

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder with limited available therapies. There is progress in developing treatments for neuropsychiatric indications including agitation, psychosis, apathy, and sleep disorders in AD. Candidate therapies progress from nonclinical/animal assessment to trials in normal volunteers (Phase 1), small Phase 2 trials in AD, and larger confirmatory Phase 3 trials. Biomarkers play an increasingly important role in selecting participants, stratifying populations, demonstrating target engagement, supporting disease modification, and monitoring safety. There are currently 121 agents in clinical trials including treatments for neuropsychiatric symptoms, cognition enhancement and, disease progression. There are 27 agents in Phase 1 trials, 65 in Phase 2 trials, and 29 in Phase 3 trials. Most of the agents in trials (80%) target disease modification. Treatments are being assessed in secondary prevention trials of cognitively normal individuals at high risk for the development of AD. There is progress in target diversification, trial designs, outcome measures, biomarkers, and trial population definitions that promise to accelerate developing new therapies for those with or at risk for AD.
Keywords: Neuropsychiatry, Alzheimer’s disease, psychosis, apathy, agitation, depression, drug development, clinical trials, cognition
Alzheimer disease (AD) is characterized by progressive cognitive and functional decline and the frequent emergence of neuropsychiatric syndromes over the course of the 10–15 year symptomatic phase of the disease(1, 2). AD is the most prevalent late-life neurodegenerative disorder and is becoming increasingly common as the world’s population ages. AD doubles in frequency every five years after the age of 60, rising from a rate of 1 percent of 60 year old’s to approximately 40% of 80 year old’s(3). There are currently 5.3 million persons with AD dementia in the US and this is projected to rise to 14 million by 2050. The corresponding economic impact will increase from its current $230 billion annually to over $1 trillion annually by 2050(4). Despite the great threat to the population posed by AD, there are limited therapies available and a high rate of failure in developing new drugs for this disorder(5).
Two classes of cognitive enhancing agents are currently approved for the treatment of AD and available on the market–cholinesterase inhibitors (ChE-Is)(donepezil, rivastigmine, galantamine) and an N-methyl-D-aspartate (NMDA) receptor antagonist (memantine). There are currently no approved disease modifying agents for the treatment of AD. Therapies that prevent or delay the onset, slow the progression, or improve the symptoms of AD are needed to respond to the cognitive, functional and behavioral changes in the burgeoning AD population.
There are 121 agents currently in clinical trials for AD(6). Drug development strategies for AD and the pipeline of emerging agents for treatment of neuropsychiatric symptoms, cognitive enhancement, and disease-modification are reviewed. The pipeline of emerging therapies is described; a neuropsychiatric perspective is emphasized.
Pharmacologic Management of Neuropsychiatric Disorders
With few exceptions --- mainly involving very rare disorders --- the randomized, double-blind, placebo-controlled trial is the gold standard for the assessment and eventual approval of therapies for all medical conditions. Clinical trials provide rigorous answers to very specific questions: they address outcomes using prespecified instruments, for participants diagnosed with specific criteria, in a given stage of disease, treated for a specified period of time, with one or more doses of a test agent. Conclusions beyond these tightly specified parameter cannot be drawn from clinical trials, and trial results cannot be confidently generalized to non-trial populations.
In the practice of neuropsychiatry treatment decisions are often not informed by the narrow outcomes of a clinical trial. Patients with mixed conditions (e.g., AD and stroke), exhibiting complex combinations of neuropsychiatric symptoms (e.g., depression, psychosis, and sleep disorder), who are older or younger than those included in a trial, or who have medical conditions or take medications that were not allowed in a trial are common in neuropsychiatric practices. For this reason, “off label” prescribing is common to achieve an improved quality of life for patients in the relentlessly patient-centric approach that characterizes neuropsychiatric practice. Neuropsychiatrists and behavioral/cognitive neurologists bear a responsibility for rational pharmacology using evidence-based medicine in a broad context. In addition to data derived from clinical trials, informative principles to guide pharmacologic decision-making include the therapeutic metaphor seeking similarities between symptoms successfully treated in clinical trials and those evident in a patient whose disorder differs from those included in the trial (e.g., use of pimavanserin for the treatment of psychosis in AD after demonstration of its utility in the psychosis of Parkinson’s disease (PD)(7, 8)), and the biological extension principle that reasons that responses seen in trial participants may be recapitulated in patients with similar symptoms and a shared biology (e.g., rivastigmine was first shown to improve cognition in AD and then shown to improve cognition in PD dementia which has a similar cholinergic deficit)(9, 10). The greater the similarity between the trial patients and the patient to be treated, the greater the likelihood that the therapeutic extension will be successful.
Case reports, multiple case observations and well conducted n-of-1 trials can provide information useful for individual patient management and may guide programs for future expanded indications of approved agents(11, 12).
Based on these principles, best practices and “medicine-based evidence” evolve that define a body of information on which neuropsychiatric prescribers can call for guidance until clinical trials provide more specific information. Professional prescribing standards are comprised of extrapolations from clinical trials, carefully observed off-label treatment responses, knowledge of potential benefit and harm, appropriate informed consent, and careful documentation in the medical record. Use of psychotropic agents in AD follow these principles since there are few agents approved by the Food and Drug Administration (FDA) for behavioral indications in AD or other neurodegenerative disorders.
Currently Available Treatments for Alzheimer’s Disease
Cognitive Enhancing Agents
Currently available cognitive enhancing agents approved for treatment of AD include three ChE-Is, one NMDA antagonist (memantine), and one fixed combination of a ChE-I and memantine (Namzaric™)(13). These represent the only agents approved for the treatment of AD; no new cognitive enhancers have been approved in the US or Europe since 2003(5). They produce significant if modest improvement in cognition and in co-primary outcomes of function or global assessment.
ChE-Is and memantine have behavioral effects. ChE-Is reduce psychosis and apathy and improve mood(14, 15); memantine reduces agitation and irritability(14, 16) Cognitive enhancers have broad effects on neurochemical systems, and combined behavioral and cognitive effects are not unexpected. Studying the effects on neuropsychiatric symptoms of emerging cognitive enhancing agents will be an important aspect of their development.
Treatments for Neuropsychiatric Symptoms
Patients with central nervous systems (CNS) diseases are usually excluded from clinical trials of psychotropic drugs (e.g., patients with PD and depression would be excluded from trials of antidepressants for major depression). At the end of the typical development program for a psychotropic agent, little is known about its efficacy or safety for use in the treatment of individuals with neurological disease.
A few psychotropic agents are approved specifically for patients with central nervous system (CNS) disorders. Pimavanserin is approved for the treatment of psychosis of Parkinson’s disease (PD)(17); dextromethorphan/quinidine is approved for pseudobulbar affect across multiple neurological disorders(18); and risperidone is approved for irritability in autism(19). Suvorexant was shown to reduce insomnia in patients with AD and the prescribing instructions and package insert have been modified to include the efficacy and side effects observed in AD(20). No other agents have been approved for any neuropsychiatric syndrome in any neurological disorder. In the absence of approved treatments, the rational pharmacology approach supports treatment of agitation with antipsychotics or antidepressants(21, 22), psychosis with antipsychotics(7), depression with antidepressants(8), and apathy with stimulants(23).
Drug Development and Clinical Trials
Emerging therapies typically progress from non-clinical testing of efficacy and safety in animals to human clinical trials(24). Phase 1 consists of testing the candidate treatment in normal healthy volunteers organized into single ascending dose (SAD) cohorts followed by multiple ascending dose (MAD) cohorts. Safety, tolerability, pharmacokinetic (PK) parameters (e.g., bioavailability, half-life, maximum concentration, time to maximum concentration, presence of metabolites, effects of food on PK, drug-drug interactions with commonly used drugs, maximum tolerated dose), blood brain barrier penetration, and doses to be advanced to Phase 2 are determined in Phase 1. Programs developing drugs for AD may include at least one cohort of elderly individuals to assess the effects of aging on safety, tolerability, and PK; a few programs include cohorts of participants with AD in Phase 1/2 designs. Immunotherapy studies may include only participants with AD since immune responses could be permanently altered in healthy volunteers.
Phase 2 in AD drug development establishes proof-of-concept (POC) in participants with AD(25, 26). Critical outcomes of Phase 2 studies are determination of the doses to be advanced to Phase 3; assessment of a dose-related response on clinical measurers, biomarkers, or both; demonstration of target engagement (discussed below) to establish that there is a pharmacodynamic effect in the dose range tested; and collection of additional evidence of safety and tolerability(24). Programs for disease modifying therapies (DMTs), treatments for behavioral symptoms, and cognitive enhancement use inclusion criteria and outcomes to match their development objectives. DMT programs may accept drug-placebo differences on a biomarker as sufficient evidence of target engagement to advance a therapy to Phase 3, whereas cognitive enhancing agents and psychotropic drug development programs demonstrate efficacy on relevant cognitive or behavioral measures in Phase 2 to provide a foundation for Phase 3. Phase 2 trials typically involve 100–400 patients per arm although some may be larger or smaller depending on the objectives and design of the trial.
Phase 3 trials provide confirmatory evidence of efficacy and safety(27) and accrue data for application to the FDA for marketing approval. Trials of cognitive enhancers and DMTs of participants with AD dementia must demonstrate a drug-placebo difference on two prespecified outcomes – either cognition and a global measure or cognition and a functional measure. Cognitive outcomes address the core deficit of AD; global and functional outcomes establish the clinical meaningfulness of the intervention(28). Candidate DMTs must show an effect on biomarkers to be regarded as disease-modifying; the magnitude of the effect and repertoire of biomarkers required are not certain since no DMT is approved for AD.
DMTs being assessed in prodromal AD can be approved by demonstrating a drug-placebo difference on a single composite measure such as the Clinical Dementia Rating – Sum of Boxes(29)(CDR-SB) plus evidence of an effect on biomarkers(30).
AD has a long preclinical phase that precedes the onset of AD dementia by 15–20 years. During this time, the individuals are cognitively normal but have positive amyloid positron emission tomography (PET) or an AD-type signature of biomarker changes in the cerebrospinal fluid (CSF) with decreased levels of amyloid beta-protein (Aß) and increased levels of phosphorylated tau (p-tau) and total tau(31, 32). In addition to individuals with abnormal state markers of AD pathology (PET or CSF), those with mutations that produce autosomal dominant AD or persons who are homozygotes for the apolipoprotein e4 gene and close to the time of onset of symptomatic AD have preclinical AD and are candidates for prevention trials(33, 34). Secondary prevention trials are conducted during the preclinical phase of AD with the goal of preventing or delaying the onset of cognitive decline(35).
An AD drug development program takes at least 7.6 years to execute after the agent has undergone non-clinical characterization in animals. Phase 1 trials require on average 12.8 months to complete; Phase 2 takes 27.7 months on average; and Phase 3 typically lasts 50.9 months(36).
AD drug development is extraordinarily expensive. Investment costs for a single agent to reach the end of Phase 1 approximate $71 million, to the end of Phase 2 the cumulative cost is $126 million, and to the end of Phase 3 the investment is $413 million. Considering the cost of capitalization and the cost of failures, the total cost of developing a successful agent is calculated at $5.69 billion(36). These costs are prohibitive and require that less cost-intensive strategies be developed if a robust repertoire of therapies for all aspects of AD are to be made available to patients and clinicians.
Biomarkers in Clinical Trials
The amyloid (A), tau (T), neurodegeneration (N) framework (A/T/N) identifies the key pathology of AD and specifies biomarkers for each of the components(32)(Table 1). For A, fibrillar/plaque amyloid is identified by amyloid PET; monomeric Aß40 and Aß42 can identified in CSF; and the monomeric Aß42/40 ratio is abnormal in the blood(37, 38). There is no consensus on a measure of Aß oligomers. T biomarkers include tau PET which identifies neurofibrillary tangles; hyperphosphorylated tau (p-tau) in the CSF; and p-tau in the blood(39). Biomarkers of N include magnetic resonance imaging (MRI) evidence of cerebral and hippocampal atrophy; fluorodeoxyglucose (FDG) PET indicative of cerebral hypometabolism; CSF total tau, neurogranin, and neurofilament light (NfL); and blood NfL(40, 41). Biomarkers are indirect measures of cerebral pathology and have their own diffusion, metabolism, and excretion characteristics that affect their detection and usefulness as markers for therapy. They provide inferential but imperfect insight into brain pathology. Many other biomarkers are in development and promise to furnish a more comprehensive window on the pathology of AD and response to treatment(42).
Table 1.
A/T/N biomarkers for AD.
| Amyloid (A) | Tau (T) | Neurodegeneration (N) | |
|---|---|---|---|
| Pathology | Aℬ species: monomers; oligomers; protofibrils; plaques | Tau species: monomers; oligomers; neurofibrillary tangles | Neuronal death; synaptic loss |
| CSF | Aℬ 42 | P-tau (181, 217) | Total tau; NfL; neurogranin |
| Blood | Aℬ 42/40 | P-tau (181, 217) | NfL |
| Imaging | Amyloid PET (fibrillar, insoluble, plaque Aℬ) | Tau PET (neurofibrillary tangles) | MRI (atrophy); FDG PET (metabolism) |
Aℬ - amyloid-beta protein; CSF – cerebrospinal fluid; FDG – fluorodeoxyglucose; MRI – magnetic resonance imaging; NfL – neurofilament light chain; PET – positron emission tomography; p-tau – hyperphosphorylated tau protein
Biomarker profiles change over the course of the illness and define an AD continuum(43, 44)(Figure 1). Biomarker trajectories reflect the dynamic nature of the evolving pathology of AD with changes in Aß being identifiable first (positive amyloid PET; decreased Aß42 in the CSF), followed by alterations in CSF tau and p-tau, and in turn by cerebral atrophy on MRI and hypometabolism on FDG PET. CSF total tau and p-tau are increased early in AD and function as state markers; tau PET becomes positive later in the disease course and functions as a stage marker(45). Cognitive and functional symptoms are relatively late manifestations, occurring coincident with evidence of T abnormalities and of N on MRI and FDG PET. Given the long course of these biomarker changes (e.g., approximately 20 years from changes in Aß to onset of AD dementia), not all individuals with positive amyloid imaging will live to develop AD and counseling patients on the basis of positive biomarkers must be done with caution(46)). The ability to detect biomarker changes prior to clinical abnormalities has made it possible to design secondary prevention trials in individuals at high risk for AD.
Figure 1.
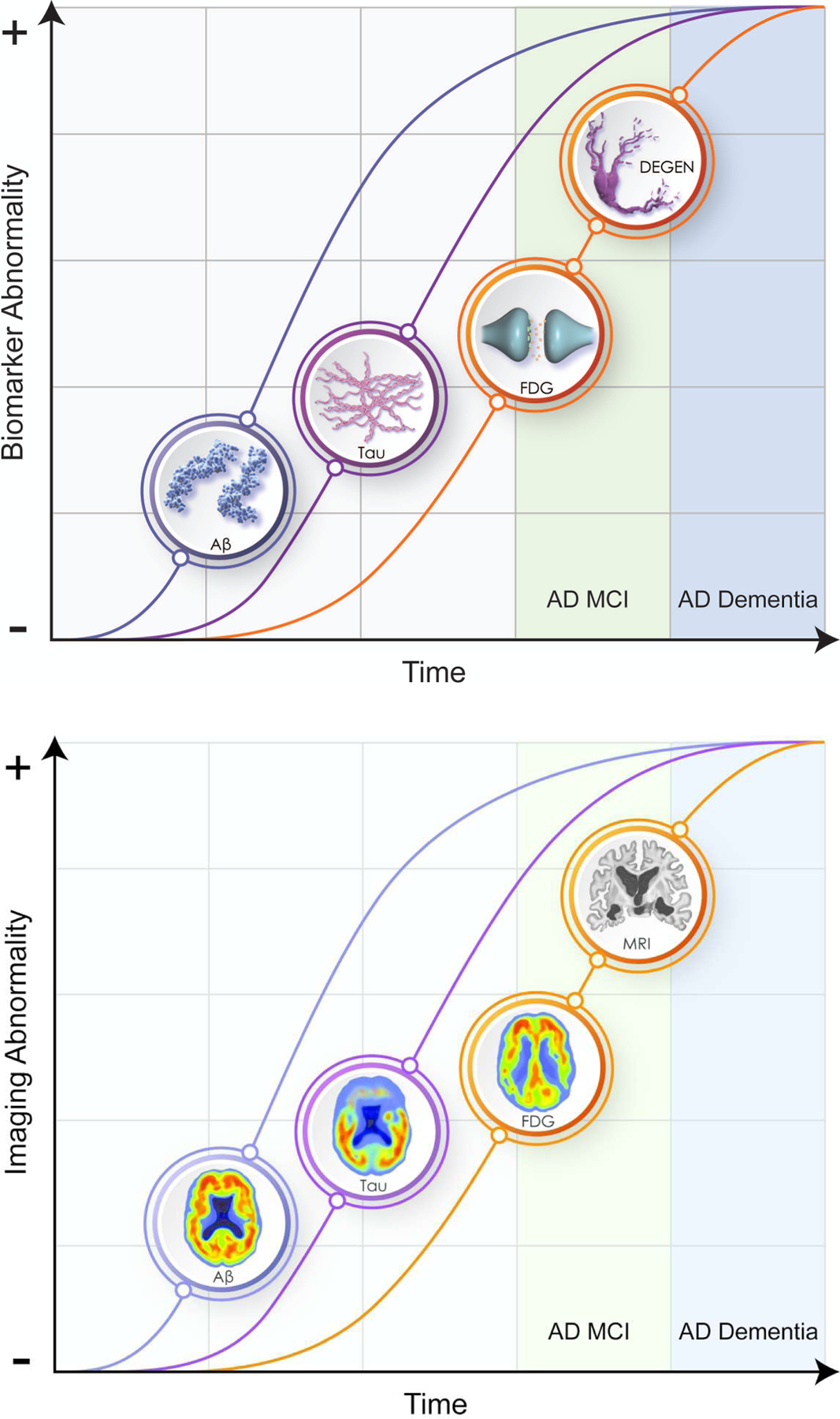
Evolution of ATN biomarkers and clinical and functional changes in the AD continuum. Panel A shows the evolution of the pathology of AD; Panel B shows the corresponding imaging abnormalities (© JLCummings; illustrator M de la Flor, PhD).
Biomarkers have transformed AD drug development (Figure 2)(47, 48). In Phase 1, biomarkers serve as safety measures to assess adverse effects in first-in-human trials. Hepatic injury revealed by elevated liver functions or adverse cardiac effects revealed by electrocardiography are examples of well-established Phase 1 biomarkers.
Figure 2.
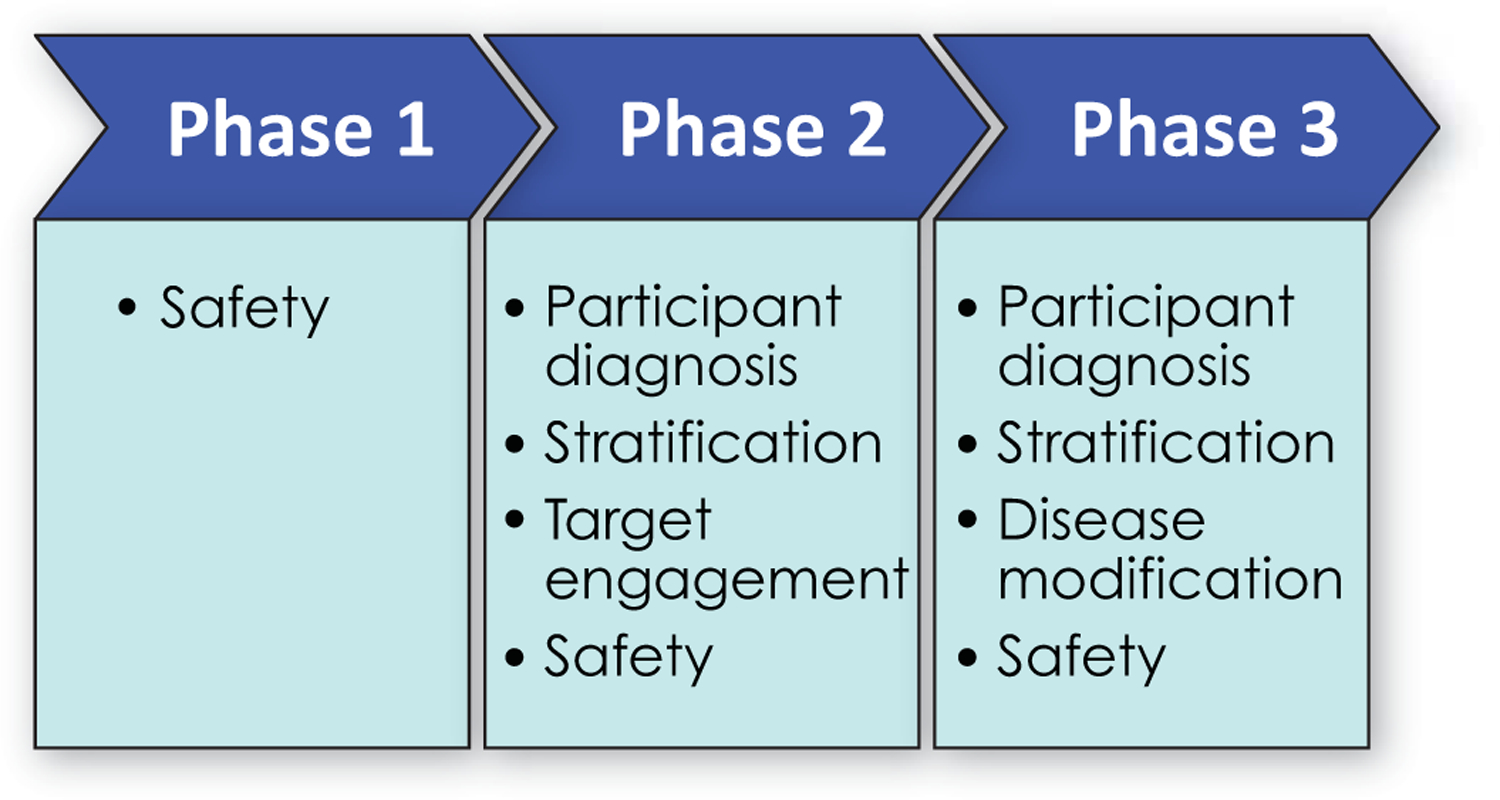
Biomarkers employed in different phases of drug development.
In Phase 2, biomarkers more specific to AD have a major role. Participant diagnosis is established by amyloid PET or CSF changes indicative of AD. Between 15 and 40% of participants referred for AD clinical trials have negative amyloid scans and are phenocopies (have the clinical phenotype of AD but not the underlying biology of AD) of the disease(49, 50). Requiring confirmation of AD-type pathology for trial participation ensures that the target biology for the intervention is present. Participants with confirmed diagnoses, decline more in the course of a trial observation period and facilitate detection of a drug-placebo difference for effective therapies(51). Plasma Aß 42/40 ratios may soon be able to replace amyloid imaging or CSF changes for trial entry or could be used to determine which potential participants are likely to be amyloid positive if scanned(37, 52, 53). The apolipoprotein E 𝜀-4 (ApoE-4) genotype is a biomarker used to stratify trial populations in either the recruitment or the analytic phase of study conduct. Some biological dimensions of AD differ in ApoE-4 carriers compared to non-carriers, and genotype could influence treatment outcomes(54). Tau PET is used to further characterize trial populations; AD participants in the preclinical period progress more rapidly to symptomatic states if they have greater tau burdens at trial baseline(38).
Target engagement biomarkers demonstrate that pharmacodynamic mediators of the therapeutic response observed in animals are present in participants with AD. Examples of biomarkers of target engagement include showing reduced Aß production using stable isotope labeled kinetics (SILK)(55), reduced CSF Aß with inhibitors of Aß-producing enzymes (56), decreased glutaminyl cyclase enzyme activity with enzyme inhibitors (57), and increased Aß fragments in plasma and CSF with gammasecretase inhibitors and modulators (58). Drug development is currently hindered by the relative lack of availability of target engagement biomarkers.
A critical role of biomarkers in Phase 3 trials is to provide evidence supportive of disease modification. Of the core biological changes of AD --- amyloid plaques and related amyloid species, tau pathology and neurofibrillary tangles, and neurodegeneration (32) --- neuronal loss is the final common denominator of disease progression. Demonstrating neuroprotection and drug-placebo difference in neurodegeneration is an essential aspect of supporting disease modification(59). MRI is one approach to demonstrating a disease modifying effect. MRI measures cerebral cortical atrophy, ventricular enlargement, and hippocampal atrophy. Loss of hippocampal volume on MRI correlates with change in hippocampal size and hippocampal neuronal number at autopsy(60, 61). NfL, neurogranin, and total tau are CSF biomarkers of N(53). They have been included in relatively few trials thus far.
Assessment of safety employs biomarkers throughout drug development. Some monoclonal antibody treatments induce amyloid-related imaging abnormalities (ARIA); these are detected by magnetic resonance imaging (MRI) collected serially during the trials(62).
Biomarkers convert clinical trials into precision drug development enterprises that identify potentially responsive participants, demonstrate target engagement, provide evidence of disease modification, and establish the safety of interventions (38).
Alzheimer’s Drug Development Pipeline
We maintain a database of information derived from the federal registry clinicaltrials.gov and review the data in the pipeline annually(6, 63–66). Registration on clinicaltrials.gov is federally mandated; all sponsors --- academic, governmental, biopharmaceutical industry, philanthropy --- who test therapeutic agents and devices must register their studies on this site. Compliance with the mandate is high, and the database is a comprehensive summary of all on-going clinical trials in the US(67–69). Trials conducted outside the US are not required to register on clinicaltrials.gov. Most development programs include trials in the US and are registered and the data available are comprehensive. The information presented here were derived from clinicaltrials.gov as of February 17, 2020.
Trials for Treatments of Neuropsychiatric Symptoms in Alzheimer’s Disease
Substantial progress is being made in clinical trials for neuropsychiatric syndromes in AD. There are currently 8 agents in clinical trials for agitation in AD including brexpiprazole, s-citalopram, lithium, dronabinol, dextromethorphan/quinidine, dextromethorphan/bupropion, mirtazapine, and prazosin(6, 21, 70). Methylphenidate is being assessed in a trial for treatment of apathy in AD(71). Two sleep agents – zolpidem and zolplicone – are being assessed for insomnia in AD; and one drug – lemborexant is being studied for its effect on irregular sleep-wake rhythm disorder (ISWRD)(72). Pimavanserin had a successful trial for dementia-related psychosis (DRP) that included AD with psychosis, PD with psychosis, dementia with Lewy bodies (DLB) with psychosis, frontotemporal lobar degeneration spectrum disorders with psychosis, and vascular dementia with psychosis. Pimavanserin will be reviewed by the FDA for the indication of DRP(73).
Together these AD drug development programs host 12 agents in clinical trials for neuropsychiatric disorders – none in Phase 1, four in Phase 2, and 8 in Phase 3 (Figure 3). These observations of the drug development pipeline suggest that the repertoire of treatments available for treatment of neuropsychiatric syndromes in AD and related neurodegenerative disorders will expand.
Figure 3.
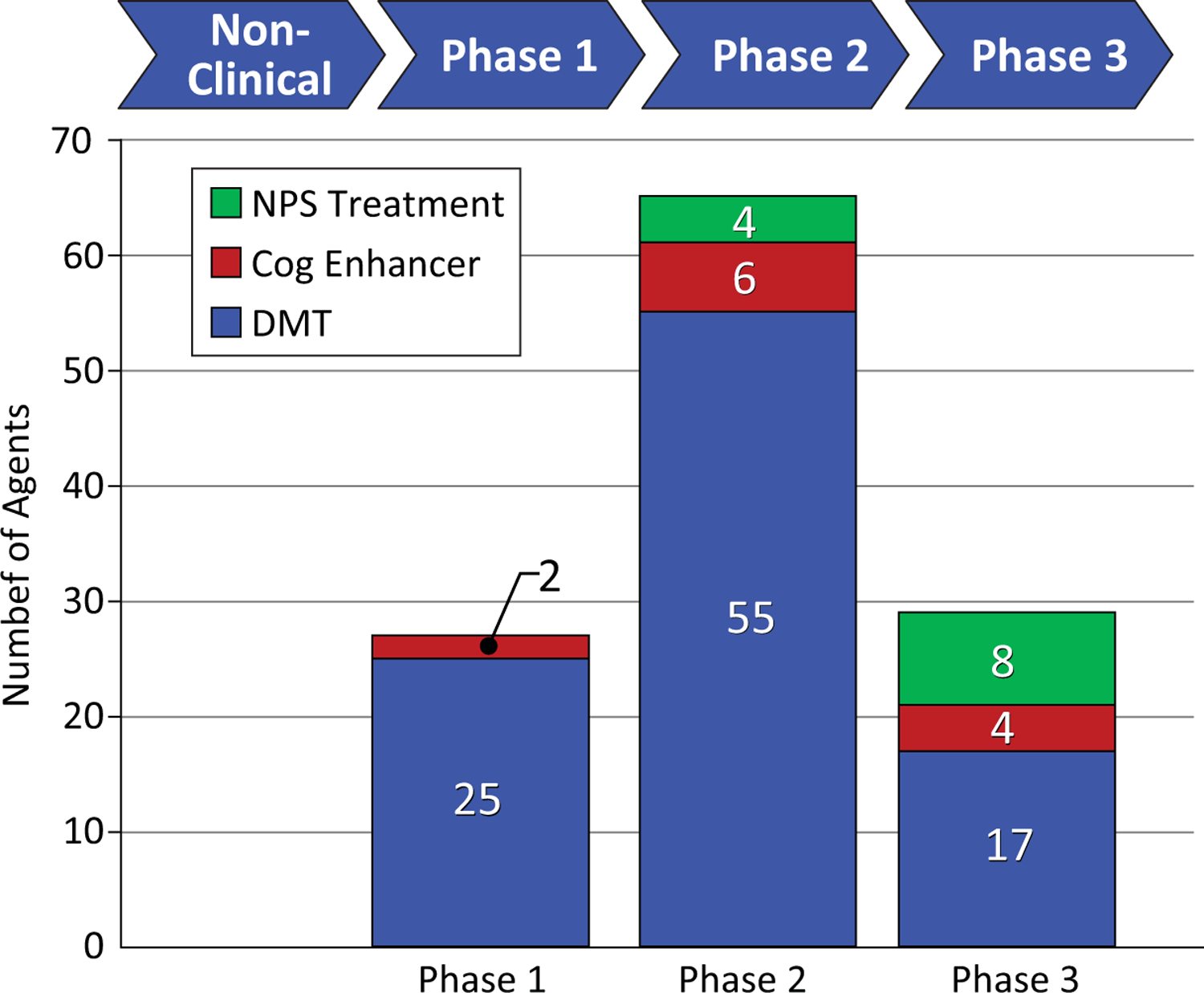
Phases of clinical trials and the number of agents in trials for Alzheimer’s disease (blue - disease modifying agents; red – cognitive enhancing agents; green – drugs for neuropsychiatric symptoms).
Progress in clinical trials for psychotropic agents is a least partially attributable to development of improved definitions of neuropsychiatric syndromes including agitation, apathy, and psychosis(74–76). Definitions facilitate construction of more homogenous trial populations, identification of appropriate outcomes, discussions with regulatory authorities, and education of clinicians concerning appropriate prescribing.
Clinical Trials for Cognitive Enhancing Agents
Several classes of cognitive enhancing agents have been assessed in recent clinical trials and found not to show a drug-placebo difference. These include nicotinic agents, histamine receptor (H3) antagonists, 11-ß-hydroxysteroid dehydrogenase inhibitors, phosphodiesterase inhibitors, norepinephrine reuptake inhibitors, and 5-HT6 antagonists(13, 65, 77–80).
Oligomannate (GV-971) was recently approved in China for the treatment of cognitive deficits in mild-moderate AD(81, 82). This agent improved cognition above baseline and promoted sustained cognitive benefit through the end of a nine month trial. Oligomannate may act through effects on the microbiome to produce both cognitive enhancing and disease-modifing effects(83).
The AD drug development pipeline currently has 2 cognitive enhancers in clinical trials in Phase 1, 6 in Phase 2, and 4 in Phase 3 (Figure 2). These agents exploit novel neurochemical pathways and seek to impact cognitive function either through indirect effects on cholinergic function or through effects on heretofore unaddressed neurochemical pathways.
Disease-Modifying Therapies
DMTs are being developed to prevent or delay the onset of cognitive decline in preclinical AD or to slow the progression of cognitive losses in prodromal AD or AD dementia(24, 84). Neuropsychiatric syndromes might be expected to be impacted by DMTs. Behavioral syndromes such as apathy, anxiety and depression occur in preclinical AD(85); neuropsychiatric syndromes emerge steadily throughout the course of the illness to more severe disorders such as agitation and psychosis(86), and amelioration of the emergence of these symptoms can be anticipated with DMTs. Emergence analyses interrogating drug-placebo differences in the emergence of new neuropsychiatric symptoms in participants who had no or few behavioral changes at baseline is the optimal approach to assessing this type of preventative neuropsychiatric effect(15).
Table 2 shows the National Institute on Aging – Alzheimer’s Association Common AD Research Ontology (CADRO; (87, 88)) listing the classes of interventions recognized for DMT drug development in AD (Table 2). The CADRO defines recognized disease processes in AD and these comprise the drug targets and related putative mechanisms of action of DMT’s. The target categories include Aß; tau protein; ApoE, lipids, and lipoprotein receptors; neurotransmitter receptors; inflammation; oxidative stress; proteostasis and proteinopathies; metabolism and bioenergetics; vascular targets; growth factors and hormones; synaptic plasticity and neuroprotection; epigenetics; neurogenesis; and “other/multi-target”. Figure 4 shows the number of agents in each phase of drug development for each target process.
Table 2.
Common Alzheimer’s Disease Research Ontology (CADRO) with classes of AD therapeutic interventions (Refolo et al, 2012; Liggins et al, 2014).
| Amyloid | Metabolism and bioenergetics |
|---|---|
| Tau | Vasculature |
| Apolipoprotein, lipids, lipoprotein receptors | Growth factors and hormones |
| Neurotransmitter receptors | Synaptic plasticity/neuroprotection |
| Inflammation | Epigenetics |
| Oxidative stress | Neurogenesis |
| Proteostasis/proteinopathies | Other/multi-target |
Figure 4.
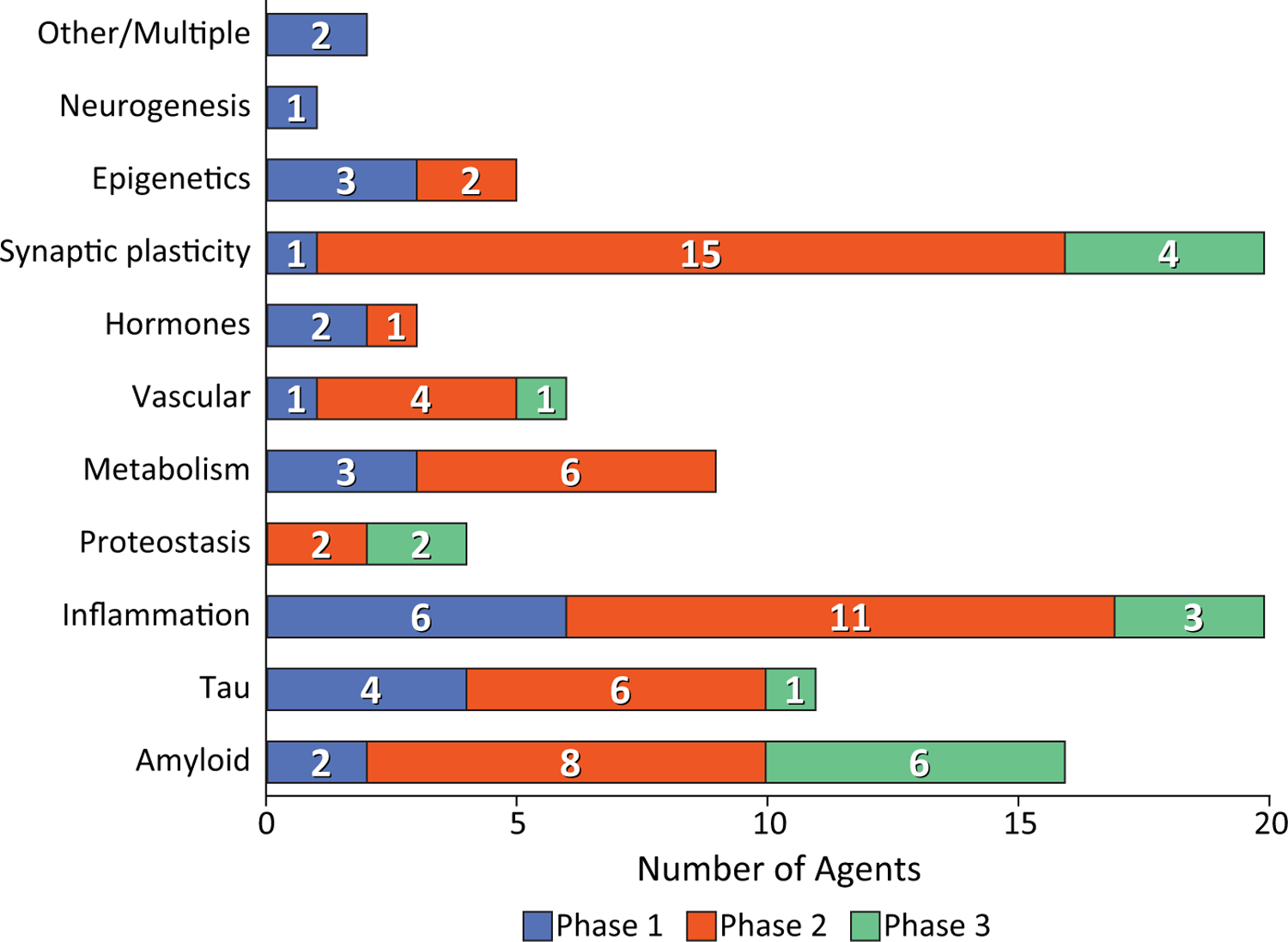
Drugs in clinical trials for Alzheimer’s disease presented by Common Alzheimer’s Disease Research Ontology (CADRO) classification of drug target (blue – Phase 1; orange – Phase 2; green – Phase 3).
In Phase 1, ninety-three percent (25 of 27) of agents in trials are DMT’s(6). Inflammation is the most common Phase 1 target (6 agents), followed by tau (4 agents), metabolism and energetics (3 agents), epigenetics (3 agents), Aß (2 agents), growth factors/hormones (2 agents), and 1 agent each for vascular targets, synaptic plasticity/neuroprotection, and neurogenesis (Figure 3). Two agents have multiple targets.
Phase 2 has the largest number of therapies in trials compared to other phases. There are 65 drugs and biological therapies, of which 55 (85%) are DMTs (Figures 2, 3). Synaptic plasticity and neuroprotection are the most common targets (15 agents); inflammation (11 agents), Aß (8 agents), metabolism and biogenergetics (6 agents), tau (6 agents) and vascular targets (4 agents) are well represented in the Phase 2 repertoire of therapies. A few sponsors are addressing proteostasis and epigenetics (2 agents each) and hormonal approaches (1 agent). Trials of DMTs are usually conducted in participants who are receiving stable therapy with ChE-Is with or without memantine. Participants are randomized to the test agent or placebo with both groups receiving the existing standard of care.
There are 29 agents being studied in Phase 3; 17 are DMTs (Figures 1,2). Six of these address amyloid targets; 4 have synaptic plasticity/neuroprotection as their target; 3 address inflammation; 2 are directed at proteostasis; and each target tau and vascular targets.
DMTs can be assessed in any phase --- preclinical, prodromal, AD dementia – where the test agent may have an effect on disease progression. Cognitive enhancing agents and psychotropic drugs must be tested in symptomatic patients in prodromal or dementia phases of AD. Of all clinical trials currently in progress, 6 involve preclinical populations, 39 are in patients with prodromal disease, and 45 are in patients with AD dementia (Figure 5).
Figure 5.
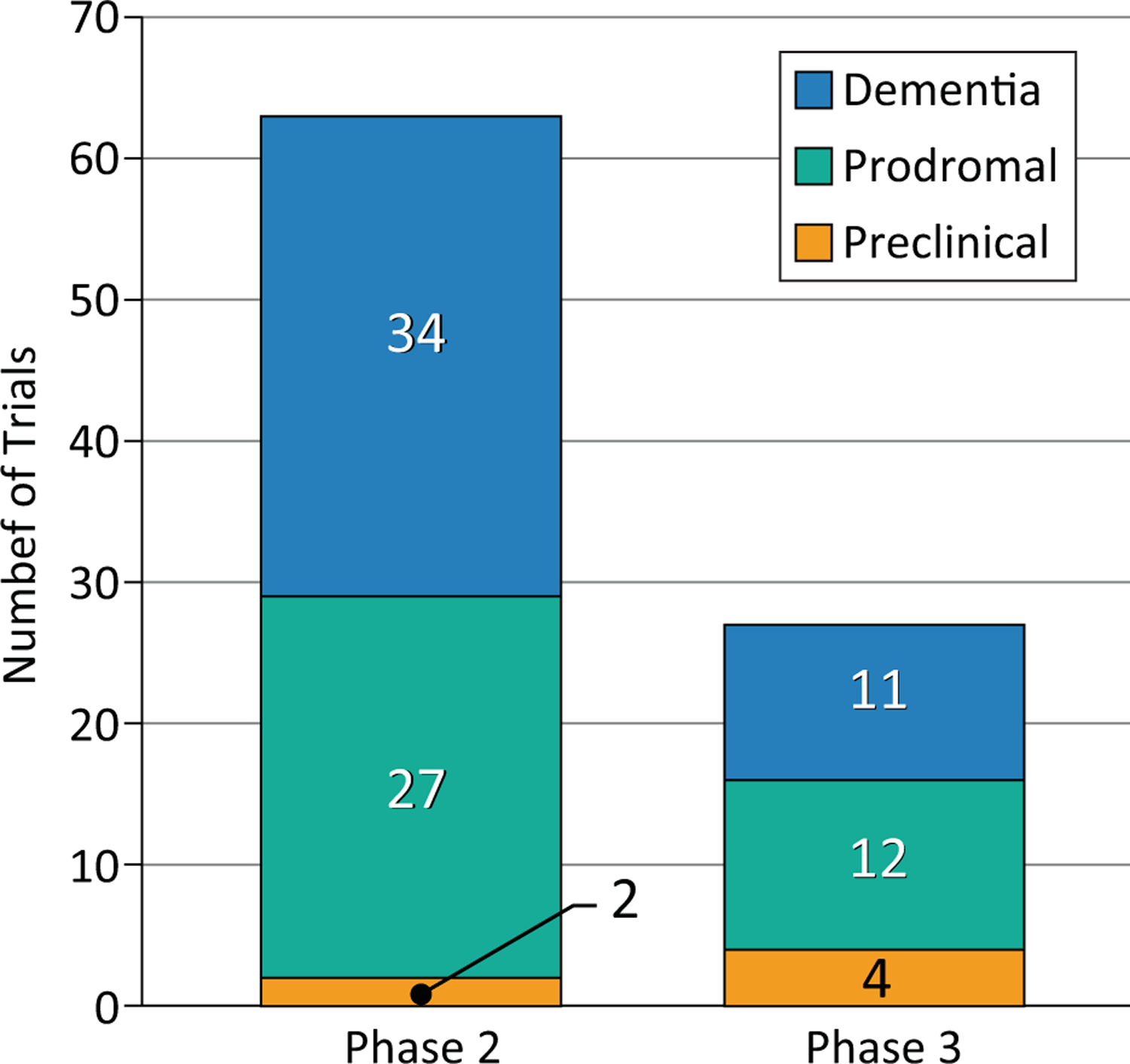
Number of trials for Alzheimer’s disease involving preclinical (gold), prodromal (aqua), and dementia (blue) populations.
Discussion
AD drug development is progressing with current clinical trials of 121 candidate therapies. Neuropsychiatric symptoms, cognitive enhancement, and disease modification are being addressed. Prevention trials are pursued in participants who are cognitively normal and at high risk for development of symptomatic AD. There is a diversification of therapeutic targets with an emphasis on amyloid, tau, inflammation, and synaptic plasticity/neuroprotection.
Innovation in clinical trial design for assessment of treatment for neuropsychiatric symptoms is evident among recently conducted trials. The sequential parallel comparative design (SPCD) used in trials of psychotropic and analgesic research was used in a development program of dextromethorphan/quinidine for the treatment of agitation in AD(89). By re-randomizing placebo non-responders to drug or placebo in the second stage of a 2-stage trial, this design allows insight into placebo responses that are robust in many agitation trials. A trial of pimavanserin for DRP used a randomized withdrawal design to demonstrate efficacy (73). This design has the advantage of placing everyone on treatment at trial entry for three months before responders are randomized to continued treatment or placebo. The outcome of the trial is relapse in the placebo group compared to relapse in the active treatment group. This trial included five types of dementia, an approach that builds on accumulating evidence that psychosis is an endophenotype with shared involvement of a common brain circuitry that transcends diagnostic categories. These trial innovations expand the toolkit of available approaches to solving challenges associated with neuropsychiatric drug development.
There are no Phase 1 agents for neuropsychiatric symptoms in the current AD pipeline. This reflects a multiplicity of convergent influences. First, most neuropsychiatric agents are developed for major depression, schizophrenia, anxiety, or sleep in non-AD development programs and then repositioned for AD after approval for a psychiatric disorder. Phase 1 of these agents is accomplished in programs devoted primarily to psychiatric conditions. Second, the specific biology of neuropsychiatric symptoms in AD is unknown and there are few avenues for initiating development of psychotropics specific to the biology of AD or other neurodegenerative disorders. Third, some Phase 1 trials are conducted outside the US, are not registered on clinicaltrials.gov, and would not be captured in our review strategy. Finally, there are too few agents entering the AD drug development pipeline for all classes of therapeutic agents. Given the 7.6 year lag between entering Phase 1 trials and exiting Phase 3 trials, the dearth of agents in Phase 1 represents a major concern for availability of new therapies for AD in the future.
Sleep disorders in AD are under-recognized and under-treated(90). There are a small number of sleep-related agents in the AD drug development pipeline. Suvorexant, a dual orexin antagonist, had a successful clinical trial for insomnia in AD(20). Lemborexant, another dual orexin antagonist, is in a clinical trial for treatment of ISWRD. Zolpidem and zoplicone are in a trial for insomnia in AD. These trials build on the improved understanding of the importance of sleep disorders in AD(90).
The majority of the AD pipeline treatments are DMTs; 59% of Phase 3 agents, 85% of Phase 2 agents, and 93% of Phase 1 agents target disease modification. Aß protein in amyloid plaques, a variety of pre-plaque amyloid species, and tau protein in the form of soluble oligomers or neurofibrillary tangles are important targets in the AD DMT pipeline. Three monoclonal antibodies --- aducanumab, gantenerumab, and BAN2401 --- have been shown to reduce brain levels of plaque amyloid and to impact CSF biomarkers indicative of neurodegeneration(91, 92). Continuing trials of these agents will determine if they produce clinical benefit.
Inflammation is increasingly recognized to play a major role in AD and other neurodegenerative disorders(93). Microglial activation and other aspects of inflammation have emerged as important targets in the AD pipeline; agents targeting inflammatory processes are the most common approaches in both Phase 1 and Phase 2 (Figure 3). Figure 6 shows the inflammatory pathways activated in AD and targeted by the anti-inflammatory agents in trials.
Figure 6.
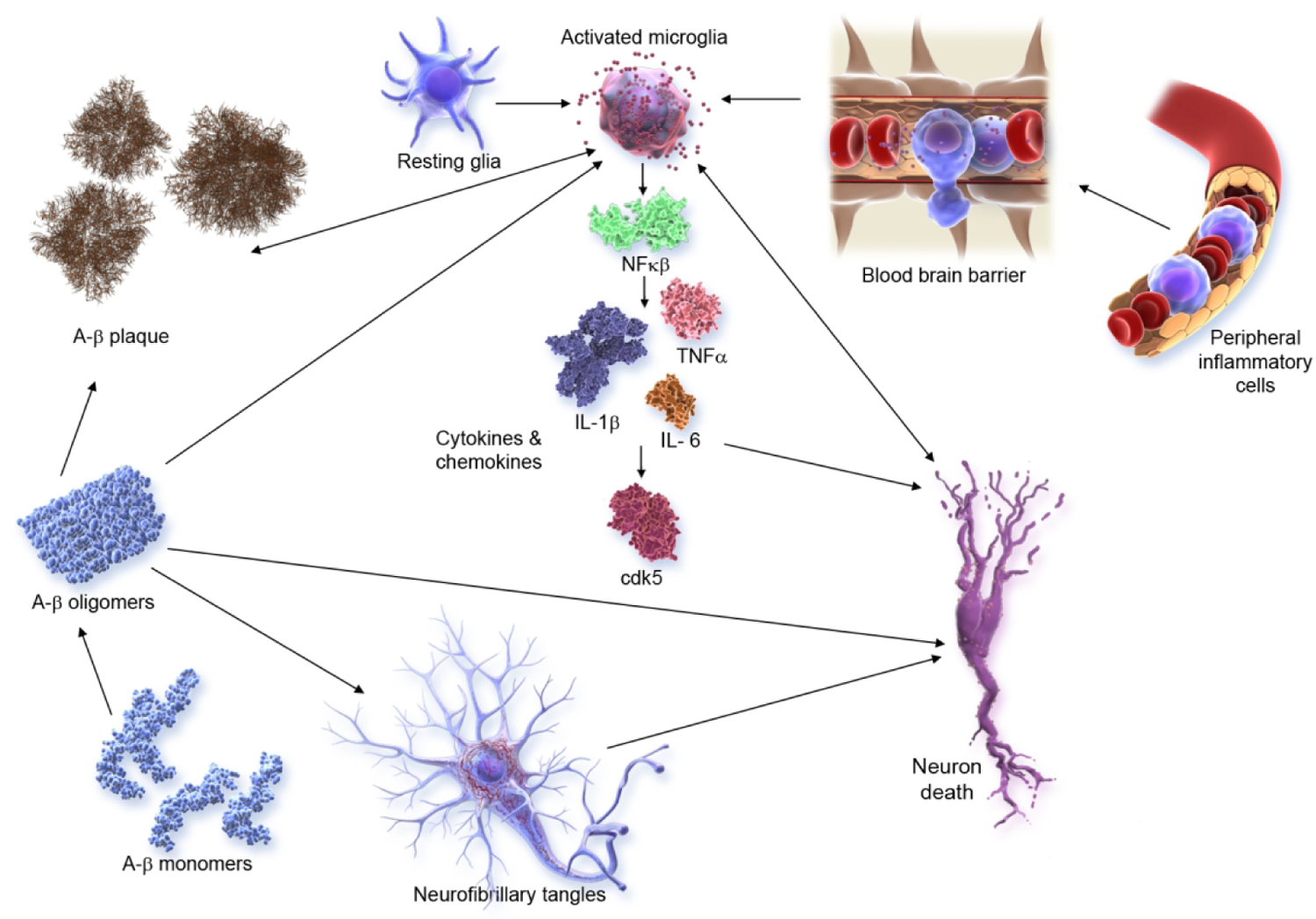
Inflammatory pathways in Alzheimer’s disease (©J Cummings; illustrator M de la Flor, PhD).
Therapies promoting synaptic plasticity and neuroprotection are well represented in Phase 2 with 15 agents of this class in clinical trials (Figure 3; Figure 7). These agents seek to promote synaptic plasticity or to protect synapses and neurons against Aß or other neurotoxins(94). Neuroprotection is the critical outcome of disease-modifying strategies(59). These interventions promote circuit function that underly cognitive and behavioral function(95). Success with these agents would be anticipated to have both cognitive and behavioral benefit.
Figure 7.
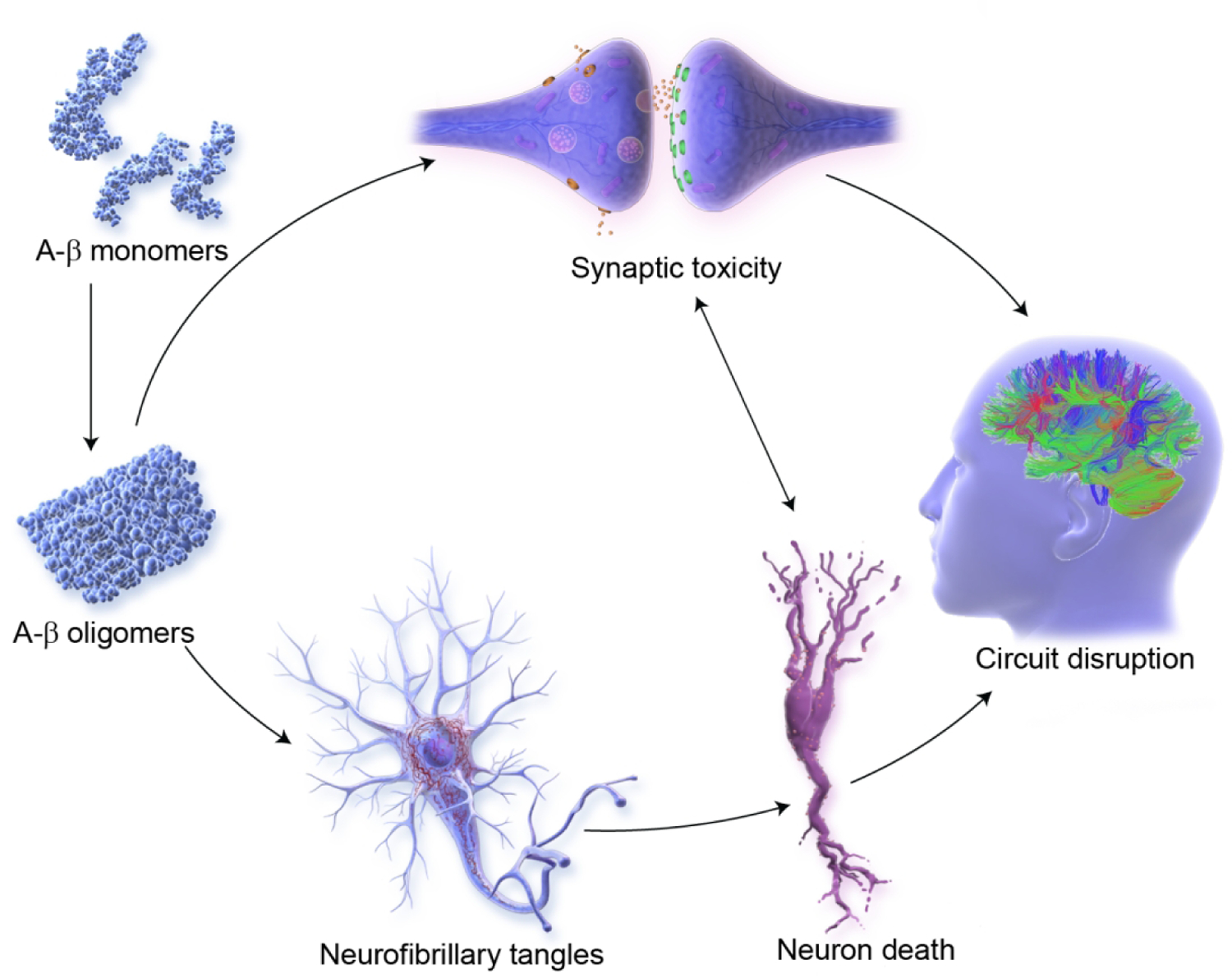
Pathways for synaptic plasticity and neuroprotection in Alzheimer’s disease (©J Cummings; illustrator M de la Flor, PhD).
The AD drug development pipeline shows the dynamic interaction between basic science and the increased understanding of the biology of AD with the development of new therapies targeting processes whose modulation may produce therapeutic benefit. This improved scientific foundation for AD drug development coupled with innovative trial designs, new biomarkers, improved clinical outcomes, and better definitions of clinical trial populations promises to accelerate delivery to new therapies to individuals with or at risk for AD.
Acknowledgments
Disclosures:
Dr. Cummings has provided consultation to Acadia, Actinogen, Alkahest, Alzheon, Annovis, Avanir, Axsome, Biogen, Cassava, Cerecin, Cerevel, Cortexyme, Cytox, EIP Pharma, Eisai, Foresight, GemVax, Genentech, Green Valley, Grifols, Karuna, Merck, Novo Nordisk, Otsuka, Resverlogix, Roche, Samumed, Samus, Signant Health, Suven, and United Neuroscience pharmaceutical and assessment companies. Dr. Cummings has stock options in ADAMAS, AnnovisBio, MedAvante, BiOasis. Dr. Cummings owns the copyright of the Neuropsychiatric Inventory. Dr Cummings is supported by Keep Memory Alive (KMA); NIGMS grant P20GM109025; NINDS grant U01NS093334; and NIA grant R01AG053798.
References
- (1).Masters CL, Bateman R, Blennow K, et al. : Alzheimer’s disease. Nat Rev Dis Primers 2015;1:15056. [DOI] [PubMed] [Google Scholar]
- (2).Scheltens P, Blennow K, Breteler MM, et al. : Alzheimer’s disease. Lancet 2016;388:505–517. [DOI] [PubMed] [Google Scholar]
- (3).Ferri CP, Prince M, Brayne C, et al. : Global prevalence of dementia: a Delphi consensus study. Lancet 2005;366:2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Alzheimer’s Association: 2019 Alzheimer’s disease facts and figures. Alzheimer Dement 2019;15:321–387. [DOI] [PubMed] [Google Scholar]
- (5).Cummings JL, Morstorf T, Zhong K: Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 2014;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cummings J, Lee G, Ritter A, et al. : Alzheimer’s disease drug development pipeline: 2020. Alzheimer Dement 2020;In press. [DOI] [PMC free article] [PubMed]
- (7).Ballard C, Banister C, Khan Z, et al. : Evaluation of the safety, tolerability, and efficacy of pimavanserin versus placebo in patients with Alzheimer’s disease psychosis: a phase 2, randomised, placebo-controlled, double-blind study. Lancet Neurol 2018;17:213–222. [DOI] [PubMed] [Google Scholar]
- (8).Cummings J, Ritter A, Rothenberg K: Advances in management of neuropsychiatric syndromes in neurodegenerative diseases. Curr Psychiatry Rep 2019;21:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bohnen NI, Kaufer DI, Ivanco LS, et al. : Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol 2003;60:1745–1748. [DOI] [PubMed] [Google Scholar]
- (10).Emre M, Aarsland D, Albanese A, et al. : Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351:2509–2518. [DOI] [PubMed] [Google Scholar]
- (11).Bradbury J, Avila C, Grace S: Practice-based research in complementary medicine: could n-of-1 trials become the new gold standard? Healthcare (Basel) 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Margolis A, Giuliano C: Making the switch: from case studies to N-of-1 trials. Epilepsy Behav Rep 2019;12:100336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Atri A: The Alzheimer’s disease clinical spectrum: diagnosis and management. Med Clin North Am 2019;103:263–293. [DOI] [PubMed] [Google Scholar]
- (14).Wynn ZJ, Cummings JL: Cholinesterase inhibitor therapies and neuropsychiatric manifestations of Alzheimer’s disease. Dement Geriatr Cogn Disord 2004;17:100–108. [DOI] [PubMed] [Google Scholar]
- (15).Cummings JL, Schneider L, Tariot PN, et al. : Reduction of behavioral disturbances and caregiver distress by galantamine in patients with Alzheimer’s disease. Am J Psychiatry 2004;161:532–538. [DOI] [PubMed] [Google Scholar]
- (16).Gauthier S, Wirth Y, Mobius HJ: Effects of memantine on behavioural symptoms in Alzheimer’s disease patients: an analysis of the Neuropsychiatric Inventory (NPI) data of two randomised, controlled studies. Int J Geriatr Psychiatry 2005;20:459–464. [DOI] [PubMed] [Google Scholar]
- (17).Cummings J, Isaacson S, Mills R, et al. : Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet 2014;383:533–540. [DOI] [PubMed] [Google Scholar]
- (18).Pioro EP, Brooks BR, Cummings J, et al. : Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol 2010;68:693–702. [DOI] [PubMed] [Google Scholar]
- (19).Anagnostou E: Clinical trials in autism spectrum disorder: evidence, challenges and future directions. Curr Opin Neurol 2018;31:119–125. [DOI] [PubMed] [Google Scholar]
- (20).Herring WJ, Ceesay P, Snyder E, et al. : Polysomnographic assessment of suvorexant in patients with probable Alzheimer’s disease dementia and insomnia: a randomized trial. Alzheimers Dement 2020;16:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Grossberg GT, Kohegyi E, Mergel V, et al. : Efficacy and safety of brexpiprazole for the treatment of agitation in Alzheimer’s dementia: two 12-week, randomized, double-blind, placebo-controlled trials. Am J Geriatr Psychiatry 2020;28:383–400. [DOI] [PubMed] [Google Scholar]
- (22).Porsteinsson AP, Keltz MA, Smith JS: Role of citalopram in the treatment of agitation in Alzheimer’s disease. Neurodegener Dis Manag 2014;4:345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rosenberg PB, Lanctot KL, Drye LT, et al. : Safety and efficacy of methylphenidate for apathy in Alzheimer’s disease: a randomized, placebo-controlled trial. J Clin Psychiatry 2013;74:810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cummings J, Ritter A, Zhong K: Clinical trials for disease-modifying therapies in Alzheimer’s disease: a primer, lessons learned, and a blueprint for the future. J Alzheimers Dis 2018;64:S3–S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gray JA, Fleet D, Winblad B: The need for thorough phase II studies in medicines development for Alzheimer’s disease. Alzheimers Res Ther 2015;7:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Greenberg BD, Carrillo MC, Ryan JM, et al. : Improving Alzheimer’s disease phase II clinical trials. Alzheimers Dement 2013;9:39–49. [DOI] [PubMed] [Google Scholar]
- (27).Vellas B, Carrillo MC, Sampaio C, et al. : Designing drug trials for Alzheimer’s disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement 2013;9:438–444. [DOI] [PubMed] [Google Scholar]
- (28).Leber P: Observations and suggestions on antidementia drug development. Alzheimer Dis Assoc Disord 1996;10 Suppl 1:31–35. [DOI] [PubMed] [Google Scholar]
- (29).Morris JC: The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- (30).Bertens D, Tijms BM, Vermunt L, et al. : The effect of diagnostic criteria on outcome measures in preclinical and prodromal Alzheimer’s disease: Implications for trial design. Alzheimers Dement (N Y) 2017;3:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sperling RA, Aisen PS, Beckett LA, et al. : Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jack CR Jr., Bennett DA, Blennow K, et al. : NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bateman RJ, Benzinger TL, Berry S, et al. : The DIAN-TU Next Generation Alzheimer’s prevention trial: adaptive design and disease progression model. Alzheimer Dement 2017;13:8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Langbaum JB, Karlawish J, Roberts JS, et al. : GeneMatch: A novel recruitment registry using at-home APOE genotyping to enhance referrals to Alzheimer’s prevention studies. Alzheimers Dement 2019;15:515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Weintraub S, Carrillo MC, Farias ST, et al. : Measuring cognition and function in the preclinical stage of Alzheimer’s disease. Alzheimers Dement (N Y) 2018;4:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Scott TJ, O’Connor AC, Link AN, et al. : Economic analysis of opportunities to accelerate Alzheimer’s disease research and development. Ann N Y Acad Sci 2014;1313:17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Schindler SE, Bollinger JG, Ovod V, et al. : High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019;93:e1647–e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Palmqvist S, Insel PS, Stomrud E, et al. : Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol Med 2019;11:e11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Karikari TK, Pascoal TA, Ashton NJ, et al. : Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol 2020;19:422–433. [DOI] [PubMed] [Google Scholar]
- (40).Mattsson N, Cullen NC, Andreasson U, et al. : Association Between Longitudinal Plasma Neurofilament Light and Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol 2019;76:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Wellington H, Paterson RW, Portelius E, et al. : Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 2016;86:829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Molinuevo JL, Ayton S, Batrla R, et al. : Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol 2018;136:821–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jack CR Jr., Knopman DS, Jagust WJ, et al. : Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jack CR Jr., Knopman DS, Jagust WJ, et al. : Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Mattsson N, Scholl M, Strandberg O, et al. : (18)F-AV-1451 and CSF T-tau and P-tau as biomarkers in Alzheimer’s disease. EMBO Mol Med 2017;9:1212–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Brookmeyer R, Abdalla N: Estimation of lifetime risks of Alzheimer’s disease dementia using biomarkers for preclinical disease. Alzheimers Dement 2018;14:981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cummings J: The Role of Biomarkers in Alzheimer’s Disease Drug Development. Adv Exp Med Biol 2019;1118:29–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Cummings J, Feldman HH, Scheltens P: The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Ther 2019;11:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Landau SM, Horng A, Fero A, et al. : Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology 2016;86:1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sevigny J, Suhy J, Chiao P, et al. : Amyloid PET Screening for Enrichment of Early-Stage Alzheimer Disease Clinical Trials: Experience in a Phase 1b Clinical Trial. Alzheimer Dis Assoc Disord 2016;30:1–7. [DOI] [PubMed] [Google Scholar]
- (51).Ballard C, Atri A, Boneva N, et al. : Enrichment factors for clinical trials in mild-to-moderate Alzheimer’s disease. Alzheimers Dement (N Y) 2019;5:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Nakamura A, Kaneko N, Villemagne VL, et al. : High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 2018;554:249–254. [DOI] [PubMed] [Google Scholar]
- (53).Hampel H, O’Bryant SE, Molinuevo JL, et al. : Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol 2018;14:639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhao N, Liu CC, Qiao W, et al. : Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol Psychiatry 2018;83:347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Paterson RW, Gabelle A, Lucey BP, et al. : SILK studies - capturing the turnover of proteins linked to neurodegenerative diseases. Nat Rev Neurol 2019;15:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kennedy ME, Stamford AW, Chen X, et al. : The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med 2016;8:363ra150. [DOI] [PubMed] [Google Scholar]
- (57).Scheltens P, Hallikainen M, Grimmer T, et al. : Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer’s disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers Res Ther 2018;10:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Portelius E, Zetterberg H, Dean RA, et al. : Amyloid-beta(1–15/16) as a marker for gamma-secretase inhibition in Alzheimer’s disease. J Alzheimers Dis 2012;31:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Cummings J, Fox N: Defining disease modifying therapy for Alzheimer’s disease. J Prev Alzheimers Dis 2017;4:109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Apostolova LG, Zarow C, Biado K, et al. : Relationship between hippocampal atrophy and neuropathology markers: a 7T MRI validation study of the EADC-ADNI Harmonized Hippocampal Segmentation Protocol. Alzheimers Dement 2015;11:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Csernansky JG, Hamstra J, Wang L, et al. : Correlations between antemortem hippocampal volume and postmortem neuropathology in AD subjects. Alzheimer Dis Assoc Disord 2004;18:190–195. [PubMed] [Google Scholar]
- (62).Sperling RA, Jack CR Jr., Black SE, et al. : Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement 2011;7:367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Cummings J, Morstorf T, Lee G: Alzheimer’s drug-development pipeline: 2016. Alzheimers Dement (N Y) 2016;2:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Cummings J, Lee G, Mortsdorf T, et al. : Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 2017;3:367–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Cummings J, Lee G, Ritter A, et al. : Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 2018;4:195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Cummings J, Lee G, Ritter A, et al. : Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement (N Y) 2019;5:272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Lassman SM, Shopshear OM, Jazic I, et al. : Clinical trial transparency: a reassessment of industry compliance with clinical trial registration and reporting requirements in the United States. BMJ Open 2017;7:e015110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Miller JE, Wilenzick M, Ritcey N, et al. : Measuring clinical trial transparency: an empirical analysis of newly approved drugs and large pharmaceutical companies. BMJ Open 2017;7:e017917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Anderson ML, Chiswell K, Peterson ED, et al. : Compliance with results reporting at ClinicalTrials.gov. N Engl J Med 2015;372:1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ehrhardt S, Porsteinsson AP, Munro CA, et al. : Escitalopram for agitation in Alzheimer’s disease (S-CitAD): methods and design of an investigator-initiated, randomized, controlled, multicenter clinical trial. Alzheimers Dement 2019;15:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Scherer RW, Drye L, Mintzer J, et al. : The Apathy in Dementia Methylphenidate Trial 2 (ADMET 2): study protocol for a randomized controlled trial. Trials 2018;19:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Abbott SM, Zee PC: Irregular sleep-wake rhythm disorder. Sleep Med Clin 2015;10:517–522. [DOI] [PubMed] [Google Scholar]
- (73).Cummings J, Ballard C, Tariot P, et al. : Pimavanserin: potential treatment For dementia-related psychosis. J Prev Alzheimers Dis 2018;5:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Cummings J, Mintzer J, Brodaty H, et al. : Agitation in cognitive disorders: International Psychogeriatric Association provisional consensus clinical and research definition. Int Psychogeriatr 2015;27:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Robert P, Lanctot KL, Aguera-Ortiz L, et al. : Is it time to revise the diagnostic criteria for apathy in brain disorders? The 2018 international consensus group. Eur Psychiatry 2018;54:71–76. [DOI] [PubMed] [Google Scholar]
- (76).Fischer CE, Ismail Z, Youakim JM, et al. : Revisiting criteria for psychosis in Alzheimer’s disease and related dementias: toward better phenotypic classification and biomarker research. J Alzheimers Dis 2020;73:1143–1156. [DOI] [PubMed] [Google Scholar]
- (77).Mohs RC, Shiovitz TM, Tariot PN, et al. : Atomoxetine augmentation of cholinesterase inhibitor therapy in patients with Alzheimer disease: 6-month, randomized, double-blind, placebo-controlled, parallel-trial study. Am J Geriatr Psychiatry 2009;17:752–759. [DOI] [PubMed] [Google Scholar]
- (78).Frolich L, Wunderlich G, Thamer C, et al. : Evaluation of the efficacy, safety and tolerability of orally administered BI 409306, a novel phosphodiesterase type 9 inhibitor, in two randomised controlled phase II studies in patients with prodromal and mild Alzheimer’s disease. Alzheimers Res Ther 2019;11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Grove RA, Harrington CM, Mahler A, et al. : A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res 2014;11:47–58. [DOI] [PubMed] [Google Scholar]
- (80).Atri A, Frolich L, Ballard C, et al. : Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials. JAMA 2018;319:130–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Syed YY: Sodium oligomannate: first approval. Drugs 2020;80:441–444. [DOI] [PubMed] [Google Scholar]
- (82).Syed YY: Correction to: sodium oligomannate: first approval. Drugs 2020;80:445–446. [DOI] [PubMed] [Google Scholar]
- (83).Wang X, Sun G, Feng T, et al. : Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res 2019;29:787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Suzuki K, Iwata A, Iwatsubo T: The past, present, and future of disease-modifying therapies for Alzheimer’s disease. Proc Jpn Acad Ser B Phys Biol Sci 2017;93:757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Donovan NJ, Locascio JJ, Marshall GA, et al. : Longitudinal Association of Amyloid Beta and Anxious-Depressive Symptoms in Cognitively Normal Older Adults. Am J Psychiatry 2018;175:530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Vik-Mo AO, Giil LM, Ballard C, et al. : Course of neuropsychiatric symptoms in dementia: 5-year longitudinal study. Int J Geriatr Psychiatry 2018;33:1361–1369. [DOI] [PubMed] [Google Scholar]
- (87).Refolo LM, Snyder H, Liggins C, et al. : Common Alzheimer’s Disease Research Ontology: National Institute on Aging and Alzheimer’s Association collaborative project. Alzheimers Dement 2012;8:372–375. [DOI] [PubMed] [Google Scholar]
- (88).Liggins C, Snyder HM, Silverberg N, et al. : International Alzheimer’s Disease Research Portfolio (IADRP) aims to capture global Alzheimer’s disease research funding. Alzheimers Dement 2014;10:405–408. [DOI] [PubMed] [Google Scholar]
- (89).Cummings JL, Lyketsos CG, Peskind ER, et al. : Effect of dextromethorphan-quinidine on agitation in patients with Alzheimer disease dementia: a randomized clinical trial. JAMA 2015;314:1242–1254. [DOI] [PubMed] [Google Scholar]
- (90).Peter-Derex L, Yammine P, Bastuji H, et al. : Sleep and Alzheimer’s disease. Sleep Med Rev 2015;19:29–38. [DOI] [PubMed] [Google Scholar]
- (91).Sevigny J, Chiao P, Bussiere T, et al. : The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016;537:50–56. [DOI] [PubMed] [Google Scholar]
- (92).Klein G, Delmar P, Voyle N, et al. : Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther 2019;11:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Nichols MR, St-Pierre MK, Wendeln AC, et al. : Inflammatory mechanisms in neurodegeneration. J Neurochem 2019;149:562–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Colom-Cadena M, Spires-Jones T, Zetterberg H, et al. : The clinical promise of biomarkers of synapse damage or loss in Alzheimer’s disease. Alzheimers Res Ther 2020;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Canter RG, Penney J, Tsai LH: The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016;539:187–196. [DOI] [PubMed] [Google Scholar]