

Abstract
Protein kinase RNA-activated (PKR) is an important and rapidly-evolving antiviral kinase. Most poxviruses contain two distinct PKR inhibitors, called E3 and K3 in vaccinia virus (VACV), the prototypic orthopoxvirus. E3 prevents PKR homodimerization by binding double-stranded RNA, while K3 acts as a pseudosubstrate inhibitor by binding directly to activated PKR and thereby inhibiting interaction with its substrate eIF2α. In our study here, we analyzed E3 and K3 orthologs from the phylogenetically distinct capripoxviruses (CaPV), which include lumpy skin disease virus, sheeppox virus, and goatpox virus. Whereas the sheeppox virus E3 ortholog did not substantially inhibit PKR, all three CaPV K3 orthologs showed species-specific inhibition of PKR, with strong inhibition of sheep, goat, and human PKR but only weak inhibition of cow and mouse PKR. In contrast, VACV K3 strongly inhibited cow and mouse PKR but not sheep, goat, or human PKR. Infection of cell lines from the respective species with engineered VACV strains that contained different K3 orthologs showed a good correlation of PKR inhibition with virus replication and eIF2α phosphorylation. Our results show that K3 orthologs can have dramatically different effects on PKR of different species and indicate that effective PKR inhibition by K3 orthologs is crucial for virus replication.
Keywords: host-pathogen interactions, poxvirus, capripoxviruses, vaccinia virus, PKR, translational regulation
Introduction
Poxviruses are double-stranded DNA viruses whose genomes can contain more than 200 genes. They exclusively replicate in the cytoplasm of their host cells. Poxviruses encompass at least two distinct subfamilies, Chordopoxvirinae and Entomopoxvirinae, which are themselves composed of at least ten and three distinct genera, respectively. The host range of poxvirus species can vary greatly, even among closely related poxviruses.1 For example, variola virus, the causative agent of human smallpox, is only capable of infecting humans. In contrast, other members of the orthopoxvirus genus such as cowpox, monkeypox, and vaccinia viruses have much broader host ranges and can infect many different species.1 The molecular basis that determines poxvirus host range is poorly understood. Unlike many other viruses, poxvirus entry into cells is species-independent because they enter their host cells by binding to ubiquitous cell surface molecules.2 Therefore, poxvirus host range is governed by events following cell entry. A group of poxvirus genes have been identified whose inactivation only affects virus replication in some host cells and are thus designated host range genes. Most identified poxvirus host range factors target antiviral host pathways.1 Not all chordopoxviruses possess orthologs of all host range genes; however, in the orthopoxvirus genus, there is a general correlation between the number of host range genes and host range.3
One important antiviral host protein is the double-stranded (ds) RNA activated protein kinase R (PKR). PKR is constitutively expressed in most vertebrate cells at moderate levels and can be induced by type I interferons in order to mount a more efficient antiviral response.4 Inactive PKR exists in a monomeric latent state and dimerizes after binding to dsRNA, which is formed during the replication of most viruses, including poxviruses. Dimerized PKR is activated by auto-phosphorylation, which leads to the phosphorylation of the alpha subunit of eukaryotic translation initiation factor 2 (eIF2α).5, 6 Phosphorylation of eIF2α converts it to an inhibitor of the guanine exchange factor eIF2B.7 This results in a general shutdown of RNA translation.8 Due to positive selection, the kinase domain of PKR has evolved much faster than the kinase domain of the other eIF2α kinases in multiple vertebrate linages.9 Positive selection throughout the PKR gene has also been detected in primates.10 The most likely explanation for these signatures of positive selection is that many viruses have evolved inhibitors of PKR that exerted selective pressure on PKR, which resulted in molecular arms races between the viruses and their hosts. It has been shown that positively selected amino acid residues contribute directly to PKR sensitivity to inhibitors from poxviruses and herpesviruses.9–11 Most poxviruses encode two PKR inhibitors, which are called K3 (encoded by K3L) and E3 (encoded by E3L) in vaccinia virus (VACV), the prototypic poxvirus.3 K3 is an eIF2α homolog and is thought to act as a pseudosubstrate inhibitor by binding to activated, phosphorylated PKR to prevent its interaction with eIF2α.12, 13 E3 contains a Z-DNA binding domain in the N-terminus and a dsRNA binding domain in the C-terminus. E3 inhibits PKR by binding dsRNA and by preventing PKR homodimerization.14, 15
VACV K3 and E3 are both host range factors. Using a VACV strain in which either E3L or K3L were deleted, E3 was found to be essential for virus replication in human HeLa cells but dispensable for infection of Syrian hamster BHK cells. In contrast, K3 was important for virus replication in BHK cells but dispensable for virus replication in HeLa cells.16 K3L-deleted VACV also showed a modest replication defect in mouse L929 cells, which was augmented after interferon treatment.12 A likely explanation for the different roles of K3L for VACV replication in human and mouse cells is that human PKR was found to be largely resistant to K3 inhibition, whereas mouse PKR was sensitive.9 The helix αG of PKR is a critical mediator of the protein-protein interaction between PKR and either eIF2α or K3.17, 18 Exchange of a single amino acid in helix αG between human and mouse PKR, at a position that has been under positive selection, rendered human PKR more sensitive and mouse PKR more resistant to K3 inhibition.9 We lack a detailed understanding of how poxvirus PKR inhibitors interact with PKR from their natural hosts. We recently described that M156, the K3 ortholog from myxoma virus, which belongs to the genus leporipoxviruses and only infects lagomorphs, showed species-specific inhibition of European rabbit PKR but did not inhibit PKR derived from seven other mammalian species. Similarly, we showed that inactivation of M156 inhibited virus replication in rabbit cells.19 However, very little is known about these PKR-inhibitor interactions in other types of poxviruses.
Capripoxviruses (CaPVs) are a distinct genus of poxviruses with substantial worldwide economic impact. Currently, three closely related species are recognized in the Capripoxvirus genus: sheeppox virus (SPPV), goatpox virus (GTPV), and lumpy skin disease virus (LSDV). Traditionally, CaPVs have been distinguished according to the host from which they have been isolated, because they cannot be distinguished by serological tests.20, 21 CaPVs that have been isolated from sheep are referred to as “SPPV”, and those isolated from goats “GTPV”. However, molecular data showed that this distinction is not absolute and that SPPV isolates sometimes cluster with GTPV and vice versa in phylogenetic analysis. LSDVs have been mainly isolated from cow. Notable exceptions include the SPPV KS-1 strain (isolated from a sheep) and sequences that have been obtained by PCR from lesions of two springboks (Antidorcas marsupialis), which all cluster with LSDV strains in phylogenetic analysis.22, 23 There is also serological evidence of CaPV infections in some wild African ruminants.24, 25 CaPV infections in sheep, goats, and cattle are responsible for serious economic losses and, because they are classified as reportable diseases by the World Organisation for Animal Health (OIE), are severe barriers to the international trade of livestock and livestock products. While all CaPVs have severe effects on their hosts, SPPV and GTPV infections in sheep and goats usually cause much higher morbidity and mortality than LSDV in cattle.20, 21.
The molecular basis for the host restriction of CaPVs, and for the differences in disease outcomes between LSDV, on one hand, and SPPV and GTPV, on the other hand, are currently unknown. Because K3 and E3 are host range determinants in VACV, we investigated if their CaPV orthologs possess similar host range functions, and whether they inhibit PKR in a species-specific manner.
Materials and methods
Cell lines
OA1 (ATCC #CRL-6538), MDOK (ATCC #CRL-1633), MDBK (ATCC #CCL-22), BT (ATCC #CRL-1390), HeLa (ATCC #CCL-2), Caprine Synovial membrane (CSM) (kindly provided by Dr. Brian Murphy),26 HeLa PKR-knock-down (kindly provided by Dr. Charles Samuel27) and RK13+E3L+K3L cells28 were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 5% or 10% (CSM cells) fetal bovine serum or 10% horse serum (BT cells). All media were supplemented with 250 μg/ml gentamycin (Quality Biologicals). HeLa PKR-knock-down cells were maintained in medium containing 1 μg/ml puromycin (Sigma). RK13+E3L+K3L cell culture medium contained 500 μg/ml geneticin and 300 μg/ml zeocin (Life Technologies).
Plasmids
All PKR and viral genes were cloned into the pSG5 expression vector (Stratagene). Construction of pSG5-hsPKRkd-res. (knock-down resistant) (human), pSG5-mmPKR (mouse) and pSG5-oaPKR (sheep) were previously described.9, 19 Bos taurus (cow) PKR was cloned from MDBK cells and the sequence was identical to NP_835210.2. Capra hircus (goat) PKR (XM_005686488.2) was synthesized (GENEWIZ) and subcloned from the pUC57 vector into pSG5 to generate pSG5-chPKR. SPPV 011 (NP_659587.1) and 034 (NP_659606.1) (both from the TU-V02127 strain29) were synthesized and subcloned into pSG5 to generate pSG5-SPPV-011 and pSG5-SPPV-034, respectively. pSG5-LSDV-014 (AAK84975.1) from the NI-2490 strain30 was generated by the introduction of three non-synonymous mutations (C25F, R27K and I85V) into pSG5-SPPV-011 by site-directed mutagenesis. pSG5-GTPV-011 (YP_001293205.1) from the Pellor strain29 was generated by the introduction of three additional non-synonymous mutations (N49D, I72V and Y86H) into pSG5-LSDV-014 by site-directed mutagenesis. SPPV 011, LSDV 014, GTPV 011, and VACV K3L (AAA48009.1) were cloned into P837-GOI-mCherry-E3L31 to generate recombinant VACV strains into the E3L locus.
Luciferase assay
Luciferase assays were performed as described.9, 19 Briefly, 24 well plates were seeded with 5 × 104 Hela PKRKD cells per well 16 hours prior to the experiment. Cells were transfected with 200 ng of the indicated PKR plasmid, the indicated amount of K3 and E3 orthologs, and 50 ng of firefly luciferase (Promega) using GenJet reagent (Signagen) at a DNA to GenJet ratio of 1:2. Empty pSG5 vector was used to maintain the DNA concentration where appropriate. At 48 hours post-transfection, cells were lysed with mammalian lysis buffer (Goldbio) and luciferin (Promega) was added following the manufacturer’s recommendations. Luciferase activity was measured using a GloMax luminometer (Promega). Experiments were conducted in triplicate for each of three independent experiments.
Viruses and infection assays
VACV vP872, which is derived from the Copenhagen strain and has K3L deleted, was kindly provided by Dr. Bertram Jacobs.12 Generation of VC-R4, a derivative of vP872 was described.31 VC-R4-SPPV 011, VC-R4-LSDV 014, VC-R4-GTPV 011, and VC-R4-VACV K3L were generated by the scarless integration of the open reading frames of the K3L orthologs into the E3L locus in VC-R4 by the same method. Resulting viruses were plaque-purified four times and the correct integrations were confirmed by PCR and Sanger sequencing.
Plaque assays were performed with confluent six-well plates of the indicated cell lines, which were infected with 50 plaque forming units (pfu), as determined on RK13+E3L+K3L cells, of each indicated virus. After one hour, the medium was aspirated and replaced with fresh medium. After 48 hours, cells were washed with PBS, and fixed and stained with 0.1% crystal violet in 20% methanol. Photos of plates were taken using an iBright Imaging System (Invitrogen).
Multiple-cycle virus replication assays were carried out in confluent six-well plates of the indicated cells, which were infected with an MOI = 0.01 of each indicated virus. After 48 hours, cells and supernatants were collected and subjected to three rounds of freezing at −80 ˚C and thawing at 37 ˚C. Lysates were sonicated for 15s at 50% amplitude (Qsonica Q500). Viruses were titered by serial dilutions on RK13+E3L+K3L cells. Infections were performed independently two times. Differences in virus production were determined by Student’s two-tailed t-test’s.
For cells of cow and sheep origins, we initially tested MDBK and MDOK cells, respectively, for permissibility to VACV infection but found no replication of wild-type VACV in those cells.
Western blots
To detect expression of K3 orthologs in transfected cells, 4 × 105 cells of Hela PKR KD cells were transfected in six well plates with indicated plasmids. After 48 hrs, lysates were subjected to Western blotting. Anti-VACV K3 and anti-CaPV K3 were custom produced by GenScript by peptide-KLH conjugates in New Zealand rabbits and immune serum was affinity-purified. The peptide sequnces were cKVIRVDYTKGYIDVNYKRM (for VACV K3L) and cIRMNKIKGYIDVKI for SPPV 011. Dilution of primary antibodies were: 1:10,000 for anti-β-actin (Sigma-Aldrich, A5441), 1:5,000 for anti-VACV K3 and 1:7,000 for anti-CaPV K3. Primary antibodies were diluted in TBST containing 5% BSA and 0.02% (w/v) sodium azide. Membranes were incubated overnight at 4˚C in the primary antibody, washed with TBST three times for 5 minutes. The membranes were incubated for 1 hr at room temperature with horseradish peroxidase (HRP) -conjugated donkey anti-rabbit or goat anti-mouse secondary antibodies (Invitrogen, A16110, 62–6520) at 1:10,000 in TBST containing 5% (w/v) nonfat milk. The membranes were then washed five times for 5 mins and proteins were detected with ECL detection reagents (GE health care). Images were taken using the iBright Imaging System (Invitrogen).
For eIF2α phosphorylation assays, six-well plates were seeded with 1 × 106 cells of the indicated type for 16 hours. Each cell line was then infected with the indicated virus at MOI = 3.0 for one hour. Inocula were then aspirated and replaced with fresh cell culture medium. After 6 hrs, the cells and supernatants were collected and centrifuged at 800 RCF for 5 minutes. Cell pellets were lysed with 1% sodium dodecyl sulfate (SDS) in PBS (VWR) and sonicated at 50% amplitude for 10 sec twice. All protein lysates were separated on 12% SDS-polyacrylamide gels and transferred to polyvinyl difluoride (PVDF, GE Healthcare) membranes. Membranes were blocked with 5% BSA in TBST (20M Tris, 150mM NaCl, 0.1% Tween 20, pH 7.4) for 1 hour. Membranes were probed with antibodies against total and phospho-eIF2α (Santa Cruz Biotechnology, sc-11386, sc-101670) at 1:3,000. Secondary antibodies were used as described above.
Results
Capripoxvirus K3 orthologs inhibit PKR in a species-specific fashion
The E3L ortholog from SPPV (034) has been previously tested for its ability to functionally replace E3L when integrated into the VACV genome. The results indicated that SPPV 034 could neither rescue VACV replication nor inhibit PKR or eIF2α phosphorylation in infected HeLa cells.32 In order to test whether SPPV 034 or the SPPV K3L ortholog 011 can inhibit PKR from its natural host, we used an established luciferase-based reporter (LBR) assay to monitor the inhibition of sheep PKR by SPPV 034 and SPPV 011.9 We co-transfected HeLa-PKRkd cells with a luciferase reporter plasmid, sheep PKR and increasing amounts of either SPPV 034 or SPPV 011. SPPV 011 showed dose-dependent inhibition of sheep PKR, demonstrating that this SPPV protein can inhibit PKR from its natural host. In contrast, sheep PKR was not substantially inhibited by SPPV 034 (Fig. 1). We extended this analysis and also tested the effects of the K3 orthologs from the other carpipoxviruses, GTPV (011) and LSDV (014) and VACV K3, as control, on human, mouse, cow, sheep and goat PKR using the LBR assay. In agreement with previous data using this assay, human PKR was only weakly inhibited by VACV K3, whereas mouse PKR was inhibited more strongly (Fig. 2).9 Interestingly, VACV K3 also showed strong inhibition of cow PKR, whereas sheep and goat PKR were largely resistant. All three CaPV K3 orthologs showed strong inhibition of sheep and goat PKR, with GTPV 011 having the strongest effect. Remarkably, none of the CaPV K3 orthologs efficiently inhibited either cow or mouse PKR, whereas human PKR was effectively inhibited by them. In contrast to sheep and goat PKR, human PKR was inhibited comparably well by all three CaPV K3 orthologs. We could not compare the expression of all K3 orthologs directly because the addition of FLAG-tags to the CaPV K3 orthologs resulted in a loss of PKR inhibition (not shown). In order to compare expression of CaPV K3 orthologs, we generated affinity-purified peptide polyclonal antibodies against a region in SPPV 011, which is highly conserved between all CaPV K3 orthologs and which was predicted to have high predicted antigenicity (Fig. 3, box). In transiently transfected HeLa-PKRkd cells all CaPV K3 orthologs were detected at comparable levels, whereas VACV K3 was not detected by this antibody (Fig. 4). In contrast VACV K3, but not the CaPV K3 orthologs were detected by an antibody that we raised against VACV K3.
Figure 1.
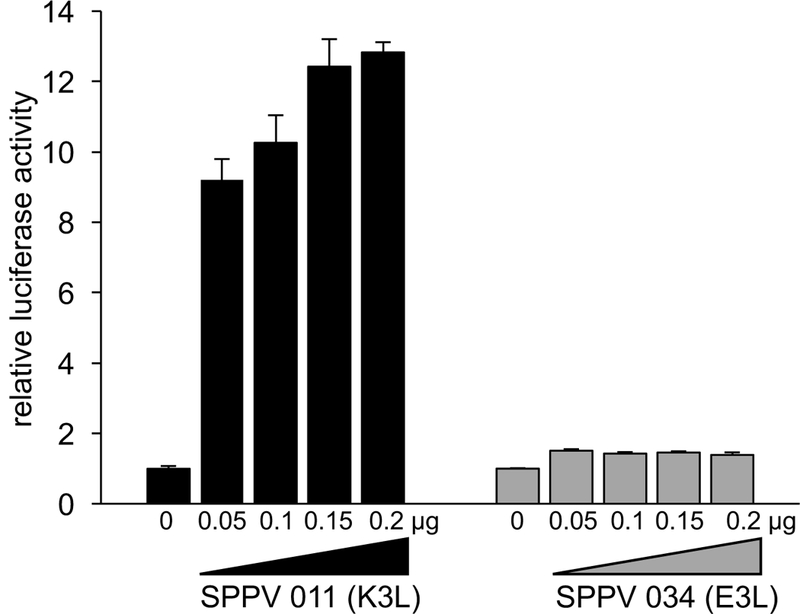
Differential sensitivity of sheep PKR to SPPV PKR inhibitors. Human HeLa-PKRkd cells were transfected with expression vectors encoding firefly luciferase (0.05 μg), sheep PKR (0.2 μg) and increasing amounts (0.05 μg, 0.1 μg, 0.15 and 0.2 μg) of either SPPV 011 (K3L ortholog) or SPPV 034 (E3L ortholog). Luciferase activity was measured 48 hours after transfection and normalized to PKR-only transfected cells to obtain relative luciferase activities. Results shown are representative of three independent experiments.
Figure 2.
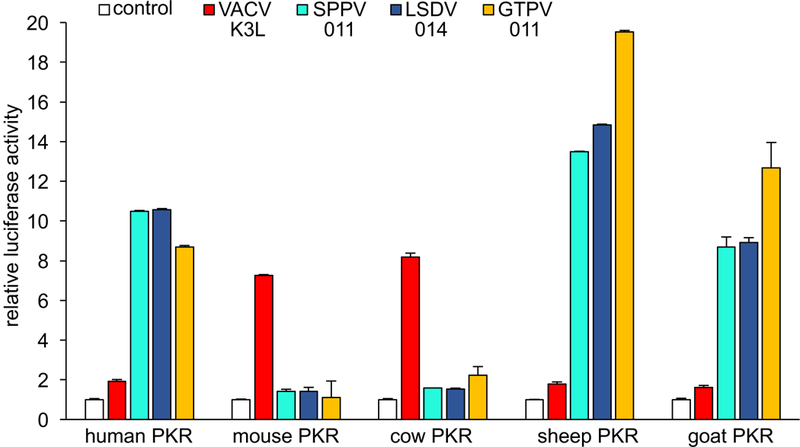
Species-specific inhibition of PKR by K3 orthologs. HeLa-PKRkd cells were transfected with expression vectors encoding firefly luciferase (0.05 μg), PKR from the indicated species (0.2 μg) and VACV K3L or CaPV K3L orthologs (0.4 μg). Luciferase activities were measured 48 hours after transfection and normalized to PKR-only transfected cells to obtain relative luciferase activities. Results shown are representative of three independent experiments.
Figure 3.

Multiple sequence alignment of CaPV K3 orthologs and VACV K3. Residues differing from SPPV 011 are highlighted in red. Sequences used to generate antibodies against SPPV 011 and VACV K3 are boxed. Sequences shown are: SPPV 011 (TU-V02127 strain), LSDV-014 (NI-2490 strain), GTPV-011 (Pellor strain), VACV K3 (WR strain).
Figure 4.
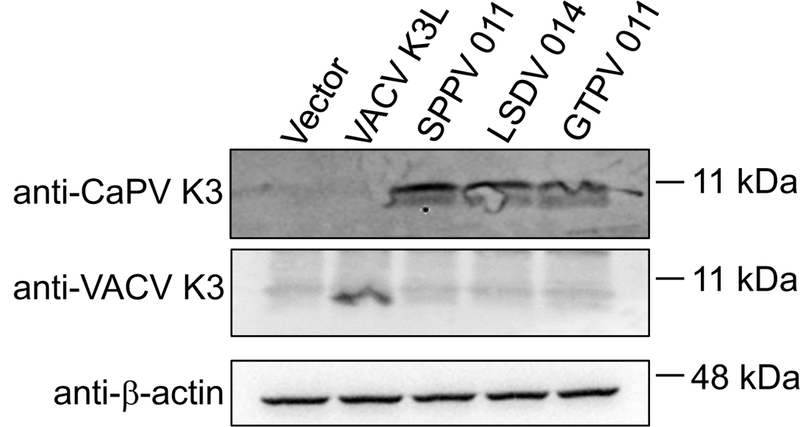
Expression of CaPV K3 orthologs and VACV K3 in transfected cells. HeLa-PKRkd cells were transfected with the indicated vectors and total protein was collected 48 hours later. Samples were separated on 12% SDS-PAGE gels and analyzed by immunoblot analysis with the indicated primary antibodies.
Correlation of PKR inhibition by K3 orthologs with virus replication
We next tested whether the species-specific inhibition of PKR by K3 orthologs that we observed in the LBR assays correlated with virus replication in cell lines from different species and focused on those of sheep, goat, cow and human origin. We developed a new strategy to generate recombinant VACV that allows the seamless and rapid integration of transgenes into the E3L locus into a VACV strain that lacks both PKR inhibitors.31 An advantage of this method is that all PKR inhibitors will be expressed from the same endogenous (E3L) promoter and thus differential expression due to the usage of different promoters is avoided. For all experiments, we used derivatives of the VACV-Copenhagen VC-2 strain. We first replaced the E3L open reading frame with that of EGFP in the vp872 strain, which has its K3L gene deleted. The resulting strain, named VC-R4, therefore contains no PKR inhibitor and can only replicate in cells that are PKR depleted, or in cells that stably express PKR inhibitors such as E3 and K3, as in the previously generated RK13+E3L+K3L cell line. We next inserted VACV K3, or the CaPV K3 orthologs SPPV 010, LSDV 014 or GTPV 011 into the E3L locus. We infected cell lines derived from sheep (OA1), goat (CSM), cow (BT) and human (HeLa), as well as RK13+E3L+K3L cells as controls with the VACV strains that express the above mentioned K3 orthologs, the E3L-expressing vP872, or VC-R4 lacking both PKR inhibitors. Six-well plates of these cells were infected with 50 plaque-forming units (pfu) of the individual viruses, as determined on RK13+E3L+K3L cells, and evaluated for plaque formation 48 hours post infection (Fig. 5). In sheep OA1 cells, plaques were seen after infection with all viruses except for VC-R4 and VC-R4+K3L. Infected goat CSM cells developed plaques after infection with all viruses except for VC-R4, although vP872 and VC-R4+GTPV011 resulted in somewhat larger plaques and infection with VC-R4+K3L yielded very small plaques. In cow BT cells, formation of large plaques was only observed after infection with vP872 and VC-R4+K3L. VC-R4+GTPV011 induced the formation of small plaques, whereas no plaques were observed after infection with the other viruses. In HeLa cells, plaque formation was observed after infection with vP872, VC-R4+SPPV010, VC-R4+LSDV014 and VC-R4+GTPV011. Thus, plaque formation in the different cell lines showed a good correlation with the LBR assay. In RK13+E3L+K3L cells, all viruses caused plaque formation. It is noteworthy that more plaques were observed in OA1 and CSM cells than in the RK13+E3L+K3L and HeLa cells, which indicates a higher plaquing efficiency in the former cell lines, although it’s unclear what governs this difference.
Figure 5.
Recombinant VACV encoding different K3 orthologs demonstrate species-specific variation in plaque formation. Indicated cells were infected with each virus for 1 hour and the overlayed with DMEM + 2% carboxymethylcellulose. Cells were stained with 0.1% w/v crystal violet to visualize plaque size two days post-infection. The pictures were all taken with the same camera and software configurations. The tested VACV strains from dark plaques RK13+E3+K3 cells, because the cells are rounded but have not sloughed off the plate at this timepoint.
Next, we tested the replication efficiency of the different VACV strains by infecting each cell line with a MOI of 0.01. We collected viruses at 48 hours post-infection and titered them on permissive RK13+E3L+K3L cells (Fig. 6). In OA1, CSM and HeLa cells, viruses containing VACV E3L, SPPV 010, LSDV 014 and GTPV 011 replicated comparably well (less than 2-fold difference) and about 1000-fold better than the virus containing VACV K3L. No plaques were observed from undiluted lysates after VC-R4 infection in these cells, which indicates no virus replication occurred (detection limit < 3.3 pfu/ml). In BT cells, the viruses containing VACV E3L or VACV K3L replicated to comparable levels. In contrast, the viruses that contain the CaPV K3L orthologs replicated more poorly, although the GTPV 011-containg virus replicated to modestly higher titers than the other viruses that contain CaPV K3L orthologs. The virus spreading assay results are consistent with results of the LBR and plaque assays. In RK13+E3L+K3L cells, all viruses replicated to comparable levels, with the exception of VC-R4, which replicated about 4-fold less efficiently. The latter observation is consistent with results obtained with VC-R2, another virus that has both E3L and K3L deleted, suggesting that PKR may not be fully inhibited in these cells, or that a different host restriction factor is modestly inhibiting VC-R4 replication.33
Figure 6.
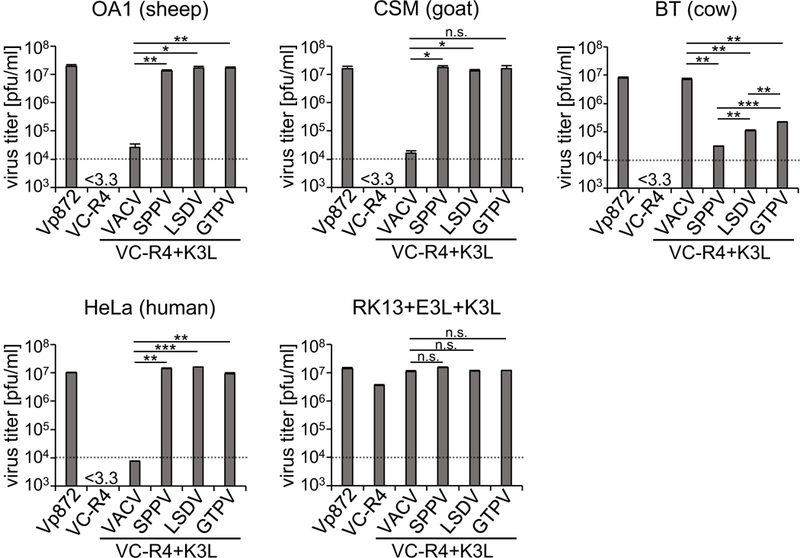
CaPV K3L orthologs alter the replication of recombinant VACV in cells derived from different species. OA (sheep), CSM (goat), BT (cow), HeLa (human), or RK13+E3+K3 cells were infected with vP872 (VACVΔK3L), VC-R4 (VACVΔE3LΔK3L), or VC-R4 recombinants containing VACV K3L or CaPV K3L orthologs at MOI = 0.01. 48 hours post-infection, virus production was determined by titering cell lysates on RK13+E3L+K3L cells. Standard deviations of two independent experiments are shown. Significant p-values as determined by the Student’s two-tailed t-test’s are indicated by asterisks: * = p < 0.05; ** = p < 0.01; *** = p < 0.005; n.s. = p > 0.05. The p-value for the comparison of VACV K3L vs GTPV K3L in CSM cells is 0.07.
PKR sensitivity and virus replication correlates with eIF2α phosphorylation in infected cells
Phosphorylation of eIF2α is the most direct readout of PKR activity. To analyze the ability of these K3 orthologs to inhibit eIF2α phosphorylation, we infected OA1, BT and HeLa cells with the different VACV strains at an MOI of 3 to ensure infection of most cells. Six hours post infection we performed Western blot analyses from whole cell lysates with antibodies targeting eIF2α phosphorylated at Ser51 (P-eIF2α) and total eIF2α. In all cells infected with VC-R4, high amounts of eIF2α phosphorylation were detected, consistent with the lack of PKR inhibitors (Fig. 7). Cells infected with vP872 showed suppressed eIF2α phosphorylation, consistent with the broad PKR inhibitory activity of E3. In OA1 and HeLa cells, infection with VC-R4+K3L resulted in little to no reduction of eIF2α phosphorylation, whereas all CaPV K3 orthologs strongly inhibited eIF2α phosphorylation. Consistent with the LBR and plaque assays, the opposite phenotype was observed in BT cells, in which only cells infected with VACV expressing K3 but not the CaPV K3 orthologs led to the inhibition of eIF2α phosphorylation.
Figure 7.
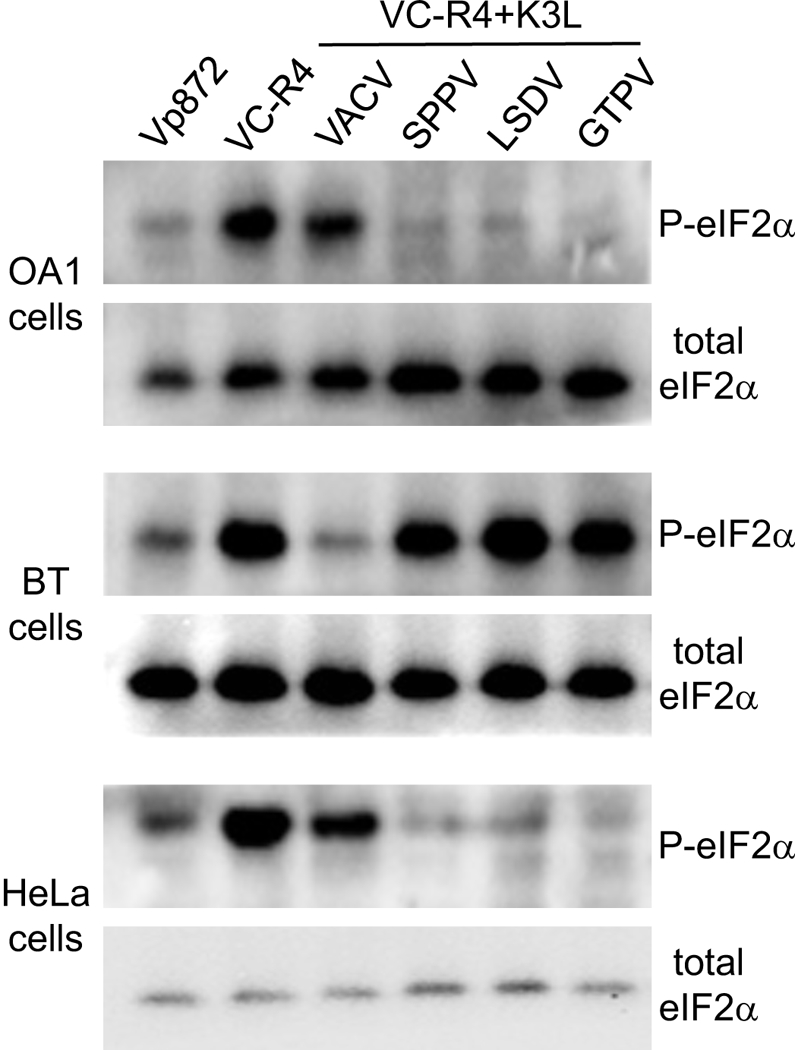
Phosphorylation of eIF2α by recombinant VACV expressing VACV K3L or CaPV K3L orthologs in cells derived from various species. OA, BT and HeLa cells were infected with the indicated viruses at MOI = 3. Cells were lysed six hours post-infection and analyzed by immunoblotting with primary antibodies detecting either P-eIF2α or total eIF2α.
Discussion
The molecular mechanisms that influence the host range, cell tropism and virulence of viruses are incompletely understood, impeding the threat assessment of newly emerging and re-emerging viruses. CaPVs have substantial differences in host tropism, and equally remarkable differences in disease outcomes between LSDV on one hand and SPPV and GTPV on the other hand. However, the molecular bases for these differences are currently unknown. One of the initial host barriers that viruses must overcome in order to establish a productive infection is PKR. Here, we analyzed how PKR inhibitors from CaPVs interact with PKR from different species, including their natural hosts, in comparison to the relatively well-studied VACV PKR inhibitor K3. Our data showed that the SPPV K3 ortholog 011 but not the SPPV E3 ortholog 034 inhibited sheep PKR. The inability of SPPV 034 to inhibit sheep PKR extends earlier results that showed that SPPV 034 did not efficiently inhibit human PKR.32 The inability of SPPV 034 to inhibit two divergent PKRs, including one from its natural host, suggests that SPPV 034 may be broadly unable to inhibit PKR. Therefore, SPPV may have to rely solely on its K3 ortholog to inhibit PKR for effective replication. If this observation is found to be broadly true, it would make SPPV E3 substantially different from the well-studied orthopoxvirus E3, which has a broad antagonistic activity against PKR from many species.
Our results show that the CaPV K3 orthologs inhibited PKR in a species-specific fashion, with strong inhibition of sheep, goat and human PKR but only weak inhibition of cow and mouse PKR. In contrast, VACV K3 only poorly inhibited sheep, goat and human PKR, but efficiently inhibited cow and mouse PKR. It is striking that bovine PKR was only weakly inhibited by all CaPV K3 orthologs, including LSDV 014. VACV containing any of the CaPV K3L orthologs showed a severe replication defect in bovine-derived BT cells (30 to 500-fold, depending on the specific ortholog) in comparison to VACV containing K3L or E3L, but still showed some replication. VACV+LSDV 014, for example, replicated to about 10-fold higher levels, as compared to input virus. The weak inhibition of cow PKR by LSDV 014 might still allow sufficient replication in LSDV infected cows and might at the same time allow cows to control the infection more efficiently than sheep and goats do after infection with SPPV and GTPV. The differential inhibition of cow PKR (weak inhibition) and sheep and goat PKR (good inhibition) by CaPV K3 orthologs generally correlates with the disease severity of LSDV infection in cow and SPPV and GTPV infection in sheep and goats.20, 21 The higher inhibition (about 41%) of cow PKR by GTPV 011 as compared to SPPV and LSDV 011 in the reporter assays also correlated with the formation of larger plaques in VACV-GTPV 011-infected goat and cow cell lines as compared to cells infected with VACV expressing the K3 orthologs from SPPV and LSDV (Fig. 5). However, genetic manipulation of PKR inhibitors in CaPVs would be necessary to study a causal relationship between levels of PKR inhibition and virulence. Nevertheless, the dramatic differences in sensitivity between the three different members of the Bovidae family shows that the sensitivities of antiviral proteins to viral inhibitors from relatively closely related species can vary dramatically.
The weak inhibition of human PKR and strong inhibition of mouse PKR by VACV K3L confirms earlier observations.9 It is interesting that VACV K3 inhibited cow PKR as well as mouse PKR, and that VACV expressing VACV K3 was the only virus that replicated to high viral titers and strongly inhibited eIF2α phosphorylation in BT cells. Cows are frequently infected by escaped VACV vaccine strains in India (also called buffalopox virus) and Brazil.34–36 It is possible that the high sensitivity of cow PKR to K3 inhibition contributes to the frequent infections of cow with feral VACV.
Interestingly, GTPV 011 inhibited sheep, goat and cow PKR approximately 41% to 46% better than SPPV 011 and LSDV 014 in the LBR assay (Fig. 2). This observation also correlated with the formation of larger plaques in VACV-GTPV011-infected goat and cow cell lines as compared to cells infected with VACV expressing the K3 orthologs from SPPV and LSDV (Fig. 5). In contrast, GTPV 011 did not inhibit human PKR any better than the other CaPV K3 orthologs. In the LBR assay, sheep PKR appeared more sensitive to CaPV K3 orthologs than goat and human PKR, the biological relevance of which is not clear. VACV strains expressing any CaPV K3L ortholog replicated essentially as well as Vp872, which contains VACV E3L, in sheep, goat and human cell lines and did not show substantial differences in eIF2α phosphorylation in infected cells (Figs. 6 and 7). The comparable replication of vP872 and VACV containing any CaPV K3 ortholog in OA1, CSM and HeLa cells or VACV K3 in BT cells is noteworthy because it indicates that a K3 ortholog that efficiently inhibits PKR can functionally substitute for VACV E3. VACV E3 is a multifunctional protein that inhibits other antiviral proteins with dsRNA capabilities, including 2′−5′-OAS in the RNaseL pathway, in addition to PKR.37–39 Our findings indicate that PKR is most likely the primary target of E3 in respect to virus replication in the analyzed cells because K3 orthologs that effectively inhibited PKR completely rescued virus replication. While we have not ruled out the possibility that CaPV K3L has evolved to inhibit multiple dsRNA-mediated host restriction factors, based on the known mechanism that orthopoxvirus K3 acts as a PKR pseudosubstrate, this possibility is unlikely. Furthermore, our interpretation that PKR is the primary target of E3 is in agreement with a report that showed that PKR knock-down in HeLa cells rescued replication of an E3L-deficient VACV.40
It was previously shown that PKRs from hominids were resistant to VACV K3 inhibition in a yeast assay and it was suggested that this resistance was due to positive selection exerted by K3-like inhibitors in the evolutionary past.10 The data presented here raise the interesting possibility that the proposed evolved resistance of human PKR against a relative of VACV K3 might have come at the price of making it more sensitive to other more distantly related PKR inhibitors such as CaPV K3 orthologs. Because PKR is an essential host barrier that poxviruses have to overcome in order to establish productive infections, such a trade-off in susceptibility to different PKRs could be a prerequisite for a successful cross-species transmission. It should be noted that productive CaPV infections have not been described in humans and our data indicate that host restriction factors other than PKR are likely responsible for this. However, we speculate that poxviruses that contain potent inhibitors of an accidental host species’ PKR will have a higher potential to successfully overcome this species barrier than a poxvirus that could not already inhibit that species’ PKR. The striking dichotomy of the tested PKR’s sensitivity to VACV K3 and the CaPV K3 orthologs indicates a relatively constrained evolutionary landscape of PKR because evolved PKR variants must still be able to interact with eIF2α. Therefore, selection for PKR variants with increased resistance against one PKR antagonist might render it more sensitive to other PKR antagonists that bind to overlapping regions.
The effects of differential PKR inhibition have so far been addressed in yeast assays and cultured cells, in which yeast growth, eIF2α and PKR phosphorylation, mRNA translation or virus replication have been used as read-outs for PKR inhibition.9–11, 19, 41 Currently, we have very limited information about the effects of differential PKR inhibition from in vivo infection models. It seems clear that no or ineffective PKR inhibition would result in abortive infection in vivo. But it is unclear what the effects of modest inhibition of PKR, that still allows for some virus replication, such as a CaPV K3 ortholog in virus infection of cow cells, as opposed to strong inhibition such as in the case of a CaPV K3 ortholog in virus infection of sheep and goat cells, will have during infection of a whole organism. It is interesting to note that a K3L deficient VACV in an intratracheal infection model showed a migration defect and accumulated to extremely high titers at the site of infection in an intratracheal infection model.42 Differential inhibition of PKR might therefore have other effects in addition to affecting virus replication, including virus dissemination. Infection of cow, sheep and goats with CaPVs that contain differentially potent PKR antagonists might prove to be important models to study the effects of differential PKR inhibition in highly relevant animal models.
Acknowledgments
This work was supported by grant AI114851 (to S.R.) from the National Institute of Allergy and Infectious Diseases, National Institutes of Health. S.R. accepts responsibility for the integrity of the data analyzed.
Footnotes
Competing Interests
The authors declare no competing interests
Note after submission
When we were in the final stages of writing this manuscript, a related manuscript by Zhao et al., was published online.43 Zhao et al. reported that cow PKR was sensitive to LSDV K3 but not to vaccinia virus K3, whereas our results showed high sensitivity of cow PKR to vaccinia virus K3 but only weak sensitivity to LSDV K3. Zhao et al. described that goat PKR was sensitive to GTPV K3 but not to LSDV and SPPV K3, whereas, in our experiments, goat PKR was sensitive to all three CaPV K3 orthologs, with GTPV K3 showing about 40% better inhibition. Our and the Zhao et al. study is comparable concerning the sensitivity of human and sheep PKR to the K3 orthologs.
One possible explanation for the discrepancies between the two studies is that Zhao et al. used epitope-tagged PKR and K3 homologs and we used untagged proteins. Our unpublished results showed that epitope tagging can impact PKR activity; in the case of SPPV 011 (mentioned in our manuscript), C-terminal FLAG-tagging abolished its inhibitory activity completely. Also, Zhao et al. did not specify the sequences of PKR and K3 orthologs used in their experiment; this might be important information, as there are intra-species variants of both PKR and some K3 orthologs (two variants for GTPV 011; all current SPPV 011 and LSDV 014 sequences are identical on the protein level). For cloning cow PKR, Zhao et al. used the same cell line as we (MDBK cells) and therefore different alleles, as an explanation for the different results, is not very likely. Another difference is that Zhao et al. employed a transient knock-down of human PKR in HeLa cells, whereas we used HeLa cells with a robust stable knock-down of PKR.
Going forward, it will be important to determine whether these discrepancies really are due to an artifact of epitope tagging or if there is a true difference mediated by genotypic differences. However, with the data available now, we cannot make this distinction.
References
- 1.Haller SL, Peng C, McFadden G, et al. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol. 21: 15–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moss B 2006. Poxvirus entry and membrane fusion. Virology. 344: 48–54. [DOI] [PubMed] [Google Scholar]
- 3.Bratke KA, McLysaght A & Rothenburg S. 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol. 14: 406–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfaller CK, Li Z, George CX, et al. 2011. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr Opin Immunol. 23: 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu S & Kaufman RJ. 1997. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem. 272: 1291–1296. [DOI] [PubMed] [Google Scholar]
- 6.Rothenburg S, Georgiadis MM & Wek RC. 2016. “Evolution of eIF2α Kinases: Adapting Translational Control to Diverse Stresses”. In Evolution of the Protein Synthesis Machinery and Its Regulation. Hernández G & Jagus R, Eds.: 235–260. Cham: Springer International Publishing. [Google Scholar]
- 7.Rowlands AG, Panniers R & Henshaw EC. 1988. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J Biol Chem. 263: 5526–5533. [PubMed] [Google Scholar]
- 8.Krishnamoorthy T, Pavitt GD, Zhang F, et al. 2001. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol. 21: 5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothenburg S, Seo EJ, Gibbs JS, et al. 2009. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat Struct Mol Biol. 16: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elde NC, Child SJ, Geballe AP, et al. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature. 457: 485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpentier KS, Esparo NM, Child SJ, et al. 2016. A Single Amino Acid Dictates Protein Kinase R Susceptibility to Unrelated Viral Antagonists. PLoS Pathog. 12: e1005966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beattie E, Tartaglia J & Paoletti E. 1991. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology. 183: 419–422. [DOI] [PubMed] [Google Scholar]
- 13.Dar AC & Sicheri F. 2002. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol Cell. 10: 295–305. [DOI] [PubMed] [Google Scholar]
- 14.Chang HW, Watson JC & Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A. 89: 4825–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romano PR, Zhang F, Tan SL, et al. 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol Cell Biol. 18: 7304–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langland JO & Jacobs BL. 2002. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology. 299: 133–141. [DOI] [PubMed] [Google Scholar]
- 17.Dar AC, Dever TE & Sicheri F. 2005. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 122: 887–900. [DOI] [PubMed] [Google Scholar]
- 18.Seo EJ, Liu F, Kawagishi-Kobayashi M, et al. 2008. Protein kinase PKR mutants resistant to the poxvirus pseudosubstrate K3L protein. Proc Natl Acad Sci U S A. 105: 16894–16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng C, Haller SL, Rahman MM, et al. 2016. Myxoma virus M156 is a specific inhibitor of rabbit PKR but contains a loss-of-function mutation in Australian virus isolates. Proc Natl Acad Sci U S A. 113: 3855–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Babiuk S, Bowden TR, Boyle DB, et al. 2008. Capripoxviruses: an emerging worldwide threat to sheep, goats and cattle. Transbound Emerg Dis. 55: 263–272. [DOI] [PubMed] [Google Scholar]
- 21.Tuppurainen ESM, Venter EH, Shisler JL, et al. 2017. Review: Capripoxvirus Diseases: Current Status and Opportunities for Control. Transbound Emerg Dis. 64: 729–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamien CE, Le Goff C, Silber R, et al. 2011. Use of the Capripoxvirus homologue of Vaccinia virus 30 kDa RNA polymerase subunit (RPO30) gene as a novel diagnostic and genotyping target: development of a classical PCR method to differentiate Goat poxvirus from Sheep poxvirus. Vet Microbiol. 149: 30–39. [DOI] [PubMed] [Google Scholar]
- 23.Tuppurainen ES, Pearson CR, Bachanek-Bankowska K, et al. 2014. Characterization of sheep pox virus vaccine for cattle against lumpy skin disease virus. Antiviral Res. 109: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedger RS & Hamblin C. 1983. Neutralising antibodies to lumpy skin disease virus in African wildlife. Comp Immunol Microbiol Infect Dis. 6: 209–213. [DOI] [PubMed] [Google Scholar]
- 25.Fagbo S, Coetzer JA & Venter EH. 2014. Seroprevalence of Rift Valley fever and lumpy skin disease in African buffalo (Syncerus caffer) in the Kruger National Park and Hluhluwe-iMfolozi Park, South Africa. J S Afr Vet Assoc. 85: e1–e7. [DOI] [PubMed] [Google Scholar]
- 26.Klevjer-Anderson P & Cheevers WP. 1981. Characterization of the infection of caprine synovial membrane cells by the retrovirus caprine arthritis-encephalitis virus. Virology. 110: 113–119. [DOI] [PubMed] [Google Scholar]
- 27.Zhang P & Samuel CE. 2007. Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J Virol. 81: 8192–8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahman MM, Liu J, Chan WM, et al. 2013. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 9: e1003465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tulman ER, Afonso CL, Lu Z, et al. 2002. The genomes of sheeppox and goatpox viruses. J Virol. 76: 6054–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tulman ER, Afonso CL, Lu Z, et al. 2001. Genome of lumpy skin disease virus. J Virol. 75: 7122–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vipat S, Brennan G, Haller SL, et al. 2018. Rapid, seamless generation of recombinant poxviruses using host-range and visual selection. bioRxiv. 467514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myskiw C, Arsenio J, Hammett C, et al. 2011. Comparative analysis of poxvirus orthologues of the vaccinia virus E3 protein: modulation of protein kinase R activity, cytokine responses, and virus pathogenicity. J Virol. 85: 12280–12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hand ES, Haller SL, Peng C, et al. 2015. Ectopic expression of vaccinia virus E3 and K3 cannot rescue ectromelia virus replication in rabbit RK13 cells. PLoS One. 10: e0119189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moussatche N, Damaso CR & McFadden G. 2008. When good vaccines go wild: Feral Orthopoxvirus in developing countries and beyond. J Infect Dev Ctries. 2: 156–173. [DOI] [PubMed] [Google Scholar]
- 35.Singh RK, Balamurugan V, Bhanuprakash V, et al. 2012. Emergence and reemergence of vaccinia-like viruses: global scenario and perspectives. Indian J Virol. 23: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matos ACD, Rehfeld IS, Guedes M, et al. 2018. Bovine Vaccinia: Insights into the Disease in Cattle. Viruses. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rivas C, Gil J, Melkova Z, et al. 1998. Vaccinia virus E3L protein is an inhibitor of the interferon (i.f.n.)-induced 2–5A synthetase enzyme. Virology. 243: 406–414. [DOI] [PubMed] [Google Scholar]
- 38.Perdiguero B & Esteban M. 2009. The interferon system and vaccinia virus evasion mechanisms. J Interferon Cytokine Res. 29: 581–598. [DOI] [PubMed] [Google Scholar]
- 39.Liu R & Moss B. 2016. Opposing Roles of Double-Stranded RNA Effector Pathways and Viral Defense Proteins Revealed with CRISPR-Cas9 Knockout Cell Lines and Vaccinia Virus Mutants. J Virol. 90: 7864–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang P, Jacobs BL & Samuel CE. 2008. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J Virol. 82: 840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Child SJ, Brennan G, Braggin JE, et al. 2012. Species specificity of protein kinase r antagonism by cytomegalovirus TRS1 genes. J Virol. 86: 3880–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice AD, Turner PC, Embury JE, et al. 2011. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J Virol. 85: 550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Z, Zhu X, Wu N, et al. 2018. Species-specific inhibition of capripoxvirus replication by host antiviral protein kinase R. Ann N Y Acad Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]