

Abstract
o-Benzynes can be utilized to construct heterocyclic motifs using various nucleophilic and cycloaddition trapping reactions. Acridines have been synthesized by capture of C,N-diarylimines with benzynes generated by classical methods (i.e., from ortho-elimination of precursor arene compounds), although in poor yields. We report here that these imines can be trapped by benzynes generated by the hexadehydro-Diels–Alder (HDDA) reaction in an efficient manner to produce 1,4-dihydroacridine products. These dihydroacridines were subsequently aromatized using MnO2 to provide structurally complex acridines.
Keywords: HDDA-benzynes, benzazetidines, azoquinone methides, dihydroacridines, acridines
Benzynes generated by the hexadehydro-Diels–Alder (HDDA) reaction are shown to capture C,N-diarylimines to produce 1,4-dihydroacridine products. The reaction is thought to proceed via benzazetidine intermediates that then undergo electrocyclic ring-opening and -closing (and final rearomatization) to give the dihydroacridines. These were easily aromatized with MnO2 to provide structurally complex acridines.
Graphical Abstract
Introduction
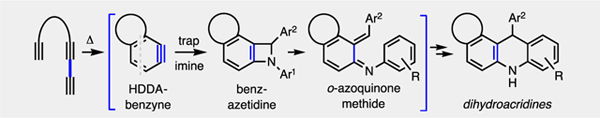
o-Benzynes and related arynes have been used as versatile intermediates for construction of various aromatic heterocycles.1 In a few instances, six-membered nitrogen-containing heterocycles of the acridine family have been produced, at least to small extents, by trapping of a classically generated aryne (i.e., one formed by way of an ortho-elimination of a precursor aromatic compound) by an imine. The previously observed reaction pathways may be roughly placed into two categories: a) a net [2+2] process by way of an initial, transient zwitterion (Scheme 1a); b) initial [4+2] cycloaddition between the benzyne (acting as a dienophile) and the N-aryl imine moiety (acting as the diene) (Scheme 1b).
Scheme 1.

Previous studies of reactions of benzynes with imines in either (a) a net [2+2] pathway or (b) a [4+2] pathway
The first reports of reactivity between o-benzynes and C,N-diaryl imines came from the laboratories of M. Yoshida2 (1975) and Storr3 (1984), which independently reported the reaction of N-benzylideneaniline [2, R1 = R2 = Ph (= 2a)] with o-benzyne (1) generated from benzenediazonium-2-carboxylate. In both studies the dihydrophenanthridine 7a was isolated (8% or 6% yield) and in the latter the dihydroacridine 4b (5%) was also obtained. These were rationalized as arising from competitive [2+2]- vs. [4+2]-cycloaddition of o-benzyne with 2a via intermediates 3b (through 3d) or 6a and 6b, respectively. The major isolated product in both of these original studies (18% and 16%, respectively) was the diamine 4a.
More recently, additional modes of reactivity between o-benzynes and imines have been studied. Some have focused on trapping of the presumed 1,3-zwitterion 3a, which arises via imine nitrogen attack on the electrophilic benzyne. In 2006 H. Yoshida and coworkers4 demonstrated the ability to access benzoxazinones 5b via trapping of the 1,3-zwitterion by carbon dioxide using imines in which the carbon-bound group is an alkyl moiety. In 2015 Hwu et al.5 showed the diastereoselective formation of imidazolidines 5a and other N-heterocycles via trapping of the intermediate 1,3-dipole 3e, formed via intramolecular proton transfer within 3a. Finally, in 2017 researchers in the Tian laboratory6 showed that 3a may be trapped by protic carbon nucleophiles in a three-component fashion to form products of the type 5c.
In 2017 H. Yoshida and coworkers7 showed that trapping of the aza-ortho-quinone methide intermediate 3c to form 2:1 imine:benzyne adducts 4c was possible when benzyne was used in molar excess. This mode of reactivity prevails when the aryl moieties are more highly substituted, presumably slowing the electrocyclization of 3c to 3d.
Finally, in addition to M. Yoshida and Storr’s initial isolation of phenanthridine adducts, other groups have sought to exploit the [4+2] pathway to access this class of product more efficiently. In 2006 Wang et al.8 showed that benzyne could be used in a 3-component process to access phenanthridines 7c with high efficiency using an electron-poor benzaldehyde and an electron-rich aniline. Similarly, in 2016 researchers in the He group9 were able to isolate 7b in a three-component process using Kobayashi10 arynes, an aldehyde ester, and an aniline. In 2015 Coquerel and coworkers11 reported the formation of isoquinolines 7d derived from an imine containing a nitrogen-bound pyrazole moiety, and, through a computational study, suggested that an electron-rich aromatic group bound to the imine nitrogen favors [4+2] cycloaddition over a [2+2] pathway.
To summarize, reported examples of benzynes reacting with imines to give acridine-like products have shown low selectivity and efficiency. Given the interest in acridine compounds more broadly as well as the fact that hexadehydro-Diels–Alder (HDDA)-derived benzynes12 often lead to outcomes complementary to those from classical benzynes, we have explored the reactions of several polyyne substrates with various imine trapping agents and report the results of those studies here.
Results and Discussion
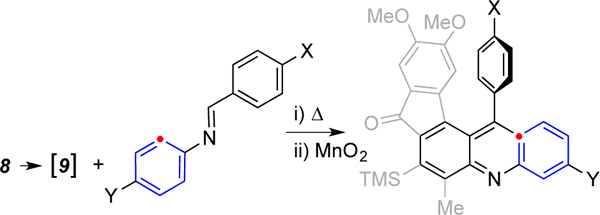
We first examined (Scheme 2) the reaction between the HDDA benzyne 9, generated by warming triyne 8, with N-benzylidene-aniline (2a). This resulted in formation of the dihydroacridine 11 in a remarkably clean reaction, which contrasts significantly with the lower efficiencies of previous reactions giving rise to dihydroacridines (cf. Scheme 1a). Compound 11 was subsequently oxidized with MnO2 to afford the acridine derivative 12a in 68% yield.
Scheme 2.

Reaction of the HDDA-generated benzyne 9 proceeds nearly exclusively to the 1,4-dihydroacridine 11, presumably via the [2+2]-benzazetidine 10. Compound 11 was subsequently oxidized to acridine 12a.
To demonstrate generality of this reaction with different types of electronically modified aryl substituents in the imines, we explored the reactions between benzyne 9 and imines 2b-2g (Table 1). For each entry, only the acridine product that results from subsequent MnO2 oxidation of the intermediate dihydroacridine is shown (see Supporting Information for characterization of the dihydro-intermediates SI-1–SI-6). The reaction showed relatively broad tolerance of electronic perturbation of the aryl substituent on both the carbon and nitrogen atoms composing the imine. That is, imines 2b-g all provided the corresponding dihydroacridines and, then, the acridines 12b-g (Table 1). Only in the instance of the most electron-poor imine 2f, was the yield of the initial dihydroacridine low. This is consistent with the view that the initial event in the engagement of benzyne with imine is nucleophilic attack by the nitrogen atom to generate the zwitterion 3a (Scheme 1a).
Table 1.
Reactions of triyne 8 with several C,N-diaryl imines.
 | ||
|---|---|---|
| entry | imine | product [step ia: yield%, step iib: yield%] |
| 1 | 2a, X = H, Y = H | 12a [step ia: 98%, step iib: 68%] |
| 2 | 2b, X = NO2, Y = H | 12b [step ia: 68%, step iib: 90%] |
| 3 | 2c, X = OMe, Y = H | 12c [step ia: 78%, step iib: 86%] |
| 4 | 2d, X = H, Y = NO2 | 12d [step ia: 62%, step iib: 82%] |
| 5 | 2e, X = H, Y = OMe | 12e [step ia: 100%, step iib: 97%] |
| 6 | 2f, X = NO2, Y = NO2 | 12f [step ia: 19%, step iib: 56%] |
| 7 | 2g, X = OMe, Y = OMe | 12g [step ia: 88%, step iib: 89%] |
yield of the 1,4-dihydroacridine from the HDDA reaction
yield of the acridine adduct following MnO2 treatment
As a final example, this with a different type of imine, the 1-naphthyl derivative 13 gave the more highly annulated benzoacridine derivative 14 (Scheme 3). This suggests that additional analogs with yet more extended conjugation can also be accessed.
Scheme 3.

Reaction of triyne 8 with imine 13.
ayield of the 1,4-dihydroacridine from the HDDA reaction
byield of the acridine adduct following MnO2 treatment
Amidines are amino-substituted analogs of imines. N,N-Dimethyl-N’-phenylamidine (15) efficiently engaged the benzyne 9 (Scheme 4). It gave rise directly to the acridine derivative 17a, which has no substituent at C9 of the acridine. However, this reaction was accompanied by the formation of a similar amount of the dimethylamine-trapped benzyne product 17b. Presumably the dihydroacridine 16, arising from a [2+2] pathway directly analogous to that depicted for 8 to 11 (Scheme 2), underwent an elimination event under the reaction conditions to produce 17a. That process can be envisioned to proceed by a direct (and possibly unimolecular) loss of dimethylamine, which, once released, then competitively trapped benzyne 9 (red arrows). Alternatively, a second copy of 9 could engage the tertiary aliphatic amine in intermediate 16 to give the 1,3-zwitterion13 18 (shown in a truncated form for simplicity), which could then collapse directly to one molecule each of 17a and 17b. Although of limited preparative value, this trapping reaction with an amidine provides interesting mechanistic insights.
Scheme 4.

Reaction of amidine 15 with aryne 9 generated from triyne 8.
To establish that the reaction is not limited to benzyne 9, we have trapped the HDDA benzynes derived from the precursor tetraynes 19a and 19b with N-benzylideneaniline (2a) (Scheme 5). The symmetrical tetrayne, which previously has been shown to react with various nucleophiles to give a pair of constitutional isomers with relatively little preference, produced only the single regioisomeric isoindoline derivative 20a in 42% yield along with the oxidized acridine 21a (19%), presumably from air oxidation. Reaction of 19b and 2a produced the expected indolinoquinoline 21b in 51% yield over two steps. The formation of a single benzyne (i.e., 22b; see dashed lines in 19b) from this unsymmetrical tetrayne precursor was first observed14 by Lee and coworkers (and on many subsequent occasions) and is consistent with a computational study15 that addressed exactly that point. To summarize, the reactions of both 19a/b with 2a proceeded via the corresponding benzazetidine 23a/b and azo-quinonemethide species 24a/b en route to the 1,4-dihydroacridines 20a/b.
Scheme 5.

Reactions of tetraynes 19a/b with imine 2a to give acridines 21a/b.
Conclusions
In summary, these results establish that trapping of thermally generated, polycyclic benzyne derivatives with C,N-diaryl imines leads to dihydroacridine derivatives, which can be further and readily oxidized to their acridine analogs. These reactions proceed considerably more efficiently than those reported earlier for imine trapping of benzyne itself (from benzenediazonium-2-carboxylate thermolysis). In no case have we observed products arising from initial [4+2] cycloaddition, as has been seen in previous studies.2,3,8,9,11 This work represents another instance in which the arynes generated through the HDDA-cycloisomerization reaction, which are performed in a purely thermal environment, has allowed for the formation of the trapping products in a much cleaner,16 if not unique,17 manner.
Experimental Section
General Experimental Protocols
13C and 1H NMR spectra were recorded on a Bruker HD-500, AV-500, AV-400, or AX-400 spectrometer. Proton chemical shifts are referenced to TMS (δ 0.00 ppm) in CDCl3 solutions and to the residual CHD5 (δ 7.16 ppm) in benzene-d6 solutions. A non-first order multiplet, doublet, or doublet of doublets in a 1H NMR spectrum is denoted as a ‘nfom’, ‘nfod’, or ‘nfodd’, respectively. For the latter two, the coupling constant is listed as an apparent value (Japp), because the spacing between the two major lines for the population of molecules having magnetically equivalent protons (ca. 50%) is actually the value of Jo + Jp). Resonances are reported in the following format: chemical shift (ppm) [multiplicity, coupling constant (s) (in Hz), integral value (to the nearest integer), and assignment of the substructural environment within the structure]. First-order coupling constants were analyzed using methods we have published elsewhere.18,19 The 13C NMR chemical shifts are taken from the “1D” spectrum where possible, although some were deduced from HMBC correlations. Carbon chemical shifts are referenced to δ 77.16 ppm in CDCl3 solutions and to δ 128.06 for C6D6 solutions. Infrared spectra were recorded using a Bruker Alpha II Spectrometer. Samples were prepared as thin films on a diamond window in the attenuated total reflectance (ATR) mode. Absorption maxima are given in cm−1. The high-resolution mass spectrometry (HRMS) measurements were made in the ESI mode using a Thermo Orbitrap Velos instrument (mass accuracy of ≤3 ppm). An external calibrant was used (Pierce™ LTQ) and the samples were directly injected into the ion source. Medium pressure liquid chromatography (MPLC) was often used to purify newly synthesized materials. Hand-packed silica gel columns (normal-phase, 25–200 psi, 20–40 μm, 60 Å pore size, Teledyne RediSep Rf Gold®) were used. The apparatus consisted of a Waters HPLC pump (model 510), a Gilson (111 UV) detector, and a Waters (R401) differential refractive index detector. Preparative flash chromatography was performed on columns packed with Agela silica gel (230–400 mesh). Thin layer chromatography (TLC) was performed on silica-gel coated, plastic-backed plates that were visualized by UV light and/or by a solution of potassium permanganate and heating. The indicated reaction temperature refers to the temperature of the external cooling or heating bath. HDDA reactions, including those performed at temperatures higher than the boiling point of the reaction solvent, were done in a screw-top culture tube that was capped with an inert, Teflon®-lined closure. Poly-yne substrates 820, 19a21, and 19b21 were synthesized according to reported methods. N-Benzylideneaniline (2a) was prepared according to a reported procedure.22
A. General Procedure for trapping of HDDA-generated arynes with imines
The polyyne precursor (1 equiv) and the imine (1–3 equiv) were combined in a screw-capped culture tube. 1,2-Dichloroethane was added (0.05 M) and the resulting solution was placed in an oil bath maintained at 90 °C and allowed to react overnight. Subsequently, the solvent was removed under reduced pressure, and the crude material was purified using MPLC with the elution solvent mixture indicated for each compound.
B. General Procedure for oxidation of 1,4-dihydroacridines to their respective acridines
A scintillation vial or a culture tube was charged with a stir bar and the respective 1,4-dihydroacridine (1 equiv). Chloroform or dichloromethane (0.005 M) was added, along with MnO2 (ca. 10–20 equiv). The resulting slurry was allowed to stir at ambient temperature until the reaction was observed to be complete by TLC analysis. The reaction mixture was filtered through Celite® and the filtrate was concentrated in vacuo.
C. General Procedure for one-pot synthesis of acridines from HDDA-generated benzynes and imines
The HDDA polyyne precursor (1 equiv) and the imine (1–3 equiv) were added to a screw-cap culture tube. 1,2-Dichloroethane was added (0.05 M), and the resulting solution was placed in an oil bath maintained at 90 °C and allowed to react overnight. MnO2 (ca. 10–20 equiv) and a stir bar were added. The slurry was then stirred at ambient temperature until the reaction was observed to be complete by TLC. The reaction mixture was filtered through Celite® and the filtrate was concentrated in vacuo.
10,11-Dimethoxy-6-methyl-13-phenyl-7-(trimethylsilyl)-5,13-dihydro-8H-indeno[1,2-a]acridin-8-one (11)
Following general procedure A, 1-(4,5-dimethoxy-2-(penta-1,3-diyn-1-yl)phenyl)-3-(trimethylsilyl)prop-2-yn-1-one (8, 0.024 g, 0.074 mmol, 1 equiv), (E)-N,1-diphenylmethanimine (2a, 0.015 g, 0.083 mmol, 1.1 equiv), and dichloroethane (2 mL) were used to prepare the 1,4-dihydroacridine 11. Purification of the crude product by MPLC (2:1 hexanes:EtOAc) yielded 11 (0.038 g, 0.076 mmol, 98%) as an orange crystalline solid. 1H NMR (500 MHz, CDCl3): δ 7.45 (d, J = 7.8, 1.4 Hz, 1H, H1), 7.29 (nfod, Japp = 7.5 Hz, 2H, ArHo), 7.22 (nfodd, Japp = 7.7, 7.7 Hz, 2H, PhHm), 7.126 (ddd, J = 7.4, 7.4, 1.5 Hz, 1H, H3), 7.125 (tt, J = 7.4, 1.5 Hz, 1H, ArHp), 7.10 (s, 1H, H9), 7.09 (s, 1H, H12), 6.95 (dd, J = 7.4, 7.4, 1.1 Hz, 1H, H2), 6.81 (dd, J = 7.9, 1.3 Hz, 1H, H4), 6.48 (s, 1H, NH), 5.77 (s, 1H, H13), 3.90 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 2.45 (s, 3H, ArCH3), and 0.46 (s, 9H, Si(CH3)3). 13C NMR (126 MHz, CDCl3): 193.6, 153.0, 149.0, 144.7, 143.1, 142.6, 141.2, 137.6, 137.5, 133.8, 129.1, 128.70, 128.67, 127.8, 127.1, 127.0, 125.3, 123.9, 122.3, 118.8, 115.2, 107.8, 106.7, 56.6, 56.2, 44.7, 18.2, and 3.2. HRMS (ESI-TOF): Calcd for C32H32NO3Si+ [M+H+]+ requires 506.2146; found 506.2150. IR (neat): 3440, 3390, 3059, 3001, 2924, 2853, 2359, 2342, 2069, 2034, 1976, 1961, 1944, 1694, 1605, 1577, 1546, 1489, 1465, 1432, 1416, 1375, 1343, 1324, 1299, 1283, 1259, 1245, 1215, 1173, 1158, 1091, 1045, 1027, 991, 936, 873, 854, 797, 771, 751, 727, 699, 676, 645, 630, 613, 599, 575, 519, 493, 449, and 408 cm−1. mp: 249–250 °C.
10,11-Dimethoxy-6-methyl-13-phenyl-7-(trimethylsilyl)-8H-indeno[1,2-a]acridin-8-one (12a)
Following general procedure C, 1-(4,5-dimethoxy-2-(penta-1,3-diyn-1-yl)phenyl)-3-(trimethylsilyl)prop-2-yn-1-one (8, 0.010 g, 0.030 mmol, 1 equiv), (E)-N,1-diphenylmethanimine (2a, 0.007 g, 0.038 mmol, 1.3 equiv), MnO2 (xs), and dichloroethane (2 mL) were used to prepare acridine 12a. Purification of the crude product yielded acridine 12a (0.011 g, 0.022 mmol, 71%) as a purple crystalline solid. 1H NMR (500 MHz, CDCl3): δ 8.26 (d, J = 8.6 Hz, 1H, H4), 8.14 (d, J = 8.9 Hz, 1H, H1), 7.78 (nfod, Japp = 7.2 Hz, 2H, PhHo), 7.77 (br dd, J = 8.8, 6.8 Hz, 1H, H3), 7.56 (nfodd, Japp = 7.5, 7.5 Hz, 2H, PhHm), 7.53 (tt, J = 6.9, 1.7 Hz, 1H, PhHp), 7.48 (br dd, J = 8.6, 6.6 Hz, 1H, H2), 7.04 (s, 1H, H9), 5.64 (s, 1H, H12), 3.83 (s, 3H, C10OCH3), 3.51 (s, 3H, C11OCH3), 3.06 (s, 1H, ArCH3), and 0.51 (s, 9H, Si(CH3)3). 13C NMR (126 MHz, CDCl3): 195.4, 153.0, 150.6, 149.0, 148.0, 146.9, 145.1, 143.0, 140.1, 138.6, 137.3, 136.1, 132.8, 130.9, 130.3, 129.5, 129.0, 126.7, 126.5, 125.2, 125.0, 120.8, 109.3, 106.5, 56.7, 56.1, 20.7, and 2.7. HRMS (ESI-TOF): Calcd for C32H30NO3Si+ [M+H+]+ requires 504.1989; found 504.1983. IR (neat): 3062, 2956, 2922, 2852, 2357, 2171, 2099, 2041, 2023, 1984, 1758, 1709, 1595, 1579, 1498, 1463, 1442, 1418, 1405, 1376, 1336, 1285, 1250, 1224, 1180, 1144, 1109, 1059, 1026, 1012, 972, 896, 842, 816, 799, 764, 734, 702, 671, 642, 619, 604, 568, 544, 519, 507, and 412 cm−1. mp: 96–97 °C.
The online Supporting Information contains experimental procedures for all reactions, analytical characterization data for all new compounds, and copies of 1H and 13C NMR spectra as well as selected 2D NMR spectra.
Supplementary Material
Acknowledgments
The U.S. Department of Health and Human Services, National Institutes of General Medical Sciences (R35 GM127097) supported this research. Some NMR spectral data were obtained using an instrument funded through the NIH Shared Instrumentation Grant program (S10OD011952). Mass spectrometry was performed in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center at the University of Minnesota, funded in part by a Cancer Center Support Grant (CA-77598).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].(a) Dubrovskiy AV, Markina NA, Larock RC, Org. Biomol. Chem 2013, 11, 191–218. [DOI] [PubMed] [Google Scholar]; (b) Peña D; Pérez D, Guitián E, Heterocycles 2007, 74, 89–100. [Google Scholar]
- [2].Nakayama J, Midorikawa H, Yoshida M, Bull. Chem. Soc. Jpn 1975, 48, 1063–1064. [Google Scholar]
- [3].Fishwick CWG, Gupta RC, Storr RC, J. Chem. Soc. Perkin 1 1984, 1, 2827–2829. [Google Scholar]
- [4].Yoshida H, Fukushima H; Ohshita J, Kunai A, J. Am. Chem. Soc 2006, 128, 11040–11041. [DOI] [PubMed] [Google Scholar]
- [5].Swain SP, Shih Y, Tsay S, Jacob J, Lin C, Hwang KC, Horng J, Hwu JR, Angew. Chem. Int. Ed 2015, 54, 9926–9930. [DOI] [PubMed] [Google Scholar]
- [6].Xu J, Li S, Wang H, Xu W, Tian S, Chem. Commun 2017, 53, 1708–1711. [DOI] [PubMed] [Google Scholar]
- [7].Yoshida H, Kuriki H, Fujii S, Ito Y, Osaka I, Takaki K, Asian J. Org. Chem 2017, 6, 973–976. [Google Scholar]
- [8].Shou W, Yang Y, Wang Y, J. Org. Chem 2006, 71, 9241–9243. [DOI] [PubMed] [Google Scholar]
- [9].Reddy RS, Lagishetti C, Chen S, Kiran INC, He Y, Org. Lett 2016, 18, 4546–4549. [DOI] [PubMed] [Google Scholar]
- [10].Himeshima Y, Sonoda T, Kobayashi H, Chem. Lett 1983, 12, 1211–1214. [Google Scholar]
- [11].Castillo J, Quiroga J, Abonia R, Rodriguez J, Coquerel Y, Org. Lett 2015, 17, 3374–3377. [DOI] [PubMed] [Google Scholar]
- [12].(a) Miyawaki K, Suzuki R, Kawano T, Ueda I, Tetrahedron Lett. 1997, 38, 3943–3946. [Google Scholar]; (b) Bradley AZ, Johnson RP, J. Am. Chem. Soc 1997, 119, 9917–9918. [Google Scholar]; (c) Hoye TR, Baire B, Niu D, Willoughby PH, Woods BP, Nature 2012, 490, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Baire B, Niu D, Willoughby PH, Woods BP, Hoye TR, Nature Protoc. 2013, 8, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Diamond OJ, Marder TB, Org. Chem. Front 2017, 4, 891–910. [Google Scholar]
- [13].(a) Ross SP, Baire B, Hoye TR, Org. Lett 2017, 19, 5705–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Arora S, Palani V, Hoye TR, Org. Lett 2018, 20, 8082–8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yun SY, Wang KP, Lee NK, M. P., Lee D, J. Am. Chem. Soc 2013, 135, 4668–4671. [DOI] [PubMed] [Google Scholar]
- [15].Chen M, He CQ, Houk KN, J. Org. Chem 2019, 84, 1959–1963. [DOI] [PubMed] [Google Scholar]
- [16].E.g., Niu D, Hoye TR, Nature Chem. 2014, 6, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].(a) E.g., Ross SP, Hoye TR, Nature Chem. 2017, 9, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shen H, Xiao X, Haj MK, Willoughby PH, Hoye TR, J. Am. Chem. Soc 2018, 140, 15616–15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hoye TR, Hanson PR, Vyvyan JR, J. Org. Chem 1994, 59, 4096–4103. [Google Scholar]
- [19].Hoye TR. Zhao H, J. Org. Chem 2002, 67, 4014–4016. [DOI] [PubMed] [Google Scholar]
- [20].Arora S, Zhang J, Pogula V, Hoye TR, Chem. Sci 2019, 10, 9069–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen J, Palani V, Hoye TR, J. Am. Chem. Soc 2016, 138, 4318–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Malig TC, Yu D, Hein JE, J. Am. Chem. Soc 2018, 140, 9167–9173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.