

Abstract
Introduction
Inflammatory markers have long been observed in the brain, cerebrospinal fluid (CSF), and plasma of Alzheimer's disease (AD) patients, suggesting that inflammation contributes to AD and might be a therapeutic target. However, non‐steroidal anti‐inflammatory drug trials in AD and mild cognitive impairment (MCI) failed to show benefit. Our previous work seeking to understand why people with the inflammatory disease rheumatoid arthritis are protected from AD found that short‐term treatment of transgenic AD mice with the pro‐inflammatory cytokine granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) led to an increase in activated microglia, a 50% reduction in amyloid load, an increase in synaptic area, and improvement in spatial memory to normal. These results called into question the consensus view that inflammation is solely detrimental in AD. Here, we tested our hypothesis that modulation of the innate immune system might similarly be used to treat AD in humans by investigating the ability of GM‐CSF/sargramostim to safely ameliorate AD symptoms/pathology.
Methods
A randomized, double‐blind, placebo‐controlled trial was conducted in mild‐to‐moderate AD participants (NCT01409915). Treatments (20 participants/group) occurred 5 days/week for 3 weeks plus two follow‐up (FU) visits (FU1 at 45 days and FU2 at 90 days) with neurological, neuropsychological, blood biomarker, and imaging assessments.
Results
Sargramostim treatment expectedly changed innate immune system markers, with no drug‐related serious adverse events or amyloid‐related imaging abnormalities. At end of treatment (EOT), the Mini‐Mental State Examination score of the sargramostim group increased compared to baseline (P = .0074) and compared to placebo (P = .0370); the treatment effect persisted at FU1 (P = .0272). Plasma markers of amyloid beta (Aβ40 [decreased in AD]) increased 10% (P = .0105); plasma markers of neurodegeneration (total tau and UCH‐L1) decreased 24% (P = .0174) and 42% (P = .0019), respectively, after sargramostim treatment compared to placebo.
Discussion
The innate immune system is a viable target for therapeutic intervention in AD. An extended treatment trial testing the long‐term safety and efficacy of GM‐CSF/sargramostim in AD is warranted.
Keywords: activities of daily living, Alzheimer's disease, amyloid, cytokine, glial fibrillary acidic protein, granulocyte‐macrophage colony‐stimulating factor, granulocytemacrophage colony stimulating factor, interleukin‐6, innate immune system, lymphocyte, Mini‐Mental State Examination, monocyte, neuroinflammation, neutrophil, Pittsburgh compound B positron emission tomography, sargramostim, tau, tumor necrosis factor‐alpha, ubiquitin C‐terminal hydrolase L1
1. INTRODUCTION
Inflammation has been implicated in Alzheimer's disease (AD) pathogenesis and provides a promising target for therapeutic intervention. 1 , 2 , 3 , 4 For example, patients with rheumatoid arthritis (RA) have a reduced risk of developing AD, 5 which was originally attributed to their use of non‐steroidal anti‐inflammatory drugs (NSAIDs). 5 However, NSAIDs showed no benefit in clinical trials with either AD or mild cognitive impairment (MCI) participants. 6 , 7
We focused on the innate immune system and hypothesized instead that intrinsic factors associated with RA pathogenesis itself may underlie its AD protective effect(s). We found that only 20 days of subcutaneous treatment with granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), which is upregulated in RA patients’ blood, reduced AD pathology by >50%, increased the activation of microglia, and completely reversed the cognitive impairment of transgenic AD mice. 8 GM‐CSF treatment also improved cognition in wild‐type (WT) aged mice. 8 These effects of GM‐CSF in both AD and WT mice have since been replicated by other groups. 9 , 10 Finally, in a retrospective study, we found that leukemia patients who underwent short‐term treatment with recombinant human GM‐CSF (rHuGM‐CSF/sargramostim) plus recombinant human granulocyte colony‐stimulating factor (rHuG‐CSF/filgastrim) as supportive therapy after high‐dose chemotherapy and hematopoietic cell transplantation (HCT) showed significantly improved cognition at 6 months compared to HCT patients who received G‐CSF alone or no treatment, 11 also indicating that GM‐CSF may generally improve cognition by mechanisms separate from an effect on AD pathology.
As will be discussed, GM‐CSF is a hematopoietic and innate immune system modulator and pro‐inflammatory cytokine whose beneficial effects on cognition may derive from several mechanisms. For example, in neurological injury and disease, GM‐CSF has been found to have anti‐apoptotic effects on neurons; to promote neurogenesis and arteriogenesis; to reduce the formation of glial scars; and, very recently, to show reduced levels in the cerebrospinal fluid (CSF) of AD patients. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29
These results prompted us to test our hypothesis that GM‐CSF/sagramostim will stimulate the innate immune system, improve cognition, and reduce pathology in participants with mild‐to‐moderate AD.
2. METHODS
We carried out a randomized, double‐blind, placebo‐controlled phase II trial to test the safety and efficacy of sargramostim treatment in mild‐to‐moderate AD participants, defined by clinical diagnosis and a screening Mini‐Mental State Examination (MMSE) score of 10 to 26 inclusive (NCT01409915; see Methods in supporting information).
Participants who met eligibility criteria (Table 1) were randomized to receive either sargramostim (20 participants, standard Food and Drug Administration dosage 250 mcg/m2/day subcutaneous injection [SC], 5 days/week for 3 weeks) or placebo (20 participants, SC saline, 5 days/week for 3 weeks).
TABLE 1.
Participant characteristics at baseline visit (see also Figure S1)
| Demographics | ||
|---|---|---|
| Treatment group | Sargramostim/leukine (250 μg/m2/day, SC) | Placebo/saline (SC) |
| Number of participants | 20 | 20 |
| Number, Females, (%) | 12 (60) | 11 (55) |
| Mean age (yrs), (SD) | 67.10 (6.57) | 70.15 (6.42) |
| Mean weight (kg), (SD) | 75.3 (15.8) | 76.9 (13.2) |
| Race/ethnicity, number, (%) | ||
| Asian/Pacific Islander | 0 (0) | 0 |
| Black/African American | 0 (0) | 1 (5) |
| White | 20 (100) | 19 (95) |
| Hispanic/Latino | 0 (0) | 0 |
| Other | 0 (0) | 0 |
| Mean educational level (yrs), (SD) | 15.70 (2.92) | 15.80 (2.71) |
| Mean assessment score, (SD) | ||
| MMSE | 17.10 (4.58) | 20.75 (4.97) |
| ADAS‐Cog13 | 42.87 (12.73) | 36.20 (12.01) |
| ADCS ADL | 56.50 (12.30) | 62.75 (8.98) |
| CDR ‐ Sum of Boxes | 7.10 (3.32) | 6.10 (2.67) |
| Trail Making Test A | 101.50 (46.17) | 84.85 (48.84) |
Abbreviations: ADAS‐Cog13, Alzheimer's Disease Assessment Scale Cognitive subscale; ADCS ADL, Alzheimer's Disease Activities of Daily Living; CDR, Clinical Dementia Rating; MMSE, Mini‐Mental State Examination; SC, subcutaneous; SD, standard deviation.
At the baseline visit, each participant underwent neurological and cognitive assessments, safety and biomarker blood draws, and a magnetic resonance imaging (MRI) scan before being administered their first dose of sargramostim or placebo at the University of South Florida School of Medicine Clinic (3 participants) or in the Colorado Clinical and Translational Studies Institute (CCTSI; 37 participants). Participants returned daily, 5 days/week for 3 weeks, for SC sargramostim or placebo treatment, and for follow‐up visits at 45 days (FU1) and 90 days (FU2) after the end of treatment (EOT). Neuropsychological tests, MMSE, ADAS‐Cog memory subscale (ADAS delayed word recall + ADAS word recognition + ADAS orientation + ADAS word recall avg 30 ), Alzheimer's Disease Assessment Scale Cognitive subscale (ADAS‐Cog13), Alzheimer's Disease Cooperative Study Activities of Daily Living Scale (ADCS‐ADL), Clinical Dementia Rating scale Sum of Boxes (CDR‐SB), and Trail Making Test A (TRAILS‐A) were performed at baseline, EOT, FU1, and FU2. Safety/biomarker blood draws, MRI, and, for the last half of the study, amyloid‐positron emission tomography (PET) images were collected. Study data were managed using REDCap electronic data capture tools at the University of Colorado. 31
3. RESULTS
3.1. Safety (primary endpoint)
Data and Safety Monitoring Board (DSMB) assessments of study events found no drug‐related serious adverse events (SAEs), including no amyloid‐related imaging abnormalities (ARIAs, determined by MRI visual reads for microhemorrhages or edema; Table S1 in supporting information). The most common sargramostim‐associated AEs were dermatological (sixteen for sargramostim vs. five for placebo), gastrointestinal (eight for sargramostim vs. five for placebo), and headache (eight for sargramostim vs. two for placebo), as expected for this medication. 32
HIGHLIGHTS
There were no granulocyte‐macrophage colony‐stimulating factor (GM‐SCF)–attributable serious adverse events, amyloid‐related imaging abnormalities.
Innate immune system blood cells and cytokines were changed by GM‐CSF/sargramostim.
Mini‐Mental State Examination scores improved at end of treatment and 45‐day follow‐up with GM‐CSF/sargramostim treatment compared to placebo.
Plasma amyloid beta 40 increased by 10% with GM‐CSF/sargramostim treatment.
Plasma total tau and UCH‐L1 decreased by 24%, 42% respectively with GM‐CSF/sargramostim treatment.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and meeting abstracts and presentations. The pathophysiology of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) is well studied but only in Alzheimer's disease (AD) biology by us and not at all in clinical studies. These relevant citations are appropriately cited.
Interpretation: The findings of our clinical trial led to the conclusion that GM‐CSF is safe and well tolerated and can improve cognition and normalize plasma biomarkers of neuropathology (A+T+N) in AD participants. Although consistent with pre‐clinical findings currently in the public domain, the finding of a clinical treatment effect and its speed and extent were novel and unexpected.
Future directions: The article indicates that a larger, longer trial of GM‐CSF/sargramostim in AD is warranted.
3.2. Innate immune system activation
To confirm the expected effects of GM‐CSF/sargramostim on the innate immune system, 33 , 34 complete blood counts (CBC) with differential were performed (Figure 1A, B, and C; Table S2‐5 in supporting information), which showed that monocytes, lymphocytes, and neutrophils increased after sargramostim treatment. The Meso‐Scale Discovery (MSD) system was used to assess changes in inflammatory cytokines. For example, sargramostim treatment resulted in statistically significant increases in interleukin (IL)‐6, IL‐10, and tumor necrosis factor alpha (TNF‐α); a statistically significant decrease in IL‐8 (Figure 1D); and a statistically significant decrease in the albumin/globulin ratio (Figure 1E), illustrating the immunomodulatory activities of GM‐CSF.
FIGURE 1.
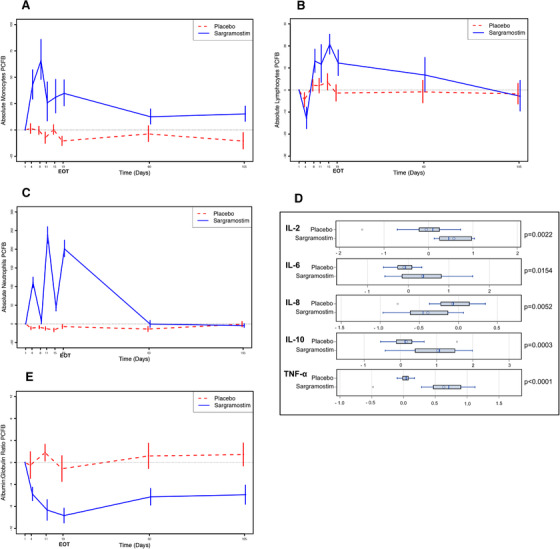
Increases in markers of the innate immune system in sargramostim‐treated participants compared to placebo‐treated participants. Complete blood counts (CBC) with differential were used to determine the effects of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF)/sargramostim treatment on cells of the innate immune system. Absolute numbers of monocytes (A), lymphocytes (B), and neutrophils (C) were all statistically significantly increased during the treatment phase (15 injections over 3 weeks) in the sargramostim group compared to the placebo group (P = .0005, P = .0512, and P < .0001, respectively) at the end of treatment (EOT) (see Tables S2‐4). The shorter half‐life of neutrophils is revealed by the fact that when a weekend intervened after an injection of GM‐CSF/sargramostim, the absolute neutrophil counts dropped, but then increased again during active treatment. D, The Meso‐Scale Discovery method was used to determine changes in plasma inflammatory cytokines with sargramostim treatment. At EOT compared to baseline, sargramostim treatment leads to statistically significant increases in interleukin (IL)‐2 (P = .0022), IL‐6 (P = .0154), IL‐10 (P = .0003), and tumor necrosis factor alpha (TNF‐α; P < .0001), and a decrease in IL‐8 (P = .0052). Shown are natural log transformed values. There were no significant changes from baseline in any inflammatory markers at either follow‐up 1 or follow‐up 2 visits (data not shown). E, The ratio of plasma albumin to globulin is often used to assess inflammation and its acute phase response. As expected, sargramostim stimulated a drop in the albumin/globulin ratio compared to placebo (P = .0029 at EOT). Graphs plotted as means +/– standard error of the mean.
3.3. Efficacy (secondary/exploratory endpoints)
Although this trial was small and thus inherently underpowered, several secondary/exploratory outcomes were statistically significant. Analyses for potential efficacy revealed a statistically significant positive treatment effect of sargramostim on the MMSE, which was selected as an outcome measure based on its superior sensitivity to temporal changes. 35 Specifically, at EOT, the mean MMSE total score change in the sargramostim group was 1.45 units higher relative to baseline (P = .0074; Figure 2A; Table S6 in supporting information). The difference in mean change from baseline in MMSE total scores between the sargramostim and placebo groups was 1.80 (P = .0370) at EOT and 1.75 (P = .0272) at FU1 (45 days after EOT).
FIGURE 2.
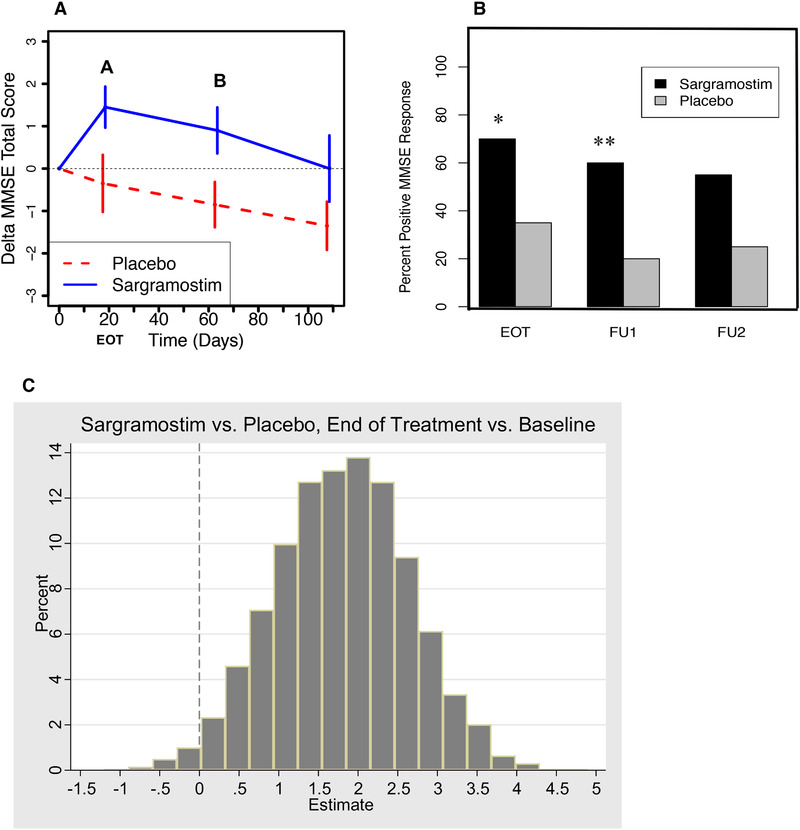
Improvement in Mini‐Mental State Examination (MMSE) scores in sargramostim‐treated participants compared to placebo‐treated participants. A, Mixed model analysis of MMSE data from the 20 sargramostim‐treated participants and 20 placebo‐treated participants shows a statistically significant improvement in the sargramostim group at the end of treatment (EOT; A) compared to baseline (1.45, 95% confidence interval [CI]: [0.44, 2.46], P = .0074) and compared to the placebo group (1.80, 95% CI: [0.12, 3.49], P = .0370), and at the first follow‐up visit (FU1; B) at 45 days after the EOT, compared to the placebo‐treated group (1.75, 95% CI: [0.21, 3.29], P = .0272). Shown are the results +/– standard error of the mean [SEM], setting the baselines at 0. See Table S6 for full statistical analyses. B, The improvement in total MMSE score occurred in: (a) 14 of 20 (70%) sargramostim‐treated participants, and 7 of 20 (35%) placebo‐treated participants (P = .0267) at the EOT, (b) 12 of 20 (60%) sargramostim‐treated participants and 4 of 20 (20%) placebo‐treated participants (P = .0098) at the 45‐day follow‐up visit (FU1), and (c) 11 of 20 (55%) sargramostim‐treated participants and 5 of 20 (25%) placebo‐treated participants (P = .0528; non‐significant trend) at the 90‐day follow‐up visit (FU2). (* P < .05; ** P < .01) Combining the results to identify an overall treatment effect shows that 16 of 20 (80%) sargramostim‐treated participants can be considered “responders” in that they showed a higher MMSE score compared to baseline at either the EOT or at the 45‐day follow‐up visit, compared to only 7 of 20 (35%) placebo‐treated participants (P = .0040). Difference in proportion by treatment group was tested with Chi‐square/Fisher's exact association test (see Table S6 for full statistical analyses). C, To assess the effect of sargramostim treatment on the participants throughout the study, the 20 sargramostim‐treated participants and 20 placebo‐treated participants were each divided into 10,000 random subsamples of 10 participants each, and for each simulated data set, a model was run, generating estimates for the means and contrasts. For > 98% of the simulated data sets, the estimate for change from baseline to the EOT within the sargramostim group, and the estimates for the treatment effects on change from baseline to EOT and change from baseline to FU1 were greater than zero. The mean MMSE scores were calculated and graphed as a delta distribution. The distribution was approximately Gaussian and the mean and median delta was 1.8, as was the delta of the entire 20 sargramostim‐treated participants and 20 placebo‐treated participants analyzed in (A) and (B), showing that the statistically significant difference between the sargramostim‐treated participants and the placebo‐treated participants at the EOT reflected all parts of the trial. Because we reported interim study results at symposia in 2018, we tested for a potential subsequent bias and found no significant difference between the mean total MMSE scores of participants enrolled during the first and second halves of the trial. Graphs plotted as means +/– SEM. (* P < .05; ** P < .01)
The improvement in total MMSE score occurred in: (1) 70% of sargramostim‐treated participants compared to 35% of placebo‐treated participants at EOT (P = .0267), (2) 60% of sargramostim‐treated participants compared to 20% of placebo‐treated participants at FU1 (P = .0098), and (3) 55% of sargramostim‐treated participants compared to 25% placebo‐treated participants at FU2 (trend; P = .0528; Figure 2A). Combining the results to identify an overall treatment effect, 80% of sargramostim‐treated participants were “responders” in that they showed a higher MMSE score compared to baseline at either EOT or at FU1, compared to only 35% of placebo‐treated participants (P = .0040).
Study participants were randomized, and although the mean MMSE scores at screening were not significantly different between the sargramostim and placebo groups, they were at baseline (–3.65, 95% confidence interval [CI]: [–6.71, 0.59], P = .0207). Correcting for baseline scores as covariates is not recommended, 36 and indeed such an alternative model showed no statistically significant effect of baseline MMSE on its change from baseline at any time point, which justifies using the unadjusted cell means model. Nonetheless, using baseline adjustment models, the treatment effect was only marginally statistically non‐significant at EOT (1.60, 95% CI: [–0.21, 3.40], P = .0808) and was statistically significant at FU1 (1.80, 95% CI: [0.15, 3.46], P = .0338).
To further investigate the sargramostim treatment effect, subsamples of 10 participants from each group were randomly drawn 10,000 times and MMSE scores analyzed, which showed a sargramostim treatment effect in each part of the entire study (Figure 2C).
The ADAS‐Cog13 assessment showed no statistically significant sargramostim effect compared to baseline or compared to the placebo group at EOT. However, at FU1, ADAS‐Cog13 showed a statistically significant increase (worsening) in the sargramostim group compared to baseline (P = .0009) and compared to the placebo group (P = .0147), and returned to non‐significance at FU2 (Figure 3A; Table S7 in supporting information). Such a rebound effect after halting treatment has been observed with other AD drugs. 37
FIGURE 3.
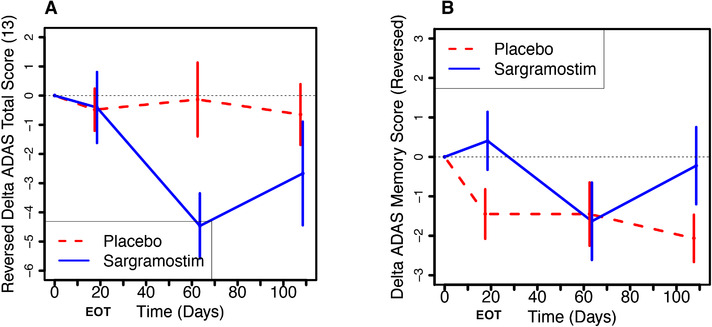
Changes in Alzheimer's Disease Assessment Scale Cognitive subscale (ADAS‐Cog13) score and its memory domain subscale in sargramostim‐treated participants compared to placebo‐treated participants. A, There were no significant changes in ADAS‐Cog13 scores between sargramostim‐treated participants and placebo‐treated participants during the treatment phase (through the end of treatment [EOT] ≈day 19). Note that the scores shown on the Y‐axis have been inverted so as to mirror Figure 1 and show improved cognition in the upward direction. However, at follow‐up 1 (FU1), 45 days after the EOT, the ADAS‐Cog13 showed a statistically significant increase (worsening) in the sargramostim group compared to baseline (4.46, 95% confidence interval [CI]: [2.11, 6.82], P = .0009), and compared to the placebo group (4.33, 95% CI: [0.90, 7.76], P = .0147) and then returned to the level of the placebo group at the 90‐day follow‐up visit. The treatment effect was stronger when baseline ADAS was adjusted for (5.54, 95% CI: [2.31, 8.78], P = .0013), and there was a statistically significant effect of baseline on expected change score at the 45‐day follow‐up (–0.173 per scale unit, 95% CI: [–0.298, –0.049], P = .0077). B, To compare to the memory‐predominant Mini‐Mental State Examination (MMSE) measure, the memory domain subscale of ADAS‐Cog13 (ADAS delayed word recall + ADAS word recognition + ADAS orientation + ADAS word recall avg) was analyzed. At the EOT, which is when the MMSE showed a statistically significant improvement (Figure 2), the ADAS‐Cog13 Memory Subscale showed a statistically trending improvement from baseline in the sargramostim group compared to placebo (–1.84, 95% CI: [–3.82, 0.11], P = .0632). Unlike the MMSE, however, the ADAS‐COG13 memory subscale showed no statistically significant improvement within the sargramostim group between baseline and the EOT. The treatment effect was attributable to the worsening of the placebo group. Nor was there a treatment effect for the ADAS‐Cog13 Memory Subscore at the 45‐day follow up. Although there was no statistically significant baseline effect on change scores for the EOT, thus justifying the uncorrected cell means models, 18 the statistical trend for the treatment effect of the ADAS‐Cog13 memory subscale at the EOT disappeared when baseline was adjusted for. Shown are the results +/– standard error of the mean, with the baselines set to 0. See Tables S7 and S8 for full statistical analyses.
The memory domain subscale of ADAS‐Cog1330 at EOT (when the MMSE showed a statistically significant improvement) showed a nearly statistically significant trending improvement in the sargramostim group compared to placebo (P = .0632), suggesting that the sargramostim treatment effect is primarily on the memory domain (Figure 3B; Table S8 in supporting information).
The ADCS‐ADL showed a small, non‐significant improvement in the sargramostim group at EOT (P = .3485; Figure S2A; Table S9 in supporting information), but was statistically significantly correlated with MMSE (see below). CDR‐SB and TRAILS‐A measures showed no statistically significant effects of sargramostim treatment (Figure S2B,C; Tables S10,11 in supporting information).
3.4. Plasma biomarkers of AD neuropathology
To assess changes in brain pathology according to the National Institute on Aging and Alzheimer's Association (NIA‐AA) Research Framework, 38 the plasma biomarkers amyloid beta (Aβ)40, Aβ42, total tau, plasma ubiquitin C‐terminal hydrolase L1 (UCH‐L1), glial fibrillary acidic protein (GFAP), and neurofilament light (NfL) were measured (SIMOA platform, Quanterix). In the sargramostim group at EOT, mean plasma Aβ40 level, which is reduced in AD, 39 , 40 showed an 8.4% increase from baseline (P = .0127), and was 10% higher compared to the placebo‐treated group (P = .0105; Figure 4A; Table S12 in supporting information), potentially indicating less sequestration of mono/oligomeric Aβ in the brain. Using the N4PB assay, total tau, whose plasma levels are increased in AD, reflecting both tau pathology and neurodegeneration, 40 , 41 decreased by 17% in the sargramostim‐treated group compared to baseline (P = .0327) and decreased by 24% (P = .0174) compared to the placebo‐treated group baseline change (Figure 4B; Table S13 in supporting information). At EOT, another independent measure of neurodegeneration, UCH‐L1, 40 , 42 , 43 decreased by 40% in the sargramostim‐treated group compared to baseline (P = .0017), and by 42% in the sargramostim‐treated group compared to the placebo‐treated group (P = .0019; Figure 4C; Table S14 in supporting information). The N3PA measures of tau were more variable than the N4PB measures and showed a smaller, non‐significant difference between the sargramostim‐treated group and the placebo‐treated group at EOT.
FIGURE 4.
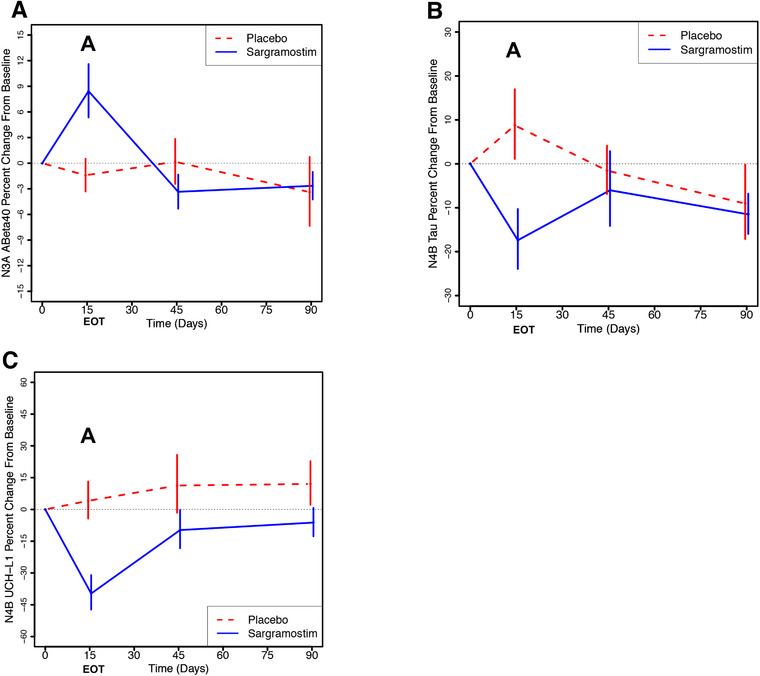
Sargramostim treatment reduces biomarkers of neuropathology compared to placebo‐treated participants. Plasma biomarkers of Alzheimer's disease amyloid and tau pathology and neurodegeneration were assessed using the SIMOA platform (Quanterix). A, Amyloid beta (Aβ)40 data from N3PA plates are plotted (+/– standard error of the mean [SEM]) and show a statistically significant increase in the sargramostim group at the end of treatment (A), compared to baseline (P = .0127) and compared to the change in placebo from baseline (P = .0105). B, Assessed using N4PB plates, total tau levels decreased 17% compared to baseline (P = .0327) and 24% (P =.0174) compared to the change from baseline in the placebo group (A). C, At the end of treatment, another measure of neurodegeneration, plasma ubiquitin C‐terminal hydrolase L1 (UCH‐L1), decreased 40% compared to baseline (P = .0017) and 42% compared to the change from baseline in the placebo group (P = .0019). Shown are the results +/– SEM, with the baselines set to 0. See Tables S12‐14 for full statistical analyses
3.5. Amyloid imaging
For the second half of the study, amyloid‐PET imaging was used as an inclusion criterion. All 16 participants diagnosed as having mild‐to‐moderate AD at screening, who were then assessed by amyloid‐PET imaging, were found to be amyloid‐positive, thus validating the clinical inclusion criteria.
We compared the amyloid‐PET imaging data at baseline and at FU1 for the remaining 16 substudy participants (Amyvid‐PET: two sargramostim‐treated, six placebo‐treated; 11C‐PiB‐PET: five sargramostim‐treated, three placebo‐treated [Text S2 in supporting information]). There were no statistically significant changes in standardized uptake value ratio (SUVr) for each ligand separately or when combined by Centiloid scale conversion 44 or by the method of Properzi et al. 45 (Tables S15‐S20 in supporting information). However, the powers of the tests are limited by the small number of participants.
3.6. Correlation analyses
Although correlations do not allow conclusions to be drawn regarding potential mechanisms, it is notable that at EOT, MMSE changes from baseline for all participants correlated to their log changes in immune cells, including neutrophils (Figure 5A; Pearson coefficient = 0.409; P = .0098) and lymphocytes (Figure 5B; Pearson coefficient = 0.353; P = .0276). Similarly, changes in plasma GFAP and plasma NfL from baseline were highly correlated at all time points (Pearson coefficient = 0.752, 0.693, 0.663 at EOT, FU1, and FU2 respectively; all P < .0001; Table S21 in supporting information; Panel 5C shows EOT correlation), indicating that neuronal damage and gliosis are linked in the brains of participants with mild‐to‐moderate AD.
FIGURE 5.
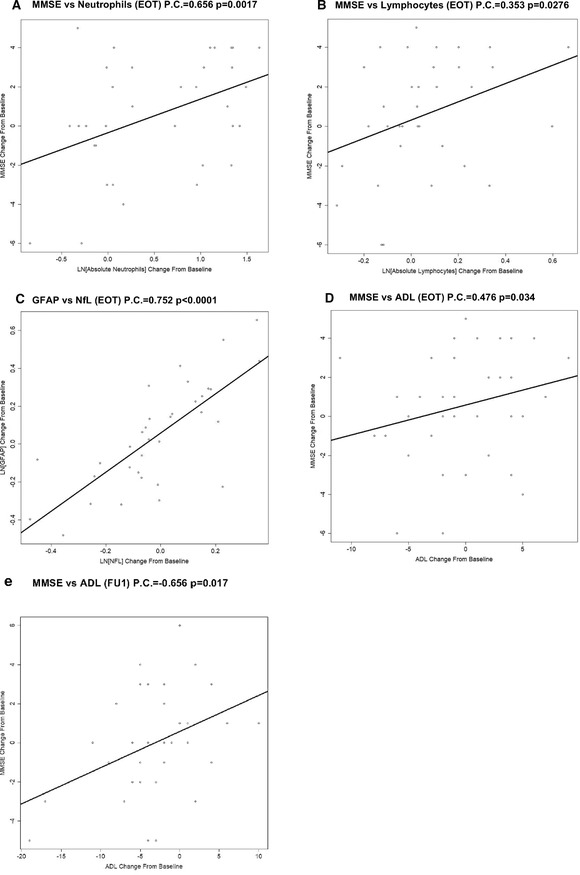
Correlations among and between changes in behavior and biomarkers of Alzheimer's disease. A, In addition to the effect of sargramostim treatment on both Mini‐Mental State Examination (MMSE) and immune system cells shown in Figure 1, we assessed the correlations between these measures (Table S21). The Pearson correlation between change in MMSE and change in absolute neutrophil counts was statistically significant at the end of treatment (EOT; 0.409; P = .0098). B, The correlation between change in MMSE and change in absolute lymphocyte counts was also statistically significant at the EOT (0.353; P = .0276). These results show that the improvement in cognition measured by MMSE is correlated with (and possibly caused by) the increase in innate immune system stimulation and its downstream effects. C, There was a very strong correlation between changes in plasma glial fibrillary acidic protein, a measure of astrocyte activation, and plasma neurofilament light, a measure of neuronal damage, from baseline at all time points for all participants (Pearson coefficient = 0.752, 0.693, 0.663 at the EOT, follow‐up 1 [FU1], follow‐up 2 [FU2]; all P < .0001; Panel C shows the correlation for the EOT), indicating a likely mechanistic link between neuronal damage and brain glial inflammation in AD. D, E, Within the sargramostim‐treated group, changes in MMSE were positively correlated with changes in Activities of Daily Living at the EOT (Pearson coefficient = 0.476; P = .034, Panel D) and at FU1 (0.656; P = .017, Panel E), the time points that showed a statistically significant treatment effect of sargramostim on MMSE.
Although ADCS‐ADL measures alone showed only a weak, non‐statistically significant beneficial effect of sagramostim treatment compared to placebo, there was a strong positive correlation between sargramostim‐associated changes in MMSE and changes in ADCS‐ADL at EOT (Figure 5D; Pearson coefficient = 0.476; P = .034) and at FU1 (Figure 5E; Pearson coefficient = 0.656; P = .017; Table S21), which corresponded to the times that showed a treatment effect of sargramostim on MMSE. Thus, the beneficial effect of GM‐CSF/sargramostim treatment on MMSE is partially mirrored in another, quite different measure of cognitive function.
4. DISCUSSION
The results of this first phase II trial of the innate immune system modulator GM‐CSF/sargramostim in participants with clinically diagnosed mild‐to‐moderate AD allow the following major conclusions:
4.1. Primary endpoint: safety and innate immune system activation
Short‐term sargramostim treatment in mild‐to‐moderate AD participants increased innate immune cell and cytokine measures and was safe and well tolerated.
4.2. Secondary, exploratory endpoints
-
2.
The mean change in an objective measure of cognition, the MMSE total score, was improved (increased) in the sargramostim group at EOT compared to baseline and compared to the placebo group and remained improved compared to placebo at FU1.
-
3.
Changes in MMSE were correlated with changes in ADCS‐ADL at EOT and at FU1.
-
4.
Changes in plasma GFAP were highly correlated with changes in NfL, suggesting a link between neuronal and glial damage in AD.
-
5.
The ADAS‐Cog13 memory subscale scores trended to improvement at EOT, mirroring the sargramostim treatment‐associated cognitive improvement shown by the MMSE. The mean ADAS‐Cog13 change score showed no statistically significant treatment effect at EOT, but was temporarily worse in the sargramostim group 45 days after EOT, suggesting a likely rebound effect. The finding that sargramostim led to improvement in MMSE and a trend improvement in the memory subscale of the ADAS‐Cog13, but not in other neuropsychological tests, suggests that its primary beneficial effect was on memory.
-
6.
Statistically significant changes in plasma measures of amyloid, tau, and neurodegeneration (Aβ40, total tau, and UCH‐L1, respectively) toward normal with sargramostim treatment compared to baseline and/or to placebo and multiple statistically significant correlations at EOT suggest a link between the modulated innate immune system, reduced neuropathology, and improved cognition resulting from sargramostim treatment.
-
7.
PET measures of amyloid load in a small substudy showed no significant differences between the scans (baseline and FU1).
Although these results suggest that short‐term GM‐CSF/sargramostim treatment leads to innate immune system activation, cognition/memory improvement, and partial normalization of blood measures of amyloid and tau pathology and of neuronal damage in participants with mild‐to‐moderate AD, the main conclusion of the study is that additional longer, larger trials of GM‐CSF/sargramostim in AD are warranted.
Unlike most AD drugs, which have been designed to target a specific step in the AD pathogenic pathway, GM‐CSF/sargramostim is different because it has broader targets: hematopoiesis and the innate immune system. The significant increase in Aβ40 and the reductions in total tau and UCH‐L1 in the current study are consistent with GM‐CSF/sargramostim having an AD‐modifying effect.
The previous findings that GM‐CSF treatment improves cognition in aged WT mice, 8 , 10 in the Dp16 mouse model of Down syndrome (DS), which lacks amyloid deposition or other AD pathology (manuscript under review), and in leukemia patients with chemotherapy‐associated cognitive decline, 11 indicate that GM‐CSF can also improve cognition independent of AD pathology, which may contribute to the improved MMSE performance observed after sargramostim treatment. Indeed, GM‐CSF treatment has been reported to be beneficial for many neurological injuries and diseases without associated AD pathology. For example, in animal models of stroke, spinal cord injury, traumatic brain injury, and retinal degeneration, GM‐CSF crosses the blood‐brain barrier and is both neuroprotective and anti‐apoptotic, stimulates arteriogenesis and blood flow, induces leptomeningeal collateral growth, decreases infarct volume, improves/restores locomotor function, and promotes axon preservation/regeneration. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 GM‐CSF also induces proliferation of neural stem cells 24 , 25 , 26 , 27 and plays a key role in neuronal plasticity. 28 In Parkinson's disease (PD) models, GM‐CSF treatment prevents dopaminergic neuronal cell death and improves locomotor function. 46 , 47 , 48 A placebo‐controlled trial that treated PD participants with sargramostim for 56 consecutive days met its primary endpoint of safety and tolerability, and showed modest improvements in motor function. 49
In addition to providing evidence that GM‐CSF/sargramostim may have both disease‐modifying and cognition‐enhancing activities in AD, the results of this trial also shed light on the role of neuroinflammation and the innate immune system in AD, an area of active investigation and debate. 3 The post mortem observation of inflammatory proteins and cells in human AD brains and the finding that pro‐inflammatory proteins in AD animal models affect brain pathology and cognitive decline suggest that inflammation plays an integral role in the AD pathogenic pathway. 3 , 50 However, given the failure of NSAID trials in participants with AD or MCI, 7 our findings that sargramostim treatment increased immune cell numbers and cytokine levels, which are traditionally considered to reflect inflammation, and that sargramostim treatment improved one measure of cognition and also normalized several biomarkers of AD pathology, suggests that targeted modulators of neuroinflammation may be a better therapeutic strategy than general anti‐inflammatory drugs.
The sargramostim effect on changes in MMSE scores at EOT, which persisted at FU1 (45 days after EOT), suggests that GM‐CSF has longer‐term effects than its plasma half‐life (terminal elimination half‐life of 1.4 hours 32 ) might predict. Whether the stable cognitive benefit is due to continued glial‐based reductions of AD pathology, or due to other neuronal benefits outlined above, remains unknown. Taken together, the potential broad‐reaching effects of GM‐CSF/sargramostim on the innate immune system and on neuronal function represent a novel approach to AD therapy.
4.3. Strengths and weaknesses
The strengths of this study include its randomized, double‐blind, placebo‐controlled design with multiple neurological, neuropsychological, cell, cytokine, AD pathology biomarkers, and neuroimaging assessments, which allow conclusions regarding the safety of sargramostim treatment, plus inferences about its potential efficacy and underlying mechanisms in AD. The main weaknesses are the short treatment period, small sample size, especially in the amyloid‐PET substudy, and non‐memory cognitive measures, showing no direct treatment effects.
5. CONCLUSION
The data presented herein show that GM‐CSF/sargramostim treatment was safe and well‐tolerated and provided measurable disease‐modifying and memory‐enhancing benefits to participants with mild‐to‐moderate AD. Inasmuch as AD is still not successfully treated using other interventions that reverse markers of both cognition and pathology, our findings indicate the need for a larger, longer trial.
CONFLICTS OF INTEREST
Drs. Potter and Boyd are two of the inventors on several U.S. patents owned by the University of South Florida, but not licensed. As of Feb.1 2021, Dr. Boyd is an employee of Partner Therapeutics.
Supporting information
Supplementary information
ACKNOWLEDGMENTS
We would like to especially thank the participants in this study who volunteered their time for comprehensive evaluations, including test article or placebo injections, blood draws, and MRI scans, and amyloid‐PET scans. We would also like to thank the members of the University of Colorado Alzheimer's and Cognition Center's Clinical Research Team, including P.S. Pressman, MD; S.K. Holden, MD; B. McConnell, MD, PhD; J. Bateman, MD; J. Mrozewski, MD; Z. Macchi, MD; I. Kletenik, MD; Ramesh Karki, MS; and other members of the radiopharmacy team, the University of Colorado Hospital Investigational Drug Service, the CCTSI, the DSMB, and T. Ferrey, BS, of Bioclinica. Research support was provided by the States of Colorado and Florida, the University of Colorado School of Medicine, the University of Colorado Hospital, the Global Down Syndrome Foundation, the Linda Crnic Institute for Down Syndrome, an Alzheimer's Association Part the Cloud Grant, the Dana Foundation, Don and Sue Fisher, the Hewit Family Foundation, the Sprout Foundation, Marcy and Bruce Benson, Les Mendelson, and other generous philanthropists. This project was also supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR002535. Its contents are the authors’ sole responsibility and do not necessarily represent official NIH views. Additionally, this project used the University of Colorado Anschutz Brain Imaging Center (BIC), which is supported by an NIH High‐End Instrumentation Grant S10OD018435 (Tregallas, PI).
Potter H, Woodcock JH, Boyd TD, et al. Safety and efficacy of sargramostim (GM‐CSF) in the treatment of Alzheimer's disease. Alzheimer's Dement. 2021;7:e12158. 10.1002/trc2.12158
Huntington Potter, Jonathan H. Woodcock and Timothy D. Boyd contributed equally in this study.
REFERENCES
- 1. Potter H. Beyond beta protein—the essential role of inflammation. Prominent Press; 2001. [Google Scholar]
- 2. Andreasson KI, Bachstetter AD, Colonna M, et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem. 2016;138:653‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frost GR, Jonas LA, Friend LiYM. Foe or Both?. Immune Activity in Alzheimer's Disease. 2019;11:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nichols MR, St‐Pierre MK, Wendeln AC, et al. Inflammatory mechanisms in neurodegeneration. J Neurochem. 2019;149:562‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McGeer PL, Schulzer M, McGeer EG. Arthritis and anti‐inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425‐432. [DOI] [PubMed] [Google Scholar]
- 6. McGeer PL, Rogers J, McGeer EG. Inflammation, anti‐inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271‐276. [DOI] [PubMed] [Google Scholar]
- 7.ADAPT_FS_Research_Group. Follow‐up evaluation of cognitive function in the randomized Alzheimer's disease anti‐inflammatory prevention trial and its follow‐up study. Alzheimers Dement. 2015;11:216‐225.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boyd TD, Bennett SP, Mori T, et al. GM‐CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. Journal of Alzheimer's disease : JAD. 2010;21:507‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiyota T, Machhi J, Lu Y, et al. Granulocyte‐macrophage colony‐stimulating factor neuroprotective activities in Alzheimer's disease mice. J Neuroimmunol. 2018;319:80‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Castellano JM, Mosher KI, Abbey RJ, et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature. 2017;544:488‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jim HS, Boyd TD, Booth‐Jones M, Pidala J, Potter H. Granulocyte macrophage colony stimulating factor treatment is associated with improved cognition in cancer patients. Brain disorders & therapy. 2012;1:1000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buschmann IR, Busch HJ, Mies G, Hossmann KA. Therapeutic induction of arteriogenesis in hypoperfused rat brain via granulocyte‐macrophage colony‐stimulating factor. Circulation. 2003;108:610‐615. [DOI] [PubMed] [Google Scholar]
- 13. Nakagawa T, Suga S, Kawase T, Toda M. Intracarotid injection of granulocyte‐macrophage colony‐stimulating factor induces neuroprotection in a rat transient middle cerebral artery occlusion model. Brain Res. 2006;1089:179‐185. [DOI] [PubMed] [Google Scholar]
- 14. Schneider UC, Schilling L, Schroeck H, Nebe CT, Vajkoczy P, Woitzik J. Granulocyte‐macrophage colony‐stimulating factor‐induced vessel growth restores cerebral blood supply after bilateral carotid artery occlusion. Stroke. 2007;38:1320‐1328. [DOI] [PubMed] [Google Scholar]
- 15. Todo K, Kitagawa K, Sasaki T, et al. Granulocyte‐macrophage colony‐stimulating factor enhances leptomeningeal collateral growth induced by common carotid artery occlusion. Stroke. 2008;39:1875‐1882. [DOI] [PubMed] [Google Scholar]
- 16. Kong T, Choi JK, Park H, et al. Reduction in programmed cell death and improvement in functional outcome of transient focal cerebral ischemia after administration of granulocyte‐macrophage colony‐stimulating factor in rats. Laboratory investigation. J Neurosurg. 2009;111:155‐163. [DOI] [PubMed] [Google Scholar]
- 17. Theoret JK, Jadavji NM, Zhang M, Smith PD. Granulocyte macrophage colony‐stimulating factor treatment results in recovery of motor function after white matter damage in mice. Eur J Neurosci. 2016;43:17‐24. [DOI] [PubMed] [Google Scholar]
- 18. Chung J, Kim MH, Yoon YJ, Kim KH, Park SR, Choi BH. Effects of granulocyte colony‐stimulating factor and granulocyte‐macrophage colony‐stimulating factor on glial scar formation after spinal cord injury in rats. J Neurosurg Spine. 2014;21:966‐973. [DOI] [PubMed] [Google Scholar]
- 19. Huang X, Kim JM, Kong TH, et al. GM‐CSF inhibits glial scar formation and shows long‐term protective effect after spinal cord injury. J Neurol Sci. 2009;277:87‐97. [DOI] [PubMed] [Google Scholar]
- 20. Shultz SR, Tan XL, Wright DK, et al. Granulocyte‐macrophage colony‐stimulating factor is neuroprotective in experimental traumatic brain injury. J Neurotrauma. 2014;31:976‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelso ML, Elliott BR, Haverland NA, Mosley RL, Gendelman HE. Granulocyte‐macrophage colony stimulating factor exerts protective and immunomodulatory effects in cortical trauma. J Neuroimmunol. 2015;278:162‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schallenberg M, Charalambous P, Thanos S. GM‐CSF protects rat photoreceptors from death by activating the SRC‐dependent signalling and elevating anti‐apoptotic factors and neurotrophins. Graefes Arch Clin Exp Ophthalmol. 2012;250:699‐712. [DOI] [PubMed] [Google Scholar]
- 23. Legacy J, Hanea S, Theoret J, Smith PD. Granulocyte macrophage colony‐stimulating factor promotes regeneration of retinal ganglion cells in vitro through a mammalian target of rapamycin‐dependent mechanism. J Neurosci Res. 2013;91:771‐779. [DOI] [PubMed] [Google Scholar]
- 24. Kim JK, Choi BH, Park HC, et al. Effects of GM‐CSF on the neural progenitor cells. Neuroreport. 2004;15:2161‐2165. [DOI] [PubMed] [Google Scholar]
- 25. Kruger C, Laage R, Pitzer C, Schabitz WR, Schneider A. The hematopoietic factor GM‐CSF (granulocyte‐macrophage colony‐stimulating factor) promotes neuronal differentiation of adult neural stem cells in vitro. BMC Neurosci. 2007;8:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang X, Choi JK, Park SR, et al. GM‐CSF inhibits apoptosis of neural cells via regulating the expression of apoptosis‐related proteins. Neurosci Res. 2007;58:50‐57. [DOI] [PubMed] [Google Scholar]
- 27. Choi JK, Kim KH, Park H, Park SR, Choi BH. Granulocyte macrophage‐colony stimulating factor shows anti‐apoptotic activity in neural progenitor cells via JAK/STAT5‐Bcl‐2 pathway. Apoptosis. 2011;16:127‐134. [DOI] [PubMed] [Google Scholar]
- 28. Krieger M, Both M, Kranig SA, et al. The hematopoietic cytokine granulocyte‐macrophage colony stimulating factor is important for cognitive functions. Sci Rep. 2012;2:697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Taipa R, das Neves SP, Sousa AL, et al. Proinflammatory and anti‐inflammatory cytokines in the CSF of patients with Alzheimer's disease and their correlation with cognitive decline. Neurobiol Aging. 2019;76:125‐132. [DOI] [PubMed] [Google Scholar]
- 30. Verma N, Beretvas SN, Pascual B, Masdeu JC, Markey MK. New scoring methodology improves the sensitivity of the Alzheimer's disease assessment scale‐cognitive subscale (ADAS‐Cog) in clinical trials. Alzheimers Res Ther. 2015;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. PartnerTherapeuticsInc. Leukine [package insert]. Lexington, MA2018.
- 33. Damiani G, McCormick TS, Leal LO, Ghannoum MA. Recombinant human granulocyte macrophage‐colony stimulating factor expressed in yeast (sargramostim): a potential ally to combat serious infections. Clin Immunol. 2020;210:108292. [DOI] [PubMed] [Google Scholar]
- 34. Borriello F, Galdiero MR, Varricchi G, Loffredo S, Spadaro G, Marone G. Innate immune modulation by GM‐CSF and IL‐3 in health and disease. Int J Mol Sci. 2019;20:834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Evans S, McRae‐McKee K, Wong MM, Hadjichrysanthou C, De Wolf F, Anderson R. The importance of endpoint selection: how effective does a drug need to be for success in a clinical trial of a possible Alzheimer's disease treatment?. Eur J Epidemiol. 2018;33:635‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roberts C, Torgerson DJ. Understanding controlled trials: baseline imbalance in randomised controlled trials. Bmj. 1999;319:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rogers SL, Doody RS, Mohs RC, Friedhoff LT. Donepezil improves cognition and global function in Alzheimer disease: a 15‐week, double‐blind, placebo‐controlled study. Donepezil study group. Arch Intern Med. 1998;158:1021‐1031. [DOI] [PubMed] [Google Scholar]
- 38. Jack CR Jr, Bennett DA, Blennow K, Research Framework NIA‐AA. Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐β biomarkers for Alzheimer's disease. Nature. 2018;554:249‐254. [DOI] [PubMed] [Google Scholar]
- 40. Janelidze S, Stomrud E, Palmqvist S, et al. Plasma β‐amyloid in Alzheimer's disease and vascular disease. Sci Rep. 2016;6:26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lue LF, Sabbagh MN, Chiu MJ, et al. Plasma levels of Aβ42 and tau identified probable Alzheimer's Dementia: findings in two cohorts. Front Aging Neurosci. 2017;9:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Öhrfelt A, Johansson P, Wallin A, et al. Increased cerebrospinal fluid levels of ubiquitin carboxyl‐terminal hydrolase l1 in patients with Alzheimer's disease. Dement Geriatr Cogn Dis Extra. 2016;6:283‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang KK, Yang Z, Sarkis G, Torres I, Raghavan V. Ubiquitin C‐terminal hydrolase‐L1 (UCH‐L1) as a therapeutic and diagnostic target in neurodegeneration, neurotrauma and neuro‐injuries. Expert Opin Ther Targets. 2017;21:627‐638. [DOI] [PubMed] [Google Scholar]
- 44. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1‐15 e1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Properzi MJ, Buckley RF, Chhatwal JP, et al. Nonlinear Distributional Mapping (NoDiM) for harmonization across amyloid‐PET radiotracers. Neuroimage. 2019;186:446‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim NK, Choi BH, Huang X, et al. Granulocyte‐macrophage colony‐stimulating factor promotes survival of dopaminergic neurons in the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine‐induced murine Parkinson's disease model. Eur J Neurosci. 2009;29:891‐900. [DOI] [PubMed] [Google Scholar]
- 47. Mangano EN, Peters S, Litteljohn D, et al. Granulocyte macrophage‐colony stimulating factor protects against substantia nigra dopaminergic cell loss in an environmental toxin model of Parkinson's disease. Neurobiol Dis. 2011;43:99‐112. [DOI] [PubMed] [Google Scholar]
- 48. Kosloski LM, Kosmacek EA, Olson KE, Mosley RL, Gendelman HE. GM‐CSF induces neuroprotective and anti‐inflammatory responses in 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine intoxicated mice. J Neuroimmunol. 2013;265:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gendelman HE, Zhang Y, Santamaria P, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double‐blind phase 1 clinical Parkinson's disease trial. NPJ Parkinsons Dis. 2017;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Potter H, Wisniewski T. Apolipoprotein e: essential catalyst of the Alzheimer amyloid cascade. Int J Alzheimers Dis. 2012;2012:489428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information