

Abstract
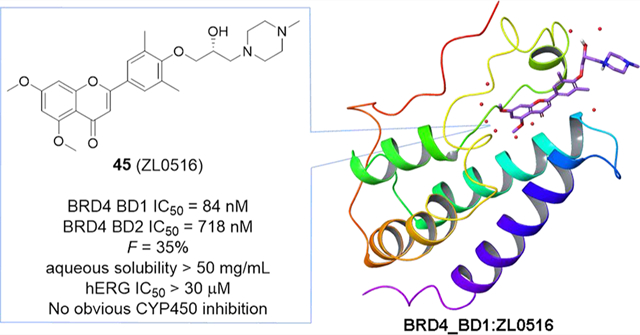
Bromodomain-containing protein 4 (BRD4) represents a promising drug target for anti-inflammatory therapeutics. Herein, we report the design, synthesis, and pharmacological evaluation of novel chromone derivatives via scaffold hopping to discover a new class of orally bioavailable BRD4-selective inhibitors. Two potent BRD4 bromodomain 1 (BD1)-selective inhibitors 44 (ZL0513) and 45 (ZL0516) have been discovered with high binding affinity (IC50 values of 67–84 nM) and good selectivity over other BRD family proteins and distant BD-containing proteins. Both compounds significantly inhibited the expression of Toll-like receptor-induced inflammatory genes in vitro and airway inflammation in murine models. The cocrystal structure of 45 in complex with human BRD4 BD1 at a high resolution of 2.0 Å has been solved, offering a solid structural basis for its binding validation and further structure-based optimization. These BRD4 BD1 inhibitors demonstrated impressive in vivo efficacy and overall promising pharmacokinetic properties, indicating their therapeutic potential for the treatment of inflammatory diseases.
Graphical Abstract
INTRODUCTION
Epigenetic modifications on DNA, histones, and other nuclear proteins regulate gene expression, affect cellular differentiation, and contribute to human diseases.1,2 Lysine acetylation (KAc) is one of the most broadly studied post-translational modifications occurring on histone proteins. This highly dynamic process is regulated by opposing actions of histone acetyl transferases (HATs) and histone deacetylases (HDACs).3 Histone acetylation also provides binding sites for proteins, especially BD-containing proteins, to promote chromatin reorganization and transcription. There are 61 structurally homologous BDs present in 46 different proteins in the human proteome. BRD4 belongs to the BD and extraterminal (BET) family consisting of four members (BRD2, BRD3, BRD4, and BRDT). Similar to other subfamilies of BD-containing proteins, BET members function in epigenetic regulation of gene expression through binding to the KAc recognition pocket on histone tails and nonhistone proteins.4–6 BET family proteins, especially BRD4, have emerged as a promising epigenetic target for human diseases and conditions, including cancers,7 inflammations,8–14 HIV infection,15–17 heart failure,18 and CNS disorders.19
A number of BET inhibitors with different chemotypes (e.g., azepines, 3,5-dimethylisoxazoles, pyridones, and diazobenzene) have been discovered and developed as depicted in Figure 1.4,20 Azepine (+)-JQ1 (1, Figure 1) has been the most widely used BRD4 inhibitor, and its analogue I-BET762 (2) has been advanced into phase II human clinical trials for neoplasms.21,22 RVX-208 (3),23 a BET inhibitor selective for the second BD, has been enrolled into phase III clinical trials for high-risk cardiovascular disease patients with type 2 diabetes mellitus and low levels of high-density lipoprotein. Despite the fact that compound 3 demonstrated tolerability and safety, it failed to meet the primary endpoint—reduction in major adverse cardiovascular events (MACE).24 Both ABBV075 (4) and BMS986158 (5) are developed as pan-BET inhibitors in clinical trials for cancer therapy.25,26 It has been reported that BD2 inhibition (e.g., 3) only modestly affects BET-dependent gene transcription and BD1-selective inhibitors are urgently needed for elucidating bromodomain-specific functions. To date, the availability of BD1-selective inhibitors is very limited, let alone BRD4 BD1 specific ones.23 To improve the selectivity and extend the application of BRD4 inhibitors, we recently designed and synthesized a series of diphenyldiazene BRD4 inhibitors (e.g., ZL0454, 6 in Figure 1) that suppress the Toll-like receptor 3 (TLR3)-induced expression of proinflammatory genes (IL-6, ISG54, and Groβ) in human small airway epithelial cells in vitro, as well as TLR3-induced airway inflammation and neutrophilia in mouse models in vivo.8,9,12,13 Although compound 6 displayed good BRD4 inhibition (IC50 values of 49 and 35 nM against BRD4 BD1 and BRD4 BD2, respectively) and selectivity over other family members, its pharmacokinetic (PK) profile is not satisfactory with poor oral bioavailability (F = 0.46%) and metabolic stability as well as poor aqueous solubility (12.8 μg/mL at pH = 7).8 To further improve the physicochemical properties of BRD4 inhibitors based on our first-generation lead compound 6, two approaches were utilized by either replacing the N=N linker with moieties that have more favorable metabolic stability or tuning its core structure to alternative privileged scaffolds with better oral bioavailability. Given our proof-of-concept study of BRD4 in airway inflammation (3 as a positive control displaying moderate in vivo efficacy) and the clinically validated safety profile of compound 3, we believe through dedicated drug design and structural optimization, new analogues with unique scaffolds based on compound 3 could be achieved to significantly improve their oral bioavailability and the in vivo efficacy for the treatment of inflammatory diseases. Herein, we report our application of scaffold hopping to discover a new class of potent, selective, and orally available BRD4 inhibitors. The binding modes of them with BRD4 BD1 have been validated by the cocrystal structural analysis of the inhibitor in complex with human BRD4 BD1 protein.
Figure 1.
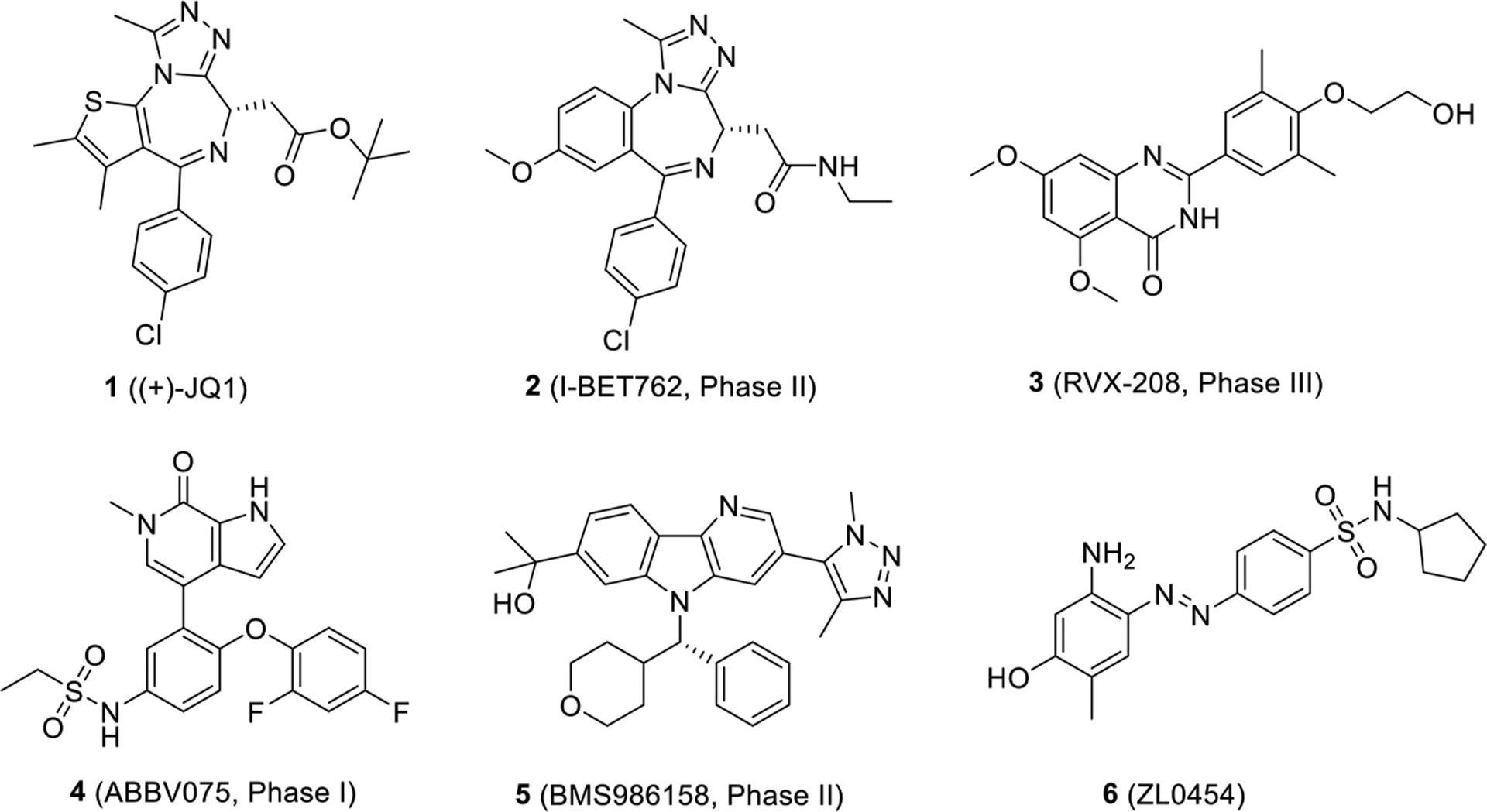
Chemical structures of representative BET inhibitors 1–5 and recently identified BRD4-selective inhibitor 6.
RESULTS AND DISCUSSION
Design.
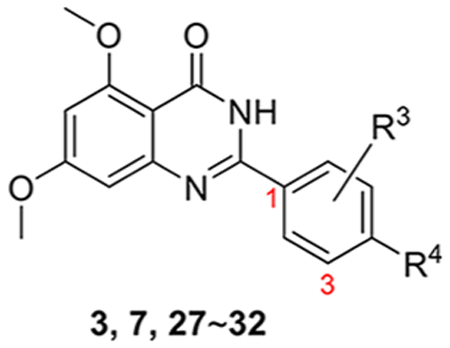
As depicted in Figure 2, the cocrystal structure of compound 3 in complex with BRD4 BD1 (PDB code: 4MR4) exhibited the lactam of the quinazolin-4-one core, especially the carbonyl oxygen atom, and formed critical H-bonds with Asn140 and Tyr97. The 3,5-dimethylphenol moiety of compound 3 does not reach the tryptophan-proline-phenylalanine (WPF) shelf but extends to the upper area of four helix bundle. Interestingly, its analogue RVX-OH (7, Figure 2) lacking a 2-hydroxyethyl group was found to interact with BRD4 BD1 in a completely reversed conformation.23 The quinazolin-4-one core extends to the WPF shelf area while the oxygen atom of the phenyl group inserted in the critical site forms direct H-bonds with Asn140 and an indirect interaction with Tyr97, exactly like compound 6. Such substantial difference may explain why compound 3 is BD2 selective while compound 7 has no preference between BD1 and BD2. Given that its close analogue compound 3 is clinically proven to be both safe and metabolically stable, we proposed to optimize substituents on the quinazolin-4-one core of compound 7 (Series I) and the 3,5-dimethylphenol moiety of compound 3 (Series II) to explore whether we can improve the BRD4 BD1 selectivity and achieve a paradigm shift in its clinical application from cardiovascular disease to various inflammatory diseases such as chronic obstructive pulmonary disease (COPD). To accomplish this objective, we used a scaffold hopping strategy to replace the quinazolin-4-one core with the chromen-4-one scaffold through fine tuning (Series III) to discover structurally novel BRD4 inhibitors with enhanced potency and BD selectivity as well as an improved drug metabolism pharmacokinetic (DMPK) profile. For series I compounds, they are expected to form the critical interactions with Asn140 and Tyr97 through free −OH like compound 7, and substitutions on the quinazolin-4-one core can reach the WPF shelf. The relevant structure–activity relationship (SAR) will be helpful in exploring the compatibility and possibility to improve potency and selectivity at this site. For series II and III compounds, we emphasized the investigations on the side chain, which is oriented to a short sequence from Lys91 to Asp 96 in a ZA loop which is believed critical for bromodomain selectivity.27 The short sequence for BRD4 BD1 is KLNLPD (91–96), while it is ALGLHD for BRD4 BD2, KLGLPD for BRD2 BD1, and ALGLHD for BRD2 BD2. They are not highly conserved, especially Asn93, which is unique for BRD4 BD1. We envisioned that incorporating proper side chains on the single phenyl ring of series II and III that can critically interact with KLNLPD of the BRD4 BD1 domain may achieve a better BD1 selectivity of BRD4.
Figure 2.
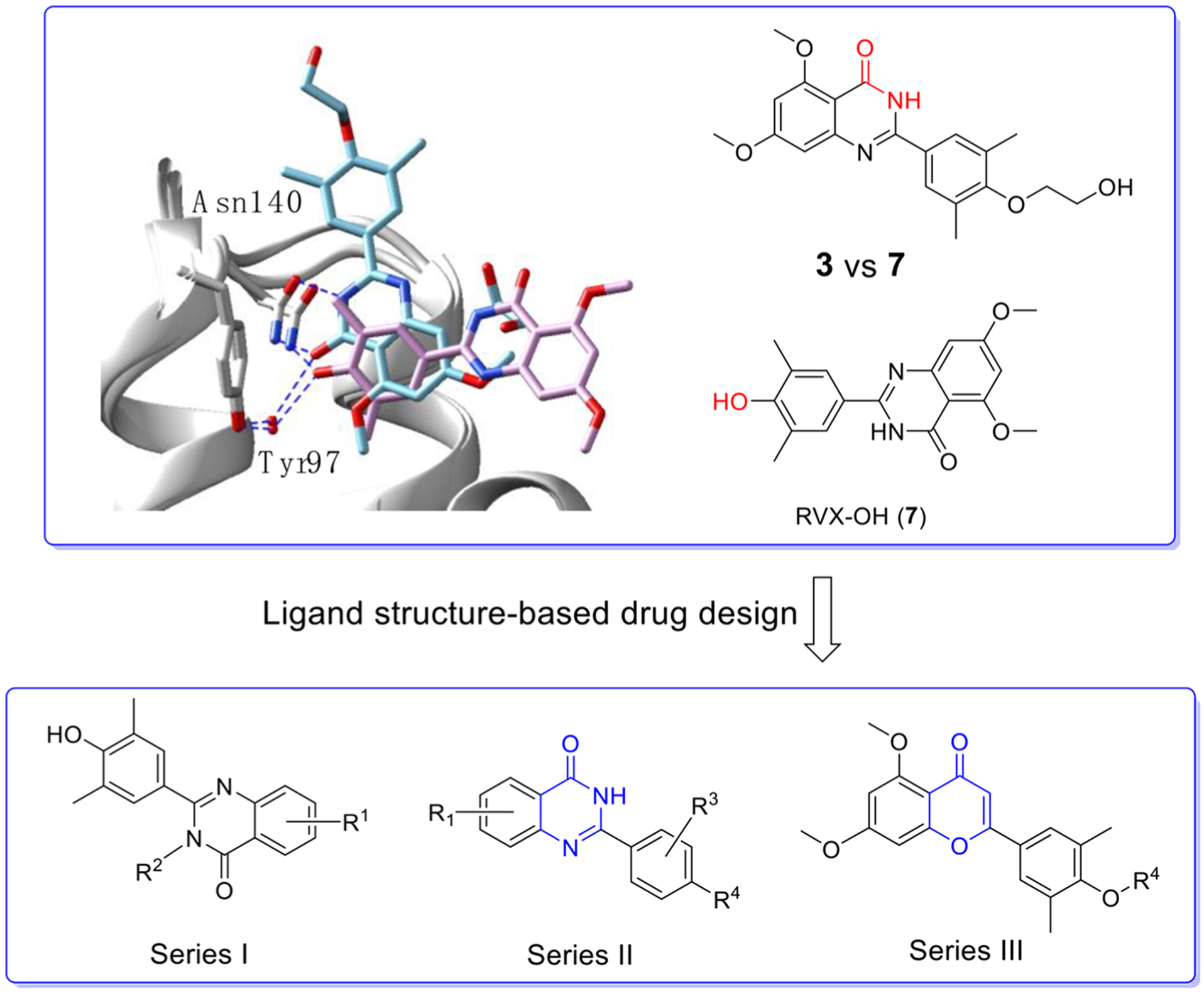
Design of three series of BRD4 inhibitors via a ligand structure-based drug design and scaffold hopping strategy. First panel: overlay of cocrystal structures of compounds 3 (in light blue, PDB code: 4MR4) and 7 (in pink, PDB code: 4MR3) with BRD4 BD1. Residues Asn140 and Tyr97 are labeled. Second panel: designed three series of novel compounds to explore the SAR.
Chemistry.
The first series of compounds 10–16 were obtained from the reaction of 4-hydroxy-3,5-dimethylbenzalde-hyde 9 with various 2-aminobenzamides 8 via an efficient, metal-free, and I2-mediated C–N bond formation method (Scheme 1).28 To explore the space tolerance around the amide moiety of 7, a methyl group was introduced through two strategies. 4-Nitro-isotoic anhydride 17 was substituted by CH3NH2 to afford intermediate 18 which was readily transformed into desired compound 19 with a methyl group on the amide. After construction of the quinazolin-4-one core via allylic-protected benzaldehyde 20, compound 21 was methylated by CH3I and deprotected in the presence of Pd(PPh3)4 and K2CO3 smoothly to give compound 22.
Scheme 1.
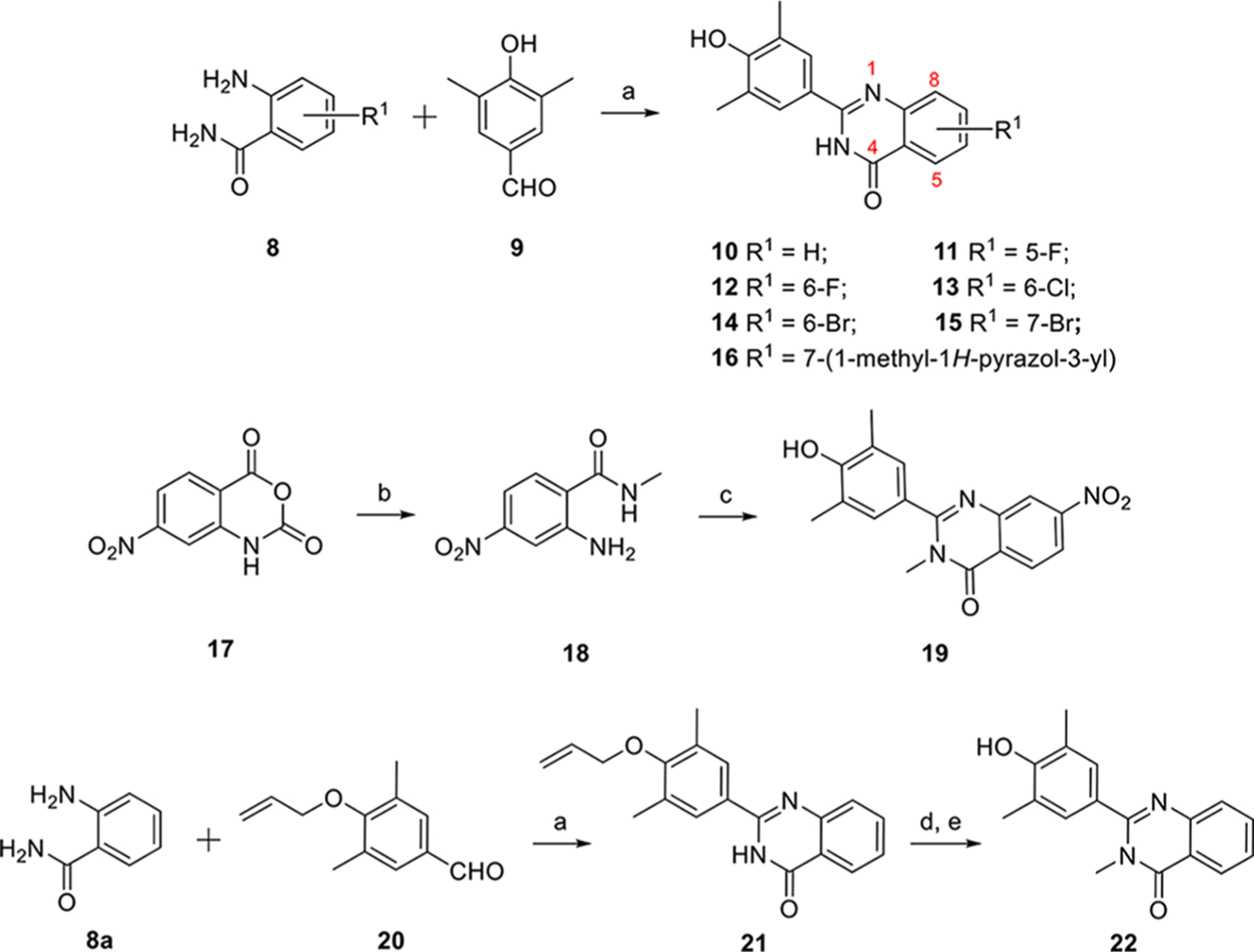
Synthesis Procedure of Series Ia aReagents and conditions: (a) I2, EtOH, reflux, 4 h, 89% ~ quant. (b) CH3NH2, THF, DIPEA; (c) 2-aminobenzamide, I2, EtOH, 57% for two steps; (d) CH3I, NaH, DMF, rt, overnight, 94%; (e) Pd(PPh3)4, K2CO3, CH3OH, reflux, 4 h, 72%.
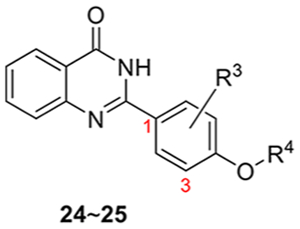
The synthesis of the second series of compounds is depicted in Scheme 2. Compounds 24 and 25 were obtained following a similar procedure to that of compounds 10–16 from commercially available starting material 2-aminobenzamide 8a and various benzaldehydes 23. Compounds 27–31 were obtained through the reaction of 2-amino-4,6-dimethoxybenzamide 8b and substituted benzaldehydes 26 (commercially available or slight modification on 4-hydroxy-3,5-dimethylbenzaldehyde 9). Addition of 1-methylpiperazine with 31 produced 32 in a yield of 35%. Compound 3 was also resynthesized as the reference compound using the same strategy in a yield of 82% for two steps (Scheme 3), while the original synthesis method of compound 3 was reported to take eight steps with an overall yield of 5% and included harsh conditions like high temperature (170 °C) and two gases in pure form (HCl and NH3).23
Scheme 2.
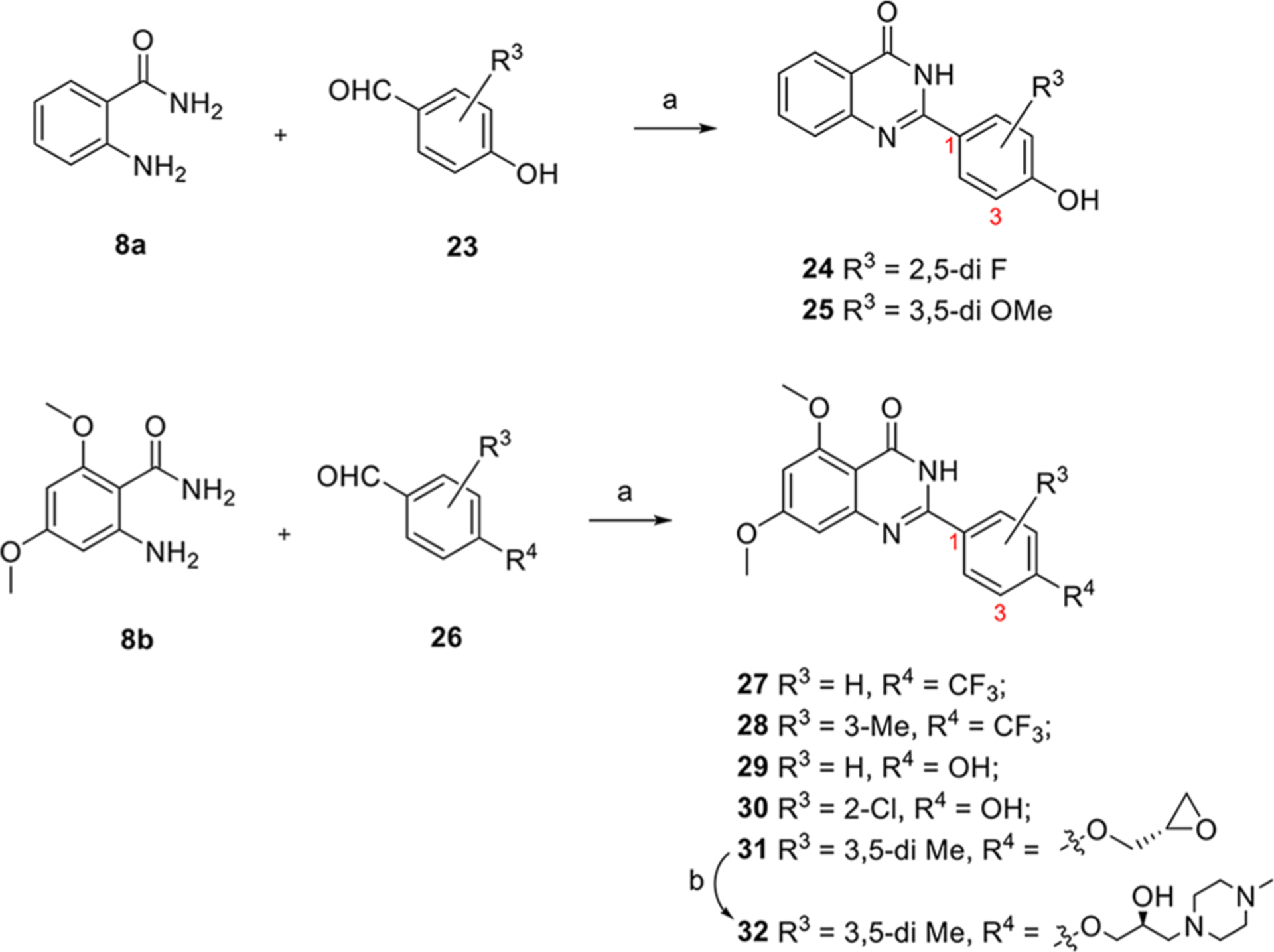
Synthesis Procedure of Series IIa aReagents and conditions: (a) I2, EtOH, reflux, 4 h, 86% ~ quant. (b) 1-methylpiperazine, K2CO3, DMF, 90 °C, overnight, 35%.
Scheme 3.
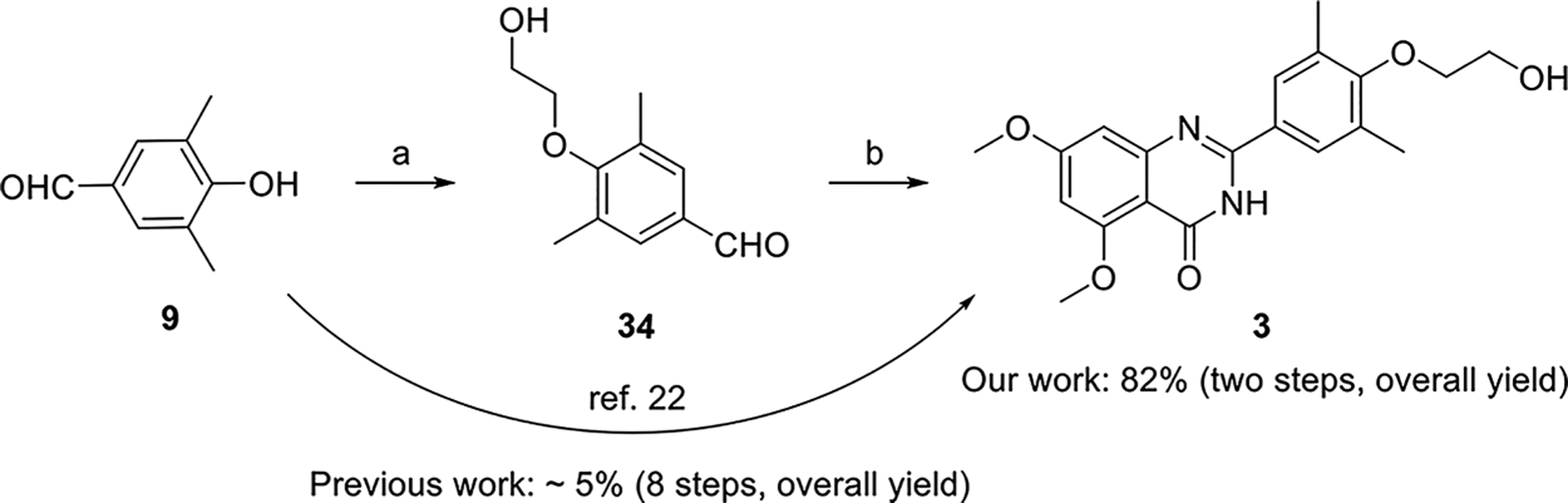
Resynthesis of Compound 3a aReagents and conditions: (a) 2-bromoethan-1-ol (33), K2CO3, CH3CN, reflux, overnight. (b) 8b, I2, EtOH, reflux, 2 h, 82% (two steps).
The synthesis procedure of series III containing a new chromone core is depicted in Scheme 4. Aldol reaction of reagents 20 and 35 gave intermediate 36 which was cyclized under I2 in DMSO leading to compound 37. Compound 37 was deprotected in presence of Pd(PPh3)4 and K2CO3 affording critical intermediate 38. Compound 38 was substituted by 2-bromoethan-1-ol (33) to produce compound 39 which is exactly the same as compound 3 except the core. More derivatives 42–49 were obtained via a substitution reaction and cyclic addition. Compound 50 was obtained through transformation of −OH (44) into −F under diethylaminosulfur trifluoride (DAST) in a yield of 80%.
Scheme 4.
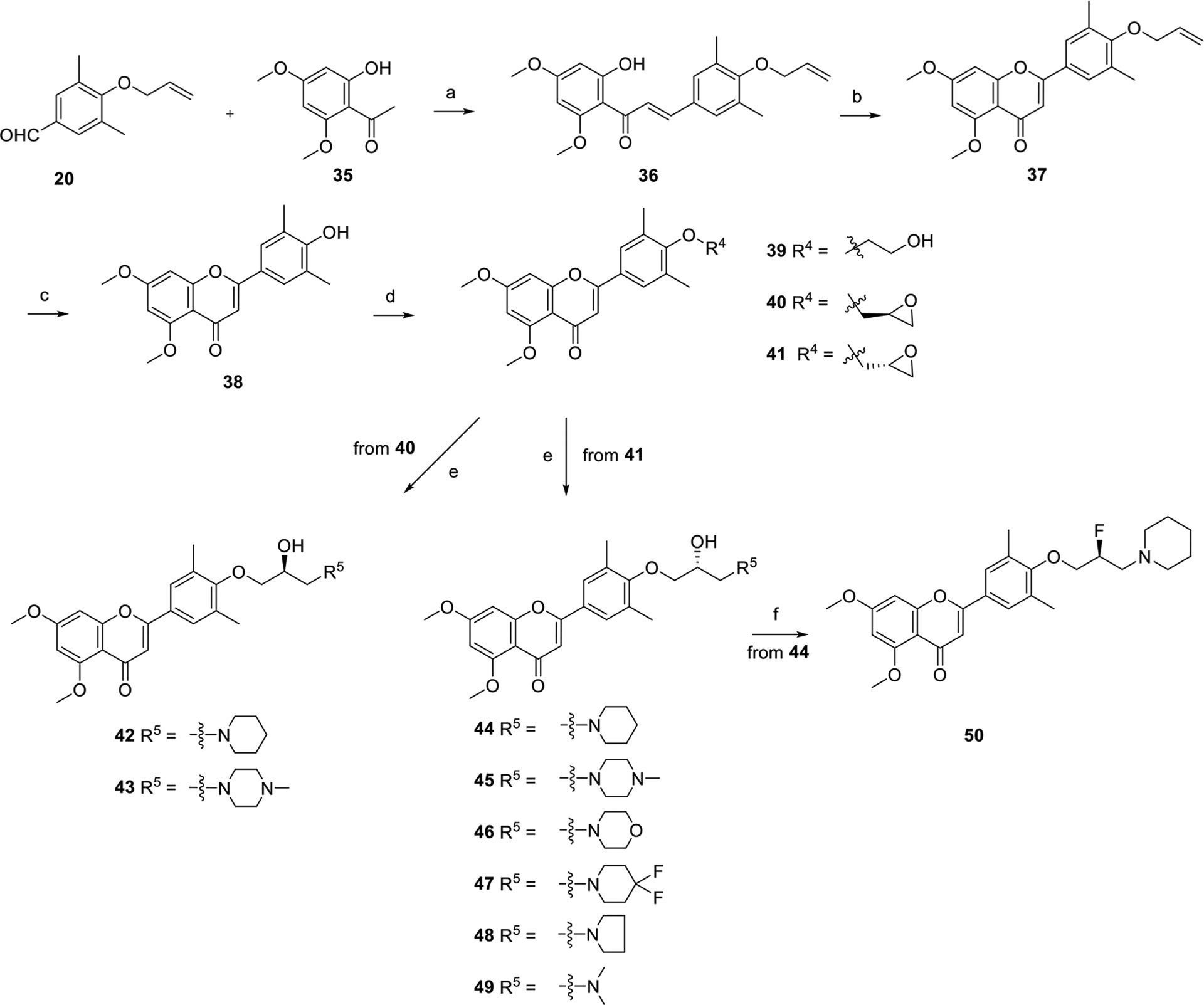
Synthesis Procedure of Series IIIa aReagents and conditions: (a) 50% KOH, EtOH, rt, overnight, used directly for next step. (b) I2, DMSO, 140 °C, 4 h, 36%. (c) Pd(PPh3)4, K2CO3, CH3OH, 90 °C, 7 h, 75%. (d) R4–Br, K2CO3, DMF, 80 °C, overnight, 55–72%. (e) R5–H, K2CO3, DMF, 80 °C, overnight, 18–65% for two steps. (f) DAST, CH2Cl2, rt, overnight, 80%.
Inhibitory Activities against TLR3-Induced Expression of Inflammatory Genes In Vitro.
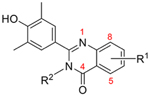
All the newly synthesized compounds were first evaluated for inhibitory activity of TLR3 signaling in human small airway epithelial cells (hSAECs) at a concentration of 10 μM, an assay to demonstrate cellular permeability and BRD4 inhibitory efficacy for further studies. TLR3-inducible expression of CIG5 and IL-6 genes was determined by quantitative real-time PCR (qRT-PCR). Percentages of inhibition (%) were calculated based on the level of mRNA expression in poly(I:C)-stimulated cells without compounds.8 For the first series of compounds, 7 was tested as the positive control. Different halogens at diverse positions and bulky substituents (e.g., 1-methyl-1H-pyrazol-3-yl and NO2) on the quinazolin-4-one core and a methyl group on the amide were explored. Despite the finding that compound 7 displayed impressive inhibitory activities of CIG5 and IL-6 mRNA induction with inhibition rates of 86 and 75%, respectively, none of its newly synthesized analogues showed obvious improvement (Table 1). This suggests that the electron-donating methoxy groups on the phenyl ring are critical for the cellular effects.
Table 1.
Inhibitory Rates of Series I Compounds on TLR3-Induced Expression of Inflammatory Genes in hSAECs at a Concentration of 10 μMa
 |
||||
|---|---|---|---|---|
| compounds | R1 | R2 | CIG5 (%) | IL-6 (%) |
| 7 | 5-OMe, 7-OMe | H | 86 ± 11 | 75 ± 8.6 |
| 10 | H | H | −26 ± 3.1 | 81 ± 8.9 |
| 11 | 5-F | H | 33 ± 4.8 | 62 ± 7.2 |
| 12 | 6-F | H | −4.5 ± 0.6 | 62 ± 7.1 |
| 13 | 6-Cl | H | 37 ± 4.9 | 53 ± 5.6 |
| 14 | 6-Br | H | 43 ± 6.1 | 25 ± 3.4 |
| 15 | 7-Br | H | 51 ± 6.3 | 15 ± 1.9 |
| 16 | 7-(1-methyl-1H-pyrazol-3-yl) | H | 71 ± 8.5 | −24 ± 2.8 |
| 19 | 7-NO2 | CH3 | −69 ± 7.4 | 86 ± 9.1 |
| 22 | H | CH3 | 18 ± 2.3 | 55 ± 6.5 |
Inhibitory rates (%) are reported as mean ± SD of expression in the compound divided by that in solvent (×100). Data are derived from three independent measurements (n = 3).
The second series of compounds were tested using compounds 3 and 7 as the positive controls, which had similar cellular activities (Table 2). We first validated the necessity of electron-donating methoxy groups on the quinazolin-4-one core. Consistent with the results of the first series of small molecules, compounds 24 and 25 without methoxy groups exhibited obvious decreased activity. Substituents on the single phenyl ring were then explored and −OH group at 4-position (29) was more favored than −CF3 (27 and 28). Replacement of the 3,5-dimethyl group with H (29) or 2-Cl (30) displayed slightly declined inhibitory activities against both poly(I:C)-induced CIG5 and IL-6 expression. Furthermore, substituents on 4-OH were investigated, and both compounds 31 and 32 showed equal or better inhibition than 3, indicating that this position is tolerable for structural optimization.
Table 2.
Inhibitory Rates of Series II Compounds on TLR3-Induced Expression of Proinflammatory Genes at a Concentration of 10 μMa
 |
 |
|||
|---|---|---|---|---|
| Compounds | R3 | R4 | CIG5 (%) | IL-6 (%) |
| 3 | 3,5-di Me | 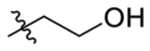 |
84 ± 8.6 | 65 ± 7.3 |
| 7 | 3,5-di Me | OH | 86 ± 9.7 | 75 ± 8.4 |
| 24 | 2,5-di F | H | 2.2 ± 0.37 | 59 ± 6.8 |
| 25 | 3,5-di OMe | H | 61 ± 7.3 | 59 ± 7.5 |
| 27 | H | CF3 | 45 ± 4.2 | −33 ± 4.1 |
| 28 | CF3 | CF3 | −21 ± 2.9 | 45 ± 5.7 |
| 29 | H | OH | 81 ± 9.2 | 63 ± 6.8 |
| 30 | 2-C1 | OH | 42 ± 4.8 | 66 ± 7.5 |
| 31 | 3,5-di Me | 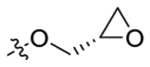 |
87 ± 9.9 | 83 ± 8.4 |
| 32 | 3,5-di Me | 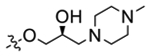 |
83 ± 8.8 | 80 ± 9.2 |
Inhibitory rates (%) are reported as the geometric mean of expression in the compound divided by that in solvent (×100). Data are derived from three independent measurements.
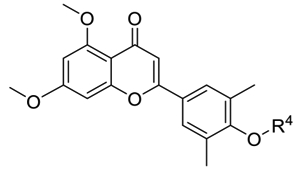
The third series of compounds were obtained through a scaffold hopping approach (Table 3). Compound 39 exhibited significantly improved inhibition of poly(I:C)-induced CIG5 and IL-6 expression (inhibitory rates of 97 and 98%, respectively) compared to compound 3, suggesting that the new chromone core appears to be superior to the quinazolin-4-one scaffold. Based on the SAR of series II compounds, the side chain on 4-OH was found to be tolerant to modification. Thus, we tried various hydrophilic side chains containing a heterocyclic ring capable of increasing the aqueous solubility of the small molecules and constraining its conformation at the same time. Compared to 32, compounds 43 and 45 with the new chromone core displayed a better inhibitory activity (99% against both CIG5 and IL-6). Compounds incorporating piperidine (42 and 44) and pyrrole (48) exhibited similar potent effects to 1-methylpiperazine (43 and 45), while compounds with moieties of morpholine (46), 4,4-difluoropiperidine (47), and dimethylamine (49) displayed substantially decreased inhibitory activities. R- or S- configuration of the −OH group on the side chain was found to make no significant differences (42 vs 44 and 43 vs 45). Fluorine is widely used to improve the metabolic stability in the drug discovery process29 and the replacement of −OH of 44 with −F (50) showed slightly reduced inhibition against poly(I:C)-induced CIG5 and IL-6 expression compared to that of compound 44.
Table 3.
Inhibitory Rates of Series III Compounds on TLR3-Induced Expression of Proinflammatory Genes at a Concentration of 10 μMa
 |
|||
|---|---|---|---|
| Compounds | R4 | CIG5 (%) | IL-6 (%) |
| 3 |  |
84 ± 8.8 | 65 ± 5.7 |
| 37 |  |
83 ± 7.7 | NTb |
| 39 | 97 ± 10.5 | 98 ± 11.4 | |
| 42 | 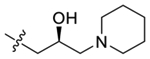 |
100 ± 9.8 | 100 ± 11 |
| 43 | 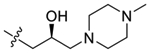 |
99 ± 11 | 99 ± 9.2 |
| 44 | 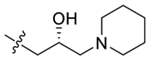 |
99 ± 12 | 100 ± 11 |
| 45 | 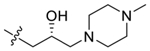 |
91 ± 10 | 97 ± 9.3 |
| 46 | 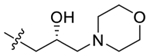 |
42 ± 5.1 | 82 ± 9.6 |
| 47 | 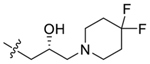 |
63 ± 7.5 | 88 ± 9.3 |
| 48 | 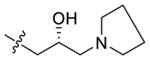 |
92 ± 11 | 99 ± 11 |
| 49 | 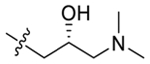 |
−19 ± 2.5 | 82 ± 7.7 |
| 50 |  |
85 ± 9.3 | 90 ± 8.6 |
Inhibitory rates (%) are reported as the geometric mean of expression in the compound divided by that in solvent (×100). Data are derived from three independent measurements.
NT: Not tested.
IC50 values were calculated for selected active compounds 39, 42–45, and 48 displaying potent inhibition against both genes with inhibitory rates over 90% (Table 4). Compound 32 with the quinazolin-4-one core was also included for comparison. Positive control 1 exhibited IC50 values of 0.92 and 1.02 μM against poly(I:C)-induced CIG5 and IL-6 expression, respectively. Compounds 32 and 39 displayed two to fourfold decreased IC50 values which were comparable to compound 3. Compounds 42–45 exhibited significantly improved effects with submicromolar cellular IC50 values, while compound 48 displayed IC50 values around 1 μM comparable to that of reference compounds 1 and 3. The most potent compound 45 showed excellent IC50 values of 280 and 310 nM against poly(I:C)-induced CIG5 and IL-6 expression, respectively (Table 4).
Table 4.
IC50 Values of Selected Compounds against TLR3-Induced Expression of Inflammatory Genesa
| compd | CIG5 (μM) | IL-6 (μM) |
|---|---|---|
| 1 | 0.95 ± 0.11 | 1.02 ± 0.10 |
| 3 | 1.66 ± 0.15 | 3.29 ± 0.41 |
| 32 | 2.6 ± 0.22 | 3.5 ± 0.33 |
| 39 | 2.6 ± 0.21 | 2.8 ± 0.32 |
| 42 | 0.58 ± 0.49 | 0.54 ± 0.47 |
| 43 | 0.83 ± 0.07 | 0.75 ± 0.08 |
| 44 | 0.52 ± 0.05 | 1.1 ± 0.12 |
| 45 | 0.28 ± 0.03 | 0.31 ± 0.02 |
| 48 | 1.48 ± 0.17 | 0.97 ± 0.09 |
IC50 values are reported as the mean concentration (μM) derived from three independent measurements. Each titration curve was generated from at least eight different concentrations.
Binding Affinities of Selected Compounds toward BET BDs.
Compounds 42, 43, 44, and 45 were selected for further studies to determine binding affinities with BET BDs and non-BET proteins via time-resolved fluorescence energy transfer (TR-FRET) assay (Table 5). Results of positive controls 1 and 3 were consistent with the reported data, which reflected the reliability of our assay. Compounds 42–45 showed about a 10-fold increase in potency over compound 3 for BRD4 BD1 binding. Compounds 42 and 43 are BRD4-selective without preference for BRD4 BD1 and BRD4 BD2, while both compounds 44 and 45 exhibited a good selectivity for BRD4 BD1 over other BET BDs, including BRD4 BD2. Besides, compound 45 displayed good selectivity over non-BET bromodomain-containing proteins (Table S1). Meanwhile, no obvious off-target effects of 45 were observed from the NIMH psychoactive drug screening program (PDSP) using a wide panel of target screening (Table S2).
Table 5.
Binding Affinities of Selected Compounds for the BET BDs and Non-BET Protein CBP (nM)a
| BDs | 1 | 3 | 42 | 43 | 44 | 45 |
|---|---|---|---|---|---|---|
| BRD4 BD1 | 92 ± 7.8 | 1142 ± 98 | 86 ± 7.8 | 127 ± 14 | 67 ± 5.4 | 84 ± 7.3 |
| BRD4 BD2 | 62 ± 5.9 | 135 ± 14 | 93 ± 8.5 | 139 ± 12 | 684 ± 7.5 | 718 ± 69 |
| BRD2 BD1 | 78 ± 7.2 | 5780 ± 463 | 1205 ± 116 | 1286 ± 113 | 791 ± 67 | 886 ± 75 |
| BRD2 BD2 | 52 ± 5.5 | 251 ± 186 | 1196 ± 97 | 1437 ± 156 | 845 ± 73 | 914 ± 81 |
| BRD3 BD1 | 81 ± 7.4 | 3962 ± 317 | 1906 ± 173 | 2631 ± 197 | 2395 ± 198 | 3122 ± 285 |
| BRD3 BD2 | 69 ± 5.7 | 203 ± 19 | 1719 ± 156 | 1962 ± 159 | 2081 ± 186 | 2495 ± 217 |
| BRDT BD1 | 183 ± 16 | 4836 ± 457 | 2502 ± 195 | 2765 ± 237 | 2869 ± 257 | 3592 ± 293 |
| BRDT BD2 | 217 ± 23 | 708 ± 69 | 2411 ± 207 | 2545 ± 236 | 2317 ± 206 | 3176 ± 276 |
| CBP | 9600 ± 785 | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
Binding affinity was measured using a TR-FRET assay with the isolated recombinant BD. Reported as mean ± SD from three separate assay runs (n = 3).
Cocrystal Structure Analysis of Compound 45 in Complex with Human BRD4 BD1.
To gain more insight into the binding modes of compound 45 with BRD4 BD1 and validate the efficiency of the scaffold hopping strategy, we have successfully solved the high-resolution cocrystal structure of 45 in complex with BRD4 BD1 at 2 Å (PDB code: 6UWU; Figure 3, Figure S1, and Table S3). Similar to the binding mode shown by compound 3, the new inhibitor 45 occupied the classical KAc recognition site very well with the aid of several water molecules (Figure 3A). The carbonyl O atom interacted with Asn140 directly and formed an indirect hydrogen bond with Tyr97 mediated by a water molecule. One methoxy group on the chromone core was accommodated in the pocket by a hydrogen bond mediated with water, while the other methoxy group extended to the WPF shelf area which was consistent with SAR analysis, indicating the critical role of −OMe moieties. The 1-methylpiperazine was excluded from the pocket and reached the solvent area consistent with the SAR and perfectly explaining why side chain modifications were tolerated (Figure 3B). The chiral −OH on the side chain was in proximity of a water molecule where it formed a water-mediated hydrogen bond with Asn93 (Figure 3C). The equivalent residue of Asn93 on BRD4 BD2 is glycine, and thus this hydrogen bond interaction was missing for BRD4 BD2 with 45. Similarly, the equivalent residues of Asn93 are glycine for BRD2 BD1, glycine for BRD2 BD2, glutamic acid for BRD3 BD2, glutamine for BRDT BD1, and glycine for BRDT BD2. This may explain the selectivity of 45 over bromodomains of BET family members. Although BRD3 BD1 has the same asparagine, the overall ZA loop shift (as far as 5–8 Å) is obvious, thereby making the KAc recognition pocket much larger than BRD4 BD1 (Figure S2). Therefore, it is likely more difficult for the compound adhering to the residues. This speculation is consistent with the previously reported results from the Morelli group.27 Overlay analysis of the crystal structures of 3 and 45 with BRD4 BD1 demonstrated a highly conserved pose except for the flexible side chain on 4-OH (Figure 3D). These findings validated and supported the high reliability and feasibility of our scaffold hopping strategy.
Figure 3.
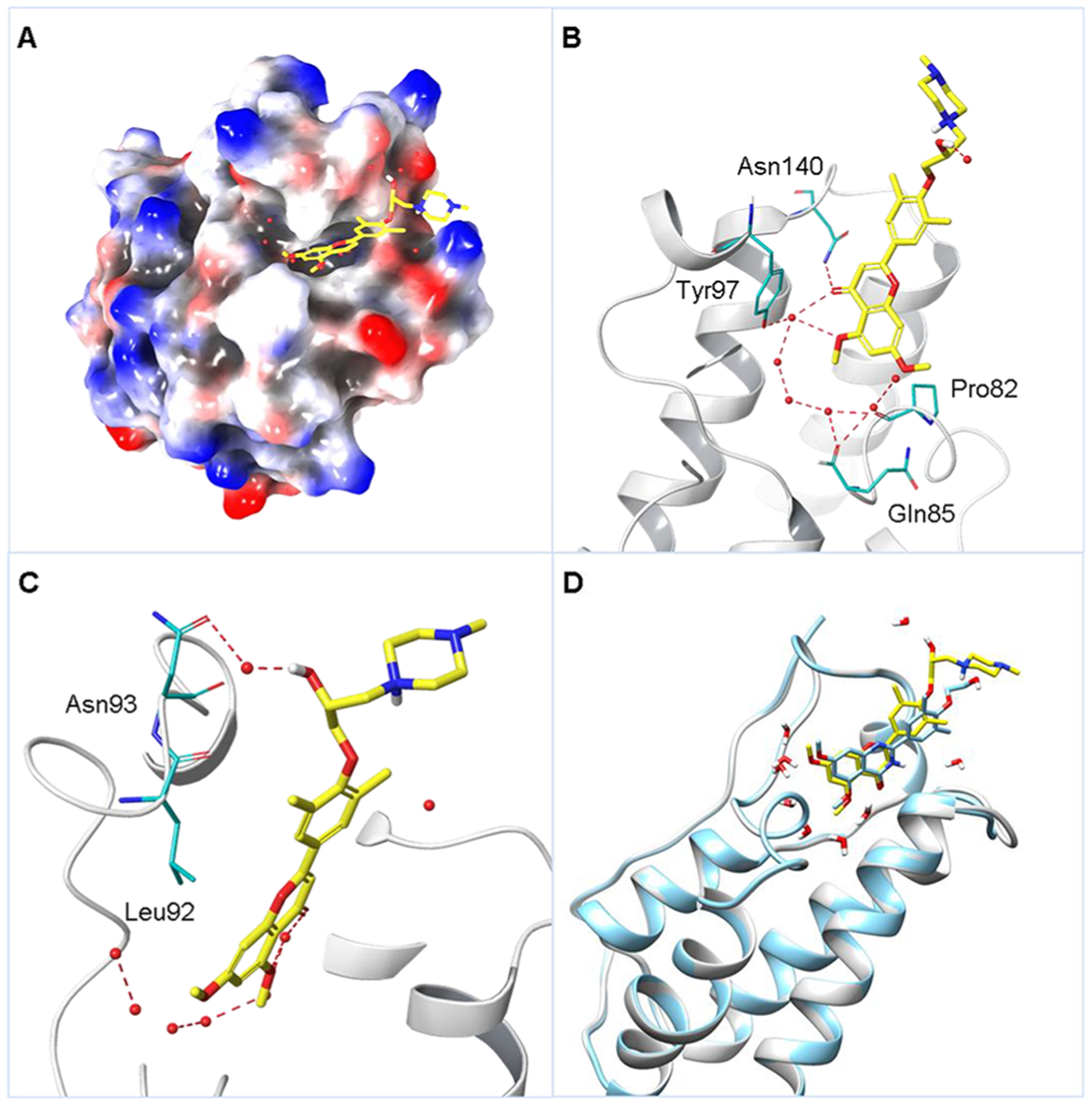
(A) Crystal structure of 45 (in yellow) in cocomplex with human BRD4 BD1 (PDB code: 6UWU) in representation of the electrostatic potential surface. (B) Critical interactions of 45 with BRD4 BD1. Residues Asn140, Tyr97, and Pro82 are highlighted in light blue and hydrogen bonds are colored in red. (C) Zoomed in details around the chiral −OH. Residues Asn93 and Leu92 are highlighted in light blue. (D) Overlay analysis of 3 (in light blue, PDB code: 4MR4) and 45 (in yellow) in cocomplex with human BRD4 BD1 in ribbon representation.
Drug-like Properties of BRD4 BD1 Inhibitors 44 and 45.
With promising in vitro activities in hand, we then studied the in vivo PK profiles of compounds 44 and 45. As listed in Table 6 and depicted in Figure S3, these inhibitors displayed similar but overall favorable PK parameters (3 to 4 h of half-life, maximum concentration around 600 ng/mL, and AUC values of about 5000 ng·h/mL). The ultimate oral bioavailability of compounds 44 and 45 are 38 and 35%, respectively, indicating that both compounds are orally available. Moreover, both compounds have an excellent aqueous solubility with concentrations >50 mg/mL. Furthermore, compound 45 is metabolically stable in the liver microsome stability assay and CYP450 inhibition study. The clearance rates were 25 and 5.6 μL/min/mg for mouse liver microsomes (MLM) and human liver microsomes (HLM), respectively. The unbound fractions of compound 45 are 12 and 17% in mouse and human plasma protein binding, respectively, which is reasonable to drive the pharmacological effects. In addition, no obvious inhibitory effects were found on either hERG or CYP450 enzymes including CYP 3A4, 1A2, 2C9, 2D6, 2C19, 2C8, or 2B6 at a concentration of 10 μM (Table 7). Taken together, these BRD4 BD1 inhibitors demonstrated impressive overall drug-like properties to support their further clinical development.
Table 6.
In Vivo PK Profiles of Compounds 44 and 45 in Ratsa
| 44 | 45 | ||
|---|---|---|---|
| i.v. (10 mg/kg) | bt1/2 (h) | 3.5 ± 0.71 | 4.1 ± 0.43 |
| cCmax (ng/mL) | 2861 ± 426 | 2090 ± 265 | |
| dAUC0-t (ng·h/mL) | 6763 ± 590 | 7033 ± 2161 | |
| eCL (L/h/kg) | 1.48 ± 0.13 | 1.48 ± 0.38 | |
| p.o. (20 mg/kg) | t1/2 (h) | 2.4 ± 0.55 | 2.8 ± 0.27 |
| Cmax (ng/mL) | 575.1 ± 179 | 605.5 ± 182 | |
| AUC0-t (ng·h/mL) | 5171 ± 3447 | 4966 ± 1772 | |
| fF (%) | 38.2 | 35.3 |
Compounds were formulated in 10%DMSO/60%PEG400/30% Saline and administrated intravenously (i.v.) or per os (p.o.).
t1/2, half-life.
Cmax, maximum plasma concentration achieved.
AUC0‑t, total exposure following a single dose.
CL, total clearance.
F, oral bioavailability.
Table 7.
In Vitro Liver Microsome Stability Assay, Cytochrome P450 Enzymes, and hERG Inhibition Assaya
| compound | 45 |
|---|---|
| MLM (CL, μL/min/mg) | 25 |
| HLM (CL, μL/min/mg) | 5.6 |
| mouse plasma protein binding (free fraction, %)b | 12 |
| human plasma protein binding (free fraction, %) | 17 |
| CYP 3A4(MDZ)c | 9.5 ± 5.6 |
| CYP 3A4(Testo)d | 8.2 ± 2.1 |
| CYP 1A2 | 0.0 ± 0.0 |
| CYP 2C9 | 1.9 ± 0.25 |
| CYP 2D6 | 0.0 ± 0.0 |
| CYP 2C19 | 1.7 ± 0.29 |
| CYP 2C8 | 17 ± 3.0 |
| CYP 2B6 | 27 ± 4.3 |
| hERG (IC50, μm) | > 30 |
Inhibitory rates (%) on the CYP450 enzyme at a concentration of 10 μM.
Concentration is 2 μM
Substrate is midazolam (MDZ).
Substrate is testosterone (Testo).
In Vivo Efficacy of Selected BRD4 BD1 Inhibitors 44 and 45.
Next, compounds 44 and 45 were investigated in our established murine model of TLR3-induced acute airway inflammation.8,12 Both oral administration (p.o.) and intraperitoneal administration (i.p.) were employed to compare their in vivo efficacy. The vehicle alone and poly (I:C)-treated groups were tested as healthy and pathological controls, respectively. Histologically, we observed that poly(I:C) induced a profound neutrophilic inflammation around the small- and medium-sized airways, which was completely inhibited by either inhibitor at a dose of 10 mg/kg via p.o. or i.p. administration, respectively (Figure 4A). Meanwhile, elevated secretion of total cells and neutrophils (Figure 4B) was suppressed, and inflammatory cytokines (IL-6, KC, MCP-1, and RANTES, Figure 4C) in bronchoalveolar lavage fluid (BALF) were also significantly blocked. We also measured inflammatory gene expression in lung tissue (Figure 4D). Administration of poly(I:C)-induced a 15- to 20-fold increase of inflammatory genes IL-6, KC, ISG54, and CIG5 in lung tissue. Compounds 44 and 45 were found to inhibit these increases significantly. These results validated the potent in vivo efficacy of our newly designed compounds achieved from the scaffold hopping approach. To further validate their in vivo efficacy actually induced by inhibiting BRD4 rather than any other off-target effects, we examined the level of H3K122Ac, which is verified as a BRD4 biomarker (Figure 5).10,12,13 Immunofluorescence staining of H3K122Ac demonstrated that poly(I:C) induced significant increase of BRD4 activities in both hSAECs and lung tissue of mice, and this poly(I:C)-induced effect was effectively blocked by BRD4 inhibitors 44 and 45 through the downregulation of H3K122Ac.
Figure 4.
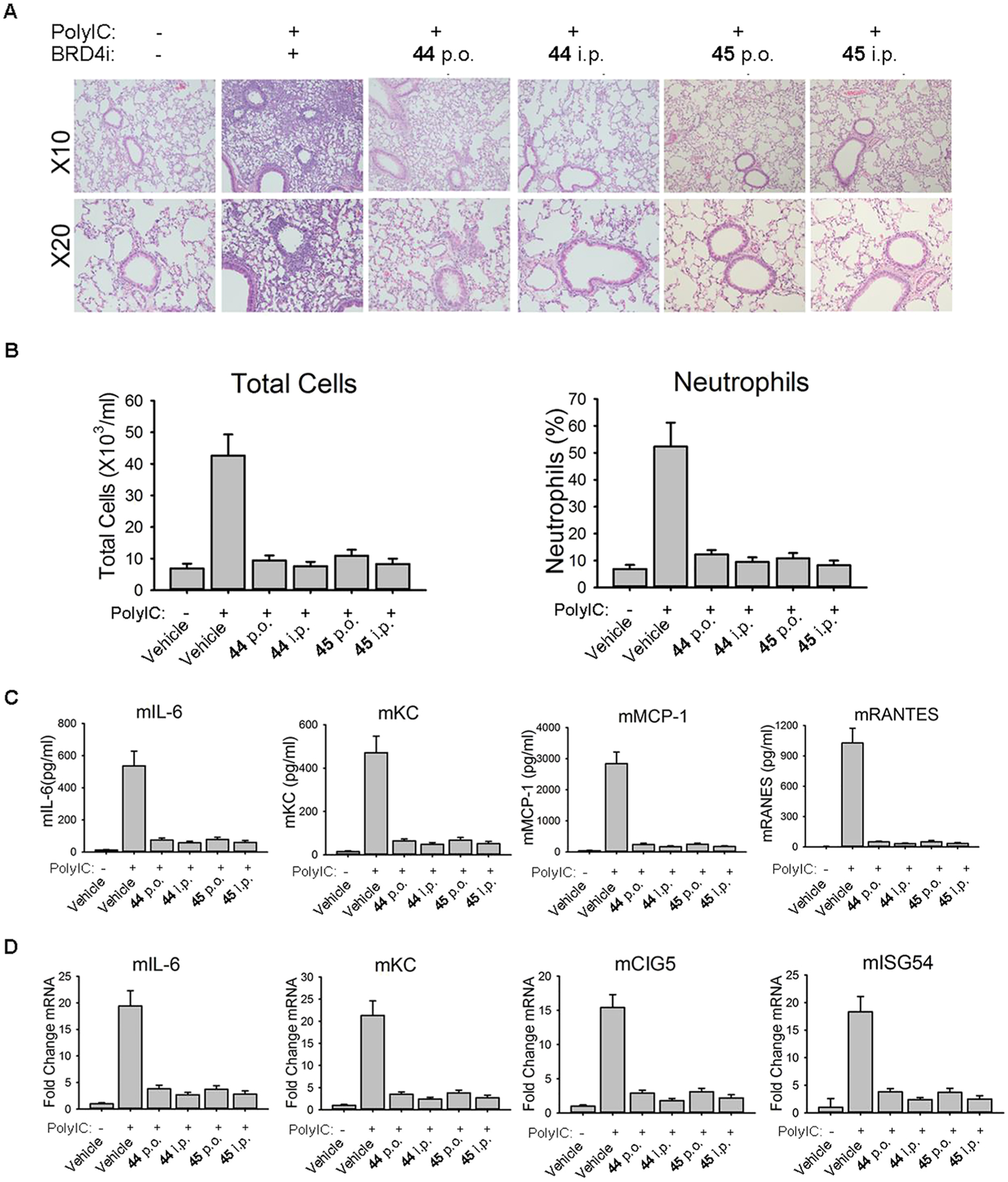
In vivo efficacy of BRD4 BD1 inhibitors 44 and 45 in poly(I:C)-induced airway inflammation of mice. (A) C57BL6/J mice were pretreated (i.p. or p.o.) with 44 and 45 at a dose of 10 mg/kg before intranasal (i.n.) poly(I:C) stimulation. Lung histology images of Hematoxylin and Eosin (H&E) staining were performed on paraffin-embedded lung sections and were shown at a magnitude of × 10 and × 20, respectively. (B) BRD4 inhibitors 44 and 45 blocked poly(I:C)-induced secretion of inflammatory cells in BALF. (C) BRD4 inhibitors blocked poly(I:C)-induced secretion of inflammatory cytokines in BALF. (D) BRD4 inhibitors blocked poly(I:C)-induced inflammatory gene expression in lung tissue.
Figure 5.
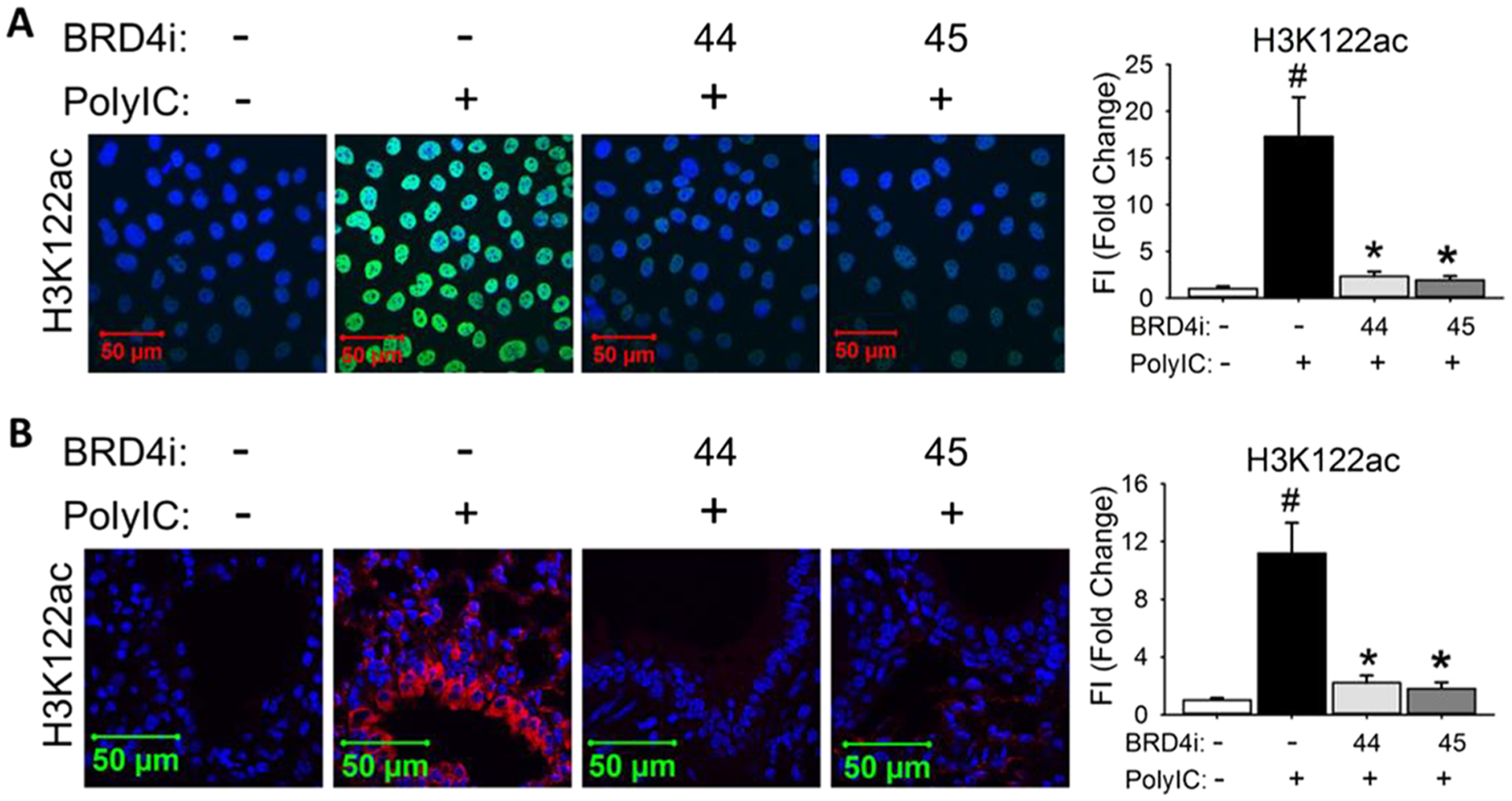
BRD4 inhibitors 44 and 45 block poly(I:C)-induced H3K122Ac (A) in hSAECs and (B) in lung tissue of mice. (A) Immunofluorescence staining of H3K122Ac (green) was performed in hSAECs. (B) Immunofluorescence staining of H3K122Ac (red) was performed on paraffin-embedded lung sections of mice. Right panel, quantification of total fluorescence intensity shown as fold changes on immunofluorescence staining of H3K122Ac. #p < 0.01, compared with control; *p < 0.01 compared with poly(I:C) only, n = 5.
CONCLUSIONS
In summary, we have discovered a series of novel chromone derivatives as potent and selective BRD4 BD1 inhibitors via a scaffold hopping strategy. Among them, compounds 44 (ZL0513) and 45 (ZL0516) displayed potent cellular effects with submicromolar IC50 values, nanomolar binding affinity of 67–84 nM with BRD4 BD1 and good selectivity as well as significant in vivo efficacy in blocking poly(I:C)-induced airway inflammation and neutrophilia of mice. The cocrystal structure of 45 in complex with human BRD4 BD1 validated its classical KAc recognition site with critical hydrogen bonds to Asn140 and Tyr97. At the same time, both BRD4 BD1 inhibitors 44 and 45 demonstrated excellent DMPK properties including bioavailability >35%, aqueous solubility, metabolic stability, and weak to low CYP450 enzymes and hERG inhibitory effects. Collectively, the findings support these BRD4 BD1 inhibitors with impressive overall profiles of in vivo efficacy and drug-like properties as promising advanced lead compounds for further optimization and extensive clinical development toward epigenetic therapeutics for the treatment of inflammatory diseases.
EXPERIMENTAL SECTION
General Chemistry Information.
All commercially available starting materials and solvents were of reagent grade and used without further purification unless otherwise specified. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was carried out by employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 or Bruker-300 (1H, 600 and 300 MHz; 13C, 150 and 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained on a Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: the nano ESI spray voltage was 1.8 kV, capillary temperature was 275 °C, and the resolution was 60,000; ionization was achieved by positive mode. Purities of the final compounds were established by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20 AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 10% acetonitrile in water to 100% acetonitrile in water in 20 min followed by 30 min of the last-named solvent (0.1% TFA was added into both acetonitrile and water). All biologically evaluated compounds were >95% pure.
2-(4-Hydroxy-3,5-dimethylphenyl)quinazolin-4(3H)-one (10).
To a solution of 2-aminobenzamide (136 mg, 1.0 mmol) and 4-hydroxy-3,5-dimethylbenzaldehyde (180 mg, 1.2 mmol) in 10 mL EtOH, I2 (279 mg, 1.1 mmol) was added. After reflux for 4 h, 5% Na2S2O4 was added to quench the reaction. Compound 10 (266 mg, quant.) was obtained through filtration as a white solid. HPLC purity 95.7% (tR = 13.13 min). 1H NMR (300 MHz, DMSO-d6) δ 12.10 (s, 1H), 8.11 (d, J = 7.9 Hz, 1H), 7.86 (s, 2H), 7.79 (t, J = 7.6 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 2.25 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 162.7, 157.0, 152.8, 149.4, 134.9, 128.5, 127.4, 126.3, 124.7, 123.4, 121.0, 17.1. HR ESI-MS (M + H)+ m/z = 267.1134 (calcd for C16H15N2O2: 267.1127).
5-Fluoro-2-(4-hydroxy-3,5-dimethylphenyl)quinazolin-4(3H)-one (11).
Compound 11 (71 mg, quant.) was obtained as a white solid following the procedure of 10. HPLC purity 95.4% (tR = 13.97 min). 1H NMR (300 MHz, DMSO-d6) δ 12.15 (s, 1H), 9.00 (s, 1H), 7.85 (s, 2H), 7.79–7.67 (m, 1H), 7.49 (d, J = 8.1 Hz, 1H), 7.17 (dd, J = 10.9, 8.2 Hz, 1H), 2.24 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 162.8, 160.0, 159.3, 157.3, 153.7, 151.7, 135.4, 135.3, 128.6, 124.7, 123.7, 122.9, 112.6, 112.4, 110.4, 17.18. HR ESI-MS (M + H)+ m/z = 285.1029 (calcd for C16H14FN2O2: 285.1039).
6-Fluoro-2-(4-hydroxy-3,5-dimethylphenyl)quinazolin-4(3H)-one (12).
Compound 12 (74 mg, quant.) was obtained as a white solid following the procedure of compound 10. HPLC purity 96.1% (tR = 14.52 min). 1H NMR (300 MHz, DMSO-d6) δ 12.27 (s, 1H), 8.95 (d, J = 2.8 Hz, 1H), 7.83 (s, 2H), 7.76 (dq, J = 9.0, 5.7, 4.1 Hz, 2H), 7.67 (dd, J = 8.5, 2.5 Hz, 1H), 2.24 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 162.1, 161.6, 158.4, 157.0, 152.2, 146.4, 130.4, 130.3, 128.5, 124.7, 123.5, 123.3, 123.2, 122.2, 122.1, 111.0, 110.7, 17.1. HR ESI-MS (M + H)+ m/z = 285.1031 (calcd for C16H14FN2O2: 285.1039).
6-Chloro-2-(4-hydroxy-3,5-dimethylphenyl)-3-methylquinazolin-4(3H)-one (13).
Compound 13 (322 mg, quant.) was obtained as a yellow solid following the procedure of compound 10. 1H NMR (300 MHz, DMSO-d6) δ 12.29 (s, 1H), 8.97 (s, 1H), 8.02 (d, J = 2.6 Hz, 1H), 7.84 (s, 2H), 7.78 (dd, J = 8.7, 2.5 Hz, 1H), 7.68 (d, J = 8.7 Hz, 1H), 2.24 (s, 6H). 13C NMR (75 MHz, DMSO) δ 161.8, 157.2, 153.2, 148.2, 134.9, 130.4, 129.7, 128.6, 125.2, 124.7, 123.2, 122.2, 17.1. HR ESI-MS (M + H)+ m/z = 301.0749 (calcd for C17H16ClN2O2: 301.0744).
6-Bromo-2-(4-hydroxy-3,5-dimethylphenyl)quinazolin-4(3H)-one (14).
Compound 14 (384 mg, quant.) was obtained as a white solid following the procedure of compound 10. HPLC purity 98.4% (tR = 17.23 min). 1H NMR (300 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.99 (s, 1H), 8.17 (d, J = 2.4 Hz, 1H), 7.91 (dd, J = 8.7, 2.4 Hz, 1H), 7.85 (s, 2H), 7.63 (d, J = 8.7 Hz, 1H), 2.24 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 161.6, 157.2, 153.3, 148.6, 137.7, 130.0, 128.6, 128.6, 128.3, 124.7, 124.7, 123.2, 123.1, 122.6, 118.5, 17.09, 17.05. HR ESI-MS (M + H)+ m/z = 345.0225 (calcd for C16H14BrN2O2: 345.0239).
6-Bromo-2-(4-hydroxy-3,5-dimethylphenyl)-3-methylquinazolin-4(3H)-one (15).
Compound 15 (340 mg, 94%) was obtained as a brown solid following the procedure of compound 10. HPLC purity 96.1% (tR = 17.54 min). 1H NMR (300 MHz, DMSO-d6) δ 8.00 (dd, J = 8.4, 1.9 Hz, 1H), 7.86 (d, J = 4.3 Hz, 3H), 7.59 (d, J = 8.4 Hz, 1H), 2.29–2.16 (m, 6H). 13C NMR (75 MHz, DMSO-d6) δ 162.4, 157.3, 154.2, 150.7, 129.7, 129.1, 128.7, 128.4, 128.3, 124.7, 123.1, 120.1, 17.1. HR ESI-MS (M + H)+ m/z = 345.0236 (calcd for C17H16BrN2O2: 345.0239).
2-(4-Hydroxy-3,5-dimethylphenyl)-7-(1-methyl-1H-pyrazol-5-yl)quinazolin-4(3H)-one (16).
Compound 16 (70 mg, 89%) was obtained as a brown solid following the procedure of compound 10. HPLC purity 95.8% (tR = 14.74 min). 1H NMR (300 MHz, DMSO-d6) δ 12.25 (s, 1H), 8.98 (s, 1H), 8.18 (d, J = 8.1 Hz, 1H), 7.89 (s, 2H), 7.81 (s, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.53 (d, J = 2.1 Hz, 1H), 6.61 (d, J = 2.3 Hz, 1H), 3.97 (s, 3H), 2.25 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 162.4, 157.2, 153.5, 149.8, 142.1, 138.6, 136.3, 128.6, 126.9, 126.2, 124.7, 123.3, 120.4, 107.2, 38.4, 17.1. HR ESI-MS (M + Na)+ m/z = 369.1333 (calcd for C20H19N4O2: 369.1327).
2-(4-Hydroxy-3,5-dimethylphenyl)-3-methyl-7-nitroquinazolin-4(3H)-one (19).
To a solution of 7-nitro-2H-benzo[d][1,3]-oxazine-2,4(1H)-dione (416 mg, 2.0 mmol) in 20 mL THF, DIPEA (516 mg, 4.0 mmol) and CH3NH2 (544 mg, 8.0 mmol) were added at room temperature. After 2 h, the solution was poured into a 0.5 M HCl solution and extracted by CH2Cl2. The organic phase was dried over anhydrous Na2SO4 and concentrated to get crude 2-amino-N-methyl-4-nitrobenzamide 18 which was used directly in the next step. Compound 19 (186 mg, 57% for two steps) was obtained from 2-amino-N-methyl-4-nitrobenzamide following the procedure of 10 as a yellow solid. HPLC purity 98.8% (tR = 16.66 min). 1H NMR (300 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.31 (d, J = 8.6 Hz, 2H), 8.17 (d, J = 8.8 Hz, 1H), 7.27 (s, 2H), 3.43 (s, 3H), 2.23 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 161.5, 159.1, 155.7, 151.5, 147.9, 129.1, 128.9, 125.8, 124.6, 124.3, 122.5, 120.3, 35.1, 17.0. HR ESI-MS (M + H)+ m/z = 326.1147 (calcd for C17H16N3O4:326.1141).
2-(4-(Allyloxy)-3,5-dimethylphenyl)quinazolin-4(3H)-one (22).
Compound 21 was obtained as a white solid following the procedure of 10. To a solution of compound 21 (153 mg, 0.5 mmol) and CH3I (142 mg, 1.0 mmol) in 3 mL of DMF, NaH (24 mg, 1.0 mmol) was added. After 4 h, the mixture was poured into iced water and extracted by CH2Cl2. The combined organic layer was concentrated and purified by PTLC to give 2-(4-(allyloxy)-3,5-dimethylphenyl)-3-methylquinazolin-4(3H)-one as a white solid. To a solution of 2-(4-(allyloxy)-3,5-dimethylphenyl)-3-methylquinazolin-4(3H)-one (100 mg, 0.31 mmol) in 5 mL of CH3OH, Pd(PPh3)4 (11 mg, 0.01 mmol) and K2CO3 (171 mg, 1.24 mmol) were added. After reflux for 4 h, the mixture was poured into a 0.5 M HCl solution and extracted by CH2Cl2. The organic phase was dried over anhydrous Na2SO4 and concentrated to get a crude product which was purified by silica gel column chromatography (CH2Cl2/MeOH = 20/1 to 10/1) to provide the desired product (62 mg, 72%) as a gray solid. HPLC purity 98.6% (tR = 15.6 min). 1H NMR (300 MHz, DMSO-d6) δ 8.15 (d, J = 7.9 Hz, 1H), 7.86–7.75 (m, 1H), 7.64 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 8.1 Hz, 1H), 7.25 (s, 2H), 3.42 (s, 3H), 2.23 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 162.4, 157.0, 155.7, 155.7, 147.7, 134.6, 129.0, 127.5, 126.8, 126.5, 126.1, 124.6, 120.2, 34.7, 17.1. HR ESI-MS (M + H)+ m/z = 281.1282 (calcd for C17H17N2O2: 281.1290).
2-(2,5-Difluoro-4-hydroxyphenyl)quinazolin-4(3H)-one (24).
Compound 24 (120 mg, 88%) was obtained as a light yellow solid following the procedure of 10. HPLC purity 95.7% (tR = 6.74 min). 1H NMR (300 MHz, DMSO-d6) δ 12.29 (s, 1H), 8.14 (d, J = 8.1 Hz, 1H), 7.83 (t, J = 7.8 Hz, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.61 (dd, J = 11.4, 6.9 Hz, 1H), 7.53 (t, J = 7.7 Hz, 1H), 6.89 (dd, J = 11.8, 7.2 Hz, 1H). 13CNMR (75 MHz, DMSO-d6) δ 162.0, 158.3, 155.0, 149.5, 149.2, 149.1, 146.3, 143.3, 135.0, 127.7, 127.2, 126.3, 121.4, 117.8, 117.5, 117.5, 112.5, 112.4, 106.0, 105.6. HR ESI-MS (M + H)+ m/z = 335.0794 (calcd for C14H9F2N2O2: 335.0843).
2-(4-Hydroxy-3,5-dimethoxyphenyl)quinazolin-4(3H)-one (25).
Compound 25 (153 mg, quant.) was obtained as a white solid following the procedure of 10. HPLC purity 95.3% (tR = 14.06 min). 1H NMR (300 MHz, DMSO-d6) δ 12.37 (s, 1H), 9.11 (s, 1H), 8.13 (dd, J = 8.0, 1.6 Hz, 1H), 7.86–7.77 (m, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.58 (s, 2H), 7.52–7.43 (m, 1H), 3.89 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 162.9, 152.5, 149.3, 148.3, 139.6, 135.0, 127.6, 126.4, 126.3, 122.6, 121.1, 105.9, 56.7. HR ESI-MS (M + Na)+ m/z = 381.1105 (calcd for C16H14N2NaO4: 381.1163).
5,7-Dimethoxy-2-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (27).
Compound 25 (75 mg, 86%) was obtained as a white solid following the procedure of 10. HPLC purity 96.6% (tR = 17.17 min). 1H NMR (300 MHz, DMSO-d6) δ 12.23 (s, 1H), 8.35 (d, J = 8.2 Hz, 2H), 7.90 (d, J = 8.1 Hz, 2H), 6.79 (s, 1H), 6.58 (s, 1H), 3.88 (d, J = 10.8 Hz, 6H). HR ESI-MS (M + H)+ m/z = 351.0947 (calcd for C17H14F3N2O3: 351.0957).
5,7-Dimethoxy-2-(3-methyl-4-(trifluoromethyl)phenyl)-quinazolin-4(3H)-one (28).
Compound 28 (80 mg, 88%) was obtained as a white solid following the procedure of 10. HPLC purity 97.5% (tR = 17.99 min). 1H NMR (300 MHz, DMSO-d6) δ 12.14 (s, 1H), 8.27–8.00 (m, 2H), 7.80 (d, J = 8.3 Hz, 1H), 6.78 (s, 1H), 6.57 (s, 1H), 3.88 (d, J = 11.0 Hz, 6H), 2.52 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 164.8, 161.4, 160.1, 153.0, 152.2, 136.9, 136.4, 131.6, 126.3, 126.0, 101.9, 98.6, 56.5, 56.1, 19.3. HR ESI-MS (M + H)+ m/z = 365.1101 (calcd for C18H16F3N2O3: 365.1113).
2-(4-Hydroxyphenyl)-5,7-dimethoxyquinazolin-4(3H)-one (29).
Compound 29 (84 mg, quant.) was obtained as a yellow solid following the procedure of 10. HPLC purity 95.4% (tR = 13.88 min). 1H NMR (300 MHz, DMSO-d6) δ 11.78 (s, 1H), 10.10 (d, J = 3.3 Hz, 1H), 8.21–7.91 (m, 2H), 7.06–6.82 (m, 2H), 6.69 (q, J = 2.2 Hz, 1H), 6.48 (q, J = 2.3 Hz, 1H), 4.05–3.65 (m, 6H). 13C NMR (75 MHz, DMSO-d6) δ 164.6, 161.4, 161.0, 160.3, 153.6, 153.2, 129.9, 129.9, 123.3, 115.7, 115.7, 104.9, 101.4, 97.7, 56.4, 56.0. HR ESI-MS (M + H)+ m/z = 299.1021 (calcd for C16H15N2O4: 299.1032).
2-(2-Chloro-4-hydroxyphenyl)-5,7-dimethoxyquinazolin-4(3H)-one (30).
Compound 30 (94 mg, quant.) was obtained as a yellow solid following the procedure of 10. HPLC purity 97.9% (tR = 13.82 min). 1H NMR (300 MHz, DMSO-d6) δ 11.91 (s, 1H), 10.29 (s, 1H), 7.42 (d, J = 7.9 Hz, 1H), 6.90 (s, 1H), 6.83 (s, 1H), 6.69 (s, 1H), 6.55 (s, 1H), 3.97–3.73 (m, 6H). 13C NMR (75 MHz, DMSO-d6) δ 164.7, 161.5, 160.0, 159.8, 153.7, 153.3, 132.6, 132.3, 124.8, 116.6, 114.7, 105.3, 101.6, 98.3, 56.5, 56.1. HR ESI-MS (M + H)+ m/z = 333.0630 (calcd for C16H14ClN2O4: 333.0642).
(S)-2-(3,5-Dimethyl-4-(oxiran-2-ylmethoxy)phenyl)-5,7-dimethoxyquinazolin-4(3H)-one (31).
Compound 31 (215 mg, 94%) was obtained as a white solid following the procedure of 10. HPLC purity 95.6% (tR = 17.17 min). 1H NMR (300 MHz, DMSO-d6) δ 11.80 (s, 1H), 7.88 (s, 2H), 6.72 (d, J = 2.2 Hz, 1H), 6.50 (d, J = 2.2 Hz, 1H), 5.59 (d, J = 3.7 Hz, 1H), 3.86 (d, J = 12.2 Hz, 6H), 3.79 (s, 2H), 3.49 (dd, J = 10.2, 3.9 Hz, 1H), 3.40 (s, 1H), 2.31 (s, 6H). 13C NMR (75 MHz, DMSO-d6) δ 164.7, 161.4, 160.2, 158.3, 153.5, 152.9, 131.1, 128.8, 127.9, 105.1, 101.6, 98.0, 75.2, 69.4, 56.4, 56.1, 16.6, 12.1. HR ESI-MS (M + H)+ m/z = 383.1615 (calcd for C21H23N2O5: 383.1607).
(S)-2-(4-(2-Hydroxy-3-(4-methylpiperazin-1-yl)propoxy)-3,5-dimethylphenyl)-5,7-dimethoxyquinazolin-4(3H)-one (32).
Compound 32 (17 mg, 35%) was obtained as a white solid following the procedure of 39. HPLC purity 96.1% (tR = 12.62 min). 1H NMR (300 MHz, MeOD) δ 7.66 (s, 2H), 6.75 (d, J = 2.3 Hz, 1H), 6.50 (d, J = 2.3 Hz, 1H), 4.14 (dd, J = 7.7, 4.5 Hz, 1H), 3.91 (s, 6H), 3.82 (t, J = 4.2 Hz, 2H), 2.66 (dd, J = 13.3, 8.3 Hz, 10H), 2.35 (d, J = 2.8 Hz, 9H).13C NMR (75 MHz, MeOD) δ 165.4, 161.6, 161.2, 158.6, 153.7, 153., 131.4, 128.1, 127.8, 104.1, 100.3, 97.4, 74.3, 67.6, 60.3, 55.0, 54.8, 54.4,52.7, 44.4, 15.3. HR ESI-MS (M + Na)+ m/z = 505.2426 (calcd for C26H35N4O5: 505.2427).
(E)-3-(4-(Allyloxy)-3,5-dimethylphenyl)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one (36).
To a solution of 17 (1406 mg, 7.4 mmol) and 1-(2-hydroxy-4,6-dimethoxyphenyl)ethan-1-one 35 (1450 mg, 7.4 mmol) in 20 mL EtOH, 50% KOH (829 mg, 14.8 mmol) was added. After stirring at room temperature overnight, the mixture was concentrated and extracted with CH2Cl2. The organic extract was washed with saturated NaHCO3 (aq), brine, and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was used directly in the next step.
2-(4-(Allyloxy)-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (37).
To a solution of the above residue 36 (1.5 g, 5 mmol) in 10 mL of DMSO, I2 (130 mg, 0.5 mmol) was added. After stirring at 140 °C for 4 h, the mixture was poured into H2O and extracted with CH2Cl2. The organic extract was washed with saturated NaHCO3 (aq), brine, and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified using a silica gel column (CH2Cl2/CH3OH = 50:1) to give the desired product 37 (650 mg, 36%) as a brown foam. HPLC purity 96.4% (tR = 21.53 min). 1H NMR (300 MHz, CDCl3) δ 7.50 (s, 2H), 6.57 (s, 1H), 6.55 (d, J = 2.3 Hz, 1H), 6.34 (d, J = 2.2 Hz, 1H), 6.19–6.02 (m, 1H), 5.44 (dd, J = 17.2, 1.5 Hz, 1H), 5.28 (dd, J = 10.4, 1.2 Hz, 1H), 4.35 (d, J = 5.6 Hz,2H), 3.91 (d, J = 9.8 Hz, 6H), 2.34 (s, 6H). 13C NMR (75 MHz,CDCl3) δ 177.6, 163.9, 160.8, 160.7, 159.9, 158.6, 133.6, 131.8, 126.8, 126.6, 117.6, 109.2, 108.4, 96.1, 92.8, 73.2, 56.3, 55.7, 16.6. MS (M + H)+ m/z 367.2. HR ESI-MS (M + H)+ m/z = 367.1548 (calcd for C22H23O5: 367.1545).
2-(4-Hydroxy-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (38).
To a solution of 35 (630 mg, 1.72 mmol) in 10 mL of CH3OH, Pd(PPh3)4 (60 mg, 0.05 mmol) and K2CO3 (949 mg, 6.88 mmol) were added. After stirring at 90 °C for 7 h, the mixture was poured into 1 N HCl and extracted with n-BuOH. The organic extract was concentrated and CH2Cl2 was added to the precipitate. The precipitate was filtered to get 36 (420 mg, 75%) as a yellow solid. HPLC purity 97.4% (tR = 17.96 min). 1H NMR (300 MHz, DMSO-d6) δ 7.64 (s, 2H), 6.86 (d, J = 2.1 Hz, 1H), 6.57 (s, 1H), 6.49 (d, J = 2.1 Hz, 1H), 3.90 (s, 3H), 3.82 (s, 3H), 2.25 (s, 6H). 13C NMR (75 MHz, DMSO- d6) δ 176.2, 164.0, 160.9, 160.6, 159.6, 157.1, 126.7, 125.3, 121.6, 108.7, 106.6, 96.7, 93.8, 56.6, 49.0, 17.2. MS (M + H)+ m/z 327.1. HR ESI-MS (M + H)+ m/z = 327.1234 (calcd for C19H19O5: 327.1232).
2-(4-(2-Hydroxyethoxy)-3,5-dimethylphenyl)-5,7-dime-thoxy-4H-chromen-4-one (39).
To a solution of 38 (20 mg, 0.061 mmol) in 5 mL of DMF, K2CO3 (25 mg, 0.184 mmol) and 2-bromoethanol (15 mg, 0.123 mmol) were added. The mixture was allowed to heat at 80 °C overnight. Then the mixture was poured into water and extracted by CH2Cl2. The organic extract was washed with saturated NaHCO3 (aq), brine, and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified using a silica gel column (CH2Cl2/CH3OH = 20:1) to give the desired product 39 (12 mg, 55%) as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.53 (s, 2H), 6.58 (d, J = 5.8 Hz, 2H), 6.38 (s, 1H), 3.96 (t, J = 10.0 Hz, 10H), 2.37 (s, 7H). 13C NMR (75 MHz, CDCl3) δ 177.6, 164.0, 160.9, 160.6, 159.9, 158.1, 131.6, 127.0, 126.7, 109.2, 108.5, 96.1, 92.8, 73.3, 62.2, 56.4, 55.8, 16.5. MS (M + H)+ m/z 371.1. HR ESI-MS (M + H)+ m/z = 371.1483 (calcd for C21H23O6: 371.1495).
(S)-2-(3,5-Dimethyl-4-(oxiran-2-ylmethoxy)phenyl)-5,7-dimethoxy-4H-chromen-4-one (40).
Compound 40 (420 mg, 72%) was obtained as a white solid following the procedure of 39. 1H NMR (300 MHz, CDCl3) δ 7.54 (s, 2H), 6.59 (d, J = 3.6 Hz, 2H), 6.38 (d, J = 2.3 Hz, 1H), 4.13 (dd, J = 11.2, 3.0 Hz, 1H), 3.94 (d, J = 10.6 Hz, 6H), 3.79 (dd, J = 11.1, 6.1 Hz, 1H), 3.39 (q, J = 3.3 Hz, 1H), 2.92 (t, J = 4.6 Hz, 1H), 2.74 (dd, J = 5.0, 2.6 Hz, 1H), 2.38 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 177.5, 163.9, 160.9, 160.5, 159.9, 158.2, 131.6, 127.2, 126.7, 109.3, 108.6, 96.1, 92.8, 77.2, 73.3, 56.4, 55.7, 50.4, 44.5, 16.5.
(R)-2-(3,5-Dimethyl-4-(oxiran-2-ylmethoxy)phenyl)-5,7-dimethoxy-4H-chromen-4-one (41).
To a solution of 38 (100 mg, 0.31 mmol) in 5 mL acetone, (R)-(-)-epichlorohydrin (285 mg, 3.1 mmol) and K2CO3 (211 mg, 1.53 mmol) were added. The mixture was refluxed for 24 h and then poured into H2O. The solution was extracted with CH2Cl2 (20 mL × 3). The organic layer was washed with saturated NaHCO3 (aq), brine, and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated to give a crude solid, which was used directly in the next step without further purifications.
(S)-2-(4-(2-Hydroxy-3-(piperidin-1-yl)propoxy)-3,5-dime-thylphenyl)-5,7-dimethoxy-4H-chromen-4-one (42).
Compound 42 (55 mg, 44%) was obtained as a white solid following the procedure of 39. HPLC purity 98.2% (tR = 16.17 min). 1H NMR (300 MHz, DMSO-d6) δ 7.68 (s, 2H), 6.81 (s, 1H), 6.62 (s, 1H), 6.47 (s, 1H), 4.83 (s, 1H), 3.95 (d, J = 5.2 Hz, 1H), 3.89 (s, 3H), 3.82 (s, 3H),3.79–3.69 (m, 2H), 2.42 (tt, J = 13.8, 8.3 Hz, 6H), 2.31 (s, 6H), 1.48 (q, J = 5.5 Hz, 4H), 1.37 (d, J = 6.1 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 176.0, 164.1, 160.7, 160.1, 159.6, 158.7, 131.8, 126.9, 126.3, 108.8, 107.9, 96.7, 93.7, 75.4, 67.6, 61.8, 56.48, 56.4, 55.2, 26.1, 24.4, 16.5. HR ESI-MS (M + H)+ m/z = 468.2392 (calcd for C27H34NO6: 468.2390).
(S)-2-(4-(2-Hydroxy-3-(4-methylpiperazin-1-yl)propoxy)-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (43).
Compound 43 (32 mg, 24%) was obtained as a white solid following the procedure of 39. HPLC purity 98.6% (tR = 14.01 min). 1H NMR (300 MHz, DMSO-d6) δ 7.69 (s, 2H), 6.83 (d, J = 2.2 Hz, 1H), 6.63 (s, 1H), 6.53–6.42 (m, 1H), 4.55 (s, 1H), 3.98–3.93 (m, 1H), 3.89 (s, 3H), 3.82 (s, 3H), 3.76 (dd, J = 9.2, 4.5 Hz, 2H), 2.53 (s, 1H), 2.38 (d, J = 43.1 Hz, 15H), 2.13 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 176.0, 164.1, 160.7, 160.1, 159.6, 158.7, 131.9, 126.9, 126.3, 108.8, 108.0, 96.7, 93.7, 75.3, 74.3, 67.7, 61.0, 56.5, 56.4, 55.3, 53.8, 46.2, 16.5. HR ESI-MS (M + H)+ m/z = 483.2493 (calcd for C27H35N2O6: 483.2495).
(R)-2-(4-(2-Hydroxy-3-(piperidin-1-yl)propoxy)-3,5-dime-thylphenyl)-5,7-dimethoxy-4H-chromen-4-one (44).
To a solution of 41 (48 mg, 0.164 mmol) in 2 mL of EtOH and 2 mL of DMF, piperidine (139 mg, 1.64 mmol) and K2CO3 (226 mg, 1.64 mmol) were added. The mixture was refluxed for 24 h and then poured into H2O. The solution was extracted with CH2Cl2 (20 mL × 3). The organic layer was washed with saturated NaHCO3 (aq), brine, and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated to give a solid residue. The crude product was purified by PTLC (CH2Cl2:CH3OH = 20:1) to give 44 (13 mg, 18% for two steps) as a white solid. HPLC purity 98.6% (tR = 16.8 min). 1H NMR (300 MHz, MeOD) δ 7.50 (s, 2H), 6.63 (s, 1H), 6.44 (d, J = 21.6 Hz, 2H), 4.00– 3.77 (m, 9H), 2.73 (s, 6H), 2.32 (s, 6H), 1.63 (d, J = 43.9 Hz, 6H). 13CNMR (75 MHz, MeOD) δ 178.4, 164.9, 161.5, 160.5, 159.8, 158.5, 131.7, 126.5, 126.2, 107.8, 106.6, 95.9, 92.7, 74.4, 66.9, 61.0, 55.2, 54.6, 24.8, 23.3, 15.3. HR ESI-MS (M + H)+ m/z = 468.2390 (calcd for C27H34NO6: 468.2386).
(R)-2-(4-(2-Hydroxy-3-(4-methylpiperazin-1-yl)propoxy)-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (45).
To a solution of 41 (48 mg, 0.164 mmol) in 5 mL of DMF, 1-methylpiperazine (164 mg, 1.64 mmol) and K2CO3 (226 mg, 1.64 mmol) were added. The mixture was refluxed for 24 h and then poured into H2O. The solution was extracted with CH2Cl2 (20 mL × 3). The organic layer was washed with saturated NaHCO3 (aq), brine, and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated to give a crude solid. The crude product was purified by PTLC (CH2Cl2:CH3OH = 20:1) to give 45 (16 mg, 20% for two steps) as a white solid. HPLC purity 96.8% (tR = 14.8 min). 1H NMR (300 MHz, CDCl3) δ 7.45 (s, 2H), 6.49 (d, J = 6.4 Hz, 2H), 6.29 (s, 1H), 4.02 (s, 1H), 3.82 (s, 8H), 2.49 (dd, J = 39.7, 24.1 Hz, 10H), 2.25 (d, J = 6.1 Hz, 9H). 13C NMR (75 MHz, CDCl3) δ 182.,5168.4, 165.4, 164.7, 163.9162.3, 135.7, 130.8, 112.6, 111.7, 100.2, 96.8, 78.1, 70.6, 64.3, 59.9, 59.7, 58.6, 56.6, 49.3, 20.2. MS (M + H)+ m/z 483.2. HR ESI-MS (M + H)+ m/z = 483.2482 (calcd for C27H35N2O6: 483.2495).
(R)-2-(4-(2-Hydroxy-3-morpholinopropoxy)-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (46).
Compound 46 (76 mg, 65%) was obtained as a white solid following the procedure of 42. HPLC purity 95.6% (tR = 15.30 min). 1H NMR (300 MHz, MeOD) δ 7.42 (d, J = 3.3 Hz, 2H), 6.55 (d, J = 2.3 Hz, 1H), 6.41 (s, 1H), 6.34 (d, J = 2.4 Hz, 1H), 4.14 (dd, J = 7.7, 4.6 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.80 (dd, J = 4.8, 3.0 Hz, 2H), 3.74 (t, J = 4.7 Hz, 3H), 3.67 (t, J = 4.8 Hz, 1H), 3.33 (t, J = 1.7 Hz, 1H), 2.61 (td, J = 8.5, 7.7, 4.0 Hz, 5H), 2.29 (s, 6H). 13C NMR (75 MHz, MeOD) δ 178.3, 164.7, 161.3, 160.4, 159.6, 158.5, 131.6, 126.3, 126.0, 107.8, 106.5, 95.8, 92.6, 74.3, 67.4, 66.5, 61.0, 55.1, 55.0, 54.0, 15.3. HR ESI-MS (M + H)+ m/z = 470.2187 (calcd for C26H32NO7: 470.2179).
(R)-2-(4-(3-(4,4-Difluoropiperidin-1-yl)-2-hydroxypropoxy)-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (47).
Compound 47 (75 mg, 60%) was obtained as a white solid following the procedure of 42. HPLC purity 97.6% (tR = 16.00 min). 1H NMR (300 MHz, CDCl3) δ 7.51 (s, 2H), 6.60–6.52 (m, 2H), 6.35 (d, J = 2.3 Hz, 1H), 4.10 (dd, J = 8.9, 4.6 Hz, 1H), 3.92 (d, J = 9.6 Hz, 6H), 3.84 (d, J = 5.6 Hz, 2H), 2.85–2.75 (m, 2H), 2.65 (td, J = 8.5, 5.6 Hz, 4H), 2.35 (s, 6H), 2.12–1.95 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 177.5, 163.9, 160.8, 160.5, 159.8, 158.1, 131.6, 127.0, 126.7, 124.8, 121.6, 118.4, 109.2, 108.4, 96.1, 92.8, 77.3, 74.1, 66.8, 59.5, 56.4, 55.7, 50.4, 50.4, 50.3, 34.4, 34.0, 33.7, 16.5. HR ESI-MS (M + H)+ m/z = 504.2189 (calcd for C27H32F2NO6: 504.2198).
(R)-2-(4-(2-Hydroxy-3-(pyrrolidin-1-yl)propoxy)-3,5-dime-thylphenyl)-5,7-dimethoxy-4H-chromen-4-one (48).
Compound 48 (30 mg, 27%) was obtained as a white solid following the procedure of 42. HPLC purity 99.2% (tR = 15.85 min). 1H NMR (300 MHz, MeOD) δ 7.49 (s, 2H), 6.62 (d, J = 2.3 Hz, 1H), 6.47 (s, 1H), 6.39 (d, J = 2.3 Hz, 1H), 4.15 (dd, J = 8.8, 4.5 Hz, 1H), 3.97–3.75 (m, 9H), 2.88 (dd, J = 12.6, 4.2 Hz, 1H), 2.74 (t, J = 6.2 Hz, 4H), 2.32 (s, 6H), 1.86 (p, J = 3.1 Hz, 4H). 13C NMR (75 MHz, MeOD) δ 178.3, 164.8, 161.4, 160.5, 159.7, 158.6, 131.6, 126.4, 126.1, 107.8, 106.6, 95.9, 92.7, 74.5, 68.9, 58.7, 55.11, 55.06, 54.3, 22.9, 15.3. HR ESI-MS (M + H)+ m/z = 454.2223 (calcd for C26H32NO6: 454.2230).
(R)-2-(4-(3-(Dimethylamino)-2-hydroxypropoxy)-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (49).
Compound 49 (20 mg, 38%) was obtained as a white solid following the procedure of 42. HPLC purity 95.8% (tR = 15.67 min). 1H NMR (300 MHz, DMSO-d6) δ 7.72 (s, 2H), 6.86 (d, J = 2.3 Hz, 1H), 6.65 (s, 1H), 6.50 (d, J = 2.3 Hz, 1H), 4.97 (d, J = 4.7 Hz, 1H), 4.66 (t, J = 5.5 Hz, 1H), 4.12 (q, J = 5.1 Hz, 1H), 3.90 (s, 3H), 3.83 (s, 3H), 3.80–3.68 (m, 2H), 3.56–3.44 (m, 6H), 3.17 (d, J = 5.0 Hz, 1H), 2.32 (s, 6H). 13CNMR (75 MHz, DMSO-d6) δ 176.1, 164.2, 160.7, 160.2, 159.7, 158.9, 131.9, 127.0, 126.3, 108.8, 108.0, 96.8, 93.8, 74.4, 71.2, 63.1, 56.5, 56.5, 49.1, 46.4, 16.6. HR ESI-MS (M + H)+ m/z = 428.2064 (calcd for C24H30NO6: 428.2073).
(S)-2-(4-(2-Fluoro-3-(piperidin-1-yl)propoxy)-3,5-dimethylphenyl)-5,7-dimethoxy-4H-chromen-4-one (50).
To a solution of 44 (10 mg, 0.021 mmol) in 1 mL CH2Cl2, DAST (5.2 mg, 0.032 mmol) was added. The mixture was allowed to stir at room temperature overnight and then concentrated. The residue was purified by PTLC and 50 (8 mg, 80%) was obtained as a white solid. HPLC purity 95.9% (tR = 16.99 min).1H NMR (300 MHz, CDCl3) δ 7.55 (s, 2H), 6.67–6.52 (m, 2H), 6.39 (d, J = 2.3 Hz, 1H), 4.91 (d, J = 4.5 Hz, 1H), 4.76 (d, J = 4.6 Hz, 1H), 3.95 (d, J = 10.8 Hz, 8H), 3.22–3.03 (m, 1H), 2.73 (d, J = 4.7 Hz, 4H), 2.38 (s, 6H), 1.60 (d, J = 6.3 Hz, 4H), 1.49 (d, J = 5.7 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 177.55, 163.9, 160.9, 160.6, 159.9, 158.4, 131.7, 127.0, 126.7, 109.3, 108.5, 96.1, 92.8, 82.6, 80.4, 68.9, 68.9, 64.5, 64.3, 56.4, 55.7, 51.6, 26.6, 24.6, 16.6. HR ESI-MS (M + H)+ m/z = 470.2330 (calcd for C27H33FNO5: 470.2343).
Crystollagraphy.
Crystals of human BRD4 BD1 in complex with the ligand compound 45 were prepared via hanging drop vapor diffusion at 20 °C. Human BRD4 BD1 protein solution (10 mg/mL) with 2 mM compound 45 was preincubated on ice for 10 min prior to being mixed in 1:1 ratio (protein: reservoir solution) with 100 mM HEPES and 15% (w/v) PEG 3350 at pH 7.5. Orthorhombic crystals grew within 3 days and were subsequently cryoprotected with 100 mM HEPES and 30% (w/v) PEG 3350 at pH 7.5 containing 2 mM compound 45. X-ray diffraction data were collected at beamline 22-ID at the Advanced Photon Source (Argonne National Labs). The diffraction data was processed and integrated using the iMOSFLM v7.2.2,30 and AIMLESS, and CTRUNCATE31 programs were used for scaling and anisotropy correction, respectively. The phase information was obtained by molecular replacement using a single chain of the human BRD4 BD structure (PDB 5KU3) as a search model. Iterative cycles of manual model building and refinement were performed within Phenix v1.16–354932 and COOT v0.8.9.133 software.
Determination of Aqueous Solubility.
Solubility in water for 44 and 45 was determined by HPLC analysis according to a previously published protocol.34,35 First, 1– 2 mg of 44 and 45 were weighed and added to 1 mL of water, respectively. Then, 10–15 mg of44 and 45 were weighed and added to 0.1–0.6 mL of water. The suspensions were shaken at 25 °C for 24 h and then centrifuged and the supernatants were filtered. Aliquots (5 μL) of the supernatants were injected into the HPLC system equipped with a C18 reverse-phase column under the same condition, which was described in the general Experimental Section. One-point calibration36 was done by injecting 5 μL aliquots of the corresponding buffer solutions of 44 or 45 with known concentrations.
Cell Culture.
Immortalized hSAECs were previously described.37,38 hSAECs were grown in SAGM small airway epithelial cell growth medium (Lonza, Walkersville, MD) in a humidified atmosphere of 5% CO2. Poly(I:C) was obtained from Sigma (St. Louis, MO) and used at 10 μg/mL in the cell culture. Compound 1 was purchased from Tocris and compound 3 was either purchased from Cayman Chemical (Ann Arbor, Michigan) or resynthesized in house. Compounds were dissolved in DMSO and added at the indicated concentrations.
Quantitative Real-Time PCR (Q-RT-PCR).
For gene expression analyses, 1 μg of RNA was reverse transcribed using Super Script III as previously described. The cDNA product (1 μL) was amplified using SYBR Green Supermix (Bio-Rad) and indicated gene-specific primers. The reaction mixtures were subjected to 40 cycles of 15 s at 94 °C, 60 s at 60 °C, and 1 min at 72 °C in an iCycler (Bio-Rad). Quantification of relative changes in gene expression was calculated using the ΔΔCt method and expression as the fold change between experimental and control samples was normalized to internal control cyclophilin (PPIA).39
In Vitro Efficacy of BRD4 Inhibitors on Poly(I:C)-Induced Innate Immune Response.
hSAECs were first pretreated with a series of final concentrations of BRD4 inhibitors from 0.01 nM to 100 μM for 24 h and were then added to poly(I:C) at 10 μg/mL for another 4 h prior to harvesting the cells. The harvested cells were first washed with PBS twice and then the total RNA was extracted using acid guanidinium phenol extraction (Tri Reagent; Sigma). The total RNA was further reverse transcribed for gene expression analysis by Q-RTPCR. The inhibitory effect of BRD4 inhibitors on poly(I:C)-induced innate immune gene expression was compared with that of poly(I:C) alone and the inhibitory percentage of each treatment was obtained.8 For compounds 1, 3, 32, 34, 39, 42–45, and 48, in vitro efficacy of these BRD4 inhibitors on poly(I:C)-induced innate immune response were presented as the IC50 values of these compounds. Compounds were dissolved in DMSO and further diluted with cell culture medium to appropriate concentrations.
Time-Resolved Fluorescence Energy Transfer (TR-FRET) Assays.
384-well plate-based commercial TR-FRET Assay kits (Cayman Chemical, Ann Arbor, Michigan) were used to determine the binding ability of the tested BRD4 inhibitors to the BRD4 and BRD2 BDs (BD) using the two recombinant BRD4 BDs or BRD2 BDs by TR-FRET assays. A series of concentrations of BRD4 inhibitors from 0.01 nM to 100 μM were added into a 384-well test plate and mixed with other reaction components based on the instructions from the vendor followed by incubation for 1 h at room temperature. The commercially available BRD inhibitor compounds 1 and 3 were used as the controls. The plates were read in time-resolved format by exciting the sample at 340 nm and reading emissions at 620 and 670 nm, using a 100 μs delay and a 500 μs window at a Tecan M1000 proreader. A plot of the TR-FRET ratio 670 nm emission/620 nm emission versus inhibitor concentration on semilog axes results in a sigmoidal dose–response curve typical of competitive assays. These data were further calculated with the IC50 values of the tested BRD4 inhibitors to the BDs of BRD2 and BRD4 as well as other relevant target proteins, respectively.8,12
In Vivo Efficacy of BRD4 Inhibitors on Poly(I:C)-Induced Acute Airway Inflammation.
Animal experiments were performed according to the NIH Guide for Care and Use of Experimental Animals and approved by the University of Texas Medical Branch (UTMB) Animal Care and Use Committee (approval no. 1312058A). Male C57BL6/J mice (12 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed under pathogen-free conditions with food and water ad libitum. C57BL/6 mice were pretreated in the absence or presence of the indicated BRD4 inhibitors (10 mg/kg body weight, via the intraperitoneal route) 1 day prior to poly(I:C) stimulation. The next day, animals (5 mice per group) were given another dose of the BRD4 inhibitor immediately followed by intranasal (i.n.) administration of phosphate-buffered saline (PBS, 50 μL) or poly(I:C) (300 μg dissolved in 50 μL PBS). After 1 day, the mice were euthanized. The bronchoalveolar lavage fluid (BALF) and lung tissues of the treated mice were collected for further analysis. Compounds were first dissolved in DMSO and further diluted in 10% hydroxypropyl β-cyclodextrin in PBS to appropriate concentration prior to intraperitoneal administration.8,12
Evaluation of Airway Inflammation.
Cellular recruitment into the airway lumen was assessed in the BALF. The lungs were perfused twice with 1 mL of sterile PBS (pH 7.4) to obtain the BALF. Total cell counts were determined by trypan blue staining 50 μL of BALF and counting viable cells using a hemocytometer. Differential cell counts were performed on cytocentrifuge preparations (Cytospin 3; Thermo Shandon, Pittsburgh, Pa) stained with Wright-Giemsa. A total of 300 cells were counted per sample using light microscopy. Formalin-fixed lungs were embedded in paraffin, sectioned at 4 μm thickness, and stained with hematoxylin and eosin or Masson’s trichrome. Microscopy was performed on a NIKON Eclipse Ti System.8,12
Cytokine Bio-Plex Assay.
BALF samples were centrifuged (800 × g for 5 min at 4 °C) and the cytokines quantitated in the supernatant using the Bio-Plex Pro Mouse Cytokine Assay (Bio-Rad, Hercules, CA) with recombinant cytokine standards (in triplicate). Readings were performed on a Bio-Plex 200 system (Bio-Rad). Data were analyzed using Bio-Plex Manager Software Version 6.0 Build 617 (Bio-Rad).8,12
Immunofluorescence Confocal Microscopy (IFCM).
Cultured hSAECs were plated on rat tail collagen-treated cover glasses. Afterward, the cells were first preincubated with compound 44 or 45 at a final concentration of 10 μM for 24 h and were then added to poly(I:C) at 10 μg/mL for another 4 h prior to harvesting the cells. The cells were fixed with 4% paraformaldehyde in PBS and incubated with 0.1 M ammonium chloride for 10 min. The cells were permeabilized with 0.5% Triton-100 followed by incubation in blocking buffer (5% goat serum, 0.1% IGEPAL CA-630, 0.05% NaN3, and 1% BSA) and incubated with anti-H3K122Ac (Abcam, Cambridge, MA) in incubation buffer (0.1% IGEPAL CA-630, 0.05% NaN3, and 2% BSA) overnight at 4 °C. After washing, the cells were stained with Alexa Fluor 488-conjugated goat antirabbit IgG (Life Technologies, Carlsbad, CA), respectively, in incubation buffer for 1 h, then counterstained with nuclear marker DAPI (Sigma-Aldrich, St. Louis, MO), and visualized with a Nikon fluorescence confocal microscope, magnification 63 ×.10,12
Formalin-fixed, paraffin-embedded lung sections were rehydrated using serial concentrations of ethanol. Antigen retrieval was performed with antigen unmasking solution based on recommendations from Abcam (TE buffer, pH 9.0). Paraffin-embedded sections were blocked using 0.1% Triton-X and 5% normal goat serum and incubated with rabbit anti-acetyl H3K122 Ab (Abcam, Cambridge, MA) overnight at 4 °C. Normal antirabbit IgG was used as a staining specificity control. After washing, the cells were stained with Alexa Fluor 568-conjugated goat antirabbit IgG (Life Technologies) in incubation buffer for 1 h, then counterstained with nuclear marker DAPI (Sigma-Aldrich, St. Louis, MO), and visualized with a LSM510 fluorescence confocal microscope, magnification 63 ×.10,12
Supplementary Material
ACKNOWLEDGMENTS
This work was supported, in part, by NIH grants (NIAID AI062885, UL1TR001439) (A.R.B.), UTMB Technology Commercialization Program, and Sanofi Innovation Awards (iAwards) (A.R.B., J.Z., and B.T.), John D. Stobo, M.D. Distinguished Chair Endowment Fund (J.Z.), Crohn’s & Colitis Foundation Entrepreneurial Investing (EI) Initiative award (J.Z., A.R.B., and B.T.), and a research fellowship award (Z.L.) from the Crohn’s & Colitis Foundation of America. Core laboratory support was provided by the UTMB Histopathology Core. We want to thank Dr. Lawrence C. Sowers at the Department of Pharmacology, Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance, and Dr. Xuemei Luo at UTMB mass spectrometry core with funding support from UT system proteomics network for the HRMS analysis.
ABBREVIATIONS USED
- KAc
lysine acetylation
- HATs
histone acetyl transferases
- HDAC
histone deacetylase
- BET
bromodomain and extraterminal domain
- BRD4
bromodomain-containing protein 4
- MACE
major adverse cardiovascular events
- TLR3
toll-like receptor 3
- hSAECs
human small airway epithelial cells
- PK
pharmacokinetic
- WPF
tryptophan-proline-phenylalanine
- THF
tetrahydrofuran
- DIPEA
N,N-diisopropylethylamine
- NaH
sodium hydride
- Pd(PPh3)4
tetrakis(triphenylphosphin)-palladium
- DAST
diethylaminosulfur trifluoride
- qRT-PCR
quantitative real-time PCR
- TR-FRET
time-resolved fluorescence energy transfer
- i.v.
intravenously
- p.o.
per os
- MLM
mouse liver microsomes
- HLM
human liver microsomes
- t1/2
half-life
- Cmax
maximum plasma concentration
- AUC0‑t
total exposure following single dose
- VSS
volume of distribution at steady state
- CL
total clearance
- F,
oral bioavailability
- p.o
oral administration
- i.p.
intraperitoneal administration
- i.n.
intranasal
- BALF
bronchoalveolar lavage fluid
- SAR
structure-activity relationship
- IFCM
immunofluorescence confocal microscopy
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00035
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00035.
Cocomplex crystal structures of 45 with human BRD4 BD1 protein; superimposition of BRD4 BD1 with BRD3 BD1; plasma concentration curves of compound 45 in male SD rats after IV and PO administration; BROMOscan profiling of compound 45; target panel assays by NIMH/NIH psychoactive drug screening program (PDSP) for compound 45; data collection and refinement statistics for the crystal analysis of the BRD4 inhibitor 45 cocomplexed with human BRD4 BD1; representative HPLC analysis; and copies of 1H and 13C NMR spectra of all new compounds (PDF)
Molecular formula strings and some data (CSV)
Accession Codes
The PDB code for BRD4 BD1 protein cocomplexed with 45 is 6UWU. The authors will release the atomic coordinates and experimentaldata upon article publication.
The authors declare no competing financial interest.
Contributor Information
Zhiqing Liu, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
Haiying Chen, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Pingyuan Wang, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Yi Li, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Eric A. Wold, Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
Paul G. Leonard, Core for Biomolecular Structure and Function, MD Anderson Cancer Center, Houston, Texas 77054, United States
Sarah Joseph, Core for Biomolecular Structure and Function, MD Anderson Cancer Center, Houston, Texas 77054, United States.
Allan R. Brasier, Institute for Clinical and Translational Research (ICTR), University of Wisconsin-Madison School of Medicine and Public Health, Madison, Wisconsin 53705, United States;
Bing Tian, Department of Internal Medicine and Sealy Center for Molecular Medicine, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
Jia Zhou, Chemical Biology Program, Department of Pharmacology and Toxicology, Sealy Center for Molecular Medicine, and Institute for Translational Sciences, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
REFERENCES
- (1).Berdasco M; Esteller M Clinical Epigenetics: Seizing Opportunities for Translation. Nat. Rev. Genet 2019, 20, 109–127. [DOI] [PubMed] [Google Scholar]
- (2).Rakyan VK; Down TA; Balding DJ; Beck S Epigenome-wide Association Studies for Common Human Diseases. Nat. Rev. Genet 2011, 12, 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kuo MH; Allis CD Roles of Histone Acetyltransferases and Deacetylases in Gene Regulation. BioEssays 1998, 20, 615–626. [DOI] [PubMed] [Google Scholar]
- (4).Liu Z; Wang P; Chen H; Wold EA; Tian B; Brasier AR; Zhou J Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem 2017, 60, 4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Filippakopoulos P; Knapp S Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation. Nat. Rev. Drug Discov 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- (6).Cochran AG; Conery AR; Sims RJ 3rd. Bromodomains: a New Target Class for Drug Development. Nat. Rev. Drug Discov 2019, 18, 609–628. [DOI] [PubMed] [Google Scholar]
- (7).Belkina AC; Denis GV BET Domain Co-regulators in Obesity, Inflammation and Cancer. Nat. Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu Z; Tian B; Chen H; Wang P; Brasier AR; Zhou J Discovery of Potent and Selective BRD4 Inhibitors Capable of Blocking TLR3-Induced Acute Airway Inflammation. Eur. J. Med. Chem 2018, 151, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhao Y; Tian B; Sun H; Zhang J; Zhang Y; Ivannikov M; Motamedi M; Liu Z; Zhou J; Kaphalia L; Calhoun WJ; Maroto R; Brasier AR Pharmacoproteomics Reveal Novel Protective Activity of Bromodomain Containing 4 Inhibitors on Vascular Homeostasis in TLR3-Mediated Airway Remodeling. J. Proteomics 2019, 205, 103415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tian B; Hosoki K; Liu Z; Yang J; Zhao Y; Sun H; Zhou J; Rytting E; Kaphalia L; Calhoun WJ; Sur S; Brasier AR Mucosal Bromodomain-Containing Protein 4 Mediates Aeroallergen-Induced Inflammation and Remodeling. J. Allergy Clin. Immun 2019, 143, 1380–1394.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nicodeme E; Jeffrey KL; Schaefer U; Beinke S; Dewell S; Chung CW; Chandwani R; Marazzi I; Wilson P; Coste H; White J; Kirilovsky J; Rice CM; Lora JM; Prinjha RK; Lee K; Tarakhovsky A Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tian B; Liu Z; Yang J; Sun H; Zhao Y; Wakamiya M; Chen H; Rytting E; Zhou J; Brasier AR Selective Antagonists of the Bronchiolar Epithelial NF-kB-Bromodomain-Containing Protein 4 Pathway in Viral-Induced Airway Inflammation. Cell Rep 2018, 23, 1138–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tian B; Liu Z; Litvinov J; Maroto R; Jamaluddin M; Rytting E; Patrikeev I; Ochoa L; Vargas G; Motamedi M; Ameredes BT; Zhou J; Brasier AR Efficacy of Novel Highly Specific Bromodomain-Containing Protein 4 Inhibitors in Innate Inflammation-Driven Airway Remodeling. Am. J. Respir. Cell Mol. Biol 2019, 60, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Brasier AR; Zhou J Validation of the Epigenetic Reader Bromodomain-Containing Protein 4 (BRD4) as a Therapeutic Target for Treatment of Airway Remodeling. Drug Discovery Today 2020, 25, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li Z; Guo J; Wu Y; Zhou Q The BET Bromodomain Inhibitor JQ1 Activates HIV Latency Through Antagonizing BRD4 Inhibition of Tat-Transactivation. Nucleic Acids Res 2013, 41, 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Niu Q; Liu Z; Alamer E; Fan X; Chen H; Endsley J; Gelman BB; Tian B; Kim JH; Michael NL; Robb ML; Ananworanich J; Zhou J; Hu H Structure-Guided Drug Design Identifies a BRD4-Selective Small Molecule That Suppresses HIV. J. Clin. Invest 2019, 129, 3361–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhu J; Gaiha GD; John SP; Pertel T; Chin CR; Gao G; Qu H; Walker BD; Elledge SJ; Brass AL Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Rep 2012, 2, 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Duan Q; McMahon S; Anand P; Shah H; Thomas S; Salunga HT; Huang Y; Zhang R; Sahadevan A; Lemieux ME; Brown JD; Srivastava D; Bradner JE; McKinsey TA; Haldar SM BET Bromodomain Inhibition Suppresses Innate Inflammatory and Profibrotic Transcriptional Networks in Heart Failure. Sci. Transl. Med 2017, 9, eaah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Korb E; Herre M; Zucker-Scharff I; Darnell RB; Allis CD BET Protein BRD4 Activates Transcription in Neurons and BET Inhibitor JQ1 Blocks Memory in Mice. Nat. Neurosci 2015, 18, 1464–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang G; Smith SG; Zhou MM Discovery of Chemical Inhibitors of Human Bromodomains. Chem. Rev 2015, 115, 11625–11668. [DOI] [PubMed] [Google Scholar]
- (21).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chung C-W; Coste H; White JH; Mirguet O; Wilde J; Gosmini RL; Delves C; Magny SM; Woodward R; Hughes SA; Boursier EV; Flynn H; Bouillot AM; Bamborough P; Brusq JM; Gellibert FJ; Jones EJ; Riou AM; Homes P; Martin SL; Uings IJ; Toum J; Clement CA; Boullay AB; Grimley RL; Blandel FM; Prinjha RK; Lee K; Kirilovsky J; Nicodeme E Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med. Chem 2011, 54, 3827–3838. [DOI] [PubMed] [Google Scholar]
- (23).Picaud S; Wells C; Felletar I; Brotherton D; Martin S; Savitsky P; Diez-Dacal B; Philpott M; Bountra C; Lingard H; Fedorov O; Muller S; Brennan PE; Knapp S; Filippakopoulos P RVX-208, an Inhibitor of BET Transcriptional Regulators with Selectivity for the Second Bromodomain. Proc. Natl. Acad. Sci. U. S.A 2013, 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Resverlogix announces topline results in BETonMACE phase 3 epigenetics trial. https://www.resverlogix.com/investors/news?article=647 (accessed Sep 30, 2019).
- (25).McDaniel KF; Wang L; Soltwedel T; Fidanze SD; Hasvold LA; Liu D; Mantei RA; Pratt JK; Sheppard GS; Bui MH; Faivre EJ; Huang X; Li L; Lin X; Wang R; Warder SE; Wilcox D; Albert DH; Magoc TJ; Rajaraman G; Park CH; Hutchins CW; Shen JJ; Edalji RP; Sun CC; Martin R; Gao W; Wong S; Fang G; Elmore SW; Shen Y; Kati WM Discovery of N-(4-(2,4-Difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a Potent and Orally Available Bromodomain and Extraterminal Domain (BET) Family Bromodomain Inhibitor. J. Med. Chem 2017, 60, 8369–8384. [DOI] [PubMed] [Google Scholar]
- (26).Gavai AV; Norris D; Tortolani D; Malley D; Zhao Y; Quesnelle C; Gill P; Vaccaro W; Huynh T; Ahuja V; Dodd D; Mussari C; Harikrishnan L; Kamau M; Tokarski JS; Sheriff S;Rampulla R; Wu D-R; Li J; Zhang H; Li P; Sun D; Yip H; Zhang Y; Mathur A; Zhang H; Huang C; Yang Z; Ranasinghe A; Arienzo C; Su C; Everlof G; Zhang L; Raghavan N; Hunt JT; Poss M; Vite GD; Westhouse RA; Wee S Abstract 5789: Discovery of Clinical Candidate BMS-986158, an Oral BET Inhibitor, for the Treatment of Cancer. Cancer Res 2018, 78, 5789. [Google Scholar]
- (27).Raux B; Voitovich Y; Derviaux C; Lugari A; Rebuffet E; Milhas S; Priet S; Roux T; Trinquet E; Guillemot JC; Knapp S; Brunel JM; Fedorov AY; Collette Y; Roche P; Betzi S; Combes S; Morelli X Exploring Selective Inhibition of the First Bromodomain of the Human Bromodomain and Extra-terminal Domain (BET) Proteins. J. Med. Chem 2015, 59, 1634–1641. [DOI] [PubMed] [Google Scholar]
- (28).Tian X; Song L; Li E; Wang Q; Yu W; Chang J Metal-Free One-Pot Synthesis of 1,3-Diazaheterocyclic Compounds via I2-Mediated Oxidative C–N Bond Formation. RSC Adv 2015, 5, 62194–62201. [Google Scholar]
- (29).Pettersson M; Hou X; Kuhn M; Wager TT; Kauffman GW; Verhoest PR Quantitative Assessment of the Impact of Fluorine Substitution on P-Glycoprotein (P-gp) Mediated Efflux, Permeability, Lipophilicity, and Metabolic Stability. J. Med. Chem 2016, 59, 5284–5296. [DOI] [PubMed] [Google Scholar]
- (30).Battye TGG; Kontogiannis L; Johnson O; Powell HR; Leslie AGW iMOSFLM: a New Graphical Interface for Diffraction-Image Processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Evans PR; Murshudov GN How Good Are My Data and What is the Resolution? Acta Crystallogr. D Biol. Crystallogr 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ding C; Zhang Y; Chen H; Yang Z; Wild C; Chu L; Liu H; Shen Q; Zhou J Novel Nitrogen-Enriched Oridonin Analogues with Thiazole-Fused A-Ring: Protecting Group-Free Synthesis, Enhanced Anticancer Profile, and Improved Aqueous Solubility. J. Med. Chem 2013, 56, 5048–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chen H; Yang Z; Ding C; Chu L; Zhang Y; Terry K; Liu H; Shen Q; Zhou J Discovery of O-Alkylamino Tethered Niclosamide Derivatives as Potent and Orally Bioavailable Anticancer Agents. ACS Med. Chem. Lett 2013, 4, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kiselev E; DeGuire S; Morrell A; Agama K; Dexheimer TS; Pommier Y; Cushman M 7-Azaindenoisoquinolines as Topoisomerase I Inhibitors and Potential Anticancer Agents. J. Med. Chem 2011, 54, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tian B; Li X; Kalita M; Widen SG; Yang J; Bhavnani SK; Dang B; Kudlicki A; Sinha M; Kong F; Wood TG; Luxon BA; Brasier AR Analysis of the TGFβ-induced program in primary airway epithelial cells shows essential role of NF-κB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genomics 2015, 16, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tian B; Zhao Y; Sun H; Zhang Y; Yang J; Brasier AR BRD4 mediates NF-κB-dependent epithelial-mesenchymal transition and pulmonary fibrosis via transcriptional elongation. Am. J. Physiol. Lung Cell Mol. Physiol 2016, 311, L1183–L1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tian B; Zhao Y; Kalita M; Edeh CB; Paessler S; Casola A; Teng MN; Garofalo RP; Brasier AR CDK9-Dependent Transcriptional Elongation in the Innate Interferon-Stimulated Gene Response to Respiratory Syncytial Virus Infection in Airway Epithelial Cells. J. Virol 2013, 87, 7075–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.