

Abstract
Alcohol use disorder (AUD) is a leading cause of morbidity and mortality. Despite AUD’s substantial contributions to lost economic productivity and quality of life, there are only a limited number of approved drugs for treatment of AUD in the United States. This chapter will update progress made on the epigenetic basis of AUD, with particular focus on histone post-translational modifications and DNA methylation and how these two epigenetic mechanisms interact to contribute to neuroadaptive processes leading to initiation, maintenance and progression of AUD pathophysiology. We will also evaluate epigenetic therapeutic strategies that have arisen from preclinical models of AUD and epigenetic biomarkers that have been discovered in human populations with AUD.
Keywords: Epigenetics, Addiction, Alcohol, Histone post-translational modifications, Alcohol Use Disorder, Alcoholism, DNA, DNA methylation
1. Introduction
Alcohol use disorder (AUD) is the third leading preventable cause of death in the United States and accounts for over $249 billion dollars in lost economic costs yearly (Sacks et al., 2015). Alcohol attributable deaths account for 88,000 deaths yearly in the US (Centers for Disease Control and Prevention, 2013). Despite this considerable health and socioeconomic burden, therapeutic treatment options for AUD are limited relative to some other drugs of abuse and other psychiatric and health diseases. AUD is a complex psychiatric disorder that involves various phases of the addiction cycle and changes in many different signaling pathways and behavioral outputs (Koob and Volkow, 2016; Pandey et al., 2017). Epigenetics is a critical mechanism that controls gene expression, a growing body of literature demonstrates that it is important in regulating many different facets of AUD, and drugs that work on an epigenetic level may be viable treatment strategies for AUD (Berkel and Pandey, 2017; Pandey et al., 2017). This chapter will review the progress that has been made in epigenetic mechanisms that underlie AUD with specific focus on histone posttranslational modifications and also briefly on DNA methylation.
1.1. Conceptual Framework of AUD
AUD can be assessed as a conceptual framework with three major stages: 1) compulsion to seek and obtain a drug, 2) a binge period viewed as a loss of control in limiting drug intake, and 3) negative affect or withdrawal after the pharmacological effects of the drug are no longer active (Koob and Volkow, 2016). In both clinical populations and in animal models of alcohol abuse, researchers can distinctly identify, define, and test each of these three stages (Koob and Volkow, 2016; Kwako et al., 2016). (Figure 1).
Figure 1. Overview of the cycle of alcohol addiction.
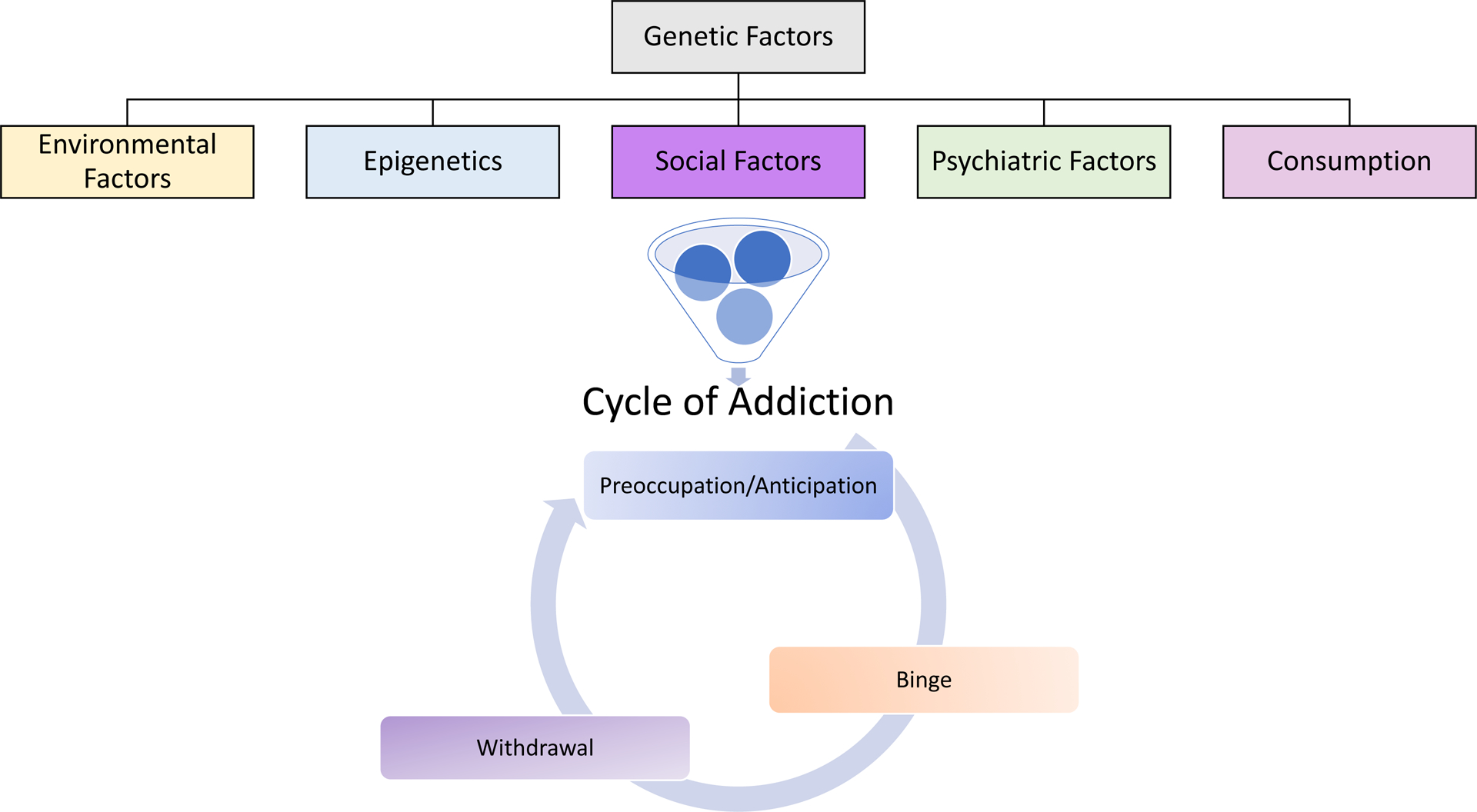
Prior to the first drink, several factors increase the risk of developing an AUD. These include genetic factors such as polymorphisms in aldehyde dehydrogenase, environmental factors such as education or social status, social factors such as if family members or friends consume alcohol, psychiatric factors such as comorbid disorders, (e.g. stress, anxiety, depression), or consumption factors such as age of onset. The cycle of addiction framework suggests that as the development of AUD occurs, individuals go through cycles of preoccupation and anticipation, binge consumption and then withdrawal, and that continued and persistent moves through this cycle eventually lead to a net affective disorder, where alcohol consumption is less for the rewarding effects and more to alleviate the negative effects (Koob and Volkow, 2016).
1.2. Risk Factors for AUD
Several risk factors have been identified that might play a role in the development of AUD. This includes comorbid psychiatric disorders, genetic factors, environmental factors, personality factors, alcohol consumption history, and age of onset.
Most individuals with AUD suffer from a comorbid psychiatric disorder, such as depression, schizophrenia, PTSD, and anxiety disorders (Kessler et al., 1996; Regier, 1990). For example, individuals with anxiety disorder are at a higher risk to also be diagnosed with alcohol dependence (Kessler et al., 1996; Regier, 1990). Much work has been done to differentiate comorbid psychiatric disorders and symptomology that occurs in response to heavy alcohol consumption and withdrawal. For example, acute anxiety induced during withdrawal after alcohol exposure typically resolves itself within a few days to weeks following detoxification, treatment, and abstinence (Anthenelli and Schuckit, 1993; Brown et al., 1991; Kranzler, 1996). Nonetheless, pre-existing anxiety disorder or the development of an anxiety disorder during withdrawal after alcohol use appears to form a vicious cycle of addiction and promote alcohol drinking behaviors (Pandey et al., 2017).
Genetic factors are also considered to increase the risk of alcohol dependence, as family and twin studies suggest that heritability of AUD is between 40 and 60% (Prescott and Kendler, 1999). A recent large genome-wide association study evaluated 274,424 individuals and found 10 genomic loci associated with AUD diagnosis (Kranzler et al., 2019). Similar to findings in previous studies, this study found that AUD was significantly associated with polymorphisms in the alcohol dehydrogenase 1C (ADH1C) gene (Kranzler et al., 2019). A second study also using a large number of individuals with alcohol use disorder (n = 173,296) found six significant loci that were associated with alcohol consumption (ADH1B, KLB, BTF3P13, GCKR, SLC39A8 and DRD2) (Thompson et al., 2020). Alcohol metabolism genes have long been shown to be associated with alcohol dependence (Li et al., 2012). However, identification of a genetic phenotype that drives AUD is unlikely, suggesting that other factors are also involved. Studies are also emerging that integrate the interaction of genetic with epigenetic factors that may be involved in pathophysiology of AUD (Krishnan et al., 2014; Starkman et al., 2012)
Not surprisingly, epidemiological studies have identified that binge consumption of alcohol (5+ drinks per day) greatly increases the chance for developing an AUD (Caetano et al., 1997) and increases the risk of associated morbidity and mortality (Rehm et al., 2006). The differences in alcohol consumption patterns and habits have received renewed attention, with one study observing that about ~15% of outbred rats will consume alcohol over a sugar reward (Augier et al., 2018), which is similar to the finding that only 5.8% of individuals in the United States qualify as having an AUD (Substance Abuse and Mental Health Services Administration & Center for Behavioral Health Statistics and Quality., 2014). This suggests that individual personality traits contribute to the development and severity of AUD. One study using the Cloninger Personality Inventory has found that individuals with higher scores for “search for novelty” and “avoidance of damage” have worse prognosis outcomes (e.g. higher number of relapses) when treated for AUD (Álvarez et al., 2018). A meta review looking specifically at binge drinking finds that “high impulsivity” and “high sensation seeking”, as well as “anxiety sensitivity”, “neuroticism” “extraversion” and “low conscientiousness” are personality traits attributed to individuals who binge drink (Adan et al., 2017).
Another risk factor that appears to be crucial in the development of AUD is the age of onset. It has been shown that individuals who begin drinking during adolescence are 5–7 times more likely to develop an AUD later in life (Grant et al., 2001). One of the largest predictors of adolescent alcohol consumption is peer influences, especially from high-intensity friendships (e.g. best friend) (Cruz et al., 2012). Other predictors involved are emotional stress and risk taking behavior, especially in individuals who rapidly escalate and begin drinking heavily (Colder et al., 2002). Recently, tremendous progress has been made to better understand the neurobiology of persistent effects of adolescent alcohol exposure in adult psychopathology (Crews et al., 2019; Kyzar et al., 2016)
Taken together this suggests that numerous social, psychological, and economic circumstances dictate risk for developing an AUD. The way different risk factors interact and contribute to outcomes of AUD may be better understood by deciphering some of the molecular and epigenetic processes that drive behavioral changes, which could also be useful for the development of suitable biomarkers that may improve the diagnosis and treatment of AUD.
2. Epigenetics and gene expression
Epigenetics is broadly defined as changes to gene expression that aren’t due to mutations to the underlying sequence of DNA (e.g. not a single nucleotide polymorphisms, truncations, or recombination events) (Allis and Jenuwein, 2016). There are several different mechanisms that control gene expression. The most broadly studied are changes in chromatin structure that permit or restrict gene transcription. Changes in three-dimensional chromatin structure open or close chromatin to allow the binding of RNA polymerase II (RNAPII) to facilitate gene transcription (Kim et al., 2005). Chromatin is tightly condensed within the nucleus and consists of a base unit of a nucleosome which is ~146 bp of DNA wrapped around an octamer of histones consisting of 2 copies of each isoform (H2A, H2B, H3 and H4) (Kouzarides, 2007). In general, open chromatin, or the lack of tightly packed nucleosomes, is associated with active transcription and is called permissive (Lee et al., 2004). Changes in chromatin structure can occur in response to an external stimulus such as alcohol consumption or withdrawal (Pandey et al., 2017). Changes can also occur because of predefined developmental programs, such as the ones that determine stem cell differentiation (Boyer et al., 2006). These processes are highly organized, and disruption to these processes either through somatic mutations or through aberrant signaling cascades can result and potentiate human disease (Jiang et al., 2004).
Several methodologies are available for testing open and closed regions in chromatin structure, including assay for transposase-accessible chromatin using sequencing (ATAC-seq) (Buenrostro et al., 2013), circularized chromosome conformation capture (4C) (Simonis et al., 2006), and chromosome conformation capture (3C) (Dekker, 2002), Hi-C (which is a sequencing method of 4C and 3C) (Lieberman-Aiden et al., 2009) and older methods such as formaldehyde-assisted isolation of regulatory elements (FAIRE-seq) (Giresi et al., 2007), and DNAseI-seq (Boyle et al., 2008). ATAC-seq has been applied recently to single-cells to better understand regulatory chromatin structure as it applies to cell-type heterogeneity within the brain (and other tissues) (Cusanovich et al., 2018). ATAC-seq has been used to determine glutamatergic gene dysregulation in human heroin abusers (Egervari et al., 2017) and is likely to be used more as a method to better understand chromatin regulatory mechanism that are involved in alcohol and other drugs of abuse.
2.1. Histone post-translational modifications
The most studied epigenetic process is histone post-translational modifications (PTMs) which involve covalent modifications to histone tails which change the charge of the nucleosome and allow for more permissive or restrictive transcription. These processes include acetylation (lysine [K] residues), methylation (arginine [R] and K residues), phosphorylation (serine [S] and threonine [T] residues), citrullination, ubiquitination, sumoylation, ADP ribosylation and proline isomerization (Kouzarides, 2007; Strahl and Allis, 2000). PTMs to histone tails are often combinatorial, but, broadly, histone acetylation (Bernstein et al., 2005) and phosphorylation are associated with permissive or open chromatin and upregulate gene transcription while sumolyation, ubiquitination, and citrullination are associated with repressive chromatin and downregulated gene transcription (Kouzarides, 2007; Strahl and Allis, 2000). Histone methylation is more complex, with H3K4 and H3K36 methylation being involved in activation and di- and tri-methylation at H3K9, H3K27, and H4K20 being involved in transcription repression (Boyer et al., 2006; Kouzarides, 2007; Strahl and Allis, 2000). Recent studies have identified other forms of histone PTMs that are enriched in neurons, such as serotonylation (Farrelly et al., 2019) which is a permissive marker of chromatin and interacts with H3K4me3. Another recent study has discovered that dopaminylation can occur at H3 glutamine 5 and is involved in cocaine-induced transcriptional plasticity in the brain (Lepack et al., 2020). Over 400 histone PTMs have been described, and the use of mass spectrometry is currently one of the best methods to discover these new variants and combinations (Huang et al., 2014). Mass spectrometry combined with sequencing technology can be used to better understand these new or previously undescribed marks and their impact on chromatin structure and gene expression (Sidoli et al., 2019). Finally, it is highly likely that there are still undiscovered histone PTMs and those may also have considerable impact to regulating chromatin structure and overall effects on gene transcription.
The most common area for histone PTMs to be associated with is within ±2kb of the gene transcriptional start site (TSS) (Barski et al., 2007; Bernstein et al., 2005; Kim et al., 2005; Wang et al., 2008). Differential association patterns of histone PTMs can be used to predict the function of the regulatory element, for example, high H3K4me1, H3K27ac, and low H3K4me3 is often used as a mark of enhancer regions whereas active promoters contain high H3K4me3 and low H3K4me1 (Heintzman et al., 2007). Other regulatory marks, such as bivalent promoters, contain both activating H3K4me3 and repressive H3K27me3 and are thought to be poised in pluripotent stem cells to allow for efficient differentiation (Mikkelsen et al., 2007). Further, H3K36me3 is often found at both coding and non-coding transcripts (Mikkelsen et al., 2007), but the absence of H3K4me3 is associated with only non-coding RNAs (Guttman et al., 2009). Adding further complexity, different transcript variants of histones can contribute to differential gene expression and presence of PTMs, and this process was recently identified to occur in the brain (Maze et al., 2015). The combinatorial control and identification of different histone PTMs is still an area of immense study, and chromatin immunoprecipitation sequencing (ChIP-seq) remains the gold-standard for probing these different mechanisms (Robertson et al., 2007). Updated methods such as “Cut and Run” that are based on a hybrid antibody-nuclease strategy vastly increase resolution and decrease background and could allow for deeper insight into epigenetic regulation by both histone PTMs and transcription factors (Skene and Henikoff, 2017). To date, there are some instances of single cell ChIP-seq (Grosselin et al., 2019); although this technique has yet to be widely employed, it remains a target goal in the field of epigenetics for better understanding of tissue and cell-type (e.g. brain) heterogeneity in both normal and disease states.
2.2. Readers, Writers, and Erasers of Histone Post-translational Modifications
Histone PTMs are regulated by three different protein family classifications based on function, readers, writers, and erasers. Readers identify histone PTMs and bind to them, writers add histone PTMs, and erasers remove histone PTMs. In some cases, reading motifs are combined with writing or erasing catalytic ability. These three classifications of proteins work in concert to tightly control gene transcription and are the target of continued drug development not only in psychiatric disorders but in other human diseases as well (Arrowsmith et al., 2012) (Figure 2).
Figure 2. Overview of histone acetylation and methylation writers, erasers, and readers.
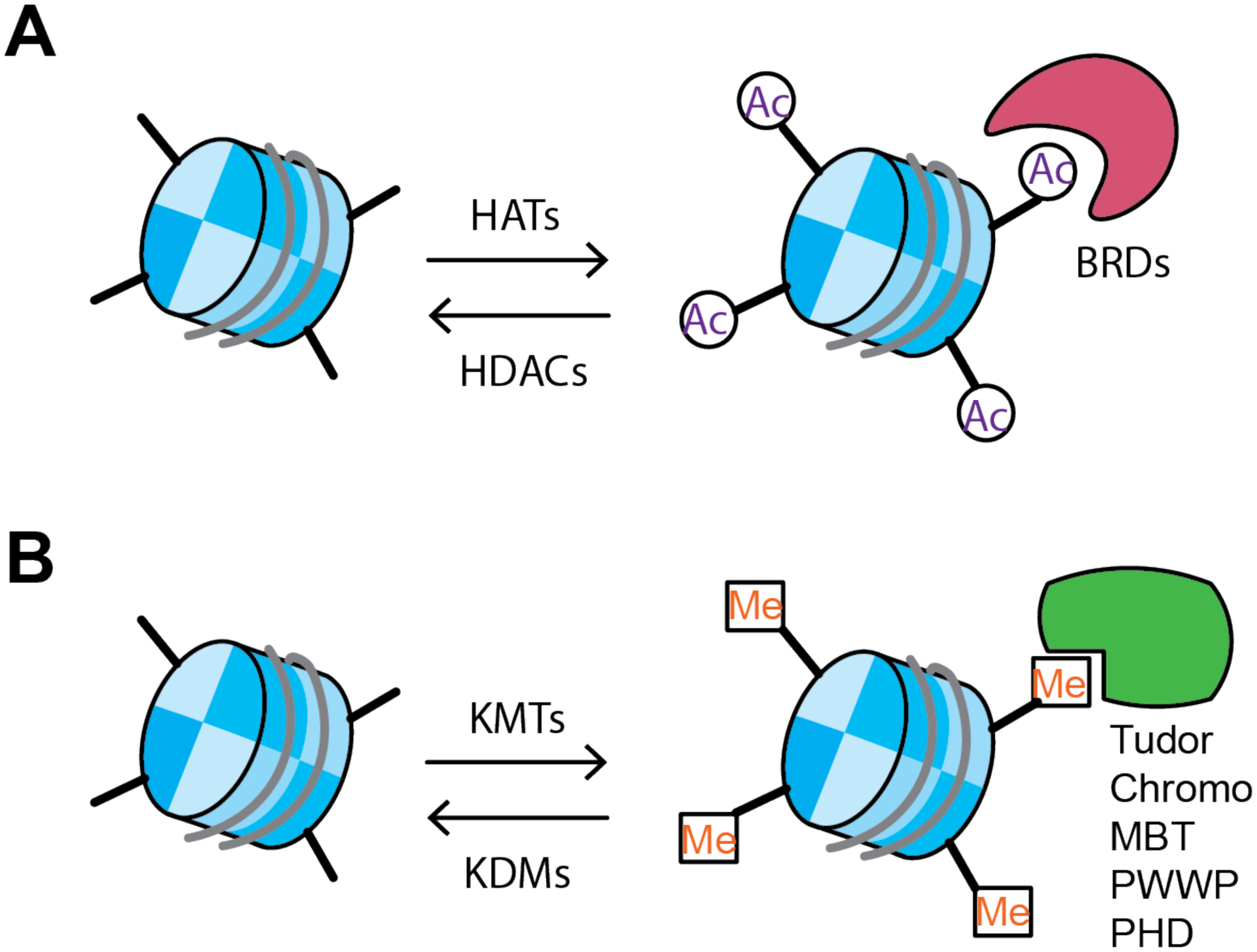
A) Histone acetylation is catalyzed by histone acetylation transferases (HATS), such as CBP, P300, and members of the Myst family, known as the writers. Histone acetylation is removed by histone deacetylases (HDACs / SIRTs) which are divided into four subgroups, Class I HDACs (HDAC1–3, 8), Class IIA HDACs (HDAC4, 5, 7, 9), Class IIB HDACs (HDAC6, 10), and Class III (SIRT1–7) and Class IV (HDAC11). HDACs have different cellular and tissue distribution and function and are known as the erasers. Histone acetylation is bound by readers, the most commonly studied are bromodomain containing proteins (BRDs) of the Bromo- and Extra-terminal domain (BET) family. B) Histone methylation is catalyzed by histone methyltransferases (KMTs) which tend to be highly selective for the specific lysine residue they methylate and there exist over 100 different KMTs. Histone methylation is removed by histone demethylases (KDMs) which are separated into two broad families, flavin adenine dinucleotide dependent amine oxidase and FE(II) and a α-ketoglutarate-dependent hydroxylase (commonly known as the JmJC KDMs). Histone methylation is bound by a diverse group of methylation readers that are defined by their structural motifs such as the Tudor, Chromo, malignant brain tumor (MBT), Pro-Trp-Trp-Pro motif (PWWP) and plant homeodomain (PHD) families. Similar to KMTs, these proteins tend to bind to specific lysine residues with a defined number of methylation (e.g. mono- vs di-).
2.2.1. Readers of Histone Post-translational Modifications:
Readers bind histone PTMs and are involved in recruitment of transcription factors or other regulatory proteins to facilitate gene transcription or repression. They also act to recruit other histone PTM modifying proteins, such as writers or erasers to modulate gene transcription. One of the most important functional readouts is that they are involved in chromatin remodeling, by mobilizing nucleosomes in and out of chromatin structure (Musselman et al., 2012). Readers can be divided into several subdivisions based on structural motifs: bromodomain-containing proteins, Tudor domains, chromodomains, MBT domains, PWWP domains, and PHD-containing proteins among others (Arrowsmith et al., 2012). We will briefly describe some of the most well-studied readers. Although orphan readers and their reader families will likely continue to be discovered and described, they are outside the scope of this review.
2.2.1.1. Bromodomain-containing proteins:
Bromodomain-containing proteins, specifically the bromodomain and extra terminal domain family (BET) family, are a group of 61 proteins with a ~110 amino acid protein domain that recognize acetylated lysine residues (Arrowsmith et al., 2012; Winston and Allis, 1999). The BET family recognizes histone lysine acetylation, is involved in transcription activation, and comprises of BRD2, BRD3, BRD4, and BRDT. BRD proteins facilitate transcription through a number of different mechanisms, including binding to positive elongation factor (P-TEFb) that leads to elongation and RNAPII transcription of mRNA (Jang et al., 2005) and fine tuning the transcription of both coding and noncoding enhancer RNAs (Kanno et al., 2014). Small molecule inhibitors for bromodomain-containing proteins have shown promise for treating cancer (Filippakopoulos et al., 2010). Cocaine self-administration increases BRD4 expression but not BRD2 and BRD3 expression, and the use of the BET inhibitor JQ1 reduced cocaine condition place preference in mice (Sartor et al., 2015). This suggests that BET family proteins may also play a role in regulating addiction and AUD, although to date this is still an emerging line of study.
2.2.1.2. Tudor domain proteins:
Tudor domain-containing proteins contain approximately sixty amino acids with 4–5 antiparallel β-strands that form a barrel like structure. Tudor domain-containing proteins act as histone methylation readers and have been found to recognize H3K4me3, H3K9me2, H3K36me3, and H4K20me3 (Lu and Wang, 2013). There are approximately 30 proteins in this family, including, JMJD2, 53BP1, SGF29, Spindlin1, UHRF1, PHF1, OHF19, LBR, and TDRD3 (which recognizes methylated arginine residues) (Lu and Wang, 2013). Functional activities of this family are involved in DNA methylation, transcription repression and activation, non-homologous end joining, DNA damage detection and repair, and rRNA gene expression (Lu and Wang, 2013). Importantly, the UHRF1, which binds H3K9me3, has been shown to be important in maintenance of DNA methylation by interactions with DNMT1 (Rothbart et al., 2012) and epigenetic inheritance of DNA methylation (Rothbart et al., 2013). Since DNA methylation is well described as being involved in addiction, this suggests that Tudor domain-containing proteins may also be involved in regulating addiction and AUD behaviors.
2.2.1.3. Chromodomain proteins:
Chromatin organization modifier domain (chromodomain)-containing proteins were historically associated with chromatin silencing (James and Elgin, 1986). There are approximately 55 chromodomain containing proteins identified in humans (Blus et al., 2011). The canonical motif for chromodomains is based off the HP1 which is a fold consisting of three anti-parallel β-strands and an α-helix (Ball et al., 1997). Chromodomain containing proteins are categorized into two groups: canonical (based on HP1’s structure), which includes polycomb proteins [Cbx1–9], Su(Var) 3–9, CMT1–3, and CDY among others, and noncanonical, which includes CHD1–8, RBBP1, HRP1 and 3, among others (Blus et al., 2011). HP1 and polycomb proteins recognize H3K9me and H3K27me via the ARKS/T amino acid motif (Blus et al., 2011; Kaustov et al., 2011), while other members of the chromodomain family recognize other forms histone methylation (Blus et al., 2011). The noncanonical chromodomain proteins are based off chromo-ATPase/Helicase-DNA-binding (CHD) proteins which contain double chromodomains in the N-terminal, a SNF2-type helicase, and are involved in chromatin remodeling (Woodage et al., 1997). These proteins are involved in a number of different processes, including transcription elongation (Sims et al., 2007) and neural stem cell formation (Bajpai et al., 2010). CHD7 uniquely recognizes H3K4me1 at enhancers (Bajpai et al., 2010). CHD8 was identified in large-scale exome sequencing as being an autism candidate gene (O’Roak et al., 2012), suggesting that chromo-domain proteins play a role in regulating brain activity and may be involved in critical periods of brain development.
2.2.1.4. MBT domain proteins:
The malignant brain tumor (MBT) domain-containing proteins recognize mono- and di-methylated lysine (Bonasio et al., 2010; Li et al., 2007). The MBT family contains ~30 proteins and is defined by a two structural motifs, a 30–50 n-terminal formed by one alpha and one 310 helix and a longer c-terminal motif containing five β strands arranged in a barrel topology, a 310 helical turn connecting β3 and β4, an α-helix, and a final sixth β-strand (Wang et al., 2003). Proteins included in this family are L3MBTL1–4, SCMH1, and SCML2 (Bonasio et al., 2010). MBT proteins are involved in transcription repression (Bonasio et al., 2010).
2.2.1.5. PWWP domains.
The PWWP domain is a 80–130 amino acid protein that contains Pro-Trp-Trp-Pro and contains two motifs, a β-barrel composed of 5 antiparallel β-strands and C-terminal helix which can contain two to six α helices (Hong Wu et al., 2011). Proteins in this family include DNA methyltransferase 3A and B, Pdp1–2, BRPF1–3, among others (Rona et al., 2016). The PWWP domain allows members of this family to bind non-specifically to DNA (Qiu et al., 2002; Rona et al., 2016) and bind H3K36me3 (Vermeulen et al., 2010), with the exception of Pdp1 (which binds H3K20me1) (Wang et al., 2009). PWWP domain containing proteins are involved in a number of biological processes including DNA repair, transcriptional regulation, DNA methylation and histone modification (Qin and Min, 2014; Rona et al., 2016).
2.2.1.6. PHD-containing proteins:
The plant homeodomain (PhD) zinc finger proteins read DNA sequences, and this is modulated by the methylation state of both lysine and arginine residues (Li et al., 2007; Sanchez and Zhou, 2011). The PHD finger is a 50–80 amino acids motif that consists of two-strand anti-parallel β-sheet and an α-helix (which is not present in all PHD family members) that has two zinc atoms anchored to the Cys4-His-Cys3 motif in a cross-brace topology (Li et al., 2007). PhD proteins recognize H3K4 methylation state (Li et al., 2007; Sanchez and Zhou, 2011), H3R2 (Chignola et al., 2009) and in one case H3K14 (Sanchez and Zhou, 2011). PhD proteins represent some of the best examples from the “reader” class of proteins in combinatorial control of transcription owing to their ability to read multiple different post-translational modifications at once (Sanchez and Zhou, 2011).
2.2.2. Writers of Histone Post-translational Modifications:
2.2.2.1. Histone Acetyltransferases (HATs):
HATs are enzymes that acetylate lysine residues on histones by converting Acetyl-CoA to form ε-N-acetyl lysine. Histone acetylation can occur on H2A (K5 and K9), H2B (K5, K12, K15, K16, K20, and K120), H3 (K4, K9, K14, K18, K23, K27, K36 and K56). Most HATs are located in the nucleus (Type A) while Type B HATS are located in the cytoplasm (Lee and Workman, 2007; Roth et al., 2001). The three most studied families of HATs are the MYST family (consisting of MOZ, YBF2/SAS3, SAS2, Tip60), GNAT, and EP300 (consisting of CREB Binding Protein [CBP] and P300) (Arrowsmith et al., 2012; Roth et al., 2001). Most HATs contain lysine acetylation recognition domain, the GNAT type A motif, (R/Q-X-X-G-X-G/A) is found in both in the GNAT and MYST families while the EP300 family contains multiple acetylation recognition motifs including a bromodomain (Roth et al., 2001). HATs demonstrate a degree of histone lysine specificity, for example Gcn5 and PCAF demonstrate selectivity for H3 over H4 while P300 and CBP broadly acetylate all four histone isoforms (Roth et al., 2001). Downstream, acetyl-CoA is produced by two enzymes, acetate-dependent acetyl-CoA synthetase 2 (ACSS2) and citrate-dependent ATP-citrate lyase (ACL); a recent report has demonstrated that ACSS2 is highly expressed in the hippocampus, binds chromatin, and is involved in hippocampal memory consolidation (Mews et al., 2017). HATs binding to DNA and chromatin can also be used as markers of active transcription and for the identification of unique regulatory mechanisms, for example, P300 associates with active super enhancer regions that drive cellular senescence (Sen et al., 2019). HATs are known to be involved in synaptic plasticity and behaviors, for example P300 is required for generating and maintaining long term memories (Chatterjee et al., 2013; Oliveira et al., 2011).
2.2.2.2. Histone Lysine Methyltransferases (KMTs):
Histone lysine methylation, in contrast to histone acetylation, has a more diverse regulatory role on the control of transcription. Histone lysine methylation can be mono-, di-, or tri-methylated, and the number of different methyl groups present can produce either an activating or repressive effect on gene transcription. Methylation occurs on at H1B (K26), H3 (K4, K9, K23, K27, K36, K45, K63, and K79), and H4 (K12 and K59) and is catalyzed by proper orientation of the lysine within a binding pocket and requires S-Adenosyl methionine as a co-factor (Dillon et al., 2005). In the human genome, homology predictions suggest that there are over 100 lysine methyltransferases, although not all of these proteins have been shown to methylate histones (Dillon et al., 2005; Husmann and Gozani, 2019). These proteins are identified by their possession of either a SET domain or a seven-beta-strand domain (Dillon et al., 2005; Husmann and Gozani, 2019). In contrast to HATs, KMTs tend to be highly selective and only methylate a specific lysine residue (for example, EZH2 trimethylates H3K27). Although there is redundancy by different KMTs that methylate the same residue (Dillon et al., 2005; Husmann and Gozani, 2019), as is the case for H3K36me2, where NSD1–3 and ASH1L both participate to catalyze H3K36me2 from H3K36me1. The more commonly studied class, SET domain containing, can be broken down into approximately 7 families: SUV39, SET1, SET2, RIZ, SYMD, EZ, SUV-4–20, and orphans (e.g. SET7/9 and SET8) (Dillon et al., 2005). The SET domain consists of 130 amino acids, and variations within this region confer lysine specificity (Dillon et al., 2005). Certain members of the SET family are involved in repressive transcription, such as G9A, which synthesizes H3K9me1 and H3K9me2, or EZH2, which catalyzes H3K27me3. G9A has been shown to be involved in cognition and adaptive behavior (Schaefer et al., 2009), cocaine addiction (Maze et al., 2010), and chronic pain (Laumet et al., 2015).
2.2.3. Erasers of Histone Post-translational Modifications:
2.2.3.1. Histone deacetylases (HDACs):
There are four classes of histone deacetylases, Class I HDACs (HDACs 1, 2, 3 and 8), are found almost exclusively in the nucleus, and are characterized by homology in their catalytic site. Class II HDACs (4, 5, 6, 7, 9 and 10) are found in both the nucleus and the cytoplasm and are characterized by homology in their C-terminal catalytic and N-terminal regulatory domains. HDAC11 has similar homology in the catalytic region of both Class I and Class II HDACs but classified as class IV HDAC (Marks et al., 2003). Class III HDACs are the sirtuin family, which are conserved nicotinamide adenine dinucleotide-dependent deacetylases that play an important role in cellular senescence and aging (Satoh et al., 2017). The sirtuin family has 7 members that often are involved in gene repression, although they play an important role for deacetylating non-histone proteins that are located in both the nucleus and the cytoplasm (Yamamoto et al., 2007). The removal of acetylation from either histones or other molecular substrates requires a Zn2+ in the case of class I & III or the presence of NAD+ (Finnin et al., 1999; Yamamoto et al., 2007). HDACs inhibitors are among the few drugs that target epigenetic proteins that have been successfully translated into the clinic. Vorinostat (also known as suberoyl anilide hydroxamic acid, SAHA) is used to treat cutaneous T-cell lymphoma (Kavanaugh et al., 2010). HDACs and HDAC inhibitors have been shown to be involved in learning, memory, and addiction (Guan et al., 2009; Krishnan et al., 2014; Kumar et al., 2005).
2.2.3.2. Lysine demethylases (KDMs):
Histone lysine demethylases are divided into two families, the flavin adenine dinucleotide dependent amine oxidase (e.g. LSD1) (Shi et al., 2004) and FE(II) and a α-ketoglutarate-dependent hydroxylase (also known as JmjC or JHDM family) (Tsukada et al., 2006). There are 27 different JmjC members, 15 of which have been shown to have histone demethylase activity (Tsukada et al., 2006). Similar to KMTs, KDMs have significant specificity for lysine they demethylate (Hyun et al., 2017). LSD1 and the JARID1 family demethylate H3K4me1 and H3K4me2 (Christensen et al., 2007; Shi et al., 2004). JARID1C mutations have been shown to cause X-linked mental retardation, implying an important role for controlling normal brain development (Jensen et al., 2005). PHF8 recognizes H3K4me3 via its PHD domain and removes H3K9me1 and H3K9me2 (Loenarz et al., 2010). Interestingly, H3K4me3 (a mark of active transcription) and H3K9me2 (a repressive mark) are seen to be mutually exclusive (Barski et al., 2007; Wang et al., 2008). KDM3 and KDM4 also demethylate H3K9 (Hyun et al., 2017). KDM6A-C is involved in demethylating H3K27, which largely functions to unsilence chromatin, particularly in critical periods of development (Agger et al., 2007; De Santa et al., 2007). JHDM1 and JHDM3 are responsible for demethylating H3K36me (Hyun et al., 2017). In addition to the KDMs mentioned above, other KDMs are still being discovered and classified. A role for KDMs in regulating behavior and synaptic plasticity has been described (Wang et al., 2015).
3. Histone Acetylation Mechanisms in Alcohol Use and Dependence
Many years of research have established that histone acetylation is important in regulating the effects of alcohol exposure on gene expression (Tables 1–4).
Table 1.
Effects of acute alcohol exposure on histone acetylation mechanism.
| Study | Species | Exposure | Region | Genes | Mark(s) | Findings |
|---|---|---|---|---|---|---|
| (Pandey et al., 2008a) | Sprague Dawley rats | 1g/kg i.p. | CeA, MeA | CBP, NPY | H3K9/14ac, H4K8ac | - Decreased HDAC activity |
| - Increased H3K9ac and H4K8ac | ||||||
| - Decreased anxiety-like behavior | ||||||
| (Finegersh and Homanics, 2014) | C57Bl/6 mice | 3 g/kg i.p. | Cortex, Hipp. | Gad1, Hdac2, Hdac3 Mt1, | H3K9/14ac, H3K4me3, H3K27me3 | - Increased global H3K4me3 in cortex and hippocampus |
| Mt2, Egr1 | - Changes at various promoters | |||||
| (López-Moreno et al., 2015) | Wistar rats | 3 g/kg i.g. | Amg, PFC | Hdacs | - Increased Hdac1, Hdac2, and Hdac5 in amygdala after one binge | |
| (López-Moreno et al., 2015) | Humans | Blood | Hdacs | - All HDACs elevated except HDAC10 and HDAC9 | ||
| (Sakharkar et al., 2012) | Sprague Dawley rats | 1 g/kg i.p & 1 or 2 g/kg next day | CeA, MeA | NPY | H3K9ac, H4K8ac | - Tolerance prevents increased histone acetylation, anxiolysis, and NPY expression, can be reversed by TSA treatment |
Abbreviations: i.p. = intraperitoneal, i.g. = intragastric, CeA = central nucleus of the amygdala, MeA = medial nucleus of the amygdala, hipp = hippocampus, PFC = prefrontal cortex.
Table 4.
Effects of fetal alcohol exposure on histone acetylation mechanism.
| Study | Species | Exp. | Region | Genes | Mark(s) | Findings |
|---|---|---|---|---|---|---|
| (Govorko et al., 2012) | Sprague Dawley rats | Liquid diet | Arcuate area / Hypo. | Cbp, Hdac2 | H3K9ac | Decrease of Cbp and increase Hdac2, decreased H3K9ac |
| (W. Guo et al., 2011) | Long-Evans | Vapor | Cereb. | CBP | H3K914ac, H3K23ac H4ac | Decreased CBP, H3K9/14ac, H3K23ac, and H4ac |
| (Subbanna et al., 2015) | C57BL/6 | i.p. 2.5 g/kg | NeocortexHipp | Cb1 | H4K8ac | Increased H4K8ac at Cb1 gene |
| (Veazey et al., 2015) | C57BL/6 | i.p. 2.9 g/kg | Cortex | 24 genes | H3K9ac | Increased H3K9ac associated with promoters of growth factor genes in offspring effected by craniofacial deformities |
Abbreviations, Exp. = exposure, i.p. = intraperitoneal, vapor = ethanol vapor, cereb = cerebellum, hipp = hippocampus
3.1. Acute Ethanol Exposure:
An early study examining the role of epigenetic marks after alcohol exposure demonstrated that acute ethanol increased H3 and H4 acetylation, increased CREB (cAMP-responsive element binding) and CBP in the central nucleus of the amygdala (CeA), and decreased HDAC activity in the amygdala which was associated with the anxiolytic effects of ethanol (Pandey et al., 2008a). These effects were associated with increases of neuropeptide Y (NPY) which has long been known to be involved in anxiety and alcohol use disorders (Pandey, 2003; Zhang et al., 2010). In the cerebral cortex, acute ethanol injection in adult C57BL/6J mice decreases expression of Hdac2 and Hdac11 and decreases H3K9/14ac associated with Hdac2 but not Hdac11 promoters but does not lead to global changes in H3K9/14ac (Finegersh and Homanics, 2014). Similar decreases in Hdac2 were not observed in the hippocampus, suggesting brain region specific regulation of HDAC expression by acute ethanol (Finegersh and Homanics, 2014). Another group evaluated self-administered binge ethanol consumption and found that Hdac2 was upregulated after a single binge session in the amygdala, suggesting that the method of ethanol exposure is important in determining regulation of HDACs by ethanol (López-Moreno et al., 2015). In humans, an acute dose of ethanol broadly increases all 11 HDACs except HDAC9 and HDAC10 in the blood (López-Moreno et al., 2015), suggesting tissue and species specific regulation of HDACs by ethanol. Until recently, decreased HDAC expression was believed to be the primary mechanism driving increased histone acetylation in response to acute ethanol, but a recent study has demonstrated that acute ethanol can also act through a metabolic pathway in which ethanol is broken down through a three step pathway resulting in increased acetate that is then available to be catalytically added to histones by chromatin-bound acetyl-CoA synthetase (ACSS2). This has relevance to the development of AUD, as a knockdown of ACSS2 prevents ethanol conditioned place preference in mice (Mews et al., 2019). Acute ethanol clearly causes changes in histone acetylation, and these changes are likely to occur through a complex interplay of HATs and HDACs.
3.2. Development of Tolerance to Effects of Ethanol:
Tolerance to the subjective effects of ethanol have been shown to be involved in the development of AUD (Schuckit, 1996). Ethanol tolerance can occur after either a single, continuous, and/or repeated exposure and is usually described as a loss of sensitivity to the hypnotic (such as the commonly used loss of right reflex assay) or behavioral effects of ethanol (Berger et al., 2004). Multiple different studies have demonstrated that there is a strong genetic component that occurs in rats (Kurtz et al., 1996), mice (Crabbe et al., 2012), and humans (Heath et al., 1999). Using a rapid tolerance model, it was found that a second acute dose of ethanol 24 hours after first dose of ethanol prevents anxiolytic effects, decreases in HDAC activity, and increases in H3K9, H4K8 acetylation and NPY expression in the CeA and MeA (Sakharkar et al., 2012), suggesting a strong epigenetic component to the development of rapid tolerance as well. Treatment with TSA can reverse the rapid tolerance phenotype, suggesting that HDACs are important from the initial stages of alcohol addiction where alcohol is rewarding to the progression to alcohol dependence where alcohol acts to relieve the negative affect induced by continued cycles of alcohol withdrawal (Koob and Volkow, 2016; Pandey et al., 2017). Interestingly, while using a model of ethanol behavioral induction sensitization in support of incentive salience sensitization theory, repeated exposure to ethanol in DBA/2J mice demonstrates that acute ethanol challenge after 10 days of ethanol exposure decreases HDAC2 activity in the nucleus of the striatum but increases it in to the cytosol; conversely, in the PFC, HDAC activity is increased in the nucleus but decreased in the cytosol of sensitized mice (Botia et al., 2012). The striatum is heavily involved in the rewarding properties of ethanol, whereas the PFC is involved in executive decisions about the rewards of ethanol (Koob and Volkow, 2016), suggesting that epigenetic reprogramming in these areas through a histone acetylation mechanism can be used to help delineate different responders to ethanol reward. Other studies in drosophila have suggested that CBP is involved in the development of tolerance, as mutating nejire, a CBP homolog, blocks increased histone acetylation at alcohol responsive genes, resulting in lack of induction of at these alcohol tolerance genes and prevention of alcohol tolerance (Ghezzi et al., 2017).
3.3. Chronic Ethanol Exposure:
Chronic ethanol and withdrawal, in general, decreases histone acetylation. One of the earliest studies on epigenetics and chronic ethanol exposure and withdrawal observed an increased anxiety-like phenotype, that corresponded with decreased H3K9 and H4K8 acetylation, decreased CBP expression, and decreased NPY expression (Pandey et al., 2008a). The use of the broad spectrum histone deacetylase inhibitor Trichostatin A (TSA) prevented changes in both increased anxiety-like behavior induced by withdrawal and decreases in H3K9 and H4K8 acetylation and NPY, BDNF and Arc expression (Pandey et al., 2008a; You et al., 2014). Interestingly, TSA treatment was able to normalize deficits in dendritic spines in the CeA and MeA during ethanol withdrawal after chronic exposure in rats (You et al., 2014). Similar increases in HDAC2 and decreases in H3K9ac were found in the hippocampus and could be reversed using SAHA. This drug treatment was able to attenuate depression-like behaviors related to ethanol withdrawal (Chen, Zhang et al., 2019).
Other models of chronic alcohol dependence have also observed changes in histone acetylation and HDACs. For example, a recent study by Bohnsack et al screened all 11 HDACs and discovered that both HDAC2 and HDAC3 were upregulated in the cortex, and this corresponded with decreases in H3Kac, H3K9ac and H3K14ac (Bohnsack et al., 2018). Similar to other studies, inhibition with TSA prevented anxiety-like behavior and alcohol consumption (Bohnsack et al., 2018). Hdac3 was also upregulated by alcohol withdrawal via vapor in a mice in a different study (Hashimoto et al., 2017). Chronic ethanol has long been known to cause decreases in GABAAR receptor function that are associated with severe withdrawal symptoms, including seizures. Most acute withdrawal symptoms are treated by the use of benzodiazepines in the clinic (Kumar et al., 2009). Interestingly, several studies have demonstrated that the use of TSA or SAHA can prevent changes in GABAAR hypofunction by increasing the most prevalent GABAAR subunit, GABAAR α1 (Arora et al., 2013; Bohnsack et al., 2018; Hughes et al., 2019; You et al., 2018).
Other studies using a strain of rat with high alcohol consumption (P rats) found that P rats have increased HDAC2 but not HDAC1, 3, 4, 5, and HDAC6 in the CeA and medial nucleus of the amygdala (MeA) and this corresponded with decreased H3K9ac in both amygdala subregions (Moonat et al., 2013). ChIP assays revealed decreases in H3K9/14 acetylation associated with the Arc and Bdnf-IV promoter (Moonat et al., 2013) that correlated with decreased Arc and BDNF protein levels and changes in spine density (Moonat et al., 2011). Interestingly, these deficits could be reversed by acute injection of ethanol (Moonat et al., 2011), by the infusion of HDAC2 siRNA directly into the CeA (Moonat et al., 2013), or by the use of TSA (Sakharkar et al., 2014). Arc, activity-regulated cytoskeleton-associated protein, is known to be a regulator of synaptic plasticity (Shepherd and Bear, 2011) and is involved in alcohol dependence (Pandey et al., 2008b; You et al., 2014). Another study evaluating alcohol self-administration in mice using a drinking in the dark two bottle choice paradigm found that TSA, SAHA, and another HDAC inhibitor that targets type I HDACs, MS275, prevents alcohol intake with no effect on saccharin consumption (Warnault et al., 2013). A study using rats in an operant paradigm (fixed ratio 3) found that administration of SAHA prevented alcohol seeking but not sucrose seeking (Warnault et al., 2013). In both mouse and rat NAc, H4 acetylation was decreased after alcohol consumption (Warnault et al., 2013).
Conversely, HATs have been shown to be downregulated in the amygdala during withdrawal after chronic ethanol exposure (Pandey et al., 2008a). One important HAT activity that is regulated by chronic ethanol consumption is the CREB pathway, which involves the HATs P300 and CBP. Phosphorylation of CREB is required for the recruitment of CBP (Chrivia et al., 1993) and CBP’s role in learning and memory is due to its HAT activity (Korzus et al., 2004). Ethanol withdrawal after chronic ethanol exposure causes a decrease in CREB phosphorylation and CBP levels in the CeA and MeA (Pandey, 2003; Pandey et al., 2008a, 2003). Inhibition of CREB in the CeA induces anxiety-like behavior and causes escalated drinking in NP rats, whereas activation of CREB in the CeA prevents anxiety-like and alcohol drinking behaviors in P rats (Pandey,Zhang, Roy,& Xu 2005). Further, haplodeficient CREB mice (+/−) have decreased NPY and BDNF expression and also consume more alcohol but not sucrose (Pandey,Roy, Zhang,& Xu, 2004). Another study in alcohol dependent rats using a vapor paradigm found decreased P300 and Myst3 in the dmPFC (Barbier et al., 2017), while a mouse study also using vapor found decreased Hat1 and Atf1 (Hashimoto et al., 2017). Taken together, these studies suggest that HAT expression and activity is modulated by ethanol exposure and plays an important role in alcohol related behaviors. Chronic studies from multiple laboratories and in multiple brain regions suggest that HDACs are elevated after chronic ethanol exposure and that this drive decreased histone acetylation and gene expression that regulate neuroplasticity or neurotransmission. Further treatment with HDAC inhibitors has been shown to prevent drinking and anxiety-like behavior, suggesting these treatments may be useful in the treatment of AUD (Pandey et al., 2017).
3.4. Adolescent Alcohol Exposure:
Adolescent alcohol exposure causes long lasting effects that persist until adulthood, and these changes have been shown to regulated by histone acetylation mechanisms (Kyzar,Floreani,Teppen, &Pandey, 2016). Exposure to ethanol during adolescence increases the risk 5–7 times of developing an alcohol use disorder later in life (Viner and Taylor, 2007) and also increases the risk of comorbid anxiety and stress disorders (Keyes et al., 2012; Wells et al., 2004). One study examined the role of HDACs after adolescent alcohol exposure and withdrawal during adolescence and found increases in HDAC activity in the amygdala which corresponded to an increase of HDAC2 and HDAC4 (Pandey et al., 2015). There was also a subsequent decrease in H3K9ac in the CeA and MeA and an anxiogenic phenotype similar to what is seen in adult rats (Pandey et al., 2015). This anxiogenic phenotype persists until adulthood even without further ethanol exposure and may be driven by an increase in HDAC2 and decreased H3K9ac (Pandey et al., 2015). Similar to what has been observed in previous studies using chronic ethanol in adult animals, both Arc and BDNF are also decreased in adulthood after adolescent alcohol exposure (Moonat et al., 2013, 2011; Pandey et al., 2015, 2008b). Similar increases in HDAC2 and decreases H3K9ac were also observed in the hippocampus (Sakharkar et al., 2016). This phenotype can be prevented by the administration of TSA reverting both the anxiogenic phenotype and an escalated drinking phenotype (Pandey et al., 2015) and preventing decreased neurogenesis in the hippocampus (Sakharkar et al., 2016). Other studies have used non-epigenetic targeting drugs, such as the cholinesterase inhibitor, to prevent increases in H3K27ac at the promoter of the Fmr1 gene in the hippocampus after adolescent alcohol exposure (Mulholland et al., 2018). When evaluating the role HATs after AIE, it was found that both Cbp, P300 and Creb1 were all downregulated in the CeA and MeA after adolescent alcohol exposure in adulthood (Zhang et al., 2018) and probably contributed to decreased H3K9ac observed in other studies (Pandey et al., 2015). Interestingly, deficits in the expression of Cbp, P300, and Creb1 appear to be regulated by decreases in histone acetylation at the promoters of these genes in the adult amygdala after adolescent ethanol exposure (Zhang et al.,2018). Taken together, this suggests that both withdrawal from repeated ethanol exposure in adolescence leads to an upregulation of HDACs, leading to decreased histone acetylation and compensatory HAT and synaptic plasticity gene expression, and that many of these effects persist until adulthood, where they drive alcohol dependent related behaviors such as anxiety and alcohol consumption. Similar to findings in tolerance and chronic ethanol adult animal models, the use of histone deacetylase inhibitors can reverse many of these phenotypes in adulthood after adolescent alcohol, which may imply their use in early-onset alcohol use disorders in humans.
3.5. Fetal Alcohol Exposure:
Fetal Alcohol Spectrum Disorder (FASD) disrupts normal fetal development, and several studies in rodents have shown that this can be mediated through epigenetic mechanisms (Haycock, 2009). Exposure to ethanol from gestational day 7–21 via liquid ethanol diet caused a decrease in H3K9ac in the arcuate area of the hypothalamus, a decrease of Cbp mRNA, and an increase in Hdac2 mRNA in the offspring of Sprague Dawley rats (Govorko et al., 2012). Another study using a model that mimics human third trimester fetal development in rats [postnatal day (PND) 2–12] found that exposure to ethanol vapor for 4hrs a day leads to a decrease in CBP protein and H3K9/14ac, H3K23ac, and H4 acetylation from PND 2 to 10 in the cerebellum of Long-Evans rats (W. Guo et al., 2011). In contrast, other studies using C57BL/6J mice found that injections at pnd7 increased H4K8ac at cannabinoid receptor type 1 (Cb1) gene in the neocortex and hippocampus, and that Cb1 activity was increased after ethanol exposure (Subbanna et al., 2015). Knockout of Cb1 prevented ethanol-induced decreases in spontaneous alternation performance deficits in adult mice and social deficits in mice (Subbanna et al., 2015). Another study evaluating growth factor genes at gd17 after ethanol injection at gd7 found that H3K9ac was increased at Ascl1, Dlx1, Dlx2, HoxA6, HoxA7, Msx2, Vdr, and decreased at Smarca2 (Veazey et al., 2015). A human study that evaluated multiple different histone acetylation marks over different developmental time points found changes in various acetylation marks at these different time points, which could be informative to the understanding of temporal regulation of histone acetylation in fetal alcohol spectrum disorder (Jarmasz et al., 2019). Taken together these studies indicate that histone acetylation may play a role in fetal alcohol exposure, but changes depend on brain region, ethanol exposure paradigm, gene evaluated, and age of offspring evaluated.
In summary, histone acetylation mechanisms clearly play a role in effects produced by acute, developmental, and chronic alcohol exposure. Currently, the role of histone acetylation readers, such as the BET family, is understudied in alcohol use disorders. Future studies should attempt to determine if changes also occur in this class of proteins as well. Additional research is needed to determine if these also play a critical role in regulating relapse. Further, there is a wide body of research that demonstrates the role of histone acetylation in many different brain regions, suggesting that interactions between alcohol and other psychiatric or neurological disorders may also be involved. This has been well described for anxiety-like behavior in animals. Finally, additional human studies are needed to validate findings from rodent models and establish a causal role for histone acetylation marks of single or multiple genes in given brain circuitry in driving the pathophysiology of AUD (Figure 3).
Figure 3. Effects of alcohol exposure on histone acetylation.
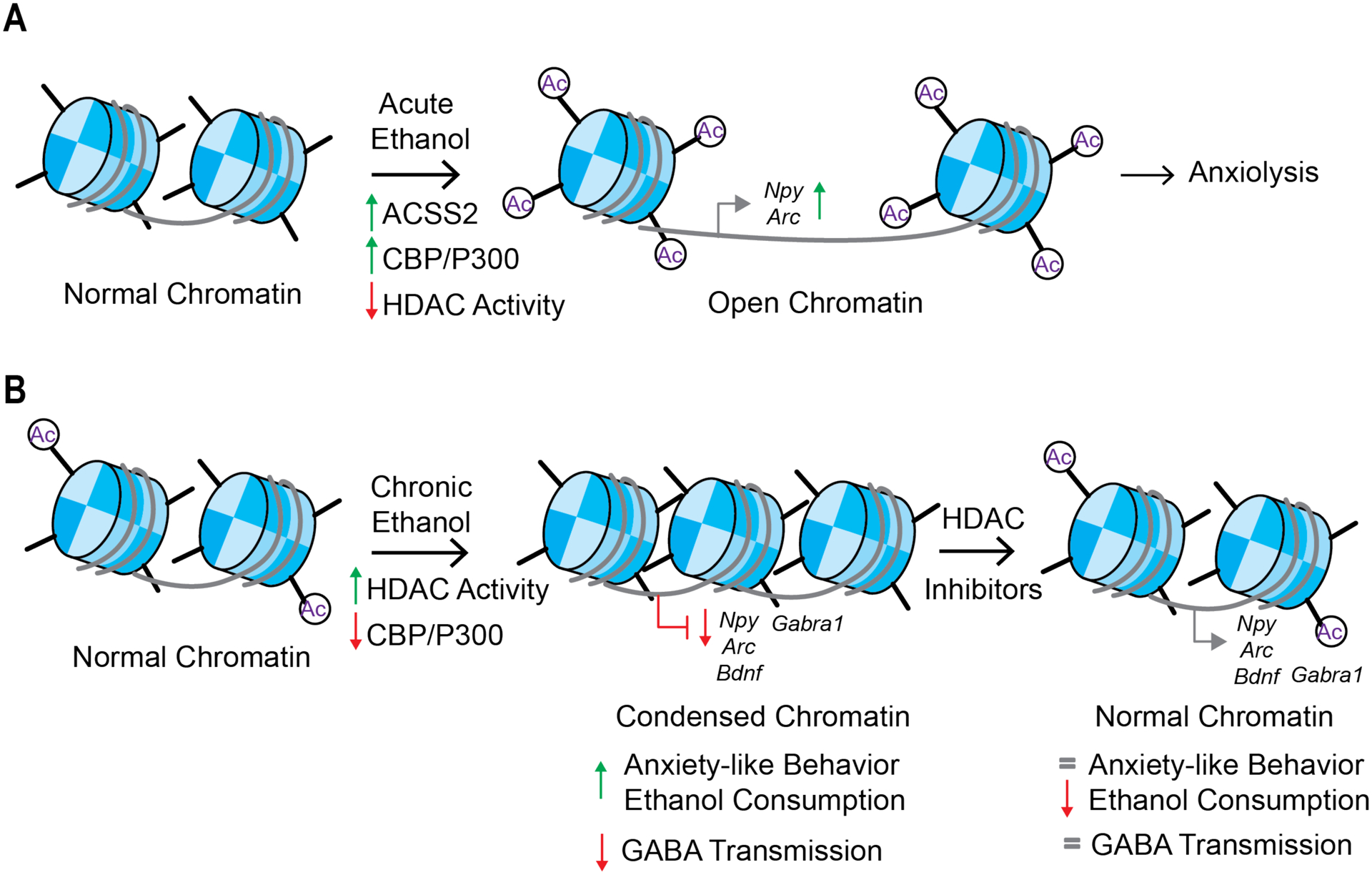
A) Acute ethanol causes an increase in ACSS2 activity which is responsible for converting acetate to acetyl-CoA, a perquisite for histone acetylation, as well as increasing expression of the histone acetyltransferases CBP and P300 and decreasing histone deacetylase activity (HDAC). This results in an open chromatin structure which increases neuropeptide y (Npy) and activity-regulated cytoskeleton-associated protein (Arc), which could explain some of the anxiolytic effects of ethanol. B) Chronic alcohol and withdrawal increases HDAC activity and decreases CBP and P300, leading to a condensed chromatin structure. This corresponds to a decrease of Npy, Arc, brain derived neurotrophic factor (BDNF) and gamma aminobutyric acid alpha1 subunit (Gabra1). Functionally, in prefrontal cortex pyramidal neurons and the VTA, there is a decrease in GABA transmission and behaviorally both anxiety-like behavior and alcohol consumption are elevated. Treatment with HDAC inhibitors, such as Trichostatin A (TSA) restores chromatin and gene expression to a normal level, decreases ethanol consumption and prevents anxiety-like behavior and decreases GABA transmission. Some of these effects are observed when alcohol is given during key stages of development such as prenatal or adolescence.
4. Histone Methylation Mechanisms in Alcohol Abuse and Dependence
Studies in histone methylation in the context of alcohol abuse and dependence have begun to catch up to histone acetylation, leading to a clearer picture of how different histone PTMs interact to drive gene expression. Unlike histone acetylation, histone methylation can be both repressive (e.g. H3K27me3) or activating (e.g. H3K4me3), and the readers, writers, and erasers tend to have more specificity to a certain residue or methylation state. Further, histone methylation readers and writers show a strong degree of selectivity, which may make them a more appropriate target for treatment of AUD (Tables 5–7).
Table 5.
Effects of chronic alcohol exposure on histone methylation mechanism.
| Study | Species | Exp. | Region | Genes | Mark(s) | Findings |
|---|---|---|---|---|---|---|
| (Barbier et al., 2017) | C57Bl/6 | Vapor | dmPFC | HTS | H3K9me1 | - Decreased Prdm2 and H3K9me1 |
| - Knockdown of PRDM2 increases alcohol self-administration | ||||||
| (Johnstone et al., 2019) | Wistar rats | Vapor | NAc, PFC, DS, Cereb. | HTS | H3K27me3 | - Increased KDM6B, decreased H3K27ac in NAc |
| - Differentially expressed genes identified by H3K27me3 ChIP-seq in NAc involved in immune signaling pathways | ||||||
| (Johnstone et al., 2019) | Human | NAc | KDM6B | - KDM6B increased in human alcoholics in the NAc | ||
| (Hashimoto et al., 2017) | WP and WSP mice | Vapor | PFC | HTS | - Multiple changes in chromatin modifying enzymes due to strain and alcohol exposure | |
| (Ponomarev et al., 2012) | Human | CeA, BLA, superior frontal cortex | HTS | H3K4me3, H3K4me2, H3K4me1 | - MLL, MLL4, and SETD1A upregulated in all three brain regions and increase of H3K4me3 | |
| (Zhou et al., 2011) | Human | Hipp, | HTS | H3K4me3 | - Differences in H3K4me3 in AUD at 729 gene promoters |
Abbreviations, WP = withdrawal protective, and WSP = withdrawal seizure prone, vapor = ethanol vapor, CeA = central nucleus of the amygdala, MeA = medial nucleus of the amygdala, BLA = basolateral nucleus of the amygdala, dmPFC = dorsomedial PFC, cereb = cerebellum, DS = dorsal striatum, hipp = hippocampus, HTS = high throughput sequencing
Table 7.
Effects of fetal alcohol exposure on histone methylation mechanism.
| Study | Species | Exp. | Region | Genes | Mark(s) | Findings |
|---|---|---|---|---|---|---|
| (Govorko et al., 2012) | Sprague Dawley rats | Liquid alcohol diet | Hypo. | Set7, G9A, Setdb1 | H3K4me2, H3K4me3, H3K9me2 | - Decreased H3K4me2, H3K4me3, and Set7 in the arcuate area of the hypothalamus |
| - Increased H3K9me2, G9A and Setdb1 in the arcuate area of the hypothalamus | ||||||
| (Bekdash et al., 2013) | Sprague Dawley rats | Liquid alcohol diet | Hypo. | Set7/9A, G9A, Setdb1 | H3K4me2/3, H3K9me2, H3K9Ac | - Choline administration prevents decreases in H3K4me2/3 and Set7/9 |
| - Choline administration prevents increases in H3K9me2 and G9A and Setdb1 | ||||||
| (Subbanna et al., 2013) | C57BL/6 | i.p. 2.5 g/kg | Hipp. NeoCtx | G9A, CC3, and Tau | H3K9me2, H3K27me3 | - Increased G9A expression, H3K9me2 and H3K27me3, and G9A activity in neocortex and hippocampus |
| - Treatment with G9A inhibitor prevents increases in Tau and CC3 protein | ||||||
| (Subbanna et al., 2014) | C57BL/6 | i.p. 1 g/kg | Hipp. NeoCtx | G9A, CC3, and Tau | H3K9me2, H3K27me3 | - Increased G9A, expression H3K9me2 and H3K27me3 and G9A activity in neocortex and hippocampus |
| - Treatment with G9A inhibitor prevents increases in Tau and CC3 protein | ||||||
| (Subbanna et al., 2015) | C57BL/6 | i.p 2.5 g/kg | Hipp. NeoCtx | Cb1 | H3K9me2 | - Decreased H3K9me2 associated with Cb1 promoter |
| (Veazey et al., 2015) | C57BL/6 | i.p 2.9 g/kg | Cortex | 24 genes | H3K4me3, H3K27me3, H3K9me2 | - Broadly, H3K27me3 was decreased and H3K9me2 was increased associated with genes in animals with craniofacial deformities |
Abbreviations, Exp = exposure, i.p. = intraperitoneal, Hyp = hypothalamus, hipp = hippocampus, NeoCtx = Neocortex.
4.1. Human AUD Studies:
One study found that MLL, MLL4, and SETD1A, which are all methyltransferases, were upregulated in CeA, basolateral amygdala (BLA), and superior frontal cortex in postmortem individuals with AUD. In all three brain regions there was also a significant increase in H3K4me3 but not H3K4me2 nor H3K4me1 (Ponomarev et al., 2012). A second study, using ChIP-seq, found differential changes in H3K4me3 at 729 gene promoters in the postmortem hippocampus of individuals with AUD. Interestingly, 250 differences in H3K4me3 associated with gene promoters were also associated with cocaine addiction, suggesting a shared pathway between different drugs of abuse (Zhou et al., 2011). Other studies in an early onset AUD human cohort have indicated that there are significant increases in H3K27me3 in the amygdala at the BDNF and ARC loci, through recruitment of EZH2 to gene regulatory elements, which correspond to decreased Arc and BDNF expression (Bohnsack et al., 2019). Interestingly, EZH2 appears to be recruited by BDNF-AS, a long non-coding RNA (Bohnsack et al., 2019). Consistent with dysregulated H3K27me3, another study found increased KDM6B in the anterior cingulate cortex of individuals with AUD (Johnstone et al., 2019). There is growing evidence that histone methylation and corresponding enzymes play a critical role in AUD in humans.
4.2. Acute Ethanol Exposure:
Acute ethanol decreases G9A and H3K9me2 expression in both the CeA and MeA but not in the BLA and is correlated with anxiolytic effects in rats. Second acute ethanol challenge after 24 hrs of first exposure increases G9A and H3K9me2 back to baseline levels. G9A and H3K9me2 occupancy in the gene body of Npy decreases by acute ethanol, and this is again rescued by second acute ethanol challenge after 24 hrs of first exposure. These changes are associated with development of rapid ethanol tolerance (RET) to anxiolytic effects of ethanol. Treatment with G9A specific inhibitor, UNC0642 prevents the development of RET by inhibiting G9A, increasing H3K9me2, and NPY expression in the CeA and MeA but not in BLA (Berkel et al., 2019). This suggests that acute ethanol leads to an open chromatin structure that is reversed by a subsequent acute ethanol dose the next day, similar to what was observed in earlier studies that evaluated histone acetylation (Sakharkar et al., 2012). These epigenetic modifications in the amygdala induced by acute ethanol appear to play an important role in the development of rapid tolerance to anxiolytic effects of ethanol.
4.3. Chronic Ethanol Exposure:
Chronic alcohol has been shown to change histone methylation in several models of alcohol dependence. In one model using chronic intermittent vapor, it was found that in the dmPFC alcohol causes a decrease in Prdm2 and decreased global H3K9me1 (Barbier et al., 2017). PRDM2, while broadly expressed, is most highly expressed in the prefrontal cortex, suggesting that it may regulate ethanol’s changes in a tissue specific manner (Barbier et al., 2017). Using ChIP-seq, it was identified that the majority of the decreased H3K9me1 was located in gene bodies where it was proposed to be involved in elongation (Barbier et al., 2017). The same study also evaluated PRDM2 knockdown and found an increase in alcohol self-administration and compulsive drinking in a quinine adulteration assay but no changes in sucrose consumption, measures of locomotion, or pain threshold (Barbier et al., 2017). Using the same ethanol vapor exposure paradigm, another study found that Kdm6b mRNA was decreased in both the nucleus accumbens (NAc) and in the dmPFC (Johnstone et al., 2019). KDM6B removes repressive H3K27me3 and is involved in preventing polycomb-repressive gene silencing (De Santa et al., 2007). When the authors evaluated protein levels of KDM6B and H3K27me3 in the NAc of dependent rats, they found increased KDM6B and decreased H3K27me3 (Johnstone et al., 2019). ChIP-seq analysis of H3K27me3 revealed that the majority of pathways with decreased H3K27me3 were involved in immune signaling, specifically the IL-6 pathway (Johnstone et al., 2019), suggesting that increased KDM6B upregulates these immune pathways and that this contributes to the dependence phenotype. In support of this hypothesis, there is a large body of work that suggests that immune signaling within the brain helps drive many different facets of AUD (Coleman and Crews, 2018). A final study, again using ethanol vapor in an inbred mouse line, found that ethanol exposure and withdrawal downregulated Smyd3, Setdb1, Setdb3, Ezh2, and Suz12, while Setd7 and Dot1l were upregulated in the PFC (Hashimoto et al., 2017). It has become increasingly clear from these studies which employ RNA-seq and qPCR arrays that many different histone methylation genes are dysregulated after chronic ethanol exposure and withdrawal.
4.4. Adolescent Alcohol Exposure:
Adolescent alcohol exposure causes long-lasting changes in histone methylation, and these changes persist until adulthood. One mark, H3K27me3, appears to be particularly important in regulating developmental processes in the brain (Gallegos et al., 2018; Mohn et al., 2008). In adult rats, knockdown of KDM6B in the CeA recapitulates the anxiety-like phenotype and molecular changes, such as decreased Arc expression and increased H3K27me3 at the Arc SARE site, that are induced by adolescent alcohol exposure in adulthood (Kyzar et al., 2019b). Increased H3K27me3 is conserved in humans, and it has been shown that the PRC2 complex, specifically EZH2, is involved in repressing the Arc SARE site (Bohnsack et al., 2019). Of particular note, changes in H3K27ac and increases in H3K27me3 at the Arc SARE decreases enhancer RNAs that are transcribed from regions flanking the Arc SARE site, preventing the release of negative elongation factor from the Arc promoter and inhibiting Arc expression in adult amygdala after adolescent alcohol exposure (Kyzar et al., 2019b).
Adolescent alcohol exposure also increases global H3K9me2, another repressive histone methylation mark in the CeA and MeA in adulthood (Kyzar et al., 2017). This process likely occurs due to downregulation of the neuron-specific variant of LSD1 + 8a, another lysine demethylase (Kyzar et al., 2017). Decreased LSD1+8a can be rescued by the acute injection of ethanol in adulthood, which also restores deficits in Bdnf-IV expression and prevents increased H3K9me2 associated with the Bdnf-IV promoter (Kyzar et al., 2017). Decreased Bdnf-IV expression has previously been suggested to be involved in the increased anxiety-like phenotype and drinking phenotype that occurs in adult rats after adolescent alcohol exposure (Pandey et al., 2015). LSD1+8A is a target of the microRNA-137 (miR-137) and adolescent alcohol exposure increases miR-137 expression (Kyzar et al., 2019a). Inhibition of miR-137 in the CeA of adult animals that have been exposed to ethanol during adolescence prevents decreases in LSD1, LSD1+8A, and Bdnf-IV expression, and prevents increased H3K9me2 at the Bdnf-IV promoter as well as attenuates anxiety-like and alcohol drinking behaviors (Kyzar et al., 2019a). Reversing changes induced by adolescent alcohol exposure on H3K9me2 can be performed using non-therapeutic options, as one study found that exercise in late adolescence (pnd 56 to 95) after alcohol exposure (pnd 25 to 55) prevents increases of H3K9me2 at the ChAT promoter and restores ChAT expression in the basal forebrain (Vetreno et al., 2019). Exercise also prevented reversal learning deficits in the Morris water maze (Vetreno et al., 2019).
H3K36me3 is a mark that is commonly found in gene bodies, where it acts to repress gene expression in order maintain stable gene expression as organisms age (Pu et al., 2015). DBA2/J mice who underwent a gavage binge model in adolescence developed last memory deficits in object recognition tests and had a greater sensitivity to ethanol in adulthood (Wolstenholme et al., 2017). Male but not female mice, had decreased H3K36me3 but no changes in H3K9 methylation and had 1912 transcripts that were differently expressed due to ethanol in the PFC (Wolstenholme et al., 2017).
Adolescent alcohol exposure appears to drive a compacted chromatin structure that is associated with decreases in synaptic plasticity genes, such as Arc and immune signaling genes. In some cases, these changes appear to be conserved between rats and humans, underlying the utility of using rodent models to discover epigenetic mechanisms of early-onset alcohol use disorder.
4.5. Fetal Alcohol Exposure:
Histone methylation, like histone acetylation has also been evaluated in the context of fetal alcohol exposure. One study using ethanol liquid diet in Sprague Dawley rats from gd7 to gd21 found a decrease in H3K4me2 and H3K4me3 in the arcuate area of the hypothalamus (Govorko et al., 2012) in adult offspring which corresponded with an decreased Set7 mRNA expression. Conversely, there was an increase in repressive H3K9me2 which also corresponded with an increase in both G9a and Setdb1 mRNA expression (Govorko et al., 2012). Changes in in H3K4me2, H3K4me3, and H3K9me2 can be normalized by co-administering choline (Bekdash et al., 2013). Another study using C57Bl/6J mice found that pnd7 ethanol injections (2.5 g/kg) increased H3K9me2 and H3K27me3 in the hippocampus and neocortex 4–24hr after the first injection of ethanol (Subbanna et al., 2013). Consistent with these increases was an upregulation of G9A protein and activity in both these brain regions (Subbanna et al., 2013). Treatment with the G9A inhibitor, Bix 01294 prevented the upregulation of Tau and CC3 protein by ethanol exposure in both the neocortex and hippocampus (Subbanna et al., 2013), suggesting that pharmacological inhibition of ethanol-induced G9A activation could prevent neurodegenerative effects of developmental ethanol exposure. A follow up study indicated that the effects observed at the higher dose also occur at low doses (1g/kg) (Subbanna et al., 2014). Interestingly, while there are global increases of H3K9me2 in both the hippocampus and neocortex, there is decreased H3K9me2 associated with the Cb1 promoter which corresponds to increased Cb1 activity in both brain regions (Subbanna et al., 2015).
Another study evaluating 22 promoters of growth factor signaling genes after gd7 injections in C57BL/6J mice found decreases in repressive H3K27me3 but increases in repressive H3K9me2 and only modest changes in H3K4me3 in GD17 mice (Veazey et al., 2015). Importantly, these effects were more profound in mice that exhibited craniofacial dysmorphology and mid line brain defects (Veazey et al., 2015), which are commonly associated with fetal alcohol spectrum disorder congenital malformations.
Changes in histone methylation appear to involve an increase in repressive histone methylation that occurs in numerous different brain regions and exposure paradigms in rodent models of FASD.
In summary, changes in histone methylation are often associated with long-term changes in repressive gene transcription. H3K27me3 is associated with dosage compensation (the silencing of one of two maternal X chromosomes), which probably is one of the most well characterized examples of long-term gene silencing (Heard, 2006). Various studies from the alcohol field demonstrate that H3K9me2 and H3K27me3 are involved in developmental processes that are disrupted by alcohol exposure during gestation, early development, or adolescence. Alternatively, chronic alcohol consumption appears to increase repressive histone methylation marks, suggesting again that timing of alcohol exposure plays an important role in regulating the epigenetic response to ethanol. Future directions should focus on whether there is a role for histone methylation readers, as this is a large untapped area that could help further understanding of epigenetic regulation that occurs after alcohol exposure. (Figure 4).
Figure 4. Effects of alcohol exposure on histone methylation.
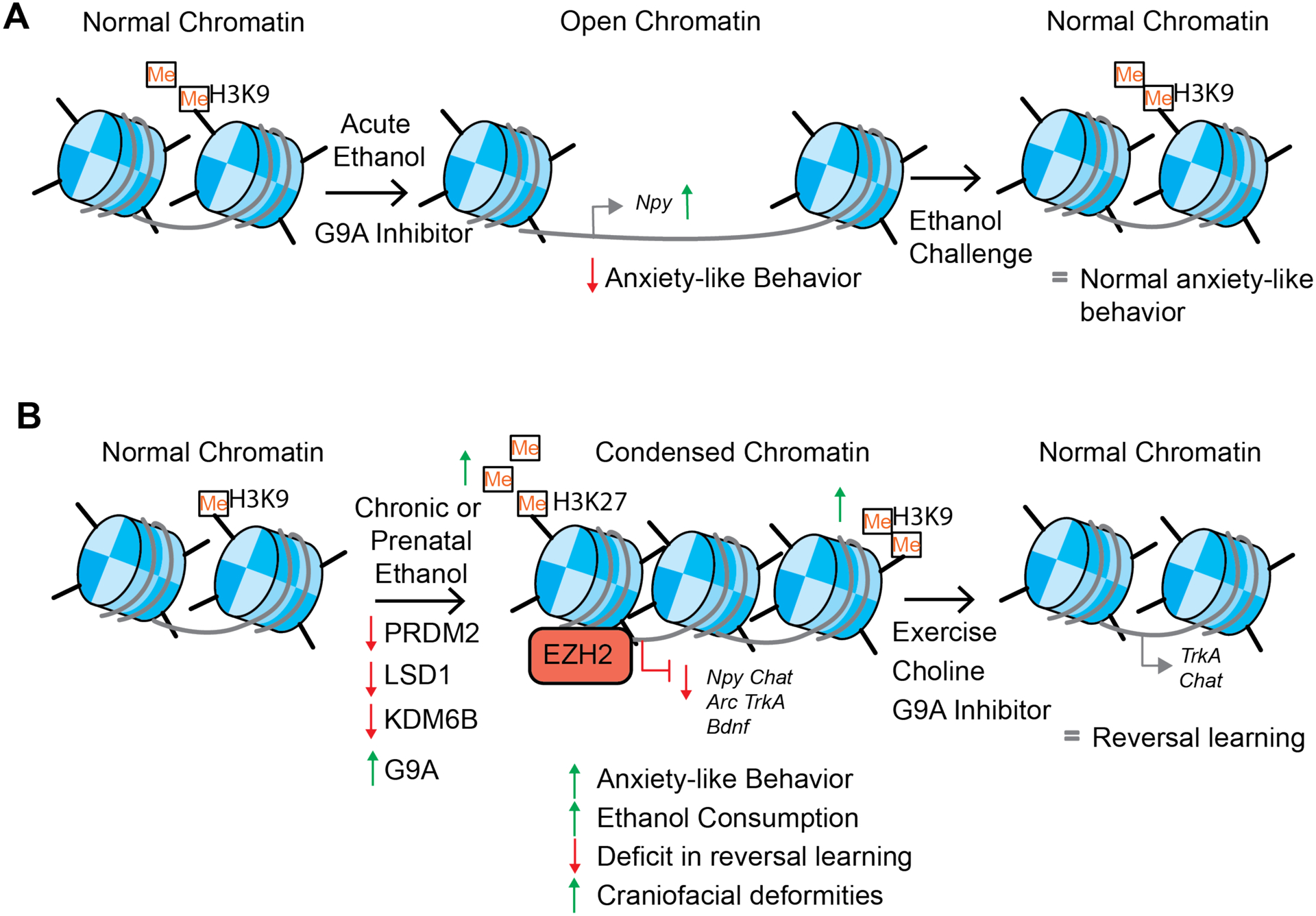
A) Acute ethanol decreases G9A expression and H3K9me2 and leads to open chromatin and increased NPY expression and decreased anxiety-like behavior. Rapid ethanol tolerance by administering an ethanol challenge restores normal H3K9me2 and normal anxiety-like behavior. G9A inhibitors decrease H3K9me2 and lead to decreased anxiety-like behavior. B) Chronic ethanol primarily causes changes via repressive histone methylation (e.g. H3K9me2, H3K9me3, and H3K27me3) however some human and rodent studies have observed an increase in H3K4me3. Ethanol decreases the expression of PRDM2, LSD1 and KDM6B. LSD1 demethylates H3K9me2 and H2K9me1 while KDM6B demethylates H3K27me3. G9A, conversely, methylates H3K9 to H3K9me1 and H3K9me2. The net result is condensed chromatin and decreased gene transcription of neuropeptide y (Npy), activity-regulated cytoskeleton-associated protein (Arc), brain derived neurotrophic factor (Bdnf), choline acetyltransferase (Chat), and tropomyosin receptor kinase A (TrkA). This is associated with anxiety-like behavior, alcohol consumption, deficits in reversal learning, and craniofacial deformities (when given during prenatal development). Exercise, choline or G9A inhibitors can reverse these changes. In ethanol naïve animals, knockdown of PRDM2, LSD1, and KDM6B leads to increased anxiety-like behavior and/or drinking. In humans, EZH2 is shown to bind to regulatory elements of ARC and BDNF and increase repressive H3K27me3.
5. DNA Methylation and Gene Expression
DNA methylation is another well-studied epigenetic modification that occurs most commonly at cytosine residues (Greenberg and Bourc’his, 2019). Adenine residues can also be methylated, and this was recently found to occur in both prokaryotes and mammals (Wu et al., 2016). Methylation of cytosine residues occurs on the 5th carbon (5mC) and is mostly found in CpG dinucleotides (Greenberg and Bourc’his, 2019), and in some tissues, 70–80% of all CpGs are methylated (Li and Zhang, 2014). In the brain, approximately ~6–8% of CpG islands are methylated (Illingworth et al., 2008). Additionally, DNA methylation appears to mostly be symmetrical so that both complementary CpGs are methylated (Greenberg and Bourc’his, 2019). In contrast to histone modifications, which are rapidly reversible, DNA methylation is generally regarded as a stable epigenetic mark (Reik, 2007). The functional outcomes of DNA methylation include transcription repression either through inhibiting transcription factor binding (Iguchi-Ariga and Schaffner, 1989) or changes in chromatin structure (Lorincz et al., 2004), genomic imprinting (Li et al., 1993), X-chromosome inactivation (Mohandas et al., 1981), allowing permissive gene transcription when found in gene bodies (Lister et al., 2009), and cell fate decisions (Cedar and Bergman, 2012), which are important in embryogenesis (Smith et al., 2012) and brain development (Lister et al., 2013). Interestingly, during development, germline DNA methylation disappears and then is re-established in the developing embryo (Cedar and Bergman, 2012). DNA methylation has been shown to interact with the transcriptome, controlling both RNA splicing (Shukla et al., 2011) and eRNAs controlling neuronal DNA methylation (Savell et al., 2016). In the brain, DNA methylation has been shown to play important roles in regulating many brain functions including synaptic plasticity (Day and Sweatt, 2010; Martinowich, 2003), response to neural activity (Junjie U Guo et al., 2011), fear conditioning (Halder et al., 2016; Lubin et al., 2008), memory formation (Day and Sweatt, 2010; Halder et al., 2016; Miller and Sweatt, 2007), reward learning (Day et al., 2013), neurogenesis (Ma et al., 2009), and addiction (Deng et al., 2010). It is clear from many years of research that DNA methylation contributes heavily to both cellular mechanisms and higher order behaviors. Importantly, owing to its inherent stability, DNA methylation signatures have been suggested as a biomarker for psychiatric and other diseases (Levenson, 2010). Currently, there are three primary methods to measure DNA methylation on a genome-wide level, sodium bisulfite conversion followed by sequencing, differential enzymatic cleavage of DNA and immunoprecipitations or methyl capture using a methyl-CpG binding domain (Laird, 2010). These technologies have been implemented on a single-cell level to determine cell type specific changes in DNA methylation (Farlik et al., 2015).
5.1. DNA Methylation Writers:
DNA methylation is catalyzed by a family of enzymes called DNA methyltransferases (DNMTs) which contains three members: DNMT1, which is involved in maintenance of DNA methylation by recognizing hemimethylated DNA and then methylating the complementary strand (Hermann et al., 2004), and DNMT3A and DNMT3B, which are involved in both de novo methylation and maintenance activities (Okano et al., 1999, 1998). DNMT3A and DNMT3B contain a conserved c-terminal DNMT domain (MTase) and two chromatin recognition domains (ADD and PWWP) (Greenberg and Bourc’his, 2019). Regulation of DNMT3A and DNMT3B gene repression occurs through interactions with H3K4me3, where increasing numbers of K4 methylation act to inhibit DNMT binding through the ADD domain (Ooi et al., 2007). H3K4me3 is commonly found in promoters of actively transcribed genes (Soares et al., 2017). In contrast, the PWWP domain recognizes H3K36me3 and acts to guide DNA methylation in gene bodies, which is believed to be permissive to gene transcription (Dhayalan et al., 2010; Lister et al., 2009). DNMT1 binds to the E3 ubiquitin-protein ligase UHRF1 which recognizes H3K9me2 and H3K9me3 through TUDOR and PHD domains (Greenberg and Bourc’his, 2019; Nady et al., 2011). A third isoform of DNMT3, DNMT3L, is a catalytically inactive variant which stimulates DNMT3A and DNMT3B activity by direct binding but doesn’t bind to DNA by itself (Greenberg and Bourc’his, 2019; Ooi et al., 2007; Suetake et al., 2004). Both DNMT3A and DNMT3B are required for normal development, as knockout of either gene results in defects culminating in death (Okano et al., 1998). Mutations in DNMTs are associated with a number of neurological disorders, including dementia (DNMT1) (Klein et al., 2011), overgrowth syndrome with intellectual disability (DNMT3A) (Childhood Overgrowth Consortium et al., 2014), and syndromes with chromosome instability, immunodeficiency and intellectual disability (DNMT3A) (Xu et al., 1999).
5.2. DNA Demethylation Erasers:
DNA demethylation occurs through oxidation by a family of proteins called ten-eleven translocation methylcytosine dioxygenases (TET) and is composed of TET1–3. All three TET proteins contain a catalytic C-terminal domain, while only TET1 and TET3 contain a CXXC domain which binds unmethylated CpG islands (Greenberg and Bourc’his, 2019). TET demethylation activities occur through oxidation of 5mC to 5-hydroxymethylcytosine (5-hmC) that is then further converted to 5-formylcytosine and 5-carboxylcytosine (Ito et al., 2011; Tahiliani et al., 2009). Alternatively, oxidation products can be repaired by thymine-DNA glycosylase, which requires double stranded breaks (He et al., 2011). 5-hmC is much more highly expressed in the brain, suggesting that there is continuous active DNA demethylation, unlike development demethylation which is controlled by temporal expression of DNMT and TET proteins (Globisch et al., 2010; Kriaucionis and Heintz, 2009). 5-hmC likely contributes to gene activation, as opposed to gene repression, and is predominantly found in intragenic regions and enhancers (Junjie U. Guo et al., 2011; H. Wu et al., 2011).
5.6. DNA Methylation Readers:
There are two classes of DNA methylation readers, the first are the methyl-CpG binding domain proteins (MDB) and the second are the unrelated methyl-CpG binding zinc-finger proteins (Kaiso) (Bogdanović and Veenstra, 2009). The MDB family contains MDB1–4 and MeCP2 and mostly are involved in transcription repression through interactions with HDAC complexes (e.g. Sin3A and CoREST) (Bogdanović and Veenstra, 2009). Most members of the MDB family preferentially bind methylated DNA with the exception of MDB3 (Saito and Ishikawa, 2002). MeCP2 mutations result in Rett Syndrome (Amir et al., 1999). In a mouse model of Rett Syndrome, it was observed that there was decreased Bdnf expression, and rescue of Bdnf using an overexpression transgene increased lifespan and locomotor deficits (Chang et al., 2006).
6. DNA Methylation Mechanisms in Alcohol Abuse and Dependence
DNA methylation is well recognized as being important in several different neuronal processes. In the past decade, DNA methylation has arisen as one of the most prominent epigenetic modifications disrupted by alcohol abuse and dependence.
6.1. DNA Methylation Studies in Preclinical Models of AUD
In an intermittent two-bottle choice paradigm, it was found that mice that consumed alcohol had increased DNMT1 in the NAc compared to mice who had only consumed H2O (Warnault et al., 2013). Pharmaceutical inhibition of DNMT with 5-azacytidine decreased alcohol intake with no effect on saccharin or water intake (Warnault et al., 2013). Conversely, in C57BL/6, a high drinking strain of mice, there is no difference in Dnmt1, Dnmt3a, or Dnmt3b compared to the DBA/2J mice, a low drinking strain (Gavin et al., 2016). An evaluation of Growth arrest and DNA-damage-inducible beta transcripts (Gadd45b) in the nucleus accumbens of both C57 and DBA mice found that C57 mice intrinsically had lower levels of Gadd45b and higher levels of 5mC and 5hmC, and acute ethanol normalizes GADD45B protein (Gavin et al., 2016). GADD45B promotes DNA demethylation and is involved in developmental processes such as adult neurogenesis (Ma et al., 2009). GADD45B (+/−) mice consume more alcohol than wild type littermates (Gavin et al., 2016).
Adolescent alcohol exposure causes a decrease of Dnmt3b transcripts during acute ethanol intoxication with no effect on Dnmt1 nor Dnmt3a either 1hr after exposure or 24hrs in the amygdala after exposure during withdrawal (Sakharkar et al., 2019). Global DNMT activity is decreased 1hr after adolescent alcohol exposure but greatly increases 24hr into withdrawal (Sakharkar et al., 2019), suggesting a bimodal response to acute and withdrawal effects of ethanol (Sakharkar et al., 2019). Conversely, Gadd45a, Gadd45b and Gadd45g are all upregulated in the amygdala after 1 hr of ethanol exposure in adolescence (Sakharkar et al., 2019). Adolescent alcohol exposure has no effect on Tet1 expression in the amygdala, suggesting that DNA methylation likely occurs through a base excision pathway, as opposed to a oxidation pathway (Greenberg and Bourc’his, 2019). Furthermore, adolescent ethanol exposure produces an increase in DNA methylation of Bdnf-IV and Npy gene promoters in the adult amygdala, which is normalized by treatment with 5-azacytidine treatment. These changes are associated with attenuation of adolescent alcohol exposure induced anxiety-like and alcohol drinking behaviors in adulthood (Sakharkar et al., 2019).
Fetal alcohol exposure causes changes in DNA methylation. In a Sprague-Dawley rat model that consumed liquid ethanol diet from GD7–21, it was discovered in the F1 generation that the Pomc has decreased expression in both male and female rats in the arcuate nucleus of the hypothalamus that corresponded to an increase in methylation at the Pomc gene (Govorko et al., 2012). These changes appeared to be cell-type specific, as there was decreased 5-hmethylcystosine in β-endorphin producing neurons in the arcuate area of the hypothalamus but an increase in corticotropin releasing hormone neurons, and this potentially could explain the observation that there was no change in global 5-methylcytosine (Govorko et al., 2012). Treatment with the DNMT inhibitor, 5-azadeoxycytidine restored normal DNA methylation at the Pomc gene and normal Pomc expression (Govorko et al., 2012). Further, it was discovered that these changes are transmitted through the male germline (Govorko et al., 2012). Caution should be taken using 5-azacytidine and other DNA orthologues during development, as these can induce neurodegeneration and neurobehavioral deficits when given during development (Subbanna et al., 2016).
6.2. Human DNA Methylation Studies
DNA methylation studies in alcoholic postmortem human brain have been done in a variety of different brain regions. In one study using 10 control and 10 AUD postmortem human cortex subjects, it was identified that 14,004 genes shared equal methylation status, while 3,806 genes differed in methylation status compared to controls (Manzardo et al., 2012). One of the largest differences in methylation between AUD subjects and controls was HIST2H2AC, indicating that there may be differences in histone isoforms and that this may contribute to changes in epigenetic regulation (Manzardo et al., 2012). A caveat to this study is that the sample size is extremely small and only promoters were analyzed. Another study evaluating human postmortem amygdala in 16 male AUD and 16 male controls found that there were 1812 CpGs in 1099 genes that had different methylation status. Further analysis found that 106 of the 1812 CpGs were mapped to 93 genes that had differential gene expression, including AUD associated genes such as (GABRA1, GRIK3, and GRIN2C) (Wang et al., 2016). Interestingly, there was no difference in CpG methylation in 7 female AUD subjects compared to 7 female controls (Wang et al., 2016). This study also further found that in male AUD subjects approximately 34.7% of hypermethylated CpGs were located in the promoter, 41.9% were located in gene bodies, 6.8% in 3’ UTR and 16.6% located in other regions (Wang et al., 2016), consistent with the idea that regulation of gene expression by alcohol through a DNA mechanism is not limited to just promoter regions. A larger study evaluating genome-wide methylation in AUD subjects in the PFC used 25 paired control and AUD subjects and found 5254 differentially methylated CpGs (Gatta et al., 2019). Using Ingenuity Pathway Analysis, the authors determined that nuclear receptor subfamily 3 group C member 1 (NR3C1) was a central hub gene (Gatta et al., 2019). NR3C1 is a member of the glucocorticoid family, which regulate glucocorticoid-response genes and are important in both stress and AUD (Heilig and Koob, 2007). Analysis of DNMT expression found decreases in DNMT1 and DNMT3A, decreases in TET1–3 and increases in GADD45B, suggesting that DNA methylation is highly dysregulated in the PFC due to AUD (Gatta et al., 2019). Another study found differentially methylated locations in the long non-coding RNA growth arrest specific 5 (GAS5) gene in blood, increased gene expression of GAS5 in human postmortem amygdala, and that this gene was highly associated with morning cortisol and stress responsiveness (Lohoff et al., 2020). GAS5 is involved in regulating glucocorticoid signaling, and this agrees with other data suggesting that glucocorticoid signaling is disrupted in alcohol use disorder (Gatta et al., 2019; Lohoff et al., 2020).
In the cerebellum of AUD patients, there was also decreased TET1 expression with no effects on DNMT1, DNMT3A or DNMT3B (Gatta et al., 2017). In the cerebellum, there was also a decrease of GABRD expression which was associated with increased DNA methylation, suggesting that DNA methylation may regulate GABAA receptor expression (Gatta et al., 2017).
6.3. DNA Methylation as a Biomarker of AUD
Owing to the stability of DNA methylation, it has been suggested that DNA methylation be used as either a biomarker of AUD or as a diagnostic tool to better subdivide AUD patients. One study applied a genome-wide approach to 13,317 blood samples from 13 different cohorts and identified 590 differentially methylated CpGs (Liu et al., 2018). It was found that higher alcohol intake drove higher levels of methylation in blood samples (Liu et al., 2018). In individuals with European ancestry, it was found that 144 CpGs are associated with current heavy alcohol intake (≥42g per day for males or ≥28g per day for females), which performed better than previous studies (Liu et al., 2018). Another study using 1549 blood samples found that self-reported alcohol use is a poor reporter of hazardous alcohol drinking and didn’t correlate with significant changes in CpG methylation; however, correlating DNA methylation changes with phosphatidylethanol (an objective marker of alcohol consumption) generated 83 phosphatidylethanol associated CpGs including 23 that were previously known (Liang et al., 2020). This demonstrates that DNA methylation can be used as a biomarker for AUD and that it is important to have good measures of alcohol consumption when designing biomarker studies.
In summary, DNA methylation remains a critical area of study in AUD owing in part to its stability, ability to pass through a germline, and its use as a biomarker. Future studies will have to expand on existing studies to determine which changes that can be detected in the blood also persist in the brain and are indicative of severity of AUD symptoms or stage. Nonetheless, DNA methylation studies at the genome-wide level in human postmortem PFC of AUD and control subjects clearly suggest abnormal epigenetic processes regulating stress mechanisms, most likely via decreased glucocorticoid receptor gene expression (Gatta et al., 2019). Also, DNMT inhibitors have the ability to attenuate alcohol drinking behaviors and other phenotypes such as anxiety-like behaviors in a preclinical model of AUD (Sakharkar et al., 2019; Warnault et al., 2013).
7. New Technologies for Studying Histone Posttranslational Modifications and Epigenetic Regulation
Recent advances in the field have led to the ability to more accurately dissect the role of individual histone marks at discrete locations in the genome. Previously, this involved the use of engineered transcription activator-like effector nucleases (TALENs), which involved protein engineering and was costly and labor intensive (Joung and Sander, 2013). More recently, the advent of CRISPR Cas9 technology has vastly simplified this process and has been demonstrated to be useful both in vitro and in vivo (Sandoval et al., 2020) (Table 8).
Table 8.
dCas9 constructs for use in target epigenetic engineering.
| Study | Cas9 | Effector | Target | Effect | Addgene |
|---|---|---|---|---|---|
| (Hilton et al., 2015) | S. pyogenes | P300 | Histone Acetylation | Gene Activation | 61357 |
| (Kearns et al., 2015) | S. pyogenes | LSD1 | Demethylates H3K4 and 9 me1, 2 | Gene Repression | 92362 |
| (Liu et al., 2016) | S. pyogenes | TET1 | DNA demethylation | Gene Activation | 84475 |
| (Liu et al., 2016) | S. pyogenes | DNMT3A | DNA methylation | Gene Repression | 84570 |
| (Kwon et al., 2017) | S. pyogenes | HDAC3 | Histone Acetylation | Gene Repression | 98591 |
| (Chen, Lin et al., 2019) | S. pyogenes | HDAC8 | Histone Acetylation | Gene Repression | n/a |
| (O’Geen et al., 2017) | S. pyogenes | EZH2 | Writer of H3K27me3 | Gene Repression | 100086 |
| (O’Geen et al., 2017) | S. pyogenes | G9A | Writer of H3K9me3 | Gene Repression | 100089 |
7.1. CRISPR Cas9
The discovery of clustered regularly interspaced short palindromic repeats (CRISPR) in the bacterial genome and Cas nucleases lead to its use as a programmable and scalable tool to modify the mammalian genome (Cong et al., 2013; Jinek et al., 2012). The use of this system over previous iterations was preferable because it simplified the protein-DNA interaction and engineering required with previous systems like TALENs and instead relied on RNA-DNA interactions. The core system has two components consisting of a small guide RNA (sgRNA) which targets complementary DNA sequences (Sandoval et al., 2020) and a Cas nuclease which binds to the sgRNA and cleaves DNA at this location (Sandoval et al., 2020). The ability to make targeted changes to DNA quickly led to an expansion of this technology (Sandoval et al., 2020) including the development of dead Cas systems that had been engineered to repressor or activator domains, base editors, proof-reading domains, fluorophores, inducible systems, optogenetic strategies, or expanded to include RNA editing Cas systems as well (Sandoval et al., 2020). For this review, we will focus exclusively on a subset of these strategies that involves using a dead Cas9 system, which is a mutant variant that contains D10A and H840A changes that lead to the Cas9 having a lack of endonuclease activity (Jinek et al., 2012).
Early iterations of dCas9 used viral proteins in order to control transcription, such as tetrameric herpes simplex viral protein 16 (VP64), which activated gene transcription or transcription factors such as Krüppel-associated box domain of Kox1 (KRAB) to repress gene transcription (Gilbert et al., 2013). Other groups have harnessed the power of using epigenetic modulating proteins to make epigenetic changes at precise genomic locations, leading to the ability to probe whether specific epigenetic changes at these loci are responsible for driving transcriptional and higher-order process changes. One of the first was dCas9-P300, which combines dCas9 to the P300 histone acetyltransferase in a fusion protein and can be used in combination with sgRNAs to selectively increase histone acetylation at distinct location (Hilton et al., 2015; Klann et al., 2017). Most studies have used this approach to increase H3K27Ac at enhancer regions in order to determine the role of the enhancers at that region (Hilton et al., 2015; Klann et al., 2017). In particular, one study demonstrated in vitro that increasing H3K27ac at cFos enhancers recruited BRD4 and that this drove cFos expression and function (Chen, Lin et al., 2019). This technology has been used to study histone acetylation regulated by alcohol. One study using dCas9-P300 demonstrated that Gabra1 expression could be restored in primary cortical neurons in vitro in a chronic ethanol exposure paradigm by increasing histone acetylation at the Gabra1 promoter (Bohnsack et al., 2017).
Several other epigenetic modulating dCas9 constructs have been described. One example is dCas9-LSD1, which can be used to increase histone demethylation at targeted genomic loci and to determine the role of LSD1 in regulating gene expression at either promoters or enhancers through its substrate H3K4me2 (Kearns et al., 2015). When targeted to a known enhancer, the target gene, Tbx3, was decreased, but when targeted toward the promoter region no decrease was observed, suggesting that regulation via LSD1 occurs primarily at the enhancer to control gene expression (Kearns et al., 2015). The same study evaluated the use of dCas9-KRAB and found that targeting both the promoter and the enhancer reduced Tbx3 expression and increased H3K27me3 (Kearns et al., 2015).
To date, epigenetic modulating fusion proteins have been constructed for DNA methylation, including dCas9-Tet1 (Liu et al., 2016; Xu et al., 2016) and dCas9-DNMTs (Liu et al., 2016), histone deacetylation, such as HDAC3 (Kwon et al., 2017), HDAC8 (L.-F. Chen et al., 2019), histone methylation, such as SUV, G9A, and EZH2 (O’Geen et al., 2017), and transcription factors such as CREB (Lorsch et al., 2019). With the growing focus on epigenetics and CRISPR Cas technology, it is likely that more epigenetic regulator dCas9 constructs will be developed in the future.
8. Conclusions
Over the past two decades, AUD research has continued to expand the role of the epigenome and epigenetics in regulating the acute, chronic, and developmental effects of ethanol. The increasing use of sequencing, single-cell, and genome editing technologies continues to help expand the role of epigenetics in regulating both the acute and chronic effects of ethanol. The hope is that this will eventually lead to tractable epigenetic drug targets that modulate histone acetylation/methylation or DNA methylation mechanisms to treat AUD (Figure 5).
Figure 5. A unifying epigenetic mechanism of the effects of alcohol consumption on different stages of the addiction cycle that lead to alcohol use disorder.
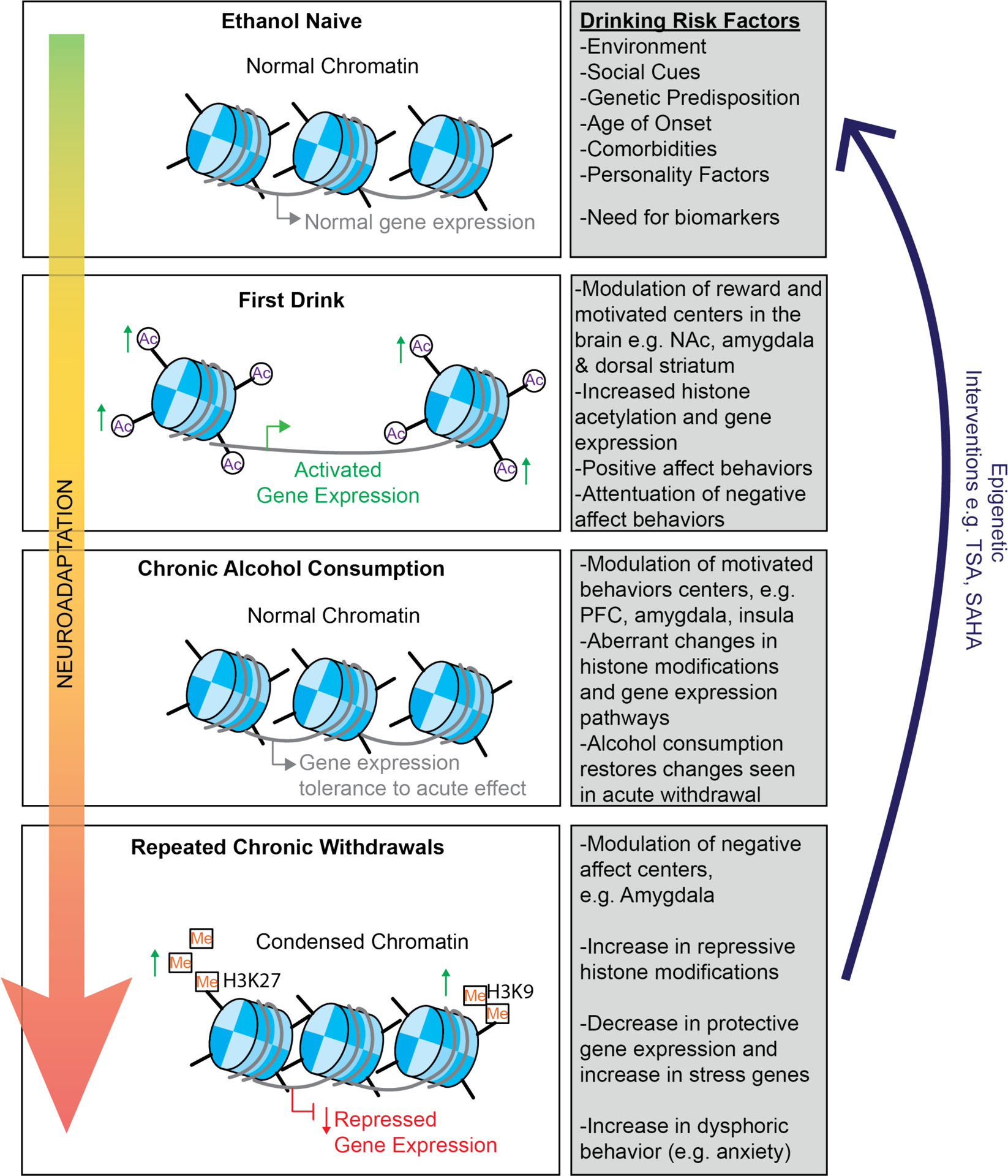
Preexisting risk factors can lead to the initiation of alcohol consumption. The first drink or alcohol experience modulates reward and motivational centers of the brain such as the nucleus accumbens (NAc), dorsal striatum, and amygdala which underlie the rewarding or positive affect of alcohol consumption and attenuate negative affect. These are associated with increased histone acetylation and gene expression. During a second stage, the move to chronic alcohol consumption causes changes in motivation and executive centers of the brain such as the insula and there is tolerance to the rewarding effects of ethanol and the epigenetic effects such as increased gene activation. In the third stage, repeated chronic consumption and withdrawal activates negative affect centers such as the amygdala and the bed nucleus of the stria terminalis (BNST) and causes decreased histone acetylation, increased repressive histone methylation and repressed gene expression which correspond with an increase in dysphoric behaviors such as anxiety. Acute ethanol challenge can restore both the epigenetic changes and behavioral changes to a more baseline levels. Importantly, these effects are also observed when ethanol is consumed during critical periods of development, such as during gestation or adolescence. Treatment with epigenetic drugs or genetic strategies can restore normal epigenetic pathways and functions and corresponding behaviors. This ultimately suggests that drugs that work on an epigenetic level could be useful for the treatment of alcohol use disorder.
Table 2.
Effects of chronic alcohol exposure on histone acetylation mechanism.
| Study | Species | Exp. | Region | Genes | Mark(s) | Findings |
|---|---|---|---|---|---|---|
| (Pandey et al., 2008a) | Sprague Dawley rats | Liquid alcohol diet | CeA, MeA | CBP, NPY | H3K9ac, H4K8ac | - Increased HDAC activity - Decreased H3K9ac, H4K8ac, and NPY |
| - Increased anxiety-like behavior | ||||||
| - TSA administration prevents changes | ||||||
| (Bohnsack et al., 2018) | Sprague Dawley and Wistar rats | i.g. (5g/kg), I2BC (20%) | Cortex, PFC | GABA (A)-R subunits, all HDACs | H3ac, H3K9ac, H3K14ac | - Decreased H3ac, H3K9ac, and H3K14ac in the cortex, increased HDAC2 and HDAC3 |
| - Decreased GABA(A)R α1 and decay tau | ||||||
| - TSA prevents molecular and behavioral effects | ||||||
| (Arora et al., 2013) | C57Bl/6 | i.p 3.5g/kg | VTA | GABA (A)-R, HDAC2 | H3K9ac | - Decreased H3K9ac and GABA(A)-R α1 and increased HDAC2 |
| - Decreased GABA(A) signaling, restored by TSA | ||||||
| (Moonat et al., 2013; Sakharkar et al., 2014) | P rats | Vs. NP | CeA, MeA | HDAC1 HDAC2 HDAC3 HDAC4 HDAC5 HDAC6 Arc, Bdnf-IV NPY |
H3K9ac | - Treatment with TSA decreases drinking - HDAC2 knockdown in CeA prevents anxiety-like and alcohol drinking behaviors |
| -HDAC2 knockdown increases H3K9/14ac associated with Arc and BDNF-IV promoter | ||||||
| (Warnault et al., 2013) | C57/BL6 and Long Evans | I2BC, Operant | NAc | Dnmt1 | H4ac | - Decreased H4ac and increased Dnmt1 in NAc |
| (Barbier et al., 2017) | C57BL/6 | Vapor | dmPFC | p300, Myst | - Decreased P300 and Myst |
Abbreviations: P rats = selectively bred alcohol preferring rats, NP = non-alcohol preferring rats, Exp. = exposure, i.p. = intraperitoneal, i.g. = intragastric, I2BC = intermittent two bottle choice, CeA = central nucleus of the amygdala, MeA = medial nucleus of the amygdala, PFC = prefrontal cortex, NAc = nucleus accumbens, dmPFC = dorsalmedial PFC.
Table 3.
Effects of adolescent alcohol exposure on histone acetylation mechanism.
| Study | Species | Exp. | Region | Genes | Mark(s) | Findings |
|---|---|---|---|---|---|---|
| (Pandey et al., 2015) | Sprague Dawley rats | Intermittent, 2g/kg i.p. | CeA, MeA | Hdac2, Hdac4, Bdnf-I, Bdnf-IV, Arc | H3K9ac, H3K9/14ac | - Decreased H3K9ac and increased HDAC2 and HDAC4. - Increased HDAC activity |
| - TSA prevents molecular effects, anxiety and alcohol consumption | ||||||
| (Sakharkar et al., 2016) | Sprague Dawley rats | Intermittent, 2g/kg i.p. | Hipp. | Ki-67, DCX, Bdnf-I, Bdnf-IV, CBP | H3K9ac, H3K9/14ac | - Decreased H3K9ac and CBP in CA1–3 regions, -Increased HDAC activity |
| - TSA prevents anxiety, restores markers of neurogenesis and decreases of H3K9/14ac at BDNF-I and BDNF-IV promoters | ||||||
| (Mulholland et al., 2018) | Sprague Dawley rats | 5g/kg i.g. | Hipp. | Fmr1 | H3K9/14ac,H3K27ac | - Donepezil prevents increases of Fmr1 mRNA and increased H3K27ac at Fmr1 gene |
| (Bohnsack et al., 2019) | Humans | Earlyage of onset | Amgydala | BDNF, Arc | H3K27ac | - Decreased Arc and BDNF expression and decrease in H3K27ac at regulatory elements |
| (Zhang et al., 2018) | Sprague Dawley rats | Intermittent 2g/kg i.p. | CeA, MeA | CBP, P300, CREB | H3K9/14Ac | - Decreased CBP, P300, and CREB expression |
| - Decreased H3K9/14ac at promoters of CBP, P300, and CREB. |
Abbreviations: Exp. = exposure, i.p. = intraperitoneal, i.g. = intragastric, CeA = central nucleus of the amygdala, MeA = medial nucleus of the amygdala, Hipp = hippocampus.
Table 6.
Effects of adolescent alcohol exposure on histone methylation.
| Study | Species | Exp | Region | Genes | Mark(s) | Findings |
|---|---|---|---|---|---|---|
| (Kyzar et al., 2019b) | Sprague Dawley rat | i.p. 2g/kg | Amg | KDM6B, Arc | H3K27me3 | - Decreased KDM6B - Increased H3K27me3 |
| - Decreased Arc expression | ||||||
| - KDM6B knockdown in CeA causes anxiety-like behavior | ||||||
| (Bohnsack et al., 2019) | Human | Amg | BDNF, Arc, EZH2 | H3K27me3 | - Increases in EZH2 and H3K27me3 associated with Arc and BDNF regulatory elements in early onset AUD | |
| (Kyzar et al., 2017) | Sprague Dawley rat | i.p. 2g/kg | Amg | Histone methyltransferases | H3K4me2, H3K9me2 | - Decreased LSD1 and LSD1+8A in CeA and MeA and increased H3K9me2 in CeA and MeA |
| (Kyzar et al., 2019a) | Sprague Dawley rat | i.p. 2g/kg | Amg | BDNF-IV, LSD1+8a | H3K9me2 | - Decreased LSD1 and increased H3K9me2 at BDNF-IV promoter |
| - Knockdown of LSD1 in CeA causes anxiety-like behavior | ||||||
| (Vetreno et al., 2019) | Wistar | i.g 5g/kg | Hipp | ChAT | H3K9me2 | - Increased H3K9me2 at Chat and Trka promoter |
| - Exercise prevents changes in H3K9me2 at Chat and Trka promoter | ||||||
| - Exercise prevents reversal learning deficits | ||||||
| (Wolstenholme et al., 2017) | DBA/2J | i.g. 4 g/kg | PFC | HTS | H3K9me1–3 and H3K36me1–3 | - Decrease of global H3K36me1–3 in males but not females |
| - Changes in myelin related genes | ||||||
| Increased ambulation and decreased sensitivity to alcohol |
Abbreviations, Exp = exposure, i.p. = intraperitoneal, i.g. = intragastric, PFC = prefrontal cortex, hipp = hippocampus, Amg = amygdala, HTS = high throughput sequencing
9. Acknowledgement and Disclosure:
This is supported by NIH-NIAAA grants (P50AA022538, UO1AA019971, U24AA024605, and RO1AA010005) and by the VA Merit (I01 BX004517) and Senior Research Career Scientist award to SCP and F32AA027410 to JP.
Footnotes
Authors report no conflict of interest.
References
- Adan A, Forero DA, Navarro JF, 2017. Personality traits related to binge drinking: A systematic review. Front. Psychiatry 8, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K, Cloos PAC, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K, 2007. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449, 731–734. [DOI] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T, 2016. The molecular hallmarks of epigenetic control. Nature Reviews Genetics 17, 487–500. [DOI] [PubMed] [Google Scholar]
- Álvarez A, Ávila J, Palao D, Montejo Á, 2018. Influence of Personality Traits on the Severity of Alcohol Use Disorders. JCM 7, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY, 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Anthenelli RM, Schuckit MA, 1993. Affective and anxiety disorders and alcohol and drug dependence: Diagnosis and treatment. Journal of Addictive Diseases 12, 73–87. [DOI] [PubMed] [Google Scholar]
- Arora DS, Nimitvilai S, Teppen TL, McElvain MA, Sakharkar AJ, You C, Pandey SC, Brodie MS, 2013. Hyposensitivity to gamma-aminobutyric acid in the ventral tegmental area during alcohol withdrawal: Reversal by histone deacetylase inhibitors. Neuropsychopharmacol 38, 1674–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M, 2012. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 11, 384–400. [DOI] [PubMed] [Google Scholar]
- Augier E, Barbier E, Dulman RS, Licheri V, Augier G, Domi E, Barchiesi R, Farris S, Nätt D, Mayfield RD, Adermark L, Heilig M, 2018. A molecular mechanism for choosing alcohol over an alternative reward. Science 360, 1321–1326. [DOI] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang C-P, Zhao Y, Swigut T, Wysocka J, 2010. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LJ, Murzina NV, Broadhurst RW, Raine ARC, Archer SJ, Stott FJ, Murzin AG, Singh PB, Domaille PJ, Laue ED, 1997. Structure of the chromatin binding (chromo) domain from mouse modifier protein 1. The EMBO Journal 16, 2473–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier E, Johnstone AL, Khomtchouk BB, Tapocik JD, Pitcairn C, Rehman F, Augier E, Borich A, Schank JR, Rienas CA, Van Booven DJ, Sun H, Nätt D, Wahlestedt C, Heilig M, 2017. Dependence-induced increase of alcohol self-administration and compulsive drinking mediated by the histone methyltransferase PRDM2. Mol Psychiatry 22, 1746–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K, 2007. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Bekdash RA, Zhang C, Sarkar DK, 2013. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in β-endorphin-producing POMC neurons of the hypothalamus. Alcohol Clin Exp Res 37, 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger KH, Heberlein U, Moore MS, 2004. Rapid and chronic: Two distinct forms of ethanol tolerance in drosophila: Alcoholism: Clinical & Experimental Research 28, 1469–1480. [DOI] [PubMed] [Google Scholar]
- Berkel TDM, Pandey SC, 2017. Emerging role of epigenetic mechanisms in alcohol addiction. Alcohol Clin Exp Res 41, 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel TDM, Zhang H, Teppen T, Sakharkar AJ, Pandey SC, 2019. Essential role of histone methyltransferase G9a in rapid tolerance to the anxiolytic effects of ethanol. International Journal of Neuropsychopharmacology 22, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, Gingeras TR, Schreiber SL, Lander ES, 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181. [DOI] [PubMed] [Google Scholar]
- Blus BJ, Wiggins K, Khorasanizadeh S, 2011. Epigenetic virtues of chromodomains. Critical Reviews in Biochemistry and Molecular Biology 46, 507–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanović O, Veenstra GJC, 2009. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 118, 549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack JP, Hughes BA, O’Buckley TK, Edokpolor K, Besheer J, Morrow AL, 2018. Histone deacetylases mediate GABAA receptor expression, physiology, and behavioral maladaptations in rat models of alcohol dependence. Neuropsychopharmacology 43, 1518–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack JP, Patel VK, Morrow AL, 2017. Ethanol exposure regulates Gabra1 expression via histone Deacetylation at the Promoter in Cultured Cortical Neurons. Journal of Pharmacology and Experimental Therapeutics 363, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack JP, Teppen T, Kyzar EJ, Dzitoyeva S, Pandey SC, 2019. The lncRNA BDNF-AS is an epigenetic regulator in the human amygdala in early onset alcohol use disorders. Translational Psychiatry 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Lecona E, Reinberg D, 2010. MBT domain proteins in development and disease. Seminars in Cell & Developmental Biology 21, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botia B, Legastelois R, Alaux-Cantin S, Naassila M, 2012. Expression of ethanol-induced behavioral sensitization is associated with alteration of chromatin remodeling in Mice. PLoS ONE 7, e47527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R, 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353. [DOI] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE, 2008. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Irwin M, Schuckit MA, 1991. Changes in anxiety among abstinent male alcoholics. J. Stud. Alcohol 52, 55–61. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano R, Tam T, Greenfield T, Cherpitel C, Midanik L, 1997. DSM-IV alcohol dependence and drinking in the U.S. population: A risk analysis. Annals of Epidemiology 7, 542–549. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y, 2012. Programming of DNA methylation patterns. Annu. Rev. Biochem 81, 97–117. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention, 2013. Alcohol and Public Health: Alcohol-Related Disease Impact (ARDI). Average for United States 2006–2010 Alcohol-Attributable Deaths Due to Excessive Alcohol Use. [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R, 2006. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49, 341–348. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Mizar P, Cassel R, Neidl R, Selvi BR, Mohankrishna DV, Vedamurthy BM, Schneider A, Bousiges O, Mathis C, Cassel J-C, Eswaramoorthy M, Kundu TK, Boutillier A-L, 2013. A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. Journal of Neuroscience 33, 10698–10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L-F, Lin YT, Gallegos DA, Hazlett MF, Gómez-Schiavon M, Yang MG, Kalmeta B, Zhou AS, Holtzman L, Gersbach CA, Grandl J, Buchler NE, West AE, 2019. Enhancer histone acetylation modulates transcriptional bursting dynamics of neuronal activity-inducible genes. Cell Reports 26, 1174–1188.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W-Y, Zhang H, Gatta E, Glover EJ, Pandey SC, Lasek AW, 2019. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol 78, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chignola F, Gaetani M, Rebane A, Org T, Mollica L, Zucchelli C, Spitaleri A, Mannella V, Peterson P, Musco G, 2009. The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Research 37, 2951–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childhood Overgrowth Consortium, Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, del Vecchio Duarte S, Zachariou A, Hanks S, O’Brien E, Aksglaede L, Baralle D, Dabir T, Gener B, Goudie D, Homfray T, Kumar A, Pilz DT, Selicorni A, Temple IK, Van Maldergem L, Yachelevich N, van Montfort R, Rahman N, 2014. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 46, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Agger K, Cloos PAC, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K, 2007. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128, 1063–1076. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH, 1993. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Colder CR, Campbell RT, Ruel E, Richardson JL, Flay BR, 2002. A finite mixture model of growth trajectories of adolescent alcohol use: Predictors and consequences. Journal of Consulting and Clinical Psychology 70, 976–985. [DOI] [PubMed] [Google Scholar]
- Coleman LG, Crews FT, 2018. Innate immune signaling and alcohol use disorders, in: Grant KA, Lovinger DM (Eds.), The Neuropharmacology of Alcohol. Springer International Publishing, Cham, pp. 369–396. [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F, 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Colville AM, Kruse LC, Cameron AJ, Spence SE, Schlumbohm JP, Huang LC, Metten P, 2012. Ethanol tolerance and withdrawal severity in high drinking in the dark selectively bred mice. Alcohol Clin Exp Res 36, 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Robinson DL, Chandler LJ, Ehlers CL, Mulholland PJ, Pandey SC, Rodd ZA, Spear LP, Swartzwelder HS, Vetreno RP, 2019. Mechanisms of persistent neurobiological changes following adolescent alcohol exposure: NADIA consortium findings. Alcohol Clin Exp Res 43, 1806–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JE, Emery RE, Turkheimer E, 2012. Peer network drinking predicts increased alcohol use from adolescence to early adulthood after controlling for genetic and shared environmental selection. Developmental Psychology 48, 1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, Lee C, Regalado SG, Read DF, Steemers FJ, Disteche CM, Trapnell C, Shendure J, 2018. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Childs D, Guzman-Karlsson MC, Kibe M, Moulden J, Song E, Tahir A, Sweatt JD, 2013. DNA methylation regulates associative reward learning. Nat Neurosci 16, 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD, 2010. DNA methylation and memory formation. Nat Neurosci 13, 1319–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G, 2007. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094. [DOI] [PubMed] [Google Scholar]
- Dekker J, 2002. Capturing chromosome conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim I-H, Wetsel WC, West AE, 2010. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci 13, 1128–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A, 2010. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem 285, 26114–26120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SC, Zhang X, Trievel RC, Cheng X, 2005. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 6, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egervari G, Landry J, Callens J, Fullard JF, Roussos P, Keller E, Hurd YL, 2017. Striatal H3K27 acetylation linked to glutamatergic gene dysregulation in human heroin abusers holds promise as therapeutic target. Biological Psychiatry 81, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schönegger A, Klughammer J, Bock C, 2015. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Reports 10, 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrelly LA, Thompson RE, Zhao S, Lepack AE, Lyu Y, Bhanu NV, Zhang B, Loh Y-HE, Ramakrishnan A, Vadodaria KC, Heard KJ, Erikson G, Nakadai T, Bastle RM, Lukasak BJ, Zebroski H, Alenina N, Bader M, Berton O, Roeder RG, Molina H, Gage FH, Shen L, Garcia BA, Li H, Muir TW, Maze I, 2019. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE, 2010. Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegersh A, Homanics GE, 2014. Acute ethanol alters multiple histone modifications at model gene promoters in the cerebral cortex. Alcohol Clin Exp Res 38, 1865–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP, 1999. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193. [DOI] [PubMed] [Google Scholar]
- Gallegos DA, Chan U, Chen L-F, West AE, 2018. Chromatin regulation of neuronal maturation and plasticity. Trends in Neurosciences 41, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta E, Auta J, Gavin DP, Bhaumik DK, Grayson DR, Pandey SC, Guidotti A, 2017. Emerging role of one-carbon metabolism and DNA methylation enrichment on δ-containing GABAA receptor expression in the cerebellum of subjects with alcohol use disorders (AUD). International Journal of Neuropsychopharmacology 20, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta E, Grayson DR, Auta J, Saudagar V, Dong E, Chen Y, Krishnan HR, Drnevich J, Pandey SC, Guidotti A, 2019. Genome-wide methylation in alcohol use disorder subjects: Implications for an epigenetic regulation of the cortico-limbic glucocorticoid receptors (NR3C1). Mol Psychiatry. 10.1038/s41380-019-0449-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin DP, Kusumo H, Zhang H, Guidotti A, Pandey SC, 2016. Role of growth arrest and DNA damage-inducible, beta in alcohol-drinking behaviors. Alcohol Clin Exp Res 40, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi A, Li X, Lew LK, Wijesekera TP, Atkinson NS, 2017. Alcohol-induced neuroadaptation is orchestrated by the histone acetyltransferase CBP. Front. Mol. Neurosci 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS, 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD, 2007. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Research 17, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, Koch S, Brückl T, Biel M, Carell T, 2010. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 5, e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorko D, Bekdash RA, Zhang C, Sarkar DK, 2012. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biological Psychiatry 72, 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Stinson FS, Harford TC, 2001. Age at onset of alcohol use and DSM-IV alcohol abuse and dependence: a 12-year follow-up. J Subst Abuse 13, 493–504. [DOI] [PubMed] [Google Scholar]
- Greenberg MVC, Bourc’his D, 2019. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 20, 590–607. [DOI] [PubMed] [Google Scholar]
- Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, Dahmani A, Lameiras S, Reyal F, Frenoy O, Pousse Y, Reichen M, Woolfe A, Brenan C, Griffiths AD, Vallot C, Gérard A, 2019. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet 51, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Guan J-S, Haggarty SJ, Giacometti E, Dannenberg J-H, Joseph N, Gao J, Nieland TJF, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai L-H, 2009. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Junjie U, Ma DK, Mo H, Ball MP, Jang M-H, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming G, Gao Y, Song H, 2011. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 14, 1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Junjie U., Su Y, Zhong C, Ming G, Song H, 2011. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Crossey EL, Zhang L, Zucca S, George OL, Valenzuela CF, Zhao X, 2011. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE 6, e19351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES, 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman R-U, Rajput A, Centeno TP, van Bebber F, Capece V, Vizcaino JCG, Schuetz A-L, Burkhardt S, Benito E, Sala MN, Javan SB, Haass C, Schmid B, Fischer A, Bonn S, 2016. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci 19, 102–110. [DOI] [PubMed] [Google Scholar]
- Hashimoto JG, Gavin DP, Wiren KM, Crabbe JC, Guizzetti M, 2017. Prefrontal cortex expression of chromatin modifier genes in male WSP and WSR mice changes across ethanol dependence, withdrawal, and abstinence. Alcohol 60, 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycock PC, 2009. Fetal alcohol spectrum disorders: the epigenetic perspective1. Biology of Reproduction 81, 607–617. [DOI] [PubMed] [Google Scholar]
- He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song C-X, Zhang K, He C, Xu G-L, 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, 2006. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes & Development 20, 1848–1867. [DOI] [PubMed] [Google Scholar]
- Heath AC, Madden PAF, Bucholz KK, Dinwiddie SH, Slutske WS, Bierut LJ, Rohrbaugh JW, Statham DJ, Dunne MP, Whitfield JB, Martin NG, 1999. Genetic differences in alcohol sensitivity and the inheritance of alcoholism risk. Psychol. Med 29, 1069–1081. [DOI] [PubMed] [Google Scholar]
- Heilig M, Koob GF, 2007. A key role for corticotropin-releasing factor in alcohol dependence. Trends in Neurosciences 30, 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B, 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Hermann A, Goyal R, Jeltsch A, 2004. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem 279, 48350–48359. [DOI] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA, 2015. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nature Biotechnology 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Sabari BR, Garcia BA, Allis CD, Zhao Y, 2014. SnapShot: Histone Modifications. Cell 159, 458–458.e1. 10.1016/j.cell.2014.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes BA, Bohnsack JP, O’Buckley TK, Herman MA, Morrow AL, 2019. Chronic ethanol exposure and withdrawal impair synaptic GABA A receptor-mediated neurotransmission in deep-layer prefrontal cortex. Alcohol Clin Exp Res 43, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmann D, Gozani O, 2019. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol 26, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun K, Jeon J, Park K, Kim J, 2017. Writing, erasing and reading histone lysine methylations. Exp Mol Med 49, e324–e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi-Ariga SM, Schaffner W, 1989. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes & Development 3, 612–619. [DOI] [PubMed] [Google Scholar]
- Illingworth R, Kerr A, DeSousa D, Jørgensen H, Ellis P, Stalker J, Jackson D, Clee C, Plumb R, Rogers J, Humphray S, Cox T, Langford C, Bird A, 2008. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol 6, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y, 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TC, Elgin SC, 1986. Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell. Biol 6, 3862–3872. 10.1128/MCB.6.11.3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong H-S, Brady JN, Ozato K, 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Molecular Cell 19, 523–534. [DOI] [PubMed] [Google Scholar]
- Jarmasz JS, Stirton H, Basalah D, Davie JR, Clarren SK, Astley SJ, Del Bigio MR, 2019. Global DNA methylation and histone posttranslational modifications in human and nonhuman primate brain in association with prenatal alcohol exposure. Alcohol Clin Exp Res. 43(6), 1145–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns J-P, Van Esch H, Kleefstra T, Hamel B, Moraine C, Gécz J, Turner G, Reinhardt R, Kalscheuer VM, Ropers H-H, Lenzner S, 2005. Mutations in the JARID1C gene, which Is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. The American Journal of Human Genetics 76, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Bressler J, Beaudet AL, 2004. Epigenetics and human disease. Annu. Rev. Genom. Hum. Genet 5, 479–510. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone AL, Andrade NS, Barbier E, Khomtchouk BB, Rienas CA, Lowe K, Van Booven DJ, Domi E, Esanov R, Vilca S, Tapocik JD, Rodriguez K, Maryanski D, Keogh MC, Meinhardt MW, Sommer WH, Heilig M, Zeier Z, Wahlestedt C, 2019. Dysregulation of the histone demethylase KDM6B in alcohol dependence is associated with epigenetic regulation of inflammatory signaling pathways. Addiction Biology e12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD, 2013. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Kanno Y, LeRoy G, Campos E, Sun H-W, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D, Siebenlist U, O’Shea JJ, Ozato K, 2014. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 21, 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, Wasney GA, Li Z, Vedadi M, Schapira M, Min J, Arrowsmith CH, 2011. Recognition and specificity determinants of the human Cbx chromodomains. J. Biol. Chem 286, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh SA, White LA, Kolesar JM, 2010. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. American Journal of Health-System Pharmacy 67, 793–797. [DOI] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R, 2015. Functional annotation of native enhancers with a Cas9–histone demethylase fusion. Nat Methods 12, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Nelson CB, McGonagle KA, Edlund MJ, Frank RG, Leaf PJ, 1996. The epidemiology of co-occurring addictive and mental disorders: Implications for prevention and service utilization. American Journal of Orthopsychiatry 66, 17–31. [DOI] [PubMed] [Google Scholar]
- Keyes KM, Hatzenbuehler ML, Grant BF, Hasin DS, 2012. Stress and alcohol: epidemiologic evidence. Alcohol Res 34, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B, 2005. A high-resolution map of active promoters in the human genome. Nature 436, 876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB, Crawford GE, Reddy TE, Gersbach CA, 2017. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nature Biotechnology 35, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CJ, Botuyan M-V, Wu Y, Ward CJ, Nicholson GA, Hammans S, Hojo K, Yamanishi H, Karpf AR, Wallace DC, Simon M, Lander C, Boardman LA, Cunningham JM, Smith GE, Litchy WJ, Boes B, Atkinson EJ, Middha S, B Dyck PJ, Parisi JE, Mer G, Smith DI, Dyck PJ, 2011. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 43, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND, 2016. Neurobiology of addiction: a neurocircuitry analysis. The Lancet Psychiatry 3, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M, 2004. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42, 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T, 2007. Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Kranzler HR, 1996. Evaluation and treatment of anxiety symptoms and disorders in alcoholics. J Clin Psychiatry 57 Suppl 7, 15–21; discussion 22–24. [PubMed] [Google Scholar]
- Kranzler HR, Zhou H, Kember RL, Vickers Smith R, Justice AC, Damrauer S, Tsao PS, Klarin D, Baras A, Reid J, Overton J, Rader DJ, Cheng Z, Tate JP, Becker WC, Concato J, Xu K, Polimanti R, Zhao H, Gelernter J, 2019. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun 10, 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N, 2009. The Nuclear DNA Base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan HR, Sakharkar AJ, Teppen TL, Berkel TDM, Pandey SC, 2014. The epigenetic landscape of alcoholism. International Review of Neurobiology 115, 75–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi K-H, Renthal W, Tsankova NM, Theobald DEH, Truong H-T, Russo SJ, LaPlant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ, 2005. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48, 303–314. [DOI] [PubMed] [Google Scholar]
- Kumar S, Porcu P, Werner DF, Matthews DB, Diaz-Granados JL, Helfand RS, Morrow AL, 2009. The role of GABAA receptors in the acute and chronic effects of ethanol: a decade of progress. Psychopharmacology 205, 529–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz DL, Stewart RB, Zweifel M, Li T-K, Froehlich JC, 1996. Genetic differences in tolerance and sensitization to the sedative/hypnotic effects of alcohol. Pharmacology Biochemistry and Behavior 53, 585–591. [DOI] [PubMed] [Google Scholar]
- Kwako LE, Momenan R, Litten RZ, Koob GF, Goldman D, 2016. Addictions neuroclinical assessment: A neuroscience-based framework for addictive disorders. Biological Psychiatry 80, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon DY, Zhao Y-T, Lamonica JM, Zhou Z, 2017. Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat Commun 8, 15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyzar EJ, Bohnsack JP, Zhang H, Pandey SC, 2019a. MicroRNA-137 drives epigenetic reprogramming in the adult amygdala and behavioral changes after adolescent alcohol exposure. eNeuro 6, ENEURO.0401–19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyzar EJ, Floreani C, Teppen TL, Pandey SC, 2016. Adolescent alcohol exposure: Burden of epigenetic reprogramming, synaptic remodeling, and adult psychopathology. Frontiers in Neuroscience 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyzar EJ, Zhang H, Pandey SC, 2019b. Adolescent alcohol exposure epigenetically suppresses amygdala Arc enhancer RNA expression to confer adult anxiety susceptibility. Biological Psychiatry 85, 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyzar EJ, Zhang H, Sakharkar AJ, Pandey SC, 2017. Adolescent alcohol exposure alters lysine demethylase 1 (LSD1) expression and histone methylation in the amygdala during adulthood. Addict Biol 22, 1191–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW, 2010. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet 11, 191–203. [DOI] [PubMed] [Google Scholar]
- Laumet G, Garriga J, Chen S-R, Zhang Y, Li D-P, Smith TM, Dong Y, Jelinek J, Cesaroni M, Issa J-P, Pan H-L, 2015. G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nat Neurosci 18, 1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-K, Shibata Y, Rao B, Strahl BD, Lieb JD, 2004. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet 36, 900–905. [DOI] [PubMed] [Google Scholar]
- Lee KK, Workman JL, 2007. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 8, 284–295. [DOI] [PubMed] [Google Scholar]
- Lepack AE, Werner CT, Stewart AF, Fulton SL, Zhong P, Farrelly LA, Smith ACW, Ramakrishnan A, Lyu Y, Bastle RM, Martin JA, Mitra S, O’Connor RM, Wang Z-J, Molina H, Turecki G, Shen L, Yan Z, Calipari ES, Dietz DM, Kenny PJ, Maze I, 2020. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science 368, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson VV, 2010. DNA methylation as a universal biomarker. Expert Review of Molecular Diagnostics 10, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhao H, Gelernter J, 2012. Further clarification of the contribution of the ADH1C gene to vulnerability of alcoholism and selected liver diseases. Hum Genet 131, 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Beard C, Jaenisch R, 1993. Role for DNA methylation in genomic imprinting. Nature 366, 362–365. [DOI] [PubMed] [Google Scholar]
- Li E, Zhang Y, 2014. DNA methylation in mammals. Cold Spring Harbor Perspectives in Biology 6, a019133–a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Fischle W, Wang W, Duncan EM, Liang L, Murakami-Ishibe S, Allis CD, Patel DJ, 2007. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Molecular Cell 28, 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Justice AC, So-Armah K, Krystal JH, Sinha R, Xu K, 2020. DNA methylation signature on phosphatidylethanol, not on self-reported alcohol consumption, predicts hazardous alcohol consumption in two distinct populations. Mol Psychiatry. 10.1038/s41380-020-0668-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR, 2013. Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905–1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR, 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Marioni RE, Hedman ÅK, Pfeiffer L, Tsai P-C, Reynolds LM, Just AC, Duan Q, Boer CG, Tanaka T, Elks CE, Aslibekyan S, Brody JA, Kühnel B, Herder C, Almli LM, Zhi D, Wang Y, Huan T, Yao C, Mendelson MM, Joehanes R, Liang L, Love S-A, Guan W, Shah S, McRae AF, Kretschmer A, Prokisch H, Strauch K, Peters A, Visscher PM, Wray NR, Guo X, Wiggins KL, Smith AK, Binder EB, Ressler KJ, Irvin MR, Absher DM, Hernandez D, Ferrucci L, Bandinelli S, Lohman K, Ding J, Trevisi L, Gustafsson S, Sandling JH, Stolk L, Uitterlinden AG, Yet I, Castillo-Fernandez JE, Spector TD, Schwartz JD, Vokonas P, Lind L, Li Y, Fornage M, Arnett DK, Wareham NJ, Sotoodehnia N, Ong KK, van Meurs JBJ, Conneely KN, Baccarelli AA, Deary IJ, Bell JT, North KE, Liu Y, Waldenberger M, London SJ, Ingelsson E, Levy D, 2018. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry 23, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R, 2016. Editing DNA methylation in the mammalian genome. Cell 167, 233–247.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenarz C, Ge W, Coleman ML, Rose NR, Cooper CDO, Klose RJ, Ratcliffe PJ, Schofield CJ, 2010. PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nε-dimethyl lysine demethylase. Human Molecular Genetics 19, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohoff FW, Roy A, Jung J, Longley M, Rosoff DB, Luo A, O’Connell E, Sorcher JL, Sun H, Schwandt M, Hodgkinson CA, Goldman D, Momenan R, McIntosh AM, Adams MJ, Walker RM, Evans KL, Porteous D, Smith AK, Lee J, Muench C, Charlet K, Clarke T-K, Kaminsky ZA, 2020. Epigenome-wide association study and multi-tissue replication of individuals with alcohol use disorder: evidence for abnormal glucocorticoid signaling pathway gene regulation. Mol Psychiatry. 10.1038/s41380-020-0734-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Moreno JA, Marcos M, Calleja-Conde J, Echeverry-Alzate V, Bühler KM, Costa-Alba P, Bernardo E, Laso F-J, Rodríguez de Fonseca F, Nadal R, Viveros MP, Maldonado R, Giné E, 2015. Histone deacetylase gene expression following binge alcohol consumption in rats and humans. Alcohol Clin Exp Res 39, 1939–1950. [DOI] [PubMed] [Google Scholar]
- Lorincz MC, Dickerson DR, Schmitt M, Groudine M, 2004. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol 11, 1068–1075. [DOI] [PubMed] [Google Scholar]
- Lorsch ZS, Hamilton PJ, Ramakrishnan A, Parise EM, Salery M, Wright WJ, Lepack AE, Mews P, Issler O, McKenzie A, Zhou X, Parise LF, Pirpinias ST, Ortiz Torres I, Kronman HG, Montgomery SE, Loh Y-HE, Labonté B, Conkey A, Symonds AE, Neve RL, Turecki G, Maze I, Dong Y, Zhang B, Shen L, Bagot RC, Nestler EJ, 2019. Stress resilience is promoted by a Zfp189-driven transcriptional network in prefrontal cortex. Nat Neurosci 22, 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wang GG, 2013. Tudor: a versatile family of histone methylation ‘readers.’ Trends in Biochemical Sciences 38, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD, 2008. Epigenetic regulation of bdnf gene transcription in the consolidation of fear memory. Journal of Neuroscience 28, 10576–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DK, Jang M-H, Guo JU, Kitabatake Y, Chang M. -l., Pow-anpongkul N, Flavell RA, Lu B, Ming G. -l., Song H, 2009. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzardo AM, Henkhaus RS, Butler MG, 2012. Global DNA promoter methylation in frontal cortex of alcoholics and controls. Gene 498, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P, Miller T, Richon VM, 2003. Histone deacetylases. Current Opinion in Pharmacology 3, 344–351. [DOI] [PubMed] [Google Scholar]
- Martinowich K, 2003. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science 302, 890–893. [DOI] [PubMed] [Google Scholar]
- Maze I, Covington HE, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ, 2010. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Wenderski W, Noh K-M, Bagot RC, Tzavaras N, Purushothaman I, Elsässer SJ, Guo Y, Ionete C, Hurd YL, Tamminga CA, Halene T, Farrelly L, Soshnev AA, Wen D, Rafii S, Birtwistle MR, Akbarian S, Buchholz BA, Blitzer RD, Nestler EJ, Yuan Z-F, Garcia BA, Shen L, Molina H, Allis CD, 2015. Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87, 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Donahue G, Drake AM, Luczak V, Abel T, Berger SL, 2017. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, Alexander DC, Riesche SL, Heller EA, Nestler EJ, Garcia BA, Berger SL, 2019. Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE, 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD, 2007. Covalent Modification of DNA Regulates Memory Formation. Neuron 53, 857–869. [DOI] [PubMed] [Google Scholar]
- Mohandas T, Sparkes R, Shapiro L, 1981. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science 211, 393–396. [DOI] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D, 2008. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Molecular Cell 30, 755–766. [DOI] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Pandey SC, 2011. The role of amygdaloid brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein and dendritic spines in anxiety and alcoholism. Addiction Biology 16, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC, 2013. Aberrant histone deacetylase2–mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biological Psychiatry 73, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Teppen TL, Miller KM, Sexton HG, Pandey SC, Swartzwelder HS, 2018. Donepezil reverses dendritic spine morphology adaptations and Fmr1 epigenetic modifications in hippocampus of adult rats after adolescent alcohol exposure. Alcohol: Clin Exp Res 42, 706–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Lalonde M-E, Côté J, Kutateladze TG, 2012. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol 19, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nady N, Lemak A, Walker JR, Avvakumov GV, Kareta MS, Achour M, Xue S, Duan S, Allali-Hassani A, Zuo X, Wang Y-X, Bronner C, Chédin F, Arrowsmith CH, Dhe-Paganon S, 2011. Recognition of multivalent histone states associated with heterochromatin by UHRF1 protein. J. Biol. Chem 286, 24300–24311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Geen H, Ren C, Nicolet CM, Perez AA, Halmai J, Le VM, Mackay JP, Farnham PJ, Segal DJ, 2017. dCas9-based epigenome editing suggests acquisition of histone methylation is not sufficient for target gene repression. Nucleic Acids Research 45, 9901–9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E, 1999. DNA Methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. [DOI] [PubMed] [Google Scholar]
- Okano M, Xie S, Li E, 1998. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19, 219–220. [DOI] [PubMed] [Google Scholar]
- Oliveira AMM, Estevez MA, Hawk JD, Grimes S, Brindle PK, Abel T, 2011. Subregion-specific p300 conditional knock-out mice exhibit long-term memory impairments. Learning & Memory 18, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin S-P, Allis CD, Cheng X, Bestor TH, 2007. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE, 2012. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H, & Xu T, 2004. Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. Journal of Neuroscience 24, 5022–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, 2003. Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends in Pharmacological Sciences 24, 456–460. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Kyzar EJ, Zhang H, 2017. Epigenetic basis of the dark side of alcohol addiction. Neuropharmacology 122, 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H, 2003. The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats: Alcohol: Clin Exp Res 27, 396–409. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Sakharkar AJ, Tang L, Zhang H, 2015. Potential role of adolescent alcohol exposure-induced amygdaloid histone modifications in anxiety and alcohol intake during adulthood. Neurobiol Dis 82, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A, 2008a. Brain Chromatin Remodeling: A Novel Mechanism of Alcoholism. Journal of Neuroscience 28, 3729–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Xu T, 2005. Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. Journal of Clinical Investigation,115,2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Ugale R, Prakash A, Xu T, Misra K, 2008b. Effector immediate-early gene Arc in the amygdala plays a critical role in alcoholism. Journal of Neuroscience 28, 2589–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD, 2012. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. Journal of Neuroscience 32, 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott CA, Kendler KS, 1999. Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins. AJP 156, 34–40. [DOI] [PubMed] [Google Scholar]
- Pu M, Ni Z, Wang M, Wang X, Wood JG, Helfand SL, Yu H, Lee SS, 2015. Trimethylation of Lys36 on H3 restricts gene expression change during aging and impacts life span. Genes Dev. 29, 718–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Min J, 2014. Structure and function of the nucleosome-binding PWWP domain. Trends in Biochemical Sciences 39, 536–547. [DOI] [PubMed] [Google Scholar]
- Qiu C, Sawada K, Zhang X, Cheng X, 2002. The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat. Struct Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier DA, 1990. Comorbidity of mental disorders with alcohol and other drug abuse: Results from the epidemiologic catchment area (ECA) study. JAMA 264, 2511. [PubMed] [Google Scholar]
- Rehm J, Greenfield TK, Kerr W, 2006. Patterns of drinking and mortality from different diseases—an overview. Contemporary Drug Problems 33, 205–235. [Google Scholar]
- Reik W, 2007. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432. [DOI] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S, 2007. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4, 651–657. [DOI] [PubMed] [Google Scholar]
- Rona GB, Eleutherio ECA, Pinheiro AS, 2016. PWWP domains and their modes of sensing DNA and histone methylated lysines. Biophys Rev 8, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD, 2001. Histone acetyltransferases. Annu. Rev. Biochem 70, 81–120. 10.1146/annurev.biochem.70.1.81 [DOI] [PubMed] [Google Scholar]
- Rothbart SB, Dickson BM, Ong MS, Krajewski K, Houliston S, Kireev DB, Arrowsmith CH, Strahl BD, 2013. Multivalent histone engagement by the linked tandem Tudor and PHD domains of UHRF1 is required for the epigenetic inheritance of DNA methylation. Genes & Development 27, 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI, Barsyte-Lovejoy D, Martinez JY, Bedford MT, Fuchs SM, Arrowsmith CH, Strahl BD, 2012. Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat Struct Mol Biol 19, 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD, 2015. 2010 National and state costs of excessive alcohol consumption. American Journal of Preventive Medicine 49, e73–e79. [DOI] [PubMed] [Google Scholar]
- Saito M, Ishikawa F, 2002. The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J. Biol. Chem 277, 35434–35439. [DOI] [PubMed] [Google Scholar]
- Sakharkar AJ, Kyzar EJ, Gavin DP, Zhang H, Chen Y, Krishnan HR, Grayson DR, Pandey SC, 2019. Altered amygdala DNA methylation mechanisms after adolescent alcohol exposure contribute to adult anxiety and alcohol drinking. Neuropharmacology 157, 107679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Vetreno RP, Zhang H, Kokare DM, Crews FT, Pandey SC, 2016. A role for histone acetylation mechanisms in adolescent alcohol exposure-induced deficits in hippocampal brain-derived neurotrophic factor expression and neurogenesis markers in adulthood. Brain Struct Funct 221, 4691–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Baxstrom K, Shi G, Moonat S, Pandey SC, 2014. Effects of histone deacetylase inhibitors on amygdaloid histone acetylation and neuropeptide Y expression: a role in anxiety-like and alcohol-drinking behaviours. Int. J. Neuropsychopharm 17, 1207–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC, 2012. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: A role in Rapid tolerance to the anxiolytic effects of ethanol. Alcohol: Clin Exp Res 36, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R, Zhou M-M, 2011. The PHD finger: a versatile epigenome reader. Trends in Biochemical Sciences 36,364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval A, Elahi H, Ploski JE, 2020. Genetically engineering the nervous system with CRISPR-Cas. eNeuro 7, ENEURO.0419–19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor GC, Powell SK, Brothers SP, Wahlestedt C, 2015. Epigenetic readers of lysine acetylation regulate cocaine-induced plasticity. Journal of Neuroscience 35, 15062–15072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Imai S, Guarente L, 2017. The brain, sirtuins, and ageing. Nat Rev Neurosci 18, 362–374. [DOI] [PubMed] [Google Scholar]
- Savell KE, Gallus NVN, Simon RC, Brown JA, Revanna JS, Osborn MK, Song EY, O’Malley JJ, Stackhouse CT, Norvil A, Gowher H, Sweatt JD, Day JJ, 2016. Extra-coding RNAs regulate neuronal DNA methylation dynamics. Nat Commun 7, 12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, Tarakhovsky A, Greengard P, 2009. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 64, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuckit MA, 1996. An 8-Year Follow-up of 450 Sons of alcoholic and control subjects. Arch Gen Psychiatry 53, 202. [DOI] [PubMed] [Google Scholar]
- Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z, Frederick B, Chen Q, Luense LJ, Garcia BA, Dang W, Johnson FB, Adams PD, Schultz DC, Berger SL, 2019. Histone acetyltransferase p300 induces de novo super-enhancers to drive cellular senescence. Molecular Cell 73, 684–698.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF, 2011. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci 14, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Yujiang, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Yang Shi, 2004. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S, 2011. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Lopes M, Lund PJ, Goldman N, Fasolino M, Coradin M, Kulej K, Bhanu NV, Vahedi G, Garcia BA, 2019. A mass spectrometry-based assay using metabolic labeling to rapidly monitor chromatin accessibility of modified histone proteins. Sci Rep 9, 13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W, 2006. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat Genet 38, 1348–1354. [DOI] [PubMed] [Google Scholar]
- Sims RJ, Millhouse S, Chen C-F, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D, 2007. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription post initiation factors and pre-mRNA splicing. Molecular Cell 28, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S, 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A, 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares LM, He PC, Chun Y, Suh H, Kim T, Buratowski S, 2017. Determinants of histone H3K4 methylation patterns. Molecular Cell 68, 773–785.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkman BG, Sakharkar AJ, Pandey SC, 2012. Epigenetics-beyond the genome in alcoholism. Alcohol Res 34, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD, 2000. The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Subbanna S, Nagre NN, Shivakumar M, Basavarajappa BS, 2016. A single day of 5-azacytidine exposure during development induces neurodegeneration in neonatal mice and neurobehavioral deficits in adult mice. Physiol. Behav 167, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Nagre NN, Shivakumar M, Umapathy NS, Psychoyos D, Basavarajappa BS, 2014. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 258, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Nagre NN, Umapathy NS, Pace BS, Basavarajappa BS, 2015. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. International Journal of Neuropsychopharmacology 18, pyu028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbanna S, Shivakumar M, Umapathy NS, Saito M, Mohan PS, Kumar A, Nixon RA, Verin AD, Psychoyos D, Basavarajappa BS, 2013. G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiology of Disease 54, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration & Center for Behavioral Health Statistics and, Quality., 2014. The NSDUH Report: Substance Use and Mental Health Estimates from the 2013 National Survey on Drug Use and Health: Overview of Findings and Detailed Tables. [PubMed]
- Suetake I, Shinozaki F, Miyagawa J, Takeshima H, Tajima S, 2004. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J. Biol. Chem 279, 27816–27823. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A, 2009. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Cook J, Choquet H, Jorgenson E, Yin J, Kinnunen T, Barclay J, Morris AP, Pirmohamed M, 2020. Functional validity, role, and implications of heavy alcohol consumption genetic loci. Sci. Adv 6, eaay5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y, 2006. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. [DOI] [PubMed] [Google Scholar]
- Veazey KJ, Parnell SE, Miranda RC, Golding MC, 2015. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenetics & Chromatin 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, Mann M, 2010. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967–980. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Bohnsack JP, Kusumo H, Liu W, Pandey SC, Crews FT, 2019. Neuroimmune and epigenetic involvement in adolescent binge ethanol-induced loss of basal forebrain cholinergic neurons: Restoration with voluntary exercise. Addiction Biology 25: e12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viner RM, Taylor B, 2007. Adult outcomes of binge drinking in adolescence: findings from a UK national birth cohort. J Epidemiol Community Health 61, 902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Xu H, Zhao H, Gelernter J, Zhang H, 2016. DNA co-methylation modules in postmortem prefrontal cortex tissues of European Australians with alcohol use disorders. Sci Rep 6, 19430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Telese F, Tan Y, Li W, Jin C, He X, Basnet H, Ma Q, Merkurjev D, Zhu X, Liu Z, Zhang J, Ohgi K, Taylor H, White RR, Tazearslan C, Suh Y, Macfarlan TS, Pfaff SL, Rosenfeld MG, 2015. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat Neurosci 18, 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WK, Tereshko V, Boccuni P, MacGrogan D, Nimer SD, Patel DJ, 2003. Malignant Brain Tumor Repeats. Structure 11, 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Reddy B, Thompson J, Wang H, Noma K, Yates JR, Jia S, 2009. Regulation of Set9-mediated H4K20 methylation by a PWWP domain protein. Molecular Cell 33, 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh T-Y, Peng W, Zhang MQ, Zhao K, 2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnault V, Darcq E, Levine A, Barak S, Ron D, 2013. Chromatin remodeling — a novel strategy to control excessive alcohol drinking. Transl Psychiatry 3, e231–e231. 10.1038/tp.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JE, Horwood LJ, Fergusson DM, 2004. Drinking patterns in mid-adolescence and psychosocial outcomes in late adolescence and early adulthood. Addiction 99, 1529–41. [DOI] [PubMed] [Google Scholar]
- Winston F, Allis CD, 1999. The bromodomain: a chromatin-targeting module? Nat. Struct Biol 6, 601–604. [DOI] [PubMed] [Google Scholar]
- Wolstenholme JT, Mahmood T, Harris GM, Abbas S, Miles MF, 2017. Intermittent ethanol during adolescence leads to lasting behavioral changes in adulthood and alters gene expression and histone methylation in the PFC. Front. Mol. Neurosci 10, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS, 1997. Characterization of the CHD family of proteins. Proceedings of the National Academy of Sciences 94, 11472–11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y, 2011. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes & Development 25, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Hong, Zeng H, Lam R, Tempel W, Amaya MF, Xu C, Dombrovski L, Qiu W, Wang Y, Min J, 2011. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE 6, e18919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, Liu Y, Byrum SD, Mackintosh SG, Zhong M, Tackett A, Wang G, Hon LS, Fang G, Swenberg JA, Xiao AZ, 2016. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 532, 329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G-L, Bestor TH, Bourc’his D, Hsieh C-L, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Péquignot E, 1999. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191. [DOI] [PubMed] [Google Scholar]
- Xu X, Tao Y, Gao X, Zhang L, Li X, Zou W, Ruan K, Wang F, Xu G, Hu R, 2016. A CRISPR-based approach for targeted DNA demethylation. Cell Discov 2, 16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Schoonjans K, Auwerx J, 2007. Sirtuin Functions in Health and Disease. Molecular Endocrinology 21, 1745–1755. [DOI] [PubMed] [Google Scholar]
- You C, Vandegrift BJ, Zhang H, Lasek AW, Pandey SC, Brodie MS, 2018. Histone deacetylase inhibitor suberanilohydroxamic acid treatment reverses hyposensitivity to γ-aminobutyric acid in the ventral tegmental area during ethanol withdrawal. Alcohol Clin Exp Re 42, 2160–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC, 2014. Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int. J. Neuropsychopharm 17, 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kyzar EJ, Bohnsack JP, Kokare DM, Teppen T, Pandey SC, 2018. Adolescent alcohol exposure epigenetically regulates CREB signaling in the adult amygdala. Scientific Reports 8, 10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sakharkar AJ, Shi G, Ugale R, Prakash A, Pandey SC, 2010. Neuropeptide Y signaling in the central nucleus of amygdala regulates alcohol-drinking and anxiety-like behaviors of alcohol-preferring rats. Alcohol: Clin Exp Res 34, 451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yuan Q, Mash DC, Goldman D, 2011. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proceedings of the National Academy of Sciences 108, 6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]