteins and clinical trials are currently underway to assess efficacy in patients.
Keywords: Adoptive cell therapy, CAR T cells, immunotherapy, cellular immunotherapy, tumor microenvironment
Introduction
CAR T cells have demonstrated impressive clinical response against certain types of leukemia and lymphoma1,2. However, despite the high rates of complete response induced by CAR T cells in these malignancies, obstacles remain to improve and expand CAR T cell efficacy to other cancer types, overcome toxicities related to therapy, and increase the durability response1,3. Many of these challenges to effective therapy can be addressed by co-modifying CAR T cells to secrete different classes of proteins. These modifications allow CAR T cells to be morphed into living “micropharmacies” capable of producing proteins that, when secreted, can work in an autocrine or paracrine manner to improve CAR T cell phenotypes and/or function in the tumor microenvironment (TME)4. Expression of genes alongside the CAR construct allows for the expression of proteins, even those not naturally made by T cells. Also referred to as “armored” CAR T cells, these cells can secrete proteins that antagonize suppressive signals in the TME. Alternatively, armored CAR T cells can secrete agonists that can activate and improve of the function of CAR T cells or other proinflammatory cells in the TME.
Localized secretion of proteins by CAR T cells at the tumor site is a major advantage associated with this approach. Systemic infusion of proinflammatory molecules can lead to significant toxicities. CAR T cells are engineered to traffic to and proliferate at site of tumor and result in localized delivery of these immunomodulatory agents to the TME.
As a mode of drug delivery, CAR T cells have the ideal physiological functions and pharmacokinetics. These cells precisely localize to the tumor area, activate, and then expand and contract with tumor burden. Proteins engineered to be secreted by CAR T cells are delivered to the tumor area in concentrations correlated with tumor size. These cells also have structurally large volume/surface area, allowing for the expression of more therapeutic cargo compared to exosomes or nanoparticles4. Therefore, CAR T cell-mediated drug delivery of therapeutic agents allows for potentially cheaper and more efficacious combination immunotherapies.
Diverse classes of proteins can be secreted by armored CAR T cells for a variety of functions. This review discusses the different classes and their intended mechanism to modulate the TME.
Cytokines
Inhibitory signals in the TME create immunosuppressive conditions which arrest endogenous immune functions. Armored CAR T cells co-modified to secrete cytokines have demonstrated promise in preclinical studies in hematological and solid tumors. CAR T cells expressing cytokines are also referred to as T cells redirected for universal cytokine killing (TRUCKS)5. The cytokines secreted by these cells are usually those not endogenously expressed by T cells but are required by T cells for optimal function. These cytokines can stimulate the CAR T cells that secrete them in an autocrine fashion, leading to enhanced T cell proliferation, survival, and anti-tumor responses. The localized delivery of the immunostimulatory cytokines by CAR T cells can also promote the recruitment of proinflammatory endogenous immune cells that can aid in combating antigen escape to CAR T cell therapy (Figure 1).
Figure 1. Localized delivery of secreted proteins by CAR T cells can alter the tumor microenvironment.
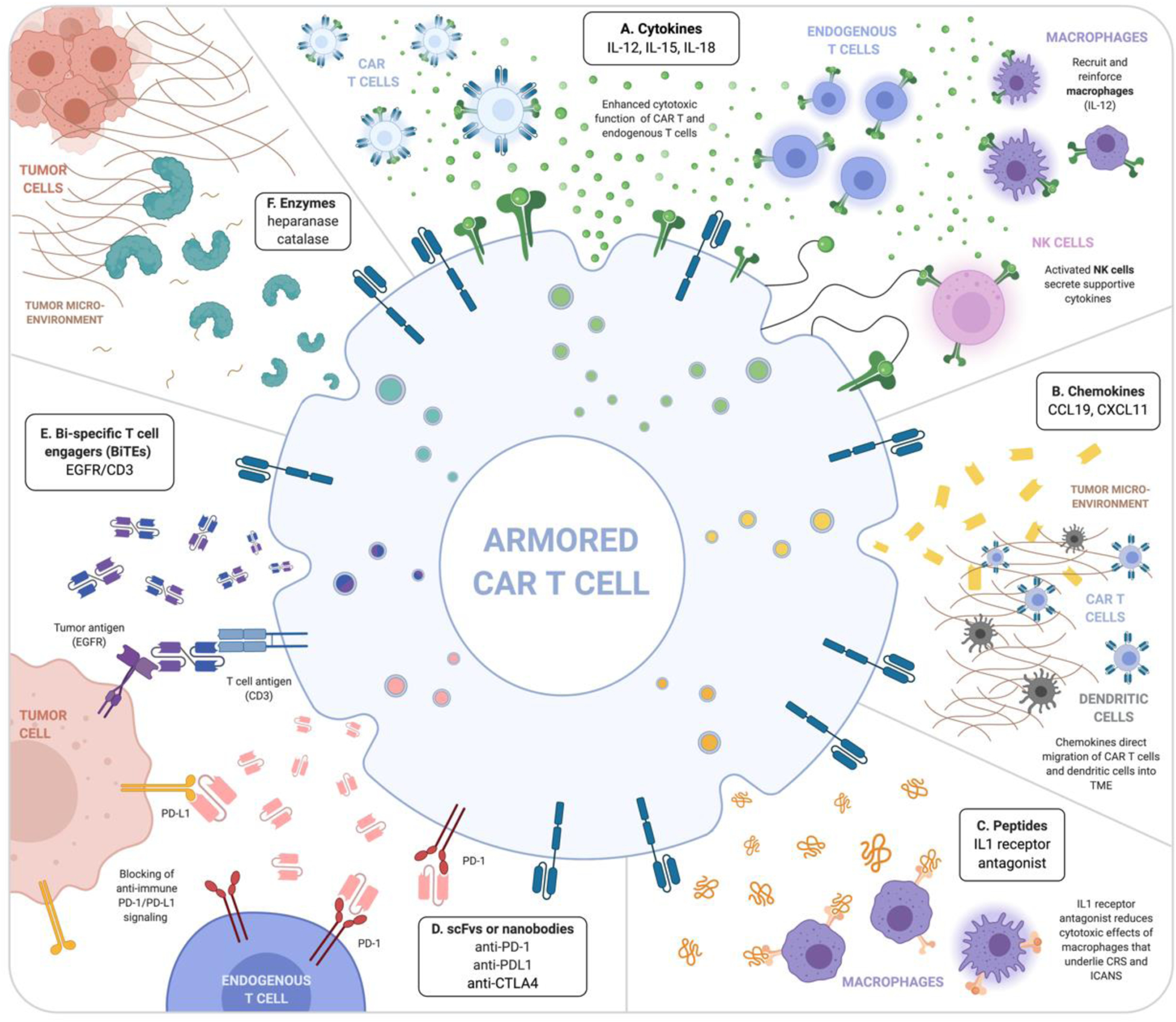
A) Cytokines (i.e. IL-12, IL-15, IL-18) enhance self and other CAR T cell function as well as endogenous T cells, NK cells, and macrophages. B) Chemokines alone or in conjunction with cytokines can direct migration of CAR T cells and dendritic cells into the tumor microenvironment (TME). C) Peptides, such as IL-1 receptor antagonist, can work to inhibit undesirable cytotoxic effects from macrophages that mediate life-threatening complications such as CRS and ICANS. Antibody-based molecules such as D) scFvs and nanobodies can target and inhibit anti-immune surface signaling molecules, such as PD-1or PD-L1. E) Bi-specific T cell engagers (BiTEs) bind a T cell antigen, often CD3, and a tumor antigen (i.e. EGFR) to bring the cells into close proximity to enhance effector function. F) Enzymes, such as heparanase and catalase, digest extracellular matrix present in the TME to allow accessibility to the tumor cells. Created with BioRender.com.
IL-12
Interleukin (IL)-12 is a pleiotropic, pro-inflammatory cytokine produced by dendritic cell, macrophages, neutrophils and B cells and plays a vital role in the immune response to microbial pathogens through T and NK cell-mediated immunity6. In addition to increasing the cytotoxic and cytokine producing capacity of T and NK cells, IL-12 can inhibit or reprogram suppressive cells in the TME6. While IL-12 has demonstrated potent anti-tumor effects in preclinical models of cancer, systemic infusion of recombinant IL-12 in patients has lethal dose-limiting toxicities and minimal efficacy at tolerated doses7,8,9. Localized delivery of the IL-12 to the TME by CAR T cells is a promising approach as it may allow for the effective anti-tumor effects of IL-12 therapy but with a reduction in systemic exposure and toxicities. Furthermore, IL-12 expression by armored CAR T cells can be engineered to secrete inducible or lower levels of IL-12, further decreasing the risk of systemic toxicities10,11. Localized IL-12 secretion by CAR T cells has been demonstrated to work in an autocrine manner to enhance CAR T cell function and in a paracrine manner to enhance activation of local immune cells while reducing immunosuppression in the TME.
CAR T cells that secrete IL-12 benefit from this modification in a number of ways. In a “conditioning-free” syngeneic model of CAR T cell therapy, armored anti-CD19 CAR T cells that constitutively express IL-12 demonstrated increased cytotoxicity against tumor cells compared to parental CAR T cells through increased expression of interferon gamma (IFN-γ, granzyme B, and perforin12. These CAR T cells also exhibited enhanced expansion and resistance to regulatory T cell (Treg)-mediated suppression12. In vivo, IL-12-secreting armored CAR T cells had superior engraftment and eradicated established systemic hCD19+ tumor cells12. In this model, autocrine IL-12 stimulation of both CD4+ and CD8+ CAR T cells was needed for the elimination of tumor. Subsequent studies of IL-12-sereting CAR T cells in a solid tumor model further demonstrated enhanced CAR T cell proliferation and persistence, as well as higher systemic IFN-γ levels and increased anti-tumor efficacy in tumor-bearing mice as compared to parental CAR T cells13,14. Furthermore, the IL-12 armored CAR T cell were more resistant to programmed death receptor ligand 1 (PD-L1) induced dysfunction14 and exhibited an enhanced pro-inflammatory gene expression profile15. Dramatic expansion of CAR T cells after IL-12 expression led to the ability to generate CAR T cells from small numbers of cells isolated from umbilical cord blood samples16. These armored CAR T cells retained a unique central-memory effector phenotype, exhibited increased anti-tumor efficacy in vitro, and significantly enhanced the survival of CD19+ tumor-bearing mice16.
IL-12 secreted by CAR T cells can also enhance the proliferation of endogenous T cells in the TME. In a xenograft mouse model of gastric cancer, CAR T cells directed against ICAM-1 were co-modified to express IL-12 downstream of the NF-κB-NFAT promoter17. Inducible IL-12 expression by CAR T cells resulted in higher levels of IFN-γ and TNFα and enhanced reduction of tumor burden as compared to parental CAR T cells. Analysis of blood from these mice revealed significant expansion of CD3+ T cells in sample groups treated with IL-12-secreting CAR T cells as compared to parental CAR T cells17. Interestingly, this expansion was not limited to transduced CAR T cells, as the total percentage of T cells was increased in these mice, suggesting paracrine effects of IL-12 on all T cells17.
IL-12 locally secreted by CAR T cells can have effects on the cells of the TME. In syngeneic mouse models of ovarian carcinoma, IL-12 secreting armored CAR T cells mediated the depletion of tumor associated macrophages14. Carcinoembryonic antigen 18-targeting CAR T cells modified to secrete IL-12 demonstrated increased penetration of metastatic tumors such as brain and inner organ metastases. These cells recruited and activated innate immune cells such as macrophages in mice xenografted with CEA-positive colon cancer cell lines19. In this model, macrophage numbers increased after treatment and were shown to be critical in the anti-tumor response against antigen positive and negative tumors19. IL-12 can also decrease or reprogram suppressive cells in the TME. VEGFR-2-targeted CAR T cells secreting IL-12 reduced immunosuppression in the TME by decreasing myeloid suppressor cell subsets expressing VEGFR-220. These armored CAR T cells penetrated solid tumors, proliferated, and persisted for extended periods in multiple mouse models of well-vascularized solid cancers and increased the anti-tumor effects of endogenous immune cells expressing IL-12 receptors20. Finally, Glypcian-3-specific T cells modified to express IL-12 in an inducible manner had superior anti-tumor efficacy in immunocompetent hosts without the need for preconditioning and decreased infiltration of Treg in established tumors21.
The success of these preclinical studies has led to the clinical translation of IL-12 secreting CAR T cells (Table 1) and supported the commencement of clinical trials testing the efficacy of MUC16ecto-targeting, IL-12 armored CAR T cells in patients with ovarian cancer18 (NCT02498912). Another study is testing Nectin/FAP-targeted CAR T cells expressing IL-7, CCL19, or IL-12 in Nectin4-positive advanced malignant solid tumors (NCT03932565). It is important to note that given the toxicities associated with prior IL-12 therapies, the clinical translation of these CAR T cells is not without danger. For example, a clinical trial testing localized delivery of IL-12 by tumor-infiltrating lymphocytes in patients with metastatic melanoma demonstrated liver dysfunction, high fevers, and life-threatening hemodynamic instability with increasing cell doses11. While this trial did not specifically use CAR T cells, it suggests that researchers still need to proceed with caution when delivering IL-12 locally to the TME.
Table 1.
Clinical trial currently open with CAR T cells modified to locally secrete proteins.
| Armor | Target antigen | Disease | Clinical Trial ID, Phase | Sponsor | |
|---|---|---|---|---|---|
| CYTOKINES | IL-12 | MUC16ecto | Solid Tumors | NCT02498912, Phase 1 | Memorial Sloan Kettering Cancer Center |
| EGFR | Metastatic Colorectal Cancer | NCT03542799, Phase 1 | Shenzhen Second People’s Hospital | ||
| IL-15 | GD2 | Neuroblastoma | NCT03721068, Phase 1 | UNC Lineberger Comprehensive Cancer Center | |
| GD2 | Neuroblastoma | NCT03294954, Phase 1 | Baylor College of Medicine | ||
| GPC3 | Liver Cancer, Rhabdomyosarcoma, Malignant Rhabdoid Tumor, Liposarcoma, Wilms Tumor, Yolk Sac Tumor | NCT04377932, Phase 1 | Baylor College of Medicine | ||
| mbIL-15 | CD19 | Lymphomas and Leukemias | NCT03579888, Phase 1 | M.D. Anderson Cancer Center | |
| ANTIBODY-BASED MOLECULES | PD-1 | Mesothelin | Advanced Solid Tumor | NCT03615313, Phase 1/2 | Shanghai Cell Therapy Research Institute |
| CD19 | Lymphoma, B-Cell | NCT04163302, Phase 2 | Chinese PLA General Hospital | ||
| BCMA | Multiple Myeloma | NCT04162119, Phase 2 | Chinese PLA General Hospital | ||
| Mesothelin | Colorectal, Ovarian cancer | NCT04503980, Phase 1 | Shanghai Cell Therapy Group Co., Ltd | ||
| Mesothelin | Non-small-cell Lung Cancer, Mesothelioma | NCT04489862, Phase 1 | Wuhan Union Hospital, China | ||
| PD-L1 | CD22 | Solid Tumor, Cervical Cancer, Sarcoma, NSCLC | NCT04556669, Phase 1 | Hebei Senlang Biotechnology Inc., Ltd. | |
| PD-1 or PD-L1 | EGFRviii | Glioblastoma Multiforme of Brain | NCT03170141, Phase 1 | Shenzhen Geno-Immune Medical Institute | |
| Sarcoma, Osteoid Sarcoma, Ewing Sarcoma | NCT03356782, Phase 1/2 | Shenzhen Geno-Immune Medical Institute | |||
| COMBINATIONS | IL-7/CCL19 | CD138, Integrin B7, CS1, CD38, BCMA | Refractory/recurrent multiple myeloma | NCT03778346, Phase 1 | The Sixth Affiliated Hospital of Wenzhou Medical University |
| CD19 | B Cell lymphoma | NCT03929107, Phase 2 | Wenbin Qian | ||
| CD19 | Diffuse Large B-cell Lymphoma | NCT04381741, Phase 1 | Second Affiliated Hospital, School of Medicine, Zhejiang University | ||
| IL-7/CCL19 or IL-12 | Nectin4 | Nectin4+ Advanced Malignant Solid Tumor | NCT03932565, Phase 1 | The Sixth Affiliated Hospital of Wenzhou Medical University | |
| IL-7/CCL19 +/− PD-1, CTLA4, Tigit | GPC3, TGFβ | Hepatocellular Carcinoma, Squamous Cell Lung Cancer | NCT03198546, Phase 1 | Second Affiliated Hospital of Guangzhou Medical University |
All trials accessed at clinicaltrials.gov. CAR, chimeric antigen receptor; MUC16ecto, glycosylated mucin 16 extracellular portion; EGFR, epidermal growth factor receptor; GD2, disialoganglioside; mb, membrane-bound; GPC3, glypican-3; PD-1, programmed cell death protein 1; BCMA, B-cell maturation antigen; PD-L1, programmed death-ligand 1; NSCLC, non-small cell lung cancer; CCL19, chemokine ligand 19; TGFβ, transforming growth factor beta; CTLA4, cytotoxic T lymphocyte associated protein 4; BiTE, bispecific T cell engager;
IL-15
IL-15 is a pleiotropic cytokine produced mainly by monocytes, macrophages and dendritic cells that plays a role in innate and adaptive immunity22. IL-15 signaling in T cells and NK cells results in differentiation, lymphocyte homeostasis, and survival22. Preclinical research in murine and rhesus macaque models suggested an anti-tumor role for IL-15. In clinical trials, recombinant IL-15 therapy in patients with hematologic and solid tumors led to significant increases in circulating T cells and NK cells but did not translate to clinical efficacy23. Furthermore, high doses of IL-15 are associated with toxicities such as thrombocytopenia and hypotension22. Similar to the rational for armored CAR T cells secreting IL-12, CAR T cells secreting IL-15 are intended to deliver potent signaling locally to the TME.
IL-15 leads to robust expansion of T cells. This phenotype has been observed in CAR T cells that have been modified to secrete IL-15. CD19-targeting CAR T cells modified to secrete IL-15 demonstrated a 10-fold expansion post antigen stimulation in vitro and up to a 15-fold increase in in vivo expansion in SCID-lymphoma xenograft mouse models when compared to parental CD19-targeting CAR T cells24. These CAR T cells exhibited reduced cell death, lower PD-1 receptor expression, and increased anti-tumor efficacy24,25. In orthotopic glioma and metastatic neuroblastoma xenografted mouse models, IL-13Ra2-targeting or GD2-targeting CAR T cells modified to secrete IL-15 demonstrated enhanced survival and anti-tumor activity26,27. A CLL-1-targeting, IL-15 armored CAR T cell was less terminally differentiated and had increased expansion of AML cell lines in vitro28.
IL-15 expression by CAR T cells has led to severe acute toxicity in preclinical models. For example, treatment with CAR T cells modified to secrete IL-15 led to acute toxicities related to high levels of TNFα, IL-15, and IL-2 in xenograft mouse models of aggressive AML28. These toxicities were managed successfully with anti-TNFα antibody treatment and an inducible caspase-9 safety switch to arrest T cell expansion. These strategies led to an increase in tumor-free survival of mice treated with IL-15-secreting CAR T cells28.
In addition to toxicities associated with treatment, uncontrolled proliferation is a major concern with armored CAR T cells constitutively expressing IL-15. Logarithmic growth and persistence in the absence of exogenous cytokine support have been observed in mature human T lymphocytes following transduction with a retroviral vector to promote IL-15 expression29. Additionally, transgenic mouse models engineered to over-express IL-15 exhibited proliferation of NK and CD8+ T lymphocytes that led to the development of leukemia30.
Given the association of constitutive IL-15 expression with uncontrolled proliferation and toxicity, researchers have developed methods to provide IL-15 signaling to CAR T cells without effecting other cell types. T cells engineered to co-express a CD19-targeting CAR and a membrane-bound chimeric IL-15 exhibited improved persistence and anti-tumor activity in xenograft leukemia mouse models and retained a phenotype with similarities to T memory stem cells31. Furthermore, engineering a CD19-targeting CAR T cell with an IL-15 receptor signaling domain fused with the CD28 co-stimulatory domain resulted in increased rates of proliferation cytotoxic activity and effector cytokine secretion as well as causing complete remission of one ALL patient after treatment with a CAR T cell infusion32. Finally, preclinical studies with nanogel engineering strategies have successfully loaded large dosages of IL-15 onto CAR T cells for tumor site delivery without systemic toxicities33.
Clinical use of IL-15 secreting CAR cells has been limited to CAR-expressing NK cell or NKT cells. A clinical trial using cord blood-derived NK cells modified to express CD19-targeted CAR and secrete IL-15 in patients with CD19+ lymphoid tumors demonstrated a 64% complete response rate without major toxic effects34. Another trial tested the safety and anti-tumor response of GD2-specific NKT cells modified to secrete IL-15 in children with relapsed or resistant neuroblastoma35. No dose-limiting toxicities occurred in this trial and the armored CAR NKT cells induced an objective response in 1 out of the 3 patients treated. Current ongoing clinical trials are testing the efficacy of IL-15 expressing conventional CAR T cells (Table 1).
IL-18
IL-18 is a proinflammatory cytokines released from epithelial cells or macrophages that facilitates IFN-γ production, contributes to multiple immune regulatory effects on T cells, and can alter the phenotypes of T cells and NK cells36. IL-18 has been shown to stimulate both innate and adaptive immunity alone or in conjunction with other cytokines such as IL-12 and IL-1336. IL-18 has demonstrated antitumor activity in preclinical models of cancer36. However, pro-IL-18 is required to be cleaved by proteases in order to converted into the active form of cytokine36. IL-18 binding protein (IL-18 BP) is a soluble factor found in the TME that can inhibit the function of IL-1837. These factors may play a role in why systemic infusion of recombinant IL-18 led to no major toxicities or efficacy38,39.
CAR T cells modified to secrete IL-18 exhibit increased expansion, persistence, and survival in mouse models of hematological and solid malignancies40,41,42. These armored CAR T cells can also alter the inhibitory conditions of the TME and recruit endogenous anti-tumor immune cells. This includes recruitment of endogenous immune cells to the TME and suppression of Treg cells and anti-inflammatory M2-polarized macrophages40,41,42. Further engineering of IL-18 has resulted in a form of IL-18 resistant to IL-18BP binding37, and if secreted by CAR T cells may be more effective in the TME.
However, as IL-18 may play a pathogenic role in autoimmune diseases43,44, caution is warranted with clinical translation of IL-18-secreting CAR T cells.
Other cytokines
Armored CAR T cells that secrete IL-21, IL-7, IL-33, or IL-36γ have demonstrated preclinical efficacy. IL-21 is mainly secreted by activated CD4+ T cells and NK T cells and has broad proinflammatory effects on the immune cells that express IL-21 Receptor45. These include B, T, and NK cells, dendritic cells, and macrophages. IL-21-secreting CAR T cells were superior to CAR T cells modified to express IL-2 or IL-15 in clearing systemic in a xenograft model of lymphoma46. These armored CAR T cells had effector memory phenotypes and long term persistence in mice. In a separate model of solid and hematologic malignancies, IL-21 armored CAR T cells exhibited increased CAR T cell expansion and reduced apoptosis in vitro as well as increased tumor infiltration and tumor growth reduction in vivo46.
IL-7 is mainly produced by epithelial and endothelial cells and is critical for every stage of T cell development47. Similar to IL-21-secreting CAR T cells, CAR T cells modified to constitutively secrete IL-7 were superior than IL-2 or IL-15 secreting CAR T cells in eradicating lymphoma in xenograft mice45. These armored CAR T cells demonstrated long term persistence and central memory phenotype. A separate study engineered CAR T cells to express both IL-7 and the chemokine CCL19 to mimic the function of T-zone reticular cells and recruit T cells and dendritic cells to the TME. These armored CAR T cells increased the numbers of endogenous T cells and dendritic cells in the TME and lead to a regression of pre-established solid tumors in mice48. This was due to a collaborative anti-tumor effort by these cells that resulted in memory responses.
Similar to IL-18, IL-33 and IL-36 are members of the IL-1 superfamily of cytokines that pay a role in immune regulation. Damaged tissue cells and activated immune cells release IL-33 to trigger inflammation and cell-mediated responses in damaged tissues49. Tumor cells down-regulate IL-33, suggesting a role for IL-33 in the inhibition of tumor progression or immune avoidance50. In preclinical models, IL-33-secreting armored CAR T cell have been used to deliver this cytokine to the TME50. Similar to IL-33, IL-36 is produced by epithelial cells in response to tissue damage. It binds to epithelial cells as well as dendritic cells, monocytes, neutrophils and T cells to initiate and amplify an immune response51. Armored CAR T cells modified to secrete IL-36γ exhibit significantly enhanced CAR T cell expansion, persistence, and anti-tumor function in vivo, as compared to second generation CAR T cells51. IL-36γ delivered to the TME also activated endogenous antigen presenting cells and T cells to mediate a secondary anti-tumor response.
Chemokines
Chemokine proteins are the largest subfamily of cytokines and are divided into four highly conserved groups: C, CC, CXC, and CX3C. Produced by stromal, tumor or immune cells, chemokines function as signals for immune cell trafficking52. Chemokines play an important role in the TME by regulating tumor cancer progression and metastasis53. Strategies to equip CAR T cells to express chemokines in the TME aim to enhance immune cell homing and anti-tumor function (Figure 1).
As mentioned in the previous section, Adachi et al. modified CAR T cells to secrete IL-7 and CCL19 in order to mimic the function of T-zone reticular cells48. CCL19 is highly expressed in the thymus and lymph nodes and functions to attract dendritic cells. In this preclinical model, the armored CAR T cells indeed recruited endogenous dendritic cells and T cells to the TME, which resulted in superior anti-tumor efficacy48.
CXCL11 expression in leukocytes, endothelial cells, and other cell types is induced by IFNγ54. CXCL11 is chemotactic for activated T cells. Intratumoral delivery of CXCL11 with oncolytic vaccinia virus along with CAR T cell therapy demonstrated potent anti-tumor efficacy and enhanced T cell infiltration in a mouse xenograft model55. However, CAR T cells modified to express CXCL11 in the TME did not demonstrate improved T cell trafficking to the tumor55. While both approaches resulted in increased levels of CXCL11 at the tumor site, the oncolytic virus strategy may prime the TME to be a more favorable environment for CAR T cells and enhance antitumor potential55,56,57. Furthermore, endogenous chemokine receptors on CAR T cells may act as a sink for the expressed chemokine.
A complementary strategy to chemokine expression by CAR T cells is the surface expression of chemokine receptors to enhance T cell migration to the tumor site. While this approach does not result in a secreted protein, it has demonstrated enhanced CAR T cell migration to the TME in preclinical models58. Clinically, armored CAR T cells that express combinations of chemokines and cytokines are currently being tested for safety in phase I trials (Table 1).
Antibodies
While antibodies are naturally produced by B cells, recent advances in technology have led to the engineering and characterization of smaller antibody-like molecules that retain similar functions as antibodies but can be produced by different cell types. These molecules include single chain variable fragments (scFv), nanobodies, and BiSpecific T cell Engagers (BiTE). When expressed by armored CAR T cells these molecules can function as agonists or antagonist in the TME and can also broaden target recognition (Figure 1).
Secretion of Antibody-based molecules to overcome suppressive TME
Antibodies that block the interaction of suppressive ligands on tumors, such as PD-L1, and inhibitory receptors on T cells, such as programmed death receptor (PD-1), are broadly referred to as immune checkpoint inhibitors. Engineering CAR T cells to secrete these inhibiting antibodies locally in the TME is an attractive approach as it overcomes the hurdle of tumor site localization and may decrease the toxicities associated with systemic administration. Furthermore, these agents can enhance the efficacy of CAR T cells against immunosuppressive tumors59. One example of this platform is an armored CAR T cell modified to secrete PD-1-blocking scFv that demonstrated efficacy against PD-L1+ tumors in syngeneic and xenogeneic mouse models of leukemia and ovarian cancer60. This approach demonstrated that checkpoint inhibiting scFv secreted by CAR T cells function in an autocrine manner to prevent suppression of CAR T cells, as well as a paracrine manner to block suppression of endogenous bystander T cells. Finally, localized delivery of checkpoint blockade to the TME by CAR T cells was demonstrated, as no PD-1-blocking scFv was detected systemically in tumor-bearing mice treated with scFv-secreting CAR T cells60.
Additional studies utilizing armored CAR T cells secreting checkpoint inhibitors to PD-1 mainly focused on testing this approach in xenograft models and the autocrine function of this strategy. Anti-CD19 CAR T cells modified to secrete anti-PD-1 nanobodies produced more inflammatory cytokines after antigen-dependent stimulation in artificial CD19-expressing solid tumor cells61. These armored CAR T cells exhibited enhanced antitumor activity in a xenograft mouse model. Recently, Zhou et al. showed that the PD-1 blocking enhanced the long-term cytotoxic capacity of EGFR-targeting CAR T cells in vitro against gastric cancer cells and significantly improved anti-tumor efficacy in a xenograft mouse model.62. Similar results of enhanced antitumor efficacy were also demonstrated with anti-mesothelin CAR T cells modified to secrete anti-PD-1 scFv63.
Suarez et al. adopted a slightly different strategy to develop CAR T cells expressing checkpoint inhibitors by targeting PD-L1 on tumor cells. This strategy has the advantage of blocking inhibition of any effector cells in the TME. Anti-carbonic anhydrase IX (CAIX)-directed CAR T cells modified to secrete anti-PD-L1 antibodies demonstrated reduced tumor growth and volume in xenograft models of clear cell renal cell carcinoma64. Armored CAR T cell secretion of anti-PD-L1 antibodies with IgG1 or IgG4 Fc domains were additionally tested and the anti-PD-L-1 IgG1 isotype recruited NK cells to the tumor site.
CTLA-4 is a checkpoint molecule expressed by activated T cells. When bound to CD80/CD86 on antigen presenting cells or tumor cells, CTLA-4 suppresses T cell function. CAR T cells modified to secrete an anti-CTLA-4 minibody demonstrated superior efficacy to second generation CAR T cells in syngeneic models of mouse and canine glioma65. These data were used to design a pre-clinical trial to evaluate the safety of anti-IL-13Ra2 CAR T cells that secrete anti-CTLA-4 minibody in dogs with spontaneous glioma.
CD47 is another checkpoint molecule that has been targeted by armored CAR T cells that secrete antibody-based molecules. CD47 is ubiquitously expressed on many cell types and overexpressed in certain cancers. Expression of CD47 on cells delivers a signal to macrophages to prevent phagocytosis. Blockade of this signal can result in clearance of tumor cells. Anti-PD-L1 CAR T cells modified to secrete anti-CD47 single-domain antibody fragment nanobodies in the TME demonstrated blockade of CD47 signaling and improved engagement of the innate immune system66.
Clinical applications of antibody-based molecules secreted from CAR T cells with checkpoint blockade are currently being tested against solid tumors and leukemia (Table 1). These are all testing CAR T cells at secrete antibody-based proteins against the checkpoint molecules PD-1 or PD-L1.
Secretion Antibody-based molecules to prevent antigen escape
Antigen escape is a currently a major hurdle to the durability of response to current CAR T cell therapy. One way to overcome this is to increase the number of antigens recognized by CAR T cells.
Bispecific T cell engagers (BiTE) are one way to direct T cells to cancer cells in the TME. Composed of two scFv-based binding domains, BiTE bind to CD3 on T cells and a tumor-associated antigen on cancer cells. In 2017, the FDA approved blinatumomab, an anti-CD3/CD19 BiTE for patients with relapsed or refractory B-cell acute lymphoblastic leukemia. Designing CAR T cells to secrete BiTE in the TME is a promising approach to increase the number of antigens being targeted on tumor cells and prevent antigen escape. For example, although EFGRvIII is a gliobastoma-specific tumor antigen, heterogenous expression of this antigen on glioblastoma results in antigen-negative relapse of disease in mice treated with anti-EGFRcIII targeting CAR T cells67. However, when these CAR T cells were modified to a secret BiTE specific for wild-type EGFR, antigen escape was prevented in mouse models of glioblastoma. BiTE secreted by CAR T cells were capable of redirecting CAR T cells as well as bystander T cells to eliminate heterogenous tumors. This dual-targeted platform demonstrated the engagement of bystander T cells in vitro and a complete and durable response in vivo glioma mouse model67. Localized delivery of the BiTE is a key aspect to this therapy, as systemic delivery of a BiTE specific for wildtype EGFR would result in on-target, off-tumor toxicity of normal tissues.
Enzymes
In addition to immunosuppressive ligands and receptors, the structure and oxidative stress environments of some solid tumors can prevent optimal T cell function. CAR T cells can be modified to secrete particular enzymes in the TME to locally alter the structure or stress (Figure 1).
For example, a stroma-rich extracellular matrix (ECM) surrounds many solid tumors and prevents immune cell penetration, which decreases anti-tumor efficacy during immunotherapy. Armored CAR T cells have been engineered to locally deliver ECM modifying enzymes directly to the solid tumor in attempts to increase penetration. Anti-GD2 CAR T cells cultured in vivo were compared to T lymphocytes isolated from patients, and researchers found downregulation of heparinase, an enzyme capable of degrading heparan sulfate proteoglycans in the ECM, in the CAR T cells68. Anti-GD2 CAR T cells were engineered to express heparinase and resulted in degradation of the ECM and increased tumor infiltration and antitumor activity in xenografted mouse models compared to anti-GD2 CAR T cells without heparinase expression68.
Furthermore, tumor cells have high levels of oxidative stress and reactive oxygen species (ROS). ROS in the TME can impair antitumor activity by inducing oxidative stress-mediated repression of immune cells. Anti-CEA or anti-Her2 CAR T cells were engineered to coexpress a catalase enzyme to increase their antioxidative capacity69. Known as CAR-CAT T cells, these T cells demonstrated increased efficacy compared second-generation CAR T cells due to increased levels of intracellular catalase and reduced oxidative stress69. The CAR-CAT T cells also exhibited substantial bystander protection to other immune effector cells even at high H2O2 concentrations69.
Lastly, a novel application of enzyme secreting CAR T cells converts these cells to synthetic enzyme armed killer (SEAKER) cells. This approach delivers pro-drug activating enzymes to the TME with CAR T cells4,70. Systemic administration of a nontoxic pro-drug converts to potent chemotherapeutic drug only in the TME where the SEAKER cells are located, allowing for localized delivery of toxic agents.
Peptides
Peptides secreted by armored CAR T cells have widespread functions and potential applications. These applications range from enhancing CAR T cell function to combating CAR T cell therapy-associated toxicities (Figure 1).
CAR T cell activation and proliferation after infusion can lead to systemic inflammatory toxicities referred to as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). Interactions between tumor cells and CAR T cells activate myeloid cells to produce IL-1 and IL-6, levels of which determine the severity of CRS70,71. Systemic administration of an IL-1 receptor antagonist (IL-1Ra) protected against CRS and ICANS-related mortality in leukemia/lymphoma mouse models, without effecting CAR T cell efficacy71,72. Anti-CD19 CAR T cells modified to express IL-1Ra were engineered and administered to NSG mice with CRS71. Results indicated the IL-1Ra armored CAR prevented CRS in the mice without compromising the CAR T cell’s anti-tumor efficacy71.
Armored CAR T cells modified to secrete peptides have also been used to overcome immunosuppressive mediators in the TME surrounding solid tumors. Prostaglandin E2 (PGE2) and adenosine in the TME activate protein kinase A (PKA), which inhibits T-cell receptor activation by binding to the membrane protein ezrin in immune synapses. Armored CAR T cell engineered to secrete a regulatory subunit 1 anchoring disruptor (RAID) peptide competitively inhibits binding of PKA to ezrin73. This disruption increases T cell receptor signaling, cytokine release, and the anti-tumor efficacy of armored CAR T cells in synergistic mouse models73.
Peptides secreted from CAR T cells can also bind to tumor cells and restore inhibitory signals mutated on cancer cells. In germinal center lymphomas, the HVEM receptor gene is the most frequently mutated genes. This leads to the disruption of the inhibitory cell to cell interaction between HVEM and BTLA (B and T lymphocyte attenuator), resulting in B cell proliferation and the development of lymphoma. Soluble HVEM delivered by CAR T cells binds to BTLA and restores the inhibitory signal and leads to lymphoma cell death74.
Finally, peptides secreted from CAR T cells can engage the endogenous immune cells to combat cancer. Recently, Lai et al. developed CAR T cells to secrete dendritic cell32 growth factor Fms-like tyrosine kinase 3 ligand (Flt3L), which substantially promoted expansion of intertumoral conventional type 1 DC cells75. When combined with immune agonists, these armored CAR T cells inhibited tumor growth and induced epitope spreading to combat antigen-negative tumor escape75.
Discussion
CAR T cells modified to secrete proteins have great potential to enhance the anti-cancer efficacy of cellular therapies. These cells allow for local protein delivery in a controlled manner by immune effector cells. Future innovations to this platform include inducible gene expression to control protein secretion only when the CAR is activated in the TME, logic gating to determine which activation signals in the TME control protein expression, and synthetic biology circuits that can elegantly modulate protein expression by CAR T cells3,4. However, as these cells have increasing genetic modifications, suicide switches or controlled expression of the CAR construct may be needed for safe translation of these engineering strategies into patients.
There are further considerations and applications for the use of armored CAR T cells for cancer therapy. Current limitations to this technology include gene size and number of proteins that can be expressed in viral constructs. Future application with CRISPR may allow multiple proteins to be expressed and, with directed insertion, in a predictable manner. Non-genetic engineering methods such as liposome fusion or phagocytosis in addition to CAR transduction can also be employed to engineer these micropharmacies4.
CAR T cells modified to secrete proteins locally in the TME can deliver a myriad of different protein to improve the elimination of cancer cells. While clinical translations of CAR T cells modified to secrete proteins is still in its infancy (Table 1), this natural next step for CAR T cell therapy has great potential to positively impact patient care. Furthermore, with the capability of overcoming the suppressive TME, improving CAR T cell function, and eliciting an endogenous immune response, armored CAR T cells may be the key to making cellular immunotherapy effective against solid tumors.
References
- 1.Majzner RG & Mackall CL Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 25, 1341–1355, doi: 10.1038/s41591-019-0564-6 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Frigault MJ & Maus MV State of the art in CAR T cell therapy for CD19+ B cell malignancies. J Clin Invest 130, 1586–1594, doi: 10.1172/jci129208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rafiq S, Hackett CS & Brentjens RJ Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nature Reviews Clinical Oncology 17, 147–167, doi: 10.1038/s41571-019-0297-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner TJ et al. Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery. Cancers (Basel) 12, 2175, doi: 10.3390/cancers12082175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chmielewski M, Hombach AA & Abken H Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 257, 83–90, doi: 10.1111/imr.12125 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Nguyen KG et al. Localized Interleukin-12 for Cancer Immunotherapy. Frontiers in Immunology 11, doi: 10.3389/fimmu.2020.575597 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonard JP et al. Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12–Associated Toxicity and Interferon-γ Production. Blood 90, 2541–2548, doi: 10.1182/blood.V90.7.2541 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Hurteau JA, Blessing JA, DeCesare SL & Creasman WT Evaluation of recombinant human interleukin-12 in patients with recurrent or refractory ovarian cancer: a gynecologic oncology group study. Gynecol Oncol 82, 7–10, doi: 10.1006/gyno.2001.6255 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Motzer RJ et al. Randomized multicenter phase II trial of subcutaneous recombinant human interleukin-12 versus interferon-alpha 2a for patients with advanced renal cell carcinoma. J Interferon Cytokine Res 21, 257–263, doi: 10.1089/107999001750169934 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E & Hayakawa T IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther 1, 376–382, doi: 10.1006/mthe.2000.0050 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Zhang L et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther 19, 751–759, doi: 10.1038/mt.2010.313 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pegram HJ et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119, 4133–4141, doi: 10.1182/blood-2011-12-400044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koneru M, Purdon TJ, Spriggs D, Koneru S & Brentjens RJ IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 4, e994446, doi: 10.4161/2162402x.2014.994446 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeku OO, Purdon TJ, Koneru M, Spriggs D & Brentjens RJ Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Scientific Reports 7, 10541, doi: 10.1038/s41598-017-10940-8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rafiq S et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia 31, 1788–1797, doi: 10.1038/leu.2016.373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegram HJ et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 29, 415–422, doi: 10.1038/leu.2014.215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung M et al. Chimeric Antigen Receptor T Cell Therapy Targeting ICAM-1 in Gastric Cancer. Molecular Therapy - Oncolytics 18, 587–601, doi: 10.1016/j.omto.2020.08.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR & Brentjens RJ A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 13, 102, doi: 10.1186/s12967-015-0460-x (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chmielewski M, Kopecky C, Hombach AA & Abken H IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 71, 5697–5706, doi: 10.1158/0008-5472.Can-11-0103 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Chinnasamy D et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res 18, 1672–1683, doi: 10.1158/1078-0432.Ccr-11-3050 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J Immunol 203, 198–207, doi: 10.4049/jimmunol.1800033 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Waldmann TA, Dubois S, Miljkovic MD & Conlon KC IL-15 in the Combination Immunotherapy of Cancer. Front Immunol 11, 868, doi: 10.3389/fimmu.2020.00868 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waldmann TA, Miljkovic MD & Conlon KC Interleukin-15 (dys)regulation of lymphoid homeostasis: Implications for therapy of autoimmunity and cancer. Journal of Experimental Medicine 217, doi: 10.1084/jem.20191062 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoyos V et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 24, 1160–1170, doi: 10.1038/leu.2010.75 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanitis E et al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med 218, doi: 10.1084/jem.20192203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krenciute G et al. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol Res 5, 571–581, doi: 10.1158/2326-6066.Cir-16-0376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y et al. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clinical Cancer Research 25, 2915–2924, doi: 10.1158/1078-0432.Ccr-18-1811 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Hsu C et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood 109, 5168–5177, doi: 10.1182/blood-2006-06-029173 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fehniger TA et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med 193, 219–231, doi: 10.1084/jem.193.2.219 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurton LV et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proceedings of the National Academy of Sciences 113, E7788–E7797, doi: 10.1073/pnas.1610544113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nair S et al. Functional Improvement of Chimeric Antigen Receptor Through Intrinsic Interleukin-15Rα Signaling. Curr Gene Ther 19, 40–53, doi: 10.2174/1566523218666181116093857 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Tang L et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol 36, 707–716, doi: 10.1038/nbt.4181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nature Communications 11, 5902, doi: 10.1038/s41467-020-19672-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heczey A et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nature Medicine 26, 1686–1690, doi: 10.1038/s41591-020-1074-2 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Yasuda K, Nakanishi K & Tsutsui H Interleukin-18 in Health and Disease. Int J Mol Sci 20, 649, doi: 10.3390/ijms20030649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou T et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 583, 609–614, doi: 10.1038/s41586-020-2422-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robertson MJ et al. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res 14, 3462–3469, doi: 10.1158/1078-0432.Ccr-07-4740 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarhini AA et al. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer 115, 859–868, doi: 10.1002/cncr.24100 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Avanzi MP et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep 23, 2130–2141, doi: 10.1016/j.celrep.2018.04.051 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chmielewski M & Abken H CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep 21, 3205–3219, doi: 10.1016/j.celrep.2017.11.063 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Hu B et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep 20, 3025–3033, doi: 10.1016/j.celrep.2017.09.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sedimbi SK, Hägglöf T & Karlsson MC IL-18 in inflammatory and autoimmune disease. Cell Mol Life Sci 70, 4795–4808, doi: 10.1007/s00018-013-1425-y (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vidal-Vanaclocha F et al. Clinical and experimental approaches to the pathophysiology of interleukin-18 in cancer progression. Cancer Metastasis Rev 25, 417–434, doi: 10.1007/s10555-006-9013-3 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Solaymani-Mohammadi S, Eckmann L & Singer SM Interleukin (IL)-21 in Inflammation and Immunity During Parasitic Diseases. Frontiers in Cellular and Infection Microbiology 9, doi: 10.3389/fcimb.2019.00401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markley JC & Sadelain M IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood 115, 3508–3519, doi: 10.1182/blood-2009-09-241398 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Štach M et al. Inducible secretion of IL-21 augments anti-tumor activity of piggyBac-manufactured chimeric antigen receptor T cells. Cytotherapy 22, 744–754, (2020). [DOI] [PubMed] [Google Scholar]
- 47.Barata JT, Durum SK & Seddon B Flip the coin: IL-7 and IL-7R in health and disease. Nature Immunology 20, 1584–1593, doi: 10.1038/s41590-019-0479-x (2019). [DOI] [PubMed] [Google Scholar]
- 48.Adachi K et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol 36, 346–351, doi: 10.1038/nbt.4086 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Dominguez D, Zhang Y & Zhang B IL-33 in Tumor Immunity: Nothing to Sneeze At. Crit Rev Immunol 38, 453–470, doi: 10.1615/CritRevImmunol.2018026335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y & Lu B Guided delivery of the “alarming” cytokine IL-33 to tumor by chimeric antigen receptor T cells. The Journal of Immunology 198, 204.223–204.223 (2017). [Google Scholar]
- 51.Li X, Daniyan AF, Lopez AV, Purdon TJ & Brentjens RJ Cytokine IL-36γ improves CAR T-cell functionality and induces endogenous antitumor response. Leukemia, doi: 10.1038/s41375-020-0874-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagarsheth N, Wicha MS & Zou W Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 17, 559–572, doi: 10.1038/nri.2017.49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vilgelm AE & Richmond A Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Frontiers in Immunology 10, doi: 10.3389/fimmu.2019.00333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Metzemaekers M, Vanheule V, Janssens R, Struyf S & Proost P Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Frontiers in Immunology 8, doi: 10.3389/fimmu.2017.01970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moon EK et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology 7, e1395997–e1395997, doi: 10.1080/2162402X.2017.1395997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishio N et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res 74, 5195–5205, doi: 10.1158/0008-5472.Can-14-0697 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosewell Shaw A et al. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol Ther 25, 2440–2451, doi: 10.1016/j.ymthe.2017.09.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tian Y, Li Y, Shao Y & Zhang Y Gene modification strategies for next-generation CAR T cells against solid cancers. Journal of Hematology & Oncology 13, 54, doi: 10.1186/s13045-020-00890-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoon DH, Osborn MJ, Tolar J & Kim CJ Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int J Mol Sci 19, doi: 10.3390/ijms19020340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rafiq S et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nature Biotechnology 36, 847–856, doi: 10.1038/nbt.4195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li S et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin Cancer Res 23, 6982–6992, doi: 10.1158/1078-0432.Ccr-17-0867 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Zhou J-T et al. EGLIF-CAR-T Cells Secreting PD-1 Blocking Antibodies Significantly Mediate the Elimination of Gastric Cancer. Cancer Manag Res 12, 8893–8902, doi: 10.2147/CMAR.S260915 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ping Y et al. Augmenting the Effectiveness of CAR-T Cells by Enhanced Self-Delivery of PD-1-Neutralizing scFv. Frontiers in Cell and Developmental Biology 8, doi: 10.3389/fcell.2020.00803 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suarez ER et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 7, 34341–34355, doi: 10.18632/oncotarget.9114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yin Y et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol Ther Oncolytics 11, 20–38, doi: 10.1016/j.omto.2018.08.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie YJ et al. Improved Antitumor Efficacy of Chimeric Antigen Receptor T Cells that Secrete Single-Domain Antibody Fragments. Cancer Immunology Research 8, 518–529, doi: 10.1158/2326-6066.Cir-19-0734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choi BD et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 37, 1049–1058, doi: 10.1038/s41587-019-0192-1 (2019). [DOI] [PubMed] [Google Scholar]
- 68.Caruana I et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 21, 524–529, doi: 10.1038/nm.3833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ligtenberg MA et al. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J Immunol 196, 759–766, doi: 10.4049/jimmunol.1401710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bourne C et al. Mechanisms of Adoptive T Cell Micropharmacies. In Proceedings of the American Society of Gene and Cell Therapy Virtual Conference, 12–15 May 2020. Online, Abstract 31. [Google Scholar]
- 71.Giavridis T et al. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nature Medicine 24, 731–738, doi: 10.1038/s41591-018-0041-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Norelli M et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 24, 739–748, doi: 10.1038/s41591-018-0036-4 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Newick K et al. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunology Research 4, 541–551, doi: 10.1158/2326-6066.Cir-15-0263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boice M et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell 167, 405–418.e413, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lai J et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3Ldrives epitope spreading and antitumor immunity. Nat. Immunol 2020, 21, 1–13. [DOI] [PubMed] [Google Scholar]