

PRMT5 directly induces methylation of cGAS and inhibits cGAS-mediated antiviral immunity.
Abstract
Cyclic GMP-AMP synthase (cGAS) functions as an essential DNA sensor, which senses the cytoplasmic double-stranded DNA and activates the antiviral response. However, the posttranslational modification of cGAS remains to be fully understood and whether it has arginine methylation modification remains unknown. Here, we identified protein arginine methyltransferase 5 (PRMT5) as a direct binding partner of cGAS, and it catalyzed the arginine symmetrical dimethylation of cGAS at the Arg124 residue. Further investigation demonstrated that methylation of cGAS by PRMT5 attenuated cGAS-mediated antiviral immune response by blocking the DNA binding ability of cGAS. Oral administration of PRMT5 inhibitors significantly protected mice from HSV-1 infection and prolonged the survival time of these infected mice. Therefore, our findings revealed an essential regulatory effect of PRMT5 on cGAS-mediated antiviral immune response and provided a promising potential antiviral strategy by modulating PRMT5.
INTRODUCTION
Infectious diseases, especially virus infection, are still a serious threat to humanity. Upon infection, eukaryotic cells immediately respond with robust innate immune responses as a critical defense against invading pathogens. Cyclic GMP (guanosine monophosphate)–AMP (adenosine monophosphate) (cGAMP) synthase (cGAS) is the key cytosolic DNA sensor that detects the cytoplasmic microbial DNA or self DNA, and induces the type I interferon (IFN) production (1). Upon binding to the double-stranded DNA (dsDNA), cGAS synthesizes cGAMP, a second-messenger molecule that binds to the adaptor protein stimulator of IFN genes (STING) and induces a conformational change in the STING dimer. Thus, the latter induces the activation of tank-binding kinase 1 (TBK1) and the transcription factor IFN regulatory factor 3 (IRF3), which further stimulates the production of type I IFN and subsequently induces the synthesis of antiviral proteins for effective defense against pathogens.
Efficient activation of cGAS signaling is crucial to prevent virus-infected disease; thus, it must be tightly regulated to ensure effective response to DNA virus while preventing collateral damage to the immune system. However, the regulatory mechanism of the cGAS-STING pathway remained to be fully understood. Several types of posttranslational modifications (PTMs) including phosphorylation, sumoylation, acetylation, polyubiquitination, monoubiquitination, and glutamylation have been shown to directly or indirectly regulate the cGAS-STING–mediated innate immune response (2–7). Whether cGAS is modified by protein arginine methylation, an important PTM in regulating multiple intracellular signaling, remains to be clarified. Protein arginine methylation could influence the interaction between protein and DNA (8), which indicated its great potential in the regulation of cGAS, the well-recognized cytosolic DNA sensor.
Protein arginine methylation is known to be catalyzed by protein arginine methyltransferases (PRMTs) in mammals, and it is a common PTM involved in multiple biological process including cellular signaling transduction, RNA processing, chromatin remodeling, and homologous recombination–mediated DNA repair (9). PRMTs are categorized into three types: type I PRMTs (PRMT1, PRMT2, PRMT3, PRMT4/CARM1, PRMT6, and PRMT8), which generate asymmetrical dimethylated arginine; type II PRMTs (PRMT5 and PRMT9), which generate symmetrical dimethylated arginine; and type III PRMT (PRMT7), which generates monomethylated arginine. Through mass spectrometry analysis of cGAS-interacting proteins, we identified PRMT5 as a potential candidate binding protein of cGAS. PRMT5 is the major type II arginine methyltransferase that symmetrically methylates arginine residues and usually exists in a complex with its cofactor, methylosome protein 50 (MEP50). Deletion of PRMT5 is embryonic lethal in mouse because of its essential requirement for the proliferation and differentiation of several stem cell lineages (10, 11). However, the development of its specific oral inhibitor, such as EPZ015666, extensively promotes the investigation of PRMT5 (12).
The role of PRMT5 has been reported in multiple biological processes, especially in the immunity-related processes including maintenance of T cell development and T helper 17 (TH17) cell differentiation, modulation of CD4+ T cell expansion, induction of IFN signaling in T cells, regulation of B cell function and antibody production, maintenance of normal adult hematopoiesis, and regulation of cell cycle progression (13–16). The regulatory role of PRMT5 has been defined in multiple signaling transduction processes including nuclear factor κB (NF-κB) signaling, phosphatidylinositol 3-kinase (PI3K)–AKT pathway, IFN signaling, and WNT/β-catenin pathway (17–19). Detailed mechanical investigation significantly helps to decipher the working mechanism of PRMT5, for example, the arginine methylation of p65 by PRMT5 profoundly regulates NF-κB activity (17), and the symmetric arginine dimethylation by PRMT5 precisely regulates IFN signaling by pre-mRNA splicing (19). Although the function of PRMT5 in multiple biological processes has been extensively studied, its direct modification of cGAS and its regulation of cGAS-mediated immunity against DNA virus remain to be clarified.
In this study, we defined a previously unidentified effect of PRMT5 on the regulation of innate immune response against DNA virus by symmetrical arginine methylation of RGG/RG motif of cGAS. We further demonstrated that methylation of cGAS by PRMT5 could inhibit the DNA binding ability of cGAS and further suppress cGAS-mediated antiviral immune response. These findings provided insight into the tightly regulated antiviral immunity and indicated a potential manipulation strategy against virus-infected diseases by modulation of PRMT5-induced methylation.
RESULTS
PRMT5 directly interacted with cGAS
To identify the possible cGAS-interacting proteins, we did repeated mass spectrometry analysis and our data consistently demonstrated that PRMT5 was the intracellular binding partner with cGAS (table S1). The reciprocal coimmunoprecipitation (co-IP) assay in the cGAS and PRMT5 exogenously overexpressed human embryonic kidney (HEK) 293T cells further verified that PRMT5 could interact with cGAS (Fig. 1A). In situ proximity ligation assay (PLA), a technique for detection of direct protein-protein interactions (20), further revealed the direct interaction of PRMT5 with cGAS (Fig. 1B and fig. S1A). The interaction between cGAS and MEP50, the cofactor of PRMT5, was also demonstrated by co-IP assay (fig. S1B). We further did GST pull-down assay to verify their direct interactions, and the data showed that cGAS could directly bind to PRMT5, but not MEP50, in vitro (Fig. 1C and fig. S1, C and D), which indicated that cGAS interacted with the PRMT5-MEP50 complex in the cells via its direct interaction with PRMT5. Further investigation showed that PRMT5 uniquely interacted with cGAS, while it had no interactions with other molecules involved in cGAS-STING signaling, including STING, TBK1, and IRF3 (fig. S1E). The endogenous interaction between PRMT5 and cGAS was detected in the herpes simplex virus-1 (HSV-1)–infected mouse bone marrow–derived macrophages (BMDMs) by endogenous IP assay (Fig. 1D), and confocal microscopy assay further showed that PRMT5 colocalized with cGAS in the HSV-1–infected BMDMs (fig. S1F), which demonstrated that PRMT5 was a bona fide binding partner of cGAS. Further analysis of these endogenous IP and colocalization data showed that the interaction level of PRMT5 with cGAS was gradually declined along with the progression of HSV-1 infection (fig. S1, F and G), which indicated a fine-tuned mechanism in the cGAS-induced antiviral immunity mediated by PRMT5.
Fig. 1. PRMT5 interacts with cGAS.
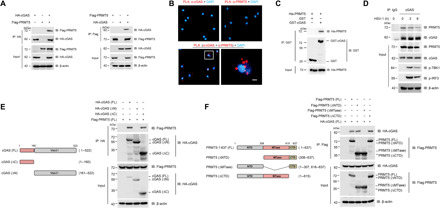
(A) Co-IP analysis of the interaction between PRMT5 and cGAS in HEK293T cells cotransfected with cGAS and PRMT5 plasmids. IP assay was done by anti-HA (left) and anti-Flag (left) antibodies to show the interaction between PRMT5 and cGAS. (B) In situ PLA analysis of the direct interaction of PRMT5 with cGAS in mouse macrophage. Scale bars, 10 μm. (C) GST pull-down analysis of purified PRMT5 incubated with GST or GST-cGAS in vitro. (D) IP analysis of the interaction between endogenous PRMT5 and cGAS in mouse BMDM cells infected with HSV-1 [multiplicity of infection (MOI) = 1] for 0, 3, and 6 hours. (E and F) Full-length (FL) and truncated mutant schematic structures of cGAS (E) and PRMT5 (F) were presented (left), and co-IP assay was performed to analyze the interaction between the full-length or truncated mutant forms of PRMT5 and cGAS (right). Data are representative of three independent experiments with similar results.
To further investigate the molecular basis of the interaction between PRMT5 and cGAS, we generated a series of domain-deleted mutants of both PRMT5 and cGAS to define their interacting domains. cGAS is composed of amino-terminal domain (N-terminal, amino acids 1 to 160) and carboxyl-terminal domain (C-terminal, amino acids 161 to 522), and PRMT5 is composed of amino-terminal domain (NTD domain, amino acids 1 to 307), SAM-dependent methyltransferase PRMT-type domain [Methyltransferase (MTase) domain, amino acids 308 to 637], and carboxyl-terminal domain (CTD domain, amino acids 616 to 637). After we cotransfected the domain-deleted mutants of these two proteins (Fig. 1, E and F), co-IP assay showed that deletion of the cGAS N-terminal domain abolished its interaction with PRMT5 (Fig. 1E), while deletion of the MTase domain of PRMT5 also ablated the interaction between PRMT5 and cGAS (Fig. 1F). Thus, these data demonstrated that the binding between PRMT5 and cGAS was a direct interaction, and the MTase domain of PRMT5 and the N-terminal domain of cGAS were the direct binding domains of these two proteins.
PRMT5 inhibits cGAS-STING pathway–triggered type I IFN production
We have demonstrated the direct interaction between PRMT5 and cGAS, and we are also interested in defining whether PRMT5 influences dsDNA-triggered cGAS-mediated antiviral immune response. PRMT5 is responsible for the maintenance of the differentiation of pluripotency in mouse embryonic stem cells, and the whole-body knockout of Prmt5 can cause embryonic lethality (11); thus, EPZ015666, a potent PRMT5-specific inhibitor, was often used for the investigation of the role of PRMT5 in different biological processes (21, 22). Our data showed that EPZ015666 treatment significantly inhibited symmetrical dimethylation of arginine (sDMA) catalyzed by PRMT5 (fig. S2A) and resulted in a significant increase in the mRNA level of Ifnb1, Cxcl10, and Mx1 (Fig. 2, A and B). We further demonstrated that EPZ015666 treatment significantly enhanced IFN-β and cGAMP production under the stimulation with IFN-stimulatory DNA (ISD), herring testis DNA (HT-DNA), and DNA virus HSV-1 (Fig. 2, C and D). However, the stimulation with cGAMP did not increase the type I IFN production (fig. S2B), which indicated that PRMT5 inhibited the activation of the cGAS-STING pathway at the upstream of cGAMP production.
Fig. 2. PRMT5 inhibits cGAS-triggered type I IFN production.
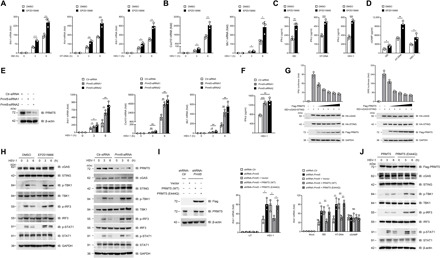
(A and B) Quantitative polymerase chain reaction (PCR) analysis of Ifnb1 (A), Cxcl10, and Mx1 (B) mRNA in mouse BMDMs pretreated with EPZ015666 (5 μM) for 48 hours followed by the indicated transfection/infection. (C and D) Enzyme-linked immunosorbent assay (ELISA) analysis of IFN-β (C) and cGAMP (D) production in EPZ015666-pretreated BMDMs followed by the indicated transfection/infection. (E and F) Mouse BMDMs were transfected with Prmt5-siRNA for 48 hours, and Ifnb1, Cxcl10, and Mx1 mRNA (E) and IFN-β production (F) after HSV-1 infection were analyzed. (G) Luciferase reporter activity analysis of IFN-β (left) and ISRE (right) in HEK293T cells with the indicated transfection. (H) Immunoblot analysis of cGAS signaling in the EPZ015666-pretreated (left) or Prmt5-siRNA–transfected (right) mouse BMDMs followed by HSV-1 infection. (I and J) The shRNA-Ctr or shRNA-Prmt5–transfected MEF cells were further transfected with PRMT5(WT) or PRMT5(E444Q). Ifb1 mRNA (I) and cGAS signaling (J) in these transfected cells was detected by real-time PCR and western blot, respectively. *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 3 mice per group, and the dots represent the value of each mouse). UT, untreated group; NS, not significant (I). Data are representative of three independent experiments with similar results [means ± SD in (A) to (G) and (I)]. DMSO, dimethyl sulfoxide.
To further verify the role of PRMT5 in cGAS-STING pathway–mediated IFN-β production, we transfected BMDMs with a couple of small interfering RNA against PRMT5 (Prmt5-siRNA) to construct the loss-of-function model. Our data showed that after successful knockdown of PRMT5 in BMDMs, the sDMA level was significantly inhibited (fig. S2C), while the mRNA level of Ifnb1, Cxcl10, and Mx1 and IFN-β production were all significantly up-regulated upon HSV-1 infection, ISD, or HT-DNA stimulation (Fig. 2, E and F, and fig. S2D), whereas simulation with cGAMP did not influence the mRNA level of Ifnb1 (fig. S2D). We further transfected a Flag-tagged PRMT5 expression plasmid into mouse embryonic fibroblasts (MEFs) to construct the gain-of-function model and showed that, under the stimulation with HSV-1, ISD, and HT-DNA, IFN-β production in the PRMT5-overexpressing MEFs was decreased, while stimulation with cGAMP had no effect on IFN-β production (fig. S2, E to G). After we cotransfected PRMT5, cGAS, and STING expression plasmids together with ISD into the HEK293T cells, both the IFN-β reporter activity and the IFN-stimulated response element (ISRE) reporter activity were significantly decreased by overexpression of PRMT5 in a dose-dependent manner (Fig. 2G). Together, both loss-of-function and gain-of-function data demonstrated that PRMT5 significantly inhibited cGAS-mediated IFN-β production upon DNA virus infection, and this effect was exerted at the level of cGAS and was upstream of cGAMP release.
To further verify the negative regulatory effect of PRMT5 on cGAS signaling, we then detected the phosphorylation levels of the cGAS downstream signaling molecules. Our data showed that after PRMT5 inhibition by its specific inhibitor EPZ015666 or by its specific siRNA, the protein levels of cGAS and STING were not altered and the phosphorylation level of TBK1, IRF3, and STAT1 was significantly increased upon HSV-1 infection (Fig. 2H), while no significant difference of their phosphorylation level was detected upon cGAMP treatment (fig. S2H). We further showed that the activation of NF-κB signaling (fig. S2I) and the production of tumor necrosis factor–α (TNF-α) and interleukin-6 (IL-6) (fig. S2, J and K) at the downstream of the cGAS-STING pathway were also significantly up-regulated after the inhibition of PRMT5. Further investigation showed that PRMT5 did not significantly influence the mRNA levels of Cgas and Sting (fig. S2L).
PRMT5 is an arginine methyltransferase; thus, we further constructed its enzymatically dead mutant, PRMT5(E444Q) (23), to define whether this regulation of cGAS signaling was related to its enzymatic activity. We transfected PRMT5(WT) or PRMT5(E444Q) into PRMT5 knockdown MEF cells, and our data showed that PRMT5(E444Q) almost completely rescued the suppression of Ifnb1 mRNA and cGAS signaling by PRMT5 under the stimulation of HSV-1, ISD, or HT-DNA (Fig. 2, I and J), which indicated that the enzymatic activity of PRMT5 played a key role in the suppression of cGAS-mediated IFN-β production. Together, our data verified that PRMT5 significantly inhibited the cGAS-STING pathway activation and type I IFN production via its enzymatic activity upon DNA stimulation.
PRMT5 catalyzed the arginine dimethylation of cGAS at the Arg124 residue
Because PRMT5 is an arginine methyltransferase responsible for most of the intracellular modification of symmetrical dimethylation of target proteins, we further tried to define whether PRMT5 induces sDMA of cGAS. We cotransfected HA-tagged cGAS and Flag-tagged PRMT5 expression plasmids into HEK293T cells and further analyzed the sDMA modification status of cGAS. IP assay showed that cGAS could be symmetrically dimethylated and exogenous overexpression of PRMT5 markedly promoted the symmetrical dimethylation of cGAS (Fig. 3A and fig. S3A), while the PRMT5(E444Q) enzymatically dead mutant abrogated the symmetrical dimethylation of cGAS induced by PRMT5 (Fig. 3B).
Fig. 3. PRMT5 mediates dimethylation of cGAS.
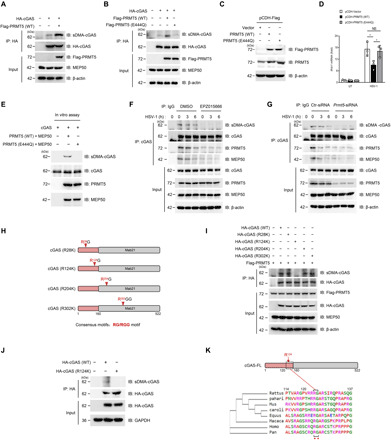
(A and B) IP analysis of cGAS dimethylation in HEK293T cells cotransfected with cGAS and PRMT5 (A) or PRMT5(E444Q) mutant (B). (C and D) MEFs were transfected with lentivirus-mediated PRMT5(WT) or PRMT5(E444Q), and stably transfected cells were selected followed by immunoblot analysis of PRMT5(C). Ifnb1 mRNA in these transfected MEFs with HSV-1 infection (n = 3 mice per group) was analyzed by quantitative PCR (D). (E) Immunoblot analysis of cGAS dimethylation in an in vitro methylation system. (F and G) IP analysis of cGAS dimethylation in the EPZ015666-pretreated BMDMs (F) or Prmt5-siRNA–transfected BMDMs (G) after infection with HSV-1 for 0, 3, and 6 hours. (H) Consensus RGG/RG motif analysis of cGAS. Red arrow indicates the position of arginine. (I) Co-IP analysis of cGAS dimethylation in HEK293T cells cotransfected with PRMT5 and cGAS(WT) or its arginine-to-lysine substitution mutants. (J) IP analysis of cGAS dimethylation in HEK293T cells transfected with cGAS(WT) or cGAS(R124K) mutant. (K) Conservation analysis of cGAS Arg124 among species by the Clustal Omega program. *P < 0.05. Data are representative of three independent experiments with similar results [means ± SD in (D)].
To further validate the dimethylation modification of cGAS by PRMT5, we constructed a knock-in cellular model by infecting the MEF cells with PRMT5(WT) or PRMT5(E444Q) overexpressed lentivirus (Fig. 3C and fig. S3B). Our data showed that overexpression of PRMT5(WT) significantly inhibited the Ifnb1 mRNA level stimulated by HSV-1 infection, while this inhibitory effect was almost completely abolished by the PRMT5(E444Q) mutant (Fig. 3D), which indicated that the inhibition of cGAS signaling upon viral infection was mediated by the arginine methyltransferase activity of PRMT5. We further verified that cGAS could be methylated by the PRMT5-MEP50 complex in an in vitro methylation system (Fig. 3E), which indicated that cGAS could be directly methylated by PRMT5.
Further investigation showed that the endogenously generated cGAS could be efficiently dimethylated by PRMT5 at quiescent cells without virus infection (fig. S3C), whereas the symmetrical dimethylation of endogenous cGAS was gradually declined after HSV-1 infection (Fig. 3, F and G). Our data further showed that cGAS dimethylation was almost completely eliminated by EPZ015666 treatment or transfection with siRNA against Prmt5 during HSV-1 infection (Fig. 3, F and G). These data indicated that cGAS could be constitutively methylated by PRMT5, while this methylation was declined to rescue cGAS activation upon virus infection.
PRMT5 is the major enzyme catalyzing arginine symmetrical dimethylation of the target proteins, and it has a preference for the consensus arginine- and glycine-rich region termed RGG/RG motifs (24, 25). Thus, we further try to determine whether cGAS harbors these similar RGG/RG motifs that could be methylated by PRMT5. We screened all arginine residues containing RGG/RG motifs in cGAS and found that cGAS contained four such motifs: R28G, R124G, R204G, and R302GG. The Arg28 and Arg124 residues were located in the N-terminal domain of cGAS, while the Arg204 and Arg302 residues were located in the C-terminal domain of cGAS (Fig. 3H). To further clarify the exact residue methylated by PRMT5, we constructed all of these arginine site mutants by altering each arginine to the lysine residue (R28K, R124K, R204K, and R302K). Arginine methylation assay showed that the substitution of the arginine at Arg124 with lysine (R124K) almost completely abolished the symmetrical arginine dimethylation of cGAS induced by PRMT5 (Fig. 3, I and J), which indicated that the Arg124 residue is the catalyzed site of PRMT5. The alignment for R124 indicated that this site was conserved among species (Fig. 3K), which suggested the biological importance of this site for the species. Further investigation showed that both the PRMT5(E444Q) mutant and the cGAS(R124) mutant reduced the binding between cGAS and PRMT5 (fig. S3, D and E). These data indicated that both the recognition of the RGG/RG motif of cGAS and the methylation modification of cGAS contributed to the interaction between PRMT5 and cGAS. Together, these data indicated that PRMT5 recognized the RGG/RG motif of cGAS and directly catalyzed the arginine dimethylation of cGAS at the Arg124 residue.
Inhibition of PRMT5-mediated dimethylation of cGAS promoted antiviral response
We next investigated whether this arginine symmetrical dimethylation of cGAS by PRMT5 had any effect on cGAS-mediated antiviral response. Our data showed that treatment with EPZ015666 significantly increased HSV-1–induced Ifnb1 expression in macrophages from wild type (WT) mice, but no significant effect of EPZ015666 on Ifnb1 mRNA was observed in macrophages from Cgas knockout (Cgas−/−) mice (Fig. 4A). We further showed that inhibition of PRMT5 by EPZ015666 and siRNA against Prmt5 had no significant influence on the cell viabilities of the investigated cells (fig. S4, A and B).
Fig. 4. Dimethylation of cGAS inhibits antiviral responses.
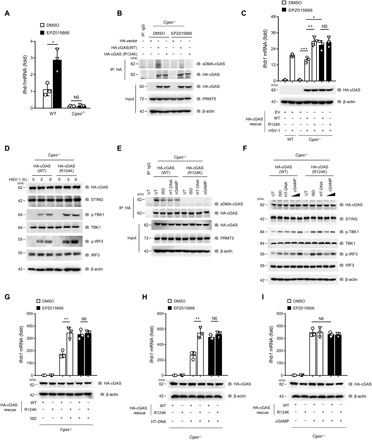
(A and B) WT or Cgas−/− BMDMs were pretreated with EPZ015666 (5 μM) for 48 hours followed by infection with HSV-1 for 6 hours. Ifnb1 mRNA (A) and dimethylation of cGAS (B) were analyzed by quantitative PCR and IP assay, respectively. (C) Quantitative PCR analysis of Ifnb1 mRNA in EPZ015666-treated Cgas−/− MEFs transfected with the indicated plasmid followed by HSV-1 infection for 6 hours. (D) Immunoblot analysis of cGAS signaling in Cgas−/− MEFs transfected with cGAS(WT) or cGAS(R124K) plasmid after HSV-1 infection for 0, 3, and 6 hours. (E) IP analysis of cGAS dimethylation in Cgas−/− MEFs transfected with cGAS(WT) or cGAS(R124K) plasmid, followed by transfection with ISD, HT-DNA, or cGAMP. (F) Immunoblot analysis of cGAS signaling in Cgas−/− MEFs transfected with cGAS(WT) or cGAS(R124K) plasmid followed by transfection with ISD, HT-DNA, or cGAMP (0.5 or 2 μg/ml). (G to I) Quantitative PCR analysis of Ifnb1 mRNA in Cgas−/− MEFs transfected with cGAS(WT) or cGAS(R124K) plasmid, followed by transfection with ISD (G), HT-DNA (H), or cGAMP (I) for 6 hours. *P < 0.05, **P < 0.01, and ***P < 0.001 (n = 3 mice per group, and dots represent the mean value per mouse). Data are representative of three experiments with similar results [means ± SD in (A), (C), and (G) to (I)].
To further verify that PRMT5 induces symmetric arginine dimethylation of cGAS at the R124 residue, we transfected equal amounts of HA-tagged cGAS(WT) or HA-tagged cGAS(R124K) mutant plasmids into the Cgas−/− MEF cells. Our data showed that transfection with the cGAS(R124K) mutant almost completely abolished the dimethylation of cGAS, and EPZ015666 treatment also almost totally eliminated the dimethylation of cGAS (Fig. 4B), which indicated the crucial role of PRMT5 in inducing dimethylation of cGAS at the R124 residue. We further showed that EPZ015666 treatment also significantly reversed the PRMT5-induced attenuation of IFN-β production (Fig. 4C), and the cGAS(R124K) mutant significantly restored HSV-1 infection–induced IFN-β signaling by RNA sequencing assay and quantitative real-time polymerase chain reaction (PCR) analysis of a panel of Interferon-stimulated genes (ISGs) (fig. S4, C and D). Moreover, the PRMT5-induced inhibition of the cGAS-STING pathway upon HSV-1 infection was also significantly rescued by the cGAS-R124K mutant as detected by the phosphorylation of TBK1 and IRF3 (Fig. 4D and fig. S4E). Together, these data indicated that PRMT5-mediated arginine dimethylation of cGAS played a crucial regulatory role in cGAS-induced antiviral immune response.
To further verify that the Arg124 residue was responsible for the symmetric arginine dimethylation of cGAS induced by PRMT5, we restored Cgas−/− MEFs with the expression of the cGAS(WT) or cGAS(R124K) mutant and further stimulated these transfected cells with ISD, HT-DNA, or cGAMP. sDMA assay showed that the cGAS(R124K) mutant almost completely abolished the PRMT5-induced symmetric arginine dimethylation of cGAS (Fig. 4E), and the activation of the cGAS-STING pathway, as well as the up-regulated Ifnb1 expression upon stimulation with ISD and HT-DNA, was significantly restored in the cGAS(R124K) mutant–transfected Cgas−/− MEFs cells, whereas cGAMP treatment did not restore the cGAS-STING pathway activation and the up-regulation of Ifnb1 mRNA expression in these transfected cells (Fig. 4, F to I). In addition, EPZ015666 treatment had no influence on the Ifnb1 mRNA level in the Cgas−/− MEF cells with cGAS(R124K) transfection (Fig. 4, G to I), which further verified that the methylation of cGAS upon cytosolic DNA stimulation was mediated by PRMT5. Together, these data demonstrated that arginine dimethylation of cGAS at the Arg124 residue by PRMT5 was crucial for the regulation of the cGAS-induced type I IFN production.
Arginine methylation of cGAS by PRMT5 inhibited the DNA binding ability of cGAS
We have demonstrated that cGAS bound with PRMT5 via its N-terminal domain, and it is recently reported that 120 to 160 amino acids in the N-terminal domain of cGAS play an essential role in recognizing the dsDNA because of its enrichment in arginine and lysine residues (26, 27). Therefore, we further try to define whether PRMT5 attenuates the activation of the cGAS signaling pathway by influencing the DNA binding ability of cGAS. Our data showed that overexpression of PRMT5 significantly reduced the amount of bound DNA with cGAS (fig. S5, A and B), which indicated that PRMT5 inhibited cGAS activation via its disruption of the DNA binding ability of cGAS.
To further demonstrate this, we transfected the biotin-labeled ISD into EPZ015666-pretreated BMDMs and further analyzed the DNA binding ability of cGAS. The streptavidin pull-down assay demonstrated that cGAS bound to higher amount of DNA after inhibition of PRMT5 by EPZ015666 or its specific siRNA, and the phosphorylation of IRF3 was also significantly increased in these PRMT5-inhibited cells (Fig. 5, A and B). Further quantitative DNA binding assay also demonstrated a higher DNA binding ability of cGAS after blocking PRMT5 by EPZ015666 or Prmt5-siRNA transfection (Fig. 5C). Moreover, we transfected fluorescein isothiocyanate (FITC)–labeled ISD into BMDMs to induce the cGAS-DNA aggregation, and confocal microscopy assay showed that inhibition of PRMT5 by EPZ015666 treatment or transfection with siRNA against Prmt5 prominently facilitated the colocalization of cGAS and FITC-ISD in the cytosol (Fig. 5, D and E), which further verified that PRMT5 could disrupt the DNA binding ability of cGAS.
Fig. 5. Dimethylation of cGAS inhibits its DNA binding ability.
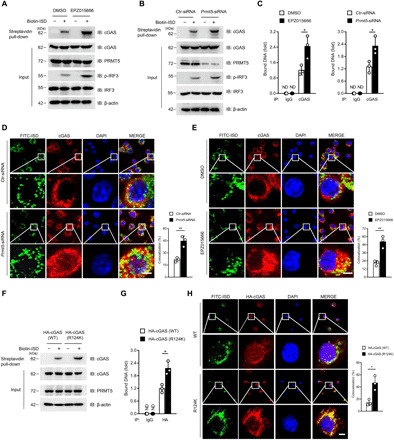
(A and B) Immunoblot analysis of ISD-bound cGAS by biotin-ISD pull-down performance in EPZ015666-treated BMDMs (A) or Prmt5-siRNA–transfected BMDMs (B). (C) Quantitative PCR analysis of the cGAS-bound DNA after IP of cGAS in the EPZ015666-pretreated BMDMs (left) or Prmt5-siRNA–transfected BMDMs (right) with further transfection with ISD for 6 hours. (D and E) Confocal microscopy assay of the colocalization of cGAS and FITC-ISD in Prmt5-siRNA–transfected BMDMs (D) or EPZ015666-treated BMDMs (E). The nucleus was stained with DAPI (blue), and the colocalization of cGAS (red) and ISD (green) was visualized as yellow fluorescence in the merged panel. Colocalization ratios of cGAS and FITC-ISD were analyzed by Manders’ coefficient values. (F and G) ISD-bound cGAS (F) and cGAS-bound DNA (G) after the biotin-ISD pull-down performance in cGAS(WT)- or cGAS(R124K)-transfected HEK293T cells were analyzed by immunoblotting and quantitative PCR, respectively. (H) Confocal microscopy analysis of the colocalization of cGAS and FITC-ISD in cGAS(WT)- or cGAS(R124K)-transfected HEK293T cells. Scale bars (bottom right), 10 μm. Statistical analysis was done with Student’s t test (n = 3 mice per group, and dots represent the mean value per mouse). *P < 0.05, **P < 0.01. Data are representative of three independent experiments with similar results [means ± SD in (C) to (E), (G), and (H)].
To further verify that the Arg124 residue was responsible for the PRMT5-mediated disruption of the DNA binding ability of cGAS, we transfected the HEK293T cells with either cGAS(WT) or cGAS(R124K) plasmid and further analyzed the DNA binding ability of cGAS. Our data showed that the amount of cGAS-bound DNA in cGAS(R124K)-overexpressing cells was significantly increased compared with that of cGAS(WT)-overexpressing cells (Fig. 5, F to H), which indicated that the cGAS Arg124 residue was responsible, at least to a great extent, for the disrupted DNA binding ability of cGAS. Together, these data indicated that PRMT5 attenuated the cGAS-mediated signaling by hindering the DNA binding ability of cGAS via the dimethylation of cGAS at the Arg124 residue.
Inhibition of PRMT5-protected mice against viral infection by enhancing type I IFN production
We have defined that PRMT5 could inhibit cGAS-mediated antiviral immune response; thus, we are also interested in demonstrating whether inhibition of PRMT5 could have any protective effect on the virus-infected mice. Thus, we pretreated mice with orally administrated EPZ015666 twice daily after selecting an optimum dosage (Fig. 6, A and B, and fig. S6, A and B) and further investigated the response of these mice to HSV-1 infection. Our data showed that EPZ015666-treated mice had higher Ifnb1 expression level in the liver, spleen, and lung tissues after HSV-1 infection (Fig. 6A). Further assay showed that the serum IFN-β production in the EPZ015666-treated mice was also significantly elevated (Fig. 6B). Moreover, the HSV-1 genome replication level, the HSV-1 titers, and the total sDMA level of the tissues from the liver, spleen, and lung of the EPZ015666-treated mice were all significantly inhibited (Fig. 6, C and D, and fig. S6C), which indicated that inhibition of PRMT5 could inhibit the methylation and further suppress the replication of HSV-1 virus in the infected mice. Hematoxylin and eosin (H&E) staining of the lung tissues showed that EPZ015666 treatment could significantly alleviate the inflammatory infiltration and tissue injury after HSV-1 infection (Fig. 6E), which further indicated that inhibition of PRMT5 could protect mice against DNA virus. When we challenged the WT and Cgas−/− mice with HSV-1 infection, the data showed that EPZ015666 treatment could prolong the survival time of the infected WT mice, while it failed to prolong the survival time of the HSV-1–infected Cgas−/− mice (Fig. 6F), which indicated that inhibition of PRMT5 protected the mice against HSV-1 infection through its regulation of cGAS activation.
Fig. 6. Blockade of PRMT5 alleviates susceptibility of mice against HSV-1 infection.
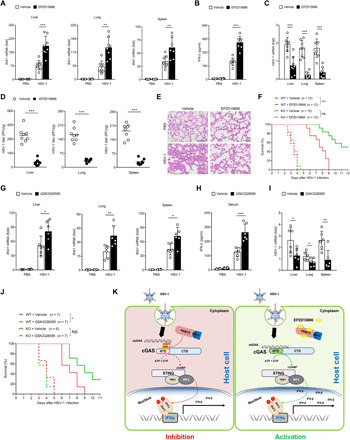
(A to E) Eight-week gender-matched BALB/c mice were pretreated with EPZ015666 or vehicle for 12 hours followed by HSV-1 infection (n = 8 mice per group), and the mice were further treated with twice daily administration of EPZ015666 (60 mg/kg, orally) or vehicle. The mouse tissue Ifnb1 mRNA (A) and serum IFN-β production (B) were analyzed by quantitative PCR and ELISA, respectively. The HSV-1 genome (C) and HSV-1 titers (D) were measured. H&E staining of lung sections was presented [(E); scale bars, 200 μm]. (F) WT or Cgas−/− mice were treated with EPZ015666, followed by HSV-1 infection (n = 12 mice per group), and the survival time of these mice was analyzed by log-rank (Mantel-Cox) test. (G to I) Eight-week gender-matched BALB/c mice were treated with GSK3326595 (40 mg/kg, orally) or vehicle, followed by HSV-1 infection (n = 6, 7 mice per group). The mouse tissue Ifnb1 mRNA (G) and serum IFN-β (H) were measured. The HSV-1 genome in mouse tissues was analyzed (I). (J) WT or Cgas−/− mice were treated with GSK3326595, followed by HSV-1 infection. The survival time of these mice was statistically analyzed. (K) Schematic working model of PRMT5-mediated arginine symmetrical dimethylation of cGAS. *P < 0.05, **P < 0.01, and ***P < 0.001. Data are representative of three independent experiments with similar results [means ± SD in (A) to (D) and (G) to (I)].
We next tried to verify the protective effect of PRMT5 inhibition on the HSV-1 challenged mice by another S-adenosylmethionine noncompetitive PRMT5 inhibitor, GSK3326595 (28–31), and our data demonstrated that inhibition of PRMT5 could enhance the Ifnb1 level and increase IFN-β production upon HSV-1 infection (Fig. 6, G and H). Further investigation demonstrated that GSK3326595 could significantly inhibit the replication of HSV-1 in mouse tissues (Fig. 6I) and further promoted the survival status of the infected mice (Fig. 6J). Thus, these data demonstrated that blockade of PRMT5 by its specific inhibitors could significantly protect mice from HSV-1 infection by abolishing the negative regulation of cGAS by PRMT5. Together, in this study, we demonstrated that PRMT5 negatively regulated cGAS-mediated antiviral immune response via the symmetric arginine dimethylation of cGAS at the Arg124 residue (Fig. 6K).
DISCUSSION
Detection of cytosolic DNA by cGAS is critical for the immune system to sense the invading pathogens, induce the downstream activation, and elicit a rapid response to fight against the pathogens. Although the fundamental principles that govern these sensors are emerging (32), the mechanisms that determine the tightly controlled innate antiviral responses remained to be fully understood. In this study, we reported the negative regulation of cGAS-mediated antiviral immune response by PRMT5 and demonstrated here the direct methylation modification of cGAS protein.
Arginine methylation is a common PTM playing a crucial role in multiple biological processes including signal transduction, transcriptional regulation, and RNA processing (9). PRMT5 belongs to the class II arginine methyltransferases, and it mainly functions as symmetrical dimethylargininase at RGG/RG motifs of the target proteins (24). It is up-regulated in various human malignancies and exacerbates the malignant progression of lymphomas, lung cancer, breast cancer, and colorectal cancer; thus, targeting PRMT5 recently becomes a hotspot for anticancer drug development (21, 33–35). However, the role of PRMT5 in cGAS-mediated antiviral immune response remained to be fully understood.
Here, we demonstrated that PRMT5 directly bound with cGAS and induced its symmetric arginine dimethylation, which significantly attenuated cGAS-mediated antiviral immune response. We further identified cGAS as an RGG/RG motif–containing protein and generated arginine-to-lysine substitutions (R28K, R124K, R204K, and R302K) in the RGG/RG motifs of cGAS to identify the specific catalytic residue by PRMT5. Our data verified that the R124 residue is the direct catalyzing site by PRMT5, and the cGAS(R124K) mutant lost the arginine methylation of cGAS and consequently abolished the PRMT5-mediated suppression of type I IFN production during virus infection, which indicated that this residue was a crucial target site responsible for the immune escape of virus. The fact that the R124 residue is a much conserved motif in different species further indicates its significant importance for the antiviral immune response mediated by cGAS.
It is reported that PRMT5 could exert its negative regulatory effect by further inducing ubiquitous modification and proteasomal degradation of its target protein (36). However, our data unexpectedly showed that the methylation of RGG/RG motif of cGAS by PRMT5 did not significantly affect the mRNA or protein level of cGAS, but rather attenuated cGAS activation. We showed that PRMT5 bound with cGAS through its N-terminal domain, and this domain could directly bind to the cytosolic DNA and further induce the full activation of cGAS (26, 27). Because it is recognized that arginine methylation played an important role in the interaction between protein and DNA (8), we further tried to define whether PRMT5 inhibited cGAS signaling by disrupting the interaction between cGAS and DNA. As expected, we verified that PRMT5 suppressed cGAS activation by inhibiting the DNA binding ability of cGAS and further demonstrated that inhibition of PRMT5 significantly rescued the antiviral immune response mediated by cGAS in vitro and in vivo. Together, our data indicated that, in quiescent cells, PRMT5 bound with cGAS and the methylated cGAS has limited capability to bind to intracellular DNA; thus, it helps to prevent the inflammation and autoimmune response caused by the abnormal presence of cytosolic DNA during stress or other types of stimuli. Upon viral infection, PRMT5 gradually dissociated with cGAS and the DNA binding ability of cGAS was enhanced, leading to the activation of cGAS signaling to fight against the invading virus. Therefore, this study indicated a fine-tuned regulatory effect on cGAS activation by PRMT5-mediated direct methylation modification of cGAS protein. Although we have provided evidence supporting a role of PRMT5-mediated dimethylation of cGAS in the antiviral immune response, the associated mechanism may require future studies to define whether other mechanisms, for example, protein demethylation and RNA splicing, are also involved in the regulatory mechanism. These related studies will constitute promising new research fields and help to thoroughly decipher the mechanism of the fine-tuned modulation of the antiviral immune response.
This study suggested a negative regulatory role of PRMT5 in cGAS-mediated type I IFN production during virus infection. Recent reports about the role of PRMT5 in the type I IFN production seemed to be controversial (19, 31, 37, 38). It was reported that PRMT5 promoted IFN signaling in T cells in a graft-versus-host disease mouse model and in the antimicrobe response (19, 37). The positive regulation of IFN signaling by PRMT5 was also reported in the mesenchymal stromal cells undergoing osteogenic differentiation (38). In contrast, a recent study reported that PRMT5 attenuated type I IFN production in a murine melanoma model (31), which is consistent with our study showing the negative regulation of IFN signaling by PRMT5. Moreover, the methylation of STAT1 (signal transducer and activator of transcription 1), the key signaling molecule downstream of IFN signaling after IFN-I bound with its receptor, was also controversially reported (39–42). These discrepancies are logical in consideration of the different modified targets and working mechanism that PRMT5 might have in distinctly originated cells through different biological processes. However, in the broad view of the role of arginine methyltransferase in the antiviral innate immunity, the related reports are consistent with our data showing the negative regulation of several PRMTs in IFN-I production, which further led to the suppression of the antiviral immune response (43–46). The PRMT5 protein may have different optimum modified target protein in different experimental system and that may cause the missing of cGAS as a potent direct modified target of PRMT5 in the previous reports (47, 48). Together, we reported here a previously unrecognized role of PRMT5 in attenuating cGAS-mediated type I IFN production against DNA virus by direct dimethylation of cGAS protein, which might have important translational implications for the treatment of virus-infected disease.
Although type I IFNs induced by cGAS signaling are critical for mediating innate immune response to fight against the invading viruses, aberrant activation of this signaling and production of these cytokines can also have pathological roles in a variety of autoimmune disorders (1, 49, 50). For example, DNA repair exonuclease 1 (TREX1) is an exonuclease that digests DNA in the cytoplasm; deletion of Trex1 in mice results in severe cGAS-dependent autoimmunity, and loss-of-function mutations of TREX1 are linked to autoimmune diseases in human (51). Thus, modulation of PRMT5-mediated suppression of cGAS signaling might also have great application potential in treating TREX1 mutation–related diseases. Because it is critical for the tight control of cGAS activation in triggering an effective immune response against infection without causing host pathology, the regulatory significance of PRMT5 in autoimmune disease and other inflammatory disorders might also be an interesting research field in the future.
In conclusion, our study demonstrated that PRMT5 induced arginine symmetrical dimethylation of cGAS at the Arg124 residue and further attenuated cGAS-mediated antiviral innate immune response. This study is the first report to identify the direct methylation modification of cGAS and provides insight into the complex mechanisms underlying cGAS-mediated antiviral immune response, which suggests a feasible therapeutic strategy for the treatment of virus-infected disease by modulating PRMT5. Meanwhile, given the fast development of the therapeutics against PRMT5 undergoing clinical trials, our findings may also have particular significance for clarifying the regulatory role of PRMT5 in the immune system and help to expand the application of therapeutic strategies targeting PRMT5.
MATERIALS AND METHODS
Plasmids, antibodies, and reagents
cGAS, STING, TBK1, and IRF3 were cloned into the pcDNA3.1 vector. PRMT5, His-PRMT5, and His-MEP50 plasmids were gifts from Q. Sun (Sun Yat-sen University). PRMT5 and cGAS mutants were generated with QuikChange site-directed mutagenesis kits (catalog no. 200518, TOYOBO, Japan) according to its protocol. The primary antibodies and their dilutions used in our study were as follows: rabbit anti-dimethyl-arginine antibody-symmetric (1:150; SYM10; Sigma-Aldrich, USA), rabbit anti-cGAS [1:1000; 31659S, Cell Signaling Technology (CST), USA], rabbit anti-sDMA (1:1000; 13222, CST, USA), rabbit anti-TBK1 (1:1000; 38066, CST, USA), rabbit anti–p-TBK1 (1:1000; 5483, CST, USA), rabbit anti–p-IRF3 (1:1000; 29047S, CST, USA), rabbit anti–p-STAT1 (1:1000; 7649, CST, USA), rabbit anti-MEP50 (1:1000; 2018, CST, USA), rabbit anti-normal immunoglobulin G (1:200; 2729, CST, USA), rabbit anti-PRMT5 (1:1000; 18436-1-AP, Proteintech, USA), rabbit anti-STING (1:1000; 19851-1-AP, Proteintech, USA), rabbit anti-IRF3 (1:1000; 11312-1AP, Proteintech, USA), mouse anti–β-actin (1:5000; 60008-1-Ig, Proteintech, USA), mouse anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:20,000; 60004-1-Ig, Proteintech, USA), rabbit anti-SNRPD3 (1:1000; ab121129, Abcam, USA), rabbit anti-Flag (1:1000; F7425, Sigma-Aldrich, USA), mouse anti-HA (1:1000, H9658, Sigma-Aldrich, USA), rabbit anti-HA (1:1000, H6908, Sigma-Aldrich, USA), and mouse anti-PRMT5 (1:200; sc-376937, Santa Cruz Biotechnology, USA). Reagents used in this study were as follows: HT-DNA (catalog no. D6898, Sigma-Aldrich, USA), cGAMP (catalog no. tlrl-cga23, InvivoGen, USA), EPZ015666 (catalog no. S7748, Selleck, USA), and GSK3326595 (catalog no. S8664, Selleck, USA). ISD, biotin-ISD, and FITC-ISD were synthesized from Sango Biotech and BGI.
IP and immunoblot analysis
Cells were lysed in the lysis buffer [20 mM tris-HCl (pH 8.0), 0.5% NP-40, 10 mM NaCl, 1 mM EDTA] containing 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and complete protease inhibitor cocktail (catalog no. 04693132001, Roche, USA), followed by centrifugation at 14,000 rpm at 4°C for 15 min. The supernatants were immunoprecipitated with protein A/G agarose (catalog no. sc-2003, Santa Cruz Biotechnology, USA), followed by incubation with anti-Flag or anti-HA antibodies at 4°C overnight. The immunoprecipitates were washed at least five times with the lysis buffer, and the protein lysate was further eluted with 5× SDS loading buffer. After heating denaturation, protein samples were analyzed by 10% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) before being transferred to polyvinylidene difluoride membrane for further detection with specific antibodies.
Isolation of MEFs and BMDMs
To obtain MEFs, mouse embryos were isolated from WT or Cgas−/− mice (BALB/c) on day 12.5 embryos. For the generation of BMDMs, bone marrow cells were isolated from the mice (BALB/c, 6 to 8 weeks) tibia and femur and plated into dishes at a density of 1 × 106/ml. The isolated cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM) containing 20% (v/v) fetal bovine serum (FBS) and 30% (v/v) L929 supernatants for 4 days before medium replacement. The cells were further cultured for another 3 days before the harvest of the adherent BMDMs.
Cells, transfection, RNA interference, and inhibitor treatment
HEK293T cells were purchased from the American Type Culture Collection, cultured with DMEM containing 10% (v/v) FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). HEK293T cells were commonly used as a model cell in consideration of its high transfection efficiency. MEFs and BMDMs were isolated and cultured as described above. All of the experiments were performed with mycoplasma-free cells. For transfection with plasmid, HEK293T cells and MEFs were plated into six-well plates or 6-cm dishes and further cultured for 24 hours before being transfected with 2 to 4 μg of plasmids via Lipofectamine 2000 (catalog no. 11668, Invitrogen, USA) according to the manufacturer’s instructions. For RNA interference, cells were transfected with control siRNA or Prmt5 siRNA by RNAiMax transfection reagent (catalog no. 13778150, Invitrogen, USA) and further cultured for 48 hours before further investigation. siRNAs were synthesized from GenePharma, and the sequence information was listed in table S3. The inhibitory effect of these siRNAs was verified by quantitative PCR and immunoblotting. For the inhibitor treatment, the investigated cells were pretreated with the PRMT5 inhibitor EPZ015666 for 48 hours before further investigation.
Mass spectrometry analysis
To identify the proteins that interact with cGAS, HEK293 cells were transfected with HA-tagged cGAS and further cultured for 48 hours. The transfected cells were then lysed with the lysis buffer, and the supernatants were collected and incubated with protein A/G agarose with or without anti-HA primary antibody at 4°C overnight for cGAS IP. The beads were washed with lysis buffer at least five times, boiled, and further transferred into 1× SDS loading buffer to run in 10% SDS-PAGE. After running, the gel was stained with Coomassie blue. Compared with the immunoprecipitated lane of the beads, the emerging bands in the HA immunoprecipitated lane were selected and cut into 1 mm by 1 mm pieces, followed by digestion and analysis by Thermo Fisher Scientific LTQ Obitrap ETD. The mass spectrometry data were analyzed using Proteome Discoverer 1.4 (Thermo Fisher Scientific), and the reliability of the identified proteins was mainly based on the value of peptide-spectral match.
Proximity ligation assay
PLA was performed with the Duolink (Sigma-Aldrich, USA) In Situ PLA technology–related kits, including In Situ Detection Reagents Orange (DUO92007), In Situ PLA Probe Anti-Rabbit MINUS (DUO92005), and In Situ PLA Probe Anti-Mouse PLUS (DUO92001). The exogenous interaction of cGAS and PRMT5 was detected in HEK293T cells cotransfected with HA-cGAS and Flag-PRMT5 plasmids, and the endogenous interaction of cGAS and PRMT5 was detected in macrophages derived from the freshly isolated mouse BMDMs. All fixation, incubation, ligation, and amplification procedures were done according to the manufacturer’s instructions.
GST pull-down assay
Glutathione S-transferase (GST)–cGAS, GST control, His-PRMT5, and His-MEP50 plasmids were transfected into BL-21 (DE3) bacteria strain (catalog no. CB106, TIANGEN, China) and incubated with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 16°C overnight. GST or His fusion proteins were purified with glutathione beads or Ni-column from BL-21 strain. After reciprocal pull-down, beads were centrifuged at 3000 rpm at 4°C for 3 min and further washed three times. After heating denaturation, protein samples were analyzed and detected according to the method in the IP and immunoblot analysis.
Lentiviral-mediated overexpression or knockdown
To establish stably overexpressing system, PRMT5 or its mutant complementary DNA (cDNA) was cloned into pCDH plasmid, followed by transfecting pCDH control, pCDH-PRMT5, or its mutant in combination with packaging plasmids (Δ8.91/VSV-G) into HEK293 cells. Lentivirus-containing supernatants were collected and added to the culture medium of MEF cells, and the successfully transfected cells were further selected by puromycin (1.5 μg/ml) for 5 days. The selected positive cells were collected and plated into six-well plates for further use. To establish lentivirus-mediated Prmt5 stable knockdown system, oligos of mouse Prmt5 (oligos were from the Sigma-Aldrich TRC library and sequences were Prmt5 shRNA: TRCN0000181891) were annealed and cloned into the pLKO.1 vector, followed by packaging lentivirus by transfection with pMD2G and pSPAX2. The virus-containing medium was collected to be put into the culture medium of the MEF cells, and the lentivirus-infected MEF cells were further selected by puromycin (1.5 μg/ml). The Prmt5 stable knockdown MEF cells and its mock control cells were cultured for further investigation.
In vitro methylation assay
cGAS, PRMT5, PRMT5(E444Q), and MEP50 proteins were expressed in vitro with the TNT Quick Coupled Transcription/Translation System (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Briefly, cGAS and histones were incubated with the PRMT5/MEP50 or PRMT5(E444Q)/MEP50 mixture in 50 μl of methylation reaction buffer (50 mM tris, 50 mM NaCl, 4 mM AdoMet, pH 8.8) at 30°C for 2 hours, and the methylation of cGAS protein was analyzed by immunoblotting with the specific antibodies.
Cell viability assay
BMDMs were plated into 96-well plates at a density of 2 × 104 cells per well, and MEFs and HEK293 cells were plated into 96-well plates at a density of 104 cells per well. The cells were treated with different doses of EPZ015666 (0.5, 1, 2.5, 5, and 10 μM) or transfected with siRNAs against Prmt5 before cell viabilities were detected at 0, 24, 48, and 72 hours with the CCK-8 Kit (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions.
Quantitative PCR and RNA sequencing analysis
Total RNA from BMDMs or MEFs was extracted with TRIzol Reagent (catalog no. 10296010, Invitrogen, USA), and the reverse transcription of 1 μg RNA samples was performed with the FastKing RT Kit (catalog no. KR116, TIANGEN, China). The quantitative PCR assay was performed with SYBR Green Master Mix (catalog no. 4367659, Applied Biosystems, USA). The primers were synthesized from Sango Biotech, and the sequence information was listed in table S2. The PCR was performed with the Bio-Rad CFX system according to the manufacturer’s instructions, and data were analyzed with Bio-Rad CFX software. Relative mRNA expression of the target genes was normalized to the internal control GAPDH, and the results of these experimental groups were further normalized to the levels of the control group (untreated cells or mock control). For RNA sequencing analysis, Cgas−/− MEFs were transfected with cGAS(WT) or cGAS(R124K) plasmid, followed by HSV-1 infection for 6 hours. The isolated RNA samples were subjected to RNA sequencing, and the data were analyzed by LC-BIO (LC-BIO, Hangzhou, China). Heatmap analysis was performed with the online OmicStudio tools (www.omicstudio.cn/tool/4).
Enzyme-linked immunosorbent assay
BMDMs or MEFs were seeded into 12-well plates and then treated with ISD, HT-DNA, cGAMP, or HSV-1 for 24 hours. BALB/c mice (8 weeks old) were treated by twice daily administration of EPZ015666 (60 mg/kg, orally) or vehicle (0.5% methylcellulose in water) followed by infection with HSV-1 [1 × 107 plaque-forming units (PFU) per mouse] for 48 hours. Supernatants from the cultured cells and sera from the HSV-1–infected mice were collected, and enzyme-linked immunosorbent assay (ELISA) was performed to detect the levels of IFN-β (catalog no. 439408, BioLegend, USA), IL-6 (catalog no. 431304, BioLegend, USA), and TNF-α (catalog no. 430907, BioLegend, USA) according to the manufacturer’s instruction. For the cGAMP assay, BMDMs were transfected with ISD or HT-DNA or infected with HSV-1 for 6 hours, and the concentration of cGAMP was measured using a cGAMP ELISA kit (catalog no. 501700, Cayman, USA) according to the manufacturer’s protocol.
Luciferase assay
For dual-luciferase reporter assay, HEK293T cells were transfected with pRL-IFN-β–Luc/pRL-TK or pRL-ISRE–Luc/pRL-TK construct together with ISD, cGAS/STING plasmids, and different doses of PRMT5 plasmids. The transfected cells were further cultured for 24 hours before luciferase detection with the Dual-Luciferase Reporter Assay System (catalog no. E1910, Promega, USA) according to the manufacturer’s instruction.
Mice
Cgas−/− mice (B6/J-Mb21d1em1Cd/Nju, T002719, Nanjing, China) and WT mice were purchased from Nanjing Biomedical Research Institute of Nanjing University. All mice were bred in specific pathogen–free conditions at the Laboratory Animal Center of Shandong University. All of the mice experiments were performed in accordance with the general guidelines of Institutional Animal Care and Use Committee.
Chromatin IP and DNA binding assay
BMDMs were transfected with ISD and further cross-linked with 1% formaldehyde at 37°C for 10 min before reactions were terminated with glycine at room temperature for 5 min. Cells were washed twice with phosphate-buffered saline (PBS), lysed, and incubated with anti-cGAS for at least 6 hours for the IP assay. The immunoprecipitated pellets were successively washed with low-salt washing buffer, high-salt washing buffer, LiCl washing buffer once, and Tris-EDTA buffer solution (TE) buffer twice, followed by elution with elution buffer (1% SDS and 0.1 M NaHCO3) for 15 min. The cGAS-ISD complex was de–cross-linked with 0.2 M NaCl at 4°C overnight. Nucleic acids were extracted with phenol-chloroform, precipitated with ethanol, and analyzed by quantitative PCR assay.
For DNA binding assay, EPZ015666-treated or Prmt5-siRNA–transfected BMDMs were further transfected with ISD for 6 hours followed by DNA binding analysis according to the reference (21). Briefly, cells were lysed and incubated with streptavidin agarose resin (catalog no. 2115, Thermo Fisher Scientific, USA) for at least 1 hour, followed by washing five times with lysis buffer, and supplemented with 1× SDS loading buffer for further immunoblotting analysis.
Virus infection and histological analysis
For virus amplification, a moderate amount of HSV-1 was incubated with Vero cells for 72 hours, all culture supernatants were aspirated, and adherent cells were scraped down as collection. The viral suspension was then obtained after repeated freezing and thawing three times, and the plaque assay was performed by the standard plaque assay to measure the titer of the virus. Lung tissues from the HSV-1–infected and uninfected mice were fixed with 10% phosphate-buffered formalin, embedded into paraffin, sectioned, stained with H&E solution, and examined by microscopy (OLYMPUS VS120) for histological analysis.
Immunofluorescence confocal microscopy
BMDMs were embedded with coverslips, fixed with 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100 for 5 min. After blocking with 1% bovine serum albumin, cells were incubated with specific primary antibodies for 2 hours, washed with PBS, and further incubated with Alexa Fluor 594–conjugated (1:200; A11012, Invitrogen, USA), Alexa Fluor 594–conjugated (1:200; A11005, Invitrogen, USA), or Alexa Fluor 488–conjugated (1:200; A11008, Invitrogen, USA) secondary antibodies for 1 hour. The nucleus was stained with DAPI (4′,6-diamidino-2-phenylindole) (catalog no. C0065, Solarbio, China) for 15 min. The stained cells were visualized and captured by confocal microscopy (Zeiss LSM 780). The colocalization proportion of cGAS and DNA or PRMT5 was further analyzed by ImageJ software and calculated by Manders’ coefficients from three visual fields with more than 150 cells.
Statistical analyses
Statistical analyses were performed with GraphPad Prism7.0 software. Student’s t test was used for statistical analysis between groups, and the log-rank (Mantel-Cox) test was used for statistical analysis of the survival curves. All tests were two-tailed, and P < 0.05 was considered statistically significant.
Acknowledgments
We thank C. Gao (Shandong University) for critically reading this manuscript. We thank D. Zhu and Y. Lv (Shandong University) for assisting us with biochemical techniques. We thank Q. Sun (Sun Yat-sen University) for PRMT5, MEP50 plasmid, and anti-sDMA; L. Zhao (Institute of Basic Medicine, Shandong Academy of Medical Sciences, China) for HSV-1; and X. Cao (Zhejiang University) for IFN-β and ISRE reporter. We thank the Translational Medicine Core Facility of Shandong University for consultation and instrument availability that supported this work. Funding: This study was supported by the National Natural Science Foundation of China (nos. 81972275 and 81672391) and the Major Innovation Project of Shandong Province (no. 2018CXGC1217). Author contributions: D.M. and L.H. designed this study, interpreted the data, and wrote the manuscript. D.M. performed most of the experiments. M.Y. and Q.W. assisted with construct mutation and animal experiments. C.S. and Y.B. assisted with confocal microscopy capture. H.S. assisted with PLA assay. Y.C. assisted with protein purification. W.J., X.S., X.M., Z.Q., Y.L., L.Z., and Y.Z. assisted with experiments and provided technical help. All authors discussed the results and commented on the manuscript. Competing interests: L.H., D.M., M.Y., Q.W., Y.L., Z.Q., and C.S. are inventors on a patent application filed by Shandong University (no. 2019109650370, filed on 11 October 2019). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabc1834/DC1
REFERENCES AND NOTES
- 1.Chen Q., Sun L., Chen Z. J., Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Dai J., Huang Y.-J., He X., Zhao M., Wang X., Liu Z.-S., Xue W., Cai H., Zhan X.-Y., Huang S.-Y., He K., Wang H., Wang N., Sang Z., Li T., Han Q.-Y., Mao J., Diao X., Song N., Chen Y., Li W.-H., Man J.-H., Li A.-L., Zhou T., Liu Z.-G., Zhang X.-M., Li T., Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 176, 1447–1460.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu M. M., Yang Q., Xie X. Q., Liao C. Y., Lin H., Liu T. T., Yin L., Shu H. B., Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45, 555–569 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Seo G. J., Kim C., Shin W. J., Sklan E. H., Eoh H., Jung J. U., TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 9, 613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seo G. J., Yang A., Tan B., Kim S., Liang Q., Choi Y., Yuan W., Feng P., Park H. S., Jung J. U., Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 13, 440–449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia P., Ye B., Wang S., Zhu X., Du Y., Xiong Z., Tian Y., Fan Z., Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 17, 369–378 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Wang Q., Huang L., Hong Z., Lv Z., Mao Z., Tang Y., Kong X., Li S., Cui Y., Liu H., Zhang L., Zhang X., Jiang L., Wang C., Zhou Q., The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLOS Pathog. 13, e1006264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bedford M. T., Clarke S. G., Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 33, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guccione E., Richard S., The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 20, 642–657 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Gkountela S., Li Z., Chin C. J., Lee S. A., Clark A. T., PRMT5 is required for human embryonic stem cell proliferation but not pluripotency. Stem Cell. Rev. Rep. 10, 230–239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tee W. W., Pardo M., Theunissen T. W., Yu L., Choudhary J. S., Hajkova P., Surani M. A., Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 24, 2772–2777 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan-Penebre E., Kuplast K. G., Majer C. R., Boriack-Sjodin P. A., Wigle T. J., Johnston L. D., Rioux N., Munchhof M. J., Jin L., Jacques S. L., West K. A., Lingaraj T., Stickland K., Ribich S. A., Raimondi A., Scott M. P., Waters N. J., Pollock R. M., Smith J. J., Barbash O., Pappalardi M., Ho T. F., Nurse K., Oza K. P., Gallagher K. T., Kruger R., Moyer M. P., Copeland R. A., Chesworth R., Duncan K. W., A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 11, 432–437 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Webb L. M., Sengupta S., Edell C., Piedra-Quintero Z. L., Amici S. A., Miranda J. N., Bevins M., Kennemer A., Laliotis G., Tsichlis P. N., Guerau-de-Arellano M., Protein arginine methyltransferase 5 promotes cholesterol biosynthesis-mediated Th17 responses and autoimmunity. J. Clin. Invest. 130, 1683–1698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Litzler L. C., Zahn A., Meli A. P., Hébert S., Patenaude A. M., Methot S. P., Sprumont A., Bois T., Kitamura D., Costantino S., King I. L., Kleinman C. L., Richard S., Di Noia J. M., PRMT5 is essential for B cell development and germinal center dynamics. Nat. Commun. 10, 22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu F., Cheng G., Hamard P. J., Greenblatt S., Wang L., Man N., Perna F., Xu H., Tadi M., Luciani L., Nimer S. D., Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Invest. 125, 3532–3544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scoumanne A., Zhang J., Chen X., PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 37, 4965–4976 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei H., Wang B., Miyagi M., She Y., Gopalan B., Huang D. B., Ghosh G., Stark G. R., Lu T., PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc. Natl. Acad. Sci. U.S.A. 110, 13516–13521 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung J., Karkhanis V., Baiocchi R. A., Sif S., Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/β-CATENIN and AKT/GSK3β proliferative signaling. J. Biol. Chem. 294, 7692–7710 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metz P. J., Ching K. A., Xie T., Delgado Cuenca P., Niessen S., Tatlock J. H., Jensen-Pergakes K., Murray B. W., Symmetric arginine dimethylation is selectively required for mRNA splicing and the initiation of type I and type III interferon signaling. Cell Rep. 30, 1935–1950.e8 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.-J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.-G., Landegren U., Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Gulla A., Hideshima T., Bianchi G., Fulciniti M., Kemal Samur M., Qi J., Tai Y. T., Harada T., Morelli E., Amodio N., Carrasco R., Tagliaferri P., Munshi N. C., Tassone P., Anderson K. C., Protein arginine methyltransferase 5 has prognostic relevance and is a druggable target in multiple myeloma. Leukemia 32, 996–1002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strobl C. D., Schaffer S., Haug T., Volkl S., Peter K., Singer K., Bottcher M., Mougiakakos D., Mackensen A., Aigner M., Selective PRMT5 inhibitors suppress human CD8+ T cells by upregulation of p53 and impairment of the AKT pathway similar to the tumor metabolite MTA. Mol. Cancer Ther. 19, 409–419 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Antonysamy S., The structure and function of the PRMT5:MEP50 complex. Subcell. Biochem. 83, 185–194 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Mersaoui S. Y., Yu Z., Coulombe Y., Karam M., Busatto F. F., Masson J.-Y., Richard S., Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J. 38, e100986 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thandapani P., O’Connor T. R., Bailey T. L., Richard S., Defining the RGG/RG motif. Mol. Cell 50, 613–623 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Wang Y., Ning X., Gao P., Wu S., Sha M., Lv M., Zhou X., Gao J., Fang R., Meng G., Su X., Jiang Z., Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46, 393–404 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Tao J., Zhang X. W., Jin J., Du X. X., Lian T., Yang J., Zhou X., Jiang Z., Su X. D., Nonspecific DNA binding of cGAS N terminus promotes cGAS activation. J. Immunol. 198, 3627–3636 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Michalak E. M., Visvader J. E., Dysregulation of histone methyltransferases in breast cancer—Opportunities for new targeted therapies? Mol. Oncol. 10, 1497–1515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castillo-Aguilera O., Depreux P., Halby L., Arimondo P. B., Goossens L., DNA methylation targeting: The DNMT/HMT crosstalk challenge. Biomolecules 7, 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerhart S. V., Kellner W. A., Thompson C., Pappalardi M. B., Zhang X. P., Montes de Oca R., Penebre E., Duncan K., Boriack-Sjodin A., Le B., Majer C., McCabe M. T., Carpenter C., Johnson N., Kruger R. G., Barbash O., Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 8, 9711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim H., Kim H., Feng Y., Li Y., Tamiya H., Tocci S., Ronai Z. A., PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 12, eaaz5683 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ablasser A., Chen Z. J., cGAS in action: Expanding roles in immunity and inflammation. Science 363, eaat8657 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Li Y., Chitnis N., Nakagawa H., Kita Y., Natsugoe S., Yang Y., Li Z., Wasik M., Klein-Szanto A. J., Rustgi A. K., Diehl J. A., PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 5, 288–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu Z., Gao S., Zhang F., Wang Z., Ma W., Davis R. E., Wang Z., Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem. J. 446, 235–241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y., Wang Z., Zhang J., Ling R., Elevated expression of protein arginine methyltransferase 5 predicts the poor prognosis of breast cancer. Tumour Biol. 39, 1010428317695917 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Hu P., Zhao H., Zhu P., Xiao Y., Miao W., Wang Y., Jin H., Dual regulation of Arabidopsis AGO2 by arginine methylation. Nat. Commun. 10, 844 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snyder K., Zitzer N. C., Gao Y., Choe H. K., Sell N. E., Neidemire-Colley L., Ignaci A., Kale C., Devine R. D., Abad M. G., Pietrzak M., Wang M., Lin H., Zhang Y. W., Behbehani G. K., Jackman J. E., Garzon R., Vaddi K., Baiocchi R. A., Ranganathan P., PRMT5 regulates T cell interferon response and is a target for acute graft-versus-host disease. JCI Insight 5, e131099 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kota S. K., Roening C., Patel N., Kota S. B., Baron R., PRMT5 inhibition promotes osteogenic differentiation of mesenchymal stromal cells and represses basal interferon stimulated gene expression. Bone 117, 37–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen K., Liu J., Liu S., Xia M., Zhang X., Han D., Jiang Y., Wang C., Cao X., Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 170, 492–506.e14 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Mowen K. A., David M., Unconventional post-translational modifications in immunological signaling. Nat. Immunol. 15, 512–520 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Komyod W., Bauer U. M., Heinrich P. C., Haan S., Behrmann I., Are STATS arginine-methylated? J. Biol. Chem. 280, 21700–21705 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Mowen K. A., Tang J., Zhu W., Schurter B. T., Shuai K., Herschman H. R., David M., Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell 104, 731–741 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Zhu J., Liu X., Cai X., Ouyang G., Zha H., Zhou Z., Liao Q., Wang J., Xiao W., Zebrafish prmt3 negatively regulates antiviral responses. FASEB J. 34, 10212–10227 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Zhu J., Liu X., Cai X., Ouyang G., Fan S., Wang J., Xiao W., Zebrafish prmt7 negatively regulates antiviral responses by suppressing the retinoic acid-inducible gene-I-like receptor signaling. FASEB J. 34, 988–1000 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Zhang H., Han C., Li T., Li N., Cao X., The methyltransferase PRMT6 attenuates antiviral innate immunity by blocking TBK1-IRF3 signaling. Cell. Mol. Immunol. 16, 800–809 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Y., Liu L., Yang S., Cao Y., Song X., Xiao J., Feng H., Black carp PRMT6 inhibits TBK1-IRF3/7 signaling during the antiviral innate immune activation. Fish Shellfish Immunol. 93, 108–115 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Musiani D., Bok J., Massignani E., Wu L., Tabaglio T., Ippolito M. R., Cuomo A., Ozbek U., Zorgati H., Ghoshdastider U., Robinson R. C., Guccione E., Bonaldi T., Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal. 12, eaat8388 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Geoghegan V., Guo A., Trudgian D., Thomas B., Acuto O., Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 6, 6758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Motwani M., Pesiridis S., Fitzgerald K. A., DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 20, 657–674 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Gao D., Li T., Li X. D., Chen X., Li Q. Z., Wight-Carter M., Chen Z. J., Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. U.S.A. 112, E5699–E5705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas C. A., Tejwani L., Trujillo C. A., Negraes P. D., Herai R. H., Mesci P., Macia A., Crow Y. J., Muotri A. R., Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell 21, 319–331.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/13/eabc1834/DC1