

Abstract
Objective:
Parenchymal arterioles (PAs) regulate perfusion of the cerebral microcirculation, and impaired PA endothelium-dependent dilation occurs in dementia models mimicking chronic cerebral hypoperfusion (CCH). Epoxyeicosatrienoic acids (EETs) are vasodilators; their actions are potentiated by soluble epoxide hydrolase (sEH) inhibition. We hypothesized that chronic sEH inhibition with trifluoromethoxyphenyl-3 (1-propionylpiperidin-4-yl) urea (TPPU) would prevent cognitive dysfunction and improve PA dilation in a hypertensive CCH model.
Methods
Bilateral carotid artery stenosis (BCAS) was used to induce CCH in twenty-week-old male stroke-prone spontaneously hypertensive rats (SHSRP), that were treated with vehicle or TPPU for 8 weeks. Cognitive function was assessed by novel object recognition. PA dilation and structure were assessed by pressure myography and mRNA expression in brain tissue was assessed by qRT-PCR.
Results:
TPPU did not enhance resting cerebral perfusion, but prevented CCH induced memory deficits. TPPU improved PA endothelium-dependent dilation but reduced the sensitivity of PAs to a nitric oxide donor. TPPU treatment had no effect on PA structure or biomechanical properties. TPPU treatment increased brain mRNA expression of brain derived neurotrophic factor, doublecortin, tumor necrosis factor-alpha, sEH, and superoxide dismutase 3,
Conclusions:
These data suggest that sEH inhibitors may be viable treatments for cognitive impairments associated with hypertension and CCH.
Introduction
Vascular cognitive impairment and dementia (VCID), the second most common cause of dementia after Alzheimer’s disease, is a spectrum of cognitive deficits with a cerebrovascular origin.1 In 2016, the World Health Organization estimated that 47.5 million people were living with dementia; this number is projected to rise to 135.5 million by 2050. While there are drugs to alleviate the symptoms of VCID, there are no reliable therapeutic options to halt its progression.2 VCID is often associated with comorbidities such as hypertension and carotid artery stenosis; this has hampered drug discovery efforts because there are no available animal models that reflect the human population at risk. Chronic cerebral hypoperfusion (CCH) is an important pathology associated with VCID 2-4, and vascular injury exacerbates other forms of dementia including Alzheimer’s disease.5 Thus, therapies aimed at improving cerebrovascular health and enhancing blood flow may have potential therapeutic effects for VCID and other mixed dementias.
Soluble epoxide hydrolase (sEH) metabolizes epoxyeicosatrienoic acids (EETs) which are arachidonic acid metabolites that are produced by several cell types including endothelial cells and astrocytes. EETs are endothelium-derived hyperpolarizing factors (EDHF) that induce vasodilation and increase cerebral perfusion.6,7 Therefore, it stands to reason that sEH inhibition has the potential to enhance blood flow to the cerebral microcirculation in VCID models and reduce cognitive deficits. Several pieces of evidence support this hypothesis.8-10 For example, reducing EET concentrations reduces blood flow in the cerebral microcirculation11 and sEH overexpression in cerebral endothelial cells impairs endothelium-dependent dilation.12 It is possible that reduced cerebral perfusion mediated by enhanced EET metabolism could drive VCID. Indeed, VCID patients have enhanced sEH activity in their brains, which indicates a role for sEH in the progression of the disease.10 The increased expression of sEH was found largely in arterioles near microinfarcts, raising the possibility that detrimental effects of sEH in the cerebral microvasculature could aid in the progression of VCID.10 The link between sEH activity and dementia has been strengthened by studies showing a correlation between the levels of sEH derived oxylipins, white matter hyperintensities, and VCID.13 Similarly, mouse studies using a cocktail of drugs to prevent the production of several vasodilators including EETs, showed that impaired vasodilation contributes to cognitive decline.14
Cerebral parenchymal arterioles (PAs) contribute to the regulation of blood flow in response to neuronal activity; a process that is essential for cognitive function.15-17 The PAs role in regulating microvascular perfusion is so important that the occlusion of a single PA induces cognitive impairment.18 Our lab established that endothelium-dependent dilation in the PAs is largely mediated by endothelium-derived hyperpolarization (EDH) or EDHF19-21 and that PA dilation is impaired in models of VCID induced by bilateral common carotid artery stenosis (BCAS).19,20 In the cerebral vasculature, sEH is expressed predominately in arterioles.10 It is possible that sEH inhibition will elevate the concentration of EETs and enhance EDHF-mediated dilation in PAs. Previous studies suggest TPPU alleviates the BCAS mediated cognitive dysfunction in normotensive mice.22 These studies focused on the potential neuronal effects of TPPU while the possible beneficial effects on vascular function of TPPU were not considered. To address appropriate co-morbidities (hypertension and carotid artery stenosis), we hypothesized that sEH inhibition in stroke prone spontaneously hypertensive rats (SHRSP), a model of human essential hypertension, with BCAS would prevent CCH-induced dilatory impairment in PAs and alleviate cognitive deficits.
Methods
Animals and surgery.
20-week-old male SHRSP from the colony housed at Michigan State University were used. BCAS surgeries were carried out as previously described.19 Briefly, rats were anesthetized with isoflurane, placed in a supine position, and both common carotid arteries were exposed. A 27-gauge blunt needle was placed alongside the left carotid artery and two 6-0 silk sutures were used to tie the carotid artery and needle firmly together. The needle was then carefully removed to induce partial occlusion, or stenosis, of the artery. The procedure was repeated on the right common carotid artery. A group of sham rats were prepared to confirm the effects of BCAS on cognitive function in SHRSP. Sham surgeries were carried out in an identical manner, but the ties were not placed on the common carotid arteries. The experimental protocol was approved by the Michigan State University Institutional Animal Care & Use Committee and was in accordance with the National Research Council's Guide for the Care and Use of Laboratory Animals (2011).
TPPU treatment.
After BCAS or sham surgeries, rats were randomized into two groups: one group received TPPU (3mg/kg/day) while the other received vehicle (1% PEG400) in their drinking water until euthanasia 8 weeks later. For the BCAS rats, 12 rats received vehicle and 11 received TPPU. For the sham surgeries, 9 rats were vehicle treated and 12 were TPPU treated.
Blood pressure measurement.
Blood pressure was measured in a subset of rats by tail-cuff plethysmography using a RTBP1001 tail-cuff blood pressure system (Kent Scientific, Torrington CT) as previously described.23
Novel object recognition test.
Novel object recognition test was carried out in an open box with opaque walls. Rats were acclimatized to the box for 15 minutes/day for three days. During the familiarization phase, the rats were allowed to explore two identical objects for 10 minutes or until the animal had explored either object for 30 seconds total. Memory function was evaluated either 90 minutes or 24 hours later by replacing one of the familiar objects with a novel object. The color, size, and texture of the familiar objects and the novel objects were similar to prevent bias. Total exploration time and the time spent exploring the novel object was recorded. Exploration took place when the rat pawed at, sniffed, or whisked with its snout directed at the object from a distance of under ~1 cm. Between each rat and each trial, 70% alcohol was used to clean the objects and the box to remove any olfactory cues. Novel exploration quotient was defined as a ratio of the time spent exploring the novel object to the total exploration time.24
Cerebral perfusion.
8 weeks after BCAS surgery, cerebral perfusion was measured with a scanning laser Doppler (PeriScan PIM 3, Perimed, Stockholm, Sweden). Rats were anesthetized and placed on a heated platform. The scanning laser Doppler was positioned approximately 18 cm above the exposed and cleaned skull, and cerebral perfusion was recorded. A total of 4 consecutive scans were performed. Mean perfusion was analyzed using the LDPIwin 3.1 software (Perimed) and expressed as mean perfusion units.20
PA isolation and cannulation.
After 8 weeks of vehicle/TPPU treatment, rats were anesthetized using isoflurane and thoracotomized. After exsanguination the brain was removed and placed in ice-cold Ca2+-free physiological saline solution (PSS, in mM: NaCl 140, KCl 5, MgCl2•7H2O 1, HEPES 10, Dextrose 10) for PA dissection. To isolate PAs, a section of brain containing the middle cerebral artery (MCA) was removed and PAs branching from the MCA were carefully dissected and transferred to a cannulation chamber. PAs were cannulated between two glass micropipettes (<40 μm outer diameter) mounted on small, 3-axis micromanipulators (MT-XYZ, Newport, Irvine, CA).25 The perfusion chamber was positioned on the stage of an inverted microscope (Leica DMIL, Wetzlar, Germany) with a 20x objective (Leitz Wetzlar objective, numerical aperture: 0.3). PAs were bathed in warm (37°C) PSS containing 1.8mM Ca2+ and pressurized at 60 mmHg until myogenic tone developed. To assess endothelium-dependent dilation, PAs were incubated with increasing concentrations of carbachol (0.1nM to 10μM) in the bath. Endothelium-independent dilation was studied by incubating PAs with the nitric oxide (NO) donor, sodium nitroprusside (SNP: 0.1nM to 10μM). Additional concentration response curves were generated to nifedipine (0.1nM to 10μM). Only one concentration-response experiment was performed on each cannulated PA, although multiple PAs can be used from each rat. PA outer diameter was continually tracked and recorded using MyoView 2.0 software (Danish Myo Technology, Aarhus, Denmark). Myogenic tone was calculated as and % dilation was defined as .
Assessment of structural and mechanical properties in PAs.
After the end of the concentration response experiments, the PAs were bathed in Ca2+-free PSS containing EGTA (2mM) and sodium nitropruside (100μM) to assess changes in passive structure. A CCD camera (Hitachi Kokusai Electric Inc., Japan) connected to a video dimension analyzer (Living Systems Instrumentation, Burlington, VT) was used to assess structural changes. Intraluminal pressure was increased from 3 to 180 mmHg in 20 mmHg increments and lumen diameter and wall thickness was measured after 5 minutes at each pressure step. Outer diameter was calculated as: . The cross-sectional area, wall-to-lumen ratio, circumferential wall stress, and passive distensibility were calculated as described previously.26
High-performance liquid chromatography coupled with tandem mass spectrometry for TPPU measurement.
Plasma levels of TPPU were assessed as described previously.27 Briefly, plasma samples were liquid-liquid extracted with 200 μL of ethyl acetate and the residues were resuspended in 50 μL of internal standard solution 12-(3-cyclohexyl-ureido)-dodecanoic acid (200 nM CUDA) for LC-MS/MS analysis to quantify relative amounts of TPPU. Samples were analyzed using an Agilent 1200 SL Series HPLC with a 2.1x150mm Eclipse plus 1.8μm particle size C18 column (Agilent, Santa Clara, CA). The liquid chromatography system was coupled with an AB Sciex 4000 QTRAP hybrid, triple-quadrupole mass spectrometer (Redwood City, CA). The solvent system consisted of water/acetic acid (999/1 v/v, solvent A) and acetonitrile/acetic acid (999/1 v/v; solvent B).
Real-time polymerase chain reaction.
Total mRNA was isolated from the brain tissue surrounding the PAs and associated with the MCAs using the Qiagen RNeasy lipid tissue kit (Qiagen Sciences). RNA concentrations and integrity were evaluated using a NanoDrop spectrophotometer. Identical amounts of RNA were reverse transcribed using a qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithesburg, MD). Real-time PCR was performed using a 96-well plate containing the cDNA and TaqMan primers and probes (Applied Biosystem, Foster City, CA) for doublecortin, synaptophysin, brain derived neurotrophic factor (BDNF), sEH, arginase 1, tumor necrosis factor-alpha (TNF-α), and superoxide dismutase 3 (SOD3). Fold changes in mRNA expression compared to the vehicle group were calculated using the 2−ΔΔCT method and β-2-microglobulin was used as an endogenous control.28 The CT (cycle threshold) was defined as the number of cycles needed for the fluorescence signal to exceed background and begin increasing exponentially. We confirmed that the expression of β-2-microglobulin was not different between the groups by comparing raw CT values (23.13 ± 0.45 vs 22.97 ± 0.10, vehicle vs TPPU p=0.607).
Statistical analyses.
Novel object recognition test, myogenic tone, and real-time PCR data were analyzed by Student’s t-test. Cerebrovascular reactivity, endothelium-dependent dilation, and passive structural properties were analyzed by two-way ANOVA followed by Sidak correction for multiple comparisons, or a non-parametric alternative. Analyses were carried out using the software GraphPad Prism 6.0 (La Jolla, CA, USA). All data is presented as mean ± SEM.
Chemicals and reagents.
TPPU was a gift from Dr. Bruce Hammock at the Department of Entomology, University of California (Davis, CA). All other chemicals and reagents were purchased from Sigma-Aldrich (Saint Louis, MO, USA).
Results
Plasma TPPU levels.
To confirm that cells in the cerebrovasculature were exposed to TPPU, we measured the concentration of TPPU in the plasma. The plasma level of TPPU after chronic administration for 8 weeks was 1276 ± 46 ng/ml in BCAS rats. Rats were weighed prior to euthanasia and TPPU administration did not affect body weight (vehicle vs TPPU: 361± 6 vs 377 ± 7 g).
Chronic TPPU administration prevented memory deficits in BCAS rats.
To determine if TPPU improves cognitive function, we used the novel object recognition test. We confirmed our own previous finding 20 that BCAS impairs memory function in SHRSP (novel exploration quotient for BCAS+vehicle vs Sham+vehicle, 90 minutes retention time: 0.48±0.06 vs 0.65±0.02 respectively; and 24 hours retention time: 0.46±0.08 vs 0.70±0.06 p<0.05). TPPU treatment improved cognitive function in the BCAS rats as evidence by an increased novel exploration quotient for retention times of 90 minutes and 24 hours (Fig 1A) compared to vehicle treated BCAS rats. The improvement in cognitive function was not linked to increased resting pial artery perfusion (Fig 1B) or a reduction in systolic blood pressure in the TPPU treated rats (Fig 1C).
Figure 1.
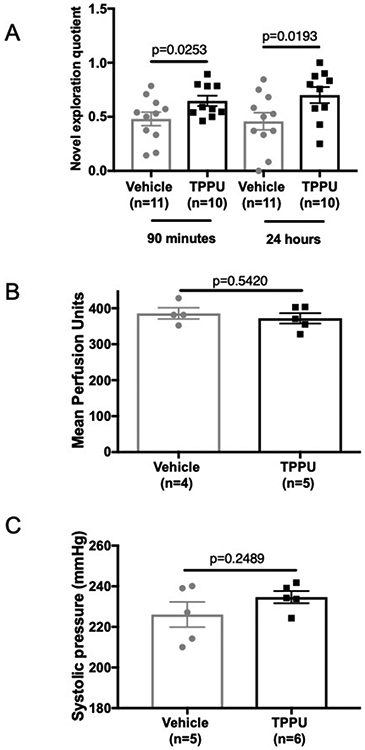
Chronic TPPU administration improves cognitive function in SHRSP with BCAS without changing cerebral perfusion or systolic blood pressure. Cognitive function, assessed by novel object testing, was improved in TPPU treated rats at both a 90 minute and 24-hour retention time (A). Perfusion was assessed by scanning laser Doppler (B) for TPPU treated) and blood pressure was measured by tail-cuff (C). Data was compared using a Student’s t-test, p values and the number of replicates are indicated on each graph.
TPPU improved dilation in PAs from BCAS rats.
TPPU inhibits sEH and should result in improved endothelium-dependent vasodilation. We utilized pressure myography to measure changes in dilation caused by TPPU treatment. Chronic TPPU treatment has no effect on myogenic tone generation in PAs (Figure 2A). As we have previously established, 8 weeks of BCAS impairs dilation in the PAs (% dilation at 10−5M of carbachol, Sham treated with vehicle compared to BCAS treated with vehicle: 23.56±3.69 vs 3.64±2.36, p<0.05) 20. Carbachol mediated dilation was improved in PAs from TPPU treated BCAS rats compared to PAs from vehicle treated BCAS rats, which had negligible dilation (Figure 2B). While maximal dilation to SNP remained unchanged, the EC50 for the NO donor was increased in PAs from TPPU treated BCAS rats (Figure 2C). The dilatory response to nifedipine, a L-type calcium channel antagonist, was unchanged between the groups (Figure 2D).
Figure 2.
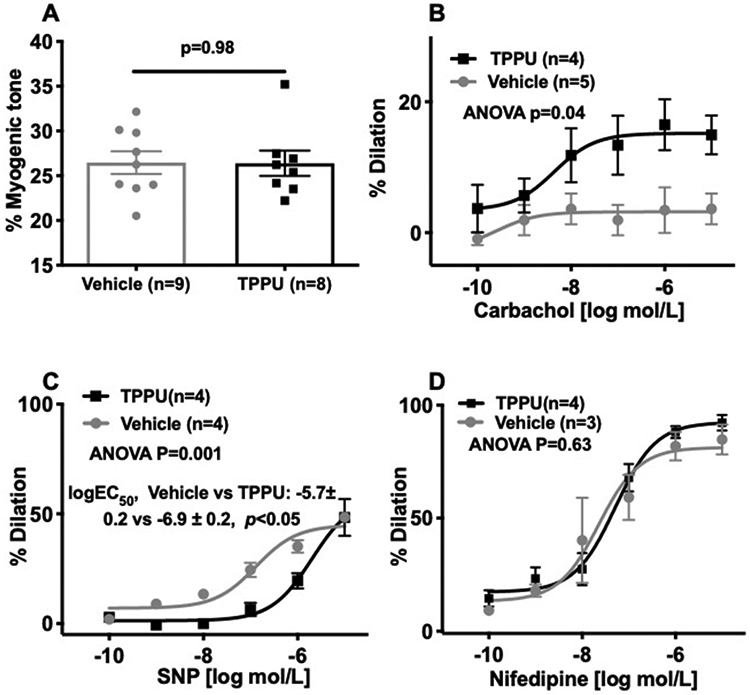
Chronic TPPU treatment improved endothelium-dependent dilation in PAs without changing myogenic tone. TPPU treatment had no effect on myogenic tone (A) but improved carbachol-mediated dilation (B). TPPU treatment reduced the sensitivity of the PAs to SNP (C) without affecting the dilation induced by calcium channel blockade (D). Concentration response curves were compares by two-way ANOVA, p values and the number of replicates are indicated on individual graphs. Myogenic tone generation was compared by a Student’s t-test, the p value and the number of replicates is indicated on the graph.
TPPU treatment had no effect on PA passive structure.
PA structure was assessed by pressure myograhy under Ca2+ free and zero flow conditions. Full pressure response curves were constructed but for clarity only the data at the physiological pressure of 60 mmHg is being shown. Chronic TPPU treatment had no effect on the lumen diameter (Fig 3A), wall thickness (Fig 3B), vessel area (Fig 3C), or the wall-to-lumen ratio (Fig 3D). TPPU treatment also had no effect on biomechanical properties of the PAs (data not shown).
Figure 3.
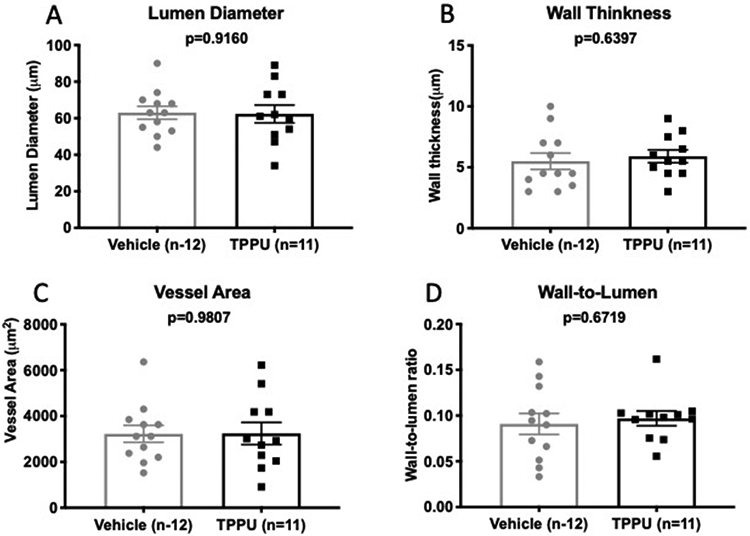
Chronic TPPU treatment after BCAS had no effect on PA structure. TPPU had no effect on the PA lumen diameter (A), wall thickness (B), vessel area (C) or wall-to-lumen ratio (D). Data was compared using an unpaired Student’s t-test, P values and replicate numbers are shown on each graph.
Chronic administration of TPPU altered mRNA expression of neural plasticity and inflammatory markers in brains from BCAS rats.
Since TPPU increases EETs and improves carbachol-mediated dilation, it stands to reason that TPPU may increase perfusion and protect cortical neurons. To test this hypothesis, we analyzed the mRNA expression of markers for neuroplasticity. The mRNA expression of doublecortin, a marker of new and immature neurons, was increased in the TPPU treated rats (Fig 4A) but the expression of synaptophysin, a synapse protein, remained unchanged (Fig 4B). Preliminary studies suggest that the mRNA expression of BDNF is increased in the TPPU treated rat (Fig 4C).
Figure 4.
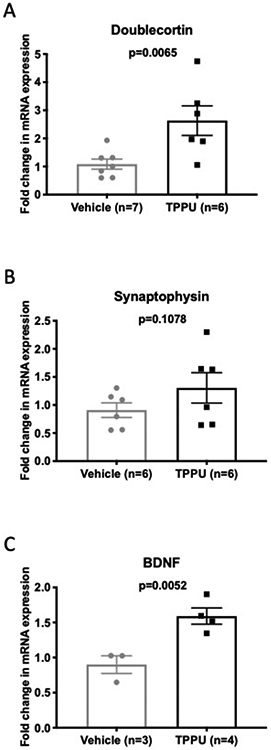
TPPU treatment increased the mRNA expression of markers of neurogenesis and neuronal support. mRNA expression was assessed in the brain by qRT-PCR using Taqman primers. The expression of doublecortin mRNA was increased (A) while the expression of synaptophysin mRNA was unchanged (B). The expression of BDNF mRNA was increased by TPPU treatment (C). Data was compared using a Mann-Whitney nonparametric t-test, p values and replicate numbers are shown on each graph.
There was increased sEH mRNA expression in the TPPU treated BCAS rats (Figure 5A). Recent studies have shown that sEH inhibition increases the number of M2 microglia in the brain after cerebral ischemia.29 To assess if this reduction in inflammation also occurs in BCAS rats with TPPU treatment we measured the mRNA expression of arginase 1, a marker of M2 microglia, and TNF-α, a proinflammatory cytokine. The expression of arginase 1 was unchanged in the TPPU treated rats (Fig 5B), but the expression of the inflammatory marker TNF-α was increased in the TPPU treated rats (Fig 5C). TPPU has also been shown to increase superoxide dismutase (SOD) gene expression in a neurodegenerative disease model.30 We found that this was also the case in the current study, with TPPU treatment increasing the mRNA expression of superoxide dismutase 3 in the BCAS rats (SOD3; Figure 5D).
Figure 5.
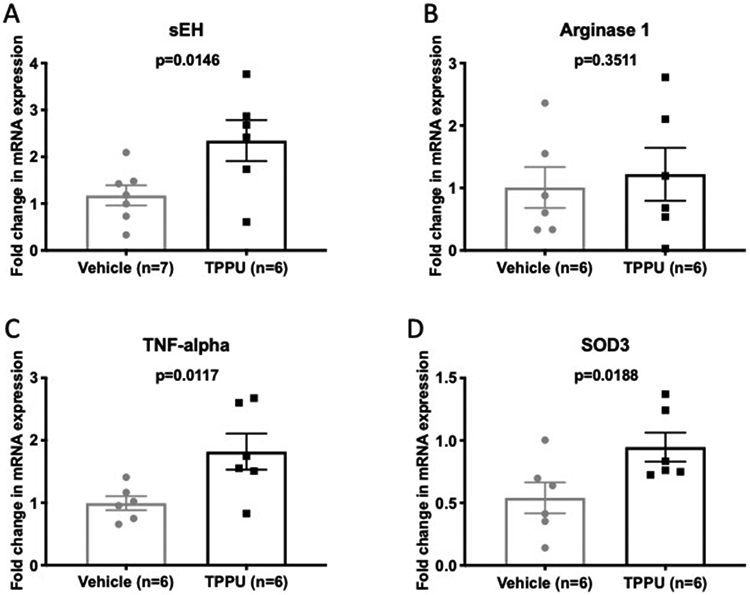
TPPU increased the mRNA expression in the brain of sEH, TNF-alpha and SOD3 without changing the expression of arginase 1. mRNA expression was assessed in the brain by qRT-PCR using Taqman primers. The expression of the mRNA for sEH was increased in TPPU treated rats (A), while the mRNA for arginase 1 was unchanged (B). The mRNA expression of TNF-alpha and SOD3 were both increased in the brains of TPPU treated rats compared to vehicle (C and D). Data was compared using an unpaired Student’s t-test, P values and replicate numbers are shown on each graph.
Chronic TPPU treatment had no effects in sham operated rats.
Chronic treatment with TPPU in Sham rats did not change the novel exploration quotient for retention times of 90 minutes and 24 hours (Fig 6A). Myogenic tone in the PAs remained unchanged between vehicle and TPPU treated Sham rats (Figure 6B). Dilation to carbachol was unchanged in the PAs from the sham rats treated with TPPU or vehicle (Figure 6C).
Figure 6.
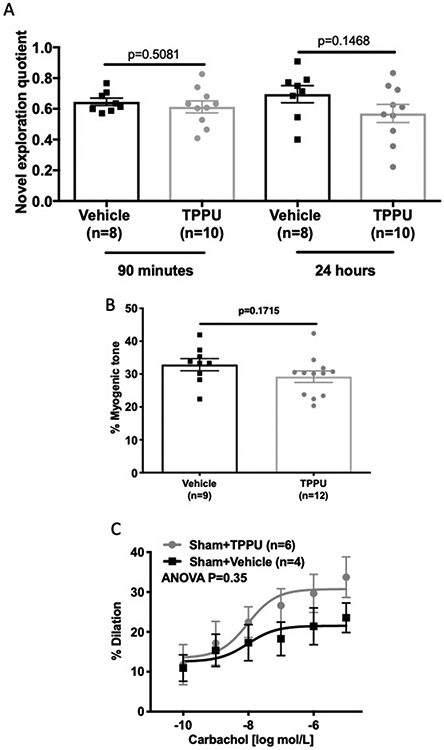
TPPU had no effect on memory function, myogenic tone, or vasodilation in sham operated rats. TPPU treatment did not improve cognitive function in sham operated SHRSP (A). TPPU treatment also had no effect on myogenic tone generation in sham rats (B) or on carbachol-mediated dilation (C). Where appropriate, data was compared using an unpaired Student’s t-test, P values are shown on each graph. The concentration response curve data was compared by two-way ANOVA and P values are shown in the graph. The number of replicates used is shown on each graph.
Discussion
sEH metabolizes EETs which are EDHFs that induce vasodilation and increase cerebral perfusion. It stands to reason that inhibition of sEH prevents EET metabolism and will enhance blood flow to the cerebral microcirculation, thereby reducing cognitive deficits in VCID models. We demonstrated, for the first time, that chronic sEH inhibition with TPPU prevents memory deficits after BCAS in a genetic model of hypertension that closely mimics the human population with essential hypertension. The improved cognitive function was associated with improved endothelial function in PAs and increased doublecortin and BDNF mRNA expression in brain tissue. These effects of TPPU treatment occurred independently of changes in blood pressure. These data suggest that sEH inhibition may be a viable treatment option to slow or stop the development of vascular dementia in hypertensive patients with significant carotid artery stenosis.
We measured plasma TPPU levels in BCAS rats. We found that the plasma concentration of TPPU was approximately 3.5μM, which is considerably higher than the reported IC50 value for TPPU (5nM).31 It should be recognized that the dose of TPPU used in the current study was higher than previously reported studies. Rats in this study received 3mg/kg/day, which equates to 25mg/L. The lower dose of 5mg/L produced significant accumulation of TPPU in the brain over 8 days, 31 therefore, it would seem that the higher dose used here would produce significant inhibition of sEH activity. Interestingly, sEH mRNA levels were increased in brain samples. Thus, it is possible that chronic sEH inhibition leads to a compensatory increase in sEH mRNA expression.
In two previous studies, we established that PA endothelium dependent dilation is impaired by CCH induced by BCAS, and that the BCAS rats have significant cognitive decline when compared to sham operated rats.19,20 One of these studies was conducted using SHRSP; the same experimental protocol as was utilized in the current study.20 The results presented here suggest that the impaired dilation post-BCAS is, at least in part, responsible for the memory deficits observed. TPPU treatment improved both endothelial and cognitive function. In vivo, EETs enhance blood flow during functional hyperemia by improving neurovascular coupling.32,33 Although we did not measure neurovascular coupling directly, TPPU treated rats displayed improved memory function after BCAS. In our ex vivo experiments, TPPU improved endothelium-dependent carbachol mediated dilation. This suggests that increased EET levels for the duration of the CCH improved endothelial function. Since EETs act upstream of various signaling cascades to induce smooth muscle cell hyperpolarization and relaxation7, dilation of PAs from TPPU treated SHRSP can occur by multiple mechanisms. At present we cannot tell if the improved dilation observed by pressure myography is due to EET stabilization, sEH inhibition, or to a more generalized improvement in endothelial health; as all situations are possible 34. Other studies utilizing treatments that improved endothelium dependent dilation and neurovascular coupling have also been shown to improve cognitive function.57 It should be noted that the improved dilation was observed in isolated arterioles in the absence of astrocytes. This suggests that enhanced EETs production from astrocytes is not the sole mechanism responsible for the improvement in vasodilation. We recognize that the findings in this study are at odds with our own previous study where we failed to show a beneficial effect of sEH inhibition with another drug, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA), on endothelium-dependent dilation in middle cerebral arteries (MCA) from SHRSP. The rats used in that study were treated with AUDA from 6-12 weeks of age and while these rats were hypertensive but they did not undergo the stenosis surgery used here. Additionally, there was little opportunity to observe an improvement in enothelium-dependent dilation with sEH inhibition because dilation in the MCA was not significantly impaired in the vehicle treated rats.35 In the current study, we analyzed the PAs instead of the MCAs, the rats we studies were significantly older, and they were exposed to the damaging effects of BCAS.
Interestingly, the PAs from TPPU treated BCAS rats were less sensitive to the NO donor, SNP, than vehicle treated BCAS rats, although the maximum dilation achieved was unchanged. EETs enhance endothelial nitric oxide synthase (eNOS) expression.36,37 Thus, it is conceivable NO production was improved in the TPPU treated BCAS rats and that this could have reduced responsiveness of soluble guanylyl cyclase in vitro38, making the vessels less sensitive to exogenous NO ex vivo.
There was no difference in myogenic tone generation in PAs from BCAS rats treated with TPPU and vehicle. Myogenic tone generation in PAs is dependent on Ca2+ influx through L-type voltage-gated Ca2+ channels.39 We assessed the function of these channels with the antagonist, nifedipine, and did not find a difference between the groups. The lack of a difference in tone generation suggests that the improvement in dilation is not merely an effect of a less constricted arteriole.
Despite observing a marked improvement in PA endothelium-dependent dilation we did not observe increased resting cerebral perfusion in the TPPU treated rats. Given that our own previous studies could not detect effects of sEH inhibition on the MCA, a pial artery35, it is perhaps not surprising that we did not detect a change in pial perfusion with TPPU. It should also be noted that the blood flow in BCAS rats improves over time following surgery. Our own studies with SHSRP rats show that the BCAS surgery causes an immediate 40% reduction in cerebral blood flow, but blood flow is not different from sham operated rats when it is assessed 8 weeks after surgery.20 This finding has been confirmed in several other models of carotid artery stenosis and occlusion.40-42 Future studies, that are out of the scope of the current study, will assess blood flow to specific brain regions including the hippocampus using arterial spin labeling MRI.
There was no change in passive structure in PAs from BCAS or sham rats with chronic TPPU treatment. The effects of sEH inhibition on cerebral artery structure are somewhat controversial. We have previously shown that sEH inhibition in SHRSP from 6-12 weeks of age has no effect on the structure of the MCA as assessed by pressure myography.35 However, using histological techniques, Simpkins et al showed that 6 weeks of sEH inhibition with AUDA reduced the wall thickness but had no effect on lumen diameter in MCAs from 12-week-old SHRSP.45 While reduced wall thickness in MCAs is associated with vascular protection against cerebral ischemia, such hypotrophic remodeling in PAs from BCAS rats could have detrimental effects. BCAS alone induces hypotrophy in PAs evidenced by a reduction in the wall thickness and wall-to-lumen ratio.20 This hypotrophy was associated with increased wall stress in PAs from BCAS rats and any further reduction in wall thickness would exacerbate this. Given the anatomy of the PAs it is unlikely that additional reductions in wall thickness are anatomically possible.
While our study focused primarily on the effects of TPPU on PAs, it is possible that enhanced neuronal plasticity played a role in the preservation of memory function. Endothelial dysfunction can lead to loss of endothelium-derived trophic signaling essential for survival and growth of neurons and oligodendrocytes.46,47 Therefore, it is possible that improved endothelial function in PAs from TPPU treated BCAS rats enabled a supportive environment for neuronal survival. Furthermore, increased mRNA levels of neurogenesis marker, doublecortin, in TPPU treated BCAS rats suggests that TPPU could enhance the formation or survival of new neurons to compensate for CCH induced neuronal death. Similarly, preliminary studies suggest that BDNF levels are increased by TPPU treatment; BDNF is critical for memory acquisition and long-term potentiation in vivo. Other studies have suggested that sEH inhibitors can support neuronal growth and survival. TPPU can prevent cell injury by modulating the BDNF-TrkB pathway in PC12 cells.48 TPPU also potentiates nerve growth factor induced neurite outgrowth in vitro49 and increases the levels of BDNF.49-52 Thus, TPPU could prevent memory impairment in BCAS rats by partially preventing PA dilatory function and enhancing neuronal growth and survival. BDNF expression is also increased in sEH−/− mice after the induction of focal cerebral ischemia and the authors also showed that 14,15 EET led to increased BDNF production from cultured astrocytes.53
It is also possible that the improved memory function and the increase in BDNF are a function of improved astrocyte health with TPPU treatment. Astrocyte mediated EETs production has been linked to neurovascular coupling54 with astrocytes also producing trophic factors and neurotransmitters and supporting and strengthening neuronalconnections.55 Recent studies have shown that increases in the number of senescent astrocytes in the brain is associated with premature cognitive decline.56 Interestingly, pharmacological inhibition vasodilator production by astrocytes impaired neurovascular coupling and produced cognitive decline in mice14. While the design of the current study did not allow for an assessment of astrocyte function, future studies will address the role that astrocytes may have played in improving cognitive function in the TPPU treated rats.
EETs are recognized to have significant anti-inflammatory properties.58 Recent studies in stroke models have shown that sEH inhibition reduces the number of pro-inflammatory M1 type macrophages in the brain. This was associated with a small but insignificant increase in the expression of M2 macrophage markers.59 In the current study, we assessed the expression of arginase 1, an M2 macrophage marker, and found no difference in arginase 1 mRNA expression between the control and the TPPU treated rats. Thus, it is possible that in the current model TPPU did not impact macrophage polarity. It should be noted that in the current study we did not attempt to separate marker expression from macrophages and microglia. We also found an unexpected increase in TNF-α mRNA levels with TPPU treatment. This finding is difficult to reconcile with the many studies which show that EETs and sEH inhibitors reduce inflammation.29,60 Additional studies using ELISA or western blotting are needed to confirm this paradoxical effect. Interestingly, the mRNA expression of SOD3, the extracellular form of SOD, was increased by TPPU treatment. Recent studies have shown that chronic cerebral hypoperfusion reduces SOD activity and that treatments which improve SOD activity also improve cognitive function.61 Interestingly, SOD2 expression appears to be regulated by EETs, with increased expression occurring with EETs treatment62, although, it is not clear if SOD3 is regulated in the same way. Although we only measured SOD3 mRNA expression, it is possible that this translates to increased activity and improved cognitive function.
TPPU did not improve memory or vascular function in Sham SHRSP despite the fact that SHRSP are a model of malignant hypertension63 and accelerated brain aging.64 Comparing the novel object recognition scores for the sham SHRSP with our previously published data in normotensive WKY rats, it does not appear that the 28-week-old SHRSP had significant non-spatial memory impairments19, thus, an improvement with treatment is unlikely. However other recent studies have shown that hypertension can exacerbate the cerebrovascular injury that occurs in atherosclerotic mice.65 Interestingly, other studies conducted in mice suggest that sEH inhibition may be an effective treatment for AD. These studies utilized an sEH knockout mouse combined with a genetic model of AD. sEH knockout not only delayed the onset of AD and almyoid β deposition, it also increased astrogliosis and increased the expression of anti-inflammatory cytokines in astrocytes. 66
Our study has a few limitations and caveats that need to be mentioned. First, our behavioral assessments were limited to novel object recognition test. More cognitive tests could have helped thoroughly characterize the cognitive profile of BCAS rats with chronic TPPU treatment. Second, we focused on vascular changes in the PAs and did not measure neurovascular coupling that is associated with cognitive function.15 We used scanning laser Doppler that has a penetration depth of 0.5 - 1 mm, on a closed skull preparation to assess perfusion. Thus, we were only able to measure perfusion in the pial circulation and not the intraparenchymal circulation. It should be noted that the rats were under isoflurane anesthesia during the analysis of perfusion. Isoflurane is a recognized cerebral artery dilator 43,44, and therefore potential differences in resting perfusion may have been masked. Finally, we only studied male mice, thus, we cannot comment on possible sex differences in the role played by sEH in the development of cognitive decline. It is also important to recognize that the SHRSP used in this study were not aged, However, SHRSP have a considerably shorter life span in comparision to normotensive rats with male rats from the colony housed at MSU having a typical lifespan between 12 – 14 months. Thus, the rats used in this study were effectively middle-aged when euthanized.
In summary, our data demonstrate that impaired memory function in SHRSP with BCAS is associated with impaired PA endothelium-dependent dilation. We also show that inhibiting sEH activity improves the dilatory function in the PAs. Even a small increase in dilatory capacity can substantially enhance blood flow to downstream capillaries during functional hyperemia given that flow is proportional to the fourth power of the radius (as per Poiseuille equation of fluid dynamics). Although our studies link vasodilation to cognitive function, it is also possible that inhibiting sEH may protect against memory deficits by promoting neurogenesis and neuronal survival.
Acknowledgements
This work was supported by National Institutes of Health Grants R35 ES030443-01 (HBD), P42 ES04699 (HBD) R01-HL-137694-01 (AMD and WKJ) and PO1-HL-070687(AMD and WKJ) and an American Heart Association Pre-doctoral fellowship 14PRE19890001(NM).
Bibliography
- 1.O’Brien JT et al. Vascular cognitive impairment. Lancet Neurol. 2, 89–98 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Hachinski V et al. National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 37, 2220–2241 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Moorhouse P & Rockwood K Vascular cognitive impairment: current concepts and clinical developments. Lancet Neurol. 7, 246–255 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Snyder HM et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement. 11, 710–717 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santisteban MM & Iadecola C Hypertension, dietary salt and cognitive impairment. J. Cereb. Blood Flow Metab 38, 2112–2128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iliff JJ, Close LN, Selden NR & Alkayed NJ A novel role for P450 eicosanoids in the neurogenic control of cerebral blood flow in the rat. Exp. Physiol 92, 653–658 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Campbell WB & Fleming I Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 459, 881–895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang X et al. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J. Biol. Chem 276, 14867–14874 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Zarriello S et al. Humble beginnings with big goals: Small molecule soluble epoxide hydrolase inhibitors for treating CNS disorders. Prog. Neurobiol 172, 23–39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson JW et al. Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat 113–115, 30–37 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alkayed NJ et al. Inhibition of brain P-450 arachidonic acid epoxygenase decreases baseline cerebral blood flow. Am. J. Physiol 271, H1541–6 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Zhang W et al. Role of endothelial soluble epoxide hydrolase in cerebrovascular function and ischemic injury. PLoS One 8, e61244 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu D et al. Soluble Epoxide Hydrolase-Derived Linoleic Acid Oxylipins in Serum Are Associated with Periventricular White Matter Hyperintensities and Vascular Cognitive Impairment. Transl. Stroke Res 1–12 (2018). doi: 10.1007/s12975-018-0672-5 [DOI] [PubMed] [Google Scholar]
- 14.Tarantini S et al. Pharmacologically-induced neurovascular uncoupling is associated with cognitive impairment in mice. J. Cereb. Blood Flow Metab 35, 1871–1881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Girouard H & Iadecola C Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol 100, 328–335 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Mogi M & Horiuchi M Neurovascular coupling in cognitive impairment associated with diabetes mellitus. Circ. J 75, 1042–1048 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Zlokovic BV Neurodegeneration and the neurovascular unit. Nat. Med 16, 1370–1371 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Shih AY et al. The smallest stroke: occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat. Neurosci 16, 55–63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matin N, Fisher C, Jackson WF & Dorrance AM Bilateral common carotid artery stenosis in normotensive rats impairs endothelium-dependent dilation of parenchymal arterioles. Am. J. Physiol. Heart Circ. Physiol 310, H1321–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matin N, Fisher C, Jackson WF, Diaz-Otero JM & Dorrance AM Carotid artery stenosis in hypertensive rats impairs dilatory pathways in parenchymal arterioles. Am. J. Physiol. Heart Circ. Physiol 314, H122–H130 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz-Otero JM et al. Mineralocorticoid receptor antagonism improves parenchymal arteriole dilation via a TRPV4-dependent mechanism and prevents cognitive dysfunction in hypertension. Am. J. Physiol. Heart Circ. Physiol 315, H1304–H1315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao J, Chen Y, Yao E & Liu X Soluble epoxide hydrolase inhibition alleviated cognitive impairments via NRG1/ErbB4 signaling after chronic cerebral hypoperfusion induced by bilateral carotid artery stenosis in mice. Brain Res. 1699, 89–99 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Pires PW, Deutsch C, McClain JL, Rogers CT & Dorrance AM Tempol, a superoxide dismutase mimetic, prevents cerebral vessel remodeling in hypertensive rats. Microvasc. Res 80, 445–452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mumby DG, Gaskin S, Glenn MJ, Schramek TE & Lehmann H Hippocampal damage and exploratory preferences in rats: memory for objects, places, and contexts. Learn. Mem 9, 49–57 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burns WR, Cohen KD & Jackson WF K+-induced dilation of hamster cremasteric arterioles involves both the Na+/K+-ATPase and inward-rectifier K+ channels. Microcirculation 11, 279–293 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baumbach GL & Hajdu MA Mechanics and composition of cerebral arterioles in renal and spontaneously hypertensive rats. Hypertension 21, 816–826 (1993). [DOI] [PubMed] [Google Scholar]
- 27.Wan D et al. In vitro and in vivo Metabolism of a Potent Inhibitor of Soluble Epoxide Hydrolase, 1-(1-Propionylpiperidin-4-yl)-3-(4-(trifluoromethoxy)phenyl)urea. Front. Pharmacol 10, 464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pires PW, Girgla SS, Moreno G, McClain JL & Dorrance AM Tumor necrosis factor-α inhibition attenuates middle cerebral artery remodeling but increases cerebral ischemic damage in hypertensive rats. Am. J. Physiol. Heart Circ. Physiol 307, H658–69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeh C-F et al. Soluble epoxide hydrolase inhibition enhances anti-inflammatory and antioxidative processes, modulates microglia polarization, and promotes recovery after ischemic stroke. Neuropsychiatr. Dis. Treat 15, 2927–2941 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lakkappa N, Krishnamurthy PT, M D P, Hammock BD & Hwang SH Soluble epoxide hydrolase inhibitor, APAU, protects dopaminergic neurons against rotenone induced neurotoxicity: Implications for Parkinson’s disease. Neurotoxicology 70, 135–145 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostermann AI et al. Oral treatment of rodents with soluble epoxide hydrolase inhibitor 1-(1-propanoylpiperidin-4-yl)-3-[4-(trifluoromethoxy)phenyl]urea (TPPU): Resulting drug levels and modulation of oxylipin pattern. Prostaglandins Other Lipid Mediat 121, 131–137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y et al. Epoxyeicosanoid Signaling Provides Multi-target Protective Effects on Neurovascular Unit in Rats After Focal Ischemia. J. Mol. Neurosci 58, 254–265 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Liu X et al. Interaction of nitric oxide, 20-HETE, and EETs during functional hyperemia in whisker barrel cortex. Am. J. Physiol. Heart Circ. Physiol 295, H619–31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Mäki-Petäjä K, Cheriyan J, McEniery C & Wilkinson IB The role of epoxyeicosatrienoic acids in the cardiovascular system. Br. J. Clin. Pharmacol 80, 28–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dorrance AM et al. An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J. Cardiovasc. Pharmacol 46, 842–848 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hercule HC et al. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler. Thromb. Vasc. Biol 29, 54–60 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Wang H et al. Up-regulation of endothelial nitric-oxide synthase by endothelium-derived hyperpolarizing factor involves mitogen-activated protein kinase and protein kinase C signaling pathways. J. Pharmacol. Exp. Ther 307, 753–764 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Papapetropoulos A et al. Downregulation of nitrovasodilator-induced cyclic GMP accumulation in cells exposed to endotoxin or interleukin-1 beta. Br. J. Pharmacol 118, 1359–1366 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pires PW, Jackson WF & Dorrance AM Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am. J. Physiol. Heart Circ. Physiol 309, H127–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li N et al. A modified bilateral carotid artery stenosis procedure to develop a chronic cerebral hypoperfusion rat model with an increased survival rate. J. Neurosci. Methods 255, 115–121 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Wang J et al. A New Rat Model of Chronic Cerebral Hypoperfusion Resulting in Early-Stage Vascular Cognitive Impairment. Front. Aging Neurosci 12, 86 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farkas E, Luiten PGM & Bari F Permanent, bilateral common carotid artery occlusion in the rat: a model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev 54, 162–180 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Matta BF, Heath KJ, Tipping K & Summors AC Direct cerebral vasodilatory effects of sevoflurane and isoflurane. Anesthesiology 91, 677–680 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Hendrich KS et al. Cerebral perfusion during anesthesia with fentanyl, isoflurane, or pentobarbital in normal rats studied by arterial spin-labeled MRI. Magn. Reson. Med 46, 202–206 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Simpkins AN et al. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am. J. Pathol 174, 2086–2095 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arai K & Lo EH An oligovascular niche: cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J. Neurosci 29, 4351–4355 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazarov O, Mattson MP, Peterson DA, Pimplikar SW & van Praag H When neurogenesis encounters aging and disease. Trends Neurosci. 33, 569–579 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Q, Song J, Meng D & Chang Q TPPU, a sEH Inhibitor, Attenuates Corticosterone-Induced PC12 Cell Injury by Modulation of BDNF-TrkB Pathway. J. Mol. Neurosci 67, 364–372 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Ren Q et al. Gene deficiency and pharmacological inhibition of soluble epoxide hydrolase confers resilience to repeated social defeat stress. Proc. Natl. Acad. Sci. USA 113, E1944–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kishi T, Hirooka Y & Sunagawa K Telmisartan protects against cognitive decline via up-regulation of brain-derived neurotrophic factor/tropomyosin-related kinase B in hippocampus of hypertensive rats. J Cardiol 60, 489–494 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Mu JS, Li WP, Yao ZB & Zhou XF Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res. 835, 259–265 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Yamada K & Nabeshima T Brain-derived neurotrophic factor/TrkB signaling in memory processes. J Pharmacol Sci 91, 267–270 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Yuan L et al. 14,15-epoxyeicosatrienoic acid promotes production of brain derived neurotrophic factor from astrocytes and exerts neuroprotective effects during ischaemic injury. Neuropathol Appl Neurobiol 42, 607–620 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Alkayed NJ et al. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke 27, 971–979 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Sidoryk-Wegrzynowicz M, Wegrzynowicz M, Lee E, Bowman AB & Aschner M Role of astrocytes in brain function and disease. Toxicol. Pathol 39, 115–123 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lye JJ et al. Astrocyte senescence may drive alterations in GFAPα, CDKN2A p14ARF, and TAU3 transcript expression and contribute to cognitive decline. Geroscience 41, 561–573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tarantini S et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol 24, 101192 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spector AA, Fang X, Snyder GD & Weintraub NL Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog. Lipid Res 43, 55–90 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Yeh C-F et al. Inhibition of soluble epoxide hydrolase regulates monocyte/macrophage polarization and improves neurological outcome in a rat model of ischemic stroke. Neuroreport 30, 567–572 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Tu R et al. Soluble epoxide hydrolase inhibition decreases reperfusion injury after focal cerebral ischemia. Sci. Rep 8, 5279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu Y, Zhang M, Chen Y, Yang Y & Zhang J-J Postoperative intermittent fasting prevents hippocampal oxidative stress and memory deficits in a rat model of chronic cerebral hypoperfusion. Eur J Nutr 58, 423–432 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Liu L et al. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J. Pharmacol. Exp. Ther 339, 451–463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davidson AO et al. Blood pressure in genetically hypertensive rats. Influence of the Y chromosome. Hypertension 26, 452–459 (1995). [DOI] [PubMed] [Google Scholar]
- 64.Rubattu S et al. Differential modulation of AMPK/PPARα/UCP2 axis in relation to hypertension and aging in the brain, kidneys and heart of two closely related spontaneously hypertensive rat strains. Oncotarget 6, 18800–18818 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Montgolfier O et al. Systolic hypertension-induced neurovascular unit disruption magnifies vascular cognitive impairment in middle-age atherosclerotic LDLr−/−:hApoB+/+ mice. Geroscience 41, 511–532 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee H-T, Lee K-I, Chen C-H & Lee T-S Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J. Neuroinflammation 16, 267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]