

Abstract
Cerebral amyloid angiopathy-related inflammation (CAA-RI) is a rare but increasingly recognized subtype of CAA. CAA-RI consists of two subtypes: inflammatory cerebral amyloid angiopathy and amyloid β (Aβ)-related angiitis. Acute or subacute onset of cognitive decline or behavioral changes is the most common symptom of CAA-RI. Rapid progressive dementia, headache, seizures, or focal neurological deficits, with patchy or confluent hyperintensity on T2 or fluid-attenuated inversion recovery sequences and evidence of strictly lobar microbleeds or cortical superficial siderosis on susceptibility-weighted imaging imply CAA-RI. The gold standard for diagnosis is autopsy or brain biopsy. However, biopsy is invasive; consequently, most clinically diagnosed cases have been based on clinical and radiological data. Other diagnostic indexes include the apolipoprotein E ε4 allele, Aβ and anti-Aβ antibodies in cerebral spinal fluid and amyloid positron emission tomography. Many diseases with similar clinical manifestations should be carefully ruled out. Immunosuppressive therapy is effective both during initial presentation and in relapses. The use of glucocorticoids and immunosuppressants improves prognosis. This article reviews the pathology and pathogenesis, clinical and imaging manifestations, diagnostic criteria, treatment, and prognosis of CAA-RI, and highlights unsolved problems in the existing research.
Keywords: Brain MRI lesions, Cerebral amyloid angiopathy, Cerebral small vessel disease, Inflammation, Review
Introduction
Cerebral amyloid angiopathy (CAA) is a common small vessel disease characterized by the deposition of amyloid β (Aβ) protein mainly in the media and adventitia of small- and medium-sized leptomeningeal and cortical blood vessels. There are two major types of CAA: one is hereditary CAA, which is associated with Down syndrome or mutations in the Aβ protein precursor (APP) gene or presenilin gene,[1] and the other one is age-related sporadic CAA. In sporadic CAA, vascular amyloid is composed of the same 39- to 43-amino acid Aβ peptide observed in the neuritic plaques of Alzheimer's disease (AD).[2] CAA is clinically diverse. It may present with symptomatic acute lobar intracerebral hemorrhage (ICH), chronic progressive cognitive decline, transient focal neurological episodes, and subacute cognitive disorder or behavioral changes caused by CAA-related inflammation (CAA-RI).[3] CAA related lobar ICH has been identified as the second most common form of spontaneous ICH following hypertensive angiopathy.[4] With the development of imaging technology, more clinical “silent” patients are identified by the classic imaging abnormalities, including multiple strictly lobar cerebral microbleeds (CMBs), cortical superficial siderosis (cSS) or cortical subarachnoid hemorrhage, and cortical atrophy.[3]
CAA-RI is now widely recognized as a relatively rare and aggressive subtype of CAA with diverse clinical presentations and characteristic radiological findings. Reid and Maloney first described CAA with vascular inflammation in a patient with AD in 1974, and subsequent cases were reported.[5] Unlike non-inflammatory CAA, acute or subacute onset of cognitive decline or behavioral changes are the most common symptom of CAA-RI.[6–8] Other features include seizures, headaches, T2-weighted white matter hyperintense (WMH) lesions on magnetic resonance imaging (MRI), and pathological evidence of inflammation against vascular Aβ, which is the hallmark of CAA.[9,10] Two pathological subtypes are now generally accepted: non-destructive perivascular inflammation (inflammatory CAA [ICAA]) and transmural or intramural inflammation (Aβ-related angiitis [ABRA]).[11] The gold standard test for diagnosis is autopsy or brain biopsy. However, biopsy is invasive; consequently, some criteria for the diagnosis of CAA-RI have been based on clinical and radiological data.[12,13] Because immunosuppressive therapy is effective for the disease, timely diagnosis and early commencement of therapy are very important.
Thus, in this review, we present the main pathological, clinical, neuroimaging, therapeutic, and prognostic features and the diagnostic criteria of CAA-RI to shed some light on its clinical practice, and then discuss issues that remain unresolved.
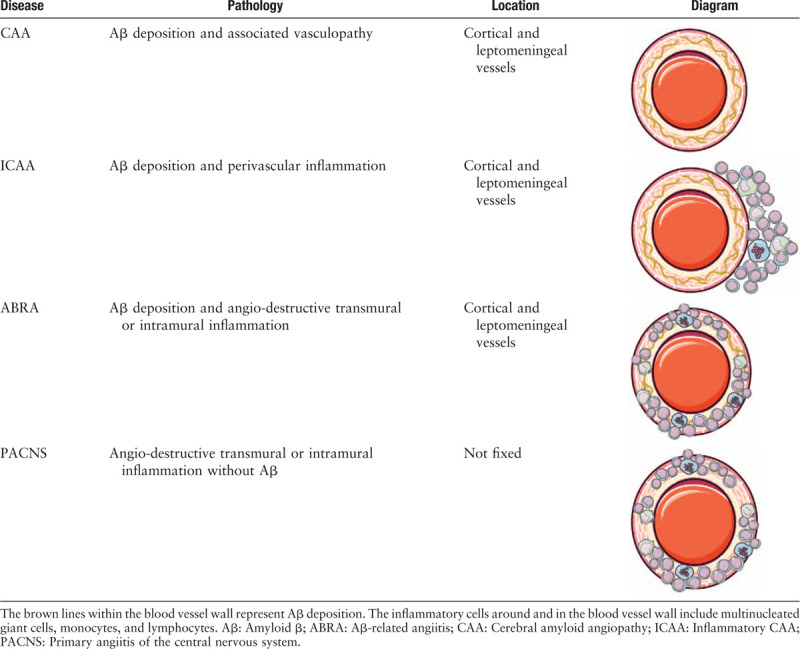
Pathology and pathogenesis
CAA-RI shares pathologic characteristics of CAA, which is Aβ deposition in the cortical or leptomeningeal vessels, with positive Congo red staining. Amyloid can be confirmed when the Congo red-stained section shows green birefringence under polarized light.[14] In addition to Aβ deposition, CAA-RI also demonstrates pronounced perivascular or transmural inflammatory infiltration. Historically, only ICAA was initially considered to be the only inflammatory form of CAA,[9] while ABRA was thought to represent coexisting primary angiitis of the central nervous system (PACNS) and CAA.[15] In fact, these two types sometimes do coexist.[16,17] However, the terms used to describe this disease are confusing. Phrases such as “CAA associated with inflammation,” “CAA-RI,” “ICAA,” and “ABRA” are used interchangeably. Some authors are consistent with the terms we have used here, while some call the two subtypes CAA-RI and ABRA.[2,17–19] In addition, some researchers still believe that CAA-RI/ICAA and ABRA are two different disease entities. The former represents the inflammatory form of CAA, while the latter is an independent disease or a subtype of PACNS associated with CAA.[20–23] In recent years, it has gradually come to be accepted that these two pathological types are essentially similar.
The mechanism underlying CAA-RI remains unclear.[24] There are three current hypotheses: (1) coexistence of vascular Aβ and vascular inflammation implies that Aβ is a bystander of angiitis; (2) inflammation promotes accumulation of Aβ in the vessel wall; (3) Aβ deposition triggers the inflammatory response.[20] Currently, most evidence favors the hypothesis that inflammation is triggered by an autoimmune response to the deposited Aβ protein. Similar clinical processes and radiological changes of CAA-RI appear in amyloid-related imaging abnormalities (ARIA), initially during the clinical trial of bapineuzumab, the monoclonal antibody for AD, and later in that of other amyloid modification therapies.[25–27] ARIA is also divided into two categories: ARIA-E, which manifests as focal or confluent vasogenic edema on fluid-attenuated inversion recovery (FLAIR) sequence images, and ARIA-H, characterized by CMBs or cSS on T2∗-weighted gradient-echo/susceptibility-weighted imaging (SWI) sequence scans, corresponding to the image hallmarks of CAA-RI. Almost half of those with ARIA-E also developed ARIA-H, with co-located lesions.[28] CAA-RI is thought to be a spontaneous ARIA, while ARIA is considered to be iatrogenic CAA-RI. AD patients who are apolipoprotein E (APOE) ε4 gene carriers are more likely to develop ARIA after anti-Aβ treatment,[25,26] in accordance with the findings in CAA-RI. Because of the similarity between CAA-RI and ARIA, the first theory seems unreasonable. In addition, it has been observed that immune activation in the parenchyma near the affected blood vessels increased significantly and the Aβ load decreased accordingly.[20] The incidence of ARIA gradually increased with an increase in the therapeutic antibody dose.[28] This strongly suggests that an immune response to Aβ is responsible for CAA-RI.
Aβ is deposited segmentally, but can be found in all those inflammation sites.[9] Cells such as CD3+, CD4+, and CD8+ T lymphocytes, CD20+ B lymphocytes, and CD68+ monocytes, including macrophages (sometimes multinucleated giant cells) in the vessel wall and reactive astrocytes can be found in the surrounding parenchyma.[19,29,30] Usually, B lymphocytes are fewer compared to T cells. Aβ engulfed in macrophages can be observed at times.[14]
Angio-destructive changes, such as fibrinoid necrosis can also be found in some of the vessel walls in patients affected by ABRA.[22] The mainstream view is that granulomatous inflammation is the pathological hallmark of ABRA, but not of ICAA.[22,31] In fact, both ICAA and ABRA can present with or without granulomatous inflammation.[32] In a systematic review, of the 142 cases with available data, 27.5% presented with both perivascular inflammation and vasculitis with granuloma formation, which is the most common pathological pattern. Perivascular and vascular inflammatory patterns without granulomas accounted for 22.5% of cases. That is, 50% of all cases showed overlap between ICAA and ABRA patterns.[18] However, these results should be carefully considered because the high proportion of granulomatous inflammation may be due to the higher biopsy rate in those cases showing more serious clinical and imaging manifestations and a tendency of malignant diseases.[22]
Moreover, ABRA was considered to be different from ICAA because it has the same vascular destructive pathological changes as PACNS. However, some studies have questioned the idea. First, ABRA has the same radiological characteristics as ICAA, which are not common in PACNS. Second, vasculitis and the vascular areas affected by Aβ co-localize. Third, Aβ was engulfed by macrophages expressing MHC class II antigens near CD4+ T cells, suggesting that Aβ plays a pathogenic role in inducing inflammation in ABRA.[14,29] Finally, in terms of clinical manifestations and prognosis, there was no difference between the two pathological subtypes of CAA-RI.[18] It can be concluded that these pathologically similar diseases constitute a spectrum from CAA to PACNS [Table 1].
Table 1.
A spectrum from CAA to PACNS: pathological differences between CAA, ICAA, ABRA, and PACNS.
Clinical manifestations
There have been few epidemiological studies on CAA-RI. A nationwide survey demonstrated that its prevalence is about 0.13 per 100,000 population in Japan.[33] Findings from several systematic reviews have shown that there is no obvious gender difference, but a slight male predominance was observed.[17,18] The main patient group is the elderly, with an average age of 67 at diagnosis; yet, this is still younger than that of CAA patients.[17] And the youngest case with pathologic evidence ever reported was 42 years old.[34]
The major clinical manifestations of CAA-RI are subacute mental disorders and behavioral or cognitive changes, headaches, seizures, and focal neurological deficits, which are different from CAA. The most recent systematic review included 213 pathologically confirmed cases of CAA-RI.[17] In this review, cognitive decline was the most common clinical manifestation, accounting for 48%, followed by seizures (32%), headache (32%), encephalopathy (27%), presenting as confusion or disturbance of consciousness, weakness (16%), and aphasia (14%). Thirteen percent of patients were affected with some forms of visual impairment.
However, many patients present with atypical symptoms other than those mentioned above, which may easily lead to an incorrect diagnosis. Some cases presented with involuntary movement,[35,36] while others had systemic diseases,[14] cerebral hernia caused by severe edema,[37] uveitis,[21] multiple malignancies,[14,15,38] extracranial vasculitis, or vascular dysplasia at baseline.[39] One patient with a history of Parkinson's disease (PD) was mistakenly thought to have developed the mental manifestation of PD when he presented with the symptoms of CAA-RI.[40] Whether the etiology of these comorbidities, such as autoimmunity, or their treatment, such as radiation therapy,[41] are related to CAA-RI requires further study. Due to these atypical symptoms, advanced imaging is very meaningful for clinical diagnosis.
Neuroimaging
Brain MRI, particularly FLAIR and T2∗/SWI sequences, is the most important imaging modality for the identification of patients suspected of CAA-RI. Findings supporting CAA-RI include patchy or confluent T2 hyperintensity of subcortical white matter lesions, which are mostly asymmetric, in addition to the presence of multiple, strictly lobar CMBs and cSS on T2∗ or SWI, which is also a typical finding in CAA [Figure 1]. WMHs sometimes extend to the cortex with a mass effect showing hyperintensity in maps of apparent diffusion coefficient suggesting vasogenic edema.[10,42,43] SWI is considered to be more reliable than T2∗ imaging, with greater reliability and sensitivity for detection of CMBs.[44,45] However, sometimes the burden of CMBs is so obvious that hypointense lesions seen on SWI can also be identified on T2 or FLAIR images. The distribution of CMBs does not follow the regional pattern of occipital dominance in non-inflammatory CAA. The incidence of multiple lobar CMBs, as well as the total number of CMBs is significantly higher in CAA-RI patients.[46,47] A possible explanation for this finding is that, once an immune response to vascular amyloid protein is generated, it affects multiple regions of brain via the spread of antibodies.[17] Multiple lobar CMBs were found on SWI or T2∗ images in most patients, but some cases of pathologically confirmed CAA-RI were without CMBs on MRI.[48,49]
Figure 1.
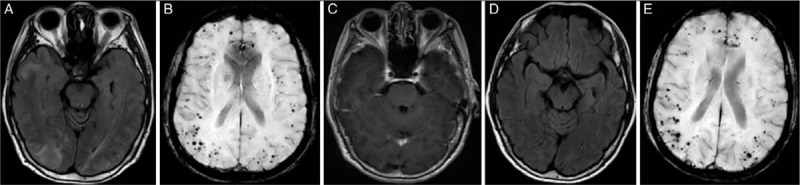
Typical images of cerebral amyloid angiopathy-related inflammation. (A) Confluent WMH. (B) Strictly lobar CMBs. (C) No enhancement was seen. After treatment with corticoids, (D) WMH faded significantly. (E) No significant changes with CMBs. CMBs: Cerebral microbleeds; WMH: White matter hyperintensity.
Gadolinium enhancement of parenchyma or leptomeninges may or may not be present [Figure 1],[43,50] although the proportion of enhancing cases in CAA-RI is significantly higher than that in non-inflammatory CAA cases.[14] Based on the criteria of leptomeningeal enhancement, the sensitivity and specificity of recognizing CAA-RI from CAA patients are reported to be 70.4% and 92.6%, respectively.[46] Two-thirds of ABRA patients and only 31.3% of ICAA patients showed contrast enhancement on MRI.[11] This phenomenon may be explained by the fact that the blood vessel wall in cases of ICAA is less destroyed than that in cases of ABRA. Leptomeningeal enhancement may be a unique imaging manifestation in some cases with confirmed CAA-RI.[50,51] In these extreme cases, brain biopsy seems to be the only choice. However, given the segmental distribution of the lesions, they may be missed by the biopsy, which will lead to a missed diagnosis. Thus, amyloid positron emission tomography (PET) might be important for the diagnosis of CAA-RI, by showing sites with markedly elevated amyloid deposition.[11,52,53]
Magnetic resonance angiography (MRA) or cerebral angiography is unremarkable in CAA-RI, due to the small caliber of the involved blood vessels, which prevents the lesion from being captured.[14] However, findings from another study have suggested that non-specific vascular changes in ABRA may be observed when medium-sized arteries are involved.[22] Nevertheless, in our experience, this is not typical and may not be meaningful in clinical practice.
Serologic and cerebrospinal fluid (CSF) tests
Blood tests may reveal signs of inflammation. An increase in inflammatory biomarkers has been observed in CAA-RI patients in different studies.[12,14,18] The erythrocyte sedimentation rate was increased in 37.5% of patients, while C-reactive protein (CRP) was elevated in 60%.[18] Sakai et al[32] reported a case of CAA-RI at the chronic stage, with persistently elevated proteinase 3-antineutrophil cytoplasmic antibody levels. This case was reminiscent of ANCA-associated vasculitis, although the relationship between proteinase 3-antineutrophil cytoplasmic antibody and the pathogenesis of CAA-RI remains unclear.
The APOE ε4 allele is currently the only confirmed risk factor for CAA-RI. Many studies have reported that APOE ε4/ε4 homozygosity is significantly correlated with CAA-RI,[47] accounting for 76.9% of CAA-RI patients.[10] The carriage rate in non-inflammatory CAA patients was only 5.1%,[10] and it is rarely seen in healthy people or stroke patients.[54] Therefore, the presence of the APOE ε4/ε4 genotype may be meaningful for the diagnosis of CAA-RI. The possible mechanism is that APOE ε4 increases Aβ deposition, and has a pro-inflammatory effect.[18] Although the APOE ε2 allele is considered a protective factor against AD, it clearly increases the risk of vascular disease. There are also cases of CAA-RI patients reported with genotype APOE ε2/ε2 and APOE ε2/ε3.[55,56] Thus, ε2 carriers may also be predisposed to CAA-RI. However, due to the relatively few ε2 alleles or genotypes detected in cases, it is difficult to determine the role of ε2 in CAA-RI in small sample studies.[55] An APOE ε4/ε4 homozygous patient with a rare SORL1 mutation has been reported.[57]SORL1 encodes a 250-kDa protein called sorting protein-related receptor with A-type repeats (SorLA), which reduces the production and deposition of Aβ peptides by regulating the processing of APP.[58,59] Thus, a variant in SORL1 may lead to dysfunction of SorLA, eventually adding to the risk of CAA-RI.
The results of lumbar puncture revealed that more than 80% of patients had increased CSF protein, 44% had pleocytosis,[17] and generally no oligoclonal bands were detected.[14] Previous studies have revealed that, compared with multiple sclerosis and healthy people, anti-Aβ autoantibodies in the CSF of CAA-RI patients increased during the acute phase, which is consistent with what was observed in ARIA, supporting the aforementioned hypothesis of an Aβ-induced immune response.[28] Antibody levels decrease after corticosteroid therapy,[2,42] indicating that anti-Aβ autoantibody may be used as a biomarker for both diagnosis and monitoring the effect of treatment. In addition, some researchers found that, compared with non-inflammatory CAA, PACNS, and healthy controls, patients with CAA-RI have relatively low levels of Aβ42 and Aβ40 in the CSF.[47,60] In the future, the significance of these indicators for the differential diagnosis of CAA-RI mimics should be studied.
Diagnostic criteria
Conclusive diagnosis of CAA-RI requires histopathological confirmation, but it is invasive and has certain risks. It is generally recommended that brain biopsy should be performed from an area with abnormal radiologic manifestations, preferably at a lesion in the cortex or leptomeninges. Biopsy obtained from the white matter showed no evidence of inflammation in one case.[61] Despite this, negative brain biopsy findings are insufficient to exclude the diagnosis of CAA-RI, because of the segmental distribution of pathological changes.
In order to make a diagnosis before histopathology, Chung et al[12] proposed the Boston criteria using clinicoradiological data in 2011. Auriel et al[13] updated the criteria in 2016, defined the WMH pattern specific for distinguishing between probable and possible CAA-RI, and proposed cSS as a marker of hemorrhage. The accuracy of the standard was verified, and yielded a sensitivity and specificity of 82% and 97% diagnosing probable CAA-RI, respectively.[13] For patients diagnosed with probable CAA-RI by means of these criteria, immunosuppressive therapy can be given empirically to avoid brain biopsy. If there is no response to corticosteroid therapy within 3 weeks, biopsy should be reconsidered to confirm the diagnosis.[13] Nevertheless, these criteria are still imperfect, as samples included in the validation trial was small. The diagnostic efficiency for possible CAA-RI is low, with a specificity of only 68%. Therefore, other biomarkers are needed to enrich the criteria. Besides, the study did not propose a specific treatment or plan for further examination for patients meeting a diagnosis of possible CAA-RI.
Diagnostic procedures in this setting include blood tests, neuroimaging, CSF analysis, and brain biopsy when necessary to make a diagnosis of CAA-RI, as well as to exclude other conditions.
Differential diagnosis
It is easy for doctors to diagnose CAA-RI when patients were APOE ε4/ε4 homozygotes with typical clinical characteristics and image. However, there are many atypical cases or cases without T2∗/SWI sequence that were initially misdiagnosed, in whom the diagnosis was later revised.[62,63] Thus, it is very important to recognize the clinical and radiological properties of CAA-RI and bear some differential diagnoses in mind; those substantial differential diagnoses should be ruled out before CAA-RI was diagnosed. It is conceivable that posterior reversible encephalopathy syndrome (PRES) is a very important differential diagnosis. Due to the potentially reversible WMH in ICAA,[43] when clinical manifestations are present and findings on conventional MRI sequences are suggestive, it must be distinguished from PRES, which also has the characteristic of bilateral confluent T2 WMH, but is often associated with hypertension or other conditions. One case was initially suspected of PRES or cerebral venous sinus thrombosis and was treated with anticoagulant and steroid. However, anticoagulation was later suspended due to cerebral hemorrhage, and the patient was finally diagnosed with CAA-RI.[64] Another patient was first diagnosed with PRES, which was responsive to anti-edema intravenous steroid and antihypertensive therapy. After several recurrences, WMH and CMBs progressed and long-term follow-up led to a diagnosis of CAA-RI.[65] Therefore, these two diseases are sometimes difficult to distinguish, and it may be necessary to observe changes during follow-up to obtain the correct diagnosis. This also reflects the importance of the SWI sequence.
Tumors including primary central nervous system lymphomas and metastases should be taken into consideration when making a diagnosis in such patients. WMH and vasogenic edema accompanied by a mass effect make brain tumors a highly suspected differentiation. Many cases have reported that patients were misdiagnosed with tumors, and the diagnosis was modified to CAA-RI when the data were retrospectively analyzed or after the biopsy results became available.[6,66] In addition, these two conditions may be present concurrently. It would be more difficult to identify patients who also have a history of tumors. Kirshner et al[8] reported a CAA-RI patient with pathologically confirmed grade III anaplastic astrocytoma. In another case, the patient had clinical and imaging characteristics of CAA-RI, but because of bicytopenia and an increase in CRP and lactate dehydrogenase, lymphoma was suspected. The biopsy result revealed intravascular large B-cell lymphoma.[67] For such patients, a clinicoradiological diagnosis only may result in missing a coexisting tumor, and thus the pros and cons of biopsy should be weighed carefully. These cases emphasize that CAA-RI is a diagnosis by exclusion. Another option is to follow the patient up closely. Since the treatment does not obviously harm the tumor, the response of the lesion to the given treatment can be observed to figure out whether it deteriorates as time goes by.
The clinical manifestations of PACNS can also mimic the pattern of CAA-RI. Thus, PACNS is on the list of differential diagnoses whenever multifocal hyperintensity is seen on FLAIR images, although it is a diagnosis of exclusion. PACNS usually occurs in younger patients (mean age, 45 years), while CAA-RI is common in slightly older people. Since there is no Aβ deposition in the blood vessels supplying the spinal cord, symptoms of myelopathy have not been reported in ICAA and ABRA; thus, PACNS is a more likely diagnosis when symptoms involving the spinal cord occur.[22] Moreover, ischemic stroke is more common in PACNS than in CAA-RI,[24] and there have been only a few cases of patients with CAA-RI presenting with ischemic stroke.[2,46,68] The most common abnormality found in PACNS is the presence of proximal or distal stenosis on MRA or conventional digital subtraction angiography; this is not commonly seen in CAA-RI.
Other differential diagnoses include viral or autoimmune encephalitis, cerebral venous thrombosis, acute disseminated encephalomyelitis (ADEM), Hashimoto encephalopathy, neurosarcoidosis, and acute toxic-metabolic leukoencephalopathy. Some of these diseases can be ruled out by T2∗ MRI or SWI. This highlights the significance of the T2∗/SWI sequences in differentiation. Although tumors, neurosarcoidosis, Hashimoto encephalopathy, ADEM, or PACNS are unlikely to be aggravated by empirical usage of corticosteroids, the treatment may obscure the diagnosis of those diseases. Hence, in such cases, close follow-up should be performed. In addition, when starting the treatment, infection needs to be ruled out first, to avoid pervasion due to corticosteroid therapy.
Treatment and prognosis
In contrast to CAA, which is currently without effective treatment, most studies have shown that empirical high-dose corticosteroids with or without additional immunosuppressive therapy can mitigate symptoms and imaging abnormalities and can improve the prognosis of CAA-RI.[69] A systematic review of both pathological subtypes revealed that, during an average follow-up period of 24 months, 55% of patients eventually end up being asymptomatic or with mild disability.[17] While another systematic review showed that the functional outcome of most patients was not ideal. About 60% of patients died or were severely disabled after immunotherapy, and there was no statistically significant difference in terms of prognosis between the two pathological types.[18] No difference in outcome was found between patients receiving mono-therapy of corticosteroid and patients receiving a combination of immunosuppressant and corticosteroid therapy.[18] The clinical and radiological manifestations may be initially relieved after glucocorticoid therapy, but can relapse after withdrawal of steroids or during dose decrease.[14] The recurrence probability of CAA-RI has differed across studies.[70] The clinical features of relapse are widely distributed, among which the decline of cognitive function and encephalopathy are the most common symptoms.[17] Steroid therapy is also effective during recurrence, but increased microbleeds may be detected with T2∗/SWI sequences in that case.[57] A reduction of CMBs was found in one case after immunotherapy, but it cannot be ruled out that the natural course of CAA-RI may include a spontaneous reduction in CMBs.[19] Spontaneous remission has been reported in some cases,[7,71] the fundamentals of which are not yet known. However, the prognosis of most untreated patients is poor.
Overall, it is believed that immunotherapy would result in better clinical outcomes in patients. Our clinical experience also supports this conclusion [Figure 1]. At present, the main recommendation is that high-dose glucocorticoids should be used. Immunosuppressants can be administered in cases showing no response to glucocorticoids or for preventing recurrence. A study has shown that more patients with ABRA (33.0%) require a combination of steroids and immunosuppressants than do patients with ICAA (12.8%), to achieve similar outcomes.[11] The most commonly used immunosuppressants are cyclophosphamide (33.9%), azathioprine (5.0%), mycophenolate mofetil (5.0%), methotrexate, immunoglobulin, and so on.[14] The dosage used is based on individual selection. There is currently no study giving recommendations on the choice of medication, dosage, and the time span of treatment. In one case, heart transplantation was performed because of sarcoid cardiomyopathy, followed by long-term use of immunosuppressants, and CAA-RI occurred during hospitalization after mycobacterial infection.[72] It is worth noting that this case involved a patient who had been using immunosuppressive agents. In addition, the treatment of infection and other comorbidities should be considered in such cases.
Future work on CAA-RI
There are still many questions related to CAA-RI that require investigation. It is not clear why only a small proportion of patients with CAA develop inflammation against Aβ. The aim of future research should focus on specific pathogenic mechanisms and inflammatory pathways to determine which types of CAA patients are prone to developing inflammation, whether other genes or alleles besides APOE ε4 are also risk factors, how they play a role in the mechanism, and so on. In addition, there is a need to determine more biomarkers by which to modify the diagnostic criteria and further improve diagnostic efficiency. Moreover, the efficacy of treatment was evaluated by observational studies; consequently, more clinical trials and even randomized clinical trials are required. It also remains unclear what should be done for those diagnosed with possible CAA-RI, and whether they still need to undergo brain biopsy. If the brain biopsy result is negative, but the patient meets the clinicoradiological diagnostic criteria, the course of action remains uncertain. In addition, CAA is a disease caused by disordered Aβ clearance, and CAA-RI is in fact the body's immune response aimed at clearing Aβ. Thus, it needs to be established whether excessive immune suppression would have an adverse effect on the long-term prognosis of patients. There is currently no long-term follow-up cohort to establish prognosis, and differences in prognoses associated with different therapies for different subtypes are worth investigating. Finally, a multi-center prospective cohort study, using unified standards for the collection of data, application of designed therapies, and follow-up strategy is necessary.
Although CAA-RI is relatively rare at present, it may become more common in future with the improvement of diagnostic techniques. When rapid progressive dementia occurs in people over 40 years of age, accompanied by headache, seizures, or focal neurological deficits, with patchy or confluent T2 or FLAIR hyperintensity and evidence of CMBs or cSS, a diagnosis of CAA-RI should be suspected. Early diagnosis and timely treatment may improve prognosis. Clinicians should have a comprehensive understanding of the disease and order an MRI with multiple sequences, including T2∗ or SWI, in patients with suspected CAA-RI, particularly in those cases whose T2/FLAIR images show hypointense dots. If only routine sequences are performed, it is easy to mistake WMH as the only image manifestation and consequently delay diagnosis and treatment. In addition to clinical symptoms and image findings, detection of genotypes, CSF biomarkers, such as anti-Aβ autoantibodies, and amyloid PET may also provide diagnostic evidence and serve as tools for evaluating treatment efficacy. However, antibody titer determination kits are currently not commercially available and are still worth developing. Amyloid PET is also unavailable in most hospitals in China. It is worth noting that CAA-RI is a diagnosis by exclusion. Thus, other differential diagnoses should be carefully ruled out. Once the diagnosis is made, glucocorticoids or even immunosuppressants should be adopted in order to improve the prognosis.
Funding
This study was supported by a grant from the National Key Research and Development Program of China (No. 2016YFC1300500-505).
Conflicts of interest
None.
Footnotes
How to cite this article: Wu JJ, Yao M, Ni J. Cerebral amyloid angiopathy-related inflammation: current status and future implications. Chin Med J 2021;134:646–654. doi: 10.1097/CM9.0000000000001427
References
- 1.Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke 2015; 17:17–30. doi: 10.5853/jos.2015.17.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salvarani C, Hunder GG, Morris JM, Brown RD, Christianson T, Giannini C. Aβ-related angiitis: comparison with CAA without inflammation and primary CNS vasculitis. Neurology 2013; 81:1596–1603. doi: 10.1212/WNL.0b013e3182a9f545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wermer MJH, Greenberg SM. The growing clinical spectrum of cerebral amyloid angiopathy. Curr Opin Neurol 2018; 31:28–35. doi: 10.1097/WCO.0000000000000510. [DOI] [PubMed] [Google Scholar]
- 4.Yeh SJ, Tang SC, Tsai LK, Jeng JS. Pathogenetical subtypes of recurrent intracerebral hemorrhage: designations by SMASH-U classification system. Stroke 2014; 45:2636–2642. doi: 10.1161/strokeaha.114.005598. [DOI] [PubMed] [Google Scholar]
- 5.Reid AH, Maloney AF. Giant cell arteritis and arteriolitis associated with amyloid angiopathy in an elderly mongol. Acta Neuropathol 1974; 27:131–137. doi: 10.1007/bf00687163. [DOI] [PubMed] [Google Scholar]
- 6.Ronsin S, Deiana G, Geraldo AF, Durand-Dubief F, Thomas-Maisonneuve L, Formaglio M, et al. Pseudotumoral presentation of cerebral amyloid angiopathy-related inflammation. Neurology 2016; 86:912–919. doi: 10.1212/WNL.0000000000002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martucci M, Sarria S, Toledo M, Coscojuela P, Vert C, Siurana S, et al. Cerebral amyloid angiopathy-related inflammation: imaging findings and clinical outcome. Neuroradiology 2014; 56:283–289. doi: 10.1007/s00234-014-1330-6. [DOI] [PubMed] [Google Scholar]
- 8.Kirshner HS, Bradshaw M. The inflammatory form of cerebral amyloid angiopathy or “cerebral amyloid angiopathy-related inflammation” (CAARI). Curr Neurol Neurosci Rep 2015; 15:54.doi: 10.1007/s11910-015-0572-y. [DOI] [PubMed] [Google Scholar]
- 9.Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol 2004; 55:250–256. doi: 10.1002/ana.10810. [DOI] [PubMed] [Google Scholar]
- 10.Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology 2007; 68:1411–1416. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- 11.Chu S, Xu F, Su Y, Chen H, Cheng X. Cerebral amyloid angiopathy (CAA)-related inflammation: comparison of inflammatory CAA and amyloid-beta-related angiitis. J Alzheimers Dis 2016; 51:525–532. doi: 10.3233/JAD-151036. [DOI] [PubMed] [Google Scholar]
- 12.Chung KK, Anderson NE, Hutchinson D, Synek B, Barber PA. Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry 2011; 82:20–26. doi: 10.1136/jnnp.2009.204180. [DOI] [PubMed] [Google Scholar]
- 13.Auriel E, Charidimou A, Gurol ME, Ni J, Van Etten ES, Martinez-Ramirez S, et al. Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol 2016; 73:197–202. doi: 10.1001/jamaneurol.2015.4078. [DOI] [PubMed] [Google Scholar]
- 14.Danve A, Grafe M, Deodhar A. Amyloid beta-related angiitis--a case report and comprehensive review of literature of 94 cases. Semi Arthritis Rheum 2014; 44:86–92. doi: 10.1016/j.semarthrit.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J, et al. Abeta-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 2005; 128:500–515. doi: 10.1093/brain/awh379. [DOI] [PubMed] [Google Scholar]
- 16.Szpak GM, Lewandowska E, Sliwińska A, Stępień T, Tarka S, Mendel T, et al. Inflammatory cerebral amyloid angiopathy: the overlap of perivascular (PAN-like) with vasculitic (Aβ-related angiitis) form: an autopsy case. Folia Neuropathol 2011; 49:335–347. [PubMed] [Google Scholar]
- 17.Corovic A, Kelly S, Markus HS. Cerebral amyloid angiopathy associated with inflammation: a systematic review of clinical and imaging features and outcome. Int J Stroke 2018; 13:257–267. doi: 10.1177/1747493017741569. [DOI] [PubMed] [Google Scholar]
- 18.Castro Caldas A, Silva C, Albuquerque L, Pimentel J, Silva V, Ferro JM. Cerebral amyloid angiopathy associated with inflammation: report of 3 cases and systematic review. J Stroke Cerebrovasc Dis 2015; 24:2039–2048. doi: 10.1016/j.jstrokecerebrovasdis.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Traschütz A, Tzaridis T, Penner AH, Kuchelmeister K, Urbach H, Hattingen E, et al. Reduction of microbleeds by immunosuppression in a patient with Aβ-related vascular inflammation. Neurol Neuroimmunol Neuroinflamm 2015; 2:e165.doi: 10.1212/nxi.0000000000000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bogner S, Bernreuther C, Matschke J, Barrera-Ocampo A, Sepulveda-Falla D, Leypoldt F, et al. Immune activation in amyloid-β-related angiitis correlates with decreased parenchymal amyloid-β plaque load. Neurodegener Dis 2014; 13:38–44. doi: 10.1159/000352020. [DOI] [PubMed] [Google Scholar]
- 21.Child ND, Braksick SA, Flanagan EP, Keegan BM, Giannini C, Kantarci OH. Amyloid-β-related angiitis presenting as a uveomeningeal syndrome. Neurology 2013; 81:1796–1798. doi: 10.1212/01.wnl.0000435560.00234.a7. [DOI] [PubMed] [Google Scholar]
- 22.Moussaddy A, Levy A, Strbian D, Sundararajan S, Berthelet F, Lanthier S. Inflammatory cerebral amyloid angiopathy, amyloid-beta-related angiitis, and primary angiitis of the central nervous system: similarities and differences. Stroke 2015; 46:e210–e213. doi: 10.1161/STROKEAHA.115.010024. [DOI] [PubMed] [Google Scholar]
- 23.Mandal J, Chung SA. Primary angiitis of the central nervous system. Rheum Dis Clin North Am 2017; 43:503–518. doi: 10.1016/j.rdc.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Moosavi B, Torres C, Jansen G. Case 232: amyloid-β-related angiitis. Radiology 2016; 280:643–647. doi: 10.1148/radiol.2016142978. [DOI] [PubMed] [Google Scholar]
- 25.Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, et al. Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 2012; 11:241–249. doi: 10.1016/s1474-4422(12)70015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salloway SP, Sperling R, Fox NC, Sabbagh MN, Honig LS, Porsteinsson AP, et al. Long-term follow up of patients with mild-to-moderate Alzheimer's disease treated with bapineuzumab in a phase III, open-label, extension study. J Alzheimers Dis 2018; 64:689–707. doi: 10.3233/jad-171157. [DOI] [PubMed] [Google Scholar]
- 27.Boncoraglio GB, Piazza F, Savoiardo M, Farina L, DiFrancesco JC, Prioni S, et al. Prodromal Alzheimer's disease presenting as cerebral amyloid angiopathy-related inflammation with spontaneous amyloid-related imaging abnormalities and high cerebrospinal fluid anti-Aβ autoantibodies. J Alzheimers Dis 2015; 45:363–367. doi: 10.3233/jad-142376. [DOI] [PubMed] [Google Scholar]
- 28.DiFrancesco JC, Longoni M, Piazza F. Anti-Abeta autoantibodies in amyloid related imaging abnormalities (ARIA): candidate biomarker for immunotherapy in Alzheimer's disease and cerebral amyloid angiopathy. Front Neurol 2015; 6:207.doi: 10.3389/fneur.2015.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melzer N, Harder A, Gross CC, Wolfer J, Stummer W, Niederstadt T, et al. CD4(+) T cells predominate in cerebrospinal fluid and leptomeningeal and parenchymal infiltrates in cerebral amyloid beta-related angiitis. Arch Neurol 2012; 69:773–777. doi: 10.1001/archneurol.2011.2441. [DOI] [PubMed] [Google Scholar]
- 30.Renard D, Collombier L, Demattei C, Wacongne A, Charif M, Ayrignac X, et al. Cerebrospinal fluid, MRI, and florbetaben-PET in cerebral amyloid angiopathy-related inflammation. J Alzheimers Dis 2018; 61:1107–1117. doi: 10.3233/JAD-170843. [DOI] [PubMed] [Google Scholar]
- 31.Porter M, Newey CR, Toth G. Teaching NeuroImages: treatment-resistant rapidly progressive amyloid β-related angiitis. Neurology 2013; 80:e187–188. doi: 10.1212/WNL.0b013e3182904cd9. [DOI] [PubMed] [Google Scholar]
- 32.Sakai K, Hayashi S, Sanpei K, Yamada M, Takahashi H. Multiple cerebral infarcts with a few vasculitic lesions in the chronic stage of cerebral amyloid angiopathy-related inflammation. Neuropathology 2012; 32:551–556. doi: 10.1111/j.1440-1789.2011.01283.x. [DOI] [PubMed] [Google Scholar]
- 33.Sakai K, Ueda M, Fukushima W, Tamaoka A, Shoji M, Ando Y, et al. Nationwide survey on cerebral amyloid angiopathy in Japan. Eur J Neurol 2019; 26:1487–1493. doi: 10.1111/ene.14031. [DOI] [PubMed] [Google Scholar]
- 34.Salvarani C, Brown RD, Jr, Calamia KT, Christianson TJ, Huston J, 3rd, Meschia JF, et al. Primary central nervous system vasculitis: comparison of patients with and without cerebral amyloid angiopathy. Rheumatology (Oxford) 2008; 47:1671–1677. doi: 10.1093/rheumatology/ken328. [DOI] [PubMed] [Google Scholar]
- 35.Fukasawa R, Shimizu S, Hirose D, Kanetaka H, Umahara T, Obikane H, et al. An individual with cerebral amyloid angiopathy-related inflammation who displayed involuntary movements. J Am Geriatr Soc 2015; 63:2644–2645. doi: 10.1111/jgs.13852. [DOI] [PubMed] [Google Scholar]
- 36.Cenina AR, De Leon J, Tay KY, Wong CF, Kandiah N. Cerebral amyloid angiopathy-related inflammation presenting with rapidly progressive dementia, responsive to IVIg. Alzheimer Dis Assoc Disord 2015; 29:347–349. doi: 10.1097/wad.0000000000000084. [DOI] [PubMed] [Google Scholar]
- 37.Ng DW, Magaki S, Terashima KH, Keener AM, Salamon N, Karnezis S, et al. Amyloid-β-related angiitis: a report of 2 cases with unusual presentations. Hum Pathol 2017; 64:191–197. doi: 10.1016/j.humpath.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Hainline C, Rucker JC, Zagzag D, Golfinos JG, Lui YW, Liechty B, et al. Tumoral presentation of homonymous hemianopia and prosopagnosia in cerebral amyloid angiopathy-related inflammation. J Neuroophthalmol 2017; 37:48–52. doi: 10.1097/wno.0000000000000474. [DOI] [PubMed] [Google Scholar]
- 39.Saliou V, Ben Salem D, Ognard J, Guellec D, Marcorelles P, Rouhart F, et al. A Collet-Sicard syndrome due to internal carotid artery dissection associated with cerebral amyloid angiopathy-related inflammation. SAGE Open Med Case Rep 2018; 6:2050313x18777176.doi: 10.1177/2050313x18777176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gera A, Witek N, Bailey M. Pearls & Oy-sters: CAA-related inflammation presents as subacute cognitive decline in a patient with Parkinson disease. Neurology 2019; 92:1116–1118. doi: 10.1212/wnl.0000000000007610. [DOI] [PubMed] [Google Scholar]
- 41.Sugihara S, Ogawa A, Nakazato Y, Yamaguchi H. Cerebral beta amyloid deposition in patients with malignant neoplasms: its prevalence with aging and effects of radiation therapy on vascular amyloid. Acta Neuropathol 1995; 90:135–141. doi: 10.1007/bf00294312. [DOI] [PubMed] [Google Scholar]
- 42.Piazza F, Greenberg SM, Savoiardo M, Gardinetti M, Chiapparini L, Raicher I, et al. Anti-amyloid beta autoantibodies in cerebral amyloid angiopathy-related inflammation: implications for amyloid-modifying therapies. Ann Neurol 2013; 73:449–458. doi: 10.1002/ana.23857. [DOI] [PubMed] [Google Scholar]
- 43.Raghavan P, Looby S, Bourne TD, Wintermark M. Cerebral amyloid angiopathy-related inflammation: a potentially reversible cause of dementia with characteristic imaging findings. J Neuroradiol 2016; 43:11–17. doi: 10.1016/j.neurad.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 44.Cheng AL, Batool S, McCreary CR, Lauzon ML, Frayne R, Goyal M, et al. Susceptibility-weighted imaging is more reliable than T2∗-weighted gradient-recalled echo MRI for detecting microbleeds. Stroke 2013; 44:2782–2786. doi: 10.1161/strokeaha.113.002267. [DOI] [PubMed] [Google Scholar]
- 45.Shams S, Martola J, Cavallin L, Granberg T, Shams M, Aspelin P, et al. SWI or T2∗: which MRI sequence to use in the detection of cerebral microbleeds? The Karolinska Imaging Dementia Study. AJNR Am J Neuroradiol 2015; 36:1089–1095. doi: 10.3174/ajnr.A4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salvarani C, Morris JM, Giannini C, Brown RD, Jr, Christianson T, Hunder GG. Imaging findings of cerebral amyloid angiopathy, Abeta-related angiitis (ABRA), and cerebral amyloid angiopathy-related inflammation: a single-institution 25-year experience. Medicine (Baltimore) 2016; 95:e3613.doi: 10.1097/MD.0000000000003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Renard D, Tatu L, Collombier L, Wacongne A, Ayrignac X, Charif M, et al. Cerebral amyloid angiopathy and cerebral amyloid angiopathy-related inflammation: comparison of hemorrhagic and DWI MRI features. J Alzheimers Dis 2018; 64:1113–1121. doi: 10.3233/JAD-180269. [DOI] [PubMed] [Google Scholar]
- 48.Liang JW, Zhang W, Sarlin J, Boniece I. Case of cerebral amyloid angiopathy-related inflammation - is the absence of cerebral microbleeds a good prognostic sign? J Stroke Cerebrovasc Dis 2015; 24:e319–322. doi: 10.1016/j.jstrokecerebrovasdis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 49.Nakaya M, Hashimoto H, Usui G, Sawada K, Shirouzu I, Oshima A, et al. Cerebral amyloid-β-related angiitis without cerebral microbleeds in a patient with subarachnoid hemorrhage. Cardiovasc Pathol 2019; 42:36–40. doi: 10.1016/j.carpath.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 50.Aghetti A, Sene D, Polivka M, Shor N, Lechtman S, Chabriat H, et al. Cerebral amyloid angiopathy related inflammation with prominent meningeal involvement. A report of 2 cases. Front Neurol 2019; 10:984.doi: 10.3389/fneur.2019.00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang P, Bucelli RC, Ferguson CJ, Corbo JC, Kim AH, Day GS. Teaching neuro: cerebral amyloid angiopathy-related inflammation presenting with isolated leptomeningitis. Neurology 2017; 89:e66–e67. doi: 10.1212/WNL.0000000000004218. [DOI] [PubMed] [Google Scholar]
- 52.Sengoku R, Matsushima S, Murakami Y, Fukuda T, Tokumaru AM, Hashimoto M, et al. 11C-PiB PET imaging of encephalopathy associated with cerebral amyloid angiopathy. Intern Med 2014; 53:1997–2000. doi: 10.2169/internalmedicine.53.1731. [DOI] [PubMed] [Google Scholar]
- 53.Carmona-Iragui M, Fernández-Arcos A, Alcolea D, Piazza F, Morenas-Rodriguez E, Antón-Aguirre S, et al. Cerebrospinal fluid anti-amyloid-β autoantibodies and amyloid PET in cerebral amyloid angiopathy-related inflammation. J Alzheimers Dis 2016; 50:1–7. doi: 10.3233/jad-150614. [DOI] [PubMed] [Google Scholar]
- 54.Mendonça MD, Caetano A, Pinto M, Cruz e Silva V, Viana-Baptista M. Stroke-like episodes heralding a reversible encephalopathy: microbleeds as the key to the diagnosis of cerebral amyloid angiopathy-related inflammation-a case report and literature review. J Stroke Cerebrovasc Dis 2015; 24:e245–250. doi: 10.1016/j.jstrokecerebrovasdis.2015.04.042. [DOI] [PubMed] [Google Scholar]
- 55.Xu YY, Chen S, Zhao JH, Chen XL, Zhang JW. A case of cerebral amyloid angiopathy-related inflammation with the rare apolipoprotein epsilon2/epsilon2 genotype. Front Neurol 2019; 10:547.doi: 10.3389/fneur.2019.00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ichimata S, Hata Y, Yoshida K, Nishida N. Autopsy of a multiple lobar hemorrhage case with amyloid-β-related angiitis. Neuropathology 2020; 40:280–286. doi: 10.1111/neup.12637. [DOI] [PubMed] [Google Scholar]
- 57.Du Y, Liu C, Ma C, Xu X, Zhou X, Zhou H, et al. Cerebral amyloid angiopathy-related inflammation: a case report presenting with a rare variant in SORL1 gene. BMC Neurol 2019; 19:97.doi: 10.1186/s12883-019-1326-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersen OM, Rudolph IM, Willnow TE. Risk factor SORL1: from genetic association to functional validation in Alzheimer's disease. Acta Neuropathol 2016; 132:653–665. doi: 10.1007/s00401-016-1615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blechingberg J, Poulsen ASA, Kjølby M, Monti G, Allen M, Ivarsen AK, et al. An alternative transcript of the Alzheimer's disease risk gene SORL1 encodes a truncated receptor. Neurobiol Aging 2018; 71:266.e11–266.e24. doi: 10.1016/j.neurobiolaging.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Renard D, Wacongne A, Ayrignac X, Charif M, Fourcade G, Azakri S, et al. Cerebrospinal fluid Alzheimer's disease biomarkers in cerebral amyloid angiopathy-related inflammation. J Alzheimers Dis 2016; 50:759–764. doi: 10.3233/JAD-150621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DiFrancesco JC, Brioschi M, Brighina L, Ruffmann C, Saracchi E, Costantino G, et al. Anti-Aβ autoantibodies in the CSF of a patient with CAA-related inflammation: a case report. Neurology 2011; 76:842–844. doi: 10.1212/WNL.0b013e31820e773c. [DOI] [PubMed] [Google Scholar]
- 62.Nouh A, Borys E, Gierut AK, Biller J. Amyloid-Beta related angiitis of the central nervous system: case report and topic review. Front Neurol 2014; 5:13.doi: 10.3389/fneur.2014.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Masrori P, Montagna M, De Smet E, Loos C. Posterior reversible encephalopathy syndrome caused by cerebral amyloid angiopathy-related inflammation. Acta Neurol Belg 2019; 119:505–507. doi: 10.1007/s13760-019-01172-w. [DOI] [PubMed] [Google Scholar]
- 64.DiFrancesco JC, Touat M, Caulo M, Gallucci M, Garcin B, Levy R, et al. Recurrence of cerebral amyloid angiopathy-related inflammation: a report of two cases from the iCAbeta international network. J Alzheimers Dis 2015; 46:1071–1077. doi: 10.3233/JAD-150070. [DOI] [PubMed] [Google Scholar]
- 65.Rajczewska-Oleszkiewicz C, Cyganek A, Stadnik A, Dziewulska D. Cerebral amyloid angiopathy-related inflammation - a case report presenting diagnostic difficulties. Neurol Neurochir Pol 2018; 52:298–305. doi: 10.1016/j.pjnns.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 66.Kotsenas AL, Morris JM, Wald JT, Parisi JE, Campeau NG. Tumefactive cerebral amyloid angiopathy mimicking CNS neoplasm. AJR Am J Roentgenol 2013; 200:50–56. doi: 10.2214/ajr.12.8500. [DOI] [PubMed] [Google Scholar]
- 67.Leclercq L, Mechtouff L, Hermier M, Cho TH, Nighoghossian N, Ducray F. Intravascular large B-cell lymphoma mimicking cerebral amyloid angiopathy-related inflammation. Rev Neurol (Paris) 2018; 174:265–266. doi: 10.1016/j.neurol.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 68.Sallèles E, Bonneville F, Delisle MB, Rigal E, Raposo N, Pariente J. Acute ischemic lesions in cerebral amyloid angiopathy-related inflammation. Rev Neurol (Paris) 2019; 175:575–577. doi: 10.1016/j.neurol.2019.01.399. [DOI] [PubMed] [Google Scholar]
- 69.Regenhardt RW, Thon JM, Das AS, Thon OR, Charidimou A, Viswanathan A, et al. Association between immunosuppressive treatment and outcomes of cerebral amyloid angiopathy-related inflammation. JAMA Neurol 2020; 77:1–10. doi: 10.1001/jamaneurol.2020.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coulette S, Renard D, Lehmann S, Raposo N, Arquizan C, Charif M, et al. A clinico-radiological study of cerebral amyloid angiopathy-related inflammation. Cerebrovasc Dis 2019; 48:38–44. doi: 10.1159/000502832. [DOI] [PubMed] [Google Scholar]
- 71.Tetsuka S, Hashimoto R. Slightly symptomatic cerebral amyloid angiopathy-related inflammation with spontaneous remission in four months. Case Rep Neurol Med 2019; 2019:5308208.doi: 10.1155/2019/5308208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nelson T, Leung B, Bannykh S, Shah KS, Patel J, Dumitrascu OM. Cerebral amyloid angiopathy-related inflammation in the immunosuppressed: a case report. Front Neurol 2019; 10:1283.doi: 10.3389/fneur.2019.01283. [DOI] [PMC free article] [PubMed] [Google Scholar]