

Abstract
A simple and robust method for electrochemical alkyl C–H fluorination is presented. Using a simple nitrate additive, a widely available fluorine source (Selectfluor), and carbon-based electrodes, a wide variety of activated and unactivated C–H bonds were converted to their C–F congeners. The scalability of the reaction was also demonstrated with a 100 gram preparation of fluorovaline.
Keywords: organic synthesis, electrochemistry, fluorination, C–H functionalization, radical
Graphical Abstract
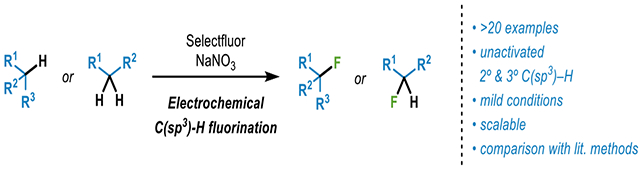
Within the realm of synthetic organic electrochemistry,1 few modern options exist for the C–H functionalization of unactivated aliphatic centers. In 2016 our lab reported a simple and inexpensive method for the oxidation of allylic C–H bonds featuring N-hydroxytetrachlorophthalimide as a mediator for hydrogen atom transfer.2 Shortly thereafter, it was found that the use of quinuclidine as mediator allowed for the oxidation of stronger C–H bonds such as unactivated methylenes.3 Based on requests from industrial collaborators in medicinal chemistry, we were compelled to extend this precedent to the problem of C–H fluorination. Although both photochemical4 and purely chemical means5 exist for accomplishing such a transformation (Figure 1A), an electrochemical alternative was pursued to determine if there was any specific advantage in terms of scalability and/or selectivity. Disclosed herein is a practical and scalable approach to C(sp3)-H fluorination that utilizes Selectfluor in a unique way when coupled to anodic oxidation in the presence of a nitrate additive.
Figure 1.

A) Electrochemical C(sp3)-H fluorination B) Reaction development.
A truncated optimization table is depicted in Figure 1B, wherein Selectfluor was chosen as a fluorine atom donor based on its wide availability. From a reactivity standpoint, one could also envisage three distinct roles for Selectfluor: as (1) its own electrolyte due to its ionic nature; (2) an electrophilic fluorine source; and (3) itself a mediator similar to quinuclidine.3 Using 1 as a model substrate, the impact of various reaction parameters was investigated (Figure 1B). The fully optimized conditions called for the use of Selectfluor (3.0 equiv.) and sodium nitrate (0.2 equiv.) in acetonitrile with a pair of reticulated vitreous carbon (RVC) electrodes at 3 mA to deliver the desired fluorinated product 2 in 62% NMR yield (54% isolated, entry 1). Not surprisingly, the reaction was found to be sensitive to oxygen, as is rationalized from the proposed radical chain mechanism (vide infra, entry 2). The reaction was confirmed to be electrochemically driven rather than initiated and required a constant supply of current (entry 3). Extensive screening revealed that the sodium nitrate was essential for the initiation as well as improving the reproducibility of this reaction (entries 4 and 5). The peculiar use of nitrate has precedent in the electrochemical literature and is known to be oxidized anodically to generate reactive radical species capable of abstracting hydrogen from substrates.6 The alternative fluorinating agent Selectfluor II (B) was also evaluated and did not improve the yield (entry 6). The likely role of Selectfluor as a mediator was supported by the fact that other electrophilic fluorinating sources (some known to capture nucleophilic radicals) failed to effect this transformation (entries 7 and 8). The use of a Ni foam cathode instead of RVC had a deleterious effect upon this reaction (entry 9, for a detailed summary of electrodes screened, see SI). Finally, increasing the current to 6 mA also resulted in lower yield (entry 10).
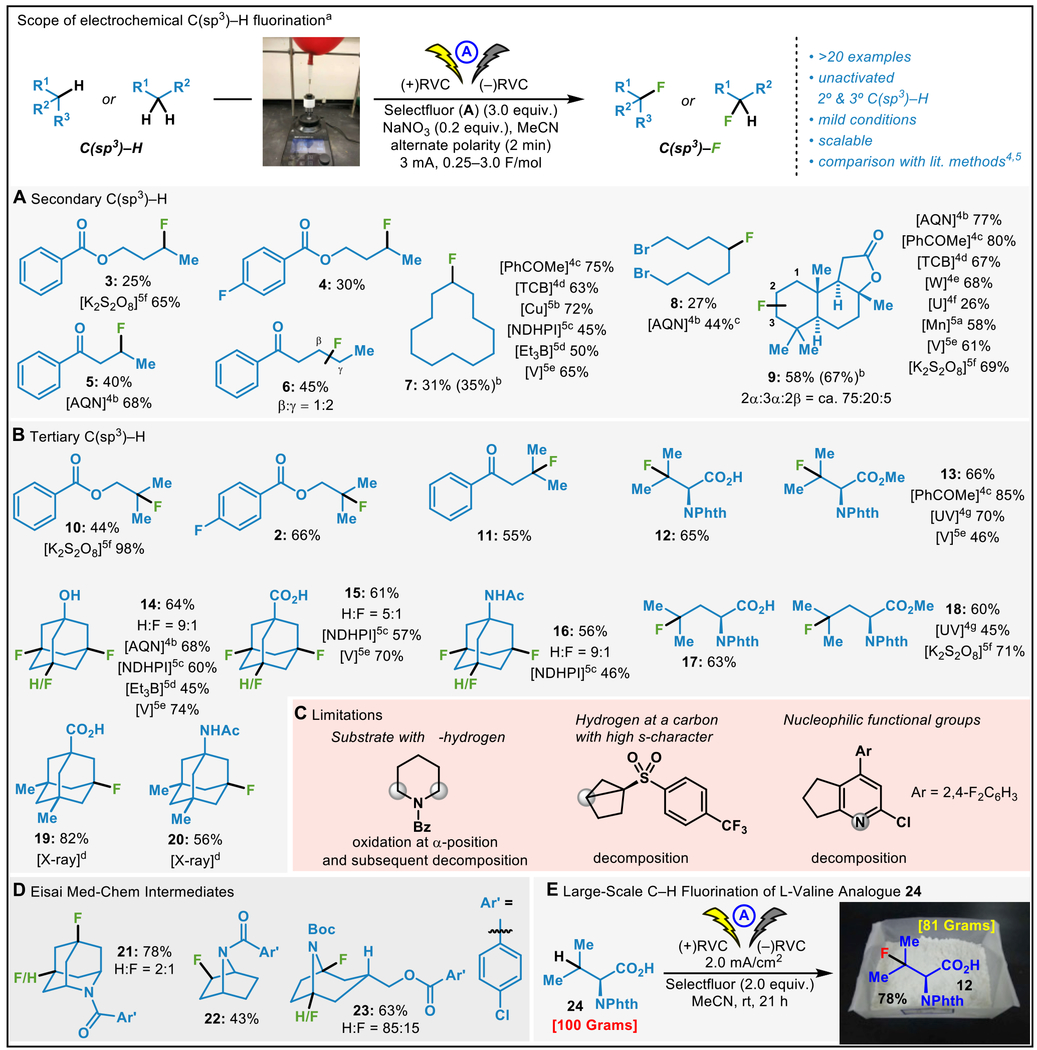
With an optimized set of conditions in hand, the utility of electrochemical C–H fluorination was explored. The generality of this transformation is shown in Table 1,7 demonstrating efficient C–H fluorination on various classes of molecules including terpenes, amino acids and pharmaceutically-relevant structures. In general, fluorination occurs at the unactivated secondary or tertiary C–H bonds that are the most distal from the electron-withdrawing group. This regioselectivity tracks with innate reactivity towards an electrophilic oxidant8 and is reminiscent of the selectivity of electrochemical unactivated C–H oxidation,3 which strongly suggests that the C–H abstraction step proceeds via a mediated radical mechanism. Within the realm of secondary systems (Table 1A), simple acyclic and cyclic alkanes were fluorinated, including those bearing esters (3 and 4), ketones (5 and 6), and even alkyl bromides (8). In the case of sclareolide, a mixture of the corresponding fluorinated compounds 9 in 58% yield. Notably, even in the absence of sodium nitrate, 9 was obtained in 67% yield. Tertiary systems (Table 1B) generally proceeded in higher yield. Thus, acyclic, amino-acid derivatives, and adamantanes were all fluorinated in synthetically useful yields. Fluorination of unsubstituted adamantanes generally led to a mixture of di-/tri-fluorinated products in reasonable yields whereas high yields of mono-fluorination were observed when only one tertiary C–H bond was available (19 and 20). Access to fluorinated amino acids (12, 13, 17, and 18) is a promising application with known uses in drug discovery contexts.9 This methodology was also field tested at Eisai where numerous building blocks were subjected to electrochemical fluorination and synthetically useful yields of valuable fluorinated products emerged (21, 22, and 23; Table 1D). In order to demonstrate the simplicity with which this chemistry could be conducted on scale, a 100 gram-scale fluorination of L-valine analogue 24 provided the corresponding fluorinated adduct 12 in 78% yield (Table 1E) without significant erosion of enantiopurity (96% ee, see SI). This was accomplished using a simple batch reactor (see SI for details) but in principle could also be easily adopted to a flow setup.
Table 1.
Electrochemical fluorination: Scope and applications.
|
Regarding the limitations of this method, several substrates delivered mixtures of isomers, showed no reaction, or decomposition under standard conditions (Table 1C). In general, yields are comparable to or slightly less than reported chemical initiation methods. The operational simplicity, reproducibility, and short reaction times are useful attributes of the present method. To further simplify the reaction conditions it is worth noting that the cases of sclareolide, adamantane, and valine did not require the use of a nitrate additive.
A proposed mechanism for electrochemical C–H fluorination is described in Figure 2. Considering the fact that this reaction is a net-redox neutral transformation and that the regioselectivity is analogous to that of other radical-based C–H functionalizations, a radical chain mechanism is proposed. Initially, a small amount of carbon radical is generated by anodic oxidation. Subsequent fluorination by Selectfluor delivers reactive radical cation 25, which can then abstract hydrogen from the substrate. Oxidative initiation was confirmed by conducting a reaction in a divided cell (without nitrate), where reaction progress was observed only in anodic chamber. As direct anodic oxidation of C–H bonds is known to be relatively inefficient, oxidation of nitrate is considered to be helpful in the initiation step. In addition to the precedent in the literature,10 cyclic voltammetry indicated that oxidation of nitrate occurred at +2.2 V in acetonitrile with respect to Ag/AgCl reference electrode (See SI). Thus, nitrate oxidation occurs before any potential direct anodic C–H abstraction.11 The precise role of nitrate in these reactions remains unclear and, as mentioned before, is not necessary for all substrates. Given that DABCO-containing fluorine sources are essential (see Figure 1), it is likely that a Selectfluor-derived DABCO species (such as 25) is an active radical mediator. The triple utility of Selectfluor as a fluorine donor, mediator, and electrolyte is a rather memorable aspect of this chemistry.
Figure 2.

Proposed mechanism.
In summary, a simple and scalable protocol for the electrochemical fluorination of unactivated C(sp3)-H bonds has been developed. The scope has been explored across a range of substrates bearing numerous types of functional groups and the ease of scale up is evidenced by the 100-gram scale fluorination of a valine derivative. As electrochemical functionalization processes become more mainstream it is likely that this method will find use alongside analogous C–H oxidation processes for both building block diversification and metabolic prediction.
Supplementary Material
Acknowledgment
The authors thank Dr. Jeremy T. Starr (Pfizer) for helpful discussions; Prof. A. L. Rheingold (UC San Diego) for X-ray crystallographic analysis; Drs. D.-H. Huang and L. Pasternack for assistance with NMR spectroscopy; Dr. J. Chen for assistance with HRMS and SFC analyses; Dr. R.R. Merchant for assistance in the preparation of the manuscript. T.H. and T.S. would like to thank Mr. So Yasui for NMR data collection and support for 21, 22, and 23; Dr. Hikaru Yoshimura for supporting chemistry of this work; Dr. Toshiki Kurokawa, Dr. Daiju Hasegawa, and Dr. Taro Terauchi for helpful discussions during the period of this work.
Funding Information
Financial support for this work was provided by the NSF (#1740656), Pfizer, AGC Inc. (to Y.T.), Nankai University (to M.C.), George E. Hewitt Foundation (to Y.K.), Fulbright Scholar Fellowship (to P.M.), JSPS (to H.N.), Swedish Research Council (VR 2017-00362 to B.K.P.), NSF GRFP (#2017237151) and Donald and Delia Baxter Fellowship (to S.H.R.).
Footnotes
Supporting Information
YES (this text will be updated with links prior to publication)
References and Notes
- (1).(a) Organic Electrochemistry, 5th ed., Revised and Expanded (Eds.: Hammerich O, Speiser B), CRC, Boca Raton, FL, 2015. For selected reviews on synthetic electrochemistry, see: [Google Scholar]; (b) Yan M; Kawamata Y; Baran PS Chem. Rev 2017, 117, 13230 and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kärkäs MD Chem. Soc. Rev 2018, 47, 5786. [DOI] [PubMed] [Google Scholar]; (d) Francke R; Little RD Chem. Soc. Rev 2014, 43, 2492. [DOI] [PubMed] [Google Scholar]; (e) Sperry JB; Wright DL Chem. Soc. Rev 2006, 35, 605. [DOI] [PubMed] [Google Scholar]; (f) Moeller KD Tetrahedron, 2000, 56, 9527. [Google Scholar]; (g) Lantaño B; Postigo A Org. Biomol. Chem 2017, 15, 9954. [DOI] [PubMed] [Google Scholar]
- (2).Horn EJ; Rosen BR; Chen Y; Tang J; Chen K; Eastgate MD; Baran PS Nature 2016, 533, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kawamata Y; Yan M; Liu Z; Bao DH; Chen J; Starr JT; Baran PS J. Am. Chem. Soc 2017, 139, 7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For a general overview of aliphatic C–H fluorination, see:; (a) Bume DD; Harry SA; Lectka T; Pitts CRJ Org. Chem 2018, 83, 8803. For specific examples, see: [DOI] [PubMed] [Google Scholar]; (b) Kee CW; Chin KF; Wong MW; Tan CH Chem. Commun 2014, 50, 8211. [DOI] [PubMed] [Google Scholar]; (c) Xia JB; Zhu C; Chen C Chem. Commun 2014, 50, 11701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bloom S; Knippel JL; Lectka T Chem. Sci 2014, 5, 1175. [Google Scholar]; (e) Halperin SD; Fan H; Chang S; Martin RE; Britton R Angew. Chem. Int. Ed 2014, 53, 4690. [DOI] [PubMed] [Google Scholar]; (f) West JG; Bedell TA; Sorensen EJ Angew. Chem. Int. Ed 2016, 55, 8923. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Egami H; Masuda S; Kawato Y; Hamashima Y Org. Lett 2018, 20, 1367. [DOI] [PubMed] [Google Scholar]
- (5).(a) Liu W; Huang X; Cheng MJ; Nielsen RJ; Goddard WA; Groves JT Science 2012, 337, 1322. [DOI] [PubMed] [Google Scholar]; (b) Bloom S; Pitts CR; Miller DC; Haselton N; Holl MG; Urheim E; Lectka T. Angew. Chem. Int. Ed 2012, 51, 10580. [DOI] [PubMed] [Google Scholar]; (c) Amaoka Y; Nagatomo M; Inoue M Org. Lett 2013, 15, 2160. [DOI] [PubMed] [Google Scholar]; (d) Pitts CR; Ling B; Woltornist R; Liu R; Lectka TJ Org. Chem 2014, 79, 8895. [DOI] [PubMed] [Google Scholar]; (e) Xia J-B; Ma Y; Chen C Org. Chem. Front 2014, 1, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhang X; Guo S; Tang P Org. Chem. Front 2015, 2, 806. [Google Scholar]
- (6).For selected examples, see:; (a) Leonard JE; Scholl PC; Steckel TP; Lentsch SE; Van De Mark MR Tetrahedron Lett 1980, 21, 4695. [Google Scholar]; (b) Shono T; Soejima T; Takigawa K; Yamaguchi Y; Maekawa H; Kashimura S Tetrahedron Lett 1994, 35, 4161. [Google Scholar]; (c) Christopher C; Lawrence S; Bosco AJ; Xavier N; Raja S Catal. Sci. Technol 2012, 2, 824. [Google Scholar]
- (7).General procedure for C(sp3)-H fluorination: To a 5 mL undivided ElectraSyn vial equipped with a stirrer bar was added substrate (0.30 mmol), Selectfluor (A, 3.0 equiv, 319 mg, 0.90 mmol), NaNO3 (0.2 equiv, 5.1 mg, 0.06 mmol), and MeCN (3.0 mL). The vial cap was screwed on, and headspace of the vial was purged with Ar using a balloon, syringe and needle assembly prior to electrolysis. Reaction parameters of ElectraSyn device were set up as follows: constant current, 3.0 mA; no reference electrode; total charge, 0.30 mmol substrate, ranging from 0.25 to 3.0 F/mol; alternate polarity, 2 min; stirring, 600 rpm. The mixture was electrolyzed under Ar atmosphere until complete or reasonable consumption of the starting material as judged by TLC. After electrolysis, the cap was removed, and the electrodes were taken out and rinsed with MeCN into the reaction mixture. The reaction mixture was poured into saturated aqueous NaHCO3 and extracted with CH2Cl2. The combined organics were washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude material was purified by silica gel column chromatography or preparative thin-layer chromatography to furnish the desired product. See SI for full details and graphical guide.
- (8).Newhouse T; Baran PS Angew. Chem. Int. Ed 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For selected examples, see:; (a) Hartman MCT; Josephson K; Szostak JW Proc. Natl. Acad. Sci 2006, 103, 4356. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nodwell MB; Yang H; Čolović M; Yuan Z; Merkens H; Martin RE; Benard F; Schaffer P; Britton R J. Am. Chem. Soc 2017, 139, 3595. [DOI] [PubMed] [Google Scholar]
- (10).Baciocchi E; Giacco T. Del; Murgia SM; Sebastiani GV J. Chem. Soc. Chem. Commun 1987, 1246. [Google Scholar]
- (11).Eberson L; Nyberg K Tetrahedron 1976, 32, 2185. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.