

ABSTRACT
The pharmacokinetic–pharmacodynamic relationship is extremely complex and tumour drug penetration is one key parameter influencing therapeutic efficacy. In the context of antibody–drug conjugates (ADCs), which has undergone many innovation cycles and witnessed many failures, this feature is being addressed by a number of alternative technologies. Immunoglobulin-based ADCs continue to dominate the industrial landscape, but smaller formats offer the promise of more-effective cytotoxic payload delivery to solid tumours, with a higher therapeutic window afforded by the more rapid clearance. To make these smaller formats viable as delivery vehicles, a number of strategies are being employed, which will be reviewed here. These include identifying the most-appropriate size to generate the larger therapeutic window, increasing the amount of functional, cytotoxic payload delivered through conjugation or half-life extending technologies or other ways of extending the dosing without inducing toxicity.
Keywords: antibody–drug conjugate, fragments, scaffolds, solid tumours
Statement of Significance: Antibody–drug conjugates are a clinically and commercially established modality of cancer therapy with five new agents approved over the last 2 years. Treating solid tumours remains a major challenge with many failures and small-format drug conjugates offer a solution to the tumour penetration issue.
INTRODUCTION
Drug penetration into solid tumours as a factor influencing efficacy has been discussed at length over the years, but is it only now being actively addressed [1]. For biological therapies in particular, the relationship between drug dosing and tumour uptake is highly complex and very often, the micro-distribution across a whole tumour does not correlate with drug dose or plasma concentration and this underappreciated variability could explain poor responses due to suboptimal concentrations of therapeutic agents in the tumour micro-environment (TME) [1,2]. This is especially true with monoclonal antibodies (MAbs), which have to overcome numerous biological barriers [3,4] such as poor vascular supply, crossing the endothelium, overcoming tumour interstitial fluid pressure, diffusing through dense stroma and passing through tight epithelial barriers (Fig. 1). This typically results in <1% of the injected dose/gram of MAb/ADC reacting the target in solid tumours in humans [4–6].
Figure 1.

Drug conjugate delivery via the tumour vasculature and penetration can be illustrated with broadly three PK profiles. (A) Conventional ADCs with MWs of > 150 kDa accumulate and penetrate into tumours over days and eliminate from the body over weeks requiring less frequent dosing, but a higher risk of off-target/cumulative toxicity. (B) A wide range of smaller (5–100 kDa), protein-based binding scaffolds such as scFv and DARPins, which have uptake and penetration kinetics lasting hours, but are eliminated more rapidly (days), reducing non-specific exposure time, but may require strategies for higher drug delivery (e.g. higher DAR, HLE, more frequent or higher dosing). (C) Very small peptidic conjugates (<5 ka) that have very rapid and more complete uptake and penetration kinetics, but are eliminated in a matter of hours also requiring strategies to improve temporal exposure.
These observations increasingly backed up by preclinical and clinical data are motivating researchers to look at smaller formats of targeted therapeutics, which (due to more rapid diffusion kinetics) are known to have superior tissue-penetrating (perfusion) properties compared with large proteins such as immunoglobulins [7]. Of course, lower molecular weight (MW) therapeutics brings with it a whole new set of issues on the positive side (e.g. reduced side effects due to decreased cross-reaction with Fc-receptors, reduced temporal exposure to normal tissues, higher tumour: plasma exposure ratio) and negative side (smaller window of bioavailability, reduced overall uptake) [8,9], so striking the balance to obtain a favourable window is key and probably none more so than in the field of antibody–drug conjugates (ADCs) [3,10].
With nine approved products and approaching 100 ADCs in clinical trials [10,11], this modality is again on an upward trend after numerous setbacks and innovation cycles. Effective treatment of solid tumours remains a significant challenge for the reasons outlined above with greater clinical successes seen in haematological cancers [11,12]. The ADC industry is firmly focused on the Immunoglobulin format with numerous approaches for refined conjugation and more a homogeneous product quality, but an evolving area is the use of smaller formats (i.e. antibody fragments or binding scaffolds smaller than 150 kDa), which promises to widen the therapeutic window by improving tumour kill efficacy whilst reducing normal organ toxicity. This review will focus on the emerging small-format ‘biologics’ from ~ 2 kDa peptide–drug conjugates to larger ~ 80 kDa immunoglobulin fragment derivatives, which will all have very different pharmacokinetic (PK) and pharmacodynamic properties (Fig. 1). For the smaller formats, the chemical linker–payload has a greater influence on these properties as it can make up 10–30% of the overall conjugate mass compared with a typical 2–3% for an IgG, therefore requires special consideration and bespoke design (Fig. 2; Table 1). This review will focus on non-radioactive and non-liposomal pharmaceutical conjugates.
Figure 2.
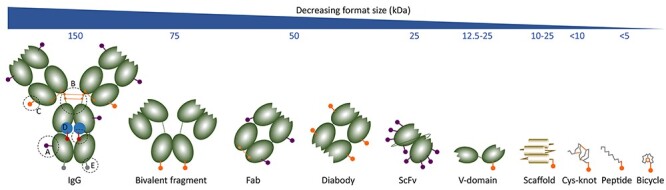
A size and format comparison of various drug conjugates. The archetypal IgG is shown with the common conjugation strategies (A) surface lysines, (B) hinge thiols, (C) site-specific thiols, (D) Fc-carbohydrate, (E) genetically engineered tag or non-natural amino acid. The same colour coding is used for the conjugation onto the alternative, smaller formats of decreasing size: bivalent antibody fragment (~75–80 kDa), Fab or diabody (~50 kDa), high-DAR ScFv (25 kDa), VH-domains (12.5–25 kDa), many types of scaffolds (10–25 kDa) and a variety of peptides.
Table 1.
A list of drug-conjugate formats in order of increasing size with examples of the targets addressed and payloads used
| Format size (kDa) | Format name | Example target | Cancer indication | Example payload | References |
|---|---|---|---|---|---|
| 1.5–2 | Bicycle (bicyclic peptides) | MMP14 EphA2 Nectin |
Breast, lung multiple solid tumours | DM1 vcMMAE | [67–69, 71] |
| ~3–5 | Pentarin | Somatostatin receptor | Neuroendocrine Liver | DM1 | [62–65] |
| ~3.5–5 | Cysteine knots | Integrin, MMP2 |
Pancreatic | Gemcitabine, MMAF, Cis-platin | [60, 61] |
| 5–6.5 | Affibody | HER2 | Breast/gastric | Idarubicin, vcMMAE Photosensitizer | [43–45, 46] |
| ~10–11 | Centyrin Adnectin | EGFR Glypican |
Multiple solid tumours Liver | vcMMAF Tubulysin | [48, 50] |
| ~15–18 | DARPIn | EpCAM | Multiple solid tumours | MMAF | [53] |
| ~15 | Abdurin | EphA2 | Prostate | vcMMAE | [56] |
| ~12.5–25 | VH (like) domains | PSMA | Prostate | DGN549 | [33] |
| ~25–27 | ScFv | HER2, EGFR CD41/61 |
Breast/gastric | Photosensitizers MMAF, vcMMAE, Auristain F vcMMAE | [21, 22, 24, 25, 28, 29] |
| ~55–60 | Diabody | CD30 | Lymphoma | MMAF | [38] |
| ~50 | Fab | CD20, HER2 | Lymphoma breast/gastric | vcMMAE PBD | [15, 18] |
| ~80 | SIP ScFv-Fc | Fibronectin, Tenascin-C FGFR-2 |
Multiple solid tumours | Cemadotin, DM1, vcMMAE | [35–37, 39] |
RECOMBINANT ANTIBODY FRAGMENTS
Recombinant antibody fragments lend themselves to a wide range of engineering approaches [13,14] to facilitate linker–payload bioconjugation such as the introduction of conjugation friendly thiols. They are normally produced in prokaryotic systems removing the glycan-conjugation option utilized by some in the ADC field. Fragments such as single-chain Fvs (scFv) and single-domain antibodies tend to be more robust and stable having been subjected to stringent selection pressures during discovery compared with Fab-fragments [14]. The resulting antibody fragment drug conjugate (FDC) at a drug:antibody ratio (DAR) of ~ 2 has the feature of carrying more payload compared with a standard ADC of DAR4 on a mass basis but has a shorter half-life and thus lower systemic bioavailability compared with an ADC.
Fab-fragments have been superseded by formats such as scFvs but examples exist of conjugates demonstrating proof-of-principle. Early conjugates with moderately potent, chemotherapy-approved payload such as paclitaxel and doxorubicin have largely been ineffective, but a trastuzumab–mono-methyl auristatin E (MMAE) FDC DAR1 with 200–500 pM potency in vitro required alternate day dosing at 20 mg/kg to see any tumour regression [15]. This high dosing requirement was also seen more recently with an anti-CD-20 Fab appended with a sortase conjugation tag used for enzymic conjugation of an MMAE payload [16]. The FDC had to be dosed at 20 mg/kg every 3 days for four doses to obtain 4/6 cures, compared with complete cures for an equivalent ADC. Notably, the FDC had ~6× lower plasma exposure as measured by the PK area under curve. The FDC, however, was better tolerated. A similar but dual-linker-payload (DAR3, cleavable and non-cleavable auristatin) was also very potent (IC50 0.7–0.9 nM) but not evaluated in vivo [17]. Trastuzumab Fab-based conjugates based on the ultra-potent pyrrolobenzodiazepine (PBD) payload class (IC50 in the low pM range) were recently described where a novel dual maleimide disulphide rebridging technology previously applied to ADCs was applied to the native cysteines in a Fab [18]. The tesirine payload has been used in several clinical-stage ADCs but was also the cause of unacceptable toxicity in the discontinued Rova-T and others subsequently [19]. This was modified to be more hydrophilic with a symmetrical dual maleimide bridge. In vitro potencies were 6–7 pM for high human epidermal growth factor receptor-2 (HER2)-expressing cells and as potent as the trastuzumab-based ADC despite the reduced avidity and possibly reduced internalization kinetics (not determined). In vivo efficacy was not explored [18].
ScFvs are artificially tethered, recombinant antibody structures but represent the preferred format for most antibody discovery programmes that utilize a display technology [13,14]. In specific applications where time-critical elimination was necessary (e.g. fast clearance ahead of a second step), they have proven useful. There are many reports on targeted photodynamic therapy where a conditionally cytotoxic photosensitizer payload is delivered to tumours but must be removed from the systemic circulation before laser illumination [20]. We and others have developed this technology and demonstrated tumour eradication in vivo with very few side effects [21,22], but the complex nature of such a two-step therapy has hampered commercial development. This has not put off some companies combining optically active payloads and conventional ADCs so that therapeutics can be simultaneously imaged and used for treatment, in a theranostic approach [23].
We later extended our work on scFv-targeted photodynamic therapy to conventional payloads with more commercial success, broadly calling them ‘FDCs’. Using particular scFv VH–VL frameworks predisposed to chemical conjugation and high payload loading, DARs of 5–10 were obtainable via lysine conjugation whilst retaining the critical biophysical properties [24,25]. Although heterogeneous in nature, stochastic high DAR FDCs have fewer permutations than lysine-conjugated ADCs. As expected, the linker–payload structure had a major impact on biophysical properties such as aggregation, binding affinity and thermal stability leading us to tailor payloads specifically to match the scFv format. Superior tumour penetration compared with ADCs has been observed and nM–pM potencies observed in vitro on cell lines using auristatin and maytansine payloads [24–27]. A key finding when developing high DAR scFv-based FDCs was that although the MW was theoretically within the range for renal excretion, the chemical–physical properties of the linker–payload became a dominating feature that altered the PK to a predominantly hepatic clearance route and a slower-than-expected systemic elimination approaching albumin-binding half-life extension (HLE) methods [26,27]. This, in turn has made FDCs a viable option with dosing now approaching that of ADCs.
We have used lysine residues to achieve the high DAR, but site-specific conjugation, more aligned to the conventional ADC field can be achieved using C-terminal cysteine thiols or dedicated conjugation tags to obtain lower DARs [10]. One example is the SNAP technology that utilizes a small, engineered DNA–alkyltransferase enzyme as a recognition and conjugation domain to link benzylguanine-modified payloads. Low nM potencies against epidermal growth factor receptor (EGFR)-expressing cells lines were seen in vitro using the scFv derived from the clinically approved panitumumab MAb [28].
Specifically focussing on the TME, Yap et al. [29] developed a scFv- based FDC targeting an integrin glycoprotein (GPIIb/IIIa: CD41/CD61), which is found in an active–high-affinity conformation on activated platelets that are increasingly thought to be involved in mediating tumour growth and metastasis in the TME. Using a sortase-recognition tag, valine–citruline (vc)-MMAE with a Gly3 linker was conjugated to a DAR1. In vivo, four doses of a 6 mg/kg scFv-GGG-vc-MMAE gave a moderate ~ 8-day tumour growth delay demonstrating proof-of-concept for this novel approach. Targeting the TME was further illustrated using a Cy5 dual-labelled conjugate [29]. An interesting twist on using scFvs was described by Wang et al. [30] aiming to capitalize on the increased macro-pinocytosis seen in ras-driven cancers such as pancreatic. An-anti-EGFR scFv recombinantly fused to domain III of human serum albumin (for HLE) and the apoprotein/carrier for the cytotoxic antibiotic lidamycin. The ~ 60 kDa conjugate effectively internalized and was highly potent across four pancreatic cancer cell lines (IC50 range 15–70 pM), although clear specificity was not shown. The concept of delivering an ADC via non-clatherin route was demonstrated and a well-tolerated, moderate tumour growth delay was shown at 0.4 mg/kg given twice [28]. Higher doses were not used. The modular design idea was exploited to build a nanobody–drug conjugate with a magnetic resonance imaging (MRI) contrast agent [30]. A biparatopic anti-EGFR nanobody was fused to a gadolinium-binding domain (imaging) and a C3 tag for payload conjugation. HLE was also incorporated through an anti-albumin nanobody. A maleimide-functionalized cis-platin chemotherapy drug was conjugated to the fusion protein’s C-terminus and Gd3+ incorporated non-covalently via dialysis. Uptake and imaging were demonstrated but, not unexpected for a relatively moderately potent drug (IC50 ~ 1 mM); only moderate potency was seen in vitro. In vivo, the conjugate was as potent as free cisplatin (in terms of platinum content), but much better tolerated. This was due to the 4–5× higher accumulation in tumours, which was further supported by the T1-weighted MRI contrast images [31].
Smaller antibody fragments such as Variable (V)-domains (Ablynx’s nanobodies: VHH-domain antibodies derived from llamas, Crescendo Biologics’ Humabodies: human VH-domains) require some sort of HLE technology to make them viable candidates. Their Humabody-Drug Conjugates (HDCs) platform is made up of 15kDa domains conjugated to a low-DAR, additionally half-life extended using albumin-binding domains. This retains the benefits of tumour penetration [32]. CB108, a biparatopic HDC against prostate-specific membrane antigen (PSMA) has been shown to be effective in vivo. A very nice study by Nessler et al [33] aimed to tease out some of the important features and benefits of smaller format drug conjugates. Low-affinity monovalent (VH1) and high-affinity, rapidly internalizing, biparatopic (VH1–VH2) HDCs were created with and without HLE domains against PSMA. These were conjugated to a DNA-alkylating payload DGN549, to a DAR1. In the absence of any drug delivery or mass transport limitations, rapid internalization led to the highest in vitro potency, but slower internalization aided tumour penetration and higher efficacy in vivo. HLE was needed for in vivo efficacy as these low-DAR conjugates would otherwise clear via renal filtration. Alexa-Fluor-680 labelling of the various formats (without the payload) confirmed the superior penetration of the VH1–HLE format, which was additionally backed up with tumour spheroid modelling data [33]. Elasmogen have a similar technology based on shark variable domains from new antigen receptors called soloMER™, which coupled with its HLE technology NDure™ [34] is being utilized to discover and develop soloMER™–drug conjugates.
Bivalent antibody-derived fragments have met with greater preclinical success as seen with small immunoproteins (SIP-Philochem) [35–37] and diabodies (Seattle Genetics) [38]. Neri’s SIP technology uses the CHε4 domain to dimerize scFvs yielding a fragment that is ~ 50% the size of an IgG, with a faster elimination time due to the absence of neonatal FcR binding. Using primarily non-internalizing, tumour neovasculature targets such as fibronectin and tenascin-C, excellent uptake and tumour/blood contrast ratios were obtained and the availability of two C-termini presented two cysteine thiol conjugation positions [35]. The aim is to destroy tumour vasculature to starve the tumour of nutrients and this removes the tumour penetration hurdle, but the payload is released extracellularly and diffuses into the nearby cells with a resulting bystander killing effect. If thiol-bearing payloads are used, practically no linker is required (‘traceless’) as long as the disulphide is hindered or buried/protected within the protein architecture to reduce the risk of inadvertent release [35]. The release mechanism is via extracellular thiols (e.g. glutathione), which is amplified upon more cells dying. Using a DM1 payload on an anti-fibronectin–EDA SIP, well-tolerated cured were seen in murine F9 teratocarcinoma animal models dosed at 7 mg/kg three times [36]. An anti-tenascin C SIP coupled to a more commonly used vc-MMAE linker–payload (DAR2) also demonstrated tumour growth inhibition at 7 mg/kg four times but was not as effecting as the IgG version that was more stable. The IgG-based ADC was again more stable in a side-by-side comparison of IgG vs. SIP using the F8 antibody and DM1 payload conjugated at a DAR2 as a C-terminal disulphide [35]. As expected, the SIP–drug conjugate accumulated into the tumour and cleared more rapidly and the 24-h uptake levels were more than four times higher for the IgG ADC. Although the payload on the ADC was ~10× more stable, the SIP conjugate was more effective on a molar basis with authors attributing this to the faster payload release leading to higher tumour payload exposure over a shorter period of time compared with the slow-release of an ADC. Toxicity, which may be higher for a less stable drug-conjugate, was not evaluated [37]. A similarly configured scFv–Fc format ADC was made from an anti-fibroblast growth factor receptor-2 (FGFR2) antibody discovered by phage display. Using the vc-MMAE payload ~nM potency was seen [39].
The most comprehensive analysis of a potent antibody FDC was described by Seattle Genetics [38] using an anti-CD30 diabody with four cysteine thiols conjugated to MMAE and mono-methyl auristatin F (MMAF) payloads with maleimide linkers. The diabody ADC (MMAF DAR ~ 4) was compared with an equivalent IgG ADC. In this example, the two formats had comparable DARs and valency. The diabody–drug conjugate had a faster blood clearance reflected by its smaller size, but the 30× lower exposure level only led to a 3× drop in in vivo efficacy (7.2 vs. 2 mg/kg needed for comparable tumour growth inhibition). Interestingly, the renal clearance expected for such fragment sizes was not evident, suggesting that the payload had a major influence diverting the conjugate to the liver for metabolism [38].
The above research and development makes observations based on therapeutic efficacy without direct evidence that tumour penetration is having a significantly positive impact. This has been difficult to quantify for drug conjugates. Direct correlations have been made between antibody size and tumour perfusion [7] but a more recent analysis in a SKOV3–HER2 model examining uptake and tumour penetration homogeneity of monovalent and bivalent nanobodies (MW ~ 15–30 kDa) size and affinity was carried out using intravital fluorescence microscopic imaging [40]. This nicely showed that the smaller format gave rapid and more homogeneous tumour uptake, during the 1- to 3-h time frame, compared with the trastuzumab IgG that was restricted to around the vasculature, and also show that a too high affinity for the nanobodies hindered penetration (binding site barrier). The IgG, as shown by many, gave higher overall uptake by 24 h.
NON-ANTIBODY SCAFFOLDS
The ‘non-antibody’ binding format field continues to thrive because they promise to solve the problems presented by conventional antibodies such as expensive manufacturing, formulation/concentration, glycosylation, thermostability and tissue penetration. These scaffolds tend to range from ~ 2 to 20 kDa (smaller than most antibody fragments), can be expressed at exceptionally high yield in Escherichia coli, selected by in vitro display, demonstrate higher stability and can be multimerized and built up according to the desired properties [41]. A few scaffold companies have published or disclosed intentions to develop SDCs, but other formats such as Anticalins, Avimers, Fynomers, Kunitz domains and Affilins have not gone down this route.
Affibody–Drug conjugates
Affibodies, based on the 6 kDa Staphylococcus protein-A, Z-domain can be engineered and displayed by phage to generate high-affinity binders. These are being developed as therapeutics by Swedish enterprise Affibody AB and are in Phase 2 clinical trials with a psoriasis therapeutic and a breast cancer positron emission tomography imaging agent [42]. No commercial affibody–drug conjugates have been disclosed, but conjugates have been described targeting HER2 (ZHER2891) with a vcMMAE payload (DAR1) with low nM potencies on high HER2-expressing cells lines [43]. Higher affinity, longer half-life, Fc-fusions had increased potency in vitro (130 pM) on SKBr3 cells [44]. More recently, the same affibody formats were coupled to the non-releasable DM1 payload (DAR1) resulting in higher in vitro potencies (270–470 pM, comparable to the trastuzumab ADC) and significant in vivo efficacy. Conjugates radiolabelled with 99Tc showed marginally higher tumour uptake at 4 h at the expense of higher blood and normal organ uptake. Doses of 8.5 mg/kg, weekly five times were needed to see well-tolerated tumour growth delay of ~ 20 days but no cures were seen in this first in vivo proof-of-principle of this scaffold format [45]. The same affibody was conjugated with photosensitizer payload pyropheophorbide-a (DAR1) with 12–23 nm IC50 potency on HER2-expressing cells. Well-tolerated cures were seen with a single injection of 20nMol of conjugate (~0.2 mg dose/8 mg/kg) upon laser illumination with the rapid clearance being optimal to allow photo-activation without skin toxicity [46].
Fibronectin type III–drug conjugates
These popular scaffolds have been reviewed extensively [47] with a number investigated as drug conjugates. Immunoglobulin-like centyrins are ~ 100-residue (11 kDa), thermal/chemical stable domains being developed by Janssen/J&J. Extensive surface cysteine scanning mutagenesis identified suitable conjugation positions and an anti-EGFR DAR1 MMAF conjugate demonstrated ~0.2 nM IC50in vitro potency [48]. No in vivo data have been presented but a bioanalytic workflow was developed for centyrin–drug conjugate analysis in tissues in a collaboration between Janssen and Immunogen [49]. Clinical-stage adnectins are also based on the fibronectin domains and are being developed by BMS. Using a tubulysin analogue payload with a cleavable cathepsin B linker against the hepatocellular carcinoma antigen glypican-3, a DAR1 (via a maleimide moiety to a C-terminal cysteine thiol) adnectin–drug conjugate was made and evaluated [50]. One candidate conjugate had high thermostability (Tm ~ 80°C), 32 nM Kd binding affinity and 0.3 nM IC50 cell-kill potency on Hep3B cells in vitro. The payload conjugation had no deleterious effect on the conformation of the adnectin structure as supported by detailed hydrogen–deuterium exchange mass spectrometry [51]. No HLE strategy was employed as the authors favoured the rapid renal clearance (half-life approx. ½ h). Quantitative biodistribution showed very specific tumour uptake with renal exposure in the first few hours but low liver and other normal organ exposure. By 7 days, it was undetectable in all tissues other than the tumour. Most impressive was the well-tolerated, complete tumour cures at 0.12 mmol/kg (~1.4 mg/kg), despite the moderate affinity and rapid clearance given three times weekly. The authors acknowledge that this observation bucks the trend seen with small-format binders and suggest that the rapid internalization of the glypican-3 target may account for these promising results. It remains to be seen if such frequent dosing remains a viable option.
DARPin–Drug conjugates
The Designed Ankyrin Repeat (DARPin) class of scaffold proteins are well-established with five clinical-stage products, one (abicipar) recently completing a Phase 3 trial in ophthalmology [52]. Drug conjugates are much further away. Using bi-orthogonal chemistry, an anti-EpCAM DARPin with a (i) C-terminal cysteine residue and a (ii) non-natural amino acid azidohomo-alanine was used to attach a half-life extending albumin-binding domain and MMAF payload (DAR1). This generated a DARPin–MMAF conjugate with an IC50 400 pM, which had extended plasma half-life (17.4 h in vivo) [53]. No in vivo or commercial developments have been disclosed, but there may be issues with this format given a recent FDC rejection setback [54].
Abdurin–Drug conjugates
Abdurins can be diversified to form libraries of binders as they are based on engineered IgG CH2 domains (~15 kDa); similar to the larger Fcabs being developed by F-Star, these retain the ability to bind to the neonatal Fc receptor and thus have an inherent extended serum half-life [55]. Recently, Abdurin–drug conjugates were described using Abzena’s CyPEG and HiPEG conjugation technologies and vcMMAE payload (DAR1). Moderate in vivo efficacy (tumour regression at 5 mg/kg, six doses) was seen in PC3 xenograft studies. A DAR2 conjugate led to some cures but significant loss of target and FcRn-binding affinity was observed in various combinations most likely due to the small size taking an impact upon chemical modification [56].
PEPTIDE–DRUG CONJUGATES
Small peptides have even faster penetration and more rapid elimination properties compared with the above examples. Their totally synthetic nature promises many benefits as drug conjugates. This topic is covered extensively by He et al [57]. There are many reports, for example with low-potency payloads such as doxorubicin. These conjugates have micromolar potencies and are not usually more potent than the free drug, but generally more specific [58]. More recent innovations with potent payloads demonstrate more promising approaches including some at the clinical stage of development.
Cystine knot–drug conjugates
Cystine knots (30–50 amino acids, also known as knottins) are at the larger end of the peptide scale, but like peptides, are amenable to scalable solid-phase synthesis and incorporation of useful stabilizing and functionalizing non-natural amino acids. They have enhanced chemical, protease and thermal stability properties compared with conventional antibody domains, due to their highly compact structure and stabilizing disulphide bridges [59]. Conjugation to cytotoxic payloads was achieved via solid-phase synthetic incorporation of a non-natural amino acid followed by azide–alkyne conjugation of a gemcitabine payload. The Knottin–drug conjugate was able to overcome drug resistance in PANC-1 pancreatic cancer cells, increasing the potency of gemcitabine 25-fold [60]. A more ‘ADC-like ‘molecule was generated using cell-free protein synthesis with click chemistry (DAR2) and an appended Fc-domain. The MMAF payload DAR2 was used resulting in potencies similar to the gemcitabine conjugates but tumour growth delay was seen in vivo at 10 mg/kg given twice/week for 3 weeks [61].
Pentarins–drug conjugates
Pentarins and bicyclic peptides represent the shorter end of the peptide scale (2–5 kDa) and are worth mentioning due to the advanced clinical stage of their drug conjugates.
The pentarin (penetrate, target) portfolio developed by Tarveda, consists of small peptides that can be made into pentarin–drug conjugates (PDC). Their lead compound, PEN-221, is a somatostatin receptor-2 (SSTR2, expressed on neuroendocrine tumours) targeted DM1 maytansine. The payload is conjugated to the disulphide-cyclized Tyr3-octreotate, which had high affinity (~51 pM) and rapid internalization. In vivo, 1–2 mg/kg PEN221 were enough to cure HCC33 (liver) and H524MD (lung) cancer tumour models given four times on a weekly schedule with maximal payload uptake achieved within 2 h [62]. Results presented at the American Society for Clinical Oncology in 2018 showed that PEN221 was well-tolerated at doses up to 18 mg every 3 weeks with evidence of efficacy in the Phase 1 arm [63]. This product is now in Phase 2 clinical trials for SSTR2-expressing neuroendocrine and lung tumours. A follow-up compound, PEN866, is a PDC carrying the SN38 payload targeting the heat-shock protein chaperone HSP90. This is currently in a Phase 1/2a clinical trial for advanced solid cancers sensitive to topoisomerase I inhibitors and recent updates at the European Society for Medical Oncology (ESMO) and American Association for Cancer Research conferences suggested good tolerability, signs of clinical efficacy [64] and promising clinical uptake and good PK profile [65].
Bicyclic peptide (Bicycle)–drug conjugates
The phage-displayable bicyclic peptide (‘bicycles’) technology discovered and developed by Heinis et al. [66] is being commercialized by Bicycle Therapeutics Ltd, including a major programme on Bicycle–drug (toxin) conjugates (BTCs). MT1-matrix metalloprotease (MMP) is overexpressed in multiple cancers including triple negative breast, non-small cell lung and soft tissue sarcoma. An anti-MT1-MMP BTC (BT1718) carrying a DM1 payload via a hindered disulphide linker has ~ 2 nM affinity, rodent cynomolgus species cross-reactivity and plasma stability of >20 h. It demonstrated efficacy in tumour models at 3 and 5 mg/kg BDC given twice weekly for 2–4 weeks. Complete cures were seen at 10 mg/kg with good tolerability as measured by body weight [67]. This product is currently in a Phase 1/2 clinical trial. An update from the ESMO identified a recommended Phase 2 dosing of 7.2 mg/m2, once weekly with demonstratable tumour uptake and signs of efficacy [68]. A follow-up clinical candidate, BT5528 addresses the ephrin A2 receptor (EphA2) receptor (target for MEDI-547, a discontinued ADC that showed severe toxicity). Using a different payload, vc-MMAE, rapid tumour uptake was seen with persistent accumulation and rapid renal clearance in xenograft models. Payload conjugation had no adverse effect on the bicyclic peptide affinity (5.7 vs. 1.9 nM) and a rapid renal clearance was observed (half-life ~ 0.4–0.6 h in rodents and non-human primates). A weekly dose of 0.5 mg/kg (equivalent to 10–15 mg/kg of a similar ADC DAR2) gave rise to tumour regressions with tumours as large as 1000 mm3 being treatable at doses of 3 mg/kg demonstration the penetration advantage over an ADC. As expected, non-cleavable variants were ineffective. A nice correlation was seen between EphA2 receptor level and tumour cure efficacy and none of the previously observed toxicities were seen when compared with a MEDI-547 equivalent ADC in rat or non-human primate toxicology studies [69]. BT5528 is in a Phase 1/II trial for solid tumours as a monotherapy and combination with checkpoint inhibitor nivolumab [70]. Other preclinical targets under commercial development include nectin-4 (BT8009) [71].
DISCUSSION
Antibody–drug conjugates are complex therapeutics to develop and those challenges remain into manufacturing. Non-IgG formats, as discussed here offer the possibilities of reduced manufacturing costs due to the easier chemistry manufacture control processes afforded by bacterial production, higher yields, lack of glycosylation and generally simplified analytics due to the smaller size. It remains to be seen if these features translate into economic or patient benefits.
Precision medicine is often a buzzword used loosely to describe tailoring a drug therapy to a patient’s genetic profile but is increasingly being used in terms of other patient characteristics. Personalized dosing schemes to improve tumour penetration could be one key element [1] and having available formats to maximize tumour penetration will add to the clinical armoury. The increasing preclinical use of payload imaging technologies such as matrix-assisted laser desorption ionization mass spectrometry imaging [72] could inform this at the preclinical animal model level. Other strategies to aid penetration, such as addition of modulators to enhance penetration (e.g. RGD (Arginine-Glutamate-Aspartate) peptides, ligands to endothelial/epithelial cells that increase vascular permeability such as NRP-1, Lys/Arg-rich peptides [73] or TEM8 targeting for targeting stroma in solid tumours [74] or LRRC15, cancer-associate fibroblasts marker [75]) will require knowledge of an additional receptor making tailoring even more complex.
Collateral exposure through non-targeted deposition within normal tissues is recognized as a key driver to ADC–payload toxicity [76,77] with the well-characterized example of dose-limiting toxicity of trastuzumab–emtansine caused by Fc-mediated binding to platelets (thrombocytopenia) [78]. Most of these small-format drug conjugates promise to overcome this due to abolished Fc-receptor binding and reduced chronic exposure, but a clear correlation between improved tolerability and conjugate size would be difficult to demonstrate given the wide variation in formats.
Tumour spheroid technology is becoming more accessible and used in the discovery workflow and evaluating penetration can help to prioritize candidates. It is acknowledged that in vitro cell kill potency (IC50) is a poor indicator of tumour cure efficacy as we and others find that it’s not necessarily that the most potent conjugates make the best in vivo candidate [33]. Shah et al [79] modelled the correlation between in vitro IC50 and in vivo ID50 and shown that 27× more ADC was needed in the plasma compared with cell culture medium to achieve tumour growth ‘stasis’. This shows that, in these models, there remain transfer barriers to solid tumour therapy and that smaller formats could make the real difference needed to address some of these difficult-to-treat solid tumours.
CONFLICT OF INTEREST STATEMENT
MD is an employee and shareholder in Antikor Biopharma Ltd and QX is an employee and shareholder in Essex Biotechnology Ltd.
Contributor Information
Mahendra P Deonarain, Antikor Biopharma Ltd, Stevenage Bioscience Catalyst, Gunnels Wood Road, Stevenage, Hertfordshire SG12FX, UK; Department of Chemistry, Imperial College London, Exhibition Road, London SW72AZ, UK.
Quinn Xue, Essex Biotechnology Ltd, Shun Tak Centre, Room 2818, China Merchants Tower, Connaught Road Central, Hong Kong 168-200, SAR China.
REFERENCES
- 1. Bartelink, IH, Jones, EF, Shahidi-Latham, SKet al. Tumor drug penetration measurements could be the neglected piece of the personalized cancer treatment puzzle. Clin Pharmacol Ther 2019; 106: 148–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert, JM, Morris, CQ. Antibody-drug conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv Ther 2017; 34: 1015–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thurber, GM, Schmidt, MM, Wittrup, KD. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv Drug Deliv Rev 2008; 60: 1421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christiansen, J, Rajasekaran, AK. Biological impediments to monoclonal antibody-based cancer immunotherapy. Mol Cancer Ther 2004; 3: 1493–501. [PubMed] [Google Scholar]
- 5. Teicher, BA, Chari, RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res 2011; 17: 6389–97. [DOI] [PubMed] [Google Scholar]
- 6. Epenetos, AA, Snook, D, Durbin, Het al. Limitations of radiolabeled monoclonal antibodies for localization of human neoplasms. Cancer Res 1986; 46: 3183–91. [PubMed] [Google Scholar]
- 7. Li, Z, Krippendorff, BF, Sharma, Set al. Influence of molecular size on tissue distribution of antibody fragments. MAbs 2016; 8: 113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidt, MM, Wittrup, KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther 2009; 8: 2861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deonarain, MP, Yahioglu, G, Stamati, Iet al. Small-format drug conjugates: a viable alternative to ADCs for solid tumours? Antibodies (Basel) 2018; 7: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gauzy-Lazo, L, Sassoon, I, Brun, MP. Advances in antibody-drug conjugate design: current clinical landscape and future innovations. SLAS Discov 2020. doi: 10.1177/2472555220912955. [DOI] [PubMed] [Google Scholar]
- 11.The Antibody Society (2020). https://www.antibodysociety.org/adc/ (accessed 01 Aug2020).
- 12. Yu, B, Liu, D. Antibody-drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J Hematol Oncol 2019; 12: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goulet, DR, Atkins, WM. Considerations for the design of antibody-based therapeutics. J Pharm Sci 2020; 109: 74–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ministro, J, Manuel, AM, Goncalves, J. Therapeutic antibody engineering and selection strategies. Adv Biochem Eng Biotechnol 2020; 171: 55–86. [DOI] [PubMed] [Google Scholar]
- 15. Badescu, G, Bryant, P, Bird, Met al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug Chem 2014; 25: 1124–36. [DOI] [PubMed] [Google Scholar]
- 16. Liu, W, Zhao, W, Bai, Xet al. High antitumor activity of sortase A-generated anti-CD20 antibody fragment drug conjugates. Eur J Pharm Sci 2019; 134: 81–92. [DOI] [PubMed] [Google Scholar]
- 17. Puthenveetil, S, Musto, S, Loganzo, Fet al. Development of solid-phase site-specific conjugation and its application toward generation of dual Labeled antibody and fab drug conjugates. Bioconjug Chem 2016; 27: 1030–9. [DOI] [PubMed] [Google Scholar]
- 18. Ruddle, BT, Fleming, R, Wu, Het al. Characterization of disulfide bond Rebridged fab-drug conjugates prepared using a dual maleimide pyrrolobenzodiazepine cytotoxic payload. ChemMedChem 2019; 14: 1185–95. [DOI] [PubMed] [Google Scholar]
- 19. Mullard, A. Cancer stem cell candidate Rova-T discontinued. Nat Rev Drug Discov 2019; 18: 814. [DOI] [PubMed] [Google Scholar]
- 20. Staneloudi, C, Smith, KA, Hudson, Ret al. Development and characterization of novel photosensitizer : scFv conjugates for use in photodynamic therapy of cancer. Immunology 2007; 120: 512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pye, H, Butt, MA, Funnell, Let al. Using antibody directed phototherapy to target oesophageal adenocarcinoma with heterogeneous HER2 expression. Oncotarget 2018; 9: 22945–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhatti, M, Yahioglu, G, Milgrom, LRet al. Targeted photodynamic therapy with multiply-loaded recombinant antibody fragments. Int J Cancer 2008; 122: 1155–63. [DOI] [PubMed] [Google Scholar]
- 23. Pye, H, Butt, MA, Reinert, HWet al. A HER2 selective theranostic agent for surgical resection guidance and photodynamic therapy. Photochem Photobiol Sci 2016; 15: 1227–38. [DOI] [PubMed] [Google Scholar]
- 24. Deonarain, MP, Yahioglu, G, Stamati, Iet al. Biological materials and uses thereof. PCT Patent WO 2016046574.
- 25. Yahioglu, G, Stamati, I, Diez-Posada, Set al. Highly-loaded Antibody-Fragment Drug Conjugates (FDCs) for solid tumour cancer therapy. Manuscript in preparation. 2020.
- 26. Deonarain MP, Stamati I, Edwards Bet al. Gastric cancer antibody fragment drug conjugates (FDCS): from concept to clinical development. Cancer Research, 2020; 80. doi: 10.1158/1538-7445.AM2020-2901. [DOI] [Google Scholar]
- 27. Deonarain, MP. Protein engineering summit Europe. Lisbon Nov-18-222019, 2019. [Google Scholar]
- 28. Woitok, M, Klose, D, Di Fiore, Set al. Comparison of a mouse and a novel human scFv-SNAP-auristatin F drug conjugate with potent activity against EGFR-overexpressing human solid tumor cells. Onco Targets Ther 10: 3313–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yap, ML, McFadyen, JD, Wang, Xet al. Activated platelets in the tumor microenvironment for targeting of antibody-drug conjugates to tumors and metastases. Theranostics 2019; 9: 1154–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang, X, Sheng, W, Wang, Yet al. A macropinocytosis-intensifying albumin domain-based scFv antibody and its conjugate directed against K-Ras mutant pancreatic cancer. Mol Pharm 2018; 15: 2403–12. [DOI] [PubMed] [Google Scholar]
- 31. Huang, H, Wu, T, Shi, Het al. Modular design of nanobody-drug conjugates for targeted-delivery of platinum anticancer drugs with an MRI contrast agent. Chem Commun (Camb) 2019; 55: 5175–8. [DOI] [PubMed] [Google Scholar]
- 32.Crescendo Biologics company website (2020). https://www.crescendobiologics.com/humabody/humabody-therapeutics (accessed 01 Aug, 2020).
- 33. Nessler, I, Khera, E, Vance, Set al. Increased tumor penetration of single-domain antibody-drug conjugates improves in vivo efficacy in prostate cancer models. Cancer Res 2020; 80: 1268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ubah, OC, Buschhaus, MJ, Ferguson, Let al. Next-generation flexible formats of VNAR domains expand the drug platform's utility and developability. Biochem Soc Trans 2018; 46: 1559–65. [DOI] [PubMed] [Google Scholar]
- 35. Bernardes, GJ, Casi, G, Trüssel, Set al. A traceless vascular-targeting antibody-drug conjugate for cancer therapy. Angew Chem Int Ed Engl 2012; 51: 941–4. [DOI] [PubMed] [Google Scholar]
- 36. Perrino, E, Steiner, M, Krall, Net al. Curative properties of noninternalizing antibody-drug conjugates based on maytansinoids. Cancer Res 2014; 74: 2569–78. [DOI] [PubMed] [Google Scholar]
- 37. Gébleux, R, Stringhini, M, Casanova, Ret al. Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int J Cancer 2017; 140: 1670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim, KM, McDonagh, CF, Westendorf, Let al. Anti-CD30 diabody-drug conjugates with potent antitumor activity. Mol Cancer Ther 2008; 7: 2486–97. [DOI] [PubMed] [Google Scholar]
- 39. Borek, A, Sokolowska-Wedzina, A, Chodaczek, Get al. Generation of high-affinity, internalizing anti-FGFR2 single-chain variable antibody fragment fused with fc for targeting gastrointestinal cancers. PLoS One 2018; 13. doi: 10.1371/journal.pone.0192194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Debie, P, Lafont, C, Defrise, Met al. Size and affinity kinetics of nanobodies influence targeting and penetration of solid tumours. J Control Release 2020; 317: 34–42. [DOI] [PubMed] [Google Scholar]
- 41. Vazquez-Lombardi, R, Phan, TG, Zimmermann, Cet al. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov Today 2015; 20: 1271–83. [DOI] [PubMed] [Google Scholar]
- 42.Affibody company website (2020). https://www.affibody.se (accessed 01 Aug, 2020).
- 43. Sochaj-Gregorczyk, AM, Serwotka-Suszczak, AM, Otlewski, J. A novel affibody-auristatin E conjugate with a potent and selective activity against HER2+ cell lines. J Immunother 2016; 39: 223–32. [DOI] [PubMed] [Google Scholar]
- 44. Sochaj-Gregorczyk, AM, Ludzia, P, Kozdrowska, Eet al. Design and in vitro evaluation of a cytotoxic conjugate based on the anti-HER2 affibody fused to the Fc fragment of IgG1. Int J Mol Sci 2017; 18: 1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Altai, M, Liu, H, Ding, Het al. Affibody-derived drug conjugates: potent cytotoxic molecules for treatment of HER2 over-expressing tumors. J Control Release 2018; 288: 84–95. [DOI] [PubMed] [Google Scholar]
- 46. Li, S, Jin, Y, Su, Yet al. Anti-HER2 affibody-conjugated photosensitizer for tumor targeting photodynamic therapy. Mol Pharm 2020; 17: 1546–57. [DOI] [PubMed] [Google Scholar]
- 47. Chandler, PG, Buckle, AM. Development and differentiation in monobodies based on the fibronectin type 3 domain. Cell 2020; 9: 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goldberg, SD, Cardoso, RM, Lin, Tet al. Engineering a targeted delivery platform using centyrins. Protein Eng Des Sel 2016; 29: 563–72. [DOI] [PubMed] [Google Scholar]
- 49. Shi, C, Goldberg, S, Lin, Tet al. Bioanalytical workflow for novel scaffold protein-drug conjugates: quantitation of total centyrin protein, conjugated centyrin and free payload for centyrin-drug conjugate in plasma and tissue samples using liquid chromatography-tandem mass spectrometry. Bioanalysis 2018; 10: 1651–65. [DOI] [PubMed] [Google Scholar]
- 50. Lipovšek, D, Carvajal, I, Allentoff, AJet al. Adnectin-drug conjugates for glypican-3-specific delivery of a cytotoxic payload to tumors. Protein Eng Des Sel 2018; 31: 159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang, RY, O'Neil, SR, Lipovšek, Det al. Conformational assessment of adnectin and adnectin-drug conjugate by hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom 2018; 29: 1524–31. [DOI] [PubMed] [Google Scholar]
- 52. Kunimoto, D, Yoon, YH, Wykoff, CCet al. CEDAR and SEQUOIA Study Groups. Efficacy and safety of abicipar in neovascular age-related macular degeneration: 52-week results of phase 3 randomized controlled study. Ophthalmology 2020; S0161-6420: 30320–1. doi: 10.1016/j.ophtha.2020.03.035. [DOI] [PubMed] [Google Scholar]
- 53. Simon, M, Frey, R, Zangemeister-Wittke, Uet al. Orthogonal assembly of a designed ankyrin repeat protein-cytotoxin conjugate with a clickable serum albumin module for half-life extension. Bioconjug Chem 2013; 24: 1955–66. [DOI] [PubMed] [Google Scholar]
- 54. Mullard, A. FDA rejects first DARPin. Nat Rev Drug Discov 2020; 19: 501. [DOI] [PubMed] [Google Scholar]
- 55. Ullman, C, Mathonet, P, Oleksy, Aet al. High affinity binders to EphA2 isolated from abdurin scaffold libraries; characterization, binding and tumor targeting. PLoS One 2015; 10: e0135278. doi: 10.1371/journal.pone.0135278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peretti, S. Abdurin-drug conjugates: a new generation of targeted therapeutics. Protein Engineering Summit (PEGS) Conference, Lisbon: Cambridge Healthtech Institute, 2017. [Google Scholar]
- 57. He, R, Finan, B, Mayer, JPet al. Peptide conjugates with small molecules designed to enhance efficacy and safety. Molecules 2019; 24: 1855. doi: 10.3390/molecules24101855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ziaei, E, Saghaeidehkordi, A, Dill, Cet al. Targeting triple negative breast cancer cells with novel cytotoxic peptide-doxorubicin conjugates. Bioconjug Chem 2019; 30: 3098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kintzing, JR, Cochran, JR. Engineered knottin peptides as diagnostics, therapeutics, and drug delivery vehicles. Curr Opin Chem Biol 2016; 34: 143–50. [DOI] [PubMed] [Google Scholar]
- 60. Cox, N, Kintzing, JR, Smith, Met al. Integrin-targeting knottin peptide-drug conjugates are potent inhibitors of tumor cell proliferation. Angew Chem Int Ed Engl 2016; 55: 9894–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Currier, NV, Ackerman, SE, Kintzing, JRet al. Targeted drug delivery with an integrin-binding knottin-Fc-MMAF conjugate produced by cell-free protein synthesis. Mol Cancer Ther 2016; 15: 1291–300. [DOI] [PubMed] [Google Scholar]
- 62. Whalen, KA, White, BH, Quinn, JMet al. Targeting the Somatostatin receptor 2 with the miniaturized drug conjugate, PEN-221: a potent and novel therapeutic for the treatment of small cell lung cancer. Mol Cancer Ther 2019; 18: 1926–36. [DOI] [PubMed] [Google Scholar]
- 63. Johnson, ML, Meyer, T, Halperin, DMet al. First in human phase 1/2a study of PEN-221 somatostatin analog (SSA)-DM1 conjugate for patients (PTS) with advanced neuroendocrine tumor (NET) or small cell lung cancer (SCLC): phase 1 results. J Clin Oncol 2018; 3615_suppl: 4097–7. [Google Scholar]
- 64. Bendell, JC, Falchook, G, Sen, Set al. Annals Oncol 2019; 30. doi: 10.1093/annonc/mdz244. [DOI] [Google Scholar]
- 65. Thomas, A, Kriksciukaite, K, Falchook, Get al. Characterization of PEN-866, a heat shock protein 90 (HSP90) binding conjugate of SN-38, in patient plasma and tumors from the first in human study. Cancer Research 2020; 80. doi: 10.1158/1538-7445.AM2020-CT156. [DOI] [Google Scholar]
- 66. Heinis, C, Rutherford, T, Freund, Set al. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol 2009; 5: 502–7. [DOI] [PubMed] [Google Scholar]
- 67. Harrison, H, Bennett, G, Blakeley, D. BT1718, a novel bicyclic peptide-maytansinoid conjugate targeting MT1-MMP for the treatment of solid tumours. Design of bicyclic peptide and linker selection Cancer Research 2017; 77. doi: 10.1158/1538-7445.AM2017-5144. [DOI] [Google Scholar]
- 68. Cook, N, Banerji, U, Evans, TRJet al. Pharmacokinetic (PK) assessment of BT1718: a phase 1/2a study of BT1718, a first in class bicycle toxin conjugate (BTC), in patients (pts) with advanced solid tumors. Annals Oncol 2019; 30. doi: 10.1093/annonc/mdz244. [DOI] [Google Scholar]
- 69. Bennett, G, Brown, A, Mudd, Get al. MMAE delivery using the Bicycle toxin conjugate BT5528. Mol Cancer Ther 2020; 19: 1385–94. [DOI] [PubMed] [Google Scholar]
- 70. Bendell J, Wang J, Bashir Bet al. BT5528-100 phase I/II study of the safety, pharmacokinetics, and preliminary clinical activity of BT5528 in patients with advanced malignancies associated with EphA2 expression. J Clin Oncol, 2020; 38. doi: 10.1200/JCO.2020.38.15_suppl.TPS3655. [DOI] [Google Scholar]
- 71. Rigby M, Bennett G, Chen Let al. BT8009, a Bicycle® Toxin Conjugate targeting Nectin-4, shows target selectivity, and efficacy in preclinical large and small tumor models. Mol Can Ther, 2019; 18. doi: 10.1158/1535-7163.TARG-19-C061. [DOI] [Google Scholar]
- 72. Schulz, S, Becker, M, Groseclose, MRet al. Advanced MALDI mass spectrometry imaging in pharmaceutical research and drug development. Curr Opin Biotechnol 2019; 55: 51–9. [DOI] [PubMed] [Google Scholar]
- 73. Corti, A, Pastorino, F, Curnis, Fet al. Targeted drug delivery and penetration into solid tumors. Med Res Rev 2012; 32: 1078–91. [DOI] [PubMed] [Google Scholar]
- 74. Szot, C, Saha, S, Zhang, XMet al. Tumor stroma-targeted antibody-drug conjugate triggers localized anticancer drug release. J Clin Invest 2018; 128: 2927–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Purcell, JW, Tanlimco, SG, Hickson, Jet al. LRRC15 is a novel mesenchymal protein and stromal target for antibody-drug conjugates. Cancer Res 2018; 78: 4059–72. [DOI] [PubMed] [Google Scholar]
- 76. Saber, H, Leighton, JK. An FDA oncology analysis of antibody-drug conjugates. Regul Toxicol Pharmacol 2015; 71: 444–52. [DOI] [PubMed] [Google Scholar]
- 77. Saber, H, Simpson, N, Ricks, TKet al. An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. Regul Toxicol Pharmacol 2019; 107: 104429. doi: 10.1016/j.yrtph.2019.104429. [DOI] [PubMed] [Google Scholar]
- 78. Uppal, H, Doudement, E, Mahapatra, Ket al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res 2015; 21: 123–33. [DOI] [PubMed] [Google Scholar]
- 79. Shah, DK, Loganzo, F, Haddish-Berhane, Net al. Establishing in vitro-in vivo correlation for antibody drug conjugate efficacy: a PK/PD modeling approach. J Pharmacokinet Pharmacodyn 2018; 45: 339–49. [DOI] [PubMed] [Google Scholar]