

ATP1A3, a mutated gene for AHC, RDP, and CAPOS, is also associated with polymicrogyria by different functional variants.
Abstract
Polymicrogyria is a common malformation of cortical development whose etiology remains elusive. We conducted whole-exome sequencing for 124 patients with polymicrogyria and identified de novo ATP1A3 variants in eight patients. Mutated ATP1A3 causes functional brain diseases, including alternating hemiplegia of childhood (AHC), rapid-onset dystonia parkinsonism (RDP), and cerebellar ataxia, areflexia, pes cavus, optic nerve atrophy, and sensorineural deafness (CAPOS). However, our patients showed no clinical features of AHC, RDP, or CAPOS and had a completely different phenotype: a severe form of polymicrogyria with epilepsy and developmental delay. Detected variants had different locations in ATP1A3 and different functional properties compared with AHC-, RDP-, or CAPOS-associated variants. In the developing cerebral cortex of mice, radial neuronal migration was impaired in neurons overexpressing the ATP1A3 variant of the most severe patients, suggesting that this variant is involved in cortical malformation pathogenesis. We propose a previously unidentified category of polymicrogyria associated with ATP1A3 abnormalities.
INTRODUCTION
Malformations of cortical development occur when neuroblast proliferation, differentiation, migration, or cortical organization is interrupted during human brain development. Polymicrogyria is a common form of malformation of cortical development and is defined by the existence of many abnormally small gyri, producing an irregular and fused cortical surface (1). Clinically, polymicrogyria causes various neurological symptoms, such as epilepsy, intellectual disability, and impaired oromotor function. Both genetic and nongenetic factors (e.g., hypoxic insults or intrauterine cytomegalovirus infection) cause this disease; however, only a minority of patients can be explained by a known genetic cause.
Na+/K+ adenosine triphosphatases (Na+/K+ ATPases) are ubiquitously expressed transmembrane ion pumps that actively export three Na+ and import two K+ using hydrolyzed adenosine 5′-triphosphate (ATP) energy. These ion pumps maintain ion gradients across the cell membrane and consist of α-, β-, and sometimes regulatory γ-subunits. The α-subunit holds cytosolic ATPase machinery and the ion-transporting membrane domain, while the β-subunit is necessary for proper trafficking to the plasma membrane and supports the functional expression and kinetic properties of the α-subunit (2). Tissue-specific isoform expressions are known among the four α- and three β-subunit isoforms that exist in humans. In the central nervous system, the α1 isoform is ubiquitously expressed, while α2 is expressed in astrocytes and α3 in neurons (3).
Dominant mutations in ATP1A3, which encodes the α3-subunit, cause characteristic functional brain diseases known as ATP1A3-related disorders, which have at least three distinct phenotypes: alternating hemiplegia of childhood (AHC) (4); rapid-onset dystonia parkinsonism (RDP) (5); and cerebellar ataxia, areflexia, pes cavus, optic nerve atrophy, and sensorineural deafness (CAPOS) (6). Dominant mutations in ATP1A3 also cause forms of developmental and epileptic encephalopathies, such as early infantile epilepsy and encephalopathy (EIEE) with or without apnea (7), relapsing encephalopathy with cerebellar ataxia (8), or fever-induced paroxysmal weakness and encephalopathy (9). Although AHC, RDP, and CAPOS have distinct neurological symptoms, they are recognized as part of a continuum and share some core clinical features (10).
Here, we report eight patients from unrelated families with de novo variants in ATP1A3, presenting with none of the features of AHC, RDP, or CAPOS. These patients had polymicrogyria with extremely severe epilepsy, intellectual disability, and sometimes progressive brain atrophy and cardiac failure. We also reveal that the specific variant localizations in ATP1A3 in our patients differ from those of AHC, RDP, or CAPOS in terms of three-dimensional protein structure and functional consequences. Furthermore, we show the possible association between an identified variant and defects in cortical architecture using embryonic mice expressing this variant. These results imply that our patients demonstrate a novel phenotype associated with ATP1A3 abnormality.
RESULTS
Clinical analysis
Clinical features are summarized in Table 1 in order of clinical severity (from patients 1 to 8). Patient 5 was Israeli, and the other patients were Japanese. No patients satisfied the diagnostic criteria for AHC, RDP, or CAPOS, but all patients had bilateral polymicrogyria of either the frontoparietal or perisylvian regions (Fig. 1 and fig. S1). Furthermore, all patients exhibited extremely severe clinical features. Of the seven patients with seizures, onset was at 1 day old in four patients and by 4 days old in three patients. Seizure types varied, including tonic-clonic, autonomic, or myoclonic seizures. Two patients had well-controlled or partially controlled seizures (patients 2 and 5), and the remaining five patients had intractable and frequent seizures. All patients had severe developmental delay, and only one achieved social smile and head control (patient 8); this patient had no seizures. At least three patients demonstrated progressive cerebral and cerebellar atrophy (patients 1, 3, and 4) (Fig. 1 and figs. S2 and S3), and one patient (patient 6) showed progressive atrophy at the cerebellar vermis only. Patient 2 underwent magnetic resonance imaging (MRI) only once, and no evaluation of brain atrophy has been performed since. Two patients (patients 1 and 2) had episodic tachycardia. This resulted in congestive heart failure in patient 1, and in patient 2, no changes in amplitude-integrated electroencephalogram were observed during the attack, suggesting that it was unlikely to be an autonomic seizure. Two patients (patients 1 and 3) had cardiac failure, which is rarely observed in typical polymicrogyria cases (see the Supplementary Materials for more clinical details).
Table 1. Clinical summary of the patients with ATP1A3 variants and polymicrogyria.
DQ, developmental quotient; EEG, electroencephalogram; CT, computed tomography; MRI, magnetic resonance imaging.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Familial occurrence | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic |
|
ATP1A3 mutation (NM_152296.4) |
c.2976_ 2978del, p.(Asp992del) de novo |
c.2976_2978del, p.(Asp992del) de novo |
c.2972_2982delins TTGCGCAT CTTCATCTG, p.(Tyr4_Ile994delins PheAlaHisLeuHisLeu) de novo |
c.2975_2983dup, p.(Ile994_ Arg995ins HisGluIle) de novo |
c.2570_2572del, p.(Phe857del) de novo |
c.2560_ 2568del, p.(Gly854_Phe856del) de novo |
c.1787G>A, p.(Cys596Tyr) de novo |
c.2684A>C, p.(Gln895Pro) de novo |
| Ethnicity | Japanese | Japanese | Japanese | Japanese | Israeli | Japanese | Japanese | Japanese |
| Age | 3 years 10 months |
1 year 4 months |
7 years 0 months | 5 years 9 months |
2 years 6 months (deceased at 3 years) |
10 years 9 months |
5 years 0 months |
6 years 10 months |
| Sex | Male | Female | Male | Female | Female | Male | Female | Male |
| Clinical diagnosis | Perisylvian polymicrogyria |
Frontoparietal polymicrogyria |
Frontoparietal polymicrogyria |
Frontoparietal polymicrogyria |
Perisylvian polymicrogyria |
Frontoparietal polymicrogyria |
Perisylvian polymicrogyria |
Perisylvian polymicrogyria |
| Gestation | 37 weeks | 35 weeks | 39 weeks | 38 weeks | 35 weeks | 37 weeks | 41 weeks | 40 weeks |
| Birth length | 40.0 cm (−2.92 SD) |
40.0 cm (−2.24 SD) |
48.0 cm (−0.46 SD) |
43.6 cm (−2.38 SD) |
Unknown | 49.5 cm (+1.01 SD) |
Unknown | 49.0 cm (−0.00 SD) |
| Birth weight | 2152 g (−1.53 SD) |
2450 g (+0.17 SD) |
2754 g (−1.01 SD) |
2622 g (−0.82 SD) |
1820 g (10th–25th percentile) |
2928 g (+0.76 SD) |
2822 g (−0.91 SD) |
3438 g (+0.68 SD) |
| Birth head circumference |
32.0 cm (−0.48 SD) |
32.0 cm (+0.47 SD) |
31.0 cm (−1.64 SD) |
31.5 cm (−1.21 SD) |
30 cm (10th percentile) |
33.0 cm (+0.17 SD) |
31.5 cm (−1.67 SD) |
33.0 cm (−0.49 SD) |
| Facial dysmorphism | No | No | Retrognathia and partial cleft palate |
No | No | No | Telecanthus and micrognathia |
No |
| Other malformations |
Hypo pigmented brownish hair |
No | Bell-shaped narrow chest, single transverse palmar crease |
No | No | No | No | No |
| Seizure onset | 30 s | Soon after birth | 3 days | 10 min | 4 days | 1 hour | 2 days | No |
| Seizure types | Tonic convulsions, clonic convulsions, eye deviation, eye blinking, and twitching of the corner of the mouth |
Generalized tonic-clonic seizures |
Right-side dominant tonic-clonic convulsions, tonic seizures, focal or generalized clonic seizures, and autonomic seizures |
Focal motor seizures and autonomic seizures |
Generalized seizures starting with increased tonus in the left hand and foot, the head and eyes turn to the left, and sometimes, the body becomes tonic and clonic |
Apneic spells and tonic seizures |
Eye opening and nystagmus with bilateral extremity quivering and facial stiffening, followed by irregular respiration and cyanosis |
No |
| EEG findings | Suppression burst at 1 month and multifocal spikes at 2 years |
Frequent multifocal sharp waves with diffuse slow waves |
Interictal: frequent sharp waves on T3 propagating to the left hemisphere; ictal: rhythmic activity at T3 or F4 to C4 |
Bilateral frontal sharp waves with decreased background activity |
Abnormal background activity and slow delta waves |
Bilateral spikes with a background of slow waves |
Multifocal epileptic discharges with a background of continuous diffuse slow waves |
Normal at 3 months |
| Prognosis of seizures |
Intractable, hourly |
Seizure-free | Intractable, daily | Intractable, hourly |
Intractable, partially controlled |
Intractable, daily |
Intractable, daily |
Seizure-free |
| Development | No social smile, no head control, DQ < 10 |
No social smile, no head control |
No social smile, no head control, DQ = 2.5 |
No social smile, no head control |
No eye contact, no head control |
No social smile, no head control, DQ < 10 |
No social smile, no head control, DQ < 10 |
No meaningful words, no rolling over |
| Intracranial calcifications on CT |
No | Yes | No | No | Unknown | No | No | No |
| Brain MRI findings |
Poly microgyria |
Poly microgyria |
Poly microgyria |
Poly microgyria |
Poly microgyria |
Poly microgyria |
Poly microgyria |
Poly microgyria |
| Progressive brain atrophy |
Yes | Unknown | Yes | Yes | Unknown | Cerebellar vermis only |
No | No |
| Laboratory findings | Hypo thyroidism |
Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| Other characteristic features |
Congestive cardiac failure during tachycardia |
Episodic tachycardia, right hearing impairment, and tracheostomy at 3 months |
Recurrent cardiac failure in early infancy |
Coagulation abnormalities induced by phenobarbital, requiring mechanical ventilation |
No | No | No | Severe spastic quadriplegia |
|
Diagnostic criteria of AHC | ||||||||
| Repeated attacks of hemiplegia or quadriparesis |
No | No | No | No | No | No | No | No |
| Relief from symptoms upon sleep |
No | No | No | No | No | No | Unknown | No |
| Involuntary movement such as choreoathetosis, dystonia, or ataxia |
No | No | No | No | No | No | No | Dystonia |
|
Supporting feature of AHC | ||||||||
| Repeated attacks of monocular nystagmus |
No | No | No | No | No | No | No | No |
|
Diagnostic criteria of RDP | ||||||||
| Parkinson’s disease | No | No | No | No | No | No | No | No |
| Clear rostrocaudal gradient of involvement |
No | No | No | No | No | No | Unknown | No |
| Prominent bulbar findings on examination |
No | No | No | No | No | No | No | No |
|
Diagnostic criteria of CAPOS | ||||||||
| Cerebellar ataxia | Unknown | Unknown (no voluntary movement) |
No | Unknown (no voluntary movement) |
No | No | Unknown | No |
| Areflexia | Yes | No | Yes | No | No | No | No | No |
| Pes cavus | No | No | No | No | No | No | No | No |
| Optic nerve atrophy | No | Unknown | Unknown | Unknown | No, but cortical blindness |
No | No | No |
| Sensorineural hearing loss |
Yes | Unknown | Unknown | Unknown | No, but normal auditory brainstem response |
No | Yes | No |
Fig. 1. Bilateral perisylvian polymicrogyria associated with de novo ATP1A3 mutations.
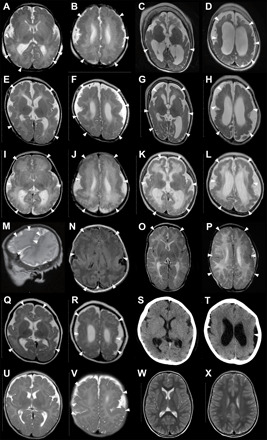
Brain MRI scans of patient 1 [(A) and (B) at 1 day old, (C) and (D) at 2 years and 10 months], patient 3 [(E) and (F) at 2 months old, (G) and (H) at 8 months], patient 4 [(I) and (J) at 3 days old, (K) and (L) at 4 months], patient 5 [(M) and (N) at 2 days old], patient 6 [(O) and (P) at 2 months old], patient 7 [(Q) and (R) at 5 months old, (S) and (T) at 3 years old], patient 8 [(U) and (V) at 2 months old], and a neurologically normal control [(W) and (X) at 3 years old]. All images are T2-weighted axial brain MRI, except for a T2-weighted sagittal MRI (M), a fluid-attenuated inversion recovery axial MRI (N), and an axial cranial CT (S and T). Brain MRI showed irregular small gyri with apparent cortical thickening, compatible with polymicrogyria, over the perisylvian cortex (arrowheads). Patients 1, 3, and 4 (A to L) showed progressive atrophic changes with dilation of the subarachnoid space and lateral ventricles.
Genetic analysis
Using whole-exome sequencing, we identified seven de novo variants in ATP1A3 (NM_152296.4) in 8 of 124 patients, a diagnostic yield of approximately 6.5%. Five variants were in-frame changes (p.Asp992del in patients 1 and 2, p.Tyr991_Ile994delinsPheAlaHisLeuHisLeu in patient 3, p.Ile994_Arg995insHisGluIle in patient 4, p.Phe857del in patient 5, and p.Gly854_Phe856del in patient 6), and two were missense (p.Cys596Tyr in patient 7 and p.Gln895Pro in patient 8) (Table 1). All variants were confirmed by Sanger sequencing (fig. S4) and were considered pathogenic on the basis of population allele frequency and in silico predictions (table S1). Six variants were novel, while the variant in patient 5 had been previously reported in a patient with nervous system abnormality (11). The variants in patients 1, 2, and 7 were novel changes at the same amino acid residues where different substitutions had previously been reported in patients with AHC [p.Asp992Tyr (4) and p.Cys596Arg (12)]. We were unable to find further clinical information on these patients in the published reports.
Structural considerations of the mutations identified in the Na+/K+ ATPase α3-subunit
To evaluate the impact of the identified mutations in the Na+/K+ ATPase α3-subunit (hereafter called the α-subunit), we mapped the mutation sites onto the crystal structure of Squalus acanthias Na+/K+ ATPase [Protein Data Bank (PDB) code: 2ZXE] (13), which Phyre2 predicts to be highly homologous to human Na+/K+ ATPase (14). The structure of S. acanthias Na+/K+ ATPase consists of the catalytic α- and β-subunits and regulatory γ-subunit (Fig. 2A). The β-subunit is essential for the correct integration of the α-subunit into the lipid bilayer and for structural stabilization of the α-subunit by preventing its degradation (15). The mutation sites in human Na+/K+ ATPase are located in the extracellular, transmembrane, and cytoplasmic regions of the α-subunit.
Fig. 2. Structural considerations of the identified mutations in human Na+/K+ ATPase α-subunits.
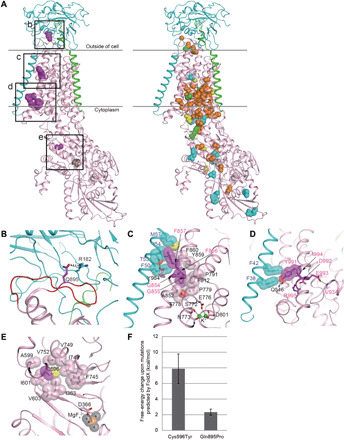
(A) The crystal structure of Na+/K+ ATPase, composed of α- (pink), β- (cyan), and γ-subunits (green), from Squalus acanthias (PDB code: 2ZXE) is shown with the residues (magenta spheres) corresponding to the identified variants in humans. The squares (b to e) correspond to the magnified areas (B to E), respectively. In the right structure, spheres indicate the residues corresponding to the previously reported variants associated with AHC (orange), RDP (cyan), both AHC and RDP (green), and other phenotypes (yellow). (B) The p.Arg182 (cyan) of the β-subunit interacting with p.Gln895 (magenta) in a loop of p.Asp882–Trp896 (red wire) of the α-subunit via hydrogen bonds (black dotted lines). The residue numbering is based on human Na+/K+ ATPase. (C) The interactions between α- and β-subunits around the p.Gly854 to Phe857 region of the α-subunit. The side chains of p.Phe856 and p.Phe857 and their interacting residues (β-subunit) are shown by sticks with translucent van der Waals spheres. The conformation of p.Met57 (β-subunit) in humans was modeled instead of leucine in the S. acanthias structure. The sulfur and oxygen atoms are shown in yellow and red, respectively. The residues interacting with K+ ions (green) are depicted as sticks. (D) The interactions between α and β subunits around the p.Tyr991-Arg995 region of the α-subunit. The p.Tyr991 and its interacting residues, p.Phe38 and p.Phe42 (β-subunit), are shown as sticks with translucent spheres. The conformation of p.Ile994 in humans was modeled instead of methionine in the S. acanthias structure. (E) A hydrophobic core involving p.Cys596 near the p.Asp366 phosphorylation site. The magnesium and fluorine atoms of a phosphate analog, MgF42−, are shown in orange and gray, respectively. (F) Free-energy changes upon the substitution of p.Cys596Tyr and p.Gln895Pro, as predicted by FoldX (31, 32).
In the extracellular region, a loop (p.Asp882–Trp896) in the α-subunit is the primary interaction site with the β-subunit (13), where the side chain of p.Gln895 (α-subunit) forms hydrogen bonds with p.Arg182 (β-subunit) (Fig. 2B). The p.Gln895Pro mutation is therefore predicted to disrupt this interaction, possibly reducing the affinity between the α- and β-subunits. FoldX predicted the p.Gln895Pro mutation to cause a moderate free-energy change (2.33 ± 0.39 kcal mol−1) (Fig. 2F), which may affect subunit interactions.
In the transmembrane sections, the mutated regions p.Gly854–Phe857 and p.Tyr991–Arg995 of the α-subunit interact with the transmembrane α helix of the β-subunit (Fig. 2A). p.Phe857 (α-subunit) is a key residue in the interaction, mainly by making van der Waals contacts with p.Phe50, p.Thr53, p.Ile54, and p.Met57 (β-subunit) (Fig. 2C). Furthermore, the Cα atoms of p.Gly854 and p.Gly855 make van der Waals contacts with the side chains of p.Tyr984 and p.Phe912, respectively, within the α-subunit (Fig. 2C). p.Tyr984 also forms a hydrogen bond with p.Thr53 (β-subunit). The p.Phe856 is involved in a hydrophobic core of the α-subunit near K+ binding sites. Thus, the identified deletion mutations of p.Gly854–Phe856 and p.Phe857 in this study are predicted to destabilize protein folding and disrupt intersubunit interactions.
In the p.Tyr991–Arg995 region of the α-subunit, the side chain of p.Tyr991, whose conformation is maintained by a hydrogen bond with p.Gln846 and proximity to p.Ile994, makes van der Waals contacts with p.Phe38 and p.Phe42 (β-subunit) (Fig. 2D). The side chains of p.Asp992 and p.Glu993 form hydrogen bonds with the side chain of p.Arg995 and the backbone amide of p.Val934, respectively. Hence, the three identified mutations in the human α-subunit (the insertion of a His-Glu-Ile tripeptide between p.Ile994 and p.Arg995, the insertion of Phe-Ala-His-Leu-His-Leu into the position of the deleted p.Tyr991–Ile994 residues, and the deletion of p.Asp992) are predicted to destabilize protein folding and disrupt interactions between the α- and β-subunits.
During the reaction cycle, Na+/K+ ATPase is autophosphorylated by ATP at p.Asp366 in the highly conserved Asp-Lys-Thr-Gly-Thr motif (16). The p.Cys596, which is close to the phosphorylation site, is involved in a hydrophobic core with many residues (p.Ile363, p.Ala599, p.Ile601, p.Val603, p.Phe745, p.Ile748, p.Val749, and p.Val752) (Fig. 2E). Thus, the p.Cys596Tyr mutation likely destabilizes protein folding and may impair catalytic activity via a structural modulation of the phosphorylation site. The FoldX-calculated free-energy change (7.88 ± 1.89 kcal mol−1) caused by the p.Cys596Tyr variant is enough to explain the destabilization of the Na+/K+ ATPase complex (Fig. 2F).
Different variant distributions between AHC, RDP, CAPOS, other phenotypes, and polymicrogyria
The distribution patterns of variants for AHC, RDP, and CAPOS are similar in that they cluster within or near transmembrane domains or are located in cytoplasmic N- or P-domains, with occasional overlaps (Figs. 2A and 3A). Functionally, these variants disrupt either Na+/K+ ion binding at/near the transmembrane domain or ATP binding and subsequent phosphorylation at cytoplasmic N- or P-domains (10). Their expression may be unstable, or they may have stable expression with reduced ATPase activity (4). The CAPOS-associated p.Glu818Lys variant affects Na+/K+ ion binding and turnover rates for ATP hydrolysis and pump currents (17). In contrast, the polymicrogyria-associated variants seemed to accumulate at the β-subunit–binding site of the α-subunit, remote from ion-binding sites (10), where AHC-, RDP-, or CAPOS-associated variants rarely exist.
Fig. 3. Previously reported and newly identified ATP1A3 variants in a schematic of Na+/K+ ATPase α3-subunits and variant distribution among the phenotypes.
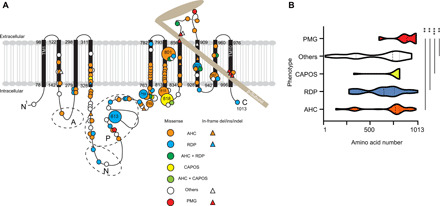
(A) The black bars represent transmembrane domains (TM1 to TM10), while the lines represent the intracellular and extracellular domains of the Na+/K+ ATPase α3-subunit. A hatchet-shaped light brown structure depicts the β-subunit. The symbols and colors used for explaining variants are described in the legend. Numbers indicate the residue number at each site. A, N, and P stand for the actuator, nucleotide binding, and phosphorylation regions, respectively. Large circles represent the commonly observed variants. (B) Graph of nontruncating variant distribution of AHC, RDP, CAPOS, others, and polymicrogyria. X axis: amino acid number; y axis: each phenotype. Dashed and dotted lines indicate the median and quartile, respectively. **P < 0.01 and ***P < 0.001 using the Kolmogorov-Smirnov test. PMG, polymicrogyria; others: phenotypes other than AHC, CAPOS, RDP, and polymicrogyria, including developmental and epileptic encephalopathy, such as EIEE with or without apnea, relapsing encephalopathy with cerebellar ataxia, or fever-induced paroxysmal weakness and encephalopathy.
To statistically compare the variant distribution among different phenotypes, we collected all positions of substituted amino acids by either missense or in-frame variants for AHC, RDP, CAPOS, and other phenotypes and compared the variant distribution using the Kolmogorov-Smirnov test for AHC versus polymicrogyria, RDP versus polymicrogyria, CAPOS versus polymicrogyria, and others versus polymicrogyria. The variant distribution in polymicrogyria was significantly different from that of all other compared phenotypes (Fig. 3B).
Functional consequences of the identified ATP1A3 variants
On the basis of the structural consideration of the identified variants, we hypothesized that polymicrogyria-associated variants might impair the interaction between the β-subunits of Na+/K+ ATPase, leading to either impaired expression, trafficking, or kinetic properties of the α-subunit.
Regarding the kinetic properties of the α-subunit, a previous study reported that variants associated with both AHC and RDP affect ATPase activity (4). Thus, we performed ATPase activity assays to validate whether the detected variants affected ATPase activity. After confirming that all constructed mutant ATP1A3 complementary DNA (cDNA) plasmids had equivalent protein expression compared with the wild-type plasmids (fig. S5), we performed ATPase activity assays. ATPase activity was significantly reduced in cells transfected with ATP1A3 cDNA plasmids with p.Asp801Asn (AHC variant), p.Ile758Ser (RDP variant), or p.Cys596Tyr (polymicrogyria-associated variant) variants but was retained in cells transfected with the other polymicrogyria-associated variants (p.Gly854_Phe856del, p.Phe857del, p.Gln895Pro, and p.Asp992del) (Fig. 4A).
Fig. 4. Functional consequences of the identified ATP1A3 variants.
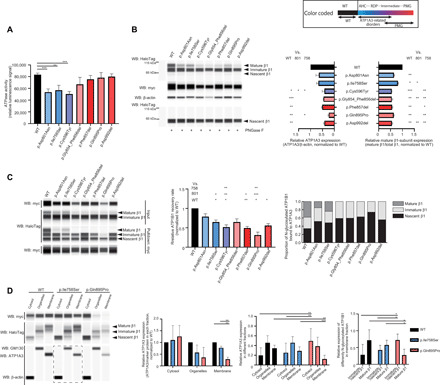
Black, blue, and purple/red bars indicate the wild-type (WT), AHC/RDP-associated, and polymicrogyria-associated variants, respectively. The upper right box shows color-coded information. Error bars, means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001. (A) ATPase activity in COS-7 cells. N = 3. (B) Expression of myc-tagged ATP1A3 and HaloTag-fused ATP1B1 in human embryonic kidney (HEK) 293T cells. Left: A representative blot. Right: The relative ATP1A3 and mature β1-subunit expression, normalized to the wild type. N = 3. WB, Western blot. (C) Coimmunoprecipitation of myc-tagged ATP1A3 and HaloTag-fused ATP1B1 in HEK293T cells. Left: A representative blot. Middle: The relative ATP1B1 recovery rate. N = 3. Right: The proportion of N-glycosylated ATP1B1 bound to ATP1A3. (D) The total HEK293T cell components cotransfected with ATP1A3 (wild type, p.Ile758Ser, or p.Gln895Pro) and ATP1B1 cDNA vectors were separated into three fractions: cytosol, organelles, and plasma membrane. Left: A representative blot. A dashed rectangle denotes Western blotting on a different cartridge. The second panel from the left: The relative ATP1A3 expression in respective fraction. N = 3. The second panel from the right: The proportion of ATP1A3 in each fraction for respective condition. N = 6. Right: The relative expression of differently N-glycosylated ATP1B1 in the plasma membrane fraction. N = 6.
Next, we performed Western blotting for ATP1A3 and ATP1B1, which is one of the proteins that forms the β-subunit of Na+/K+ ATPase. We used human embryonic kidney (HEK) 293T cells cotransfected with ATP1A3 (wild-type or mutant) and ATP1B1 (wild-type) vectors to investigate protein expression with the coexistence of both α- and β-subunits. The β-subunit is glycosylated and modified to a higher molecular weight (the mature form) from the endoplasmic reticulum to the Golgi apparatus. The mature N-glycosylated β-subunit is then folded and trafficked to the cell surface with the α-subunit (2, 18). Compared with the wild-type, all polymicrogyria-associated variants had decreased expression of ATP1A3 and mature β1-subunits. This tendency was also observed in some polymicrogyria-associated variants compared with AHC- or RDP-associated variants, suggesting impaired binding, folding, or trafficking of αβ-heterodimers (Fig. 4B).
We therefore performed coimmunoprecipitation analyses to validate the binding between ATP1A3 and ATP1B1. Using immunoprecipitated ATP1B1 signal intensity, we measured the relative ATP1B1 recovery rate normalized to that of the wild type; this measurement reflects binding efficiency between ATP1A3 and ATP1B1. Binding efficiency was significantly decreased in the p.Ile758Ser, p.Phe857del, and p.Asp992del variants compared with the wild type and significantly decreased in the p.Cys596Tyr and p.Gln895Pro variants compared even with the p.Ile758Ser variant. Of these variants, p.Gln895Pro had the lowest binding efficiency. Among the total immunoprecipitated ATP1B1 (ATP1B1 bound to ATP1A3), mature β1-subunits were detected in wild-type, p.Asp801Asn, and p.Ile758Ser variants but not in any of the polymicrogyria-associated variants. This result suggests that polymicrogyria-associated variants affected both the binding to β1-subunits and the proper folding of αβ-heterodimers, although we could not exclude the possibility that decreased binding is due to the decreased expression of some ATP1A3 variants associated with polymicrogyria (Fig. 4C).
To confirm the abnormal localization of αβ-subunits because of trafficking errors, we separated total cellular components from HEK293T cells coexpressing ATP1A3 (wild type, p.Ile758Ser, or p.Gln895Pro) and ATP1B1 (wild type) into three fractions (cytosol, organelles, and plasma membrane). We chose to validate p.Ile758Ser and p.Gln895Pro variants representing non–polymicrogyria-associated and polymicrogyria-associated ones, respectively, since they showed the significantly decreased ATP1B1 recovery rate in coimmunoprecipitation analyses in each group of variants. We then evaluated ATP1A3 abundance in each fraction and measured the relative expression of mature β1-subunits in the plasma membrane fraction. In the plasma membrane, ATP1A3 expression was significantly lower in the p.Gln895Pro variant compared with the other variants. Between the p.Gln895Pro and the other variants, there was also a significant difference in the proportion of ATP1A3 abundance in each fraction. In cells expressing the p.Gln895Pro variant, ATP1A3 was significantly higher in the cytosol and lower in the plasma membrane compared with the other variants. Furthermore, in the plasma membrane fraction, immature/nascent β1-subunits were predominant in cells expressing the p.Gln895Pro variant; in contrast, in cells expressing the other variants, mature β1-subunits were significantly more common (Fig. 4D).
This result suggests a mechanism of association between the two subunits in the Golgi and their subsequent aberrant trafficking to the membrane. To validate the cellular/subcellular localizations of the α- and β-subunits, we performed an immunofluorescence study using HEK293T cells cotransfected with ATP1A3 (either wild-type or the p.Gln895Pro mutant representing a polymicrogyria-associated variant) and ATP1B1 plasmids, with superresolution confocal microscopy. At the single-cell level, both mutant ATP1A3 and ATP1B1 localized at the cis-Golgi network (cisternae nearest the endoplasmic reticulum) and trans-Golgi network (cisternae farthest from the endoplasmic reticulum) and at the plasma membrane, with no apparent differences in localization patterns compared with wild-type ATP1A3 and ATP1B1 (Fig. 5). Thus, p.Gln895Pro-mutant ATP1A3 is expressed in the plasma membrane. These findings retain the possibility of trafficking delay or stacking through Golgi to the plasma membrane but exclude the possibility of a complete failure of their delivery to their final destination.
Fig. 5. Colocalization of ATP1A3 and ATP1B1 at the cis-/trans-Golgi apparatus and plasma membrane.
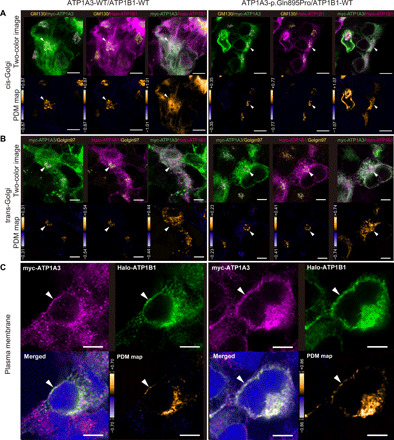
Through (A) to (C), the left and right panels show immunofluorescence under the cotransfection of wild-type ATP1A3 and wild-type ATP1B1 plasmids and p.Gln895Pro ATP1A3 and wild-type ATP1B1 plasmids, respectively. Scale bars, 5 μm. (A) ATP1A3 and ATP1B1 colocalizing at the cis-Golgi network in both panels (arrowhead). (B) ATP1A3 and ATP1B1 colocalizing at the trans-Golgi network in both panels (arrowhead). (C) ATP1A3 and ATP1B1 colocalizing at the plasma membrane in both panels (arrowhead). For (A) and (B), lower images are the pseudocolored maps of the product of the differences from the mean analysis (PDM maps) for the respective upper images. For (C), lower right panels are the PDM maps for the upper two images. In the PDM maps, positive correlation is indicated in yellow, negative correlation in blue, and random distribution in black.
To further evaluate the direct interactions between α- and β-subunits at the plasma membrane, we performed an in situ proximity ligation assay (PLA) using HEK293T cells cotransfected with ATP1A3 (either wild-type or p.Ile758Ser or p.Gln895Pro mutants) and ATP1B1 plasmids. PLA signals, derived from the interactions between α- and β-subunits, were predominantly observed at the plasma membrane. The mean PLA signal intensity of wild-type ATP1A3 and ATP1B1 expression and of p.Ile758Ser ATP1A3 and ATP1B1 expression was similar, but that of p.Gln895Pro ATP1A3 and ATP1B1 expression was apparently reduced (Fig. 6). These findings suggest that polymicrogyria-associated variants may have different functional impacts compared with previously known variants (such as p.Ile758Ser), likely through the decreased interaction of α- and β-subunits at the plasma membrane, possibly impairing proper Na+/K+ ATPase function but retaining ATPase activity via an as-yet unknown mechanism.
Fig. 6. In situ PLA fluorescence study showing decreased interaction between ATP1A3 and ATP1B1 with the polymicrogyria-associated p.Gln895Pro variant.
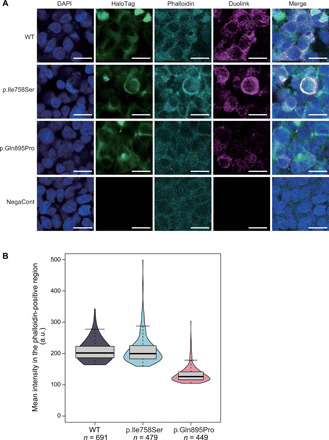
(A) The production of in situ PLA fluorescent spots (Duolink) indicates the subcellular sites where myc-tagged ATP1B1 and HaloTag-fused ATP1B1 colocalize within 40 nm. Immunofluorescence study showing that Duolink signals were substantially decreased at HaloTag-fused ATP1B1 expressed in the plasma membrane in HEK293T cells cotransfected with p.Gln895Pro ATP1A3 and ATP1B1 plasmids, compared with those with p.Ile758Ser ATP1A3/ATP1B1 and wild-type ATP1A3/ATP1B1 plasmids. Scale bars, 20 μm. DAPI, 4′,6-diamidino-2-phenylindole. (B) Mean intensity in the specified region was significantly decreased in HEK293T cells cotransfected with p.Gln895Pro ATP1A3 and ATP1B1 plasmids, while those with p.Ile758Ser ATP1A3/ATP1B1 did not appear to differ in mean signal intensity compared with wild-type ATP1A3/ATP1B1. We confirmed this tendency with replicate experiments (N = 4). a.u., arbitrary units.
Disrupted neuronal migration in embryonic mice expressing the p.Asp992del variant and impaired neural activity in primary cultured neurons
To investigate the effects of ATP1A3 variants on brain development, we examined ATP1A3 overexpression in the embryonic mouse cortex using in utero electroporation. We used plasmids expressing either one of three variants (p.Asp992del, p.Gln895Pro, or p.Ile758Ser) or wild-type ATP1A3. We chose to validate p.Asp992del and p.Gln895Pro variants as the ones associated with the most severe and mildest phenotypes among our patients with polymicrogyria, respectively, while p.Ile758Ser is representing the nonpolymicrogyria-associated variant. The four expression vectors were introduced into fetal mouse brains together with green fluorescent protein (GFP) expression vectors on embryonic day 14.5 (E14.5). Four days later, the brains were removed, and the effects on cell migration were examined. Compared with an empty GFP vector, significantly impaired cell migration was detected only when the p.Asp992del variant was expressed. This result is consistent with our identification of this variant in the two patients with the most severe forms of polymicrogyria in our cohort. No significant differences were observed when the other variants (p.Gln895Pro or p.Ile758Ser) were expressed. Together, these findings suggest that the p.Asp992del variant disrupts cell migration during cortical development and causes defects in cortical architecture (Fig. 7, A to C).
Fig. 7. Migration defects in ATP1A3 p.Asp992del variant–expressing neurons in the embryonic mouse cortex.
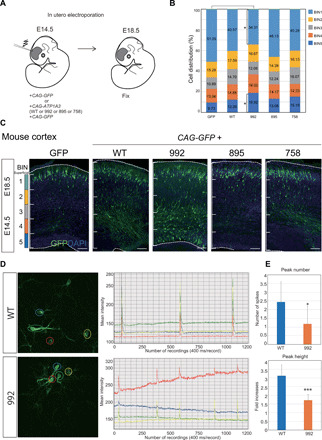
(A) Schematic of the study design using mice. (B and C) Plasmids expressing the wild-type, p.Asp992del, p.Gln895Pro, and p.Ile758Ser variants of ATP1A3 were introduced, together with GFP-expressing plasmids, into the ventricles of mice on E14.5. After in utero electroporation, the embryonic brains were collected and fixed at E18.5, and migration was quantified using cell distributions. The total cortical thickness was divided into five equal parts, named BIN1 to BIN5. The number of cells that were distributed in each BIN was counted and expressed as a percentage. Scale bars, 50 μm. *P < 0.05 (Student’s t test). (B) denotes cell distribution across the cortex as a summary, while (C) shows a representative image of the mouse cortex expressing either GFP, mutant ATP1A3, or wild-type ATP1A3. (D) Left: Representative images of monitored cultured neurons introduced with wild-type or p.Asp992del ATP1A3 plasmids together with GCaMP plasmids. Regions of interest (ROIs) were marked with four different colors to monitor the intensities of GCaMP signals. Right: Graphs of intensity plot in one recording (8 min). The same colors of ROIs are used for GCaMP signal recording. (E) Peak numbers (upper) and heights (lower) of calcium spikes of the recordings. For peak height, fold increases of GCaMP signal intensity compared to the average intensity were shown. *P < 0.05 and ***P < 0.001.
To explore the effects of the p.Asp992del variant on neural function, we introduced wild-type and p.Asp992del ATP1A3 expression vectors, together with the calcium sensor, GCaMP6 (Green fluorescent protein – calmodulin protein 6) plasmids, by in utero electroporation. We isolated cortical neurons from the electroporated embryonic brains, made primary cultures, and performed GCaMP imaging in DIV4. The results showed that calcium spikes were reproducibly observed to manifest neural circuit maturation in wild-type ATP1A3–introduced neurons. In contrast, the number and height of calcium spikes were significantly lower in the p.Asp992del ATP1A3–introduced neurons. This result suggests that the p.Asp992del variant may affect neural activity and impair neural function apart from cell migration in the patients with p.Asp992del variants (Fig. 7, D and E, and movie S1).
DISCUSSION
Here, we report eight patients with de novo ATP1A3 variants, who presented with polymicrogyria, extremely severe intellectual disability, intractable epilepsy, and occasionally cardiac failure. Table S2 presents clinical comparisons among patients with AHC, RDP, or CAPOS, and our patients with polymicrogyria. AHC, RDP, and CAPOS are considered a continuum of clinically broad functional disorders with abrupt/paroxysmal symptoms, asymmetric anatomical distribution, presence of a trigger, and typically no morphological brain abnormalities. In contrast, our patients had relatively permanent clinical features, no phasic disease course, no obvious trigger for disease onset, and morphological abnormalities in the brain. Thus, our patients present a new phenotype of ATP1A3 abnormalities.
In addition to the clinical phenotype, the localization of variants in ATP1A3 and their functional consequences were different between AHC/RDP/CAPOS and polymicrogyria, indicating different molecular mechanisms of pathogenesis, which may require different therapeutic strategies. Furthermore, embryonic mice with in utero electroporation–induced overexpression of the p.Asp992del variant showed disrupted neuronal migration during cortical development, suggesting a possible association between the identified variants and defects in brain architecture.
In patients 1, 3, and 4, who had variants at the p.Tyr991–Arg995 transmembrane region, cardiac failure and progressive cerebral and cerebellar atrophy were noted. In addition, patient 6 showed progressive atrophy in the cerebellar vermis. Considering that malformations of cortical development occur during the fetal stage, the co-occurrence of postnatal brain atrophy is not particularly common but has been observed in genetic disorders such as 22q11.2 deletion syndrome (19) and Zellweger syndrome (20). Postnatal brain atrophy might therefore be associated with ATP1A3-related polymicrogyria, although more patients are needed to confirm our findings. However, we cannot exclude the possibility that other nongenetic factors, such as antiepileptic drugs and a ketogenic diet, may have led to an atrophied appearance on brain MRI scans.
The α3-subunit is highly expressed in human heart tissue. Cardiac channelopathy (21) and cardiac symptoms known as paroxysmal autonomic disturbance (22) have been reported in AHC, and left ventricular enlargement in early childhood has been noted in CAPOS (6). However, a transient cardiac failure might be a specific complication of polymicrogyria-associated ATP1A3 variants, especially at p.Tyr991–Arg995 in transmembrane regions.
Localization of the p.Cys596Tyr variant in ATP1A3 is atypical among the polymicrogyria-associated variants; it is far from the β-subunit interaction site. The ATPase assay for this variant demonstrated decreased activity, similar to what occurs in AHC. In addition, its expression under the coexistence of ATP1B1, its binding to the β-subunit, and the trafficking of αβ-heterodimers were all impaired, similar to what was observed in other polymicrogyria-associated variants. These findings imply that p.Cys596 substitution may lead to an intermediate form between AHC and polymicrogyria; this hypothesis is supported by a previous report of a p.Cys596Arg (12) variant (which occurred at the same amino acid but substituted a different amino acid) in a patient with AHC.
A few instances of heterozygous ATP1A3 variants have been reported to accompany malformations of cortical development: two cases of EIEE and postnatal microcephaly, with p.Gly358Val (7) and p.Leu924Pro (18) variants, and a case of AHC with polymicrogyria, with p.Leu888Pro (23). These previously reported cases support the association between mutant ATP1A3 and malformation of cortical development. Furthermore, the latter case strengthens our finding that ATP1A3 variants that disrupt α- and β-subunit interactions may be associated with polymicrogyria. The p.Leu888 is near the p.Gln895Pro variant and within an extracellular loop (p.Asp882–Trp896) in the α-subunit, which is a primary interaction site with the β-subunit.
Very recently, a compound heterozygous missense variant in ATP1A3 was reported to cause congenital obstructive hydrocephalus and other structural brain anomalies (24), and homozygous ATP1A2-truncating variants have been reported to cause lethal polymicrogyria (25). ATP1A2 encodes the α2-subunit of Na+/K+ ATPase and is associated with familial hemiplegic migraine. These cases imply that Na+/K+ ATPase is associated with brain development in humans, and its total loss is likely lethal. However, no heterozygous truncating variants have been reported in patients with ATP1A3-related disorders to date, implying that a simple loss-of-function model is unlikely. In contrast, we suppose that our patients demonstrated polymicrogyria because of the dominant-negative effect of the ATP1A3 variants. This supposition is based on the results of protein structural considerations, immunoprecipitation and immunofluorescence experiments, and in situ PLA fluorescence assays, which suggested that only polymicrogyria-associated variants, and not those associated with AHC or RDP, impair the interactions between α- and β-subunits at the plasma membrane. With the decreased interaction abilities, there might also be stacking or delay of α- and β-subunits from the endoplasmic reticulum to the Golgi apparatus or some degradation of α- and β-subunits; these possibilities were suggested from the fractionation and expression analyses under cotransfection of ATP1A3 and ATP1B1 plasmids. However, these findings need to be further clarified. In addition, the decreased interaction of α- and β-subunits at the plasma membrane possibly impairs normal Na+/K+ ATPase function, although the mechanisms remain elusive (fig. S6).
The p.Gln895Pro variant, detected in the patient with the mildest clinical phenotype in our cohort, appeared to have the most severe binding and trafficking defects in our experiments. Our dominant-negative hypothesis may explain this phenomenon: There may be competition between aberrant and wild-type ATP1A3 to develop αβ-heterodimers and reach the plasma membrane. More severe trafficking errors lead to more degradation of aberrant ATP1A3; thus, more wild-type ATP1A3 can reach the plasma membrane and contribute to normal Na+/K+ ATPase functioning. A recent report on trafficking defects in αβ-heterodimers demonstrated similar experimental results and implied the possible competition of wild-type and mutant alleles, supporting our hypothesis (18).
The molecular pathogenesis of polymicrogyria remains largely unknown. Among the molecules/pathways known to cause polymicrogyria (tubulins, metabolic disorders, or the phosphatidylinositol 3-kinase–AKT–mechanistic target of rapamycin pathway), it has been proposed that mutations in genes encoding epilepsy-associated ion channel components (SCN3A, GRIN2B, and GRIN1) cause polymicrogyria by altering channel physiology during the fetal stage (26–28). This is termed “developmental channelopathy” (29), and ATP1A3 may belong to this group because of its unknown mechanisms that may relate to β-subunit malfunction (16). We found that primary cultured neurons expressing p.Asp992del variant in mice showed impaired calcium spikes leading to abnormal neural function. Figure 8 illustrates our understanding of the ATP1A3 gene as a cause of these ion channel diseases throughout the human life span.
Fig. 8. ATP1A3-related disorders as ion channel diseases from fetal to adulthood stages.
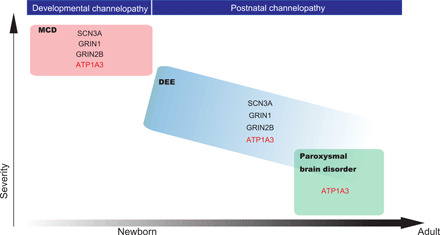
A schematic showing an overview of ATP1A3-related disorders as ion channel diseases throughout the human life span, together with other known genes, based on the latest knowledge (29). The x axis approximately (not linearly) shows the life span of a human, from newborn to adulthood. The y axis represents disease severity. MCD, malformation of cortical development; DEE, developmental and epileptic encephalopathy.
From phenotypes, genotypes, and functional analyses, we demonstrate that polymicrogyria can be associated with ATP1A3 variants. Variants in ATP1A3 may be a common cause of polymicrogyria, considering our diagnostic yield of 6.5%. Clinicians should therefore be aware of both polymicrogyria and ATP1A3-related disorders because distinguishing between these diseases may be difficult using clinical symptoms only (e.g., both epilepsy in polymicrogyria and the abrupt onset of hemiplegia or autonomic symptoms in AHC can look alike).
MATERIALS AND METHODS
Participants
In total, 124 families with polymicrogyria were recruited to the study. Experimental protocols were approved by the Committee for Ethical Issues at Yamagata University Faculty of Medicine, Showa University School of Medicine, and Yokohama City University School of Medicine. Written informed consent was obtained from all individuals or their parents. Clinical information was collected from medical records.
Experimental animals
All animals were treated in accordance with the Tokyo Metropolitan Institute of Medical Science Animals Care and Use Committee guidelines. Pregnant ICR (Institute of Cancer Research) mice were purchased from Japan SLC Inc. (Hamamatsu, Japan).
Whole-exome sequencing
As previously described, 114 of 124 families were sequenced at Yokohama City University (30). Among these 114 families, trio-based or patient-only whole-exome sequencing was performed in 54 families and 60 patients, respectively. For these 114 families, the mean depth of coverage for whole-exome sequencing was 68.98×, and 96.17% of the total coding sequence of RefSeq genes was covered with a depth of 10× reads or more, on average (88.3 to 98.1%). The other three and seven families in this study were analyzed at Hamamatsu University School of Medicine (including patient 7) and RIKEN Center for Integrative Medical Sciences (including patient 1), respectively. All variants were confirmed by Sanger sequencing.
Protein structure modeling
The structure of the human Na+/K+ ATPase α3-subunit is not available in the PDB; therefore, we used the Phyre2 program to search for a homologous structure using its amino acid sequence (RefSeq NM_152296.4) (14). Next, we used FoldX (ver.3.0) (31, 32) to calculate free-energy changes caused by the identified mutations, using the crystal structure of S. acanthias Na+/K+ ATPase (PDB code: 2ZXE) as a homologous template (listed by Phyre2). The calculation was repeated five times, and data were presented as the means ± SD. The modeled structures were drawn using PyMOL (www.pymol.org).
Vectors and transfection procedures
Human ATP1A3 (ORH26659) and ATP1B1 (FHC02644) clones were purchased from Promega (Madison, WI, USA). Human ATP1A3 cDNA was introduced into a pcDNA3.1/myc-His C vector (Invitrogen, Carlsbad, CA, USA) to express C-terminal myc-His–tagged ATP1A3. Site-directed mutagenesis using the KOD-Plus-Mutagenesis Kit (Toyobo, Osaka, Japan) allowed the generation of mutant ATP1A3 vectors (p.Asp992del, p.Phe857del, p.Gly854_Phe856del, p.Cys596Tyr, p.Gln895Pro, p.Ile758Ser, and p.Asp801Asn). For the animal experiments, wild-type and mutated ATP1A3 expression plasmids for in utero electroporation were constructed using PBCAG-EGFP vectors (33). Briefly, CAG-ATP1A3 variant vectors were assembled in a pUC57-mini plasmid backbone (Genscript Biotech Corp., Piscataway, NJ, USA) using standard restriction- and ligation-based cloning. We drove ATP1A3 expression with the strong eukaryotic CAG promoter. The inserts of ATP1A3 variants were cloned into Eco RI/Sma I enzyme sites on PBCAG-EGFP using Ligation high Ver.2 (Toyobo). For the ATPase assays, COS-7 cells were transfected with ATP1A3 expression plasmids using ViaFect Transfection Reagent (Promega). For the rest of the analyses, HEK293T cells were transfected with ATP1A3 and/or ATP1B1 expression plasmids using either ViaFect Transfection Reagent or polyethylenimine (Polysciences Inc., Warrington, PA, USA) according to the manufacturer’s protocol.
Western blotting
Capillary electrophoretic Western blotting was performed on the Wes system (ProteinSimple, San Jose, CA, USA) using a 12- to 230-kDa separation module according to the manufacturer’s protocol for size separation. Myc-tagged ATP1A3, HaloTag-fused ATP1B1, and β-actin were detected using mouse anti–Myc-tag monoclonal antibody (mAb) [MBL(MEDICAL & BIOLOGICAL LABORATORIES), Nagoya, Japan], rabbit anti-HaloTag polyclonal antibody (pAb) (Promega), mouse anti-HaloTag mAb (Promega), and mouse anti–β-actin (ab6276; Abcam, Cambridge, UK), respectively. To prepare cell lysate for ATP1A3 detection, we did not boil the lysate but stored it at 4°C overnight with urea at its final concentration of 4 M.
ATP1A3 protein blotting
HEK293T cells were transfected with ATP1A3 (wild type or mutant) cDNA vectors. Forty-eight hours after transfection, the cells were lysed with radioimmunoprecipitation assay buffer and subjected to Western blotting on the Wes system to detect ATP1A3 and β-actin signals, using anti–Myc-tag and anti–β-actin antibodies.
ATPase assay
COS-7 cells expressing wild-type or mutant alleles of ATP1A3 were assessed in the ATPase assay as described previously (4). Briefly, synthesized adenosine 5′-diphosphate (ADP) by ATPase reaction was detected using the EnzyLight ADP Assay Kit (BioAssay Systems, Hayward, CA, USA). ATPase activity was estimated by subtracting the luminescence signal after a 1-hour pretreatment with 100 μM ouabain (Sigma-Aldrich, St. Louis, MO, USA) from the luminescence reading without ouabain.
ATP1A3 and N-glycosylated β1-subunit protein blotting
Lysate from HEK293T cells cotransfected with ATP1A3 (wild type or mutant) and ATP1B1 (wild type) cDNA vectors was collected 48 hours after transfection and was subjected to Western blotting to detect ATP1A3, ATP1B1, and β-actin signals using anti–Myc-tag, anti-HaloTag, and anti–β-actin antibodies. Relative ATP1A3 expression was calculated as the band intensity of ATP1A3 normalized to that of β-actin and to wild-type ATP1A3. For ATP1B1, the molecular weight of nascent, immature (stayed at the endoplasmic reticulum), and mature (in the Golgi apparatus or trafficked to the cell surface) HaloTag-fused ATP1B1 were predicted as around 60, 80, and 100 kDa in size, respectively. We calculated the relative mature β1-subunit expression by dividing the signal intensity of mature β1-subunits by that of total β1-subunits and then normalized it to the wild type. The denatured lysate was then treated with PNGase F (N-Zyme Scientifics, Doylestown, PA, USA) for 2 hours at 37°C to cleave the N-linked oligosaccharides, to ensure that the higher molecular signals disappeared.
Immunoprecipitation
For the coimmunoprecipitation assay, wild-type or mutant myc-tagged ATP1A3 vectors and HaloTag-fused ATP1B1 vectors were cotransfected into HEK293T cells. At 48 hours after transfection, cells were lysed with Mammalian Lysis Buffer (Promega) containing Protease Inhibitor Cocktail (Promega) and PhosSTOP (Roche Diagnostics, Basel, Switzerland). The lysate was incubated with 2 μg of anti–Myc-tag mAb (MBL) at 4°C overnight and with Dynabeads Protein G (Thermo Fisher Scientific, Waltham, MA, USA) at 4°C for 2 hours. After washing with phosphate-buffered saline (PBS), the beads were resuspended with 1% SDS buffer. Inputs and immunoprecipitates were subjected to Western blotting to detect ATP1A3 and ATP1B1 using anti–Myc-tag and anti-HaloTag antibodies. Because an excess of both myc-tagged ATP1A3 and HaloTag-fused ATP1B1 was confirmed in the flow-through after immunoprecipitation by Western blotting, and equivalent amounts of anti–Myc-tag mAb were added to the lysates for immunoprecipitation, we measured the band intensities of HaloTag signals from each immunoprecipitate. We then calculated the binding efficiency between mutant ATP1A3 and wild-type ATP1B1 as the HaloTag signal intensity of the particular immunoprecipitate divided by that of wild-type ATP1A3 and wild-type ATP1B1.
ATP1A3 and ATP1B1 localization analysis in HEK293T cells
HEK293T cells were cotransfected with ATP1A3 (wild-type, p.Ile758Ser, or p.Gln895Pro) and ATP1B1 (wild-type) cDNA vectors and collected 48 hours after transfection. Using the Plasma Membrane Protein Extraction Kit (101Bio, Palo Alto, CA, USA), we separated the total cellular components into three fractions: cytosol, organelles, and plasma membrane. This was conducted according to the manufacturer’s instructions, and the three fractions were then subjected to Western blotting to detect ATP1A3 and ATP1B1 signals using anti–Myc-tag and anti-HaloTag antibodies. We also used anti–β-actin antibody, anti-GM130 (anti-Golgin subfamily A member 2) antibody [EP892Y, cis-Golgi Marker (ab52649; Abcam)], and ATP1A3 (middle) rabbit pAb (28030-1-AP; Proteintech, Rosemont, IL, USA) as marker antibodies for the respective cellular fractions. We calculated the relative ATP1A3 expression of p.Ile758Ser or p.Gln895Pro compared with wild-type variants in the cytosol, organelles, and plasma membrane as follows: The band intensities of ATP1A3 in either fraction were divided by that of either β-actin, GM130, or endogenous ATP1A3 and normalized to that of wild type. We also calculated the relative ATP1A3 expression in each of the three fractions as the band intensities of ATP1A3 in a fraction divided by that of the sum of all three fractions. In addition, we calculated the relative expression of differently N-glycosylated ATP1B1 in the plasma membrane fraction as the band intensities of mature β1-subunits divided by the band intensities of the sum of all β1-subunits (nascent, immature, and mature) in the plasma membrane fraction.
Immunofluorescence study
Forty-eight hours after transfection, HEK293T cells on glass coverslips were fixed with 4% paraformaldehyde (PFA)/PBS for 10 min. After being washed twice with PBS-glycine, the cells were permeabilized with 0.1% Triton X/PBS for 10 min. The cells were then blocked with 10% normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA, USA)/0.05% Tween 20/PBS before immunolabeling was performed using anti-HaloTag pAb [1:500, with goat anti-rabbit immunoglobulin G (IgG) (H + L) secondary antibody; Alexa Fluor 546 conjugate, Thermo Fisher Scientific; 1:500] and mouse anti–Myc-tag mAb [1:1000, with goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody; Alexa Fluor 647, Invitrogen; 1:1000], for plasma membrane evaluation. For Golgi apparatus evaluation, we performed immunolabeling with mouse anti–Myc-tag mAb [1:1000, with either goat anti-mouse IgG2b cross-adsorbed secondary antibody; Alexa Fluor 647, Invitrogen; 1:1000; or goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody; Alexa Fluor 647, Invitrogen; 1:1000], purified mouse anti-GM130 (BD BioSciences, San Jose, CA, USA; 1:250, with goat anti-mouse IgG1 cross-adsorbed secondary antibody; Alexa Fluor 488, Invitrogen; 1:1000) as a cis-Golgi marker, and golgin-97 (D8P2K) rabbit mAb [number 13192, Cell Signaling Technology, Danvers, MA, USA; 1:1000, with goat anti-rabbit IgG (H + L) secondary antibody; Alexa Fluor 488 conjugate, Molecular Probes; 1:1000] as a trans-Golgi marker. We also performed live-cell no-wash labeling for Golgi evaluation using HaloTag TMRDirect Ligand (Promega; 1:1000) overnight before the day of sample fixation, according to the manufacturer’s protocol. Maps of the product of the differences from the mean analysis (PDM maps) (34) were prepared using ImageJ 1.53 software with the plug-in “Intensity Correlation Analysis” in the MBF “ImageJ for Microscopy” Collection (https://imagej.net/plugins/mbf/) (35). Each pixel in PDM maps represents the PDM value: (intensity of channel 1 − mean intensity of channel 1) × (intensity of channel 2 − mean intensity of channel 2).
In situ PLA
We performed the in situ PLA using Duolink In Situ PLA Fluorescence (Sigma-Aldrich) following the manufacturer’s protocol. Forty-eight hours after transfection, HEK293T cells on an eight-well slide chamber were fixed with 4% PFA/PBS for 10 min and permeabilized with 0.1% Triton X/PBS for 10 min before starting the Duolink procedure. We used mouse anti–Myc-tag mAb (1:1000) and anti-HaloTag pAb (1:500) as primary antibodies, anti-mouse PLUS and anti-rabbit MINUS as Duolink PLA probes, and Detection Reagents Red for signal detection. We calculated the mean intensity of speckled signals at each of the specified regions of interest (ROIs) where both HaloTag-fused ATP1B1 and phalloidin signals were obtained (the numbers of ROIs were 691 for wild-type ATP1A3/ATP1B1, 479 for p.Ile758Ser mutant ATP1A3/ATP1B1, and 449 for p.Gln895Pro mutant ATP1A3/ATP1B1, respectively).
Microscopy and image analysis
Conventional and super-resolution confocal images were acquired using the TCS SP8 System (Leica, Wetzlar, Germany) with 20×, 63×, and 100× objective lenses (HC PL APO 20×/0.75 DRY, 63×/1.40 OIL, and 100×/1.40 OIL, respectively). Excitation was provided by a white-light laser, and the fluorescent signal was acquired using a GaAsP hybrid detector system (Leica). For the superresolution microscopy, we set the pinhole at a diameter of 0.5 to 0.75 Airy units, and acquired images were deconvoluted using Huygens software (Scientific Volume Imaging, Hilversum, Netherlands), if necessary. Images were quantified and arranged using ImageJ 1.53 software (National Institutes of Health, Bethesda, MD, USA) and Photoshop 12.0 (Adobe, San Jose, CA, USA).
In utero electroporation and migration analysis
In utero electroporation was performed as previously reported (36). Briefly, mice were deeply anesthetized with sodium pentobarbital at 50 mg/kg of body weight, and the uterine horns were exposed. A plasmid DNA solution [ATP1A3 plasmids (5 μg/μl) + CAG-EGFP (0.5 μg/μl)] in Hepes-buffered saline (pH 7.2) containing 0.01% Fast Green was injected into the lateral ventricle using a glass micropipette with an IM-31 microinjector (Narishige Scientific Instrument Lab, Tokyo, Japan). Approximately 1 to 2 and 0.5 μl of plasmid solutions were injected into the mouse brains at E14.5. The heads of E14.5 embryos (in the uterus) were placed between a tweezer-type electrode, 5 mm in diameter (LF650P5; BEX, Tokyo, Japan), and five electric pulses (35 V, 50 ms in duration, at intervals of 950 ms) were then delivered using a CUY21E electroporator (BEX). After electroporation, the uterine horns were returned to the abdominal cavity to allow the embryos to continue their development. The brains of the electroporated embryos were harvested at E18.5, fixed in 4% PFA/PBS for 20 hours, and embedded with OCT (optimal cutting temperature) compound after sucrose substitution. Frozen sections were prepared from each brain at a thickness of 20 μm. After 4′,6-diamidino-2-phenylindole staining, images were acquired using an LSM 780 confocal laser microscope (Zeiss, Jena, Germany). The GFP-positive migrating cells in each image were counted using ImageJ software. Two brains (for GFP, wild type, and the p.Ile758Ser variant) or four brains (p.Asp992del and p.Gln895Pro variants) were used for each model. Between two and four sections from each brain were analyzed (N = 5 for p.Ile758Ser, N = 6 for wild type and p.Gln895Pro, N = 7 for GFP, and N = 8 for p.Asp992del).
GCaMP imaging of primary cultured neurons overexpressed with wild-type or p.Asp992del variant of ATP1A3
GCaMP6 and red fluorescent protein plasmids were electroporated together with either wild-type or p.Asp992del ATP1A3 expression vectors at E14.5. The electroporated embryonic brains were dissected at E16.5, and dorsal cortices were excised and dissociated with papain. Cells were cultured in the polyethylenimine-coated glass-bottom dishes in minimum essential medium–base culture medium. At 4DIV, GCaMP signals of cultured neurons were monitored using spinning disk confocal scanner unit CSU-W1(YOKOGAWA, Tokyo, Japan). For each recording, 1200 images were captured every 400 ms. Intensities of fluorescent signals were analyzed by IQ3 Cell Imaging Software (Andor, Belfast, Northern Ireland). Only peak that have an intensity of >1.5 times of the average fluorescent signal value was considered valid. Two independent in utero electroporation experiments were performed, and six and four electroporated brains from wild-type and p.Asp992del variant mice were collected, respectively. We acquired the calcium imaging data from N = 9 or more observations (wild type; N = 11, p.Asp992del; N = 9) for statistical processing.
Statistical analysis
All statistical analyses were performed using GraphPad Prism8 (GraphPad Software, San Diego, CA, USA). For the ATP1A3 expression analyses, ATPase assays, ATP1A3, N-glycosylated β1-subunit protein blotting, and immunoprecipitation, statistical differences were analyzed using one-way analysis of variance (ANOVA) with post hoc Dunnett’s test for multiple comparisons. For the ATP1A3 and ATP1B1 localization analysis, we used two-way repeated-measure ANOVA with post hoc Tukey’s test for multiple comparisons. To compare ATP1A3 variant distribution among phenotypes, we first collected all positions of substituted amino acids by either missense or in-frame variants for AHC, RDP, CAPOS, and other phenotypes (including those who manifested some of the symptoms for AHC, RDP, or CAPOS but were unable to be specifically diagnosed with them) from the Human Gene Mutation Database (https://portal.biobase-international.com/cgi-bin/portal/login.cgi; as of 25 August 2020). For an amino acid where multiple patients (multiple reports) had substitutions, we counted cumulatively. We then performed the Kolmogorov-Smirnov test to compare the variant distribution tendencies of AHC versus polymicrogyria, RDP versus polymicrogyria, CAPOS versus polymicrogyria, and others versus polymicrogyria. A value of P < 0.05 was considered significant, except for the Kolmogorov-Smirnov test, which used P < 0.0125 (after Bonferroni correction) because multiple tests were performed. For the mouse experiments, statistical differences were analyzed using the Student’s t test.
Acknowledgments
We thank all the participants for cooperation in this research. We are grateful to K. Takabe, N. Watanabe, S. Sugimoto, and S. Nakamura from the Department of Human Genetics, Yokohama City University Graduate School of Medicine; K. Tanaka from the Department of Pediatrics, Yamagata University Faculty of Medicine; M. Ogawa from the Department of Pediatrics, Showa University School of Medicine; and M. Yokouchi and F. Sakurada from Gene Regulation Research, Nara Institute of Science and Technology for technical assistance. We also thank A. Ryo from Department of Microbiology, Yokohama City University Graduate School of Medicine for invaluable advice toward the functional study and B. Gardner from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript. Funding: This work was supported by the Japan Agency for Medical Research and Development (AMED) under grant numbers JP20ek0109280, JP20dm0107090, JP20ek0109301, JP20ek0109348, and JP20kk0205012 (N.Ma.); JP19ek0109297 (H.S. and M.K.); JP19lk0201069 (M.K.); JP20dm0307028, JP20km0405214, and JP20ek0109381 (A.T.); and JP18ek0109288h00021 (T.Ma.); JSPS KAKENHI under grant numbers JP17H01539 (N.Ma.), JP16H05160 (H.S.), JP19H03621 (N.Mi.), JP17K10080 (S.M.), JP17H05621 (T.Ma.), JP19K16921 (E.K.), JP20H03270 (C.O.-M.), and JP16K09975 (M.K.); Grant-in Aid for Scientific Research on Innovative Areas “Interplay of developmental clock and extracellular environment in brain formation” under grant number JP19H04795 (C.O.-M.); and intramural research grants for Neurological and Psychiatric Disorders of NCNP from the Ministry of Health, Labour and Welfare under grant numbers 30-6 (N.Ma. and M.K.) and 30-7 (N.Ma.), the Naito Foundation (C.O.-M.), the Takeda Science Foundation (N.Mi., H.S., M.N., C.O.-M., and N.Ma.), and the Suntory Foundation for Life Sciences Bioorganic Research Institute (T.Ma.). Author contributions: S.M., M.K., T.K., C.O.-M., and N.Ma. designed the study. M.K., Y.So., M.Sas., H.To., T.Ha., Y.Sa., S.O., Y.No., Mazumi Miura, Y.Ni., K.N., H.A., N.H., M.T., Y.I., N.K., K.A., A.H., M.F., H.I., G.M., N.E., and Masaki Miura performed the clinical analyses. S.M., E.K., T.Ma., H.Take., H.D., K.S., A.Y., M.N., T.Hi., A.T., K.Haman., M.Sat., T.Miy., F.M., T.T., T.Miz., N.Mi., A.S., H.S., H.Taka., and F.T. performed the genetic and functional analyses. K.Hamad. and K.O. performed the structural analyses. T.K. and C.O.-M. performed the mouse experiments and analyses. N.Ma. supervised all aspects of the study. S.M., M.K., K.Hamad., K.O., H.Take., T.K., C.O.-M., T.Ma., and N.Ma. wrote the paper. All authors reviewed the paper and approved the final manuscript before submission. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabd2368/DC1
REFERENCES AND NOTES
- 1.Parrini E., Conti V., Dobyns W. B., Guerrini R., Genetic basis of brain malformations. Mol. Syndromol. 7, 220–233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmalzing G., Gloor S., Na+/K+-pump beta subunits: Structure and functions. Cell. Physiol. Biochem. 4, 96–114 (1994). [Google Scholar]
- 3.Holm T. H., Lykke-Hartmann K., Insights into the pathology of the α3 Na+/K+-ATPase ion pump in neurological disorders; Lessons from animal models. Front Physiol 7, 209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heinzen E. L., Swoboda K. J., Hitomi Y., Gurrieri F., Nicole S., de Vries B., Tiziano F. D., Fontaine B., Walley N. M., Heavin S., Panagiotakaki E.; European Alternating Hemiplegia of Childhood (AHC) Genetics Consortium; Biobanca e Registro Clinico per l’Emiplegia Alternante (I.B.AHC) Consortium; European Network for Research on Alternating Hemiplegia for (ENRAH) for Small and Medium-sized Enterprieses (SMEs) Consortium, Fiori S., Abiusi E., Di Pietro L., Sweney M. T., Newcomb T. M., Viollet L., Huff C., Jorde L. B., Reyna S. P., Murphy K. J., Shianna K. V., Gumbs C. E., Little L., Silver K., Ptáček L. J., Haan J., Ferrari M. D., Bye A. M., Herkes G. K., Whitelaw C. M., Webb D., Lynch B. J., Uldall P., King M. D., Scheffer I. E., Neri G., Arzimanoglou A., van den Maagdenberg A. M., Sisodiya S. M., Mikati M. A., Goldstein D. B., De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat. Genet. 44, 1030–1034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Carvalho Aguiar P., Sweadner K. J., Penniston J. T., Zaremba J., Liu L., Caton M., Linazasoro G., Borg M., Tijssen M. A., Bressman S. B., Dobyns W. B., Brashear A., Ozelius L. J., Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron 43, 169–175 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Demos M. K., van Karnebeek C. D., Ross C. J., Adam S., Shen Y., Zhan S. H., Shyr C., Horvath G., Suri M., Fryer A., Jones S. J., Friedman J. M.; FORGE Canada Consortium , A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J. Rare Dis. 9, 15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paciorkowski A. R., McDaniel S. S., Jansen L. A., Tully H., Tuttle E., Ghoneim D. H., Tupal S., Gunter S. A., Vasta V., Zhang Q., Tran T., Liu Y. B., Ozelius L. J., Brashear A., Sweadner K. J., Dobyns W. B., Hahn S., Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 56, 422–430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabouraud P., Riquet A., Spitz M.-A., Deiva K., Nevsimalova S., Mignot C., Lesca G., Bednarek N., Doummar D., Pietrement C., Laugel V., Relapsing encephalopathy with cerebellar ataxia are caused by variants involving p.Arg756 in ATP1A3. Eur. J. Paediatr. Neurol. 23, 448–455 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Yano S. T., Silver K., Young R., DeBrosse S. D., Ebel R. S., Swoboda K. J., Acsadi G., Fever-induced paroxysmal weakness and encephalopathy, a new phenotype of ATP1A3 mutation. Pediatr. Neurol. 73, 101–105 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Holm R., Toustrup-Jensen M. S., Einholm A. P., Schack V. R., Andersen J. P., Vilsen B., Neurological disease mutations of α3 Na+,K+-ATPase: Structural and functional perspectives and rescue of compromised function. Biochim. Biophys. Acta 1857, 1807–1828 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Retterer K., Juusola J., Cho M. T., Vitazka P., Millan F., Gibellini F., Vertino-Bell A., Smaoui N., Neidich J., Monaghan K. G., McKnight D., Bai R., Suchy S., Friedman B., Tahiliani J., Pineda-Alvarez D., Richard G., Brandt T., Haverfield E., Chung W. K., Bale S., Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Viollet L., Glusman G., Murphy K. J., Newcomb T. M., Reyna S. P., Sweney M., Nelson B., Andermann F., Andermann E., Acsadi G., Barbano R. L., Brown C., Brunkow M. E., Chugani H. T., Cheyette S. R., Collins A., DeBrosse S. D., Galas D., Friedman J., Hood L., Huff C., Jorde L. B., King M. D., LaSalle B., Leventer R. J., Lewelt A. J., Massart M. B., Mérida M. R. II, Ptáček L. J., Roach J. C., Rust R. S., Renault F., Sanger T. D., de Menezes M. A. S., Tennyson R., Uldall P., Zhang Y., Zupanc M., Xin W., Silver K., Swoboda K. J., Alternating hemiplegia of childhood: Retrospective genetic study and genotype-phenotype correlations in 187 subjects from the US AHCF registry. PLOS ONE 10, e0127045 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shinoda T., Ogawa H., Cornelius F., Toyoshima C., Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 459, 446–450 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geering K., The functional role of beta subunits in oligomeric P-type ATPases. J. Bioenerg. Biomembr. 33, 425–438 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Kaplan J. H., Biochemistry of Na,K-ATPase. Annu. Rev. Biochem. 71, 511–535 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Roenn C. P., Li M., Schack V. R., Forster I. C., Holm R., Toustrup-Jensen M. S., Andersen J. P., Petrou S., Vilsen B., Functional consequences of the CAPOS mutation E818K of Na+,K+-ATPase. J. Biol. Chem. 294, 269–280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arystarkhova E., Haq I. U., Luebbert T., Mochel F., Saunders-Pullman R., Bressman S. B., Feschenko P., Salazar C., Cook J. F., Demarest S., Brashear A., Ozelius L. J., Sweadner K. J., Factors in the disease severity of ATP1A3 mutations: Impairment, misfolding, and allele competition. Neurobiol. Dis. 132, 104577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mudigoudar B., Nune S., Fulton S., Dayyat E., Wheless J. W., Epilepsy in 22q11.2 deletion syndrome: A case series and literature review. Pediatr. Neurol. 76, 86–90 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Weller S., Rosewich H., Gartner J., Cerebral MRI as a valuable diagnostic tool in Zellweger spectrum patients. J. Inherit. Metab. Dis. 31, 270–280 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Jaffer F., Avbersek A., Vavassori R., Fons C., Campistol J., Stagnaro M., De Grandis E., Veneselli E., Rosewich H., Gianotta M., Zucca C., Ragona F., Granata T., Nardocci N., Mikati M., Helseth A. R., Boelman C., Minassian B. A., Johns S., Garry S. I., Scheffer I. E., Gourfinkel-An I., Carrilho I., Aylett S. E., Parton M., Hanna M. G., Houlden H., Neville B., Kurian M. A., Novy J., Sander J. W., Lambiase P. D., Behr E. R., Schyns T., Arzimanoglou A., Cross J. H., Kaski J. P., Sisodiya S. M., Faulty cardiac repolarization reserve in alternating hemiplegia of childhood broadens the phenotype. Brain 138, 2859–2874 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinzen E. L., Arzimanoglou A., Brashear A., Clapcote S. J., Gurrieri F., Goldstein D. B., Johannesson S. H., Mikati M. A., Neville B., Nicole S., Ozelius L. J., Poulsen H., Schyns T., Sweadner K. J., van den Maagdenberg A., Vilsen B.; ATP1A3 Working Group , Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 13, 503–514 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurrieri F., Tiziano F. D., Zampino G., Neri G., Recognizable facial features in patients with alternating hemiplegia of childhood. Am. J. Med. Genet. A 170, 2698–2705 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Allocco A. A., Jin S. C., Duy P. Q., Furey C. G., Zeng X., Dong W., Nelson-Williams C., Karimy J. K., DeSpenza T., Hao L. T., Reeves B., Haider S., Gunel M., Lifton R. P., Kahle K. T., Recessive inheritance of congenital hydrocephalus with other structural brain abnormalities caused by compound heterozygous mutations in ATP1A3. Front. Cell. Neurosci. 13, 425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chatron N., Cabet S., Alix E., Buenerd A., Cox P., Guibaud L., Labalme A., Marks P., Osio D., Putoux A., Sanlaville D., Lesca G., Vasiljevic A., A novel lethal recognizable polymicrogyric syndrome caused by ATP1A2 homozygous truncating variants. Brain 142, 3367–3374 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Platzer K., Yuan H., Schutz H., Winschel A., Chen W., Hu C., Kusumoto H., Heyne H. O., Helbig K. L., Tang S., Willing M. C., Tinkle B. T., Adams D. J., Depienne C., Keren B., Mignot C., Frengen E., Stromme P., Biskup S., Docker D., Strom T. M., Mefford H. C., Myers C. T., Muir A. M., LaCroix A., Sadleir L., Scheffer I. E., Brilstra E., van Haelst M. M., van der Smagt J. J., Bok L. A., Moller R. S., Jensen U. B., Millichap J. J., Berg A. T., Goldberg E. M., De Bie I., Fox S., Major P., Jones J. R., Zackai E. H., Jamra R. A., Rolfs A., Leventer R. J., Lawson J. A., Roscioli T., Jansen F. E., Ranza E., Korff C. M., Lehesjoki A. E., Courage C., Linnankivi T., Smith D. R., Stanley C., Mintz M., McKnight D., Decker A., Tan W. H., Tarnopolsky M. A., Brady L. I., Wolff M., Dondit L., Pedro H. F., Parisotto S. E., Jones K. L., Patel A. D., Franz D. N., Vanzo R., Marco E., Ranells J. D., Di Donato N., Dobyns W. B., Laube B., Traynelis S. F., Lemke J. R., GRIN2B encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 54, 460–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fry A. E., Fawcett K. A., Zelnik N., Yuan H., Thompson B. A. N., Shemer-Meiri L., Cushion T. D., Mugalaasi H., Sims D., Stoodley N., Chung S. K., Rees M. I., Patel C. V., Brueton L. A., Layet V., Giuliano F., Kerr M. P., Banne E., Meiner V., Lerman-Sagie T., Helbig K. L., Kofman L. H., Knight K. M., Chen W., Kannan V., Hu C., Kusumoto H., Zhang J., Swanger S. A., Shaulsky G. H., Mirzaa G. M., Muir A. M., Mefford H. C., Dobyns W. B., Mackenzie A. B., Mullins J. G. L., Lemke J. R., Bahi-Buisson N., Traynelis S. F., Iago H. F., Pilz D. T., De novo mutations in GRIN1 cause extensive bilateral polymicrogyria. Brain 141, 698–712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith R. S., Kenny C. J., Ganesh V., Jang A., Borges-Monroy R., Partlow J. N., Hill R. S., Shin T., Chen A. Y., Doan R. N., Anttonen A. K., Ignatius J., Medne L., Bonnemann C. G., Hecht J. L., Salonen O., Barkovich A. J., Poduri A., Wilke M., de Wit M. C. Y., Mancini G. M. S., Sztriha L., Im K., Amrom D., Andermann E., Paetau R., Lehesjoki A. E., Walsh C. A., Lehtinen M. K., Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral motor development. Neuron 99, 905–913.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith R. S., Walsh C. A., Ion channel functions in early brain development. Trends Neurosci. 43, 103–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sekiguchi F., Tsurusaki Y., Okamoto N., Teik K. W., Mizuno S., Suzumura H., Isidor B., Ong W. P., Haniffa M., White S. M., Matsuo M., Saito K., Phadke S., Kosho T., Yap P., Goyal M., Clarke L. A., Sachdev R., McGillivray G., Leventer R. J., Patel C., Yamagata T., Osaka H., Hisaeda Y., Ohashi H., Shimizu K., Nagasaki K., Hamada J., Dateki S., Sato T., Chinen Y., Awaya T., Kato T., Iwanaga K., Kawai M., Matsuoka T., Shimoji Y., Tan T. Y., Kapoor S., Gregersen N., Rossi M., Marie-Laure M., McGregor L., Oishi K., Mehta L., Gillies G., Lockhart P. J., Pope K., Shukla A., Girisha K. M., Abdel-Salam G. M. H., Mowat D., Coman D., Kim O. H., Cordier M. P., Gibson K., Milunsky J., Liebelt J., Cox H., El Chehadeh S., Toutain A., Saida K., Aoi H., Minase G., Tsuchida N., Iwama K., Uchiyama Y., Suzuki T., Hamanaka K., Azuma Y., Fujita A., Imagawa E., Koshimizu E., Takata A., Mitsuhashi S., Miyatake S., Mizuguchi T., Miyake N., Matsumoto N., Genetic abnormalities in a large cohort of Coffin–Siris syndrome patients. J. Hum. Genet. 64, 1173–1186 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Guerois R., Nielsen J. E., Serrano L., Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 320, 369–387 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L., The FoldX web server: An online force field. Nucleic Acids Res. 33, W382–W388 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumamoto T., Maurinot F., Barry-Martinet R., Vaslin C., Vandormael-Pournin S., Le M., Lerat M., Niculescu D., Cohen-Tannoudji M., Rebsam A., Loulier K., Nedelec S., Tozer S., Livet J., Direct readout of neural stem cell transgenesis with an integration-coupled gene expression switch. Neuron 107, 617–630.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q., Lau A., Morris T. J., Guo L., Fordyce C. B., Stanley E. F., A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: Analysis by quantitative immunocolocalization. J. Neurosci. 24, 4070–4081 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins T. J., ImageJ for microscopy. Biotechniques 43, 25–30 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Ohtaka-Maruyama C., Hirai S., Miwa A., Heng J. I., Shitara H., Ishii R., Taya C., Kawano H., Kasai M., Nakajima K., Okado H., RP58 regulates the multipolar-bipolar transition of newborn neurons in the developing cerebral cortex. Cell Rep. 3, 458–471 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Uchitel J., Helseth A., Prange L., McLean M., Ghusayni R., Sachdev M., Hunanyan A., Mikati M. A., The epileptology of alternating hemiplegia of childhood. Neurology 93, e1248–e1259 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Sweney M. T., Silver K., Gerard-Blanluet M., Pedespan J. M., Renault F., Arzimanoglou A., Schlesinger-Massart M., Lewelt A. J., Reyna S. P., Swoboda K. J., Alternating hemiplegia of childhood: Early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics 123, e534–e541 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabd2368/DC1