

Abstract
The successful use of chimeric antigen receptor (CAR) for hematological cancer treatment has influenced the direction taken in translational research toward an increasing focus on personalized targeted immunotherapy. Thus, a growing number of labs worldwide are now interested in testing their old antibody collections in this format to broaden the spectrum of utility and improve safety and efficacy. We herein present a straightforward protocol for the identification of an antibody from a hybridoma and the design of the single chain fragment that will be placed on the extracellular part of the CAR construct. We further show how to test the expression and the activity of the construct in primary T cells. We illustrate our demonstration with two new CARs targeted against the B cell receptor, more precisely the light chains κ and λ, that represent potential alternatives to the CD19 CAR used in the treatment of B-cell malignancies.
Keywords: hybridoma, antibody, single chain variable fragment, chimeric antigen receptor
Statement of Significance
Chimeric antigen receptor molecules are becoming increasingly popular, and an easy-to-follow method to isolate and express them is herein presented.
INTRODUCTION
Chimeric antigen receptor (CAR)-expressing T cells are now used to treat leukemia and lymphoma patients who are refractory to standard therapy. These powerful cells combine the specific target recognition offered by an antibody fused with parts of the natural signaling machinery of the T-cell receptor (TCR) (Fig. 1A). CAR is usually expressed in immune effector cells (NK or T cells) isolated from the patients (autologous settings), from a donor (allogeneic settings) or in a cell line. They are genetically transformed ex vivo or in vitro, (re)-injected into the patients and expected to reach the tumor and kill it [1].
Figure 1.
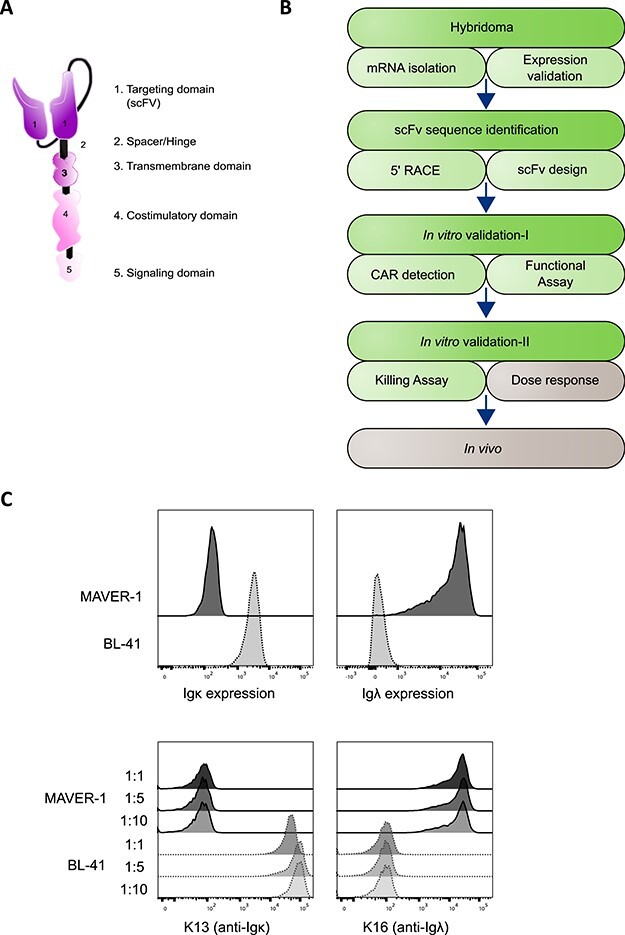
The workflow of CAR development from a hybridoma. (A) The visual depiction of a second-generation CAR design. (B) A simplified roadmap of CAR development and assessment with the focus of the article indicated by the green part of the diagram. (C) Validation of target antigen expression in MAVER-1 (Igλ+, dark gray) and BL-41 (Igκ+, light gray) cell lines with the indicates antibodies. The upper panel was stained with commercial antibodies, and lower was done using the hybridoma supernatants in duplicates at the indicated dilutions. Representative data of two independent experiments are shown.
A CAR molecule at the surface of an effector cell will, upon target recognition, form clusters and trigger endogenous signaling component recruitment. The intracellular signaling tail is a combination of domains that generate a fast response in the effector cell against the target (killing and cytokine release) and a long persistence signal to allow a durable and lasting response. Thus, a CAR is able to transform a polyclonal population of effector cells into tumor killing cells by retargeting them against a selected target. This is what was successfully achieved when patients were treated with anti-CD19 CAR T cells [2]. Here, a heterogeneous population of T cells was redirected to gain specificity against the B-cell marker CD19, which lead to the destruction of malignant B cells as well as the healthy ones. The latter has been shown to lead to complications in patients and stimulated the quest for alternative antigens, such as the ones we present here, targeting only a subgroup of the population.
Although antibodies have been sequenced for almost four decades [3] and a very comprehensive protocol was released 20 years ago to isolate antibody sequences [4], a practical protocol for the design of CAR molecules does not seem to be available. We herein present a method that can be adapted in any lab to achieve the creation of a CAR. We illustrate it by describing the isolation of the coding sequence from two validated hybridomas producing antibodies with specificity against the B cell receptor (BCR) light chain -Igκ and -Igλ chains. We will explain how the single chain variable fragment (scFv) sequence was extracted and designed to create the binding part of the CAR. Finally, we will also give insight into the preliminary tests that should be run to confirm the expression and activity of a new CAR. The complete procedure is displayed in Fig. 1B.
MATERIALS AND METHODS
5′-RACE
mRNA preparation
Pellets from 1 million cells and isolation of the total RNA were done with a starting volume of 600 μL lysis buffer, then every step of the manufacturer’s protocol was followed (Absolutely RNA Miniprep, Agilent Technologies, USA) including the DNAse I treatment. Importantly, the lysis of each pellet was divided in two and the protocol was performed in parallel to ensure that somatic mutations detected are original and not coming from the use of a Taq polymerase. Final elution was performed in 50 μL of pre-warmed buffer. All mRNAs were quality/quantity checked on a Nanodrop apparatus (Thermo Fisher, USA) and stored at −20°C until cDNA preparation.
cDNA synthesis
The reverse transcription was performed using SuperScript III Reverse Transcriptase (200 U/μL, Invitrogen, Eureka, CA, USA) with a reaction mix of Vfinal = 20.5 μL, prepared in two parts containing: 1 μL oligo-dT (50 μM, Invitrogen), 1 μL dNTPs (10 mM, Thermo Fisher), 1 μg mRNA and dH2O to a final V of 14 μL. This part was then incubated at 65°C for 5 minutes and chilled on ice, then the rest of the mix was added as follows: 4 μL first strand buffer, 1 μL DTT (0.1 M, Invitrogen), 0.5 μL RNasin (40 U/μL, Promega) and 1 μL Reverse Transcriptase. The mix was incubated at 50°C for 1 hour, then the RNasin was inactivated by 15 minutes incubation at 60°C. The mix was treated with 1 μL RNaseH (5000 U/mL, New England Biolabs, USA) at 37°C for 20 minutes, to remove the remaining mRNA. The nucleotides were precipitated by NaAc-EtOH method: 21.5 μL cDNA is mixed with 0.5 μL Glycogen (20 mg/ml, Thermo Fisher), which is used as carrier, and 1/10 V of NaAc (3 M pH 5.5, Invitrogen) and 2.5 V EtOH 100%, incubated at −20°C for minimum 30 minutes. The precipitated cDNA was spun down at 10,000g at 4°C for 10 minutes, washed once with 200 μL 70% EtOH and finally resuspended in 21 μL dH2O. One μL was used to control the presence of product on an agarose gel, 10 μL for dC tailing and 10 μL kept at −20°C.
5′dC-tailing
Samples were placed at 95°C for 1 minute and directly chilled on ice; this will break the structures formed in the DNA that might mask the site of tailing. The reaction was performed using TdT enzyme and buffer mix (400 U/μL, Roche, Switzerland): 10 μL cDNA, 4 μL TdT reaction buffer 5X, 4 μL CoCl2, 1 μL dCTP (10 mM, Thermo Fisher) and 1 μL TdT incubated for 15 minutes at 37°C, and the reaction was stopped by adding the NaAc-EtOH precipitation components as described in the previous section.
Nested PCR
One microliter of the dC-tailed cDNA was finally amplified in a serial nested PCR using the constant part-specific primers and a universal dC-annealing primer (Fig. 2B). The primers were designed in-house except pGI15_TOPO [5], mIgG2bCH1, mIgGKCrev, mIgG1CH1–2, mIgG2bCH1–2 [6], mIgG2aCH1–2 [7] and mIgGKCrev-2 [8]. Each PCR was run in these conditions: 4′ 94°C for the initial denaturation and 25 cycles (1′ 94°C, 1′ 53°C, 1′ 72°C) with the following mix: 5 μL 10X TAQ Buffer, 1.5 μL of primer (10 mM stock), 1 μL dC-tailed cDNA, 1 μL Taq DNA polymerase (Hospital collection) and dH2O added to a Vfinal = 50 μL. Both PCR products were separated on a gel; the second is the same as the first, but 1 μL of the first reaction was used as a template. The expected size should be larger than 450 bp, a smear should be visible (Fig. 2C).
Figure 2.
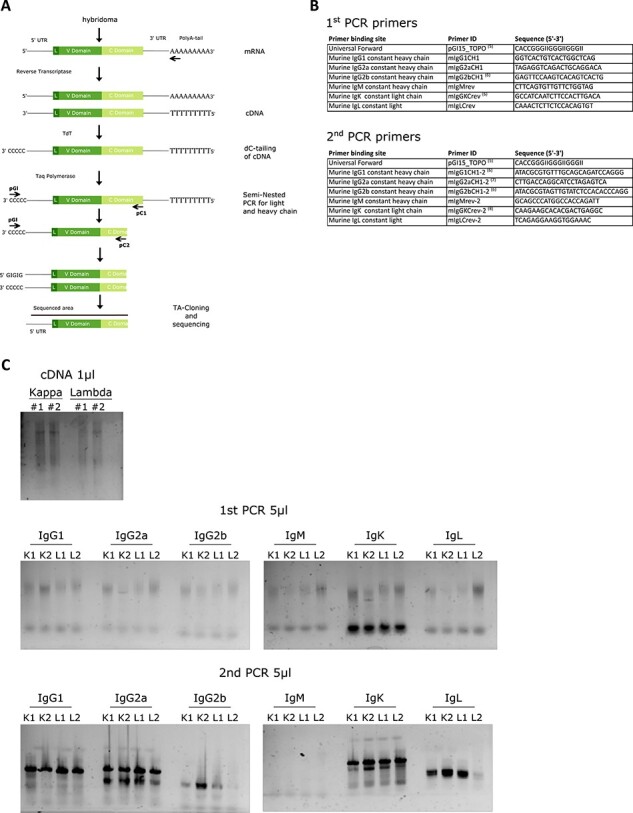
Investigating the hybridoma by 5′-RACE. (A) The depiction of 5′-RACE step by step protocol. (B) List of primers used in this study and references to already published ones. (C) Acquired gel pictures throughout the 5′-RACE protocol, each kappa and lambda hybridomas were divided into two and the protocol was followed in two replicates (e.g. named as K#1 and K#2). The top picture shows the result of the cDNA synthesis, the middle pictures the first PCR results of dC tailed samples and the bottom pictures the result of the second PCR performed with the first PCR products. The ladder information corresponding to 500 and 400 bp bands were specified as well as the primer dimer with an asterisk on each gel image.
Sequence identification, scFv design and subcloning into a signaling cassette
The PCR products were run on gel, and bands were extracted and purified using NucleoSpin® Gel and PCR Clean-up kit (Macherey-Nagel, Germany); after checking the presence of the band and estimating its concentration on a gel, it was subcloned into pGEM T/A cloning vector following manufacturer’s protocol (Promega, USA) and transformed into NEB 5-alpha competent cells (New England Biolabs, USA) and blue/white colonies were screened. Single colonies were picked, and the presence of an insert was determined using EcoRI (New England Biolabs). Insert-containing vectors were sent to sequencing (Eurofins MWG, Germany) using T7 and M13(−29) primers. The sequences were analyzed using the IMGT (http://www.imgt.org/IMGT_vquest/vquest) online tool to identify the V chains, hypervariable domains and the somatic mutations. The beginning and the end of the V domain were defined according to IMGT nomenclature, and these limits were used to design the scFv. The resulting protein sequence of the construct was constructed as follows, signal sequence (METDTLLLWVLLLWVPGSTG)-VL-(G4S)4(GGGGSGGGGSGGGGSGGGGS)-VH; it was ordered as codon optimized for production in human cells (Eurofins MWG), and restriction sites were added (NcoI/BamHI) to subclone it into a signaling tail-containing vector (pENTR-seq8843). Verified clones were recombined using the Gateway cloning system (Invitrogen) into pMP71 converted plasmid [5].
T-cell transduction, CAR detection and killing assay
T-cell transduction, cytokine staining and bioluminescence (BLI)-based killing assay have been described in details elsewhere [9]. Briefly, primary T-cell transduction with retroviral particles was performed by double spinoculation followed by expansion on CD3/CD28 beads. The staining of the CAR was performed with Biotin-SP-AffiniPure F (ab′)2 Fragment Goat Anti-Mouse IgG (Jackson ImmuResearch, USA), followed by a secondary staining with Streptavidin`-PE (Biolegend, USA). CAR-expressing T cells were co-cultured with cells positive or negative for the target antigen, and TNF-α was detected by flow cytometry using TNF-α-PE (MAb11, BD Biosciences). The samples were run on a BD FACSCanto flow cytometer (BD Biosciences, USA), and data were analyzed using FlowJo software (Treestar Inc., USA). BLI killing assay was also based on a co-culture experiment where target cells permanently expressed firefly Luciferase gene, and killing was correlated to the luminescence signal [10].
Expression validation of the target cells
Target cells were assessed for their expression status for Igκ and Igλ by flow cytometry. The first assessment was done by staining with the following commercial antibodies; anti-human Igκ-APC (MHK-49, Biolegend) and anti-human Igλ-PE (1-155-2, Thermo Fisher). The same cell lines were also stained with the hybridoma supernatants with varying dilutions, followed by a secondary staining with anti-mouse IgG-APC (Poly4053, Biolegend).
Statistical analysis
Student’s t-test was used when two groups were compared. BLI killing assay was analyzed by non-linear regression (curve fit) on GraphPad Prism® (GraphPad Software, USA).
RESULTS
Isolation of the antibody coding sequence from a hybridoma
We previously created hybridomas producing anti-human Igκ and -Igλ antibodies (clones K13 and K16, respectively). The cells were grown and supernatants were tested for antibody production and confirmation of the target specificity, as shown in Fig. 1C; we compared the target recognition of our antibodies to commercial ones. We distinguished the BCR light chain isoforms by using model cell lines known to be positive for either κ or λ.
When the hybridoma culture reached around 1 million cells, pellets were prepared, lysed in mRNA extraction buffer and frozen until use (see Materials and Methods for kit listing and detailed procedure). Fig. 2A depicts the whole cloning procedure: first total mRNA was used to synthesize cDNA using oligo-dT as a primer. After treatment with RNaseH to digest remaining mRNA, the cDNA was dC tailed on its 5′-end using TdT enzyme. The reaction was stopped after 15 minutes and the tailed cDNA extracted by precipitation. This product was checked on a gel (Fig. 2C, top panel) and used as a template for a nested PCR using GI oligo that binds the TdT synthesized C-tail and antibody specific primers (Fig. 2B) annealing to the constant domain. The first and second amplifications were run on a gel (Fig. 2C, middle and bottom panels, respectively). These PCR reactions gave a first indication on the identity of the antibody chain type. As shown both anti-Igλ and anti-Igκ possess IgG1 as heavy chain and Igκ as light chain. Since IgG1 and IgG2a sequences are related we can observe their amplification with the different sets of primers; however the sequence obtained after sequencing of the antibody will provide information on the exact IgG identity.
The PCR reactions were then subcloned: since the PCR reactions were conducted with a Taq polymerase, one-nucleotide overhang was created and could accommodate a T/A cloning system. Bacterial colonies were picked and minipreps digested to estimate the size of the insert that is expected to be higher than 450 bp, but might vary due to the tailing of differently sized mRNA. This range ensures that a piece of the 5′-UTR will be present; hence the natural start detected (Fig. 3A).
Figure 3.
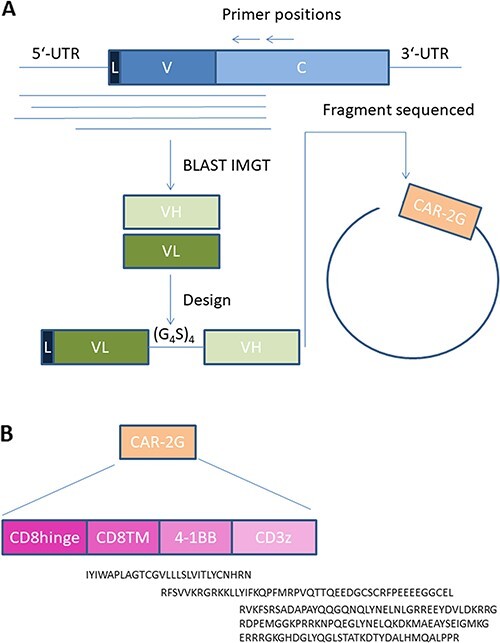
The discovery of a scFv sequence and the assembly of the CAR. (A) A simplified depiction of the acquisition of the heavy and light chain’s sequences through IMGTV database and a general scFv structure. (B) A general description of a second-generation CAR design with the protein sequences of each domains.
Identification of V-chain types and scFv design
The sequences were then identified using the IMGT database. In addition, somatic hypermutations were identified and the limit of the V chains was defined, which facilitates the further design of the V chains in the scFv (Fig. 3A). The preparation of scFv can be designed in multiple manners, but we experienced that the design starting with the light chain variable fragment (VL) including a secretion sequence (L chain) linked to the variable heavy chain (VL) by glycine serine repeats (G4S)4 was normally adaptable to all our constructs. Nevertheless, it is always an advantage to compare different designs by swapping VL and VH or testing linkers of different size if the “classical” design is not working or with low efficiency [11].
The protein sequence of the scFv fragments of the two antibodies was then ordered as a synthetic DNA, which was subsequently cloned in our system. Here the second-generation signaling tail was used (Fig. 2B). The final product of each CAR construct will thus be L-VL-linker-VH-CD8hinge-CD8TM-4-1BB-CD3ζ. The sequences of the different gene blocks are depicted under each entity and were based upon description in Uniprot database: CD8hinge and CD8TM, CD8A_HUMAN (P01732), 4-1BB, TNR9_HUMAN (Q07011), CD3ζ and CD3Z_HUMAN (P20963).
CAR expression and activity detection
The complete CAR coding sequence was then subcloned into a retroviral expression vector and particles were prepared using Gateway-compatible system as we previously described [5]. We routinely test the expression of our constructs in an easily manipulated cell line such as Jurkat cells before proceeding to primary cells. Indeed, this cell line can be transduced in one spinoculation and the transferred gene can usually be detected within 2 to 3 days. Fig. 4A shows the expression of the CARs in Jurkat cells detected by flow cytometry using specific antibodies against mouse antigen-binding fragment (Fab). From these data we confirmed that the design was leading to a correctly folded product expressed at the cell surface. We further validated this result by using a system based on primary human T cells. Here, stimulated T cells isolated from buffy coat were used as effector cells and transduced by double spinoculation (see Materials and Methods). Seven days after spinoculation, T cells were tested for CAR expression (Fig. 4B).
Figure 4.
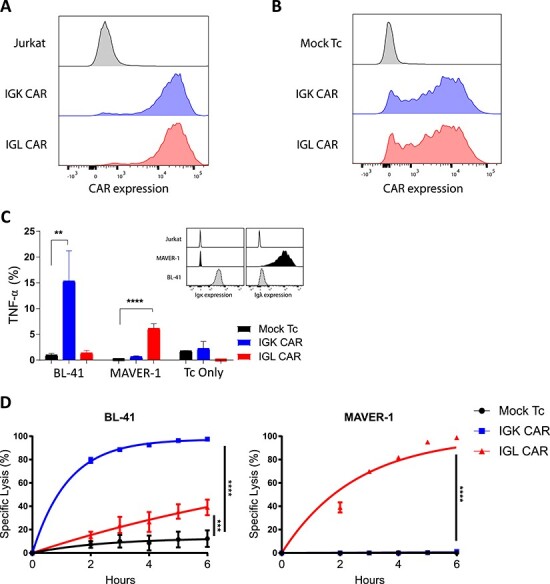
In vitro validation of CAR constructs. (A) Jurkat cells were retrovirally transduced with either IGK (blue) or IGL CAR (red). The expression was assessed by flow cytometry detection of anti-Fab antibody. Non-transduced Jurkat cells (gray) were used as baseline control, representative of N > 2. (B) Activated primary T-cells were retrovirally transduced with either IGK (blue) or IGL CAR (red). The expression was assessed by flow cytometry, and mock transduced T cells (gray) were used as a baseline control, representative of N = 2. (C) CAR expressing T cells (IGK, blue; IGL CAR, red; and mock, black) specificity to their respective targets in functional assay. Inset: staining of the target cells with specific antibodies, MAVER-1 (Igλ+, black), BL-41 (Igκ+, light gray) and as a negative control Jurkat (Ig-, white). Cytokine secretion (TNF-α) was analyzed by flow cytometry and plotted. Data represent mean ± SD of triplicates, Student t-test where **P < 0.01, ****P < 0.0001, N = 2. (D) Bioluminescence-based killing assay of CAR expressing T cells (IGK, blue; IGL CAR, red; and mock, black) against the same target cells as in (C) and plotted as percentage of lysis over time. Data represent mean ± SD of quadruplicates, Student t-test where ***P < 0.001, ****P < 0.0001, N = 2.
Analysis of CAR reactivity was performed using primary T cells in co-culture assay with target cells expressing the cognate antigen. Here BL-41 and MAVER-1 cell lines were used since their BCRs are Igκ + and Igλ+, respectively. The target cells were incubated with CAR T cells or mock-transduced T cells. The latter are used to set the baseline of the stimulation in order to distinguish specific CAR response from background. Indeed, T cells from diverse donors might react differentially against various targets; this can be explained by a difference in HLA composition between the blood donor and the target cells (alloreactivity). In addition, both CARs are here tested against the two target lines, in order to verify that the scFv has kept its original specificity. By using flow cytometry, the stimulation of cytokine (TNF-α) production in a 5-hour co-culture assay was detected by intracellular labeling of the T cells (Fig. 4C). As shown, each construct bound its expected target specifically and stimulated the effector cell upon target recognition.
It is expected that CAR will redirect effector cells against target cells to kill them, and the quantitative monitoring of the killing efficiency is sound. Chromium-51 (51Cr) release assay is still frequently used, but alternative methods avoiding the manipulation of radioactive substances are nowadays favored [10]. We routinely use BLI assay to monitor CAR-redirected effector cell efficacy. This assay is based on the presence of firefly Luciferase in target cells and the detection of its decreasing activity upon cell killing [12]. BL-41 and MAVER-1 cells were transduced to express the firefly Luciferase gene and then a pure population was isolated by FACS-based sorting. They were co-incubated with CAR-redirected T cells at the indicated ratio, and specific killing was quantified as the decrease of Luciferase signal (Fig. 4D). Importantly, every method has its limitations and the BLI killing assay can sometimes show unexpected high background killing. Here IGL CAR reacted slightly against BL-41 cells although no signal was detected by antibody staining. We have recently discussed the origin of these signals [13] and have evidence from recently performed experiments that this signal was probably due to IGL CAR-related tonic signaling rather than specific killing by IGL CAR. From these data we concluded that our novel CARs targeted against the two BCR light chains were expressed and were efficient to redirect T cells against B cells.
DISCUSSION
In this study we introduce a complete protocol for CAR preparation. We depicted the details presently being tested to conveniently extract an antibody sequence from a hybridoma and further design from this sequence a molecule that will redirect effector immune cells. We then displayed the minimal experiments required to validate that a given construct turns effector cells into a tumor cell killer. It is worth saying that due to the growing interest for using human antibodies as a source for CAR constructs, this protocol could be adapted. If humanized mice are used [14], only the primer sequences should be changed to human specific ones.
The clinical success of CD19 CAR in the treatment of B-cell malignancies has demonstrated the power of immunotherapy [2]. However, recent clinical data have shown that alternative solutions to CD19 CAR are needed; some patients become refractory to the treatment through several mechanisms directly related to the target recognition or the CAR itself [15]. Alternative B-cell CAR constructs have been identified [16–18] and some are presently being tested in the clinic [19, 20], even in alternative formats such as scFv placed in tandem on a single construct [19]. Importantly, there is growing evidence that more than one CAR molecule should be tested to identify the CAR with optimal affinity and function. Even if the original antibody has a high affinity for the target and the in vitro testing of the CAR construct is successful, the in vivo assessment might not follow this trend [21], thus more hybridomas will need to be tested. Although some of these CARs such as the CD22 CAR clone m971 [16] were isolated from phage display libraries [22], the majority of molecules still come from hybridoma, and we believe that the protocol proposed herein will help scientists to deliver the future clinical CARs.
Funding
This work was supported by the Research Council of Norway (284983), the Norwegian Cancer Society (6829007 to S.W.) and South-Eastern Norway Regional Health Authority (2016006 to H.K. and 2018591 to S.W.).
Acknowledgements
We are grateful to our colleagues from the laboratory of Translational Research and Immunomonitoring, in particular to Anne Fåne for expert technical assistance. We also thank the team of the Flow Cytometry Core Facility (Department of Core Facilities, Institute for Cancer Research, OUS).
References
- 1. June, CH, Sadelain, M. Chimeric antigen receptor therapy. N Engl J Med 2018; 379: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fesnak, AD, June, CH, Levine, BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 2016; 16: 566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dolby, TW, Devuono, J, Croce, CM. Cloning and partial nucleotide sequence of human immunoglobulin mu chain cDNA from B cells and mouse-human hybridomas. Proc Natl Acad Sci U S A 1980; 77: 6027–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doenecke, A, Winnacker, EL, Hallek, M. Rapid amplification of cDNA ends (RACE) improves the PCR-based isolation of immunoglobulin variable region genes from murine and human lymphoma cells and cell lines. Leukemia 1997; 11: 1787–92. [DOI] [PubMed] [Google Scholar]
- 5. Wälchli, S, Løset, GÅ, Kumari, Set al. . A practical approach to T-cell receptor cloning and expression. PLoS One 2011; 6: e27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lau, C, Gunnarsen, KS, Høydahl, LSet al. . Chimeric anti-CD14 IGG2/4 hybrid antibodies for therapeutic intervention in pig and human models of inflammation. J Immunol 2013; 191: 4769–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khantasup, K, Chantima, W, Sangma, Cet al. . Design and generation of humanized single-chain Fv derived from mouse hybridoma for potential targeting application. Monoclon Antib Immunodiagn Immunother 2015; 34: 404–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nilssen, NR, Frigstad, T, Pollmann, Set al. . DeltaPhage—a novel helper phage for high-valence pIX phagemid display. Nucleic Acids Res 2012; 40: e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walseng, E, Köksal, H, Sektioglu, IMet al. . A TCR-based chimeric antigen receptor. Sci Rep 2017; 7: 10713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karimi, MA, Lee, E, Bachmann, MHet al. . Measuring cytotoxicity by bioluminescence imaging outperforms the standard chromium-51 release assay. PLoS One 2014; 9: e89357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dwivedi, A, Karulkar, A, Ghosh, Set al. . Lymphocytes in cellular therapy: functional regulation of CAR T cells. Front Immunol 2018; 9: 3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown, CE, Wright, CL, Naranjo, Aet al. . Biophotonic cytotoxicity assay for high-throughput screening of cytolytic killing. J Immunol Methods 2005; 297: 39–52. [DOI] [PubMed] [Google Scholar]
- 13. Köksal, H, Dillard, P, Josefsson, SEet al. . Preclinical development of CD37CAR T-cell therapy for treatment of B-cell lymphoma. Blood Adv 2019; 3: 1230–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laffleur, B, Pascal, V, Sirac, Cet al. . Production of human or humanized antibodies in mice. Methods Mol Biol 2012; 901: 149–59. [DOI] [PubMed] [Google Scholar]
- 15. Orlando, EJ, Han, X, Tribouley, Cet al. . Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 2018; 24: 1504–6. [DOI] [PubMed] [Google Scholar]
- 16. Haso, W, Lee, DW, Shah, NNet al. . Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013; 121: 1165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scarfò, I, Ormhøj, M, Frigault, MJet al. . Anti-CD37 chimeric antigen receptor T cells are active against B and T cell lymphomas. Blood 2018; 132: 1495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vera, J, Savoldo, B, Vigouroux, Set al. . T lymphocytes redirected against the light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood 2006; 108: 3890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fry, TJ, Shah, NN, Orentas, RJet al. . CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 2018; 24: 20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramos, CA, Savoldo, B, Torrano, Vet al. . Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J Clin Invest 2016; 126: 2588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith, EL, Staehr, M, Masakayan, Ret al. . Development and evaluation of an optimal human single-chain variable fragment-derived BCMA-targeted CAR T cell vector. Mol Ther 2018; 26: 1447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiao, X, Ho, M, Zhu, Zet al. . Identification and characterization of fully human anti-CD22 monoclonal antibodies. MAbs 1: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]