

ABSTRACT
Solid tumors are inherently difficult to treat because of large regions of hypoxia and are often chemotherapy- or radiotherapy-resistant. It seems that cancer stem cells reside in hypoxic and adjacent necrotic tumor areas. Therefore, new treatments that are highly selective for tumors and can eradicate cells in both hypoxic and necrotic tumor regions are desirable. Antibody α-radioconjugates couple an α-emitting radionuclide with the specificity of a tumor-targeting monoclonal antibody. The large mass and energy of α-particles result in radiation dose delivery within a smaller area independent of oxygen concentration, thus matching key criteria for killing hypoxic tumor cells. With advances in radionuclide production and chelation chemistry, α-radioconjugate therapy is regaining interest as a cancer therapy. Here, we will review current literature examining radioconjugate therapy specifically targeting necrotic and hypoxic tumor cells and outline how α-radioconjugate therapy could be used to treat tumor regions harboring more resistant cancer cell types.
Statement of Significance
Tumor-targeting antibodies are excellent vehicles for the delivery of toxic payloads directly to the tumor site. Tumor hypoxia and necrosis promote treatment recurrence, resistance, and metastasis. Targeting these areas with antibody α-radioconjugates would aid in overcoming treatment resistance.
Keywords: antibody, alpha-radioconjugate, tumor, necrosis, hypoxia
INTRODUCTION
Tumor-selective targeting is a key criterion in devising novel cancer treatment strategies. The use of tumor-specific monoclonal antibodies (mAbs) armed with cytotoxic agents such as high-potency drugs or radionuclides has the benefit of increasing tumor cell-targeting while reducing exposure to surrounding, healthy tissues. Two mAbs, which are specific for the B-cell antigen CD20 and labeled with β-emitting radionuclides, have been approved by the US Food and Drug Administration (FDA) as antibody radioconjugates for treatment of relapsed or refractory (r/r) non-Hodgkin lymphoma (NHL). Tositumomab and Iodine-131 (131I)-labeled tositumomab was FDA-approved in 2003. However, marketing approval was withdrawn in 2014 because of poor sales, which in part reflected the success of effective alternative treatments directly available to hemato-oncologists. Ibritumomab tiuxetan labeled with Yttrium-90 (90Y) was FDA-approved in 2002 for r/r NHL and in 2009 for newly diagnosed follicular NHL responding to initial anti-cancer treatment [1]. Despite the promise of these agents in the treatment of NHL, their precise place in the therapeutic armamentarium for NHL remains to be defined [2–4]. In contrast, antibody radioconjugate therapy for radio-resistant non-hematological malignancies has had little clinical impact (reviewed by [5]) apart from the approval by the Chinese State Food and Drug Administration of 131I-labeled tumor necrosis therapy (TNT) for advanced lung cancer [6]. Although there is currently no US FDA-approved antibody α-radioconjugate therapy, there is a range of completed and ongoing clinical trials examining α-radioconjugate therapy for a number of malignancies (reviewed by [7]). The first (and only) FDA-approved α-therapy is Radium-223 (233Ra) dichloride for the treatment of patients with symptomatic skeletal metastases of castration-resistant prostate cancer [8].
The clinical problem: tumor hypoxia and recurrence after definitive chemoradiotherapy for inoperable, locally advanced cancers
Surgery often cures cancer in its earliest stages when the disease is localized and resectable. Conversely, metastatic cancer is usually incurable but can be controlled by systemic therapies including cytotoxic chemotherapy, small-molecule signal transduction inhibitors, and immune checkpoint inhibitory antibodies [9]. In between these two extremes are locally advanced, unresectable cancers such as those of the head and neck, lung, oesophagus, stomach, pancreas, bladder, cervix, rectum, or anus. Here, the standard of care is concomitant chemotherapy and radiotherapy (chemoradiotherapy), which is given with curative intent [10]. Nonetheless, despite remission in many cases, treatment failure occurs in half or more of these cases because of locoregional or distant recurrence. For example, regional-stage disease, which may be treatable with chemoradiotherapy, occurs in approximately one-third of the following cancers and has a relatively poor survival. During 2008–14 in the USA, the 5-year relative survival figures for regional-stage cancers of cervix, rectum, esophagus, larynx, oral cavity and pharynx, lung and bronchus (non-small cell), pancreas, stomach, urinary bladder, and anus were 56, 74, 24, 46, 65, 33, 12, 31, 35, and 64%, respectively [11]. Treatment failure is generally associated with intratumoral areas of hypoxia, which are a feature of many larger tumors and harbor chemotherapy- and radiotherapy-resistant cells [12, 13].
Tumor hypoxia has a complex, multifactorial origin including chaotic, dysfunctional tumor vasculature, increased oxygen demands of tumor cells, and the acidic tumor microenvironment limiting oxygenation of hemoglobin [14, 15]. Tumor hypoxia is identified as a negative prognostic and predictive factor because it underlies such important malignant processes as angiogenesis, vasculogenesis, invasiveness, metastasis, altered metabolism, and genomic instability, and it is central to the phenomena of chemotherapy- and radiotherapy-resistance [16]. Hence, clinically and regardless of treatment, tumor hypoxia is associated with cancer aggressiveness and resistance resulting in locoregional recurrence and metastasis [13]. Indeed, hypoxia-induced radiotherapy-resistance is the major factor limiting tumor control to radiotherapy [17].
Tumor necrosis—another breeding ground for treatment-resistant tumor cells
Both hypoxia and necrosis are unique pathologic features of many solid tumors [17] and are intimately associated with the cancer hallmarks of deregulated cellular energetics and tumor-promoting inflammation, respectively [18]. Tumor xenograft data indicate that as hypoxic cells die, they become necrotic and coalesce to form the necrotic core of larger tumors [19]. Abundant clinical and laboratory evidence demonstrates that necrotic cancer cells lie side by side with hypoxic cancer cells [20–24]. Unlike the temporally and spatially dynamic state of tumor hypoxia [22], necrotic tumor cells are a fixed pathologic feature of many tumors [25, 26]. Necrotic tumor cell death, which results from poor tumor vascularization and the associated areas of ischemia, is inflammatory and immunogenic. It results in infiltration of immune cells that further promote tumor growth through production of growth and angiogenic factors [27]. Furthermore, it is becoming increasing clear that cancer stem cells, also known as cancer initiating cells, reside within these necrotic and perinecrotic areas of tumors [28] and may be associated with treatment resistance. Therefore, resistant cell types living in the necrotic tumor microenvironment may particularly evade treatment with antibody radioconjugates targeting live cancer cells.
Targeted α-particle therapy and α-emitting radionuclides of medical interest
Although β-emitting radionuclides have predominantly been used for clinical antibody radioconjugate therapy, using antibodies for targeted α-particle therapy (TAT) has the potential to be more effective than β-emitting radioconjugate therapy. Alpha-particles are charged helium nuclei with high initial energies of 5–8 MeV. Alpha-particles have a short and well-defined track length with a range in tissue of 40–100 μm, which can target several cells (2–10 cells). The α-particle is characterized by a high linear energy transfer (LET), which describes the ratio between the amount of energy transferred and distance traveled by the α-particle and is usually expressed as kiloelectronvolts per micrometer (keV/μm) [29]. The dense ionization tracks of α-particles have a LET of 60–230 keV/μm, which contrasts with the sparsely ionizing photons commonly used in external beam radiotherapy (EBRT) or the electrons in antibody β-radioconjugate therapy (Table 1). The high incidence of α-particle-induced DNA damage results from greater clustering of ionizations (2 000–7 000 ion pairs/μm) compared to β-particle-induced DNA damage (5–20 ion pairs/μm).
Table 1 .
Comparison between β- and α-particles
| Properties | β-particles | α-particles |
|---|---|---|
| LTE | Low | High |
| Pathlength | mm | μm |
| Energy | 100’s to 1000’s keV | >5000 keV |
| Oxygen dependence to elicit cellular damage | High | Low |
| Decays at cell membrane to achieve 99% cell killing [30] | 1000’s | 10’s |
Consequently, α-particles have a much greater chance of producing double-stranded breaks (DSBs) in DNA [31], which, if multiple, are among the most difficult DNA lesions to repair and which are thus highly lethal [32]. In contrast, thousands of β-particle tracks of low LET radiation can be required for the same response because β-particles typically induce sublethal single-stranded DNA breaks salvageable by cellular DNA repair mechanisms [33]. As few as one to two α-particle traversals of the nucleus can produce a sufficient number of such DNA hits to kill a cell with irreparable DNA damage [34]. Therefore, α-hits can be cytotoxic in non-cycling cells including stem cells, which are supported by modeling experiments [35]. For the same reason that DNA damage from high LET radiation is not easily repaired, dose rate and fractionation of dose have relatively little impact on the cell-killing potential of α-particles [36] (refer to Table 1 for a general comparison between α-particles and β-particles).
Hence, these physico-chemical characteristics make α-particles ideally suited for cancer treatment because the high linear energy deposited in a short path limits cytotoxicity to the immediate vicinity of the α-emissions and results in high target to non-target dose ratios [33, 37, 38]. However, TAT often has a non-uniform distribution in organs and tumors, a non-uniform distribution of radioactivity, and a non-uniform distribution of absorbed dose. Consequently, the ‘hit or miss’ stochastic properties of α-particles can limit some of the therapeutic effect of TAT and as many as 20 α-particle nuclear traversals may be required to kill a cell [36].
Nevertheless, when compared to antibody β-radioconjugate therapy and EBRT in pre-clinical studies, antibody α-radioconjugate therapy was found to be more effective per absorbed radiation dose unit in the low-dose range (up to 2 Gy) in a human lymphoma xenograft model [39] and was more effective than antibody β-radioconjugate therapy at equivalent absorbed dose in a human breast cancer xenograft model [40]. Similarly, antibody radioconjugate therapy using an α-emitter in a pre-clinical multiple myeloma model was more effective than that using a β-emitter [41].
Several α-emitting radionuclides are of interest for medical applications and are listed in Table 2. Among this list, the radiometals bismuth, actinium, lead, and thorium require bifunctional chelators for conjugation to mAbs, whereas Astatine-211 (211At) requires halogenation chemistry for conjugation to mAbs. Pairs of radiometals are listed in Table 2 because the second member of the pair is the decay daughter of the first member of the pair [42] and, thus, the parent radionuclide represents an internal generator of therapeutic α-particles. Indeed, this in vivo generator concept allows for a more effective, high-dose TAT by matching the longer half-life of the parent nuclide with the relatively long biological half-life of a mAb to enable tumor targeting of shorter-lived daughter(s) with high decay energy. This enables blood clearance of the parent nuclide while the high-LET daughter accumulates at the tumor site. Consequently, the therapeutic index of TAT improves and may allow the therapy dose to be reduced [43]. Moreover, radionuclides such as Actinium-225 (255Ac) and Thorium-227 (277Th), which have extended decay chains generating 4–5 α-particles with most of the activity occurring within an hour, result in much higher relative doses to tumor than the halogen nuclide 211At but at the expense of the discharged radioactive daughters leaving the tumor site and accumulating in non-target tissues such as kidney in the case of 225Ac decay or bone in the case of 227Th decay and resulting in late toxicities.
Table 2 .
Half-lives of radionuclides of medical relevance [42]
| Radionuclide | t 1/2 |
|---|---|
| Astatine-211 (211At) | 7.2 h |
| Pb-212/Bi-212 (212Pb/212Bi) | 10.6 h/61 m |
| Ac-225/Bi-213 (225Ac/213Bi) | 10 d/46 m |
| Th-227/Ra-223 (227Th/223Ra) | 18.7 d/11.4 d |
t1/2 indicates half-life; m, minutes; h, hours; d, days.
Notwithstanding its pre-clinical effectiveness, the limited clinical application of TAT relates mainly to the restricted availability of the parent isotopes and the current high needs for developing (i) improved complexation chemistry for radiometals such as bismuth, actinium, lead, and thorium; (ii) specialized facilities for handling; and (iii) workforce, infrastructure, and logistics for administering facilities.
The geographic relationship of tumor necrosis and hypoxia
The physical relationship between tumor necrosis and hypoxia is depicted schematically in Figure 1 to show tumor blood vessels cuffed by a sheath of viable cells. The oxygen gradient is reduced from the center to the periphery of each tumor cord where tumor cells adjacent to necrotic areas would be anoxic and consequently radioresistant [20]. To indicate the distances relevant to the passage of therapeutic α-particles from necrotic to hypoxic regions, we report more detail from Gray’s seminal study [20]. In a quantitative analysis of 160 tumor areas of human bronchial squamous cell carcinoma specimens, Gray found that cords of tumor cells coursed through vascularized stroma. The tumor cords varied in diameter and often contained a concentric necrotic cord surrounded by a rim of viable proliferative tumor cells, which was limited to a thickness no greater than 180 μm by oxygen diffusion from the surrounding stroma. No tumor cord without central necrosis was more than 200 μm in radius, and no central necrosis was seen in any tumor cord of less than 160 μm in radius. The average critical radius, i.e. the minimum radius required for the tumor cord to contain central necrosis was 169 μm.
Figure 1 .
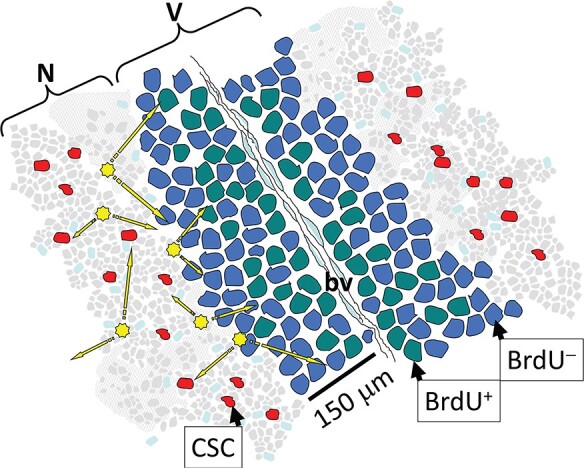
Model relating hypoxic to necrotic tumor regions. A cord of viable tumor cells (V) surrounds a blood vessel (bv) adjacent to a necrotic region (N). Cycling cells (BrdU+) lie closer to a bv than hypoxic cells, which lie closer to necrotic regions and are usually quiescent (BrdU) [83]. These necrotic regions harbor cancer stems cells (CSCs). Radiobiologically, antibody α-radioconjugates targeting hypoxic or necrotic markers can result in irradiation of hypoxic cells or CSCs within the necrotic regions (arrows).
Tumor hypoxia, high LET radiation and the oxygen enhancement ratio
Hypoxic tumor cells are inherently resistant to conventional EBRT [44], and the effectiveness of antibody β-radioconjugate therapy also depends on oxygenated tumor tissue [45]. High LET radiation, which can directly sterilize tumor cells independently of the presence of molecular oxygen, is one way to overcome the treatment resistance of hypoxic tumor cells. For example, high LET external carbon beam therapy provides some benefit in the treatment of hypoxic tumors [46] but only a handful of heavy ion accelerators currently operate for clinical use. An alternative approach is to deliver high LET radiation directly to the tumor tissue with TAT.
The DNA-damaging effects of low LET photons and electrons are indirect. The DNA lesions are oxidative alterations of DNA and other macromolecules, which are mediated by oxygen-centered radicals formed by the ionization of water surrounding the DNA. Here, oxygen ‘fixes’ the radiation damage to DNA because it reacts with the broken ends of DNA by creating stable organic peroxides, which are not easily repaired by cells [47]. In contrast, α-particles are less sensitive to the presence of molecular oxygen because the closely clustered ionizations produced by their high LET radiation generate a high density of delta or ‘knock-on’ electrons that directly damage DNA [48].
The oxygen enhancement ratio (OER) quantifies the effects of oxygen on effectiveness of therapeutic ionizing radiation and is the ratio of iso-effective radiation doses in hypoxic and oxic conditions. For low LET radiation, the OER is 3 and OER decreases with LET, approaching 1 at LET values >250 keV/μm [49, 50]. The OER reduction is generally attributed to recombination of the radiation-induced free radicals or production of ‘oxygen in the track’ [51]. Consequently, α-particle therapy targeting hypoxic and necrotic tumor areas may be especially effective against cancer stem cell subpopulations hiding in the hypoxic niches of tumors [52] and thus particularly important for treating solid tumors that are more resistant to conventional EBRT [53].
Targeting hypoxic tumor cells with carbonic anhydrase 9-specific antibody β-radioconjugates
The most commonly targeted protein for treating hypoxic tumor cells is carbonic anhydrase 9 (CAIX). CAIX is a transmembrane zinc metalloenzyme that catalyzes the hydration of carbon dioxide to bicarbonate ions and proton, with an increased expression of CAIX in cancers being required to maintain an optimal intracellular pH [54]. For this reason, CAIX is an ideal protein for targeting hypoxic cells, and radiolabeled antibody-targeting CAIX has been investigated in clinical trials (Table 3). In early clinical trials, the effect of the mouse anti-CAIX antibody G250 labeled with the β-emitting radionuclide 131I was examined in patients with metastatic clear cell renal cell carcinoma (ccRCC). This treatment resulted in 17 of 33 patients having stable disease with no major responses but with the development of human anti-mouse antibodies limiting further cycles of treatment [55]. To circumvent this, a chimeric version of G250 (cG250; girentuximab) was developed, labeled with 131I and administered to 12 metastatic ccRCC patients in whom a low dose was given to evaluate tumor uptake. Uptake in metastases was visualized in nine of the patients, of whom eight received a second dose 131I-cG250 at 1665, 2220, or 2775 MBq/m2, resulting in a partial response in one patient and stable disease lasting for 3–6 months in another patient [56]. Fractionated dosing of 131I-cG250 given at a whole-body absorbed dose of 0.5, 0.75, or 1 Gy (3–7 fractions/patient) did not increase clinical response, with 7 of the 14 patients who completed treatment having stable disease while the remaining 7 patients had disease progression [57]. Furthermore, administration of 131I-cG250 given at 2220 MBq/m2 followed 3 months later at 1110 or 1665 MBq/m2, in 3 and 16 patients, respectively, also did not increase clinical response, with 5 patients having stable disease and the remaining patients having progressive disease [58].
Table 3 .
Clinical and pre-clinical antibody radioconjugate therapies targeting hypoxic or necrotic tumor cells
| Antibody | Target antigen | Radionuclide | Target cancer | Phase | Ref. |
|---|---|---|---|---|---|
| G250 | CAIX | 131I | ccRCC | I/II | [55] |
| cG250 | CAIX | 131I | ccRCC | I | [56–58] |
| cG250 | CAIX | 177Lu | ccRCC | I, II | [59, 60] |
| chTNT-1/B | Nuclear Antigens | 131I | Colorectal cancer | I | [61] |
| chTNT-1/B | Nuclear antigens | 131I | Glioblastoma | I/II | [62] |
| chTNT-1 | Nuclear antigens | 131I | Lung cancer | II | [63] |
| chTNT-3 | Nuclear antigens | 213Bi | Prostate cancer | Pre-clinical | [64] |
| 6D2 | Melanin | 188Re | Melanoma | Pre-clinical | [65, 66] |
| 8C3 | Melanin | 213Bi | Melanoma | Pre-clinical | [67] |
| DAB4 | La/SSB | 90Y | Lymphoma, lung, prostate, pancreatic cancer | Pre-clinical | [68] |
| DAB4 | La/SSB | 177Lu | Lung cancer | Pre-clinical | [69] |
| DAB4 | La/SSB | 227Th | Lung cancer | Pre-clinical | [70] |
Treatment of ccRCC patients with cG250 labeled with Lutetium-177 (177Lu), a residualizing radionuclide compared to non-residualizing 131I, resulted in improved responses in patients who received up to 3 cycles of treatment, with 1 partial responder and 17 out of 24 patients having stable disease 3 months after the first cycle of treatment [59]. In a second, nonrandomized single-arm trial, 14 ccRCC patients received 2405 MBq/m2 177Lu-cG250, resulting in 8 patients having stable disease and 1 patient having a partial regression [60]. Of these responding patients, six patients received a second cycle of treatment, resulting in durable responses in five patients but with prolonged thrombocytopenia restricting further cycles of treatment [60]. To date, there have been no studies targeting CAIX with α-radioconjugate antibody therapy.
Targeting necrotic tumor cells with antibody β- and α-radioconjugates
Melanin is an intracellular pigment that becomes accessible in dead and dying tumor cells because of melanin release and the loss of membrane integrity thus allowing intracellular antibody targeting [65]. An IgM antibody targeting melanin, 6D2, has been labeled with the β-emitting 188-Rhenium (188Re) and been used pre-clinically to effectively treat mice bearing melanoma tumors [65, 66]. Use of an IgG antibody-targeting melanin improved tumor uptake and it was labeled with the α-emitting radioisotope, Bismuth-213 (213Bi), to treat metastatic melanoma in a syngeneic mouse model. This treatment significantly decreased lung metastases and was superior to the IgM antibody labeled with 213Bi [67]. Interestingly, the anti-melanin 213Bi-labeled IgG antibody showed equivalent efficacy to the same antibody labeled with 188Re in reducing the lung metastatic load. Moreover, this result was achieved despite the half-life of 188Re being 16.9 h compared to the short half-life of 213Bi (45 min), which may be considered less well-matched to the long circulating half-life of IgG.
Necrotic cells have been targeted using the tumor necrosis therapy (TNT) antibodies directed against nucleic acids/histone complexes that are retained in necrotic tissues, particularly solid tumors [71, 72]. Pre-clinically, these antibodies showed sustained uptake within necrotic tumor regions [72, 73]. Clinical development of a chimeric version of this antibody radiolabeled with the β-emitting nuclide, 131I (chTNT-1B, Cotara), has now been discontinued. In a phase 1 study, Cotara was well tolerated as a single intravenous infusion in 21 advanced colorectal cancer patients. Although no objective responses were observed in sentinel lesions, stabilization of sentinel lesions at 8 weeks post-infusion tended to be associated with a smaller volume of these lesions at baseline [61]. Cotara has also been administered via a convection-enhanced delivery system into the primary or recurrent glioblastoma tumors of 51 patients enrolled in a phase 1/2 study. In a subset of 11 evaluable patients who received a total radioactive dose in a ‘therapeutic window’ not associated with excessive toxicity or rapid disease progression, 1 patient had a partial response, 4 patients obtained stable disease, and 4 patients progressed. However, in this early phase study, patient numbers were too small for formal evaluation of therapeutic efficacy [62]. 131I-chTNT has received approval from the Chinese State Food and Drug Administration for the treatment of advanced lung cancer patients who had previous treatment failure with radiotherapy or chemotherapy. Patients received two treatments of 131I-chTNT, which showed favorable tumor uptake, resulting in an objective response rate of 34.6% [63].
Pre-clinically, TNT has been examined as an antibody for TAT and has been labeled with 213Bi for the treatment of a pancreatic cancer xenograft [64]. In this study, antibody radioconjugate therapy was more effective at controlling tumor growth with fewer side effects when compared to gemcitabine or cisplatin. The 213Bi decay chain results in the emission of both α- and β-particles before the long-lived 209Bi is reached. The resulting delivery of both α- and β-doses would be ideal when targeting necrotic tumors for two main reasons. First, the high-energy α-particles emanating from the necrotic tumor core would irradiate hypoxic cells within 2–3 cell diameters from the source located in the necrotic region. Second, the longer tissue range of β-particles would result in dose delivery to the well-oxygenated tumor cells, which are distant from the necrotic and hypoxia tumors cells and would not require as high a β-radio-dose for effective killing.
Necrotic tumor targeting using the monoclonal antibody DAB4 specific for the La (lupus-associated)/SSB (Sjögren Syndrome B) antigen
We have shown that the mouse monoclonal antibody DAB4 (APOMAB®) targets the La/SSB protein, which only becomes available for antibody binding in cells that have lost membrane integrity, particularly in apoptotic and necrotic cancer cells after DNA-damaging anticancer treatment, making DAB4 a tumor cell-targeting mAb [68–70, 74–77]. We have labeled this antibody with β-emitting radionuclides, 90Y and 177Lu, for pre-clinical antitumor therapy using a variety of murine and human models [68, 69]. Because of its ability to target dead cancer cells, DAB4 antibody radioconjugate therapy is more effective when given after chemotherapy, resulting in high tumor uptake of the antibody and therefore more tumor dose delivery. Our pre-clinical data [68–70, 74–77] show that DAB4 binds within the necrotic tumor areas that lie next to hypoxic areas. Therefore, the hypoxic, treatment-resistant areas of the tumor are located within microns of DAB4-binding and would therefore be within range of α-particles if DAB4 were radiolabeled with an α-emitting radionuclide such as 227Th.
In the syngeneic Lewis Lung carcinoma (LL2) cell line model, after an initial chemotherapy step, we have shown equivalent antitumor efficacy in vivo using DAB4 conjugated to either the shorter lived, high-energy, and long-range β-emitter, 90Y [68] or the longer lived, lower energy, and short-range β-emitter, 177Lu [69]. These data suggest that we may adapt antibody radioconjugate therapy to tumor volume as the reduced tumor volume resulting from chemotherapy-induced tumor cell death enables efficient β-energy deposition from 177Lu within a smaller tumor volume [78]. Similarly, we hypothesized that substituting the even longer lived, higher energy, and shorter range α-emitter 227Th for 177Lu in DAB4 radioconjugates at least maintains efficacy, if not improves it.
To this end, we used single doses of 227Th-labeled conjugates of DAB4 (227Th-DAB4) at 5, 10, or 20 kBq/kg to treat mice bearing subcutaneous LL2 tumors [70] This was the same syngeneic murine tumor model that we had employed in the previous experiments with conjugates of DAB4-labeled with 90Y [68] or 177Lu [69]. We found that single-agent 227Th-DAB4 had significant antitumor activity at doses of 10 or 20 kBq/kg. Prior chemotherapy was associated with even greater antitumor activity of 227Th-DAB4 with significant antitumor effects observed at all administered doses, even at the lowest dose of 5 kBq/kg [70]. Interestingly, the antitumor effects of low administered activities of 227Th-DAB4 were similar to those observed for the higher administered activities of 90Y-DAB4 [68] or 177Lu-DAB4 [69], which likely reflects the much greater relative biological effectiveness of α-emissions compared to β-emissions [79]. After chemotherapy, compared to 227Th-DAB4 alone, there was a greater and more prolonged tumor accumulation over a five-day period of 227Th-DAB4 rather than its first α-decay daughter, 227Ra. Hence, these data suggest that the slow rate of the first high energy α-decay in the extended 227Th chain, which occurred within the confines of a smaller post-chemotherapy tumor volume, was sufficient to exert a significant therapeutic effect. Finally, autoradiography of excised LL2 tumor sections showed that the α-emitting necrotic areas abutted the hypoxic areas marked by carbonic anhydrase 9 immunostaining [70].
Our in silico studies support this concept of necrotic cell-targeting by vectored α-emitters as means of irradiating hypoxic tumor regions. We adopted the representative necrotic and hypoxic tumor geometry first described by Thomlinson and Gray [20] to perform Monte Carlo modeling with GEANT4 software. We compared the dose deposition characteristics of the pure β-emitting radionuclide, 177Lu, with the combined α- and β-emitting radionuclide, Lead-212 (212Pb). We showed that modeled uptake of these radionuclides within a necrotic tumor core resulted in extremely localized large α-particle doses from 212Pb decay that would deposit in highly radio-resistant cells in an approximately 20–30 μm margin immediately surrounding a region of necrosis. In further modeling, when EBRT was added to α-particle therapy, chronically hypoxic cells would receive a concentrated boost with 212Pb while oxic cells would continue to receive the uniform low LET EBRT [48].
Although the α-camera can provide in situ imaging of α-particles in tissue sections [80], we adapted the Timepix pixelated semiconductor radiation detector for dosimetry of α-emissions in vitro. We demonstrated that the number of transmitted α-particles correlated with the observed DSBs and that the deposited dose fitted with that calculated using Monte Carlo code stopping range of ions in matter (SRIM) [81]. In LL2-bearing mice, which had received chemotherapy or not and which were then treated 24 h later with 227Th-labeled conjugates of DAB4, we used Timepix to image and quantify α-emissions from tumor sections ex vivo. We calculated that the number of α-hits detected by Timepix was proportional to the isotope concentrations in the tumor sections. We next determined that the α-particle energy spectrum emitted by 227Th-DAB4 from tumor sections ranged from 4 to 7.4 MeV and that a statistically significant 4-fold greater number of α-hits originated from tumor sections of mice given prior chemotherapy than those not given chemotherapy. Although most α-hits were transmitted vertically via a collimator from the tumor section through a ≈ 2 mm air gap and released their energy as a charge cluster across several pixels, other α-hits emitted at small angles were detected beyond the tumor-defined limits [82].
Finally, given that the high LET of α-particles reduces the dependence on oxygen for cell killing, the ability of α-particles to overcome hypoxic radio-resistance will critically depend on the spatial distribution of the α-emitters relative to the hypoxic region. This is mainly because the greatest energy deposition of an α-particle is toward the end of its track at the Bragg peak [36]. Using the SRIM software model in water, the Bragg peaks of α-particles emitted as 227Th decays were in the range of 35–60 μm. Although this range does not cover the entire hypoxic rim surrounding a concentric necrotic cord as suggested by Thomlinson and Gray [20], the short-ranging α-particles would traverse the most oxygen-deficient cells.
Altogether these data suggest that α-particles, which have originated from necrotic tumor regions, can penetrate into closely apposed hypoxic tumor regions and thereby contribute significantly to tumor control but only by virtue of radiation crossfire effects.
SUMMARY
In this review, we have explored the potential of targeting hypoxic and necrotic tumor cells with antibody radioconjugates. In particular, targeted delivery of α-therapy to the tumor areas of hypoxia and necrosis that harbor cancer stem cells could be effective because, compared to β-emitting radionuclides, the hypoxic conditions in the tumor microenvironment would not be expected to attenuate the dose delivery of α-emitting radionuclides. Moreover, α-particle targeting of necrotic tumor regions could also result in the effective therapeutic targeting of nearby hypoxic tumor cells, which are resistant to conventional radiotherapy. To date, there have only been pre-clinical studies using antibody α-radioconjugates targeting hypoxic or necrotic tumor cells. However, improvements in chelation chemistry as well in the production of α-radionuclides with favorable therapeutic properties may help to further this field of research.
ACKNOWLEDGEMENTS
This work was supported in part by National Health and Medical Research Council Project Grant ID1126304.
Conflict of interest. MP Brown is an inventor on APOMAB®-related patents issued and pending.
REFERENCES
- 1. Witzig, TE, Gordon, LI, Cabanillas, Fet al. . Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin's lymphoma. J Clin Oncol 2002; 20: 2453–63. [DOI] [PubMed] [Google Scholar]
- 2. Scholz, CW, Pinto, A, Linkesch, Wet al. . (90)Yttrium-ibritumomab-tiuxetan as first-line treatment for follicular lymphoma: 30 months of follow-up data from an international multicenter phase II clinical trial. J Clin Oncol 2013; 31: 308–13. [DOI] [PubMed] [Google Scholar]
- 3. Press, OW, Unger, JM, Rimsza, LMet al. . Phase III randomized intergroup trial of CHOP plus rituximab compared with CHOP chemotherapy plus (131)iodine-tositumomab for previously untreated follicular non-Hodgkin lymphoma: SWOG S0016. J Clin Oncol 2013; 31: 314–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vose, JM, Carter, S, Burns, LJet al. . Phase III randomized study of rituximab/carmustine, etoposide, cytarabine, and melphalan (BEAM) compared with iodine-131 tositumomab/BEAM with autologous hematopoietic cell transplantation for relapsed diffuse large B-cell lymphoma: results from the BMT CTN 0401 trial. J Clin Oncol 2013; 31: 1662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jain, M, Venkatraman, G, Batra, SK. Optimization of radioimmunotherapy of solid tumors: biological impediments and their modulation. Clin Cancer Res 2007; 13: 1374–82. [DOI] [PubMed] [Google Scholar]
- 6. Tomblyn, MB, Katin, MJ, Wallner, PE. The new golden era for radioimmunotherapy: not just for lymphomas anymore. Cancer Control 2013; 20: 60–71. [DOI] [PubMed] [Google Scholar]
- 7. Marcu, L, Bezak, E, Allen, BJ. Global comparison of targeted alpha vs targeted beta therapy for cancer: in vitro, in vivo and clinical trials. Crit Rev Oncol Hematol 2018; 123: 7–20. [DOI] [PubMed] [Google Scholar]
- 8. Parker, C, Nilsson, S, Heinrich, Det al. . Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med 2013; 369: 213–23. [DOI] [PubMed] [Google Scholar]
- 9. Brown, MP, Burdett, M. Targeted therapies, aspects of pharmaceutical and oncological management. Cancer Forum 2013; 37: 70–80. [Google Scholar]
- 10. Lawrence, TS, Blackstock, AW, McGinn, C. The mechanism of action of radiosensitization of conventional chemotherapeutic agents. Semin Radiat Oncol 2003; 13: 13–21. [DOI] [PubMed] [Google Scholar]
- 11. Noone, AM, Howlader, N, Krapcho, Met al. SEER Cancer Statistics Review, 1975–2015, National Cancer Institute: Bethesda, MD. https://seer.cancer.gov/csr/1975_2015/, based on November 2017 SEER data submission, posted to the SEER web site, April 2018.
- 12. Nordsmark, M, Bentzen, SM, Rudat, Vet al. . Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol 2005; 77: 18–24. [DOI] [PubMed] [Google Scholar]
- 13. Hockel, M, Vaupel, P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 2001; 93: 266–76. [DOI] [PubMed] [Google Scholar]
- 14. Vaupel, P, Harrison, L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist 2004; 9: 4–9. [DOI] [PubMed] [Google Scholar]
- 15. Ljungkvist, AS, Bussink, J, Kaanders, JHet al. . Dynamics of tumor hypoxia measured with bioreductive hypoxic cell markers. Radiat Res 2007; 167: 127–45. [DOI] [PubMed] [Google Scholar]
- 16. Wilson, WR, Hay, MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer 2011; 11: 393–410. [DOI] [PubMed] [Google Scholar]
- 17. Brown, JM, Wilson, WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 2004; 4: 437–47. [DOI] [PubMed] [Google Scholar]
- 18. Hanahan, D, Weinberg, RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–74. [DOI] [PubMed] [Google Scholar]
- 19. Ljungkvist, AS, Bussink, J, Kaanders, JHet al. . Hypoxic cell turnover in different solid tumor lines. Int J Radiat Oncol Biol Phys 2005; 62: 1157–68. [DOI] [PubMed] [Google Scholar]
- 20. Thomlinson, RH, Gray, LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer 1955; 9: 539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang, T, Civelek, AC, Li, Jet al. . Tumor microenvironment-dependent 18F-FDG, 18F-fluorothymidine, and 18F-misonidazole uptake: a pilot study in mouse models of human non-small cell lung cancer. J Nucl Med 2012; 53: 1262–8. [DOI] [PubMed] [Google Scholar]
- 22. Olive, PL, Aquino-Parsons, C, MacPhail, SHet al. . Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res 2001; 61: 8924–9. [PubMed] [Google Scholar]
- 23. Santiago, A, Eicheler, W, Bussink, Jet al. . Effect of cetuximab and fractionated irradiation on tumour micro-environment. Radiother Oncol 2010; 97: 322–9. [DOI] [PubMed] [Google Scholar]
- 24. Hoogsteen, IJ, Marres, HA, van denHoogen, FJet al. Expression of EGFR under tumor hypoxia: identification of a subpopulation of tumor cells responsible for aggressiveness and treatment resistance. Int J Radiat Oncol Biol Phys 2012; 84: 807–14. [DOI] [PubMed] [Google Scholar]
- 25. Richards, CH, Mohammed, Z, Qayyum, Tet al. . The prognostic value of histological tumor necrosis in solid organ malignant disease: a systematic review. Future Oncol 2011; 7: 1223–35. [DOI] [PubMed] [Google Scholar]
- 26. Soini, Y, Pääkkö, P, Lehto, VP. Histopathological evaluation of apoptosis in cancer. Am J Pathol 1998; 153: 1041–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vakkila, J, Lotze, MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol 2004; 4: 641–8. [DOI] [PubMed] [Google Scholar]
- 28. Heddleston, JM, Li, Z, Lathia, JDet al. . Hypoxia inducible factors in cancer stem cells. Br J Cancer 2010; 102: 789–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loeffler, JS, Durante, M. Charged particle therapy—optimization, challenges and future directions. Nat Rev Clin Oncol 2013; 10: 411–24. [DOI] [PubMed] [Google Scholar]
- 30. Humm, JL, Cobb, LM. Nonuniformity of tumor dose in radioimmunotherapy. J Nucl Med 1990; 31: 75–83. [PubMed] [Google Scholar]
- 31. Kassis, AI. Therapeutic radionuclides: biophysical and radiobiologic principles. Semin Nucl Med 2008; 38: 358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ritter, MA, Cleaver, JE, Tobias, CA. High-LET radiations induce a large proportion of non-rejoining DNA breaks. Nature 1977; 266: 653–5. [DOI] [PubMed] [Google Scholar]
- 33. Sgouros, G. Alpha-particles for targeted therapy. Adv Drug Deliv Rev 2008; 60: 1402–6. [DOI] [PubMed] [Google Scholar]
- 34. Raju, MR, Eisen, Y, Carpenter, Set al. . Radiobiology of alpha particles. III. Cell inactivation by alpha-particle traversals of the cell nucleus. Radiat Res 1991; 128: 204–9. [PubMed] [Google Scholar]
- 35. Sgouros, G, Song, H. Cancer stem cell targeting using the alpha-particle emitter, 213Bi: mathematical modeling and feasibility analysis. Cancer Biother Radiopharm 2008; 23: 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sgouros, G, Roeske, JC, McDevitt, MRet al. . MIRD Pamphlet No. 22 (abridged): radiobiology and dosimetry of alpha-particle emitters for targeted radionuclide therapy. J Nucl Med 2010; 51: 311–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brechbiel, MW. Targeted alpha-therapy: past, present, future? Dalton Trans 2007; 43: 4918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chan, HS, deBlois, E, Morgenstern, Aet al. In vitro comparison of 213Bi- and 177Lu-radiation for peptide receptor radionuclide therapy. PLoS One 2017; 12: e0181473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dahle, J, Bruland, ØS, Larsen, RH. Relative biologic effects of low-dose-rate alpha-emitting 227Th-rituximab and beta-emitting 90Y-tiuexetan-ibritumomab versus external beam X-radiation. Int J Radiat Oncol Biol Phys 2008; 72: 186–92. [DOI] [PubMed] [Google Scholar]
- 40. Abbas, N, Bruland, ØS, Brevik, EMet al. . Preclinical evaluation of 227Th-labeled and 177Lu-labeled trastuzumab in mice with HER-2-positive ovarian cancer xenografts. Nucl Med Commun 2012; 33: 838–47. [DOI] [PubMed] [Google Scholar]
- 41. Fichou, N, Gouard, S, Maurel, Cet al. . Single-dose anti-CD138 radioimmunotherapy: bismuth-213 is more efficient than lutetium-177 for treatment of multiple myeloma in a preclinical model. Front Med (Lausanne) 2015; 2: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Makvandi, M, Dupis, E, Engle, JWet al. . Alpha-emitters and targeted alpha therapy in oncology: from basic science to clinical investigations. Target Oncol 2018; 13: 189–203. [DOI] [PubMed] [Google Scholar]
- 43. Edem, PE, Fonslet, J, Kjaer, Aet al. . In vivo radionuclide generators for diagnostics and therapy. Bioinorg Chem Appl 2016; 2016: 6148357–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rockwell, S, Dobrucki, IT, Kim, EYet al. . Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med 2009; 9: 442–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Hara, JA, Blumenthal, RD, Grinberg, OYet al. . Response to radioimmunotherapy correlates with tumor pO2 measured by EPR oximetry in human tumor xenografts. Radiat Res 2001; 155: 466–73. [DOI] [PubMed] [Google Scholar]
- 46. Nakano, T, Suzuki, Y, Ohno, Tet al. . Carbon beam therapy overcomes the radiation resistance of uterine cervical cancer originating from hypoxia. Clin Cancer Res 2006; 12: 2185–90. [DOI] [PubMed] [Google Scholar]
- 47. Moeller, BJ, Richardson, RA, Dewhirst, MW. Hypoxia and radiotherapy: opportunities for improved outcomes in cancer treatment. Cancer Metastasis Rev 2007; 26: 241–8. [DOI] [PubMed] [Google Scholar]
- 48. Penfold, SN, Brown, MP, Staudacher, AHet al. . Monte Carlo simulations of dose distributions with necrotic tumor targeted radioimmunotherapy. Appl Radiat Isot 2014; 90: 40–5. [DOI] [PubMed] [Google Scholar]
- 49. Furusawa, Y, Fukutsu, K, Aoki, Met al. . Inactivation of aerobic and hypoxic cells from three different cell lines by accelerated (3)He-, (12)C- and (20)Ne-ion beams. Radiat Res 2000; 154: 485–96. [DOI] [PubMed] [Google Scholar]
- 50. Blakely, EA, Ngo, FQH, Curtis, SBet al. . Heavy-ion radiobiology—cellular studies. Adv Radiat Biol 1984; 11: 295–389. [Google Scholar]
- 51. Tinganelli, W, Durante, M, Hirayama, Ret al. . Kill-painting of hypoxic tumours in charged particle therapy. Sci Rep 2015; 5: 17016–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pignalosa, D, Durante, M. Overcoming resistance of cancer stem cells. Lancet Oncol 2012; 13: e187–8. [DOI] [PubMed] [Google Scholar]
- 53. Harrison, LB, Chadha, M, Hill, RJet al. . Impact of tumor hypoxia and anemia on radiation therapy outcomes. Oncologist 2002; 7: 492–508. [DOI] [PubMed] [Google Scholar]
- 54. Benej, M, Pastorekova, S, Pastorek, J. Carbonic anhydrase IX: regulation and role in cancer. Subcell Biochem 2014; 75: 199–219. [DOI] [PubMed] [Google Scholar]
- 55. Divgi, CR, Bander, NH, Scott, AMet al. . Phase I/II radioimmunotherapy trial with iodine-131-labeled monoclonal antibody G250 in metastatic renal cell carcinoma. Clin Cancer Res 1998; 4: 2729–39. [PubMed] [Google Scholar]
- 56. Steffens, MG, Boerman, OC, deMulder, PHet al. Phase I radioimmunotherapy of metastatic renal cell carcinoma with 131I-labeled chimeric monoclonal antibody G250 .Clin Cancer Res 1999; 5: 3268s–74s. [PubMed] [Google Scholar]
- 57. Divgi, CR, O'Donoghue, JA, Welt, Set al. . Phase I clinical trial with fractionated radioimmunotherapy using 131I-labeled chimeric G250 in metastatic renal cancer. J Nucl Med 2004; 45: 1412–21. [PubMed] [Google Scholar]
- 58. Brouwers, AH, Mulders, PF, deMulder, PHet al. Lack of efficacy of two consecutive treatments of radioimmunotherapy with 131I-cG250 in patients with metastasized clear cell renal cell carcinoma. J Clin Oncol 2005; 23: 6540–8. [DOI] [PubMed] [Google Scholar]
- 59. Stillebroer, AB, Boerman, OC, Desar, IMet al. . Phase 1 radioimmunotherapy study with lutetium 177-labeled anti-carbonic anhydrase IX monoclonal antibody girentuximab in patients with advanced renal cell carcinoma. Eur Urol 2013; 64: 478–85. [DOI] [PubMed] [Google Scholar]
- 60. Muselaers, CH, Boers-Sonderen, MJ, vanOostenbrugge, TJet al. Phase 2 study of lutetium 177-labeled anti-carbonic anhydrase IX monoclonal antibody girentuximab in patients with advanced renal cell carcinoma. Eur Urol 2016; 69: 767–70. [DOI] [PubMed] [Google Scholar]
- 61. Street, HH, Goris, ML, Fisher, GAet al. . Phase I study of 131I-chimeric(ch) TNT-1/B monoclonal antibody for the treatment of advanced colon cancer. Cancer Biother Radiopharm 2006; 21: 243–56. [DOI] [PubMed] [Google Scholar]
- 62. Patel, SJ, Shapiro, WR, Laske, DWet al. . Safety and feasibility of convection-enhanced delivery of Cotara for the treatment of malignant glioma: initial experience in 51 patients. Neurosurgery 2005; 56: 1243–52. [DOI] [PubMed] [Google Scholar]
- 63. Chen, S, Yu, L, Jiang, Cet al. . Pivotal study of iodine-131-labeled chimeric tumor necrosis treatment radioimmunotherapy in patients with advanced lung cancer. J Clin Oncol 2005; 23: 1538–47. [DOI] [PubMed] [Google Scholar]
- 64. Bryan, RA, Jiang, Z, Jandl, Tet al. . Treatment of experimental pancreatic cancer with 213-Bismuth-labeled chimeric antibody to single-strand DNA. Expert Rev Anticancer Ther 2014; 14: 1243–9. [DOI] [PubMed] [Google Scholar]
- 65. Dadachova, E, Nosanchuk, JD, Shi, Let al. . Dead cells in melanoma tumors provide abundant antigen for targeted delivery of ionizing radiation by a mAb to melanin. Proc Natl Acad Sci U S A 2004; 101: 14865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dadachova, E, Revskaya, E, Sesay, MAet al. . Pre-clinical evaluation and efficacy studies of a melanin-binding IgM antibody labeled with 188Re against experimental human metastatic melanoma in nude mice. Cancer Biol Ther 2008; 7: 1116–27. [DOI] [PubMed] [Google Scholar]
- 67. Nosanchuk, JD, Jeyakumar, A, Ray, Aet al. . Structure-function analysis and therapeutic efficacy of antibodies to fungal melanin for melanoma radioimmunotherapy. Sci Rep 2018; 8: 5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Al-Ejeh, F, Darby, JM, Brown, MP. Chemotherapy synergizes with radioimmunotherapy targeting la autoantigen in tumors. PLoS One 2009; 4: e4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Staudacher, AH, Al-Ejeh, F, Fraser, CKet al. . The La antigen is over-expressed in lung cancer and is a selective dead cancer cell target for radioimmunotherapy using the La-specific antibody APOMAB(R). EJNMMI Res 2014; 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Staudacher, AH, Bezak, E, Borysenko, Aet al. . Targeted alpha-therapy using 227Th-APOMAB and cross-fire antitumour effects: preliminary in-vivo evaluation. Nucl Med Commun 2014; 35: 1284–90. [DOI] [PubMed] [Google Scholar]
- 71. Epstein, AL, Chen, FM, Taylor, CR. A novel method for the detection of necrotic lesions in human cancers. Cancer Res 1988; 48: 5842–8. [PubMed] [Google Scholar]
- 72. Chen, FM, Epstein, AL, Li, Zet al. . A comparative autoradiographic study demonstrating differential intratumor localization of monoclonal antibodies to cell surface (Lym-1) and intracellular (TNT-1) antigens. J Nucl Med 1990; 31: 1059–66. [PubMed] [Google Scholar]
- 73. Hornick, JL, Sharifi, J, Khawli, LAet al. . A new chemically modified chimeric TNT-3 monoclonal antibody directed against DNA for the radioimmunotherapy of solid tumors. Cancer Biother Radiopharm 1998; 13: 255–68. [DOI] [PubMed] [Google Scholar]
- 74. Al-Ejeh, F, Darby, JM, Brown, MP. The La autoantigen is a malignancy-associated cell death target that is induced by DNA-damaging drugs. Clin Cancer Res 2007; 13: 5509s–18s. [DOI] [PubMed] [Google Scholar]
- 75. Al-Ejeh, F, Darby, JM, Pensa, Ket al. . In vivo targeting of dead tumor cells in a murine tumor model using a monoclonal antibody specific for the La autoantigen. Clin Cancer Res 2007; 13: 5519s–27s. [DOI] [PubMed] [Google Scholar]
- 76. Al-Ejeh, F, Darby, JM, Tsopelas, Cet al. . APOMAB, a La-specific monoclonal antibody, detects the apoptotic tumor response to life-prolonging and DNA-damaging chemotherapy. PLoS One 2009; 4: e4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Al-Ejeh, F, Staudacher, AH, Smyth, DRet al. . Postchemotherapy and tumor-selective targeting with the La-specific DAB4 monoclonal antibody relates to apoptotic cell clearance. J Nucl Med 2014; 55: 772–9. [DOI] [PubMed] [Google Scholar]
- 78. Scott, AM. Radioimmunotherapy of prostate cancer: does tumor size matter? J Clin Oncol 2005; 23: 4567–9. [DOI] [PubMed] [Google Scholar]
- 79. Howell, RW, Azure, MT, Narra, VRet al. . Relative biological effectiveness of alpha-particle emitters in vivo at low doses. Radiat Res 1994; 137: 352–60. [PMC free article] [PubMed] [Google Scholar]
- 80. Back, T, Jacobsson, L. The alpha-camera: a quantitative digital autoradiography technique using a charge-coupled device for ex vivo high-resolution bioimaging of alpha-particles. J Nucl Med 2010; 51: 1616–23. [DOI] [PubMed] [Google Scholar]
- 81. AL Darwish, R, Staudacher, AH, Li, Yet al. . Development of a transmission alpha particle dosimetry technique using A549 cells and a Ra-223 source for targeted alpha therapy. Med Phys 2016; 43: 6145. [DOI] [PubMed] [Google Scholar]
- 82. AL Darwish, R, Staudacher, AH, Bezak, Eet al. . Autoradiography imaging in targeted alpha therapy with Timepix detector. Comput Math Methods Med 2015; 2015: 612580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Petersen, C, Eicheler, W, Frömmel, Aet al. . Proliferation and micromilieu during fractionated irradiation of human FaDu squamous cell carcinoma in nude mice. Int J Radiat Biol 2003; 79: 469–77. [DOI] [PubMed] [Google Scholar]