

Recent studies have revealed the existence of numerous and complex biological roles of RNA,[1–3] eliciting the need for chemical tools that can aid in analyzing biomolecular structure and interactions.[4,5] RNA has been shown to play a central role in many important regulatory networks.[6,7] For example, interactions between non-coding RNAs (ncRNA) and proteins are crucial for transcriptional regulation,[8] chromatin modifications[9] and post-translational modifications,[10] among others.[10] Riboswitches interact with small-molecule substrates to regulate gene expression,[11] such as the thiamine pyrophosphate (TPP) riboswitch in bacteria and several eukaryotes.[12,13] Further, RNA can interact with other RNA molecules to regulate RNA splicing,[14] translation[15] and modification.[16] For example, small nucleolar RNAs (snoRNAs) have been shown to guide modifications on ribosomal RNA (rRNA) through base pairing.[16] Other prominent examples include the binding of miRNAs to messenger RNAs to control translation,[17] and the dimerization of viral RNA genomes.[18] However, it has been challenging to study such RNA-RNA interactions due to the transient nature of most of these complexes,[19–21] and many such interactions likely remain uncharacterized.
The majority of methods for studying nucleic acid interactions rely on chemical crosslinking.[22–25] The most widespread example of this is formaldehyde crosslinking,[26] which involves adducts to exocyclic amine groups; while it is routinely used for preserving RNA in clinical settings and for studying biomolecular interactions,[27] it suffers from low recovery yields.[27,28] Other amine-reactive aldehydic crosslinkers include 1,4-phenyl-diglyoxal.[29] More selective crosslinking methods have been developed more recently, including the use of modified nucleobases such as 4-thiouridine,[30] phenyl selenide uridine[31] and carbazole derivatives,[32] but the need to incorporate these modifications in the RNA of interest prior to use hinders their application in native settings. Furthermore, most of these methods form irreversible crosslinks, which can interfere with downstream analysis of crosslinked RNA complexes. This problem can be overcome by applying small molecule crosslinkers such as psoralen,[33,34] which was discovered to crosslink opposing pyrimidines in duplex nucleic acids upon irradiation with 365 nm light.[33] Exposure to 254 nm light reverses the crosslink, and this forms the basis for several recent transcriptome-wide methods for interrogation of RNA-RNA interactions like psoralen analysis of RNA interactions and structures (PARIS)[35] and ligation of interacting RNA followed by high-throughput sequencing (LIGR-seq).[36] However, psoralen crosslinking can suffer from low crosslink efficiency[37] and modest recovery yields,[38] and is biased towards pyrimidine bases.[39]
To overcome these obstacles, we describe here a new molecular approach to crosslinking transient RNA-RNA interactions. Our design involves bis-nicotinic azide reversible interaction (BINARI) probes bearing two electrophilic moieties that acylate 2’-OH groups of nearby ribonucleotides,[5] resulting in a covalent link between interacting RNA strands (Fig. 1). Phosphine-mediated reduction of azide trigger groups conveniently reverses the crosslink, enabling downstream analysis of individual components of RNA complexes. We find that crosslinks are formed in high yields of up to 84% with minimal sequence bias and are readily reversed at up to 70% yield. BINARI is also functional in complex biological media such as cell lysates. We show that RNA can be temporarily protected from nuclease degradation by BINARI crosslinking, which might prove useful for RNA handling and storage applications. Finally, the utility of BINARI is demonstrated by trapping transient RNA-RNA interactions of hairpins derived from the E. coli DsrA-rpoS RNA complex. The captured RNA-RNA complexes are readily reversed to identify the individual components. We expect that this method will find use for studying both inter- and intra-molecular RNA-RNA interactions.
Figure 1.
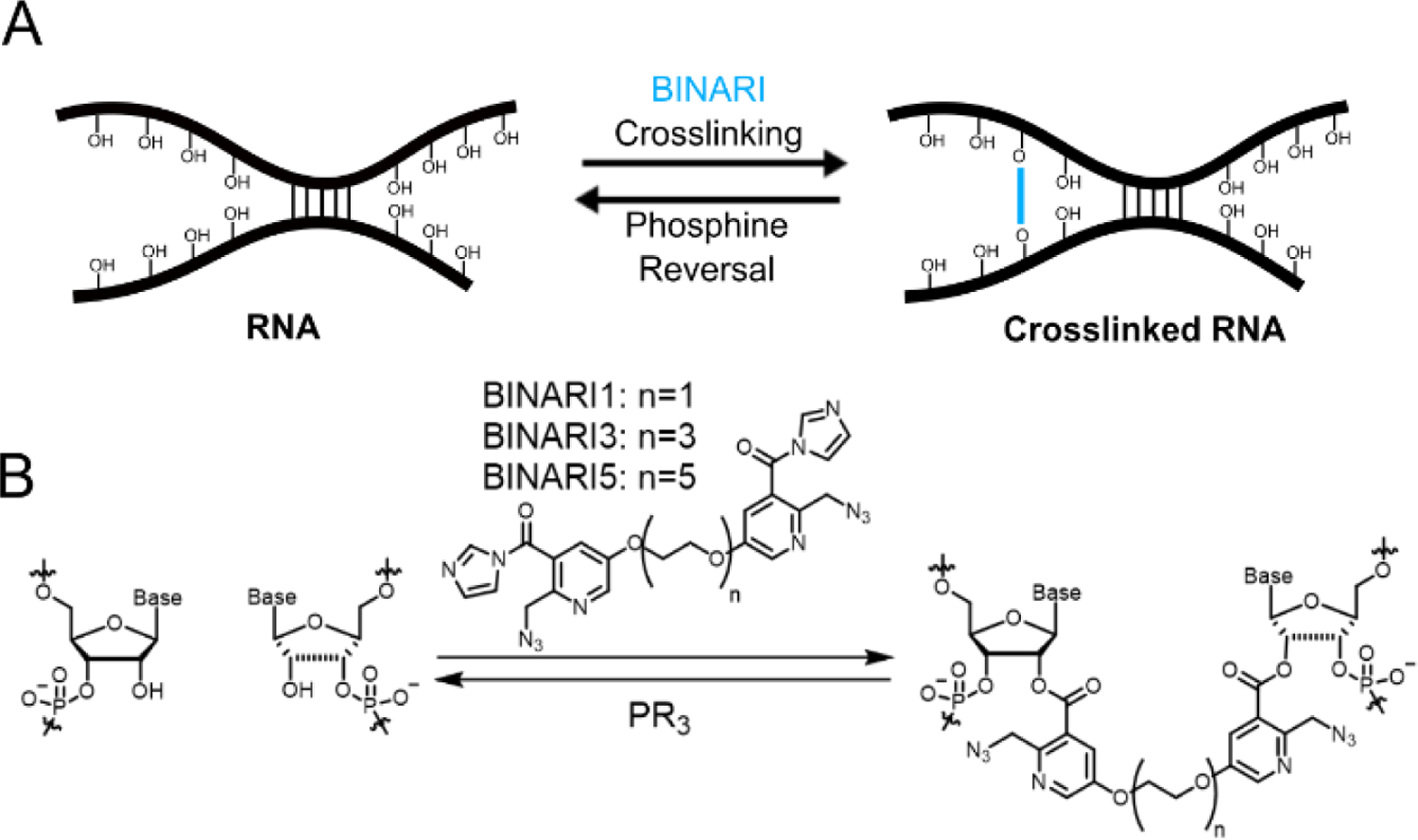
RNA crosslinking through reversible bis-acylation. A) Schematic diagram of RNA strands crosslinked with BINARI probes and later crosslink reversal upon phosphine treatment. B) Molecular structure of BINARI probes and crosslinking through bis-acylation of 2'-OH groups of spatially proximal nucleotides. Phosphine-mediated reduction of the azide groups results in intramolecular lactam formation and liberation of the crosslinked RNA strands.
Our design of a chemically reversible crosslinker relies on high-yield chemistry for 2’-OH acylation as well as a strategy for efficient reversal. We recently reported the azide-substituted RNA acylating reagent NAI-N3,[40] which was able to modify a large fraction of 2’-OH positions of model RNA strands,[41] and this reaction was shown to be reversible upon treatment with phosphines.[41] For the new design, we envisioned chemically related reversible acylating moieties that are linked via polyethylene glycol (PEG) spacers. Three crosslinkers were prepared with increasing spacer length: PEG1, PEG3 and PEG5 (Fig. 1B). The compounds were synthesized from ethyl 5-nitro-2-methyl-nicotinate (1) (Scheme 1), which was converted to phenolic compound 3 via a consecutive reduction and Sandmeyer reaction. TBDMS protection and bromination afforded compound 5, which was reacted with sodium azide and deprotected with TBAF to obtain intermediate 7. Reaction with electrophile-modified PEG linkers resulted in compounds 8a, 8b and 8c, which were converted to the final crosslinkers by hydrolyzing the esters and activating the acid with carbonyldiimidazole.
Scheme 1.
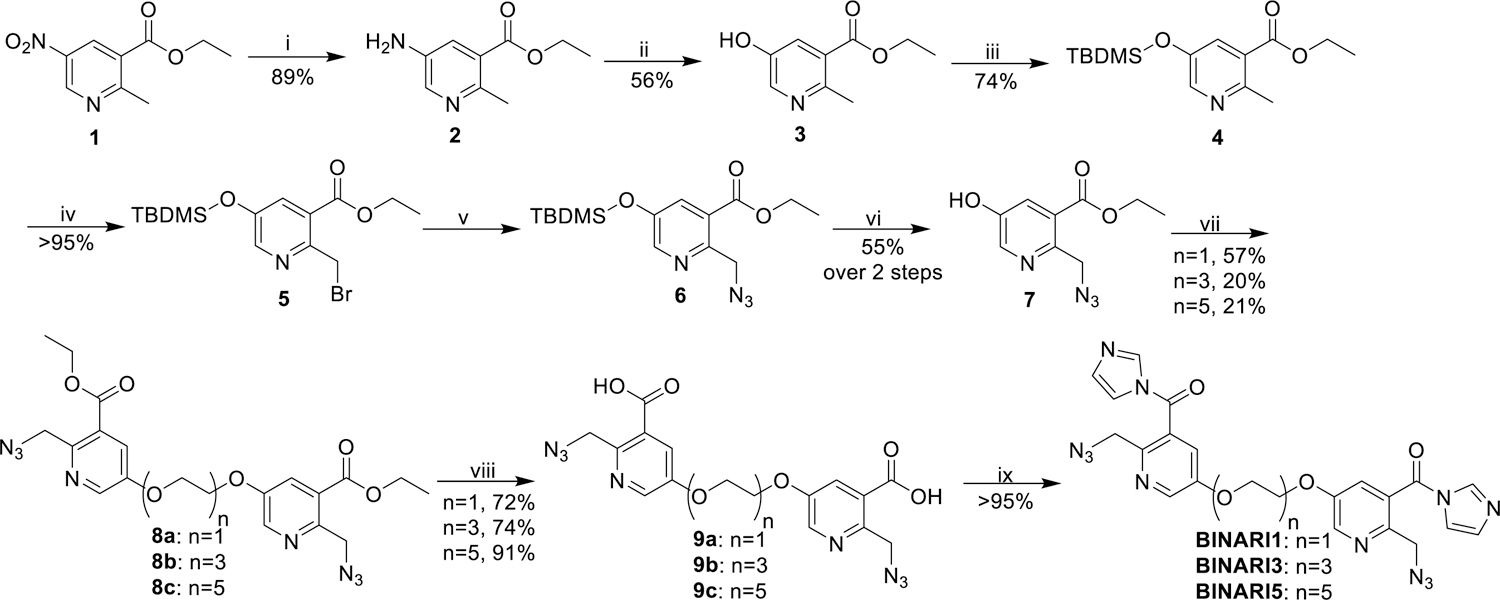
Synthesis of BINARI probes. Reagents and conditions: (i) Pd/C 10%, H2, MeOH, 40 °C, 48 h; (ii) NaNO2, H2O, H2SO4, 0 °C-100 °C, 30 min; (iii) TBDMSCl, imidazole, CH2Cl2, rt, 2h; (iv) NBS, AIBN, CCl4, 70 °C, 3 h; (v) NaN3, DMF, rt, 16 h; (vi) TBAF, THF, rt, 48 h; (vii) Tf(C2H4O2)n, DIPEA, CH2Cl2, 0 °C-rt, 2 h; (viii) NaOH, H2O, MeOH, rt, 16 h; (ix) CDI, DMSO, rt, 1 h.
An in vitro test with model RNA strands was executed to assess the performance of the crosslinkers. The partially self-complementary short RNA 1 (10 μM), which is complementary via only 6 base pairs, (Fig. 2A) was incubated with BINARI 1, 3, or 5 (100 mM) in 100 mM MOPS buffer (pH= 7.5, 6 mM MgCl2, 100 mM NaCl) for 4 hours at room temperature. RNA was purified by precipitation and the amount of crosslinking was analyzed by 20% polyacrylamide gel-electrophoresis (PAGE) (Fig. 2B), quantified by the intensity of the bands. Under these conditions BINARI 3 and BINARI 5 achieved a high degree of crosslinking into the dimeric form (84% and 75% respectively), while BINARI 1 provided a lesser 45% crosslinking yield, which may reflect less-than-optimal linker length to bridge the two reactive RNA groups (Fig. 2C and Fig. S1). These results suggest that significant distance between the two acylating groups is required for most efficient crosslinking. This is in accordance with earlier findings that acylation of RNA mainly occurs in non-base-paired regions,[5] and as such the reactive 2’-OH groups may be further apart than they are in RNA duplexes. This hypothesis is further supported by experiments showing decreased crosslinking efficiency when the optimized conditions were applied to RNA 2 (Fig. S4), which has the same base pairing sequence as RNA 1, but lacks the noncomplementary tails. The experiments indicate that crosslinking mostly occurs in non-base paired regions, but does rely on duplex formation to bring opposing strands in close proximity. When the analogous DNA variant of RNA 1 was used, no crosslinking was observed (Fig. S4), which demonstrates that BINARI is selective for RNA-RNA interactions. To conclusively demonstrate that the lower mobility band observed by PAGE is crosslinked RNA, Cy5-labeled RNA S2 was treated with BINARI 3 alone or in the presence of complementary RNA S1 (Fig. S5). Imaging using a Cy5-fluorescence filter set showed that the lower mobility band is only observed when both complementary RNA strands are present, implying that this band is indeed crosslinked RNA and not monofunctionalized RNA (Fig. S5).
Figure 2.
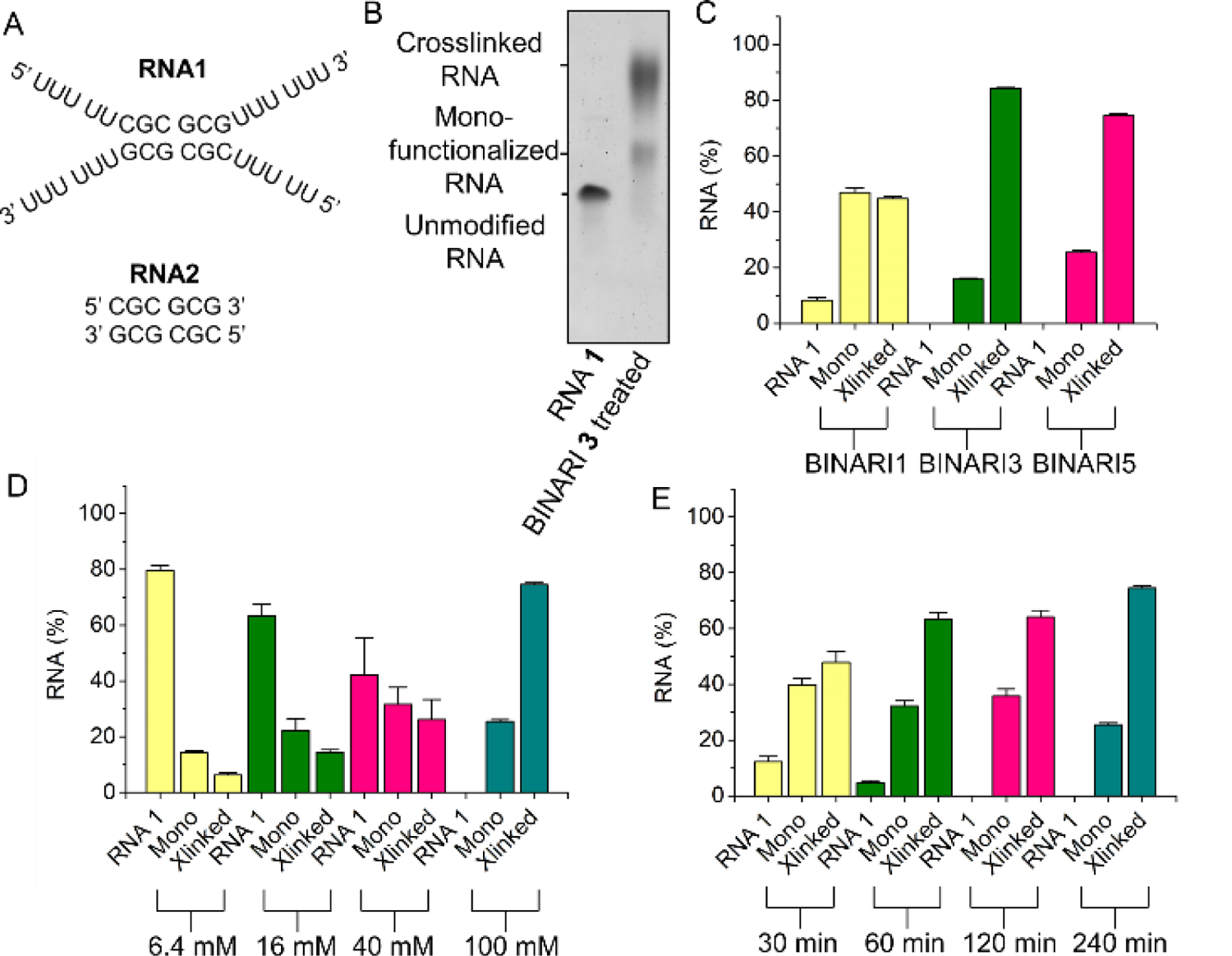
RNA crosslinking. A) Sequences of self-complementary RNA strands in this experiment. B) PAGE analysis of BINARI 3 crosslinked RNA 1. C) Crosslinking yields of the different BINARI probes with self-complementary RNA 1. D) Crosslinking yields of BINARI 5 at different concentrations. E) Crosslinking yields of BINARI 5 at different incubation times. Error bars represent s.d. of triplicate experiments.
Crosslinking conditions were optimized using BINARI 5 with the dimeric RNA 1. Testing increasing concentrations of crosslinker showed that the greatest amount of crosslinked RNA was formed, not surprisingly, at the highest concentration tested (100 mM) (Fig. 2D and Fig. S2), and increased incubation time resulted in more efficient crosslinking, which plateaued after about 4 hours (Fig. 2E and Fig. S3). Next, we tested if the crosslinked RNA complex could be reversed into the original single stranded RNA (ssRNA) upon phosphine treatment. 4-(Diphenylphosphino)benzoic acid (DPBA), triphenylphosphino-3-sulfonic acid sodium salt (TPPMS) and tris(hydroxypropyl) phosphine (THPP) (Fig. 3A) as initial candidates for reductants[41] were incubated with the crosslinked RNA 1 complex for 2 hours at room temperature and analyzed by PAGE (Fig. 3B).
Figure 3.
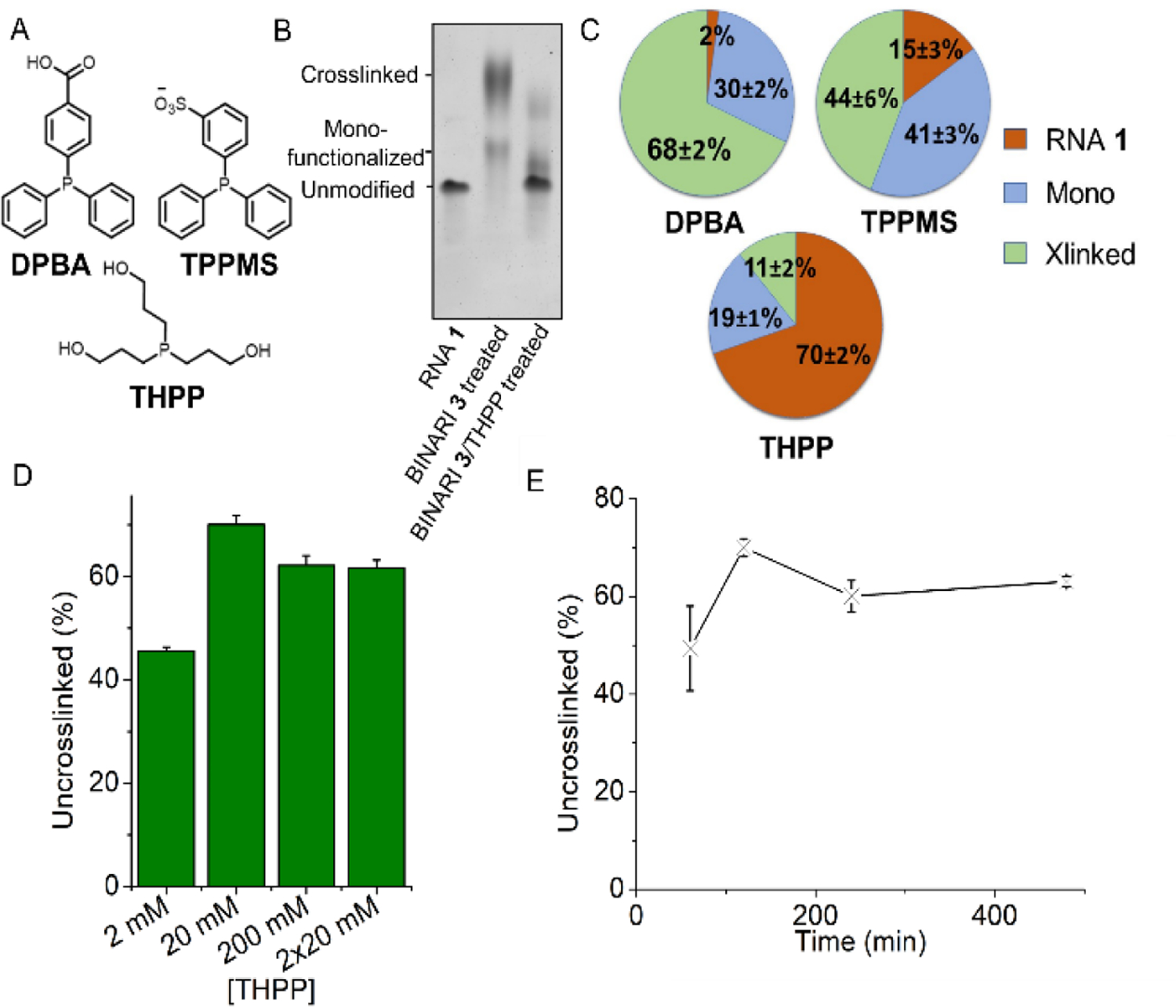
Reversal of RNA crosslinks. A) Structures of tested phosphines. B) PAGE analysis of BINARI 3 crosslinked RNA 1 and THPP reversal of crosslinked RNA 1. C) Amount of uncrosslinked- monofunctionalized and crosllinked RNA 1 obtained after incubation with each of three phosphines. D) Yields of the crosslink reversal reactions at varied concentrations of THPP. E) Amount of uncrosslinked RNA obtained as a function of incubation times with THPP. Error bars represent s.d. of triplicate experiments.
Treatment with DPBA and TPPMS resulted in low yields of 2% and 15% of unmodified RNA 1 respectively, while THPP efficiently reversed the crosslink, resulting in 70% recovery of unmodified RNA 1 (Fig. 3C and Fig. S6). The difference in reversal efficiency between the tested phosphines can potentially be explained by the more flexible nature and lower steric bulk of THPP compared to DPBA and TPPMS, which could allow for it to access sterically hindered crosslinks.
Increasing the concentration of THPP showed that maximum reversal was reached at a concentration of 20 mM (Fig. 3D and Fig. S7). Since THPP can oxidize during the reversal reaction, we checked whether a second treatment with 20 mM THPP can result in higher crosslinking reversal, which was not the case. Increasing the THPP incubation time showed that maximum reversal was reached after 2 hours (Fig. 3E and Fig. S8). The optimized conditions resulted in up to 84% crosslinking and 70% reversal, which compares favorably to known psoralen crosslink reactions that yield mostly below 50% and more typically less than 10% crosslinking[37,42] and exhibit low reversal efficiency.[43]
The ribose structure of RNA renders it susceptible to nuclease catalyzed hydrolysis.[44] This makes handling and storing RNA for biomedical applications particularly challenging. We envisioned that by applying BINARI, RNA could temporarily be stabilized and protected from nuclease degradation. Subsequent reversal of the crosslink reaction would liberate the native ssRNA for further downstream applications. Since BINARI likely locks RNA in a partly double stranded complex, it was anticipated that it could provide protection against single-strand-selective nucleases. Using the optimized crosslinking conditions, we tested RNAse T1 and S1 nuclease. Five μM RNA 1 or BINARI 3-crosslinked RNA 1 in the appropriate buffer (see SI for details) was incubated with S1 Nuclease or RNAse T1 for 10 minutes at room temperature. The reaction was stopped by the addition of 7 M urea and cooled on ice. PAGE analysis of the enzymatic reactions (Fig. 4) showed that both nucleases fully digested unmodified RNA 1. Significantly, crosslinked RNA 1 remained largely intact upon treatment with nucleases, providing evidence that the BINARI crosslink does provide protection against nuclease degradation. RNAse T1 appears to hydrolyze mono-functionalized RNA 1 as well (Fig. 4), whereas S1 nuclease mostly hydrolyzes unmodified RNA 1.
Figure 4.
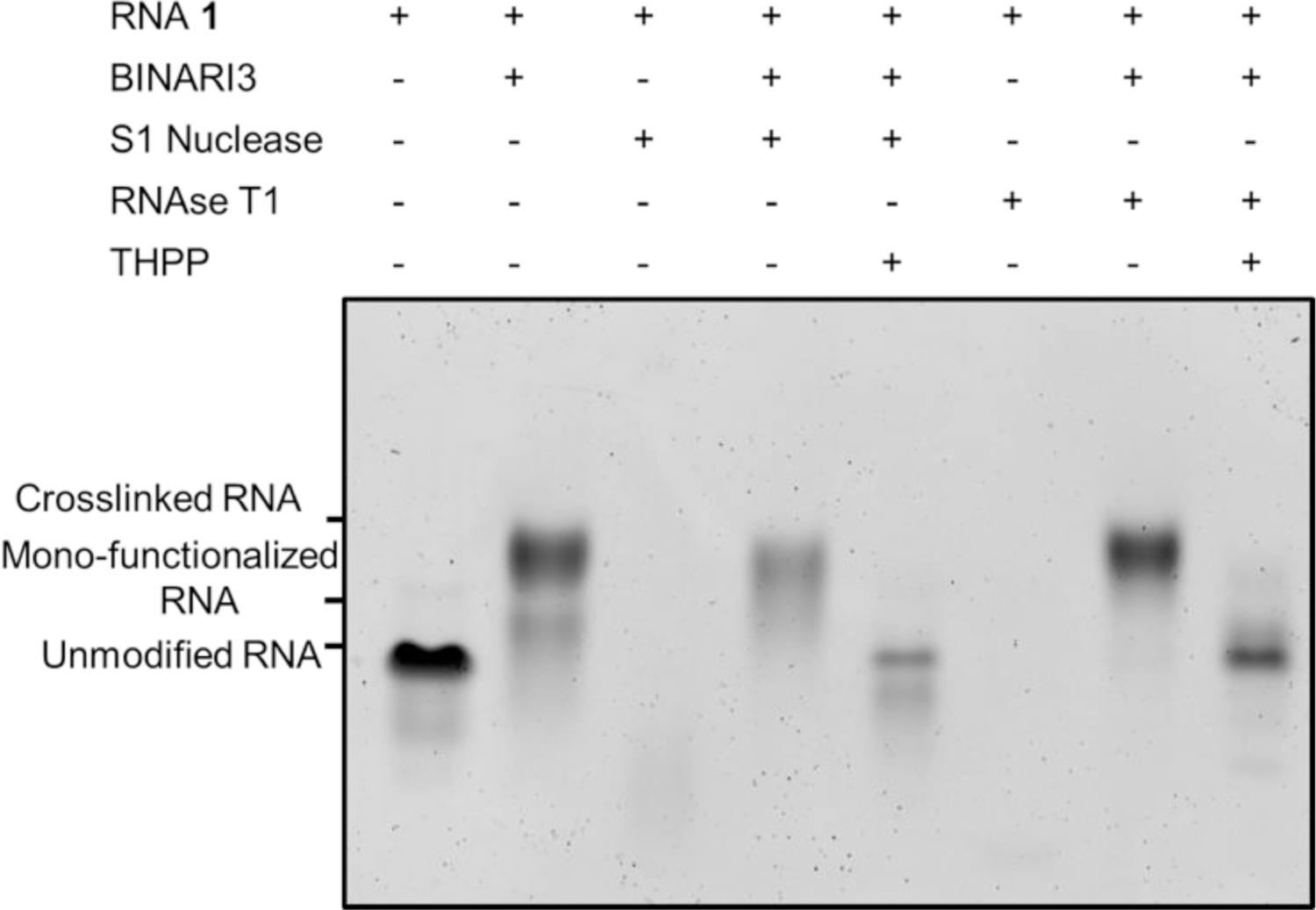
Gel electrophoretic analysis of temporary protection against nucleases. S1 nuclease and RNAse T1 fully digest RNA1, whereas BINARI 3 crosslinked RNA 1 remains largely intact. Reversal of the crosslink with THPP (20 mM) efficiently liberates native RNA 1.
Subsequent treatment of crosslinked RNA 1 with THPP (20 mM) in water liberated the native ssRNA 1 (Fig. 4).These results indicate that BINARI could potentially be used to protect RNA from nuclease degradation; future experiments will explore the applications of BINARI for RNA preservation purposes, which is highly important for clinical sample storage[28].
One outstanding challenge in RNA structural and functional analysis is to capture and analyze transient RNA complexes.[45,46] One such class of interactions are loop-loop interactions in bacterial and viral systems, which can occur when two hairpins have sequence complementarity in their loops.[47] For example, the HIV-1 virus genomic RNA is dimerized via “kissing loop” interactions.[48] A second case is DsrA RNA in Escherichia coli (E.coli), which has been shown to regulate translation of rpoS mRNA, a sigma factor, through a bulge-loop interaction, which initiates a strand-displacement reaction.[49,50] To test the utility of BINARI probes for analysis of such a transient and relatively weak RNA interaction, we applied the crosslinking approach to capture this RNA-RNA complex. It was hypothesized that BINARI can acylate 2´-OH groups adjacent to the complementary region on both sides of the complex. Three hairpin RNAs were designed based on the DsrA-rpos interaction[50] (Fig. 5A). HP1 has full complementarity to HP3 in its loop region, while HP2 has two nucleotide deletions in its loop. Importantly, none of the potential complexes have complementarity in the stem region, excluding the formation of an extended duplex such as occurs with HIV dimerization. One challenging aspect of studying transient complexes is to identify the individual components after capturing the interaction. This is made difficult in part with poorly reversible crosslinking chemistries, where the chemical modification can prevent molecular analysis, and is also particularly challenging when interactions between multiple components are possible, which would be the case in a cellular context.
Figure 5.
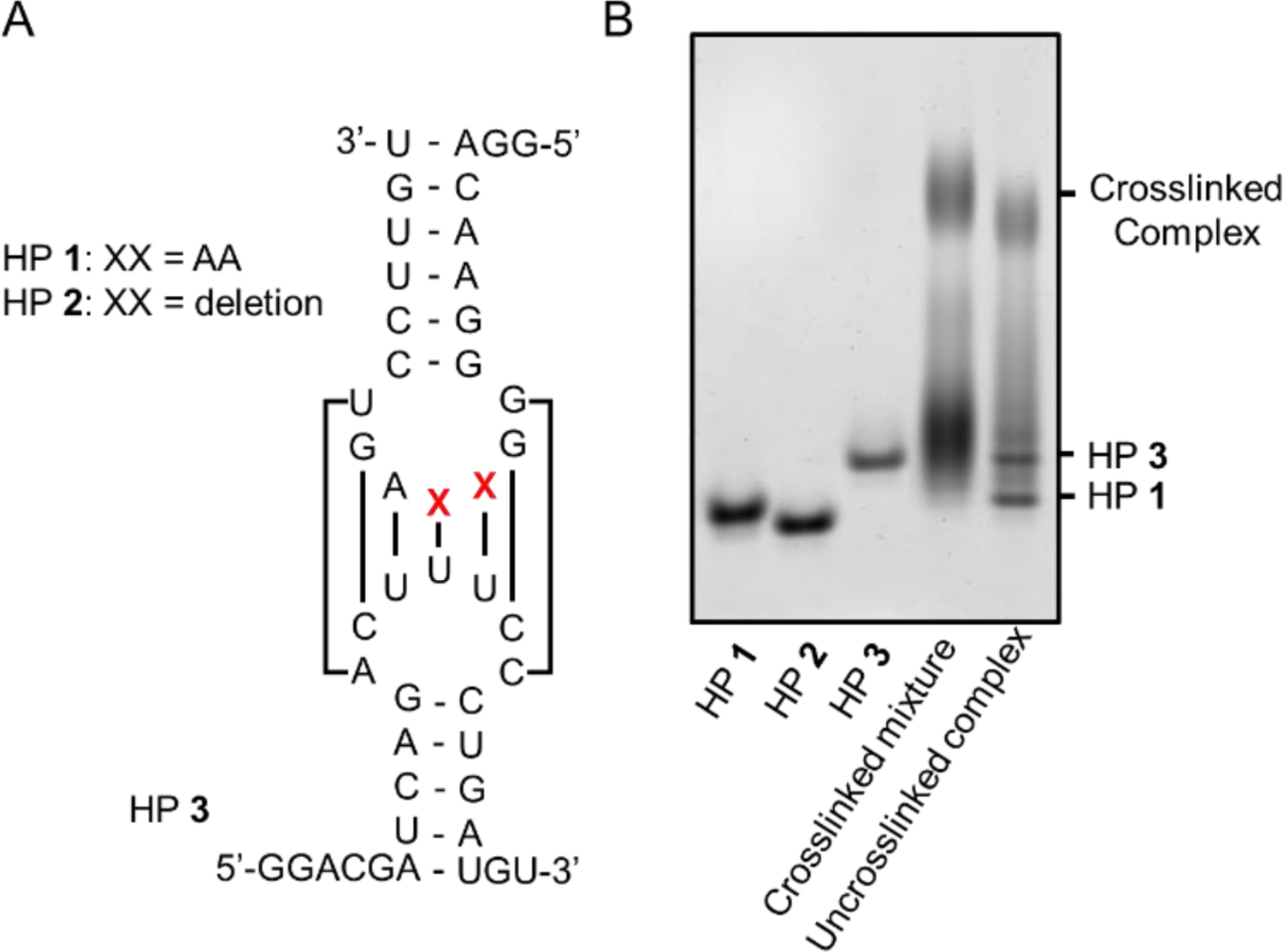
Structures of DraS-rpoS derived hairpins and analysis of their transiently formed complexes with BINARI. A) Sequence and structure of the DraS-rpoS derived hairpins HP1, HP2 and HP3. HP1 and HP3 have full complementary in their loop regions, while HP2 has a two-nucleotide deletion. B) PAGE analysis of the BINARI 3-captured RNA complex. THPP reversal reveals that the formed complex solely consists of HP1 and HP3.
To test this challenge, HP1, HP2 and HP3 were mixed at 10 μM in 100 mM MOPS buffer (pH= 7.5, 6 mM MgCl2, 100 mM NaCl) for 4 hours at room temperature with BINARI 3 (100 mM). Analysis of the reaction by 20% PAGE showed a clear band with decreased mobility (Fig. 5B), indicative of a captured RNA dimeric complex. The band was excised from the gel and the extracted RNA complex was treated with THPP (100 mM) in water for 2 hours at room temperature to reverse the crosslink. Interestingly, when the isolated and reversed complex was analyzed by PAGE, two clear bands were observed that correspond in mobility to HP1 and HP3, which is in accordance with superior sequence complementarity of HP1-HP3 compared to a potential HP2-HP3 complex as well as three other possible self-dimeric complexes. Potential crosslinking of self-dimeric complexes was ruled out by treating individual HPs with BINARI 3 (Fig. S9). This experiment illustrates the need for efficient crosslinking to capture a transient complex, and underlines the potential for employing BINARI crosslinkers to probe RNA-RNA transient and weak interactions more widely. The reversibility of the method allows for further downstream analysis.
BINARI crosslinking and reversal efficiency was compared to psoralen with two additional partially self-complementary RNA sequences. The effects of different overhanging bases neighboring a duplex (RNA 1-A) and different internal sequences (RNA S3) were examined. BINARI displayed high crosslinking yield for all three sequences, and the crosslinks were reversed successfully with THPP with relatively small sequence bias (Fig. S10). On the other hand, psoralen displayed lower crosslinking yields across all sequences and even failed to crosslink RNA 1-A, confirming psoralen’s bias for uridines (Fig. S11). Whereas BINARI uncrosslinking showed little RNA degradation, reversal of psoralen crosslinks with 254 nm UV resulted in poor recovery of the original RNA bands, likely due to UV induced degradation.[51–53] One key advantage of psoralen is its compatibility with biological environments (e.g.such as cell lysate or intact cells.[35,36] To test whether BINARI is also functional in such complex mixtures, RNA crosslinking was conducted in HeLa whole cell lysate (Fig. S12). BINARI 3 efficiently cross-linked both a fluorescently labeled self-complementary RNA (Cy5-RNA 1) and separate complementary RNAs (RNA S1 and S2) without loss of efficiency when compared to buffer. Taken together, BINARI displays superior perfomance with minimal sequence bias for crosslinking and high reversal recovery over psoralen and compatibility with cell lysates.
In summary, the BINARI probe design constitutes a new reversible crosslinking strategy for RNA. The presented approach offers superior crosslinking yields compared to existing methods with minimal sequence bias, and is selective for RNA-RNA interactions with no detectable crosslinking of duplex DNA. It is also compatible with complex biological media such as cell lysates. We have shown that BINARI-crosslinked RNA is temporarily protected from nuclease degradation and that crosslink reversal liberates the native RNA, suggesting future applications in storage and preservation of RNA. Furthermore, BINARI was applied successfully to capture transient RNA complexes of E.coli DsrA-rpos derived hairpins, and the efficient crosslink reversal reaction enabled the identification of individual RNA components of the complex. We anticipate that the future combination of this reversible crosslinking approach with high-throughput sequencing technologies could aid in mapping the RNA interactome.
Supplementary Material
Acknowledgements
We are grateful for support from the U.S. National Institutes of Health (R01GM127295 and R43ES028559).
Contributor Information
Willem A. Velema, Institute for Molecules and Materials, Radboud University, 6525 AJ Nijmegen, The Netherlands
Hyun Shin Park, Department of Chemistry, Stanford University, Stanford, CA 94305, USA.
Anastasia Kadina, Department of Chemistry, Stanford University, Stanford, CA 94305, USA.
Lucian Orbai, Cell Data Sciences, 46127 Landing Parkway, Fremont, CA 94538, USA.
Eric T. Kool, Department of Chemistry, Stanford University, Stanford, CA 94305, USA
Literature Cited
- [1].Cech TR, Steitz JA, Cell 2014, 157, 77–94. [DOI] [PubMed] [Google Scholar]
- [2].He C, Nat Chem Biol 2010, 6, 863–865. [DOI] [PubMed] [Google Scholar]
- [3].Yao R-W, Wang Y, Chen L-L, Nat. Cell Biol 2019, 21, 542–551. [DOI] [PubMed] [Google Scholar]
- [4].Li F, Dong J, Hu X, Gong W, Li J, Shen J, Tian H, Wang J, Angew. Chemie - Int. Ed 2015, 54, 4597–4602. [DOI] [PubMed] [Google Scholar]
- [5].Velema WA, Kool ET, Nat. Rev. Chem 2020, 4, 22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Waters LS, Storz G, Cell 2009, 136, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Anastasiadou E, Jacob LS, Slack FJ, Nat. Rev. Cancer 2017, 18, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Long Y, Wang X, Youmans DT, Cech TR, Sci. Adv 2017, 3, eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Han P, Chang C-P, RNA Biol 2015, 12, 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Geisler S, Coller J, Nat. Rev. Mol. Cell Biol 2013, 14, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Serganov A, Nudler E, Cell 2013, 152, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mayer G, Raddatz M-SL, Grunwald JD, Famulok M, Angew. Chemie Int. Ed 2007, 46, 557–560. [DOI] [PubMed] [Google Scholar]
- [13].Li S, Breaker RR, Nucleic Acids Res 2013, 41, 3022–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Matlin AJ, Clark F, Smith CWJ, Nat. Rev. Mol. Cell Biol 2005, 6, 386–398. [DOI] [PubMed] [Google Scholar]
- [15].Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V, Science (80-. ) 2006, 313, 1935–1942. [DOI] [PubMed] [Google Scholar]
- [16].Watkins NJ, Bohnsack MT, Wiley Interdiscip. Rev. RNA 2012, 3, 397–414. [DOI] [PubMed] [Google Scholar]
- [17].Djuranovic S, Nahvi A, Green R, Science (80-. ) 2012, 336, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dubois N, Marquet R, Paillart J-C, Bernacchi S, Front. Microbiol 2018, 9, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang SY, Lejault P, Chevrier S, Boidot R, Robertson AG, Wong JMY, Monchaud D, Nat. Commun 2018, 9, 4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gong J, Ju Y, Shao D, Zhang QC, Quant. Biol 2018, 6, 239–252. [Google Scholar]
- [21].Lu Z, Chang HY, Curr. Opin. Struct. Biol 2016, 36, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hong IS, Greenberg MM, J. Am. Chem. Soc 2005, 127, 3692–3693. [DOI] [PubMed] [Google Scholar]
- [23].Shigdel UK, Zhang J, He C, Angew. Chemie Int. Ed 2008, 47, 90–93. [DOI] [PubMed] [Google Scholar]
- [24].Liu J, Cai L, Sun W, Cheng R, Wang N, Jin L, Rozovsky S, Seiple IB, Wang L, Angew. Chemie - Int. Ed 2019, n/a, DOI 10.1002/anie.201910135. [DOI] [PMC free article] [PubMed]
- [25].Shelbourne M, Brown T, El-Sagheer AH, Brown T, Chem. Commun 2012, 48, 11184–11186. [DOI] [PubMed] [Google Scholar]
- [26].Huang H, Hopkins PB, J. Am. Chem. Soc 1993, 115, 9402–9408. [Google Scholar]
- [27].Do H, Dobrovic A, Clin. Chem 2015, 61, 64–71. [DOI] [PubMed] [Google Scholar]
- [28].Karmakar S, Harcourt EM, Hewings DS, Scherer F, Lovejoy AF, Kurtz DM, Ehrenschwender T, Barandun LJ, Roost C, Alizadeh AA, Kool ET, Nat. Chem 2015, 7, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wagner R, Garrett RA, Nucleic Acids Res 1978, 5, 4065–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen K, Lu Z, Wang X, Fu Y, Luo G-Z, Liu N, Han D, Dominissini D, Dai Q, Pan T, He C, Angew. Chemie Int. Ed 2015, 54, 1587–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sloane JL, Greenberg MM, J. Org. Chem 2014, 79, 9792–9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fujimoto K, Yamada A, Yoshimura Y, Tsukaguchi T, Sakamoto T, J. Am. Chem. Soc 2013, 135, 16161–16167. [DOI] [PubMed] [Google Scholar]
- [33].Calvet JP, Pederson T, Proc. Natl. Acad. Sci 1979, 76, 755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stafforst T, Hilvert D, Angew. Chem. Int. Ed. Engl 2010, 49, 9998–10001. [DOI] [PubMed] [Google Scholar]
- [35].Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, Mesirov JP, Cech TR, Chang HY, Cell 2016, 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sharma E, Sterne-Weiler T, O’Hanlon D, Blencowe BJ, Mol. Cell 2016, 62, 618–626. [DOI] [PubMed] [Google Scholar]
- [37].Zhen WP, Buchardt O, Nielsen H, Nielsen PE, Biochemistry 1986, 25, 6598–6603. [DOI] [PubMed] [Google Scholar]
- [38].Stadler JM, Stafforst T, Org. Biomol. Chem 2014, 12, 5260–5266. [DOI] [PubMed] [Google Scholar]
- [39].Good L, Awasthi SK, Dryselius R, Larsson O, Nielsen PE, Nat. Biotechnol 2001, 19, 360–364. [DOI] [PubMed] [Google Scholar]
- [40].Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung J-W, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, Chang HY, Nature 2015, 519, 486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kadina A, Kietrys AM, Kool ET, Angew. Chemie - Int. Ed 2018, 57, 3059–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kirsch R, Olzog VJ, Bonin S, Weinberg CE, Betat H, Stadler PF, Mörl M, Biotechniques 2019, 67, 178–183. [DOI] [PubMed] [Google Scholar]
- [43].Gasparro F, Psoralen Dna Photobiology, Boca Raton: CRC Press, 1988. [Google Scholar]
- [44].Genna V, Marcia M, De Vivo M, J. Am. Chem. Soc 2019, 141, 10770–10776. [DOI] [PubMed] [Google Scholar]
- [45].Šponer J, Bussi G, Krepl M, Banáš P, Bottaro S, Cunha RA, Gil-Ley A, Pinamonti G, Poblete S, Jurečka P, Walter NG, Otyepka M, Chem. Rev 2018, 118, 4177–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Batey RT, Rambo RP, Doudna JA, Angew. Chemie Int. Ed 1999, 38, 2326–2343. [DOI] [PubMed] [Google Scholar]
- [47].Eguchi Y, Tomizawa J, Cell 1990, 60, 199–209. [DOI] [PubMed] [Google Scholar]
- [48].Paillart JC, Skripkin E, Ehresmann B, Ehresmann C, Marquet R, Proc. Natl. Acad. Sci 1996, 93, 5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Majdalani N, Cunning C, Sledjeski D, Elliott T, Gottesman S, Proc. Natl. Acad. Sci 1998, 95, 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Salim N, Lamichhane R, Zhao R, Banerjee T, Philip J, Rueda D, Feig AL, Biophys. J 2012, 102, 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ariza-Mateos A, Prieto-Vega S, Díaz-Toledano R, Birk A, Szeto H, Mena I, Berzal-Herranz A, Gómez J, Nucleic Acids Res 2012, 40, 1748–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jericević Z, Kućan I, Chambers RW, Biochemistry 1982, 21, 6563–6567. [DOI] [PubMed] [Google Scholar]
- [53].Wurtmann EJ, Wolin SL, Crit. Rev. Biochem. Mol. Biol 2009, 44, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.