

ABSTRACT
A bispecific antibody (bsAb) can simultaneously bind two different epitopes or antigens, allowing for multiple mechanistic functions with synergistic effects. BsAbs have attracted significant scientific attentions and efforts towards their development as drugs for cancers. There are 21 bsAbs currently undergoing clinical trials in China. Here, we review their platform technologies, expression and production, and biological activities and bioassay of these bsAbs, and summarize their structural formats and mechanisms of actions. T-cell redirection and checkpoint inhibition are two main mechanisms of the bsAbs that we discuss in detail. Furthermore, we provide our perspective on the future of bsAb development in China, including CD3-bsAbs for solid tumors and related cytokine release syndromes, expression and chemistry, manufacturing and controls, clinical development, and immunogenicity.
Keywords: bispecific antibody (bsAb), format, MOA, CRS, expression, check point, immunogenicity
Statement of Significance: This review provides insight into the molecular formats, mechanisms of action, production, and clinical development of bispecific antibodies (bsAbs) discovered and developed in China.
INTRODUCTION
Able to bind two different epitopes or antigens simultaneously, bispecific antibodies (bsAbs) are a class of artificial antibodies that have been developed in the past few decades. Nisonoff et al. described the concept of bsAbs in the 1960s [1] and used pepsin to generate univalent antigen-binding fragments (Fab) that specifically inhibited antigen precipitation and speculated new technologies “to attempt to prepare an antibody of two mixed specificities.” In the following decades, the rapid development of antibody engineering technologies led to many innovations in antibody design and production. In 1983, Milstein et al. produced the first bsAbs with intact immunoglobulin G (IgG) structure by hybrid–hybrid (quadroma) technology [2], by which Catumaxomab, a murine anti-EpCAM × anti-CD3 bsAb was produced and became the first marketed bsAb drug in the world in 2009 for controlling EpCAM-positive malignant ascites [3]. Catumaxomab was developed using mouse IgG2a and rat IgG2b to form a heterodimer pairing naturally. However, the drug was withdrawn from the market in 2017 for commercial reasons. The second bsAb brought to market was blinatumomab (Blincyto®, anti-CD19 × anti-CD3 antibody). Developed by Amgen, it was launched in 2014 for treating acute lymphoblastic leukemia [4]. Blinatumomab was generated as a recombinant format in a single-chain variable fragments of two antibodies, trademarked as BiTE® (invented in 1993) [5]. The third marked bsAb, Emicizumab (anti-FIXa × anti-FX, or Hemlibra®), was launched by Roche in 2017. Emicizumab brings together FIXa and FX to mimic the function of activated factor FVIII (FVIIIa), which is reduced in patients with hemophilia A [6]. Its core technologies termed ART-Ig consist of a method to identify a common light chain, isoelectric point (pI) engineering, and Fc heterodimerization by an electrostatic steering effect [7,8]. The common light chain (LC) of Emicizumab was discovered by screening the phage library using phage display, a common technology for bsAb discovery [9].
In the past several decades, over 100 bsAb structures and more than 30 technology platforms were reported and reviewed [10–14], including CrossMab (Roche) [15], DuoBody® (Genmab) [16], Nanobody® (Ablynx) [17], DVD-IgG (Abbvie) [18], DART® (Macrogenics) [19], Modular antibody technology™ (F-star) [20], Azymetric™ (Zymeworks) [21], TandAb® (Affimed) [22], XmAb® (Xencor) [23], Biclonics® (Merus) [24], ADAPTIR™ (Aptevo therapeutics) [25], etc. In addition of two bsAbs on the market, about 110 bsAbs designed to redirect T cells to kill tumor cells or to interact with two different disease mediators such as cells surface receptors, soluble ligands, and other proteins are currently under clinical trials worldwide [26, 27]. Twenty-one of these clinical trials are undergoing in China. With a record number of molecules approved for clinical trials by the China National Medical Products Administration (NMPA) and fueled by continuous financial investments, the product pipelines of Chinese biotech companies are becoming more innovative and competitive.
Bispecific antibodies are clinically promising as the next generation of biotherapeutics for cancer, autoimmune, and infectious diseases [28]. A strong scientific rationale for engaging two or more targets in the therapeutic strategy to a specific disease is often required for improving the efficacy, safety, and drug resistance of a single-mediator targeted biologic, which can be achieved by a bsAb with potential for novel functionalities that cannot be performed by a single target antibody [27, 28]. Meanwhile, there remain multiple challenges during the development, including preventing toxicity and immunogenicity caused by novel epitopes, meeting thresholds for activating multiple molecular pathways, and ensuring the quantity and quality during bsAb production. This article provides an overall review on the designs of molecular structures, generation, and target selection of bsAbs under clinical trials, and prospects for the future development in China.
BISPECIFIC PLATFORM TECHNOLOGIES
Several platforms for bispecific constructions, including YBODY®, CRIB™, ITab™, FIT-Ig™, WuXiBody™, and SMAB™, have been developed in China.
YBODY®, developed by Wuhan YZY Biopharma Co., Ltd. (YZYBio), is an asymmetric bsAb platform. The bsAb based on this platform composes of three polypeptide chains: a heavy chain (HC), a light chain (LC), and a single chain (SC) to form three segments. The first segment is a Fab that targets tumor-associated antigen (TAA). The second is a single-chain variable fragment (scFv), which targets immune-associated antigen (IAA) [29]. The third segment is a heterodimeric fragment crystallizable (Fc) region (Fig. 1A). In this platform, two engineering technologies are applied on Fc-CH3 interface based on both Knobs-in-Holes (KiHs), which is formed by paring one large hydrophobic reside in one Fc chain with a hole-like structure of small residues in the other Fc chain [30], and electrostatic steering effects or “salt-bridge” [31]. These two Fc technologies overcome the HC-HC and the SC-SC misassembles. Furthermore, the proprietary design of scFv in YBODY® is applied for the SC and overcomes the HC-LC misassemble [29]. The integration of these three engineering technologies ensures the favorable expression of desired heterodimeric bsAb in CHO cells and downstream purification of the desired bsAb product by chromatography. A more detailed discussion is provided in section expression and Production.
Figure 1.
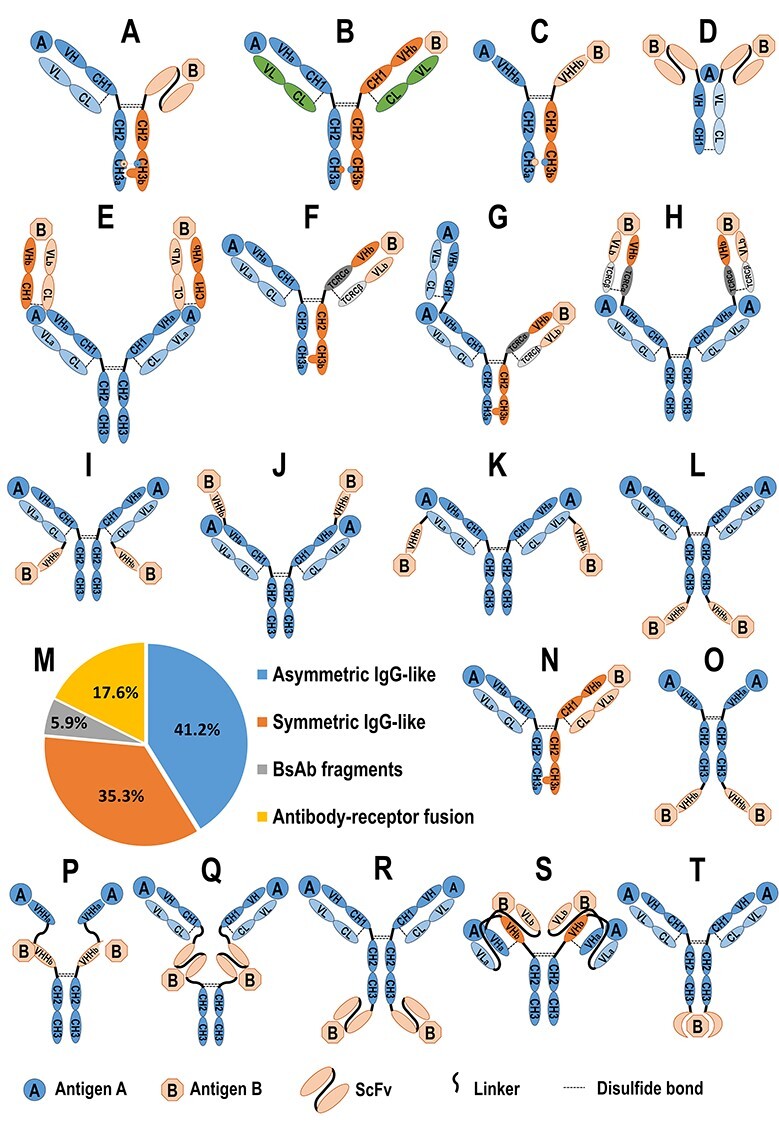
The bsAbs developed by Chinese biopharmaceutical companies in different formats. The structure diagrams of YBODY®, CRIB™, ITab™, FIT-Ig™, WuXiBody™, and SMAB™ are illustrated. (A) YBODY® composites three segments including a Fab, scFv, and Fc region, wherein the Fab targets tumor-associated antigen (TAA), scFv to IAA, and heterodimeric Fc is stabilized by KiHs and a salt bridge. (B) and (C) In CRIB™ platform, the charge network among various Fc bonds is manipulated to increase the formation of heterodimers. (D) In ITab™, a Fab domain binds to CD3 and two scFv domains bind to tumor surface antigen to form an immune synapse to recruit and activate T cells at the tumor site. (E) In tetravalent FIT-Ig™ technology, two parental antibodies are combined into one single molecule, where the Fab A is structurally fused to Fab B in tandem at its N-terminus. (F, G, and H) In WuXiBody™, TCR Cα/Cβ pair, (where TCR Cα and TCR Cβ represent the α and β chains of human TCR constant region, respectively), is used to substitute the first constant domain of the heavy chain (CH1) and the constant domain of the κ or λ light chain (CL) of one of the two Fabs, while maintaining the variable regions of heavy chain (VH) and light chain(VL) pair (VH/VL) of this Fab and the whole structure of the other Fab to be their native forms. WuXiBody can be assembled by 1 or 2 of the different Fabs connected with each Fc of a heterodimer or a homodimer to provide 1 + 1 asymmetric bivalent (F), 2 + 1 asymmetric trivalent (G) and 2 + 2 symmetric tetravalent (H) bispecific antibodies (bsAbs), respectively. (I–L) In SMAB™, a VHH is fused to the C-terminal end of each light chain (I), or to the N-terminal end of each HC (J), or to the N-terminal end of each light chain (K), or to the C-terminal end of each HC (L) to provide 2 + 2 symmetric tetravalent bsAbs. (M) 17 of 20 bsAbs, which are currently under clinical trials in China have disclosed their structural formats, among which include seven asymmetric IgG-like bsAbs (7 of 17, 41.2%), six symmetric IgG-like bsAb (6 of 17, 35.3%), one bispecific fragment (1 of 17, 5.9%), and three antibody-receptor fusion proteins (3 of 17, 17.6%). Asymmetric IgG-like structure can be sub-classified as scFv-Fab IgG (A), hetero-H+ common LC IgG (B), hetero-VHH (C), and Fab-arm exchange (N). Symmetric IgG-like structure can be sub-classified as (Fab)2-IGG (E), VHH-Fc-VHH (O), tandem VHHs-Fc (P), (Fab)2-(scFv)2-Fc (Q) where the two identical scFvs target antigen A and two Fabs target antigen B, and IgG-(scFv)2 (R); bispecific fragments structure involved scFv(s) and Fab(s) (D), and an antibody-receptor fusion protein (S) consisting a whole monoclonal antibody connected on its two C-terminal of HCs with the ex-cellular domain of the receptor.
Charge Repulsion Improved Bispecific (CRIB™) platform was developed by Alphamab Co., Ltd. (Alphamab) to manipulate the charge network among various Fc bonds (Fig. 1B and C), which greatly increased the formation of heterodimers [32].
Immune-therapy antibody (ITab™), a unique bispecific platform developed by Shanghai Generon Co., Ltd. (Generon), was designed to produce bispecific antibodies for activating T cells directly (Fig. 1D). It simultaneously binds to CD3 and a specific tumor surface antigen to form an immune synapse to recruit to and activate T cells at the tumor site [33] to release mediators that lyse the tumor cells. ITab™ antibodies can drive expansion of T cells, rendering them as serial killers of tumor cells.
Tetravalent Fabs-In-Tandem immunoglobulins(FIT-Ig™) technology, developed by EpimAb Biotherapeutics, Inc. (EpimAb), combines the functions of two parental antibodies into one single molecule (Fig. 1E) [34], which is achieved by re-arranging the DNA sequences of two monoclonal antibodies into three-gene construct(s) and by co-expressing them in mammalian cells.
WuXiBody™ platform developed by WuXi Biologics is based on the human T-cell receptor (TCR) constant region of α chain (Cα) and constant region of β chain (Cβ) to ensure correct HC/LC pairing of bispecific antibodies [35]. The platform enables almost any two mAb sequence pairs to be assembled into the bispecific construct. Its structural flexibility makes the platform convenient to build 1 + 1 asymmetric bivalent bsAbs (Fig. 1F), 2 + 1 asymmetric trivalent bsAbs (Fig. 1G), and 2 + 2 symmetric tetravalent bsAbs (Fig. 1H).
SMAB™, a bsAb designed by fusing a VHH (or single-domain antibody (SDA)) to a monoclonal antibody, was developed by Genscript Biotech using all-natural components to fulfill bivalent and multivalent purposes [35]. VHH is the antigen-binding immunoglobulin variable domain of “HC antibodies”. As it is a small, robust, and efficient antigen recognition unit, the VHH-based on SMAB™ platform has the potential to bind to “hidden” epitopes, such as on a cavity of an enzyme, within ion channels, etc., as well as being highly flexible for the construction of multivalent molecules using a “plug and play” fashion. Such VHH can be connected to the N- or C-terminal end of each peptide of a monoclonal antibody to form an SMAB (Fig. 1I–L).
Based on the above bsAb platforms, 12 patents from China have been granted so far by the patent office of the USA, among which four patents are based on YBODY® platform by YZYBio, one is based on each of FIT-Ig™ by EpimAb, ITab™ by Generon and CRIB™ by Alphamab (Table 1).
Table 1.
Bispecific antibody patents from Chinese applications granted by the United States Patent and Trademark Office [36,37]
| Number | Title | International patent classification | Assignee | Earliest publication date | Patent no. |
|---|---|---|---|---|---|
| 1 | Anti-human ovarian cancer-anti CD3 bsAb | C07K16/30AI A61K39/395AI C07K16/28AI C07K19/00AI C12N15/13AI C12N15/62AI C12N15/63AI C12P21/08AI | Beijing ABT Genetic Engineering Technology Co. Ltd. (Beijing, CN) | 17 November 2005 | US7262276 |
| 2 | Multi-specific FAB fusion proteins and methods of use | A61K39/395 C07K14/705 C07K16/28 C07K16/30 C07K16/46 | Generon (Shanghai, CN) | 20 December 2012 | US8846042 |
| 3 | Bispecific antibody | C07K16/46 C07K16/28 C07K16/32 | YZYBio (Wuhan, CN) | 28 August 2014 | US9079965 US9562110 |
| 4 | Bispecific antibodies that bind EGFR and VEGF | C07K16/22 C07K16/28 C07K16/30 | Bio-Thera Solutions Co. Ltd. (Guangzhou, CN) | 12 February 2015 | US9567403 |
| 5 | Multi-functional antibody polypeptide for cryptic epitope of epidermal growth factor receptor and T cell antigen | A61K39/00 C07K16/28 C07K14/71 | Carsgen Therapeutics Co. Ltd. (Shanghai, CN) | 2 April 2015 | US10023639 |
| 6 | Method for preparing homodimer protein mixture by using charge repulsion effect | C12P21/02 A61K39/395 C07K16/00 A61K39/00 | Alphamab (Suzhou, CN) | 1 October 2015 | US9708389 |
| 7 | Construction and application of bsAb EpCAM×CD3 | C07K16/46 C07K16/28 C07K16/30 C07K16/32 | YZYBio (Wuhan, CN) | 31 March 2016 | US9777073 |
| 8 | Construction and application of bsAb HER2×CD3 | C07K16/28 C07K16/40 C07K16/32 | YZYBio (Wuhan, CN) | 26 May 2016 | US9611325 US10118964 |
| 9 | Fabs-in-tandem immunoglobulin and uses thereof | A61K39/00 A61K39/395 A61K45/06 C07K16/00 C07K16/24 C07K16/28 C07K16/46 | Epimab (Shanghai, CN) | 6 October 2016 | US10266608 US10519251 |
| 10 | Bispecific anti-HER2 antibody | C07K16/32 A61K39/395 A61K39/00 | Mabworks (Beijing, CN) | 29 August 2017 | US9745382 US10377833 |
| 11 | Bispecific multivalent fusion proteins | A61K39/21 A61K38/00 A61K39/00 A61K47/68 A61P31/18 C07K14/73 C07K16/10 | US Dept of health (MD,US) Fudan university (Shanghai, CN) | 3 May 2018 | US10472412 |
| 12 | BsAb-mediated cancer therapy with cytokine-induced killer cell | C07K16/32 C07K16/28 A61P35/00 | YZYBio (Wuhan, CN) | 1 November 2018 | US10556964 |
EXPRESSION, PRODUCTION, AND BIOASSAY OF BSABS
The production of an IgG-like bsAb with physical and chemical properties similar to some those of an IgG can be generated following typical bioprocesses for producing a typical monoclonal antibody. However, unlike natural mAbs, technical challenges with respect to quantity, quality, and stability of bispecific antibodies have hampered the wider clinical application and acceptance of these antibodies [14]. High expression level and production yield with desired purity and biological activities of the drug product are much required during the chemistry, manufacturing, and controls (CMC) development of a bsAb.
Expression and production
The structural format of an antibody can be a key factor underlining the selection of the antibody expression systems. Fc-lacking bsAbs, including BiTE, tandem bispecific scFv and other non-IgG bsAb, can be expressed in Escherichia coli and yeast or in a mammalian cell line, including Chinese hamster ovary (CHO) and human embryonic kidney (HEK293) cells [14,38–40]. While E. coli is the most widely used host for expression due to its rapid growth, cost efficiency, high productivity, well-known and matured technology and easy genetic manipulation [41], it does not always provide the same levels of secondary structure and bioactivity of the therapeutics as mammalian cell lines [42,43]. Scientific efforts continue in developing other expression systems such as stable transgenic plants for a large-scale production due to its advantages in cost efficiency and safety [44,45], and cell-free protein synthesis system for a high-throughput protein library generation due to its efficiency and flexibility [46,47]. Yet, the “best” expression system for scFv bispecifics remains to be determined as their molecule sizes, confirmations, solubility and stability, and their amino acid sequences all impact the optimizing the expression system [14].
IgG-like bsAbs have been expressed in mammalian cells [41,48] and the CHO-based platform is more often used since it allows for advanced protein folding pathways and post-translation modifications specifically in Fc fragments. CHO cells are popular hosts of cell lines for stable expression required for robust production of a bsAb, where the expression vectors are a key factor to impact both quality and quantity of the expressed target proteins [49,50]. Glutamine synthetase (GS) inhibitor (methionine sulfoximine, MSX) to a CHO cell, or the GS-knockout CHO cell is often chosen to increase the protein expression [51]. Specifically, a bsAb with three or more genes usually requires two or more vectors to be co-transfected into the CHO cells, and the ratios of the two vectors could also impact both the quality and quantity of the expressed bsAbs. While the expression levels are independent of the integration site of the gene, there is a clear correlation between expression levels and the numbers of integrated transgene copies. Higher quantity of the target protein can be produced with higher copies of integrated transgenes [52]. Furthermore, bsAb protein instability can be significant in recombinant CHO cells, which occurs due to the instability of the cell line and long culture time [53]. The early selection of a stable cell line with constant expression of the target protein is the key for maintaining the stable production yield.
The production process of an IgG-like bsAb is similar to a conventional IgG for culture expansion and purification processes including protein A-based affinity chromatography. In the upstream process by Fed-batch method, which is mostly used for Ab production currently in worldwide including in China, cells are expanded through a series of steps from shake flasks to N-1 seed bioreactor and finally production reactor. The multiple parameters, including viable cell density (VCD), cell viability (CV), pH, temperature, dissolved oxygen (DO), and stirring speed, have to be carefully monitored and controlled during the whole process. In the downstream process, the product purity including product-related impurities (e.g. fragments and aggregations) and the process-related impurities (e.g. residual DNA and host cellular protein), and microbial contaminations (e.g. endotoxin and bioburden) should be well controlled to ensure the product quality [54]. It is also critical to ensure viral inactivation and reduction during the developed purification processes to reach the safety requirements [55].
This production strategy for a conventional IgG can be also applied to an asymmetric IgG-like bsAb with molecular size or physical property differences between left-half and right-half molecule, with more challenges to deal with than a symmetric IgG-like molecule. One of the key challenges for the production of an asymmetric bsAb is to minimize the formation of its mismatched two HCs (left–left and right–right) and mismatched heavy-light chains (Fig. 1G) in the upstream cell expression. The mismatched Fc formations can be largely overcome using Fc engineering to make the favorable interactions of the left and the right to form the heterodimer by inducing KiH and/or salt bridge [30, 56] (Fig. 1A), and scFv designed for the second half bsAb such as in the structure shown in Fig. 1A can also eliminate the potential occurrence of the mismatched heavy–light chains when two different LC are designed in one molecule (Fig. 1G). Another technology to overcome the mismatch of two LC to their HCs is to use one common LC, which can be identified by enough screening efforts for the desired affinities binding both to its two HCs and together to the two targets during the discovery phase. In each case, any small portion of mismatched impurities formed during the upstream process can be removed by the downstream chromatography, which is more favorable by differences in chemical and physical properties between the right assembled bsAb and mismatched species.
Another challenge for a bsAb production is to maintain its native structures and activities during the process development and production. While a candidate bsAb is selected during the early discovery phase based to its functional property and developability based on its efficacy, safety, PK/PD, immunogenicity, physiochemical properties, and manufacturability [26], the selected candidate should maintain its bioactivity, purity, and solubility being resistant to aggregation under the stress conditions such as heating, freeze-thaw, and shaking. Being mostly dependent on its structure platform and molecular sequences, the bioactivity and purity of the candidate bsAb or impurities due to the missed assemblies of bsAb fragments should be confirmed during cell line development, which is required for a large-scale development and production. The stability of the bsAb intermediates and the storage conditions during downstream processes should be well studied to ensure its solubility and activity in each step of the production.
Biological activity and bioassay
During bsAb development for cancer therapeutics, it is important to use a reliable method for measuring growth, death, and viability of cancer cells. Cell-based in vitro assays including CCK-8 [57,58], MTS [59,60], MTT [61,62], XTT [63], WST-1 [64], and CellTiter-Glo Luminescent Cell Viability Assay [65], have been well developed for measuring tumor cell growth and viability. All these assays are based on the measurement of substances released from the damaged or dead cells. Among these assays, CCK-8 has been widely used due to its convenience for operation and high analytical sensitivity. However, the reductant for the reaction by CCK-8 may interfere with the assay, while MTS and CellTiter-Glo Luminescent Cell Viability Assay have no such issue. For testing cytotoxic activity of a biologics against tumor cells, assays for measuring the LDH [66–68] or 51Cr [69–72] are traditionally available, and CytoTox-Glo™ Cytotoxicity Assay has been also developed [73].
A panel of biophysical and biochemical analyses have to be developed for quality study and quality control during bioprocess development and manufacture of a biologic product. A special and unique property of a bsAb is its synergistic effect of two binding arms in simultaneously targeting on two antigens to provide 1 + 1 > 2 in biological activities. The potency assays should reflect the synergistic effect based on its MOA [74]. In the case of Blinatumomab (anti-CD19 × anti-CD3) that works by targeting both CD19 and CD3 proteins on the surface of leukemia cells and T cells, respectively, measuring CD69 and CD25 levels released by the T cells (Effect) activated by the CD19+ B cells (Target) linked through the bsAb will help us evaluate the activation activity of the Blinatumomab [75] when the two cells are co-cultured in different E:T ratios with addition of the bsAb. Similarly, the cytotoxic activity of a bsAb can be measured by monitoring the death of its target cells.
For PD-1/PD-L1-based bsAb, a blockade bioassay kit (Promega) has been developed for assessment of PD-1/PD-L1 interaction using two engineered cells: a Jurkat cell with the expressions of both PD-1 and Neclear Factors of Activated T-Cells (NFAT)-inducible luciferase, and a CHO cell with the expressions of both PD-L1 and a membrane-linked agonistic anti-CD3 antibody (CHO PD-L1 CD3 cells) [76,77]. Specifically, engagement of the TCR and CD3 complex on the Jurkat cell leads to the phosphorylation and activation of PLC-g, intracellular calcium flux, and transcriptional activation of NFAT pathway. When the Jurkat and CHO cells are co-cultured, the interaction of the two cells medicated by the PD-1/PD-L1 interaction inhibits TCR signaling and NFAT-mediated luciferase activity in the Jurkat cell. Addition of a PD-1/PD-L1 blocking agent, e.g. an anti-PD-1 or anti-PD-L1 antibody, interrupts the cell–cell interaction and results in the increase of the fluorescence signal released by NFAT-mediated luciferase activity in the Jurkat cell [60]. Based on this mechanism, the bioactivities of bsAbs targeting on CD3 and PD-1/PD L1 antigens can be analyzed through luciferase activity in Jurkat-PD1-NFAT-luc T cells. This assay can be used for evaluation of PD-L1-targeted and CD3-targeted bsAbs (data not shown).
BSAB MOLECULES: THEIR ARCHITECTURES AND MOAS
As of May 2020, there are 21 bsAbs under clinical development or with Investigational New Drug (IND) approval in China (Table 2), for Immuno-Oncology (IO)-based cancer therapies. Among these antibodies, 16 bsAbs are targeting for solid tumors, and four are for hematological tumors including lymphoma and cancer-caused malignant ascites (Table 2).
Table 2.
BsAbs under clinical development in China by Chinese companies as of May 2020 [78]
| Identifier | Drug | Company | Targets | BsAb format and platform | Indication | Phase |
|---|---|---|---|---|---|---|
| CTR20171194 | M802 | YZYBio | HER2 × CD3 | scFv-Fab IgG; YBODY | HER2-positive advanced solid tumor | I |
| CTR20181212 | M701 | YZYBio | EpCAM × CD3 | scFv-Fab IgG; YBODY | Malignant ascites | I |
| CTR20182027 | AK104 | Akeso | PD-1 × CTLA-4 | IgG-(scFv)2; Tetrabody | Advanced solid tumor and advanced or metastatic gastric adenocarcinoma or gastroesophageal junction adenocarcinoma | Ib/II |
| CTR20190205 | A-319 | Generon | CD19 × CD3 | (scFv)2-Fab; ITAB | Refractory or relapsed B cell lymphoma | I |
| CTR20190853 | KN026 | Alphamab | HER2 × HER2 | Hetero H, common LC IgG; CRIB | Advanced gastric and gastroesophageal junction carcinoma with overexpression and low expression of HER2 | II |
| CTR20182404 | SHR-1701 | Hengrui | PDL1 × TGFβ | Antibody-receptor fusion | Metastatic castration resistant prostate cancer | I |
| CTR20181823 | Advanced malignant solid tumor | |||||
| CTR20181760 | MBS301 | Mabworks | HER2 × HER2 | Fab-arm exchange | HER2 high expression of locally advanced, inflammatory or early breast cancer, metastatic breast cancer, metastatic gastric cancer, etc. | I |
| CTR20190427 | KN046 | Alphamab | PD-L1 × CTLA-4 | Hetero VHH-Fc; CRIB | Advanced unresectable or metastatic esophageal squamous cell carcinoma | II |
| CTR20190197 | Triple negative breast cancer | II | ||||
| CTR20190195 | Non-small-cell lung cancer | II | ||||
| CTR20181996 | Chinese advanced solid tumor and lymphoma subjects | I | ||||
| CTR20190241 | EMB-01 | Epimab | EGFR × c-MET | F(ab)2-IgG; FIT-Ig | Advanced/metastatic solid tumors, including but not limited to non-small-cell lung cancer, colorectal cancer (no RAS-positive mutation), gastric cancer, liver cancer, and other solid tumors | I |
| CTR20190340 | IBI318 | Innovent | PD-1 × PD-L1 | Fab-arm exchange | Advanced malignant tumor | I |
| CTR20190888 | ES101 | Elpiscience | PD-L1 × CD137 | Tandem VHH-Fc | Advanced solid tumor | I |
| CTR20191955 | K193 | Lvzhu | CD19 × CD3 | F(ab)2-(scFv)2-Fc | B-cell lymphoma | I |
| CTR20191677 | IBI315 | Innovent | HER2 × PD-1 | Fab-arm exchange | Advanced malignant tumor | I |
| CTR20192612 | IMM0306 | ImmuneOnco | CD47 × CD20 | Antibody-receptor fusion | Lymphoma | I |
| CTR20192299 | HX009 | HanxBio | CD47 × PD-1 | Antibody-receptor fusion | Malignant tumors such as liver cancer, stomach cancer, and colorectal cancer | I |
| CTR20200502 | SI-B001 | Biokin | HER3 × EGFR | IgG-(scFv)2 | Locally advanced or metastatic epithelial tumor, including esophageal squamous cell carcinoma, lung squamous cell carcinoma, triple negative breast cancer, head and neck squamous cell carcinoma, colorectal cancer, etc. | I |
| CTR20200175 | IBI322 | Innovent | PD-L1 × CD47 | Not available | Solid tumors and hematological tumors | I |
| CTR20200549 | MGD013 | ZLAB | PD-1 × LAG-3 | DART | Advanced liver cancer (including hepatocellular carcinoma and intrahepatic cholangiocarcinoma) | I |
| CTR20200289 | Advanced gastric adenocarcinoma or adenocarcinoma at the gastroesophageal junction with previous treatment failed | I | ||||
| CXSL1900112 | SI-B003 | Biokin | Undisclosed | Not available | Undisclosed | IND |
| CXSL1900131 | KD6001 | Kanda | Undisclosed | Not available | Advanced malignant tumor | IND |
| CXSL1900150 | PM8001 | Biotheus | Undisclosed | Not available | Advanced solid tumor | I |
Structures of bsAbs approved for clinical trials by NMPA
The structures of 17 bsAbs among 21 projects in the clinical stage in China had been disclosed, and these can be categorized as asymmetric IgG-like bsAbs (7 of 17, 41.2%), symmetric IgG-like bsAbs (6 of 17, 35.3%), bispecific fragments (1 of 17, 5.9%), and antibody-receptor fusion bsAbs (3 of 17, 17.6%) (Fig. 1M). An IgG-like bsAb with two full-length and functional Fc chains can be expected to have half-life time, pharmacodynamics (PD) and pharmacokinetics (PK) behaviors similar to those of its parent monoclonal antibodies (mAb) when their charges, glycosylation, polyreactivity and binding activity to the FcRn receptors are maintained [79,80], while a bispecific fragment such as Blinatumomab has a shorter half-life due to its faster clearance, which provides a safety advantage to mitigate the drug-associated neurological adverse events [80,81].
Asymmetric IgG-like bsAbs
Asymmetric IgG-like bsAbs have whole Fc and two distinct arms binding to two different epitopes/antigens. The Fc region can be artificially engineered to improve efficiency of heterodimer formation using methods such as KiH, electrostatic steering [30], CRIB, or controlled Fab-arm exchange (FAE) technologies [82]. The binding domains can be derived from a Fab, an scFv, or a VHH. These asymmetric IgG-like bsAbs included (1) M802 and M701, in an scFv-Fab IgG-like structure developed by YZYBio and shown in Fig. 1A [27], were the first two IND-approved bsAb molecules in China [28,83–85]. The safety and efficacy of both projects are currently under clinical evaluation. (2) KN026 developed by Alphamab contains hetero-HCs, common LC IgG to form a full IgG molecule (Fig. 1B), in which both CRIB and common light chain were utilized [32]. (3) MBS301 developed by Mabworks [86], and both IBI318 [87] and IBI315 [88] by Innovent Biologics (Innovent), are based on FAE technology (Fig. 1N). The Fc of MBS301 is modified by afucosylation to enhance Antibody-Dependent Cellular Cytotoxicity (ADCC) [86]. (4) KN046, hetero-VHH-Fc developed by Alphamab (Fig. 1C), is based on CRIB platform and VHH-Fc fusion in which two distinct VHHs are fused to one of the heterodimeric Fc, respectively [89].
Symmetric IgG-like bsAbs
A symmetric IgG-like bsAb contains IgG-like Fc and two symmetric binding arms constructed by a combination of one or more of Fab, scFv, and VHH. Current symmetric IgG-like bsAb projects developed in China include: (1) EMB-01 (by EpimAb) is an (Fab)2-IgG-like structure (Fig. 1E) based on FIT-Ig platform technology [34]; (2) ES101 developed by Elpiscience Biopharmaceuticals Co., Ltd. (Elpiscience) is a VHH-Fc-VHH (Fig. 1O) or tandem VHH-Fc homodimer structure (Fig. 1P) in which two different VHHs are fused with one Fc [90]; (3) K193 developed by Beijing Lvzhu Biological Technology (Lvzhu) is a (Fab)2-(scFv)2-Fc structure (Fig. 1Q), in which all four binding domains are at the N-terminus of Fc [91]; (4) AK104 developed by Akeso Biopharma (Akeso) is based on a tetrabody platform technology to construct an IgG-(scFv)2 structure (Fig. 1R) in which the scFvs are fused to the two HCs at the C-terminal ends [92], and SI-B001, developed by Sichuan Biokin Pharmaceutical Co. (Biokin), is also an IgG-(scFv)2 structure [93]. MGD013 by Zai Lab Ltd. (ZLAB) is a tetravalent DART® bsAb with the hinge-region-stabilized IgG4 (Fig. 1S) [94].
Bispecific fragments
A319, developed by Generon, belongs to the bispecific fragments format, which is a fusion protein with the combination of Fab and/or scFv, such as (scFv)2-Fab (Fig. 1D), where the two identical scFv domains target antigen A and the Fab domain targets antigen B [33].
Antibody-receptor fusion
The binding interaction between a receptor and its ligand can be mimicked to the interaction between an antibody and its antigen. Therefore, a fusion protein constructed by fusing a receptor with a natural antibody has the dual binding functions as a bsAb. For instance, SHR-1701 developed by Shanghai Hengrui Pharmaceutical Co. (Hengrui) [95], IMM0306 developed by Shanghai ImmuneOnco Biopharmaceuticals Co. (ImmuneOnco) [96], and HX009 developed by Hangzhou Hanx Biopharmaceutics, Inc. (HanxBio) [97] are such structure in which a receptor is connected to the C-terminal ends of the Fc region in the whole mAb (Fig. 1T). Specifically, the receptor-portion of SHR-1701 is the extracellular domain (ECD) of TGF beta receptor II (TGFBRII), and both of IMM0306 and HX009 (Table 2) are the ECD of signal regulatory protein α (SIRPα). This antibody-receptor fusion format may have time-cost savings by adapting the natural receptor to the bsAb over the two mAbs-based bsAb for which one more mAb has to be developed. However, due to relatively lower stability of the receptor portion, the antibody-receptor fusion proteins may suffer less stability than the bsAbs based on two mAbs.
Targets and mechanisms of bsAbs
Bringing together two cells (e.g. T cell and cancer cell) or two targeted proteins is an additional and unique mechanism of a bispecific Ab, with CD3-dependent T cell engaging bsAbs being the most common in development (Fig. 2A) [26, 27]. The requirement of two targets to be connected by one molecule is easily achieved by using a bsAb. For instance, some bsAbs can redirect T cells to tumor cells by in-trans binding, while some other bsAbs by in-cis binding on the same cell. The targets and mechanisms of action of the bsAbs in China are discussed in more details below.
Figure 2.
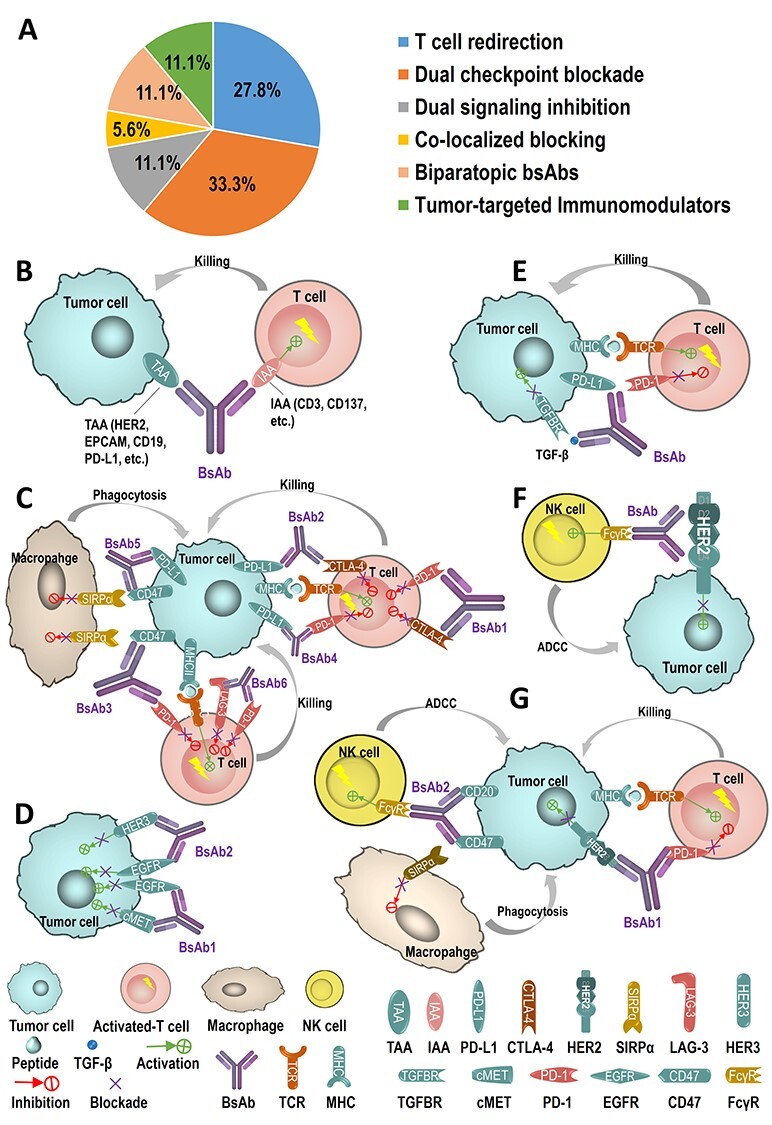
MOAs of 18 bsAbs in Chinese clinical trials. (A) Among the 18 bsAbs, five of which are by T-cell redirection (5 of 18, 27.8%), six by dual-checkpoints blockade (6 of 18, 33.3%), two by dual signaling inhibitions (2 of 18, 11.1%), one by co-localized blocking (1 of 18, 5.6%), two by biparatopic bsAbs (2 of 18, 11.1%), and two by tumor-targeted immunomodulators (2 of 18, 11.1%). (B) T-cell redirection refers to bridge T cells and tumor cells by binding to both a TAA and IAA and redirecting the cytotoxic activity of effector T cells to attack specifically to the tumor cells. Represent projects in using the MOA include M802 (HER2 × CD3, by YZYBio), M701 (EPCAM×CD3, by YZYBio), A-319 (CD19 × CD3, by Generon), K193 (CD19 × CD3, by Lvzhu), and ES101 (PD-L1 × CD137, by Elpiscience). (C) Dual checkpoints blockade is by two-checkpoint blockers integrated into one antibody to inhibit two immune checkpoints simultaneously. AK104 (PD-1 × CTLA-4, by Akeso), KN046 (PD-L1 × CTLA-4, by Alphamab), IBI318 (PD-1 × PD-L1, by Innovent), HX009 (PD-1 × CD47, by HanxBio), IBI322 (PD-L1 × CD47, by Innovent), and MGD013 (PD-1 × LAG-3) are six of these examples. (D) Dual signaling inhibitions are to target two different receptors for preventing the receptors from phosphorylation and/or from the activation of both receptor-mediated signaling pathways to inhibit tumor proliferation. EMB-01 (EGFR × c-MET, by Epimab) and SI-B001 (HER3 × EGFR, by Biokin) are two of these typical bsAbs. (E) Co-localized blocking is by inhibiting two or more tumor cell intrinsic and extrinsic pathways to raise the possibility of superior antitumor activity compared with the monotherapy. SHR-1701 (PD-L1 × TGF-β, by Hengrui) is one of such bsAbs. (F) Biparatopic bsAbs are by binding to two different epitopes of the same antigen or same receptor to enhance the antigen–antibody affinity and to improve the drug efficacy. Both KN026 (HER2 × HER2, by Alphamab) and MBS301 (HER2 × HER2, by Mabworks) bind to the D2 and D4 subdomains of HER2. (G) Tumor-targeted immunomodulators are designed for binding to both one TAA (e.g. HER2, CD20) to inhibit TAA signaling pathway and one immunomodulating receptor (e.g. PD-1, CD47) to regulate the immune system to attack the tumors. IBI315 (HER2 × PD-1, by Innovent) and IMM0306 (CD20 × CD47, by ImmuneOnco) utilize such MOAs.
T-cell redirection
The bsAbs targeting both a T-cell associated antigen and a TAA can bridge T cells and tumor cells to achieve the concept that the redirecting the cytotoxic activity of the effector T cells to specifically eliminate tumor cells (Fig. 2B). These bsAbs also activate T cells through binding of TAA and IAA such as CD3ε in TCR complex or CD137 or CD28, thereby bypassing major histocompatibility complex (MHC) restriction and causing activation in independence of the epitope specificity of the TCR [98,99].
M802 (anti-HER2 × anti-CD3) was approved for clinical trials in both China and the USA to treat advanced HER2-positive solid tumors. Human epidermal growth factor receptor 2 (HER2) plays an important role in cell proliferation, survival, differentiation, angiogenesis, cellular migration, metastatic growth, and invasion of cancer cells. The current standard treatment for HER2+ breast cancer is the anti-HER2 mAb-trastuzumab, which consists of two identical antigen-specific binding sites that bind to the juxtamembrane portion of ECD of HER2 to prevent the activation of its intracellular tyrosine kinase, and the mAb is human IgG1 isotype with Fc effector functions including ADCC, ADCP, and CDC to kill the tumor cells [100]. Similar to trastuzumab, by targeting HER2, M802 prevents the dimerization of the receptor HER2, increases endocytotic destruction of the receptor, and inhibits shedding of the ECD of HER2. The other target of M802, CD3 is the specific surface antigen of T cells. Therefore, mechanistically different from trastuzumab, M802 recruits and redirects T cells to HER2+ tumor cells through binding to CD3 and HER2, and further activates T cells to kill the tumor cells. The comparative in vitro studies showed that the cytotoxicity activity of M802 was five times stronger than that of trastuzumab on HER2-positive breast cancer cells BT-474 killed by PBMCs, and furthermore, M802 displayed significant cytotoxicity to JIMT-1 (a trastuzumab-resistant breast cancer cell line) [85], indicating a new promising therapeutics for HER2-positive and/or trastuzumab-resistant breast cancer patients.
The indication of M701 (anti-EpCAM×anti-CD3) was EpCAM+ malignant ascites. Similar to M802 by binding CD3, M701 inhibits EpCAM positive tumor growth by recruiting and activating T cells to attack the tumor cells. M701 has the biological functions and MOA similar to catumaxomab [101]. In contrast to catumaxomab, which is a rat and mice chimeric antibody, M701 was expressed in CHO cells by antibody engineering technology including humanization of the constant regions. The immunogenicity observed in catumaxomab was not detectable in M701-based in vitro and in vivo studies (internal unpublished data).
Both A-319 (developed by Generon) and K193 (developed by Lvzhu) target CD19 and CD3 are used for refractory/relapsed B cell lymphoma. CD19 is a surface protein expressed on malignant B cells. By co-binding CD3 and CD19, these two bsAbs recruit T cell activity in killing the CD19+ tumor cells. The commercially available bsAb (blinatumomab, Amgen) has the same target as A-319 and K193, and has a short plasma half-life due to its small size (about 55 kDa) [102]. As K193 has an Fc fragment and half-life extended BITE (HLE-BITE, Amgen) by fusing an Fc to a BITE construct can increase the half-life [103].
ES101 is an anti-PD-L1 × anti-CD137 bsAb, and its clinical trials are developed by Elpiscience. The indication of ES101 is an advanced solid tumor. The bsAb binds to the CD8+ T cell surface antigen CD137 (also known as 4-1BB), which is a co-stimulatory molecule, and simultaneously binds to the tumor surface antigen PD-L1 to block the PD-1/PD-L1 interaction. These two MOAs synergistically enhanced the activation and cytotoxicity of CD8+ T cells to kill the tumors and CD8+ T cells’ response to chronic viral infection [104].
Both CD3 and CD137 are T-cell surface molecules, which induce T-cell activations differently. Anti-CD3 binding to T cells causes the aggregation of CD3/TCR and transduces the signal inside to the T cells, while anti-CD137 binding to T cells generates a co-stimulatory signal to stimulate T cell expansion [105,106]. Therefore, the bsAbs based on these two antigens have different anti-tumor mechanisms. The anti-CD3 bsAb functions to activate the T cells to release the high levels of granzymes and perforins to kill the cancer cells [107]. While CD137 is expressed primarily on activated T cells and belongs to the tumor necrosis factor receptor superfamily, CD137 enhances the proliferation of CD8+ T cells, prevents activation-induced cell death, and promotes preferentially the memory formation of CD8+ T cells [106]. The anti-CD137 (anti-4-1BB) treatment may amplify the autocrine IL-2/IL-2R signaling loop leading to enhance the expansion of CD8+ T cells [108] and may activate glucose and fatty acid metabolism, which enhances the cell-cycle progression of anti-CD3-activated CD8+ T cells in vitro and the anti-apoptotic effects of CD137 signaling on these cells [109].
Dual checkpoint blockade
Cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), as an immune checkpoint, was identified by Golstein et al. in 1987 [110]. CTLA-4 acts as a physiological brake on the activated immune system to maintain immune homeostasis and contributes to the suppressive effects on T-cell functions. Ipilimumab (IgG1 isotype), a monoclonal antibody to CTLA-4, was approved by FDA as the first immunotherapeutic drug directing toward CTLA-4 inhibition with overall survival and remarkable long-term benefit in metastatic melanoma [111]. In 1992, another immune checkpoint programmed cell death protein 1 (PD-1) discovered by Honjo et al. [112] is another inhibitory receptor expressed on some immune cells and causes down regulation of the immune system by reducing the T-cell activity. PD-L1, known also as B7-H1 discovered by Chen et al. in 1999 [113], is one of the programmed death ligands of PD-1. In the cancer disease state, the interaction of PD-L1 on the tumor cells with PD-1 on T cells reduces T-cell signaling function to prevent the immune system from attacking the tumor cells. Blocking the interaction of PD-L1 with PD-1 can prevent the cancer from evading the killing of the immune system [114,115]. Both anti-PD-1 monoclonal antibodies (nivolumab, pembrolizumab, and cemiplimab) and anti-PD-L1 monoclonal antibodies (atezolizumab, durvalumab, and avelumab) approved for marketing by FDA can block PD-1/PD-L1 interaction so that the T cells are no longer inhibited and thus activates the immune response against the tumor [116–119]. In addition, several new checkpoints were reported in recent years, including LAG3, TIGIT, CD47, Tim-3, Siglec-15, and many more. Based on these checkpoints, a number of clinical trials with the combination of two checkpoint inhibitors were designed to improve the efficacy, including anti-PD-1 + anti-CTLA-4, anti-PD-L1 + anti-CTLA-4, anti-PD-L1 + anti-PD-1, and anti-PD-1 + anti-LAG3. These combinations selected based on the mechanistic differences between checkpoints and the differences in expression of checkpoints and their ligands in the tumor microenvironment showed enhanced efficacy in a given cancer subtype, but also increased the toxicity [120–123]. Therefore, it is interesting to test if the integration of two checkpoint blockers into a single molecule as a dual-checkpoint blockade (Fig. 2C) can exceed the enhanced efficacy of a combination of two mAbs, along with its reductions in production costs, product operation costs, regulatory complications, and clinical utility of a combination therapy. This dual-checkpoint blockade has been adopted into several projects listed in Table 2.
AK104, an anti-PD-1 × anti-CTLA-4 bsAb by Akeso, targets advanced solid tumors including advanced or metastatic gastric adenocarcinoma or gastroesophageal junction adenocarcinoma [92]. The bsAb binds and blocks the human negative immunoregulatory checkpoint receptors PD-1 and CTLA-4 at the same time, resulting potential immune two-checkpoint inhibitory and antineoplastic activities. Both PD-1 and CTLA-4 are expressed on TILs under the tumor microenvironment (TME) and down regulate the activation and effector functions of T cells. They play key roles in the down-regulation of the immune system and tumor evasion from host immunity. Dual checkpoint blockade of PD-1 and CTLA4 with AK104 may provide T-cell activation and proliferation higher than either of the two immune checkpoint inhibitors alone.
KN046, developed by Alphamab, is an anti-PD-L1x anti-CTLA-4 bsAb, which is currently in phase II clinical trials. The combination therapy of anti-PD-L1 mAb and anti-CTLA-4 mAb are also currently under multiple late-stage clinical trials. However, the severe autoimmune side effects induced by CTLA-4 antibody may limit its therapeutic utility. Recently, Du et al. [124,125] have demonstrated that immunotherapy-related adverse events and the cancer immunotherapeutic effects represent distinct and therapeutically separable activities of anti-CTLA-4 antibody. The former is attributable to inactivation of the CTLA-4 checkpoint by blocking its interaction with B7–1/B7–2, while the latter is due to selective depletion of regulatory T cells (Treg) in a tumor microenvironment [124–127]. In order to achieve safer and more effective CTLA-4-targeting immune therapy, the CTLA-4 checkpoint activity should be preserved rather than inhibited while enhancing the efficacy and selectivity of depleting regulatory T cells (Treg) in a tumor microenvironment through ADCC and ADCP by NK cells and macrophages, respectively [127]. Similar to this opinion, KN046 was designed to have a wild-type IgG1 Fc portion that preserves the intact Fc effector functions for depletion of regulatory T cells, and it was also developed with a proprietary CTLA-4 domain antibody to target to CTLA-4but limit the CTLA-4 binding function to the tumor site [89]. Along with the other arm binding to PD-L1, the KN046 may provide an improved safety and efficacy profiles compared to their parental mAbs [127].
IBI318, developed through a collaboration between Innovent and Eli Lilly, is a recombinant fully humanized IgG1 anti-PD-1 × anti-PD-L1 bsAb and blocks simultaneously both PD-1 and PD-L1 to enhance the formation of immune synapses, thereby enhancing potentially anti-tumor efficacy [87].
HX009 developed by HanxBio and IBI322 by Innovent are two therapeutic candidates that are CD47-related bsAbs. They synergistically activate the immune system for the treatment of malignancies such as liver cancer, gastric cancer, and colorectal cancer. CD47 is a cell surface receptor that conveys a “don’t eat me” signal to immune cells; its interaction with SIRPα leads to the inhibition of macrophage activation and protections of cancer cells from phagocytosis, which allows cancer cells to proliferate [128]. Anti-PD-1 antibodies restored large amounts of exhausted T cells and anti-CD47 antibodies stimulated macrophage phagocytosis. HX009 consists of human IgG4-Fc of anti-PD-1 mAb and ECD of SIRPα, and can achieve the anti-tumor synergistic effects by simultaneously activating innate and adaptive immune responses and also by suppressing tumor immune escape by inhibiting the immune checkpoint [97]. The other candidate bispecific therapeutic, IBI322, was showed that its binding affinity to PD-L1 was stronger than that to CD47, implying a potential therapy to PD-L1-positive cancers [129].
MGD013, an anti-PD-1 × anti-LAG-3 bsAb early developed by Macrogenics, was licensed to ZLAB for clinical trials in China. Lymphocyte activation gene 3 (LAG3), or CD223, is expressed on multiple cell types including CD4+ and CD8+ T cells, and Tregs, and is required for optimal T-cell regulation and homeostasis [130,131]. The ligands of LAG3 include the class II major histocompatibility complex (MHC) [132] and fibrinogen-like protein 1 (FGL1), which is a major LAG-3 functional ligand independent from MHC-II. While the interaction of LAG-3 and MHC negatively regulate T-cell proliferation and the development of lasting memory T cells [133], FGL1 inhibits antigen-specific T cell activation, and ablation of FGL1 in mice promotes T cell immunity [134]. LAG3, like PD-1, is also one of several immune checkpoint receptors coordinately upregulated on both Treg cells and anergic T cells. In particular, PD-1 and LAG3 are commonly co-expressed on anergic or exhausted T cells, simultaneous blockades to both checkpoints can result in an enhanced reversal of this anergic state relative to the blockade of either receptor [135]. MGD013 was showed to enhance cytokine production upon antigenic rechallenge of prior superantigen-stimulated T cells in vitro, and to increase CD8:Treg ratio and to induce loss of LAG3 surface expression on T cells in vivo [135]. Furthermore, anti-PD-1 × anti-LAG-3 bsAb may offer clinical opportunities to checkpoint naïve patients as well as to checkpoint experienced patients who have progressed on prior therapy with PD-1/PD-L1 inhibitors [94, 135, 136].
Dual signaling inhibitions
EMB-01 based on EpimAb’s proprietary platform FIT-Ig™ has showed efficacy in multiple preclinical cancer models. It targets the epidermal growth factor receptor (EGFR) and c-mesenchymal-epithelial transition factor (c-MET). Extensive evidence has demonstrated that the EGFR and c-MET signaling pathways are partially compensatory and both cross-mediate the resistance to the inhibitors of the two signal pathways [137–139]. Non-small cell lung cancer (NSCLC) is frequently caused by activating mutations in the kinase domain of EGFR, which may lead to resistance to tyrosine kinase inhibitors (TKI) with less than 1 year of average response duration [140,141]. Interestingly, resistant tumors may also develop the activation of the c-MET pathway, which provides an alternative mechanism for cancer cells to bypass the TKI block of EGFR for survival. These two mechanisms occurred simultaneously in EGFR TKI-resistant NSCLC patients [142,143]. Similar to JNJ-61186372 (anti-EGFR × anti-c-MET bsAb developed by Janssen & Genmab) with multiple mechanisms of action to inhibit primary/secondary EGFR mutations and the c-MET pathway [144], EMB-01 targets simultaneously to wild-type or certain mutant forms of both EGFR and c-MET expressed on cancer cells prevents receptor phosphorylation and the activation of both EGFR- and c-MET-mediated signaling pathways, and as a result, tumor cell proliferation is inhibited (Fig. 2D).
SI-B001, developed by Biokin, is an IgG-(scFv)2 bsAb that targets to both EGFR and human epidermal growth factor receptor 3 (HER3). HER3 forms a heterodimer with EGFR and transduces the signals through the intracellular motifs of EGFR [145], and directly activates the PI3K/AKT pathway and participates in tumorigenesis [146]. Thus, mechanistically, HER3 has the potential to synergize with EGFR (Fig. 2D). Duligotuzumab (MEHD7945A, Roche), a bsAb in clinical trials with the same targets as SI-B001, showed superior activity compared with those by mono-specific EGFR- or HER3-targeting antibodies in several SCCHN and NSCLC tumor models acquired resistance to EGFR inhibitors [147,148].
Co-localized blockage
SHR-1701, anti-PD-L1 mAb fused with TGFBRII by Hengrui, blocks both PD-L1 and TGF-β on a tumor cell. PD-L1 and TGF-β are two key components with independent and complementary immunosuppressive functions; thus, dual targeting of PD-L1 and TGF-β represents a rational of therapeutically synergistic strategy. Anti-PD-L1 and TGF-β-targeted combination therapy raises the possibility of superior antitumor activity compared with monotherapies in tumor cell-intrinsic and tumor cell-extrinsic pathways. For instance, T-helper 1 cytokine production was restored most effectively when PD-1 blockade was combined with TGF-β inhibition [149]. In addition, blocking TGF-β signaling targeted the mechanisms of resistance to PD-1/PD-L1 and sensitized tumors to anti-PD-1/PD-L1 therapies, as the up-regulation of TGF-β signaling-associated genes was linked to anti-PD-1 resistance in metastatic melanoma [150]. The TGF-β-targeted portion of SHR-1701 is the N-terminal-truncated ECD of TGFBRII [94]. The indications of SHR-1701 included multiple advanced solid tumors, with its structure and MOAs similar to those of M7824 (Merck) [151], which was designed to combine co-localized blocking of the two immuno-suppressive pathways (PD-L1 and TGF-β) and to control tumor growth by potentially both restoring and enhancing anti-tumor responses (Fig. 2E).
Biparatopic bsAbs
To improve tumor targeting and tumor retention time of monoclonal antibodies, a biparatopic bsAb (BpAb) was designed to bind two non-overlapping epitopes of the same antigen to enhance the Ab–Ag interactions [152]. From six BpAbs constructed by coupling two different Fab’ fragments from four different specific anti-CEA mAbs recognizing four CEA epitopes, it was confirmed that the apparent Ka value was increased by 8.1- to 9.5-fold as compared with their parental Fabs. The improvement in affinity led a significant higher tumor uptake of the BpAbs in 72 hours and higher tumor localization in 48 hours post-injection into mice [152].
In breast cancer, overexpression of HER2 gene occurs in only 20–25% population of patients who are classified as HER2-positive cancer in clinic. The excessive expression of HER2 often leads to constitutive receptor activation and therefore aggressive tumor growth [153]. Inhibiting HER2 activity with a monoclonal antibody has been proven to be an effective therapy for treating HER2-positive metastatic breast cancer. To date, three HER2-specific monoclonal antibodies including trastuzumab, pertuzumab, and ado-trastuzumab emtansine (T-DM1) have been approved by the FDA for treating the Her2+ breast cancers. However, not all of the treated patients respond to these therapies, which is possibly due to the high degrees of heterogeneity of HER2 expression in breast cancers or the low efficacy of current anti-HER2 therapeutics to the low levels of HER2 expressed on tumor cells [154]. Much effort has been made to improve anti-HER2 agents that can kill cancer cells expressing a broader range of HER2. Synergistic effects have been observed in the combination therapy of trastuzumab and pertuzumab [155], and in silico modeling also showed that the binding affinity towards the HER2 molecule was enhanced by the cooperative interactions between the two monoclonal antibodies [156]. Furthermore, Cryo-EM structural studies on the ternary HER2-trastuzumab-pertuzumab complex implicated an anti-HER2 biparatopic molecule design of this novel therapeutics [157] (Fig. 2F). The experimental investigation indicated that the bivalent BpAb targeting two non-overlapping epitopes on HER2 can induce HER2 receptor clustering, which in turn promotes robust internalization, lysosomal trafficking, and degradation, and the BpAb-based ADC showed a better anti-tumor activity than T-DM1 and kill both T-DM1-resistant and low HER2-expressing tumor cells [154]. These rationalized to develop an idea BpAb further to increase the clinical benefits of these two mAbs. Currently, two bsAbs in China have been developed with such HER2 × HER2 targeted: KN026 and MBS301. As an anti-HER2 Fc-based heterodimer molecule with integration of trastuzumab and pertuzumab, KN026 was demonstrated to simultaneously bind to two non-overlapping epitopes of HER2-expressing cells (BT-474, NCI-N87, and Calu-3) with a similar affinity but with more BsAb molecules bound on the tumor cells than its parental mAbs. Its efficacy was either equivalent to those of the two mAbs combination on several cells (SK-BR-3, BT474, etc.) or better than those of a few others (NCI-H2170 and HCC1419) [158]. KN026 is currently under clinical trials for both its safety and efficacy in China and in the USA. The other anti-HER2 × anti-HER2, MBS301, is an afucosylated bsAb developed by Mabworks and targets for treating HER2-positive breast cancer and gastric cancer. It simultaneously binds both D2 and D4 domains of HER2, therefore achieving the synergistic effects of both trastuzumab and pertuzumab. Specifically, the fucose-knockout bsAb enhanced its ADCC anti-tumor activity [86].
Tumor-targeted immunomodulators
TAAs, such as HER2 and CD20, are expressed generally at high levels on tumor cells and also at lower levels on healthy cells. HER2 protein was identified on cell membranes of epithelial cells in the gastrointestinal, respiratory, reproductive, and urinary tract as well as in the skin, breast, and placenta. The HER2 expressions in human breast and ovarian cancers were correlated positively to the clinical outcomes of the anti-HER2 therapy [159]. While CD20 is one B-cell surface marker involved in development and differentiation of the B cells, it is also expressed in a majority of B-cell malignancies, including chronic lymphocytic leukemia, diffuse large B-cell lymphoma, follicular lymphoma, and mantle cell lymphoma [160]. Tumor-targeted immunomodulators are designed to bind to a TAA and an immunomodulating receptor (e.g. PD-1 or CD47) (Fig. 2G) to enhance the immunotherapy to the targeted tumor.
IBI315, an anti-HER2 × anti-PD-1 bsAb, developed by Innovent in collaboration with Beijing Hanmi pharmaceutical company, targets on HER2+ solid tumor with multiple MOAs. This bsAb functions as a PD-1 blockers, a HER2 antagonist and a linker bridging a PD-1+ immune cell and a HER2+ tumor cell together. IBI315 was found to be effective in treating difficult diseases by binding to both PD-1 and HER2 [88].
IMM0306 developed by ImmuneOnco was designed to target simultaneously CD47 and CD20 antigens on the B cells but to avoid binding to human red blood cells (RBCs). Extensive in vitro characterization studies demonstrated that IMM0306 bound to both CD47 and CD20 with 3–8-folds lower affinity than each of its parent mAbs and maintained the pro-phagocytosis activity of anti-CD47 mAb to the targeted CD47-positive cells and had even stronger ADCC activity than rituximab (an anti-CD20 mAb). Intriguingly, IMM0306 had no binding activity to human RBCs and low binding activity to monkey RBCs [161].
PROSPECTS
CD3-T cell redirecting bsAbs
The approvals of catumaxomab and blinatumomab have stimulated the influx of CD3+ T cell engaging bsAbs from preclinical studies into clinical trials. More than four different CD3 bsAbs developed by Chinese biotech companies have been undergoing clinical evaluation for the treatment of hematologic malignancies or solid cancers (Table 2). Meanwhile, CAR-T, with MOAs similar to CD3+ T cell engaging bsAbs, usually requires genetically modifying patients’ own T cells to recognize and to destroy cancers; currently, there are more than 150 CAR-T programs under clinical development in China. Both bsAb and CAR-T platforms demonstrated a comparable efficacy in preclinical studies and earlier stages of clinical trials [162]. These two approaches shared similar side effects like the cytokine release syndrome (CRS) [163], and the on- and off-tumor toxicities. One of the potential challenges for CD3-targeted bsAbs and CAR-T is to reduce or to control their toxicities.
CD3 bsAbs and CRS
Several approaches in addition to a stepwise dosing and corticosteroids are used to reduce CRS induced by CD3-targeted bsAbs. In a recent clinical trial (trial identifier: NCT03075696) using CD20-TCB to treat relapsed/refractory B-cell Non-Hodgkin’s lymphoma patients, a small dose of anti-CD20 antibody to be given before the bsAb injection can prevent undesired activation of immune cells and avoid CRS [164]. It was hypothesized that initial B-cell depletion by pretreatment of obinutuzumab (anti-CD20 mAb) prior to CD20-TCB administration could reduce T-cell activation by TCB in peripheral blood or other normal tissues, thus reducing the risk of CRS and other potential side effects [164]. Similarly, in preclinical studies of anti-glypican 3 (GPC3) × anti-CD3 bsAb (ERY974, developed by Chugai), cytokine production following the high-dose ERY974 treatment was mitigated by a low-dose pre-treatment in vitro and in vivo [165]. This strategy of pretreatment by a low-dose CD3 bsAb for mitigation of CRS is currently under evaluation on the clinical trials of both M802 (anti-Her2 × anti-CD3) and M701 (anti-EpCAM × anti-CD3) (Table 2).
In another case of anti-HER2 × anti-CD3 TDB, the pre-treatment with either anti-IL6 or anti-TNF-α was found to be effective in mitigating the CRS while maintaining the cytotoxicity of the bsAb [158]. Mechanistic studies indicated that human monocytes in CAR-T therapy are the major source of IL-1 and IL-6 release [167]. Accordingly, the CRS was prevented by monocyte depletion or by blocking IL-6 receptor with tocilizumab (anti-IL6L mAb) approved by FDA for clinical CAR-T therapy [163,166,167]. During the CD3 bsAb treatment, TNF-α, IL-6, and IL-1b driven from T-cells by the bsAb are the primary factors mediating monocyte activation for systemic cytokine release, which further causes the CRS [104]. Preventing TNF-α release by anti-TNF-α or IL-6 release by anti-IL-6 from T-cells is sufficient to impair the systemic release of monocyte cytokines without affecting the antitumor efficacy by the T cells [104]. Also, systemic cytokine release is only observed upon initial exposure to CD3 bsAb not after the subsequent doses [104]. The mechanistic uncoupling of toxic cytokines and T-cell cytotoxicity in the context of CD3-bsAbs also provides a biological rationale to clinically apply a low first dose of the CD3 bsAb to mitigate toxic cytokines. These types of pretreatments of low-dose drug or using anti-TNF-α or anti-IL-6 mAb implied new clinical management of CRS for the clinical application of T cells redirecting bispecific antibodies [165].
Another approach to mitigate CRS is to reduce the binding affinity of a bsAb to CD3. Two previous studies showed that the CRS of anti-CLL-1 × anti-CD3 in cynomolgus monkeys [168] and anti-HER2 × anti-CD3 in mice [169] was dependent on the bsAbs’ binding affinity to CD3: the higher-affinity variants induced higher levels of cytokines released. A project of anti-CD38 × anti-CD3 bsAbs by Amgen also demonstrated the positive correlation between CD3 affinity and CRS [170]. In this case, three bsAbs XmAb4, AMG424, and XmAb5 were constructed with the same affinity to CD38, but 4.4, 34, and 150–230 nM of Kd to CD3, respectively, the CRS side effects were studied comparatively in cynomolgus monkeys. While the monkeys could not tolerate the XmAb4 and too low efficacy was observed by XmAb5, AMG424 with moderate affinity to CD3 was selected for further clinical trials [171]. Therefore, modulating T-cell activation by attenuating CD3-targeted binding affinity while maintaining anti-tumor activity is a promising method to improve the therapeutic window of T-cell engager bsAbs.
CD3 bsAbs for solid tumors
The majority of CD3 bsAbs are being developed for the treatment of hematological malignancies. The success of CD3 bsAbs in solid tumors has been hampered by the lack of a target molecule with sufficient tumor selectivity to avoid a dose-limiting toxicity. Glypican 3 (GPC3) is a heparan sulfate proteoglycan and cell surface oncofetal protein, which is specifically expressed in adult hepatocellular carcinoma (HCC), ovarian clear cell carcinoma, melanoma, squamous cell carcinoma of the lung, hepatoblastoma, nephroblastoma (Wilms’ tumor), and yolk sac tumor [172], as well as some pediatric cancers [173]. GPC3 has become an attractive target for next-generation cancer immunotherapy specifically for liver and kidney cancers [174]. The anti-GPC3 × anti-CD3 bsAb (ERY974) is highly effective in killing various types of GPC3-expressing tumors. ERY974 also induced a robust antitumor efficacy even against tumors with nonimmunogenic features by converting the poorly inflamed cold tumor microenvironment to a highly inflamed hot microenvironment [175,176]. The similar phenomenon was also observed by other researchers that CD3 bsAb therapy turned solid tumors into inflammatory sites but did not install long-term protective memory [177]. These data provided a rationale for testing a combinatorial approach by using a CD3 bsAb and a checkpoint blocker (e.g. anti-PD-1 or anti-PD-L1), in which the CD3 bsAb could both inhibit the tumor and also changed the tumor microenvironment to facilitate the checkpoint blocker-based immune response to the cold tumors.
BsAbs targeting two checkpoints
Targeting two immune checkpoints is a “hot” approach to construct bsAbs by Chinese biotech companies; currently, several projects such as anti-PD-1 × anti-PD-L1, anti-PD-1 × anti-CTLA-4, and anti-PD-L1 × anti-CTLA-4 are under the safety and early efficacy evaluations in clinical trials (Table 2). Theoretically, two targets incorporated into a single bsAb could yield various benefits. The biological functions of such a molecule might be enhanced by co-localization of synergistic receptors on the immune cells and tumor microenvironment, thus enhancing immune activation and minimizing toxicity. However, potential disadvantages of such bsAb may prevent the sequential administration or personalized dosing of two antibodies. As the blocking activity can be achieved by combing two mAbs in clinical settings, the mechanisms of actions of this type of bsAb are combinational rather than synergistic. Data from clinical studies should provide further evidence and rational for further optimization of such a bsAb targeting two immune checkpoints.
Clinical development
Due to the “dual-targeting” nature, a bsAb may modulate novel biology that is impossibly achieved by a combination (mixture) of two mAbs, and the clinical development strategies of bsAbs may also differ from traditional combination therapies. In current clinical trials, the studies of bsAbs are normally designed to be compared with a standard therapy or with the placebo of the tested drug based on NMPA’s guidance. When there is an approved mAb that targets to the antigen of the studied bsAb, e.g. in the case of PD-1 × PD-L1, it is suggested to plan a clinical study to compare the bsAb (e.g. PD-1 × PD-L1) with the mAb (e.g. PD-1 or PD-L1) available on the market. Such head-to-head comparison should provide the risk-benefit assessment of the bsAb. As an example, a multi-centered phase II trial to compare M7824 (anti-PD-L1 × TGF-β bsAb) versus pembrolizumab (anti-PD-1) as the first-line treatment in PD-L1+ advanced NSCLC (ClinicalTrials.gov Identifier: NCT03631706) is currently ongoing, wherein M7824 has showed remarkably increased efficacy.
Expression and CMC
The advances in protein engineering and bioprocesses based on mammalian cells for over expression and cost-effective production of a typical bsAb have allowed many start-up companies in China to bypass the early developed quadroma technology [14,178,179] or even latterly improved chimeric quadroma technology [180,181], by which the Catumaxomab was first launched on and then off market due to the instable and low-yield production and immunogenicity in clinical consequences [182,183]. The engineered CHO cell systems are commercially available and often provide higher than 3–5 g/L of a typical IgG-like bsAb.
More efforts are also focused on searching new expression systems including co-culture methodology [184] and CrossMab technology [15,185] to further resolve incorrect assembly of bsAb fragments [14]. Yet, it still remains a key goal of the field to establish idea expression system to achieve a stable bsAb production with high yield and low cost. Our opinions for how to produce a uniform bsAb with a high quality, and negligible by-products are highly dependent on the structure platform, which should be selected based on their biological and functional needs. The biology should drive the discovery of bsAbs as innovative therapeutics [26]. Along with the discovery, multiple engineering technologies can be applied. For a full IgG-like bsAb with a symmetric or asymmetric structure, knobs-in-holes technology induced between Fc/Fc chains is an efficient way to mitigate the formations of two homodimers [29,46,186]. A common light chain [8,24,187] has been approved to be effective for eliminating the mismatch between HC/LC arms for a whole IgG type, and an scFv-IgG-like bsAb [27] also do so for an asymmetric structure, specifically when attenuating binding affinity to one of the antigen is required, e.g. to CD3 in T cell engaging bsAb for mitigating CRS (see Section CD3 bsAbs and CRS).
CMC technology developed with protein A-based purification for mAb production can be adapted in general for the production of IgG-like bsAb. The advent of biosimilar developments is a driving desire to achieve a lower cost of production and to globalize biologics manufacturing [188]. High titers routinely achieved for IgG-like antibody in mammalian cell culture and high yield of an mAb production have resulted in significant evolution in process platform approaches, including the consideration of alternative expression systems [44,45], continuous biomanufacturing [188–190] and non-chromatographic separation formats [188,191]. The continuous bioprocess reduced hold steps, improved facility utilization, and reduced capital investment with less contamination risk, less deviation and high integrity. However, there are still several challenges to overcome, including high upfront investment, core technologies and experiences barrier in China, new control and validation strategies, and regulatory uncertainties [190]. Meanwhile, CDMOs booming currently in China provide well-established technology platforms for full CMC services from DNA to bioprocess development, and to manufacturing production for national and international biologic companies. To choose CDMO for CMC development and manufacturing production can bypass upfront investments including building the team and facility, and speed up the project progress as well.
Immunogenicity
One of the challenges in developing bispecific antibodies is the immunogenicity caused by new epitopes in the artificial structures of the bsAbs. Although the specific epitope determination of the anti-drug antibodies (ADA) response is not frequently required by FDA, a more general assessment of domain specificity for a multiple-domain product, such as a bsAb, was more commonly performed [74]. The domain specificity is generally started with ADA-positive samples confirmed using the whole molecule and further characterization may require multiple assays to measure immune responses to different domains of the molecule [74].
It was expected that a bsAb produced by a non-B cell might be more immunogenic than an mAb in general because its non-natural structures or intact antibody structures with additional domains could potentially provide novel epitopes leading to increased immunogenicity [192]. The more engineering operations on a bsAb architecture, the higher risk of the molecule immunogenicity. However, this could not be verified by two marked bsAbs. Blinatumomab was constructed as a non-natural structure of scFvs from anti-CD19 and anti-CD3 sequences of variable regions of murine antibodies. However, the results from clinical trials showed that only a few cases of patients with anti-blinatumomab antibodies were detectable and the impact of the immunogenicity on its PK could not be concluded [193]. The other marketed bsAb, Emicizumab, is an asymmetric IgG-like format with common light chains, which bridges FIXa and FX to restore the function of missing FVIIIa in people with hemophilia A. The bsAb was also shown with low immunogenicity (3.5% tested positive for ADA) in the phase III studies [194]. Meanwhile,10 CD3 bsAbs, despite their different product attributes and structures binding to both human and monkey antigens, were shown to be ADA positive in cynomolgus monkeys for toxicology studies [195]. Although ADA generation in the animals resulted in reduced systemic drug exposure or even loss of the exposure by the end of a one-month study, it did not interfere with overall interpretation of toxicology results in these short-term studies. There was sufficient duration of exposure (at least 2 weeks) or sufficient number of animals exposed (a subset of animals with longer than 2-week exposure) to allow for assessing the toxicities of the bsAbs. It was found that only one of the 10 bsAbs reported immunogenicity with nearly 60% of patients developing ADAs [195]. Therefore, the correlation of the immunogenicity found between animals and human patients was also not established.
It was generally accepted that higher immunogenicity comes from more artificial engineering operations: (1) a transformation of the basic domain, including Fc mutations (e.g. KiH, CRIB) and the modification of CH1 and CL (e.g. CrossMab); (2) a introduction of linkers for connecting different domains, such as a linker between VH and VL in scFv, a linker between two scFv in BiTE, etc.; (3) a deletion or addition of a sequences or a domain, which provide new surface exposed for an Ab to recognize; and (4) any change introducing aggregation of a bsAb or new post-translation modifications (e.g. O-glycosylation) on the molecule. Minimizing these structural modifications should effectively reduce the immunogenicity of the entire bsAb molecule along with humanization of the entire bsAb although the humanization of variable regions of an antibody does not always reduce the immunogenicity in humans [196]. The specific strategies include as follows: (1) in domain modification, mutate residues facing to the interface of two domains or facing to the inside of the domain and keep the natural protein surface unchanged. The molecular structure after mutation can be predicted and confirmed by computer simulation (e.g. by discovery studio software). (2) When choosing a linker, it is recommended to use common flexible linkers, such as (GGGGS)n (n = 1 ~ 3), of which low immunogenicity have been verified in clinical trials of blinatumomab [193,197] (3). The structure of a new bsAb is currently difficult to simulate by computer modeling, and a compromise method is to compare the characteristics of the new bsAb with its parental antibodies by monitor its functional activities and stabilities. In theory, the closer in the structures of a bsAb to that of a natural mAb, the lower the risk in immunogenicity of the molecule.
How to predict the immunogenicity of a candidate molecule in earlier preclinical development is still a challenge. Three assays are currently used to assess immunogenicity: (1) T-cell epitope prediction in silico, which is a computer system from Epibase (Lonza). The protein sequence was truncated into short peptides with a length of 7–13 amino acids, which are then aligned in silico with the peptide sequences recognized by TCR in database. All the protein-derived peptides are scored according to their alignment degrees with the reference peptides to determine their immunogenicity risk [198]. (2) In vitro assay to detect the immunogenicity of human-based mAb, which is a cell proliferation assay using flow cytometry to detect a slight increase in proliferating helper-T cells. PBMCs of multiple (20 or more) donors are treated with the candidate molecule, and a high- and a low-immunogenic antibody drugs. The degree of helper T-cell proliferation is analyzed to determine the positive threshold level. Based on the helper T-cell proliferation and threshold levels, the positive incidence of the candidate antibodies was obtained to assess their immunogenicity [199]. (3) In vivo assay by ADA analysis in non-human primate (NHP): the immunogenicity of a bsAb is assessed through the proportion of ADA positive individuals after drug administration. Using this assay, the relative immunogenicity of 27 mAbs in NHPs and human were studied, and 59% (16) of 27 cases were detected with ADA formations, which were comparable between NHPs and human. However, NHPs and human had different types of ADAs in response to these 16 mAbs [196]. Each of above three assays has limitations when used individually, and the integrated use of in silico and in vitro assays provides a predicted immunogenicity for selecting a candidate but not for making “go or not-go” decision during the development [200]. As ADAs could cause the failure of a bio-therapeutic development program, it is still highly required to improve and validate the evaluation assays to reduce the risk of the preclinical programs due to the immunogenicity.
Perspectives for industry of Chinese bsAbs
Prompted by the success of fast follow-up,license in/out,and collaboration of anti-PD-1/anti-PD-L1 and other IO therapeutic developments with matured pharmaceutical companies in developed countries, China is at a stage of rapid developments in biopharmaceutical innovations of therapeutic biologics, CAR-T, and regenerative medicine. BsAb is among the fastest growing class of investigational drugs. The M802 was reported with clinical potential and was approved as the first bsAb for clinical trials in China by the late 2017. Then in about two-and-half years, 21 bsAbs have been developed into clinical trials in China. Technology platforms with focus on bsAbs, such as FIT-Ig, ITab, CRIB, SMAB, Doubody, WuXiBody, and YBODY, all contribute to this new wave of innovations. Based upon the clinical results of bsAbs in China, we expect many more IND approvals from NMPA, quick indication expansion from cancers to other diseases, new combinations of bsAbs with other therapeutics or therapies, and further improved discovery platforms with defined intellectual property. Although no bsAb drugs developed in China have been approved on marketing, bsAb will maintain its rapid growth in China for the years to come.
ACKNOWLEDGEMENT
The authors thank Dr. Shouye Wang for manuscript reviewing and Brian Chi for manuscript editing assistance.
CONFLICT OF INTEREST STATEMENT
The authors are employees of Wuhan YZY Biopharma Co., Ltd that develops and commercializes antibody therapeutics including bispecific antibodies.
ABBREVIATION
ADA, anti-drug antibodies, ADC, antibody drug conjugation, ADCC, antibody-dependent cellular cytotoxicity, ADCP, antibody-dependent cellular phagocytosis, ALL, acute lymphoblastic leukemia, B7-H1, B7 homolog 1, BITE, bispecific T cell engager, BsAb, bispecific antibody, BpAb, biparatopic antibody, CAR-T, chimeric antigen receptor T-Cell immunotherapy, CCK-8, cell counting kit-8, CHO, Chinese hamster ovary cells, CLL-1, C-type lectin-like molecule-1, CMC, chemistry manufacturing and controls, c-MET, c-mesenchymal-epithelial transition factor, CPB, checkpoint blockade, CRIB, charge repulsion-improved bispecific, CRS, cytokine release syndrome, CTLA-4, cytotoxic T-lymphocyte-associated protein-4, CV, cell viability, Cα, constant region of α chain, Cβ, constant region of β chain, DO, dissolved oxygen, ECD, extracellular domain, EGFR, epidermal growth factor receptor, Fab, antigen-binding fragment, FAE, Fab-arm exchange, Fc, crystallizable fragment, FDA, the US Food and Drug Administration, FISH, fluorescence in situ hybridization, FIT-Ig, Fabs-in-tandem immunoglobulins, FIXa, factor IXa, FX, factor X, GPC3, glypican-3, GPCs, cell-surface glypicans, GS, glutamine synthetase, HC, heavy chain, HCCHN, squamous cell carcinoma of the head and neck, HER2, human epidermal growth factor receptor 2, HER3, human epidermal growth factor receptor 3, HGFR, hepatocyte growth factor receptor, HLE-BITE, half-life extended BITE, IAA, immune-associated antigen, IgG, immunoglobulin G, IND, investigational new drug, IO, immuno-oncology, ITab, immune-therapy antibody, KiH, knobs-into-holes, LC, light chain, LAG-3, lymphocyte activation gene-3, mAb, monoclonal antibody, MHC, major histocompatibility complex, MOA, mechanism of action, MSX, methionine sulfoximine, MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3 carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, MTT, 3-(4,5-dimethylazol-2-yl)-2,5-dipheny-tetrazoliumbromide, NFAT, nuclear factors of activated T-cells, NMPA, the China National Medical Products Administration, NSCLC, non-small-cell lung cancer, PD, pharmacodynamics, PD-1, programmed cell death protein 1, PD-L1, programmed death-ligand 1, PK, pharmacokinetics, RBC, red blood cell, scFv, single-chain variable fragment, SIRPα, signal regulatory protein α, TAA, tumor-associated antigen, TCR, T-cell receptor, TGFBRII, TGF beta receptor II, TGF-β, transforming growth factor β, TNF-α, T-cell-generated tumor necrosis factor-α, TKI, tyrosine kinase inhibitors, TME, tumor microenvironment, VCD, viable cell density, VHH, or single-domain antibody (SDA): antigen-binding immunoglobulin variable domain of “heavy-chain antibodies”, WST-1, 4-[3-(4-iodophenyl)-2(4-nitrophenyl)-2H-5-tetrazolio] -1,3benzene disulfonate, XTT, sodium,3-[1-[phenylamino-carbonyl]-3,4-tetrazolium]-bis(4-methoxy-6-nitro) benzene-sulfonic acid hydrate
REFERENCES
- 1. Nisonoff, A, Wissler, FC, Lipman, LN. Properties of the major component of a peptic digest of rabbit antibody. Science 1960; 132: 1770–1. [DOI] [PubMed] [Google Scholar]
- 2. Milstein, C, Cuello, AC. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983; 305: 537–40. [DOI] [PubMed] [Google Scholar]
- 3. Chelius, D, Ruf, P, Gruber, Pet al. Structural and functional characterization of the trifunctional antibody catumaxomab. MAbs 2010; 2: 309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klinger, M, Brandl, C, Zugmaier, Get al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012; 119: 6226–33. [DOI] [PubMed] [Google Scholar]
- 5. Holliger, P, Prospero, T, Winter, G. "Diabodies": small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A 1993; 90: 6444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kitazawa, T, Esaki, K, Tachibana, Tet al. Factor VIIIa-mimetic cofactor activity of a bispecific antibody to factors IX/IXa and X/Xa, emicizumab, depends on its ability to bridge the antigens. Thromb Haemost 2017; 117: 1348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Igawa, T. Next generation antibody therapeutics using Bispecific antibody technology. Yakugaku Zasshi 2017; 137: 831–6. [DOI] [PubMed] [Google Scholar]
- 8. Shiraiwa, H, Narita, A, Kamata-Sakurai, Met al. Engineering a bispecific antibody with a common light chain: identification and optimization of an anti-CD3 epsilon and anti-GPC3 bispecific antibody, ERY974. Methods 2019; 154: 10–20. [DOI] [PubMed] [Google Scholar]
- 9. McWhirter, J, MacDonald, L, Stevens, S, et al. (2018) Patent US 10143186B2; PCT/US2011/023971.
- 10. Brinkmann, U, Kontermann, RE. The making of bispecific antibodies. MAbs 2017; 9: 182–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shim, H. Bispecific antibodies and antibody–drug conjugates for cancer therapy: technological considerations. Biomolecules 2020; 10: 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suurs, FV, Lub-de Hooge, MN, deVries, EGEet al. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacology & therapeutics 2019; 201: 103–19. [DOI] [PubMed] [Google Scholar]
- 13. Spiess, C, Zhai, Q, Carter, PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67: 95–106. [DOI] [PubMed] [Google Scholar]
- 14. Wang, Q, Chen, Y, Park, Jet al. Design and production of Bispecific antibodies. Antibodies 2019; 8: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schaefer, W, Regula, JT, Bahner, Met al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci U S A 2011; 108: 11187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Labrijn, AF, Meesters, JI, deGoeij, BEet al. Efficient generation of stable bispecific IgG1 by controlled fab-arm exchange. Proc Natl Acad Sci U S A 2013; 110: 5145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hamers-Casterman, C, Atarhouch, T, Muyldermans, Set al. Naturally occurring antibodies devoid of light chains. Nature 1993; 363: 446–8. [DOI] [PubMed] [Google Scholar]
- 18. Wu, C, Ying, H, Bose, Set al. Molecular construction and optimization of anti-human IL-1alpha/beta dual variable domain immunoglobulin (DVD-Ig) molecules. MAbs 2009; 1: 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moore, PA, Zhang, W, Rainey, GJet al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011; 117: 4542–51. [DOI] [PubMed] [Google Scholar]
- 20. Kainer, M, Antes, B, Wiederkum, Set al. Correlation between CD16a binding and immuno effector functionality of an antigen specific immunoglobulin fc fragment (Fcab). Arch Biochem Biophys 2012; 526: 154–8. [DOI] [PubMed] [Google Scholar]
- 21. Von Kreudenstein, TS, Escobar-Carbrera, E, Lario, PIet al. Improving biophysical properties of a bispecific antibody scaffold to aid developability: quality by molecular design. MAbs 2013; 5: 646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reusch, U, Burkhardt, C, Fucek, Iet al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs 2014; 6: 728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moore, GL, Bernett, MJ, Rashid, Ret al. A robust heterodimeric fc platform engineered for efficient development of bispecific antibodies of multiple formats. Methods 2019; 154: 38–50. [DOI] [PubMed] [Google Scholar]
- 24. De Nardis, C, Hendriks, LJA, Poirier, Eet al. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J Biol Chem 2017; 292: 14706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pellerin, L, Chen, P, Gregori, Set al. APVO210: a Bispecific anti-CD86-IL-10 fusion protein (ADAPTIR) to induce antigen-specific T regulatory type 1 cells. Front Immunol 2018; 9: 881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nie, S, Wang, Z, Moscoso-Castro, Met al. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antibody therapeutics 2020; 23: 18–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Labrijn, AF, Janmaat, ML, Reichert, JMet al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 2019; 18: 585–608. [DOI] [PubMed] [Google Scholar]
- 28. Thakur, A, Huang, M, Lum, LG. Bispecific antibody based therapeutics: strengths and challenges. Blood reviews 2018; 32: 339–47. [DOI] [PubMed] [Google Scholar]
- 29. Zhou, P, Zhang, J, Yan, Y. (2017) Patent US 9562110B2; PCT/CN2012/084982.
- 30. Ridgway, JB, Presta, LG, Carter, P. Knobs-into-holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 1996; 9: 617–21. [DOI] [PubMed] [Google Scholar]
- 31. Gunasekaran, K, Pentony, M, Shen, Met al. Enhancing antibody fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem 2010; 285: 19637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu, T, Xu, T, Wang, X, et al. (2019) Patent US 20190031782; PCT/CN2016/070447.
- 33. Cui, Y, Huang, Z, Chen, H, et al. (2017) Patent US 20190092862; PCT/CN2017/076816.
- 34. Wu, C. (2019) Patent US 10266608B2; PCT/US2014/072336.
- 35. Li, H, Li, Y, Wang, Cet al. Highlights of 2019 protein engineering summit (PEGS) in Boston, USA: advancing antibody-based cancer therapies to the clinic. Antibody therapeutics 2019; 2: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. United States Patent and Trademark Office , Public Patent Application Information Retrieval. https://portal.uspto.gov/pair/PublicPair (20 April 2020, last accessed)
- 37. United States Patent and Trademark Office , Search for patents. http://patft.uspto.gov/netahtml/PTO/search-bool.html (20 April 2020, last accessed)
- 38. Brischwein, K, Schlereth, B, Guller, Bet al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol 2006; 43: 1129–43. [DOI] [PubMed] [Google Scholar]
- 39. Stadler, CR, Bähr-Mahmud, H, Plum, LMet al. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. OncoImmunology 2015; 5: e1091555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu, Y, Yu, M, Sun, Zet al. Generation and characterization of a Bispecific antibody targeting both PD-1 and c-MET. Protein and peptide letters 2018; 24: 1105–12. [DOI] [PubMed] [Google Scholar]
- 41. Verma, R, Boleti, E, George, AJT. Antibody engineering: comparison of bacterial, yeast, insect and mammalian expression systems. Journal of Immunological Methods 1998; 216: 165–81. [DOI] [PubMed] [Google Scholar]
- 42. Vendel, MC, Favis, M, Snyder, WBet al. Secretion from bacterial versus mammalian cells yields a recombinant scFv with variable folding properties. Arch Biochem Biophys 2012; 526: 188–93. [DOI] [PubMed] [Google Scholar]
- 43. Jain, S, Aresu, L, Comazzi, Set al. The development of a recombinant scFv monoclonal antibody targeting canine CD20 for use in comparative medicine. PLoS One 2016; 11: e0148366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Seber Kasinger, LE, Dent, MW, Mahajan, Get al. A novel anti-HIV-1 bispecific bNAb-lectin fusion protein engineered in a plant-based transient expression system. Plant biotechnology journal 2019; 17: 1646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fischer, R, Schumann, D, Zimmermann, Set al. Expression and characterization of bispecific single-chain Fv fragments produced in transgenic plants. European journal of biochemistry 1999; 262: 810–6. [DOI] [PubMed] [Google Scholar]
- 46. Xu, Y, Lee, J, Tran, Cet al. Production of bispecific antibodies in “knobs-into-holes” using a cell-free expression system. mAbs 2015; 7: 231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hunter, DJB, Bhumkar, A, Giles, Net al. Unexpected instabilities explain batch-to-batch variability in cell-free protein expression systems. Biotechnol Bioeng 2018; 115: 1904–14. [DOI] [PubMed] [Google Scholar]
- 48. Jäger, V, Büssow, K, Wagner, Aet al. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnology 2013; 13: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rajendra, Y, Balasubramanian, S, Peery, RBet al. Bioreactor scale up and protein product quality characterization of piggyBac transposon derived CHO pools. Biotechnology Progress 2017; 33: 534–40. [DOI] [PubMed] [Google Scholar]
- 50. Balasubramanian, S, Peery, RB, Minshull, Jet al. Generation of high expressing Chinese hamster ovary cell pools using the leap-in transposon system. Biotechnology Journal 2018; 13: 1700748. [DOI] [PubMed] [Google Scholar]
- 51. Fan, Z, Sun, A, Liu, Xet al. The accelerated atherogenesis of venous grafts might be attributed to aggravated concentration polarization of low density lipoproteins: a numerical study. J Biomech 2013; 46: 2388–93. [DOI] [PubMed] [Google Scholar]
- 52. Chusainow, J, Yang, YS, Yeo, JHMet al. A study of monoclonal antibody-producing CHO cell lines: what makes a stable high producer? Biotechnology and Bioengineering 2009; 102: 1182–96. [DOI] [PubMed] [Google Scholar]
- 53. Bailey, LA, Hatton, D, Field, Ret al. Determination of Chinese hamster ovary cell line stability and recombinant antibody expression during long-term culture. Biotechnology and Bioengineering 2012; 109: 2093–103. [DOI] [PubMed] [Google Scholar]
- 54. The International Conference on Harmonization (ICH) guidance : Q6B specifications: test procedures and acceptance criteria for biotechnological/biological products. https://www.fda.gov/media/71510/download (20 April 2020, last accessed)
- 55. The International Conference on Harmonization (ICH) guidance : Q5A viral safety evaluation of biotechnology products derived from cell lines of human or animal Origin. https://www.fda.gov/media/71394/download (20 April 2020, last accessed) [PubMed]
- 56. Liu, H, Saxena, A, Sidhu, SSet al. Fc engineering for developing therapeutic Bispecific antibodies and novel scaffolds. Front Immunol 2017; 8: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Han, H, Ma, J, Zhang, Ket al. Bispecific anti-CD3 x anti-HER2 antibody mediates T cell cytolytic activity to HER2-positive colorectal cancer in vitro and in vivo. Int J Oncol 2014; 45: 2446–54. [DOI] [PubMed] [Google Scholar]
- 58. Lu, L, Liu, NB, Fan, KHet al. A tetravalent single chain diabody (CD40/HER2) efficiently inhibits tumor proliferation through recruitment of T cells and anti-HER2 functions. Mol Immunol 2019; 109: 149–56. [DOI] [PubMed] [Google Scholar]
- 59. Asano, R, Nagai, K, Makabe, Ket al. Structural considerations for functional anti-EGFR x anti-CD3 bispecific diabodies in light of domain order and binding affinity. Oncotarget 2018; 9: 13884–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koopmans, I, Hendriks, D, Samplonius, DFet al. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. Oncoimmunology 2018; 7: e1466016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zeidler, R, Reisbach, G, Wollenberg, Bet al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol 1999; 163: 1246–52. [PubMed] [Google Scholar]
- 62. Zhong, YB, Zhang, XH, Chen, JXet al. Overexpression, purification, characterization, and pathogenicity of Vibrio harveyi hemolysin VHH. Infect Immun 2006; 74: 6001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang, SG, Yu, HT, Wickliffe, JK. Limitation of the MTT and XTT assays for measuring cell viability due to superoxide formation induced by nano-scale TiO2. Toxicol in Vitro 2011; 25: 2147–51. [DOI] [PubMed] [Google Scholar]
- 64. Cheng, Y, Hu, R, Jin, Het al. Effect of 14-3-3 tau protein on differentiation in BeWo choriocarcinoma cells. Placenta 2010; 31: 60–6. [DOI] [PubMed] [Google Scholar]
- 65. McDonagh, CF, Huhalov, A, Harms, BDet al. Antitumor activity of a novel Bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits Heregulin-induced activation of ErbB3. Mol Cancer Ther 2012; 11: 582–93. [DOI] [PubMed] [Google Scholar]
- 66. Leconet, W, Liu, H, Guo, Met al. Anti-PSMA/CD3 Bispecific antibody delivery and antitumor activity using a polymeric depot formulation. Mol Cancer Ther 2018; 17: 1927–40. [DOI] [PubMed] [Google Scholar]
- 67. Ma, J, Ge, J, Xue, Xet al. Targeting bladder cancer using activated T cells armed with bispecific antibodies. Oncol Rep 2018; 39: 1245–52. [DOI] [PubMed] [Google Scholar]
- 68. Iizuka, A, Nonomura, C, Ashizawa, Tet al. A T-cell-engaging B7-H4/CD3-bispecific fab-scFv antibody targets human breast cancer. Clin Cancer Res 2019; 25: 2925–34. [DOI] [PubMed] [Google Scholar]
- 69. Mack, M, Riethmuller, G, Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci USA 1995; 92: 7021–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chan, JK, Hamilton, CA, Cheung, MKet al. Enhanced killing of primary ovarian cancer by retargeting autologous cytokine-induced killer cells with bispecific antibodies: a preclinical study. Clin Cancer Res 2006; 12: 1859–67. [DOI] [PubMed] [Google Scholar]
- 71. Grabert, RC, Cousens, LP, Smith, JAet al. Human T cells armed with Her2/neu bispecific antibodies divide, are cytotoxic, and secrete cytokines with repeated stimulation. Clinical Cancer Research 2006; 12: 569–76. [DOI] [PubMed] [Google Scholar]
- 72. Lum, LG, Ramesh, M, Thakur, Aet al. Targeting cytomegalovirus-infected cells using T cells armed with anti-CD3 x anti-CMV bispecific antibody. Biol Blood Marrow Transplant 2012; 18: 1012–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harms, JS, Khan, M, Hall, Cet al. Brucella peptide cross-reactive major histocompatibility complex class I presentation activates SIINFEKL-specific T cell receptor-expressing T cells. Infect Immun 2018; 86: e00281–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. The guidance for industry : Immunogenicity testing of therapeutic protein products-developing and validating assays for anti-drug antibody detection. https://www.fda.gov/media/119788/download (20 April 2020, last accessed)
- 75. Brandl, C, Haas, C, d'Argouges, Set al. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer immunology, immunotherapy : CII 2007; 56: 1551–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Koopmans, I, Hendriks, M, vanGinkel, RJet al. Bispecific antibody approach for improved melanoma-selective PD-L1 immune checkpoint blockade. J Invest Dermatol 2019; 139: 2343–51 e3. [DOI] [PubMed] [Google Scholar]
- 77. Musielak, B, Kocik, J, Skalniak, Let al. CA-170 - a potent small-molecule PD-L1 inhibitor or not? Molecules 2019; 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Center for Drug Evaluation, the National Medical Products Authority . http://www.cde.org.cn/ (20 April 2020, last accessed)
- 79. Seifert, O, Rau, A, Beha, Net al. Diabody-Ig: a novel platform for the generation of multivalent and multispecific antibody molecules. mAbs 2019; 11: 919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Husain, B, Ellerman, D. Expanding the boundaries of biotherapeutics with Bispecific antibodies. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy 2018; 32: 441–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dahlen, E, Veitonmaki, N, Norlen, P. Bispecific antibodies in cancer immunotherapy. Therapeutic advances in vaccines and immunotherapy 2018; 6: 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Strop, P, Ho, WH, Boustany, LMet al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol 2012; 420: 204–19. [DOI] [PubMed] [Google Scholar]
- 83. Zhou, P, Wang, T, Fang, L, et al. (2017) Patent US 9777073B2; PCT/CN2014/082590.
- 84. Zhou, P, Zhang, J, Hu, L, et al. (2017) Patent US 9611325B2; PCT/CN2014/082590.
- 85. Yu, S, Zhang, J, Yan, Yet al. A novel asymmetrical anti-HER2/CD3 bispecific antibody exhibits potent cytotoxicity for HER2-positive tumor cells. J Exp Clin Cancer Res 2019; 38: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li, F, Zhang, B, Ye, P, et al. (2017) Patent US 9745382B1; PCT/CN2017/093816.
- 87. Liu, J, Song, N, Yang, Y, et al. (2018) Patent WO2018177324; PCT/CN2018/080858.
- 88. Liu, J, Song, N, Yang, Y. (2018) Patent WO2018090950; PCT/CN2017/111310.
- 89. Xu, T, Dong, Y, Wang, P. (2017) Patent US 20180291103; PCT/CN2016/092679.
- 90. Eckelman, BP, Timmer, JC, Hata, C, et al. (2017) Patent US 20170198050A1; PCT/US2017/013040.
- 91. Kong, J, Ye, Y, Zhou, P, et al. (2019) Patent US 20190284279; PCT/CN2018/085397.
- 92. Li, B, Xia, Y, Wang, ZM, et al. (2019) Patent US 20190185569; PCT/CN2017/098466.
- 93. Gao, Z, TAN, P, Kovacevich, B, et al. (2015) Patent WO2016106157A1; PCT/US2015/066951.
- 94. LaMotte-Mohs, R, Shah, K, Smith, Det al. Abstract 3217: MGD013, a bispecific PD-1 x LAG-3 dual-affinity ReTargeting (DART®) protein with T-cell immunomodulatory activity for cancer treatment. Cancer research 2016; 76: 3217–7. [Google Scholar]
- 95. Gu, J, Luo, X, Tao, W. (2018) Patent CN 201880004344.6A; PCT/CN2018/086451.
- 96. Tian, W, Li, S. (2018) Patent WO201816650; PCT/CN2018/079187.
- 97. Huang, Y, Zhang, F, Xi, G. (2019) Patent WO2019109357; PCT/CN2017/115323.
- 98. Hinner, MJ, Aiba, R-SB, Wiedenmann, Aet al. Costimulatory T cell engagement via a novel bispecific anti-CD137 /anti-HER2 protein. Journal for ImmunoTherapy of Cancer 2015; 3. [Google Scholar]
- 99. Chames, P, Baty, D. Bispecific antibodies for cancer therapy. mAbs 2014; 1: 539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hudis, CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007; 357: 39–51. [DOI] [PubMed] [Google Scholar]
- 101. Seimetz, D. Novel monoclonal antibodies for cancer treatment: the trifunctional antibody catumaxomab (removab). J Cancer 2011; 2: 309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Buie, LW, Pecoraro, JJ, Horvat, TZet al. Blinatumomab: a first-in-class Bispecific T-cell engager for precursor B-cell acute lymphoblastic leukemia. Ann Pharmacother 2015; 49: 1057–67. [DOI] [PubMed] [Google Scholar]
- 103. Arvedson, TL, Balazs, M, Bogner, P, et al. Abstract 55: Generation of half-life extended anti-CD33 BiTE® antibody constructs compatible with once-weekly dosing. 2017, doi: 10.1158/1538-7445.am2017-55. [DOI] [Google Scholar]
- 104. Vezys, V, Penaloza-MacMaster, P, Barber, DLet al. 4-1BB signaling synergizes with programmed death ligand 1 blockade to augment CD8 T cell responses during chronic viral infection. The Journal of Immunology 2011; 187: 1634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Beyersdorf, N, Kerkau, T, Hunig, T. CD28 co-stimulation in T-cell homeostasis: a recent perspective. ImmunoTargets and therapy 2015; 4: 111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li, Y, Tan, S, Zhang, Cet al. Limited cross-linking of 4-1BB by 4-1BB ligand and the agonist monoclonal antibody Utomilumab. Cell Rep 2018; 25: 909–20 e4. [DOI] [PubMed] [Google Scholar]
- 107. Li, J, Piskol, R, Ybarra, Ret al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med 2019; 11: eaax8861. [DOI] [PubMed] [Google Scholar]
- 108. Freitas, AA, Oh, HS, Choi, BKet al. 4-1BB signaling enhances primary and secondary population expansion of CD8+ T cells by maximizing Autocrine IL-2/IL-2 receptor signaling. Plos One 2015; 10: e0126765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Choi, BK, Lee, DY, Lee, DGet al. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8+ T cell proliferation. Cellular & Molecular Immunology 2016; 14: 748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Brunet, JF, Denizot, F, Luciani, MFet al. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987; 328: 267–70. [DOI] [PubMed] [Google Scholar]
- 111. Buchbinder, EI, McDermott, DF. Cytotoxic T-lymphocyte antigen-4 blockade in melanoma. Clin Ther 2015; 37: 755–63. [DOI] [PubMed] [Google Scholar]
- 112. Ishida, Y, Agata, Y, Shibahara, Ket al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992; 11: 3887–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dong, H, Zhu, G, Tamada, Ket al. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med 1999; 5: 1365–9. [DOI] [PubMed] [Google Scholar]
- 114. Syn, NL, Teng, MWL, Mok, TSKet al. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 2017; 18: e731–e41. [DOI] [PubMed] [Google Scholar]
- 115. Dong, H, Strome, SE, Salomao, DRet al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8: 793–800. [DOI] [PubMed] [Google Scholar]
- 116. Poole, RM. Pembrolizumab: first global approval. Drugs 2014; 74: 1973–81. [DOI] [PubMed] [Google Scholar]
- 117. Markham, A. Atezolizumab: First Global Approval. Drugs 2016; 76: 1227–32. [DOI] [PubMed] [Google Scholar]
- 118. Kim, ES. Avelumab: First Global Approval. Drugs 2017; 77: 929–37. [DOI] [PubMed] [Google Scholar]
- 119. Syed, YY. Durvalumab: First Global Approval. Drugs 2017; 77: 1369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Osipov, A, Zaidi, N, Laheru, DA. Dual checkpoint inhibition in pancreatic cancer: revealing the limitations of synergy and the potential of novel combinations. JAMA Oncology 2019; 5: 1438–9. [DOI] [PubMed] [Google Scholar]
- 121. Reck, M, Borghaei, H, O'Byrne, KJ. Nivolumab plus ipilimumab in non-small-cell lung cancer. Future Oncol 2019; 15: 2287–302. [DOI] [PubMed] [Google Scholar]
- 122. Winer, A, Ghatalia, P, Bubes, Net al. Dual checkpoint inhibition with Ipilimumab plus Nivolumab after progression on sequential PD-1/PDL-1 inhibitors Pembrolizumab and Atezolizumab in a patient with lynch syndrome, metastatic colon, and localized Urothelial cancer. The Oncologist 2019; 24: 1416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hassel, JC, Heinzerling, L, Aberle, Jet al. Combined immune checkpoint blockade (anti-PD-1/anti-CTLA-4): evaluation and management of adverse drug reactions. Cancer Treatment Reviews 2017; 57: 36–49. [DOI] [PubMed] [Google Scholar]
- 124. Du, X, Liu, M, Su, Jet al. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Research 2018; 28: 433–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Du, X, Tang, F, Liu, Met al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Research 2018; 28: 416–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Pol, J, Kroemer, G. Anti-CTLA-4 immunotherapy: uncoupling toxicity and efficacy. Cell Res 2018; 28: 501–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Liu, Y, Zheng, P. Preserving the CTLA-4 checkpoint for safer and more effective cancer immunotherapy. Trends in pharmacological sciences 2020; 41: 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Schurch, CM, Forster, S, Bruhl, Fet al. The "don't eat me" signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. Oncoimmunology 2017; 7: e1373235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhang, H, Deng, M, Lin, Pet al. Frontiers and opportunities: highlights of the 2(nd) annual conference of the Chinese antibody society. Antibody therapeutics 2018; 1: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ruffo, E, Wu, RC, Bruno, TCet al. Lymphocyte-activation gene 3 (LAG3): the next immune checkpoint receptor. Semin Immunol. 2019; 42: 101305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Huang, CT, Workman, CJ, Flies, Det al. Role of LAG-3 in regulatory T cells. Immunity. 2004; 21: 503–13. [DOI] [PubMed] [Google Scholar]
- 132. Baixeras, E, Huard, B, Miossec, Cet al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. 1992; 176: 327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Workman, CJ, Cauley, LS, Kim, IJet al. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004; 172: 5450–5. [DOI] [PubMed] [Google Scholar]
- 134. Wang, J, Sanmamed, MF, Datar, Iet al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. 2019; 176: 334–47e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Rotte, A, Jin, JY, Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol. 2018; 29: 71–83. [DOI] [PubMed] [Google Scholar]
- 136. Kraman M, Fosh N, Kmiecik K, et al. , Abstract 2719: dual blockade of PD-L1 and LAG-3 with FS118, a unique bispecific antibody, induces CD8+ T-cell activation and modulates the tumor microenvironment to promote antitumor immune responses, Cancer research 78(13Supplement) (2018) 2719–2719. [Google Scholar]
- 137. Engelman, JA, Zejnullahu, K, Mitsudomi, Tet al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 138. Turke, AB, Zejnullahu, K, Wu, YLet al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17: 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yano, S, Yamada, T, Takeuchi, Set al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol 2011; 6: 2011–7. [DOI] [PubMed] [Google Scholar]
- 140. Kobayashi, S, Boggon, TJ, Dayaram, Tet al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 141. Pao, W, Miller, VA, Politi, KAet al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Bean, J, Brennan, C, Shih, JYet al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007; 104: 20932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chen, HJ, Mok, TS, Chen, ZHet al. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol Oncol Res 2009; 15: 651–8. [DOI] [PubMed] [Google Scholar]
- 144. Moores, SL, Chiu, ML, Bushey, BSet al. A novel Bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor-resistant lung tumors. Cancer Res 2016; 76: 3942–53. [DOI] [PubMed] [Google Scholar]
- 145. van Lengerich, B, Agnew, C, Puchner, EMet al. EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc Natl Acad Sci U S A 2017; 114: E2836–E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Mujoo, K, Choi, BK, Huang, Zet al. Regulation of ERBB3/HER3 signaling in cancer. Oncotarget 2014; 5: 10222–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Huang, S, Li, C, Sliwkowski, MXet al. Abstract 637: MEHD7945A, an EGFR/ErbB3 dual specific antibody, overcomes acquired resistance to EGFR inhibitors in head and neck and lung tumors. Cancer Research 2011; 71: 637. [Google Scholar]
- 148. Schaefer, G, Haber, L, Crocker, LMet al. A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 2011; 20: 472–86. [DOI] [PubMed] [Google Scholar]
- 149. Topalian, SL, Drake, CG, Pardoll, DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol 2012; 24: 207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hugo, W, Zaretsky, JM, Sun, Let al. Genomic and Transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016; 165: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lan, Y, Zhang, D, Xu, Cet al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Science Translational Medicine 2018; 10: eaan5488. [DOI] [PubMed] [Google Scholar]
- 152. Robert, B, Dorvillius, M, Buchegger, Fet al. Tumor targeting with newly designed biparatopic antibodies directed against two different epitopes of the carcinoembryonic antigen (CEA). International Journal of Cancer 1999; 81: 285–91. [DOI] [PubMed] [Google Scholar]
- 153. Slamon, DJ, Clark, GM, Wong, SGet al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987; 235: 177–82. [DOI] [PubMed] [Google Scholar]
- 154. Li, JY, Perry, SR, Muniz-Medina, Vet al. A Biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 2019; 35: 948–9. [DOI] [PubMed] [Google Scholar]
- 155. Nami, B, Maadi, H, Wang, Z. Mechanisms underlying the action and synergism of Trastuzumab and Pertuzumab in targeting HER2-positive breast cancer. Cancers 2018; 10: 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Fuentes, G, Scaltriti, M, Baselga, Jet al. Synergy between trastuzumab and pertuzumab for human epidermal growth factor 2 (Her2) from colocalization: an in silico based mechanism. Breast Cancer Res 2011; 13: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Hao, Y, Yu, X, Bai, Yet al. Cryo-EM structure of HER2-trastuzumab-pertuzumab complex. PLoS One 2019; 14: e0216095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wei, H, Cai, H, Jin, Yet al. Structural basis of a novel heterodimeric fc for bispecific antibody production. Oncotarget 2017; 8: 51037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Press, MF, Cordon-Cardo, C, Slamon, DJ. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene 1990; 5: 953–62. [PubMed] [Google Scholar]
- 160. Prevodnik, VK, Lavrencak, J, Horvat, Met al. The predictive significance of CD20 expression in B-cell lymphomas. Diagn Pathol 2011; 6: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Tian, W, Li, S, Chen, Det al. Preclinical development of a bispecific antibody-trap selectively targeting CD47 and CD20 for the treatment of B cell lineage cancer. In: AACR Annual Meeting 2019. Atlanta GA: American Association for Cancer Research, 2019. Abstract 545 [Google Scholar]
- 162. Regeneron CD20xCD3 Bispecific REGN1979 Shows Positive Results in Patients with Relapsed or Refractory B-cell Non-Hodgkin Lymphoma, including in CAR-T Failures. https://newsroom.regeneron.com/node/22521/pdf (20 April 2020, last accessed)
- 163. Shimabukuro-Vornhagen, A, Gödel, P, Subklewe, Met al. Cytokine release syndrome. Journal for ImmunoTherapy of Cancer 2018; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Bacac, M, Colombetti, S, Herter, Set al. CD20-TCB with Obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin Cancer Res 2018; 24: 4785–97. [DOI] [PubMed] [Google Scholar]
- 165. Hoshino, M, Sano, Y, Kinoshita, Yet al. Differential exhaustion on cytokine release (DECREASE) by ERY974, a novel T-cell-redirecting antibody targeting glypican-3: A new type of T-cell exhaustion. In: AACR Annual Meeting 2019. Atlanta GA: American Association for Cancer Research, 2019. Abstract 2378 [Google Scholar]
- 166. Le, RQ, Li, L, Yuan, Wet al. FDA approval summary: Tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. The Oncologist 2018; 23: 943–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Norelli, M, Camisa, B, Barbiera, Get al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nature Medicine 2018; 24: 739–48. [DOI] [PubMed] [Google Scholar]
- 168. Leong, SR, Sukumaran, S, Hristopoulos, Met al. An anti-CD3/anti-CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood 2017; 129: 609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Staflin, K, Zuch de Zafra, CL, Schutt, LKet al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight 2020; 5: e33757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Moore, GL, Lee, S-H, Schubbert, Set al. Tuning T cell affinity improves efficacy and safety of anti-CD38 × anti-CD3 Bispecific antibodies in monkeys - a potential therapy for multiple myeloma. Blood 2015; 126: 1798. [Google Scholar]
- 171. Zuch de Zafra, CL, Fajardo, F, Zhong, Wet al. Targeting multiple myeloma with AMG 424, a novel anti-CD38/CD3 Bispecific T-cell-recruiting antibody optimized for cytotoxicity and cytokine release. Clin Cancer Res 2019; 25: 3921–33. [DOI] [PubMed] [Google Scholar]
- 172. Shimizu, Y, Suzuki, T, Yoshikawa, Tet al. Next-generation cancer immunotherapy targeting Glypican-3. Frontiers in oncology 2019; 9: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Taguchi, T, Nakatsura, T, Oda, Yet al. Glypican 3 expression in pediatric malignant solid tumors. European Journal of Pediatric Surgery 2014; 25: 138–44. [DOI] [PubMed] [Google Scholar]
- 174. Ortiz, MV, Roberts, SS, Glade Bender, Jet al. Immunotherapeutic targeting of GPC3 in pediatric solid Embryonal tumors. Frontiers in oncology 2019; 9: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kaur, SP, Cummings, BS. Role of glypicans in regulation of the tumor microenvironment and cancer progression. Biochemical pharmacology 2019; 168: 108–18. [DOI] [PubMed] [Google Scholar]
- 176. Ishiguro, T, Sano, Y, Komatsu, S-iet al. An anti–glypican 3/CD3 bispecific T cell–redirecting antibody for treatment of solid tumors. Science Translational Medicine 2017; 9: eaal4291. [DOI] [PubMed] [Google Scholar]
- 177. Benonisson, H, Altintas, I, Sluijter, Met al. CD3-Bispecific antibody therapy turns solid tumors into inflammatory sites but does not install protective memory. Mol Cancer Ther 2019; 18: 312–22. [DOI] [PubMed] [Google Scholar]
- 178. Suresh, MR, Cuello, AC, Milstein, C.. [17] Bispecific monoclonal antibodies from hybrid hybridomas. 1986;121:210–28. [DOI] [PubMed] [Google Scholar]
- 179. Nolan, O, O'Kennedy, R. Bifunctional antibodies: concept, production and applications. Biochimica et biophysica acta 1990; 1040: 1–11. [DOI] [PubMed] [Google Scholar]
- 180. Sedykh, SE, Prinz, VV, Buneva, VNet al. Bispecific antibodies: design, therapy, perspectives. Drug design. development and therapy 2018; 12: 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Lindhofer, H, Mocikat, R, Steipe, Bet al. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol 1995; 155: 219–25. [PubMed] [Google Scholar]
- 182. Seimetz, D, Lindhofer, H, Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM×anti-CD3) as a targeted cancer immunotherapy. Cancer Treatment Reviews 2010; 36: 458–67. [DOI] [PubMed] [Google Scholar]
- 183. Linke, R, Klein, A, Seimetz, D. Catumaxomab: clinical development and future directions. mAbs 2014; 2: 129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Spiess, C, Merchant, M, Huang, Aet al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nature Biotechnology 2013; 31: 753–8. [DOI] [PubMed] [Google Scholar]
- 185. Klein, C, Schaefer, W, Regula, JTet al. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods 2019; 154: 21–31. [DOI] [PubMed] [Google Scholar]
- 186. Wagner, EK, Wang, X, Bui, Aet al. Synergistic neutralization of pertussis toxin by a Bispecific AntibodyIn VitroandIn vivo. Clinical and Vaccine Immunology 2016; 23: 851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Krah, S, Kolmar, H, Becker, Set al. Engineering IgG-like Bispecific antibodies—an overview. Antibodies 2018; 7: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Shukla, AA, Wolfe, LS, Mostafa, SSet al. Evolving trends in mAb production processes. Bioengineering & Translational Medicine 2017; 2: 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Zydney, AL. Continuous downstream processing for high value biological products: a review. Biotechnol Bioeng 2016; 113: 465–75. [DOI] [PubMed] [Google Scholar]
- 190. Zhang, H, Deng, M, Pei, Fet al. Next-generation antibody therapeutics: discovery, development and beyond: highlights of the third annual conference of the Chinese antibody society. Antibody therapeutics 2019; 2: 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Trabbic-Carlson, K, Meyer, DE, Liu, Let al. Effect of protein fusion on the transition temperature of an environmentally responsive elastin-like polypeptide: a role for surface hydrophobicity? Protein Engineering Design and Selection 2004; 17: 57–66. [DOI] [PubMed] [Google Scholar]
- 192. FDA Guidance for industry (draft guidance): Bispecific antibody development programs https://www.fda.gov/media/123313/download (20 April 2020, last accessed)
- 193. Przepiorka, D, Ko, CW, Deisseroth, Aet al. FDA Approval: Blinatumomab. Clin Cancer Res 2015; 21: 4035–9. [DOI] [PubMed] [Google Scholar]
- 194. Paz-Priel, I, Chang, T, Asikanius, Eet al. Immunogenicity of Emicizumab in people with hemophilia a (PwHA): results from the HAVEN 1-4 studies. Blood 2018; 132: 633. [Google Scholar]
- 195. Saber, H, Del Valle, P, Ricks, TKet al. An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. Regul Toxicol Pharmacol 2017; 90: 144–52. [DOI] [PubMed] [Google Scholar]
- 196. van Meer, PJK, Kooijman, M, Brinks, Vet al. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. mAbs 2014; 5: 810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Nagorsen, D, Kufer, P, Baeuerle, PAet al. Blinatumomab: a historical perspective. Pharmacology & therapeutics 2012; 136: 334–42. [DOI] [PubMed] [Google Scholar]
- 198. Sampei, Z, Igawa, T, Soeda, Tet al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One 2013; 8: e57479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Ito, S, Ikuno, T, Mishima, Met al. In vitro human helper T-cell assay to screen antibody drug candidates for immunogenicity. Journal of Immunotoxicology 2019; 16: 125–32. [DOI] [PubMed] [Google Scholar]
- 200. Gokemeijer, J, Jawa, V, Mitra-Kaushik, S. How close are we to profiling immunogenicity risk using in Silico algorithms and in vitro methods?: an industry perspective. The AAPS Journal 2017; 19: 1587–92. [DOI] [PubMed] [Google Scholar]