

ABSTRACT
Background
Programmed cell death 1 (PD-1) is an inhibitory immune checkpoint expressed on activatedT cells. Upon the formation of T cell receptor (TCR)-pMHC complexes, concomitant PD-1 ligation to its ligands programmed death-ligand 1 (PD-L1) or programmed death-ligand 2 (PD-L2) downregulates TCR signaling and effector function. Here we describe the preclinical characterization of Sintilimab, a fully human IgG4 antibody that potently blocks PD-1 interactions with PD-L1 and PD-L2.
Methods
The binding affinity and blockade function were detected by using surface plasmon resonance (SPR), Enzyme-linked immunosorbent assay (ELISA) and flow cytometry. The biology function properties were measured with luciferase assay and mixed lymphocyte reaction assay. In vivo anti-tumor function and preclinical pharmacokinetic (PK) were identified with human PD-1 transgenic mice and non-human primates separately.
Results
Sintilimab can specifically and strongly bind to human PD-1 (hPD-1) and cynomolgus PD-1 and the affinity of Sintilimab to human PD-1 was measured at 0.3 nm via surface SPR, and displayed slow dissociation kinetics. Sintilimab can block the interaction of PD-1 to PD-L1 and PD-L2 and induce high secretion levels of interferon (IFN)-γ and interleukin (IL)-2 in primary T cell assays. In humanized hPD-1 knock-in mouse models, Sintilimab showed potent anti-tumor activity and increased tumor-infiltrating CD8/CD4 T cell and CD8/ Treg ratios. Preclinical experimentation in non-human primates following a single intravenous infusion of Sintilimab at 1, 6 and 30 mg/kg presented with no signs of drug-related toxicity, and showed typical PK characteristics of an IgG antibody.
Conclusions
Sintilimab has desirable preclinical attributes that supports its clinical development for cancer treatment.
Keywords: PD-1, Sintilimab, PD-L1, PD-L2, Cancer Immunotherapy
Statement of Significance
As one of the first five anti-PD-1 drugs with BLAs accepted by CFDA, Sintilimab has shown potent T cell stimulating activity and significant anti-tumor efficacy.
INTRODUCTION
The identification and destruction of tumors is mounted by a multitude of immune cell types with T cells acting as potent mediators of anti-tumor immunity. Anti-tumor immune responses begin at the tumor site with the uptake of tumor antigens by professional antigen-presenting cells (APCs) followed by antigen processing and presentation to T cells. These antigen-specific T cells then mobilize to destroy target tumor cells [1, 2]. Inhibitory immune checkpoints are expressed by T cells after activation and serve to limit over-exuberant immune-mediated inflammation through binding to their ligands [3].
The B7 family member programmed cell death 1 (PD-1) is an immune checkpoint expressed on activated T cells and its ligands include programmed death-ligand 1 (PD-L1) and programmed death-ligand 2 (PD-L2). Upon ligation, a negative pathway is promoted to inhibit T cell function through the downregulation of TcR/CD28 signals [4–8]. PD-L1 is expressed by APCs, but is also upregulated by tumors to directly downregulate immune effector function in the tumor microenvironment [9–16]. Thus, the PD-1/PD-L1 pathway is seen as an important mechanistic axis adopted by tumors to facilitate tumor escape [17].
Nivolumab was the first anti-PD-1 therapeutic monoclonal antibody (mAb) to show significant clinical activity in unresectable or metastatic melanoma with limited toxicity. Pembrolizumab also provided significant antitumor efficacy and has a good safety profile. These anti-PD-1 therapies were approved by the FDA in 2014, thus, in addition to Ipilimumab, supported the role of cancer immunotherapy through checkpoint blockade [18–23]. PD-L1 blocking antibodies, such as Atezolizumab [24], Durvalumab [25–28] and Avelumab [29] have also been subsequently approved for cancer.
Sintilimab is a fully human IgG4 anti-PD-1 mAb generated from yeast display technology and developed for cancer treatment. At present, Sintilimab has been received by over 400 human subjects in clinical studies for both solid and blood-borne cancers for safety and efficacy evaluation. In this report, the high binding affinity and specificity of Sintilimab to PD-1, blocking potency towards PD-L1 and PD-L2, and T cell activating effects of Sintilimab were determined in vitro. In vivo anti-tumor efficacy was assessed in human PD-1 knock-in mice. Additionally, pharmacokinetics (PK), toxicity and safety parameters were evaluated showing Sintilimab as an ideal therapeutic candidate for targeting the PD-1/PD-L1 axis for the treatment of cancer.
MATERIALS AND METHODS
Reagents
Sintilimab, Sintilimab-IgG1 and IgG4 Isotype control were generated at Innovent Biologics (Suzhou) Co., Ltd. Recombinant PD-1, CTLA-4, CD28, PD-L1 and PD-L2 murine Fc fusions were from Acro Biosystems. FBS and DMEM/F12 medium were from Hyclone. Human interleukin (IL)-2 Ready-SET-GO! ELISA kit was from eBioscience and Human interferon (IFN)-γ ELISA kit was from R&D Systems. MC38 murine colon carcinoma cell line (Lot#: HYC0116) was purchased from Obio Technology (Shanghai) Corp., Ltd. human PD-1 (hPD-1) knock-in (C57BL/6 background) mice were purchased from Shanghai Model Organisms Center, Inc. Antibodies for Fluorescence-activated cell sorting (FACS) staining against CD3, CD4, CD8 and FOXP3 were purchased from BioLegend and eBioscience.
Affinity and binding specificity to PD-1 molecules
Affinities were determined with ForteBio-based biolayer interferometry (Pall: OctetRED96). Sintilimab was biotinylated with EZ-Link Sulfo-NHS-LC-Biotin kit (Thermo Scientific), and loaded onto SA - Streptavidin biosensors at 150 μg/ml. After washing, sensors were dipped into buffer containing antigen at 5 μg/ml or the indicated concentrations, then dissociated in SD buffer (sample dilution buffer: 1× Phosphate Buffered Saline (PBS) + 0.1% BSA + 0.05% Tween-20). Data analysis was carried out on ForteBio software. Biotinylated FcγRs were loaded onto SA - Streptavidin biosensors and dipped into IgG in solution. For C1q binding, biotinylated anti-PD-1 antibody was loaded onto SA - Streptavidin biosensors and dipped into C1q.
Surface plasmon resonance (SPR) analysis was carried out using Series sensor S chips (protein A; GE Healthcare Life Sciences) for measuring affinity kinetics between IgG and hPD-1 antigen. HBS-EP buffer (running buffer; 0.01 M HEPES, 0.15 m NaCl, 3 mm EDTA, 0.05% v/v P20, pH 7.4) and regeneration buffer (10 mm glycine-HCl, pH 1.5∼2.0) were used throughout experimentation. Antibodies were diluted in running buffer to 5 μg/ml. Antigens were prepared and serially diluted 2× from 25 nm (human) or 50 nm (cynomolgus) to 0.78 nm. Antibody was immobilized onto the chip with an/tigen flowed across the chip in running buffer.
Binding to PD-1-expressing CHO and T cells
Chinese hamster ovary (CHO) stable pool overexpressed with human PD-1 (CHO-hPD-1) was engineered and used to determine the binding affinity of Sintilimab by flow cytometry at the single cell level. CHO-hPD-1 cells were seeded onto a 96-well U-bottom plate, and treated with antibody at concentrations ranging from 0.003∼50 nm. Antibodies that bound to CHO-hPD-1 cells were detected with PE-labeled goat anti-human Fc secondary antibody (SouthernBiotech, Cat: 2040-09) by flow cytometry (BD: ACCURI-C6). EC50 was calculated using GraphPad.
Blocking assay of human PD-L1 and PD-L2 to CHO-hPD-1 cells
Different concentration Sintilimab was mixed with human PD-L1 or PD-L2 (fused to murine Fc) and then the mixture was incubated with pre-fix CHO-hPD-1 cells at 37°C for 1 h, PD-L1 or PD-L2 binding to CHO-hPD-1 cells was detected with anti-mouse IgG (Fc-specific) peroxidase antibody (Sigma, Cat: A0168-1 ML) using standard ELISA through absorbance at 450 nm. IC50 was calculated on GraphPad.
Antibody-dependent cell-mediated cytotoxicity and Complement-dependent cytotoxicity assays
Antibody-dependent cell-mediated cytotoxicity (ADCC): Logarithmic expansion stage target cells (CHO-hPD-1) and effector cells (Jurkat FcgRIIIA/Nuclear factor of activated T-cells (NFAT)-RE-luc2, Promega) were mixed together in assay medium and incubated at 37°C, 5% CO2 for 6 h under indicated antibodies. After that luciferase assay reagent was added and the luminescence signal was detected on a SpectraMax i3x reader (Molecular Devices) following the addition of standard substrate.
Complement-dependent cytotoxicity (CDC): Complement solution (Complementtech A099) was added into CHO-hPD-1 cells and then incubated at 37°C, 5% CO2 for 2 h under indicated antibodies. After that, 10 μl of CCK-8 reagent (Dojindo) were added per well and incubated for3 more h. Optical density of the wells was detected using a standard reader.
Mixed lymphocyte reaction test
CD4+ T cells were isolated from peripheral blood monocyte cells (PBMC) using Dynabeads untouched human CD4+ T cells’ kit (Invitrogen, Cat: 11346D). Monocytes from another donor’s PBMCs were used to generate dendritic cells (DC) by differentiation after incubation with IL-4 (1000 U/ml) and Granulocyte-macrophage colony-stimulating factor (GM-CSF) (1000 U/ml) for 5 days, followed by maturation in media containing Tumor necrosis factor (TNF)-α (1000 U/ml), IL-1β (5 ng/ml), IL-6 (10 ng/ml) and prostaglandin E2 (PGE2) (1 μM) for 2 days. Cytokines/growth factors were purchased from R&D Systems. In each well of a 96-well U-bottom plate, 10 000 DC and 100 000 CD4+ T cells were mixed together, test antibodies were added at the given final concentrations 0.4, 2 and 10 μg/ml for 5 days before detection of IL-2 and IFN-γ secretion in supernatants with ELISA.
Luciferase reporter assay
A luciferase reporter assay (Promega) was used for mechanistic analysis of blocking PD-1 on the activation of the NFAT pathway. Briefly, Logarithmic expansion phase PD-L1-CHO-K1 cells were seeded into 96-well plate. After 16 h resting, PD-L1-CHO-K1 cells were co-cultured with NFAT-Luc2 Jurkat/PD-1 effector cells for 6 h under indicated antibodies. After 6 h, media was removed and Bio-Glo™ luciferase assay reagent was added, after which chemiluminescence was detected on a SpectraMax i3x reader (Molecular Devices) and analyzed using GraphPad.
MC38 in vivo tumor model in hPD-1 knock-in mice
MC38 cells (in PBS at 5 × 106 cells/ml) were subcutaneously (s.c.) implanted into the right flank of hPD-1 knock-in female mice (Shanghai Model Organisms Center, Inc.) with 1 × 106 cells/mouse. At 11 days post implantation, mice were randomized with mean tumor volumes calculated at approximately 83 mm3 (LxW2/2). On days 11, 14, 17 and 20 post implantation, mice were dosed intraperitoneally with PBS or test antibody. Tumor volume and body weight were measured twice a week, and mice were euthanized when body weight had >20% loss. Tumors were measured with a digital caliper.
Total growth inhibitio (%): 100% × (Tvolcontrol –Tvoltreated)/(Tvolcontrol – Tvolpredose)
Tvolcontrol – Tvoltreated: tumor volume of control mouse after dosing – tumor volume of mice treated with antibody;
Tvolcontrol – Tvolpredose: tumor volume of control mouse after dosing – tumor volume of control mice before dosing.
All animals were maintained under pathogen-free conditions in the animal facilities of GenePharma (Suzhou). All animal-related experiments were approved by the Animal Use and Care Committee of Innovent Biologics (Suzhou) Co., Ltd. Statistical significance between the groups was determined by one way ANOVA for tumor volume, and statistical difference between the treatment group vs. control was determined if P < 0.05.
Analysis of tumor-infiltrating lymphocytes
MC38 colon tumor cells (1 × 106) were implanted s.c. into the right flank of hPD-1 transgenic male C57BL/6 mice. After tumor size reached 80–120 mm3, tumor-bearing mice were randomized and dosed with 1 mg/kg of the indicated antibody (Q3-4x2). On days 8 and 14 post first dose, tumors and spleens were harvested, dissociated or digest into single-cell suspensions, and stained with CD3, CD4, CD8 and FOXP3 for flow cytometry analysis. Statistical significance between the groups was determined by unpaired two-tailed t-test analysis where statistical differences between the groups were determined if P < 0.05.
PK study in cynomolgus monkeys
A single-dose PK study of Sintilimab was conductedin cynomolgus monkeys at doses of 1, 6 and 30 mg/kg. Eighteen cynomolgus monkeys were assigned into three treatment groups and received one intravenous infusion of Sintilimab on day 0. Blood samples (∼1 ml) for PK and anti-drug antibody (ADA) analysis were collected from each animal via the femoral vein and processed for serum extraction. PK samples were collected on days 0 (pre-dose, 0.5, 1, 2, 4, 8 and 12 h post-dose), 1, 2, 3, 7, 14, 21, 28, 35 and 42, while ADA test samples were collected pre-dose (day 0), days 14 and 42 post-dose.
An indirect antigen ELISA assay was used for the detection of Sintilimab in cynomolgus monkey serum. The capture agent was recombinant human PD-1 (Sino Biological Inc.; 10377-H03H), coated onto 96-well ELISA plates. Following overnight incubation, plates were blocked followed by addition of the samples. Captured Sintilimab was detected by HRP-conjugated anti-human antibody (Sigma; A0293). Serum concentration-time profiles were used to estimate PK parameters using non-compartmental analysis (Phoenix 6.2.1.51). Total drug exposure was defined as area under the plasma concentration-time curve, clearance, volume of distribution at steady state, observed maximum serum concentration, time to reach maximum serum concentration and half-life. Each animal was analyzed separately and results were summarized as mean ± standard deviation. ADAs were detected using acid dissociation followed by a standard bridging ECL assay, detected on an MSD Sector Imager (MSD).
RESULTS
Affinity and binding specificity of Sintilimab to PD-1
Therapeutic antibody candidates against PD-1 were generated using a yeast display platform (Adimab LLC). Sintilimab was selected as the final lead molecule due to its high binding affinity and specificity to PD-1 and blocking potency of PD-1 to its ligands PD-L1 and PD-L2, and enhance T-cell function. SPR was used to detect the binding affinity of Sintilimab to hPD-1 and cynomolgus PD-1 (cynoPD-1). The KD of Sintilimab was determined to be 0.25 nm for hPD-1 and 0.446 nm for cynoPD-1 (Figure 1A–D and Table 2), showing strong human and cynomolgus antigen binding affinities, and that Sintilimab is suitable for preclinical evaluation in cynomolgus monkeys.
Figure 1.
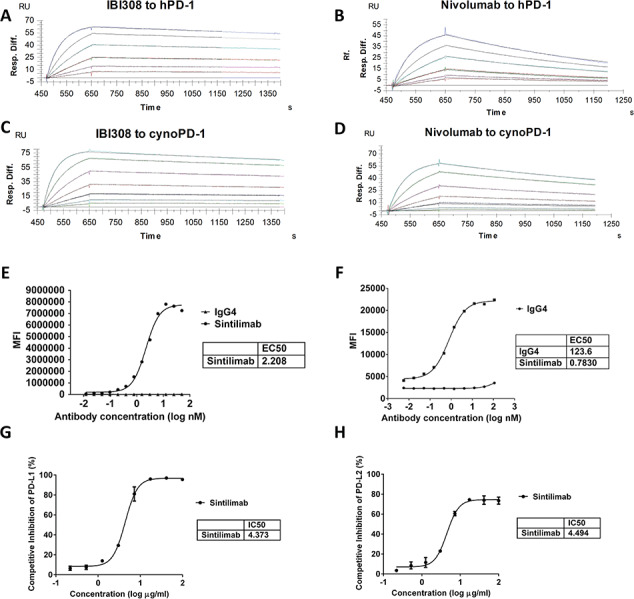
Binding affinity and ligand blocking measurements of Sintilimab and Nivolumab to PD-1 by SPR (Biacore) and cell-based assays. (A) Association and dissociation of Sintilimab to hPD-1 by SPR. (B) Association and dissociation of Nivolumab to hPD-1 by SPR. (C) Association and dissociation of Sintilimab to cynoPD-1 by SPR. (D) Association and dissociation of Nivolumab to cynoPD-1 by SPR. Antigens were serially diluted 2× from 25 nm (human) or 50 nM (cynomolgus) to 0.78 nm. (E) Affinity of Sintilimab to CHO-hPD-1 cells. (F) Affinity of Sintilimab to activated CD4+ T cells. (G–H) CHO-hPD1 cell-based ELISA with fixed concentrations of human PD-L1 (G) or PD-L2 (H). Figures show representative data from three independent experiments.
Table 2.
Affinity of Sintilimab to Fc-gamma receptors measured by Fortebio-based method. W.B. = weak binding; N.B. = no binding
| Fc interaction molecule | Affinity to Sintilimab (M) | Affinity to Sintilimab-IgG1 (M) |
|---|---|---|
| FcγRI | 1.0E-08 | 3.4E-09 |
| FcγRIIa | W.B. | 4.5E-07 |
| FcγRIIb | W.B. | W.B. |
| FcγRIIIa (V158) | W.B. | 1.7E-06 |
| FcγRIIIa (F158) | N.B. | 1.2E-06 |
| FcγRIIIb | N.B. | 2.9E-05 |
| C1q | N.B. | 1.9E-07 |
Biotinylated FcγR was loaded onto sensors and dipped into IgG for recording binding and dissociation kinetics. For C1q, biotinylated IgG was loaded onto sensors and dipped into C1q in solution. Data is representative of three independent experiments.
ForteBio-based biolayer interferometry was used to determine the binding affinity and cross-specificity of Sintilimab. The results confirmed that Sintilimab has strong binding affinity to hPD-1 and cynoPD-1. Weak binding to rabbit PD-1(rbPD-1) and no binding to murine (muPD-1), rat PD-1(rtPD-1), human CTLA-4 (hCTLA-4) and human CD28(hCD28) was detected (Supplementary Table 1). Cell-based binding experiments on CHO cells overexpressing human PD-1 (CHO-hPD-1) were conducted, the EC50 value is 2.2 nm (Figure 1E). The binding of Sintilimab and hPD-1 is confirmed through FACS staining with activated CD4+ T cells (Figure 1F). PD-L1 and PD-L2 blocking assays using recombinant PD-L1 and PD-L2 and antibody-treated CHO-hPD-1 cells in a cell-based ELISA assay showed Sintilimab could efficient inhibits hPD-1 binding to PD-L1 and PD-L2 with IC50 at 4.373 ug/ml and 4.494 ug/ml, respectively (Figure 1G and H). Through the above studies, the binding and blocking activities of Sintilimab were potent and possessed the desirable traits for further development.
Characterization of the FC domain of Sintilimab
IgG4 isotypes are known to have very low effector function and are ideal for use in therapeutic antibodies that are developed for their blocking activity [30]. As Sintilimab was discovered with the aim to block the interaction of its antigen PD-1 with PD-L1 and PD-L2, we engineered Sintilimab with an IgG4 backbone. Using ForteBio-based biolayer interferometry, Sintilimab was found to have no detectable binding to FcγRIIIa (F158) and FcγRIIIb, weak affinity to FcγRIIa, FcγRIIb, FcγRIIIa (V158), but noticeable binding to FcγRI (Table 2). An IgG1 isotype positive control of Sintilimab was produced where the VH and VL domains of Sintilimab were fused to the constant regions of IgG1 (Sintilimab-IgG1). Sintilimab-IgG1 had noticeable binding to all of the FcγRs expect for FcγRIIb (Table 2). Both Sintilimab and Sintilimab-IgG1 showed similar binding affinity to FcRn (data not shown) and only the IgG1 version of Sintilimab was detected to bind to C1q (Table 2). These data show that Sintilimab behaves as expected for an antibody in the IgG4 framework. Sintilimab exhibited no ADCC and CDC function in vitro, whereas Sintilimab-IgG1 had potent ADCC and CDC responses (Figure 2A and B). Thus, Sintilimab displays the desirable traits for a therapeutic IgG4 antibody with blocking activity.
Figure 2.
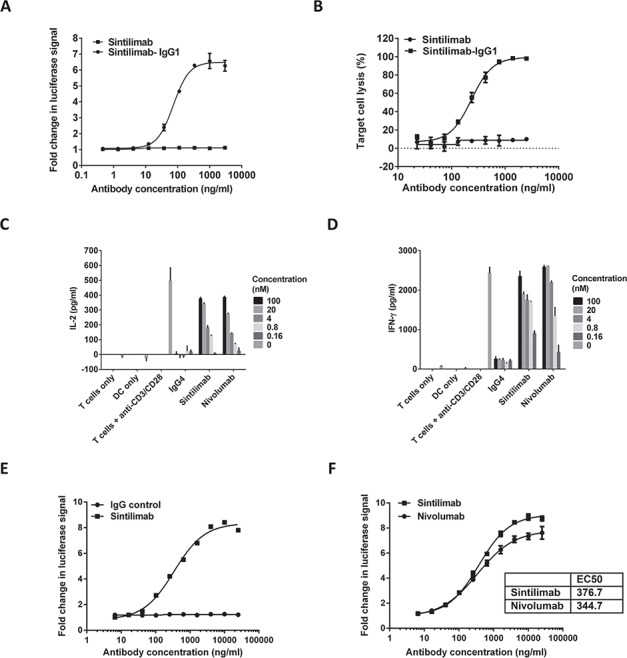
Functional activity of Sintilimab in cell-based bioassays. (A) Sintilimab tested in a luciferase-based ADCC functional assay. (B) Testing CDC activity of Sintilimab using a cell lysis assay. (C–D) MLR and effect of Sintilimab on T cell activation. moDC were generated and mixed with CD4+ T cells from a different donor for 5 days before detection of IL-2 (C) and IFN-γ (D) secretion by ELISA. (E–F) Luciferase reporter assay of IgG vs. Sintilimab, and Sintilimab vs. Nivolumab. Figures show representative data from three independent experiments.
Functional and cytokine release assays of Sintilimab
The functional activity of Sintilimab was investigated using a mixed lymphocyte reaction (MLR) assay. Monocyte-derived dendritic cells (moDCs) were generated and mixed with allogeneic whole CD4+ T cells and different concentrations of Sintilimab or Nivolumab. On day 5, Sintilimab significantly increased IL-2 and IFN-γ levels in a concentration-dependent manner, just like positive control Nivolumab (Figure 2C and D). Using a luciferase reporter system, where luciferase expression is under the control of an NFAT promoter, Sintilimab induced concentration-dependent NFAT activation (Figure 2E) with comparable potency to Nivolumab (Figure 2F). These experiments show that Sintilimab can activate T cells with high potency.
Sintilimab was also tested for their propensity to directly induce cytokine release upon their addition to PBMC. Luminex was used for testing cytokine release from antibody-treated whole blood, and showed that Sintilimab had no significant influence on cytokine release. Compared with the blank control, cytokine levels from both plasma and PBMC supernatant including GM-CSF, IFN-γ, IL-10, IL-1Ra, IL-1β, IL-2, IL-4, IL-5, IL-6 and TNF-α increased significantly after stimulation with the positive control (anti-CD3 + 10 anti-CD28 antibodies) (Supplementary Figure 1). There were no significant changes in cytokine profiles of whole blood treated with Sintilimab, these results show that Sintilimab has no significant direct agonistic effects on immune cells.
Anti-tumor efficacy in hPD-1 knock-in mice
In vivo anti-tumor efficacy of Sintilimab was evaluated in a therapeutic human PD-1 knock-in tumor mouse model of MC38 colon adenocarcinoma. Mean tumor volumes were calculated on day 31 post-tumor cell implantation (Figure 3A and Supplementary Table 2). Human IgG isotype at 5 mg/kg showed a slight effect on tumor growth inhibition (TGI). Sintilimab at 1 and 5 mg/kg exhibited significant TGI of 84 and 100% compared with vehicle control, respectively, and in these groups inhibition vs. IgG control was statistically significant. Maximal efficacy appeared to be reached at 5 mg/kg (6/8 mice tumor-free). No abnormal body weight changes or signs of toxicity throughout the study were observed.
Figure 3.
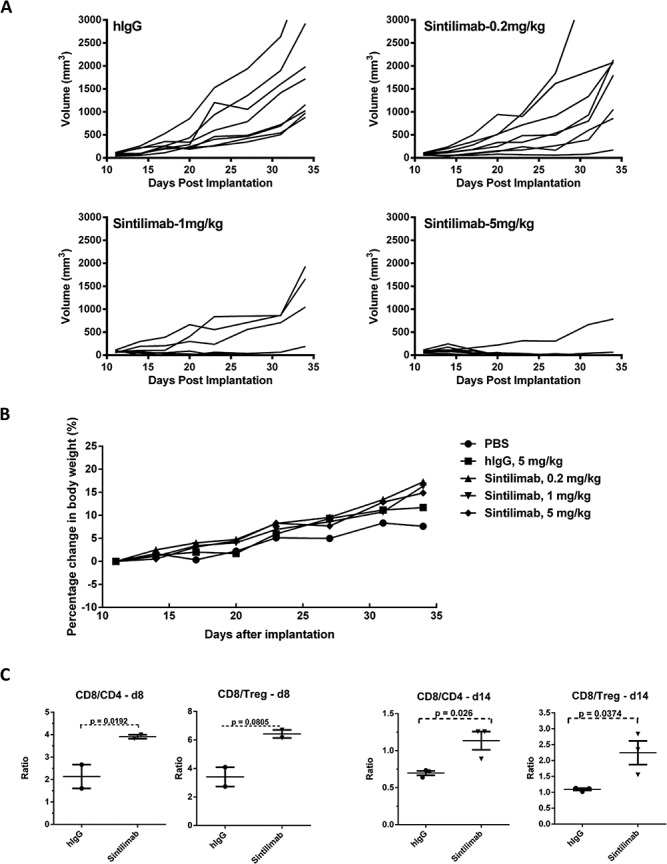
In vivo hPD-1 knock-in mouse model to test anti-tumor efficacy of Sintilimab. (A) Tumor growth inhibition (TGI) of MC38 tumors in hPD-1 knock-in mice of individual animals treated with different doses of Sintilimab. (B) Effect of Sintilimab on percentage changes in mouse body weight (mean). (C) Changes in ratios of tumor infiltrating CD4+, CD8+ and Treg cells. For d8: IgG (n = 2); Sintilimab (n = 2). For d14, n = 3 for all groups.P values were calculated using a two-tailed t-test method.
To test for immune regulation, we analyzed CD4+ and CD8+ T cell ratios after Sintilimab treatment of MC38 tumor-bearing mice on days 8 and 14 (Figure 3C). Flow cytometry was used to directly assess changes in tumor-infiltrating lymphocytes (TILs). Treatment with Sintilimab only modestly increased the frequency of effector CD8+ and CD4+ T cells amongst TIL (data not shown), whereas the ratios of CD8/CD4 and CD8/Treg cells were upregulated in groups dosed with Sintilimab when compared with the IgG control. And this phenotype was also observed in Sintilimab treated mice on day 14 after first drug dosage (Figure 3C).
Preclinical testing in cynomolgus monkeys
PK and ADA analyses of Sintilimab were undertaken in cynomolgus monkeys following single intravenous infusion at 1, 6 and 30 mg/kg dosages. As shown in Figure 4 and Supplementary Table 3, the serum concentration of Sintilimab and drug area under curve increased with the increase of the dose among 1∼30 mg/kg, and there were no obvious gender difference in systemic exposure at any dose level, PK profiles and calculated PK parameters were also not significantly different for both sexes. ADAs were detected in the serum of all animals receiving 1 mg/kg Sintilimab and in five of six animals in the 6 and 30 mg/kg dose group. Combined with the results of PK parameters and the serum concentration-time curves, serum levels of Sintilimab decreased as the doses added, suggesting that ADA-mediated elimination of Sintilimab could be responsible for this observation throughout the study.
Figure 4.
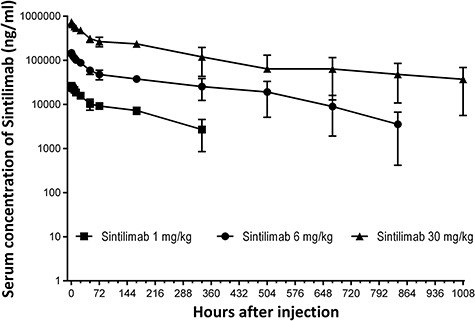
Serum concentration-time profiles following a single intravenous infusion of 1, 6 and 30 mg/kg Sintilimab into cynomolgus monkeys. n = 6 animals/group (three males, three females). The Lower Limit of Quantification (LLOQ) of Sintilimab concentration ELISA assay was 500 ng/ml, and values below the LLOQ were interpreted as non-detectable during statistical analysis.
In a 26-week toxicity study, Sintilimab was well tolerated when administrated biweekly up to 200 mg/kg. There were no drug-related deaths during the study. No drug-related clinical or pathology signals were observed (Supplementary Table 4). Based on the absence of significant toxicological finding at the highest dose administrated, no adverse effect-level would be test in further study.
DISCUSSION
Malignant tumors possess unique mechanisms for evading host immune responses. One such mechanism is the expression of inhibitory ligands by tumor cells that lead to the suppression of effector lymphocytes within the tumor microenvironment. Among the key coinhibitory molecules that are expressed by immune cells, broadly categorized as “immune checkpoints”, include CTLA-4 and PD-1[16, 31]. PD-1 is a member of the B7 family that is expressed on activated T cells and plays an important role in mitigating over-exuberant immune responses. Upon binding to its cognate ligands PD-L1 or PD-L2, PD-1 downregulates TcR/CD28 signaling, promotes T-cell anergy and apoptosis and thus leads to immune suppression. Upregulation of PD-L1 expression has also been directly correlated with immune suppression/resistance and poor prognosis in several cancer types [15].
Sintilimab is a fully human IgG4 anti-PD-1 mAb developed by Innovent Biologics (Suzhou) Co., Ltd., that was generated by yeast display technology (Adimab LLC). This study describes the in vitro and in vivo characterization and non-human primate PK/toxicology evaluation of Sintilimab. Nivolumab was used as a reference in this study and the results were equivalent to those previously reported [32]. Potent PD-1 inhibitors require strong and specific binding to PD-1 and blocking of PD-1 binding with its ligands. To this extent, we aimed to discover and develop a high affinity and potent therapeutic mAb to PD-1 with ligand blocking properties. We show, by use of SPR affinity measurements that Sintilimab binds strongly to hPD-1 with sub-nanomolar monovalent affinity (Figure 1A–D and Table 1). We also confirmed the binding and ligand blocking potency of Sintilimab using cell-based binding/blocking assays (Figure 1E–H) and specificity of Sintilimab to human and cynomolgus PD-1 (Supplementary Table 1).
Table 1.
Affinity of Sintilimab to hPD-1 and cynoPD-1 measured by SPR
| Antigen | Antibody | ka (1/Ms) | kd (1/s) | KD (M) | Chi2 |
|---|---|---|---|---|---|
| hPD-1 | Sintilimab | 7.27E+05 | 1.81E-04 | 2.50E-10 | 0.09 |
| cynoPD-1 | Sintilimab | 4.57E+05 | 2.04E-04 | 4.46E-10 | 0.12 |
Different concentrations of antigen were tested and mean kinetics values calculated and shown in the table above. Data is representative of three independent experiments.
We show obvious T cell activation effects after Sintilimab treatment as indicated by IL-2 and IFN-γ release in a dose-dependent manner via an MLR assay, where the potency of Sintilimab was comparable with Nivolumab (Figure 2C and D). However, direct T cell activation was not observed in cytokine release assays using whole blood (Supplementary Figure 1). The potency of Sintilimab was supported by results using a luciferase reporter-based system to test T cell activation, where effector cells expressed PD-1 and activator cells expressed PD-L1 (Figure 2E and F). All these observations above indicate that Sintilimab harbors the necessary functional pharmacological specifications in vitro for a blocking antibody that targets the PD-1 axis.
Human gene knock-in mice are becoming routinely used to evaluate therapeutic biologics that do not have mouse antigen cross-reactivity [33]. Here, we evaluated the anti-tumor efficacy of Sintilimab in human PD-1 knock-in mice. We found that Sintilimab had significant anti-tumor efficacy. Moreover in Sintilimab group, the CD8/CD4 and CD8/Treg ratio are increased in the TIL populations with statistical difference over the control on days 8 and 14 post antibody dosage (Figure 3C). These data support the functional mechanistic action of Sintilimab and its ability to improve the ratio of T cell effector cell populations that typically corresponds to anti-tumor immune responses (Figure 3 and Supplementary Table 2). The mechanistic significance of these findings warrants further exploration.
Following single intravenous infusion of Sintilimab into cynomolgus monkeys, serum PK profiles were similar between males and females besides the 30 mg/kg group, which may have resulted from differences in the emergence of ADAs. PK values were within acceptable range typically seen for IgGs and were similar to Nivolumab, and the emergence of ADAs in non-human primates is expected as Sintilimab is a fully human antibody (Figure 4 and Supplementary Table 3).
In summary, Sintilimab exhibited specific and high binding affinity to human PD-1, and blocked the interaction between PD-1 to PD-L1 or PD-L2. Sintilimab inducedIL-2 and IFN-γ expression in MLR assays with strong in vitro potency. Sintilimab inhibited tumor growth in vivo in a human PD-1 knock-in model and its anti-tumor efficacy coincided with expected changes in TIL signatures after administration. PK and Tox profiles that were evaluated in non-human primates showed no safety concerns regarding the selection of Sintilimab for clinical development. Thus, Sintilimab is an ideal therapeutic candidate for the treatment of cancer as a monotherapy or for future combination therapies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lihua Yuan (Sinano,CAS in Suzhou, China) for flow cytometric analysis and Adimab LLC for yeast display. This work was supported by grants from the National Health Commission of the People’s Republic of China (2014ZX09201041001).
References
- 1. Chen, DS, Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017; 541: 321–30. [DOI] [PubMed] [Google Scholar]
- 2. Chen, DS, Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39: 1–10. [DOI] [PubMed] [Google Scholar]
- 3. Baumeister, SH, Freeman, GJ, Dranoff, Get al. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol 2016; 34: 539–73. [DOI] [PubMed] [Google Scholar]
- 4. Freeman, GJ, Long, AJ, Iwai, Yet al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192: 1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Latchman, Y, Wood, CR, Chernova, Tet al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001; 2: 261–8. [DOI] [PubMed] [Google Scholar]
- 6. Blank, C, Brown, I, Peterson, ACet al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res 2004; 64: 1140–5. [DOI] [PubMed] [Google Scholar]
- 7. Riley, JL. PD-1 signaling in primary T cells. Immunol Rev 2009; 229: 114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arasanz, H, Gato-Canas, M, Zuazo, Met al. PD1 signal transduction pathways in T cells. Oncotarget 2017; 8: 51936–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iwai, Y, Ishida, M, Tanaka, Yet al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Nat Acad Sci USA 2002; 99: 12293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ghebeh, H, Mohammed, S, Al-Omair, Aet al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: correlation with important high-risk prognostic factors. Neoplasia 2006; 8: 190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thompson, RH, Kwon, ED. Significance of B7-H1 overexpression in kidney cancer. Clin Genitourin Cancer 2006; 5: 206–11. [DOI] [PubMed] [Google Scholar]
- 12. Thompson, RH, Gillett, MD, Cheville, JCet al. Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc Nat Acad Sci USA 2004; 101: 17174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohigashi, Y, Sho, M, Yamada, Yet al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res 2005; 11: 2947–53. [DOI] [PubMed] [Google Scholar]
- 14. Konishi, J, Yamazaki, K, Azuma, Met al. B7-H1 expression on non-small cell lung cancer cells and its relationship with tumor-infiltrating lymphocytes and their PD-1 expression. Clin Cancer Res 2004; 10: 5094–100. [DOI] [PubMed] [Google Scholar]
- 15. Zou, W, Wolchok, JD, Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 2016; 8: 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baksh, K, Weber, J. Immune checkpoint protein inhibition for cancer: preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Semin Oncol 2015; 42: 363–77. [DOI] [PubMed] [Google Scholar]
- 17. Topalian, SL, Drake, CG, Pardoll, DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27: 450–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brahmer, J, Reckamp, KL, Baas, Pet al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015; 373: 123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brahmer, JR, Drake, CG, Wollner, Iet al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010; 28: 3167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Topalian, SL, Hodi, FS, Brahmer, JRet al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366: 2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamid, O, Carvajal, RD. Anti-programmed death-1 and anti-programmed death-ligand 1 antibodies in cancer therapy. Expert Opin Biol Ther 2013; 13: 847–61. [DOI] [PubMed] [Google Scholar]
- 22. Brahmer, JR, Tykodi, SS, Chow, LQet al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366: 2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robert, C, Thomas, L, Bondarenko, Iet al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 2011; 364: 2517–26. [DOI] [PubMed] [Google Scholar]
- 24. Powles, T, Eder, JP, Fine, GDet al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014; 515: 558–62. [DOI] [PubMed] [Google Scholar]
- 25. Gibbons, DL, Chow, LQ, Kim, DWet al. 57O Efficacy, safety and tolerability of MEDI4736 (durvalumab [D]), a human IgG1 anti-programmed cell death-ligand-1 (PD-L1) antibody, combined with gefitinib (G): a phase I expansion in TKI-naive patients (pts) with EGFR mutant NSCLC. J Thorac Oncol 2016; 11: S79. [Google Scholar]
- 26. Mok, T, Schmid, P, Aren, Oet al. 192TiP: NEPTUNE: a global, phase 3 study of durvalumab (MEDI4736) plus tremelimumab combination therapy versus standard of care (SoC) platinum-based chemotherapy in the first-line treatment of patients (pts) with advanced or metastatic NSCLC. J Thorac Oncol 2016; 11: S140–1. [Google Scholar]
- 27. Peters, S, Antonia, S, Goldberg, SBet al. 191TiP: MYSTIC: a global, phase 3 study of durvalumab (MEDI4736) plus tremelimumab combination therapy or durvalumab monotherapy versus platinum-based chemotherapy (CT) in the first-line treatment of patients (pts) with advanced stage IV NSCLC. J Thorac Oncol 2016; 11: S139–40. [Google Scholar]
- 28. Rothschild, SI, Zippelius, A, Prince, SSet al. 129TiP: SAKK 16/14 - anti-PD-L1 antibody durvalumab (MEDI4736) in addition to neoadjuvant chemotherapy in patients with stage IIIA (N2) non-small cell lung cancer (NSCLC): a multicenter single-arm phase II trial. J Thorac Oncol 2016; 11: S112. [Google Scholar]
- 29. Kaufman, HL, Russell, J, Hamid, Oet al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol 2016; 17: 1374–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beers, SA, Glennie, MJ, White, AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood 2016; 127: 1097–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen, L, Flies, DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13: 227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang, C, Thudium, KB, Han, Met al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2014; 2: 846–56. [DOI] [PubMed] [Google Scholar]
- 33. Burova, E, Hermann, A, Waite, Jet al. Characterization of the anti-PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther 2017; 16: 861–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.