

Abstract
Current treatments targeting amyloid beta in Alzheimer’s disease (AD) have minimal efficacy, which results in a huge unmet medical need worldwide. Accumulating data suggest that brain mitochondrial dysfunction play a critical role in AD pathogenesis. Targeting cellular mechanisms associated with mitochondrial dysfunction in AD create a novel approach for drug development. This study investigated the effects of nilotinib, as a selective tyrosine kinase inhibitor, in astroglia derived from 3xTg-AD mice versus their C57BL/6-controls. Parameters included oxygen consumption rates (OCR), ATP, cytochrome c oxidase (COX), citrate synthase (CS) activity, alterations in oxidative phosphorylation (OXPHOS), nuclear factor kappa B (NF-κB), key regulators of mitochondrial dynamics (mitofusin (Mfn1), dynamin-related protein 1 (Drp1)), and mitochondrial biogenesis (peroxisome proliferator-activated receptor gamma coactivator1-alpha (PGC-1α), calcium/calmodulin-dependent protein kinase II (CaMKII), and nuclear factor (erythroid-derived 2)-like 2 (Nrf2)). Nilotinib increased OCR, ATP, COX, Mfn1, and OXPHOS levels in 3xTg astroglia. No significant differences were detected in levels of Drp1 protein and CS activity. Nilotinib enhanced mitochondrial numbers, potentially through a CaMKII-PGC1α-Nrf2 pathway in 3xTg astroglia. Additionally, nilotinib-induced OCR increases were reduced in the presence of the NF-κB inhibitor, Bay11-7082. The data suggest that NF-κB signaling is intimately involved in nilotinib-induced changes in bioenergetics in 3xTg brain astroglia. Nilotinib increased translocation of the NF-κB p50 subunit into the nucleus of 3xTg astroglia that correlates with an increased expression and activation of NF-κB. The current findings support a role for nilotinib in improving mitochondrial function and suggest that astroglia may be a key therapeutic target in treating AD.
Keywords: mitochondrial function, bioenergetics, biogenesis, Alzheimer’s disease, astroglia, nuclear factor kappa B (NF-κB), cytochrome c oxidase, citrate synthase, oxidative phosphorylation
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder, characterized by progressive cognitive impairments [1]. Approaches focusing on therapeutics that interfere with amyloid β plaques (Aβ), a classic hallmark of AD [2, 3], have failed for the most part to show efficacy for stopping or slowing cognitive decline. However, accumulating evidence demonstrates mitochondrial dysfunction early in the pathology of human diseases, cardiovascular, neurodegenerative, and cancers, which may serve as an important and primary therapeutic target [4-9]. Low doses of the anti-cancer drug nilotinib suggest cognitive benefits for patients with Parkinson's disease (PD) and related dementias [10, 11]. Nilotinib (Tasigna®, AMN107) is a c-Abl tyrosine kinase inhibitor approved by the Food and Drug Administration (FDA) for patients with chronic myeloid leukemia (CML) [12, 13]. Compared to the other c-Abl inhibitors, low dose nilotinib is more advantageous for penetrating the blood brain barrier (BBB) and degrades α-synuclein through a process of autophagy in PD models [14-17]. Safety and efficacy of nilotinib in AD patients has been tested. It has recently been reported that biomarkers of AD were changed as a function of nilotinib treatment [18]. Moreover, nilotinib enhances dopaminergic neurons and improves motor function in PD and Lewy body dementia (LBD) [10, 14]. The level of c-Abl is elevated in post-mortem brains of patients with AD and PD [19-21]. Proteomic analyses show that c-Abl phosphorylates tau tangles in AD brains [19]. In PD models, c-Abl increased oxidative stress, presumably due to the accumulation of dysfunctional mitochondria that generate and release reactive oxygen species (ROS) [21, 22]. Overexpression of glial c-Abl leads to inflammation and neurodegeneration [23, 24]. Under oxidative stress conditions, c-Abl is activated in astroglia and blocking c-Abl prevented the oxidative death of astroglia [25]. More research identified that overexpression of glial c-Abl contributes to both oxidative stress as well as inflammation in a mouse pain model [24]. Moreover, c-Abl inhibitors reduced both inflammatory activation of glia and chronic pain [26].
Astroglia, as important central nervous system (CNS) cells, play key roles in the pathogenesis of neurodegenerative disorders such as AD; excessive oxidative stress, inflammation as well as mitochondrial impairment is observed in AD. Accumulating evidence highlights the critical role of astroglia in inflammatory, oxidative stress, modulation of synaptic activity, and bioenergetic changes present in AD [27-31]. According to all of these functional properties, astroglia have become exciting targets for our study. The effect of nilotinib on metabolic activity and inflammation in astroglia, however, has not been examined.
The notion of testing nilotinib for AD may seem counterintuitive since AD involves cell loss and cancer involves unregulated cell proliferation, but the earliest deficits in the pathological progression of AD and cancer are associated with mitochondrial dysfunction; that is, before the robust appearance of neurotoxic proteins in AD and oncogene expression in cancer [4-8, 32-39].
Therefore, targeting mitochondrial bioenergetics may be a promising new strategy for treating AD [5, 40-44]. Interestingly, mitochondrial function is considered as a target for cancer therapy, but has not been exploited for AD. Reprograming metabolic activity of cancer cells led to non-apoptotic function [45]. This finding indicates that apoptotic pathway is linked to mitochondria and metabolic pathways. Therefore, the linkage between cancer and AD provides a rational for repurposing existing anticancer drugs for AD.
Previously, our laboratory documented reductions in brain metabolic activity and in the expression of mitochondrial protein subunits (Complexes I-V) in 3xTg brain samples estimated to compromise the site of oxidative phosphorylation (OXPHOS) and mitochondrial efficiency through the action of ATP synthesis [8]. Beside down-regulation of OXPHOS capacity in AD, reduced activity of other mitochondrial metabolic enzymes such as citrate synthase (CS) is associated with AD [46]. Both mitochondrial dynamics (fission and fusion) and number are significantly altered in AD models that ultimately results in mitochondrial dysfunction [47-50]. The effects of nilotinib on mitochondrial function, however, have not been examined. The regulation of mitochondrial function is also linked to nuclear factor kappa B (NF-κB) signaling, a highly-conserved pathway for immune function, mitochondrial function and previously studied in AD and cancer [43, 51-53]. Some studies reported that NF-κB signaling regulates Aβ-induced mitochondrial function and cytochrome c oxidase (COX) activity [54, 55]. Other studies confirm that the enzymatic activity of COX may be modulated by the NF-κB signaling pathway [54, 56]. NF-κB plays a crucial role in the metabolic activity of AD and cancer [53,57]. Several studies have also shown that NF-κB activity restricts activation of inflammasomes through the removal of damaged mitochondria [53, 58-60]. Whether the mitochondrial-related NF-κB signaling pathway is affected by nilotinib is not known.
Mitochondrial function is partially regulated by calcium/calmodulin-dependent protein kinase (CaMK), a downstream target of the NF-κB, signaling pathway [61]. CaMK has been shown to be a key protein in neurodegeneration, metabolic regulations, and biogenesis [62]. Peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1α), a downstream target of CaMK, has been implicated in mitochondrial biogenesis [63]. Further, PGC-1α activates transcription factor nuclear factors (Nrf) and mitochondrial transcription factor A (TFAM), known to regulate mitochondrial biogenesis [64, 65]. The effect of nilotinib on mitochondrial biogenesis in astroglia, however, has not been investigated.
The aim of this study was to test the hypothesis that nilotinib improves astroglia mitochondrial function. We performed bioenergetic profiling in AD vs. wild type (WT)/control astroglia while in the presence and absence of nilotinib. Specifically, we measured 1) oxygen consumption rates (OCR) and ATP levels; 2) enzymatic activity of COX and CS; 3) levels of OXPHOS; 4) numbers of mitochondria; 5) levels of proteins involved in mitochondrial dynamics (e.g., Mfn1, Drp1); 6) levels of proteins involved in mitochondrial biogenesis (e.g., CaMKII, PGC-1α, Nrf2, and TFAM); 7) translocation, activation, and expression levels of NF-κB subunit proteins (NF-κB p50, p105, p65, and p75) and IκB-α; and 8) effects of NF-κB inhibition on cellular respiration and ATP levels.
MATERIALS AND METHODS
Animals
We utilized both female and male pups (5-7 days) from 3xTg mice. This transgenic model of AD expresses hAPPSwe, hPS1M146V, and hTauP301L mutations, and exhibit synaptic dysfunction. As a control, female and male pups (5-7 days) derived from wild-type C57BL/6 mice were used. Mice were given ad libitum access to food and water and housed under the standard light/dark cycle (12 h light, 12 h dark at room temperature (22°C)). All animal procedures followed guidelines of the University of Manitoba Animal Care Committee, the Canadian Council of Animal Care rules, and the Institutional Animal Care and Use Committee (IACUC) standards.
Primary cultures of astroglia
Primary cortical astroglia derived from both AD (3xTg) and control (C57BL/6) pups (5-7 days) were prepared as previously described [66]. Briefly, cortical tissue was dissected and placed into a chilled dissection medium [67] composed of Ca2+ and Mg2+- free Hanks’ balanced salt solution (HBSS), pH 7.4, containing the following: 137 mM NaCl, 5.36 mM KCl, 0.27 mM Na2HPO4, 1.1 mM KH2PO4, and 6.1 mM glucose. Isolated cortical tissue was sliced using a razor. Following slicing, tissues were gently separated from each other in fresh cold dissecting media and mechanically dissociated by sequential passage through a Pasteur pipette. After dissociation, we centrifuged the cells at 1,000 rpm for 5 min, and the pellet was re-suspended in Dulbecco’s modified Eagle’s medium (DMEM)/F12 culture medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS), 0.5 mg mL-1 streptomycin, 0.5 U mL-1 penicillin (Invitrogen, Carlsbad, CA, USA), 8.39 mM HEPES, and 23.8 mM NaHCO3. Cells derived from 3xTg-AD and C57Bl/6 pups were plated on 25-cm2 flasks at a density of 6×105 cells cm-2 and grown at 37 °C using an incubator with humidified 5% CO2 and 95% air. After 12 days, the cell culture reached confluence. Cultures were then shaken gently, and any microglia/floating cells were removed, [68, 69] resulting in more than 90% pure culture of astroglial, as described previously (Supplementary Fig. 1).
Measurement of mitochondrial respiration rates
Astroglia derived from 3xTg (AD) and C57BL/6 (control) mice were cultured separately on Seahorse XF24 plates (Seahorse Biosciences, Billerica, MA). To investigate whether nilotinib increased mitochondrial respiration in 3xTg astroglia through the NF-κB signaling pathway, astroglia were treated with either Bay11-7082, a well-known NF-κB inhibitor that has pharmacological activities including neuroprotective, anticancer, and anti-inflammatory roles [70], or co-treated with nilotinib and Bay11-7082 for 24 hours. Following treatment, cells were subjected to un-buffered DMEM supplemented with 1 mM sodium pyruvate, 1 mM glucose, at pH 7.4 and incubated in a non- CO2 incubator for one hour at 37 °C. First, the basal oxygen consumption rate was measured before injection of selected mitochondrial inhibitors. Second, oligomycin (1 µM), an irreversible ATP synthase inhibitor, was added to drive a proton gradient. Third, carbonyl cyanide-p-trifluoromethoxyphenyl-hydrazone, a.k.a FCCP (1 µM), an electron transport chain (ETC) uncoupler, was added to lower the proton gradient and allow for maximum respiration of cells. Fourth, rotenone (1 µM), a Complex I inhibitor, and antimycin A (1 µM), a Complex III inhibitor, were added to cease respiration, thus blocking mitochondrial electron transfer at cytochrome c oxidase. Following the addition of rotenone and antimycin, basal respiratory data were calculated and corrected with subtraction of non-mitochondrial respiration rates. Maximal respiration rate was calculated by dividing FCCP-stimulated OCR by OCR oligomycin-stimulated OCR. Spare respiratory capacity (ability of the cell to respond to improved energy demand) was approximated by measuring the differences between basal and maximal respiration rates. Coupling efficiency is the proportion of OCR that is utilized to make ATP and was estimated as the fraction of basal mitochondrial OCR used for ATP synthesis (ATP-linked OCR/basal OCR).
For analysis, OCR readings were measured and recorded by Seahorse XF-24 software (Seahorse Biosciences, Billerica, MA). Finally, the OCR level was normalized based on protein concentrations colorometric detergent compatible (DC) protein assay kit (BioRad, Hercules, California, USA)) for each well. The OCR measures from each well were normalized to total protein levels.
Measurement of cellular ATP
ATP production was determined with a luminescent ATP detection assay kit (ab113849: Abcam, Cambridge, UK) as per manufacturer's instructions. Briefly, cells were washed with phosphate buffered saline (PBS). Then, ATP standard dilutions were made. Further, cells and ATP standard dilutions were incubated with detergent at room temperature (RT) for five minutes on an orbital shaker. After 10 minutes incubation in the dark, ATP levels were measured on a Micoplate Reader (Dynex Technologies, Denkendorf, Germany) by tracking the change of absorbance at 535/587nm.
Measurement of cytochrome c oxidase and citrate synthase enzymatic activity
The activity of cytochrome c oxidase (COX) and citrate synthase (CS) was assessed spectrophotometrically (Ultrospec 2100 pro; GE Healthcare) using ScienCell assay kits (8278 and 8318, ScienCell Research Laboratory, California, USA) on isolated mitochondria derived from control and 3xTg astroglia. The activity of COX and CS was determined spectrophotometrically by measuring absorbance at 550 nm, 412 nm, respectively.
All relative quantification for enzyme assays were analyzed based on densitometry values normalized to total protein level. Protein concentrations were measured utilizing a colorometric DC protein assay kit (BioRad, Hercules, California, USA).
Nuclear protein extraction and electrophoretic mobility shift assay (EMSA)
Nuclear protein was isolated from astroglia cultures using the NE-PER™ nuclear and cytoplasmic extraction kit (ThermoFisher Scientific, San Jose, CA, USA) as per the manufacturer’s instructions. EMSA, used to study protein: DNA interactions, was performed using the LightShift™ Chemiluminescent EMSA Kit (ThermoFisher Scientific, San Jose, CA, USA). For the EMSA, 25 μL reaction mixtures containing binding buffer (10x binding buffer, 0.05μg/μL Poly (dI•dC), 2.5% glycerol, 0.05% NP40, 5mM MgCl2, 50mM KCl, 1mM EDTA, 0.5mg/mL BSA), 15 μg nuclear protein (astroglia) and 40 fM biotinylated probe and 20 fM biotinylated probe, respectively, corresponding to a NF-κB-binding site (5′-AGTTGAGGGGACTTTCCCAGGC-3′) were incubated at room temperature for 30 minutes. For the competition reaction, 100 pM unbiotinylated probe was added to the nuclear protein/binding buffer reaction for 20 minutes at room temperature after which a 40 fM biotinylated probe was added for 30 minutes at room temperature. A total of 20 μL of reaction mixture was loaded on native 6% polyacrylamide gels prepared in 0.5× TBE and electrophoresed at 100V for 90 minutes at room temperature. Gels were then transferred onto a nylon membrane for 90 minutes at 4°C at 380mA then UV cross-linked for one minute at 120 mJ/cm2. The processing of the membrane was performed using the Chemiluminescent Nucleic Acid Detection Module Kit (ThermoFisher Scientific, San Jose, CA, USA) as per the manufacturer’s instructions. The nylon membrane was imaged with the Chemidoc™ MP (Bio-Rad, Hercules, CA, USA).
Protein extraction and Western blot analysis
Proteins were stored at -80 °C prior to Western blotting as previously described [8]. Briefly, astroglia were harvested from the cortex of C57BL/6-control and 3xTg-AD mice and then homogenized in ice-cold radioimmuno-precipitation (RIPA) buffer containing: 150 mM sodium chloride, 50 mM Tris, pH 8.0, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA), and 1% protease inhibitor cocktail (Amresco, Solon, OH, USA). Further, protein concentrations were quantified utilizing a colorometric DC protein assay kit (BioRad, Hercules, California, USA). Further, a 4X Laemmli buffer (20% β-mercaptoethanol, 16% SDS, 40% glycerol, 0.01% bromophenol blue, and 0.25M Tris, pH 6.8) was added to the samples, followed by denaturing at 50˚C for 8 minutes. Equal amounts of proteins (15 ug per well) were loaded in each well of a 10% polyacrylamide SDS-PAGE gels (Bio-Rad, Hercules, CA, USA) and electrophoresed with a Tris-glycine running buffer at 200 volts for 45 minutes. Gels were then activated using a ChemiDoc™ MP imager (Bio-Rad, Hercules, CA, USA) and transferred to a 0.2 µm nitrocellulose membranes (Bio-Rad Hercules, CA, USA) with the Trans-Blot® Turbo™ Transfer System (Bio-Rad, Hercules, CA, USA) and total protein on the membranes were measured utilizing the ChemiDoc™ MP imager. Following imaging, all membranes were blocked in Tris-buffered saline with 0.1% Tween-20 (TBS-T) with 5% milk, with the exception of membranes detecting phosphorylated proteins, which were blocked in TBS-T with 5% bovine serum albumin (BSA) instead of 5% milk, for one hour on a tilting platform at room temperature. Following blocking, membranes were then incubated at 4°C overnight with the following primary antibodies: Total OXPHOS Rodent WB Antibody Cocktail (ab110413, Abcam, Cambridge, UK; 1:1000), NF-κB p105/p50 (ab32360, Abcam, Cambridge, UK; 1:1000), NF-κB p65 (ab16502, Abcam Cambridge, UK; 1:1000), NF-κB p75 (108299, Abcam, Cambridge, UK; 1:1000), IκB-α (ab32518, Abcam, Cambridge, UK; 1:500), Drp1 (ab156951, Abcam, Cambridge, UK; 1:200), Mfn1 (ab56889, Abcam, Cambridge, UK; 1:500), c-Abl (2862, Cell signaling technology, Danvers, USA; 1:100), phospho-c-Abl (Tyr245) (2868, Cell signaling technology, Danvers, USA; 1:100), PGC-1α (PA5-38022, Thermo Fisher Scientific, Massachusetts, USA; 1:500), CaMKII (M-176, Santa Cruz, Texas, USA; 1:500), Nrf2 (ab62352, Abcam, Cambridge, UK; 1:500), and TFAM (ab131607, Abcam, Cambridge, UK; 1:500). Following primary antibody incubation, all membranes were washed with TBS-T buffer (three times; fifteen minutes each) and then incubated with either goat anti-mouse, or anti-rabbit IgG (H+L) antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA, 1:2000 dilution) prepared in TBS-T buffer with 5% milk or BSA for one hour at 4 °C. Later, membranes were washed with 1X TBS-T buffer (three times; fifteen minutes each) and subjected to enhanced chemiluminescence ECL, utilizing a clarity ECL kit (Bio-Rad, Hercules, CA, USA) for five minutes and then visualized by the ChemiDoc™ MP imager using Bio-Rad Image Lab software.
Immunohistochemistry
Immunohistochemistry was utilized to detect astroglia as described previously [8]. Briefly, cells were rehydrated using 1X TBS with 0.1% Tween 20 (TBS-T), for five minutes. Following rehydration, astroglia were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 10 min at room temperature. Further, antigen retrieval was performed for 20-30 minutes in preheated (80 °C) citrate buffer (PH 8). All cells were then washed tree times with TBS-T (each time: 5 minutes each). Further, cells were then treated with anti-glial fibrillary acidic protein (GFAP) antibody (ab16997, abcam Cambridge, Massachusetts, USA, 1:100 dilution) to detect astrocytes combined with the EXPOSE mouse specific horseradish peroxidase/3-3-diamino-benzidine chromogen solution (HRP/DAB) detection immunohistochemistry kit (ab80436, Abcam, Cambridge, Massachusetts, USA). Fixed cells were observed with an inverted microscope (Nikon, Eclipse, TE200-U). Images captured with an infinity 2-1R CCD camera (Lumenera Corp., Ottawa, Ontario, Canada).
Immunofluorescence analysis
Immunofluorescence was utilized to detect translocation of NF-κB subunits (p50, p65, p75, also called RelA, RelB, and c-Rel, respectively) in 3xTg astroglia. Briefly, astroglia were rehydrated with TBS-T, for five minutes. Following rehydration, cells were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 10 min at RT. Following the fixation procedure, antigen retrieval was performed for 30 minutes in preheated (80 °C) citrate buffer. All cells were then washed with TBS-T (three times; 5 minutes each). Cells were incubated with blocking buffer (1% bovine serum albumin (BSA) in T-BST) for 1 hour at RT. Cells were then incubated with primary antibodies: NF-κB p50 ab32360, Abcam, Cambridge, UK; 1:100; NF-κB p65: ab16502, Abcam Cambridge, UK; 1:100; and NF-κB p75: 108299, Abcam, Cambridge, UK; 1:100; anti-GFAP antibody (ab16997, abcam Cambridge, Massachusetts, USA; 1:100 dilution) to detect astroglia or Ionized calcium binding adaptor molecule 1 (Iba-1) (019-19741, wako, Saitama, Japan, 1:100 dilution) to detect microglia overnight at 4 °C. Further, cells were washed three times with TBS-T and incubated for 2 h at RT with an Alexa fluorTM 488 secondary antibody (1:500; Invitrogen, Carlsbad, CA). Further, cells were washed three times with TBS-T and were mounted using mounting medium with 4′,6-diamidino-2-phenylindole (DAPI). Positively stained cells were captured on a Carl Zeiss Axioscope-2 fluorescence microscope equipped with AxioVision3 software using 20X objective. The volume density of NF-κB subunits was quantified using ImageJ software (http://rsbweb.nih.gov/ij/).
Transmission Electron Microscopy (TEM)
Transmission electron microscopy (TEM) was used to study the fine structure of the mitochondria and to count the number of mitochondria, as described previously [60]. Briefly, astroglia derived from 3xTg mice were treated with/without nilotinib (100 nM) for 24 hours. After incubation, cells were washed with PBS and fixed 3 hours in 3% v/v glutaraldehyde in 0.1 M Sorensen’s buffer. Cells were again fixed in 1% osmium tetroxide in 0.1 M Sorensen’s phosphate buffer for two hours, and dehydrated in a 30% to 100% ethanol series and embedded with Epon resin and infiltrated for 4 days. Later, sections were polymerized at 60 °C for 30 hours; Images were collected with Morgagni 268D electron microscope (Philips, Netherlands).
Statistical analysis
Data was analyzed using two-tailed Student's t-tests (GraphPad Prism 6, GraphPad Software). A difference between or among groups was determined as statistically significant, if *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
RESULTS
Comparing the cellular bioenergetics profile between C57BL/6 and 3xTg astroglia
The mitochondrial oxygen consumption rate (OCR) was measured in astroglia derived from cortical brain regions from control (C57BL/6) and AD (3xTg) mice (Fig. 1A). At baseline levels, OCR in 3xTg astroglia was significantly decreased compared to control groups (Fig. 1B). Similarly, OCR associated with maximal respiration capacity was significantly decreased in 3xTg astroglia vs. controls (Fig. 1C). Consistent with changes in parameters associated with basal and maximal respiration, spare respiratory capacity was significantly lower in 3xTg astroglia vs. controls (Fig. 1D). However, coupling efficiency was not significantly different in wells containing 3xTg astroglia and control cells (Fig. 1E).
Figure 1.
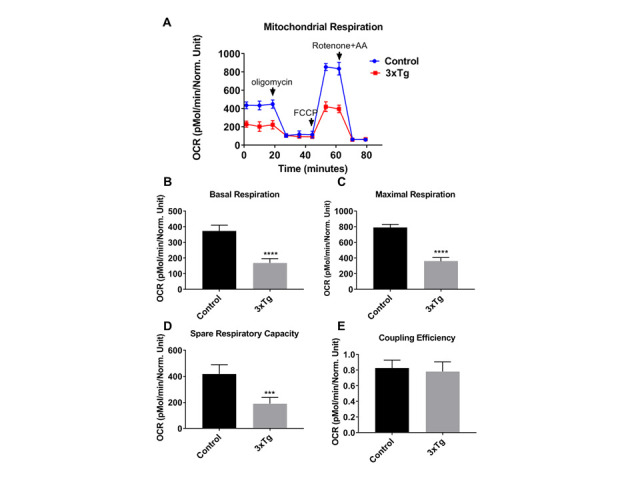
Mitochondrial respiration rates are reduced in 3xTg-AD astroglia. (A) Kinetics graph indicating real-time OCR at baseline and after addition of oligomycin, FCCP, and rotenone/antimycin. The OCR was measured in 3xTg and C57BL/6-WT astroglia utilizing the XF24 analyzer. (B) Basal respiration, (C) maximal respiration, (D) spare respiratory capacity, and (E) Coupling efficiency were calculated compared between 3xTg and control cells. Data are mean ± SD of n = 6 per group (*** = p < 0.001 or **** = p < 0.0001) analyzed by unpaired Student's t-test.
Effects of nilotinib in mitochondrial bioenergetics in C57BL/6 and 3xTg astroglia
To assess the effects of nilotinib on mitochondrial respiration, astroglia derived from C57BL/6 mice were cultured and treated with increasing concentrations of nilotinib (0, 1, 10, 100, and 1000 nM) for 24 hours. Bioenergetic parameters of basal, maximal respiration, spare respiratory capacity, and coupling efficiency were not altered in C57BL/6 astroglia as a result of nilotinib treatment (Fig. 2A-E). To further determine if the OCR rates were reflected by similar alterations in cellular energy production, total ATP production was measured. ATP levels were not altered in C57BL/6 astroglia that were treated with or without nilotinib (Fig. 2F).
Figure 2.

Nilotinib did not alter mitochondrial respiration rate or expression of total or phospho c-Abl in C57BL/6-WT astroglia. The OCR was measured in C57BL/6 astroglia utilizing the XF24 analyzer after 24 hours treatment with nilotinib (1, 10, 100, and 1000 nM). (A) Basal respiration, (B) maximal respiration, (C) spare respiratory capacity, (D) Coupling efficiency, and (E) total ATP level were calculated and compared between control and treated cells. Western blot experiments demonstrating relative levels of c-Abl and p-c-Abl in C57BL/6-WT astroglia (F) in the presence and absence of 24 hrs. 100 nM nilotinib treatment. Relative quantification for protein levels of c-Abl and p-c-Abl normalized to total protein and total c-Abl, respectively (G, H). Data are mean ± SD of n = 6 per group (* = p < 0.05) analyzed by unpaired Student's t-test.
To assess the effect of nilotinib in the cellular bioenergetics profile of AD, astroglia derived from 3xTg mice were cultured and treated with increasing concentrations of nilotinib (0, 1, 10, 100, and 1000 nM) for 24 hours. Unlike C57BL/6 astroglia, mitochondrial OCR in 3xTg astroglia was enhanced significantly at 100 nM of nilotinib after 24 hours (Fig. 3A). At baseline, OCR in 3xTg astroglia in the presence of 100 nM nilotinib was significantly increased as compared to non-treated cells (Fig. 3B). Additionally, bioenergetics parameters of maximal respiration rates as well as spare respiratory capacity were significantly increased 24 hours after nilotinib treatment (Fig. 3C-D). However, nilotinib did not affect the coupling efficiency (coupled respiration relative to basal levels) (Fig. 3E). To confirm OCR data, total ATP production was measured in 3xTg astroglia. ATP levels were significantly increased in nilotinib-treated 3xTg astroglia (Fig. 3F).
Figure 3.

Nilotinib improved mitochondrial respiration rates in 3xTg-AD astroglia. Nilotinib inhibited the expression level of phospho c-Abl in 3xTg astroglia. The OCR was measured in 3xTg-AD astroglia utilizing the XF24 analyzer after 24 hours treatment with nilotinib (1, 10, 100, and 1000 nM). (A) Basal respiration, (B) maximal respiration, (C) spare respiratory capacity, (D) coupling efficiency, and (E) total ATP level were calculated and compared between control and treated cells. Western blot experiments demonstrating relative levels of c-Abl and p-c-Abl in 3xTg-AD astroglia (F) in the presence and absence of 24 hrs. 100 nM nilotinib treatment. Relative quantification for protein levels of c-Abl and p-c-Abl normalized to total protein and total c-Abl, respectively (G, H). Data are mean ± SD of n = 6 per group (* = p < 0.05 or ** = p < 0.01 or *** = p < 0.001 or **** = p < 0.0001) analyzed by unpaired Student's t-test.
To further determine whether c-Abl levels are altered in response to the c-Abl inhibitor, nilotinib, 3xTg and control astroglia were treated with 100 nM nilotinib for 24 hours (Fig. 2G and 3G). Nilotinib did not lower total c-Abl levels in control or 3xTg astroglia (Fig. 2H and 3H). However, nilotinib (100 nM) significantly decreased the levels of phosphorylated c-Abl in 3xTg astroglia, but not control astroglia (Fig. 2I and 3I).
Effects of nilotinib in mitochondrial-associated proteins in C57BL/6 and 3xTg astroglia
Similar to the OCR findings in C57BL/6 astroglia, Western blot data revealed that the expression levels of key mitochondrial subunit proteins (Complexes I-V) were not altered with nilotinib treatment (Fig. 4A-B). Additionally, activity of COX and CS in the nilotinib-treated group was not altered compared to that of controls (Fig. 4C).
Figure 4.

Nilotinib did not alter the expression of mitochondrial Complex (I-V) protein subunits in C57BL/6-WT astroglia. Western blot experiments demonstrating relative levels of mitochondrial protein subunits in C57BL/6-WT astroglia in the presence and absence of 24 hrs. 100 nM nilotinib treatment. (A) Representative Western blot for NADH dehydrogenase beta sub complex subunit 8 of Complex I (NDUFB8), succinate dehydrogenase subunit B of Complex II (SDHB), cytochrome b-c1 complex subunit 2 of Complex III (UQCRC2), Cytochrome c oxidase subunit 1 of Complex IV (MTCO1), and ATP synthase subunit alpha of Complex V (ATP5A). (B-F) Relative quantification for protein levels of Complex I-V normalized to total protein. (G) COX activity was measured in C57BL/6-WT astroglia in presence and absence of 100 nM nilotinib treatment. Results are expressed as mean ± SD of n = 6 per group (*P ≤ 0.05) analyzed by unpaired Student's t-test.
Consistent with the mitochondrial OCR findings in 3xTg astroglia, Western blot results revealed a significant increase in the protein levels of NADH dehydrogenase beta sub-complex subunit 8 of Complex I, cytochrome b-c1 subunit 2 of Complex III, cytochrome c oxidase subunit 1 of Complex IV, as well as ATP synthase subunit alpha of Complex V in nilotinib-treated 3xTg astroglia vs. non-treated astroglia (Fig. 5A-B). The activity of CS was not altered as a function of nilotinib (Suppmentary Fig. 2). The activity of COX in the nilotinib-treated group was significantly increased compared to that of controls (Fig. 5C). However, the protein levels of Succinate dehydrogenase subunit B of Complex II were not found to be altered in nilotinib-treated 3xTg astroglia compared with non-treated ones (Fig. 5A-B).
Figure 5.

Nilotinib significantly increased the expression of mitochondrial Complex (I and III-V) protein subunits and cytochrome c oxidase (COX) activity in 3xTg-AD astroglia. Western blot experiments demonstrating relative levels of mitochondrial protein subunits in 3xTg astroglia in the presence and absence of 24 hrs. 100 nM nilotinib treatment. (A) Representative Western blot for NADH dehydrogenase beta subcomplex subunit 8 of Complex I (NDUFB8), succinate dehydrogenase subunit B of Complex II (SDHB), cytochrome b-c1 complex subunit 2 of Complex III (UQCRC2), Cytochrome c oxidase subunit 1 of Complex IV (MTCO1), and ATP synthase subunit alpha of Complex V (ATP5A). (B-F) Relative quantification for protein levels of Complex I-V normalized to total protein. (G) COX activity was measured in 3xTg-AD astroglia in presence and absence of 100 nM nilotinib treatment. Results are expressed as mean ± SD of n = 6 per group (*P ≤ 0.05**P ≤ 0.01) analyzed by unpaired Student's t-test.
To measure the effect of nilotinib in mitochondrial dynamics, the level of dynamin-related protein 1 (Drp1) and mitofusin 1 (Mfn1) proteins were measured by immunoblotting. Levels of Mfn1 and Drp1 were not significantly different as a function of nilotinib treatment in C57BL/6 astroglia (Fig. 6A-C), indicating that nilotinib did not induce changes in mitochondrial dynamics’ proteins in C57BL/6 astroglia. As well, nilotinib did not alter the level of Drp1 from 3xTg astroglia; however, nilotinib significantly increased the level of Mfn1 in 3xTg astroglia, indicating that nilotinib induces changes in mitochondrial mass (Fig. 6D-E). To further clarify the role of nilotinib on mitochondrial dynamics in 3xTg astroglia, we compared the number of mitochondria in nilotinib-treated vs. non-treated cells utilizing TEM. As shown in Fig. 7A-C, nilotinib significantly increased the number of mitochondria in 3xTg astroglia as compared to that of the non-treated group. To support the TEM data, we measured the effect of nilotinib in key regulators of mitochondrial biogenesis, including PGC-1α, CaMKII, Nrf2, and TFAM. The expression of PGC-1α, CaMKII, and Nrf2 in the nilotinib-treated 3xTg astroglia was significantly increased compared to that of controls (Fig. 7D-H). However, the protein level of TFAM was not found to be altered in nilotinib-treated 3xTg astroglia compared with non-treated cells (Fig. 7G).
Figure 6.

Nilotinib significantly altered the expression of Mfn1 in 3xTg-AD astroglia. Western blot experiments demonstrating relative levels of Mfn1 and Drp1 in C57BL/6-WT astroglia (A-C) and 3xTg astroglia (D-F) inthe presence and absence of 24 hrs. 100 nM nilotinib treatment. Relative quantification for protein levels of Mfn-1 and Drp-1 normalized to total protein. Results are expressed as mean ± SD of n = 6 per group (***P ≤ 0.001) analyzed by unpaired Student's t-test.
Figure 7.

Nilotinib increased the number of mitochondria and mitochondrial biogenesis (CAMKII, PGC1-α, and Nrf2) in 3xTg-AD astroglia. The numbers of mitochondria in 3xTg astroglia were compared in the presence and absence of nilotinib (100 nM). Representative TEM image A showing the cell body of 3xTg astroglia. Representative TEM image B showing the cell body of the nilotinib-treated 3xTg-AD astroglia. (C) The number of mitochondria per cross-sectioned cell was counted (20 cells per group counted by TEM). Results are expressed as mean ± SD of n=20 per group (****P ≤ 0.0001) analyzed by unpaired Student's t-test. M: mitochondria; Scale bars: 500 μm; Magnification: 46000x. (D-H) Western blot experiments demonstrating relative levels of CAMKII, PGC1-α, TFAM, and Nrf2 in 3xTg-AD astroglia in the presence and absence of 24 hrs. 100 nM nilotinib treatment. Relative quantification for protein levels of CAMKII, PGC1-α, TFAM, and Nrf2 normalized to total protein. Results are expressed as mean ± SD of n = 5 per group (* = p < 0.05 or ** = p < 0.01 or **** = p < 0.0001) analyzed by unpaired Student's t-test.
Effects of nilotinib in neuroinflammatory-associated proteins in C57BL/6 and 3xTg astroglia
To further investigate the molecular mechanisms underpinning the enhanced mitochondrial function with nilotinib, expression of various neuroinflammatory-associated proteins in C57BL/6 and 3xTg astroglia were semi-quantified with immunoblotting (Fig. 8A-F and Fig. 9A-F). Nilotinib significantly increased expression levels of NF-κB p50 and NF-κB p105 in C57BL/6 astroglia (Fig. 8C-E). However, the expression levels of IκB-α and NF-κB p75 in C57BL/6 astroglia were unaffected (Fig. 8B and 8F). Further, EMSA was used to verify the effect of nilotinib on NF-κB activation and binding. Nilotinib increased transcriptional activity of NF-κB in C57BL/6 astroglia (Fig. 8G).
Figure 8.

Nilotinib significantly increased the activation of NF-κB and expression of NF-κB p50/p105 subunits in C57BL/6-WT astroglia. (A) Western blot experiments demonstrating relative levels of NF-κB subunits (p50, p105, p65, and p75), and IκB-α in cultured cortical astroglia derived from C57BL/6 in the presence and absence of 24 hrs. 100 nM nilotinib treatment. (B-F) Relative quantification for protein levels of NF-κB subunits (p50, p105, p65, and p75) and IκB-α normalized to total protein. (G) Nuclear extract derived from nilotinib-treated and non-treated C57BL/6-WT astroglia were assayed for NF-κB activation by EMSA using a biotin-labeled oligonucleotide encompassing the NF-κB consensus motif. Results are expressed as mean ± SD of n = 6 per group (**P ≤ 0.01) analyzed by unpaired Student's t-test.
Figure 9.

Nilotinib significantly increased the expression of NF-κB p50/p105 subunits and activation of NF-κB in 3xTg-AD astroglia. (A) Western blot experiments demonstrating relative levels of NF-κB subunits (p50, p105, p65, and p75), and IκB-α in cultured cortical astroglia derived from 3xTg in the presence and absence of 24 hrs.100 nM nilotinib treatment. (B-F) Relative quantification for protein levels of NF-κB subunits (p50, p105, p65, and p75) and IκB-α normalized to total protein. (G) Nuclear extract derived from nilotinibtreated and non-treated 3xTg-AD astroglia were assayed for NF-κB activation by EMSA using a biotinlabeled oligonucleotide encompassing the NF-κB consensus motif. Results are expressed as mean ± SD of n = 6 per group (*P ≤ 0.05) analyzed by unpaired Student's t-test.
In addition, nilotinib increased expression levels of NF-κB p50 and NF-κB p105 subunits in 3xTg astroglia (Fig. 9C-D). In contrast, the expression levels of IκB-α, NF-κB p65, and NF-κB p75 subunits in 3xTg astroglia were unaffected (Fig. 9B, 9E, and 9F). Further, EMSA data verify that nilotinib significantly increased transcriptional activity of NF-κB in 3xTg astroglia (Fig. 9G).
Stimulation with specific inducers activate NF-κB signaling pathways resulting in the translocation of NF-κB dimers to the nucleus to activate NF-κB target genes [71]. In the current study, immunofluorescence (IF) was utilized to verify the effect of nilotinib on NF-κB translocation. IF data in 3xTg astroglia indicated that nilotinib (100 nM) treatment significantly translocated NF-κB p50 to the nucleus but had no effect on the translocation of NF-κB p65 or NF-κB p75 subunits (Fig. 10A-C).
Figure 10.

Nilotinib translocated the NF-kB p50 subunit into the nucleus of 3xTg-AD astroglia. Quantitative immunofluorescent nuclear/cytoplasmic ratios of NF-κB subunits (p50 (A), p65 (B), and p75 (C)) in 3xTg-AD astroglia. DAPI (blue) marks the nucleus. Images were captured at 100x magnification. Volume density of NF-kB subunits immunofluorescence was quantified using ImageJ software (****P ≤ 0.0001); n=5 per group analyzed by unpaired Student's t-test.
Effects of nilotinib and NF-κB inhibitor, Bay11-7082, on mitochondrial respiration in 3xTg astroglia
To investigate whether nilotinib increased mitochondrial respiration in 3xTg astroglia through the NF-κB signaling pathway, astroglia were treated with either a NF-κB inhibitor (Bay11-7082 (1, 3, 5, and 10 µM)) alone, or co-treated with nilotinib (100 nM) for 24 hours. The mitochondrial OCR in 3xTg astroglia was decreased significantly at 5 µM of Bay11-7082 after 24 hours (Fig. 11A). At basal and maximal level, OCR in 3xTg astroglia in the presence of Bay11-7082 (5 µM) was significantly decreased as compared to non-treated cells (Fig. 11B-C). Additionally, bioenergetics’ parameters of spare respiratory capacity, as well as spare coupling efficiency, were unchanged after Bay11-7082 treatment (Fig. 11D-E). Although Bay11-7082 decreased OCR level in 3xTg astroglia, co-treatment of Bay11-7082 (5 µM) and nilotinib (100 nM) did not compensate mitochondrial deficits (Fig. 12A-C). Spare respiratory and coupling efficiency were not affected with co-treatment of Bay11-7082 (5 uM) and nilotinib (100 nM) (Fig. 12D-E). To further determine if the mitochondrial respiration rates were reflected by similar alterations in cellular energy production, total ATP production was measured. ATP levels were significantly decreased in 3xTg astroglia that were treated with nilotinib or co-treated with Bay11-7082 (Fig. 12F).
Figure 11.

Bay11-7082 decreased mitochondrial respiration rates in 3xT-AD astroglia. (A) Kinetics graph indicating real-time OCR at baseline and after addition of oligomycin, FCCP, and rotenone/antimycin. The OCR was measured in 3xTg-AD astroglia utilizing the XF24 analyzer after 24 hours treatment with Bay11- 7082 (1, 3, 5, and 10 uM). (B) Basal respiration, (C) maximal respiration and (D) spare respiratory capacity, and (E) coupling efficiency were calculated compared between control and treated cells. Data are mean ± SD of n = 5 group (** = p < 0.01) analyzed by unpaired Student's t-test.
Figure 12.

Nilotinib-induced enhancements in energy metabolism appear NF-kB-dependent, as demonstrated in 3xTg-AD astroglia. The OCR was measured in 3xTg-AD astroglia utilizing the XF24 analyzer after 24 hours treatment with nilotinib (100 nM) or Bay11-7082 (5 uM) or co-treatment with nilotinib (100 nM) + Bay11- 7082 (5 uM). (A) Basal respiration, (B) maximal respiration, (C) spare respiratory capacity, (D) coupling efficiency, and (E) ATP level were calculated compared between control and treated cells. Data are mean ± SD of n= 5 per group (* = p < 0.05 or ** = p < 0.01 or *** = p < 0.001 or **** = p < 0.0001) analyzed by unpaired Student's t-test.
DISCUSSION
We report, for the first time, the effects of nilotinib treatment on brain astroglia derived from the cortex of 3xTg-AD versus C57BL/6-WT mice. The results showed cortical astroglia from 3xTg mice exhibited significant declines in parameters of mitochondrial bioenergetics as compared to the control group. This study is the first to demonstrate increased mitochondrial function of 3xTg astroglia as a result of nilotinib treatment (100 nM). Also, for the first time we provide evidence that nilotinib’s benefit in mitochondria involves an NF-κB-dependent pathway. Nilotinib has been previously shown to induce apoptosis in cancer cells at micromolar rather than nanomolar concentrations [12, 72-75]. However, data from our study indicate that nilotinib is effective in 3xTg astroglia at nanomolar concentrations. Growing evidence suggests that mitochondrial dysfunction contributes to early events in the pathogenesis of AD [7, 40, 76, 77]. Therefore, the current study has important implications for the treatment of disorders affected by dysfunctional mitochondria, including AD.
Different types of brain cells have unique features of mitochondrial function, and display explicit roles related to their metabolic function. A previous proteomic study designates that the mitochondrial function and morphology is various in different types of brain cells [78]. A better understanding of mitochondrial impairment in AD requires a cell-type specific approach. Although astroglia and neurons are linked in their energy metabolism, they are still different types of brain cell that utilize a very different metabolic profile. Neuronal cells, during periods of enhanced activity, produce the toxic fatty acids (FA) that are transported into astroglia, which can be detoxified in mitochondria instead of being dealt with in neurons [79]. Unlike astroglia, neuronal cells have a restricted capacity to up-regulate glycolysis or to counteract oxidative damage [80, 81]. Additionally, neurons rely on OXPHOS to meet their energy requirements; therefore, mitochondrial dysfunction leads to increased generation of reactive oxygen species (ROS), energy failure, and ultimately neuronal cell death. Under environmental and cellular stress conditions, astroglia have their distinct metabolic reprogramming mediated via mitochondrial dynamics, which determine the inflammatory features of glia [82, 83]. Other studies show that metabolic activity in astroglia may actually increase with age [84]. Increased metabolic activity in astroglia was linked with an age-dependent increase in inflammatory pathways, including NF-κB signaling [85, 86]. Further, inflammatory cytokines in astroglia triggered OXPHOS as well as mitochondrial biogenesis [84]. Human astroglia infected with human immunodeficiency virus (HIV) displayed mitochondrial damage, severe inflammation, and ultimately cell death. On the other hand, HIV-infected astroglia modulates inflammatory responses and regulation of mitochondrial dynamics followed by mitophagy [87]. These opposed scenarios with regard to modulation of mitochondrial dynamics regulate cell viability. Investigators have speculated that mitophagy in HIV-infected astroglia may reduce inflammation via removing damaged mitochondria that leads to cell viability [60, 87]. Interestingly, Lonskaya et al. show that nilotinib improves autophagic machinery, leading to Aβ clearance in transgenic amyloid precursor protein (APP) mice, thus suggesting therapeutic benefits for AD [88]. A current study also shows that mitochondrial OCR was significantly lower in AD astroglia as compared to control and nilotinib increased mitochondrial function and inflammatory responses. It has also been suggested that the astroglia response to nilotinib leads to increased inflammatory responses that support the functionality switch of astroglia to increase metabolic activity for energy support. Also, nilotinib increases the expression of mitochondrial dynamics, which might act as a compensatory mechanism to preserve energy by balancing proteins from one healthy mitochondrion to another mitochondrion that might be damaged.
The cumulative evidence has reported that mitochondrial dysfunction plays an important role in neurodegenerative disorders including AD. The present study demonstrates that nilotinib regulates brain bioenergetics in 3xTg astroglia. Our data show that nilotinib treatment of 3xTg astroglia increased the mitochondrial respiration rate, ATP levels, and the expression of mitochondrial complex subunit proteins. Metabolic investigations into synaptic mitochondia show that a ~25% reduction in Complex I activity and >70% reduction in Complexes III and IV activity results in depletion of ATP and OCR levels [89, 90]. Therefore, a significant portion of oxidative phosphorylation in synaptic mitochondria is regulated via the activity of Complex I and Complexes (III and IV), respectively. In the current study, nilotinib increased the expression of mitochondrial subunits (Complexes I and III-V) that may lead to increased newly-generated synapses.
A decrease in mitochondrial respiration rate in 3xTg cells is also consistent with literature indicating mitochondrial function impairment in 3xTg mouse models [91, 92]. Previously, our laboratory identified significant decreases in brain glucose uptake rates and decreases in the activity of the terminal mitochondrial enzyme (COX or Complex IV) in 3xTg cortex [8]. In the present study, nilotinib increased the activity of COX (Complex IV) in 3xTg astroglia. These results suggest that nilotinib improves the function of respiratory complexes in 3xTg astroglia. However, nilotinib had no effect on protein levels of mitochondrial complex subunits in control astroglia. Together, these data indicate that nilotinib improves mitochondrial function under pathological states.
Additionally, increase in mitochondrial oxidative metabolism is paralleled by increase in the level of COX suggesting an enhanced mitochondrial biogenesis [93]. Moreover, several reports link mitochondrial bioenergetics to mitochondrial dynamics [94-96]. Additionally, impaired mitochondrial function is consistent with altered mitochondrial dynamics in AD [49, 77, 97-101]. Some studies report increased levels of mitochondrial fission protein Drp1 and decreased levels of mitochondrial fusion protein Mfn2 in AD brain [102, 103], which lead to loss of respiratory capacity, mitochondrial damage and thus a decreased energy metabolism [5, 104, 105]. Other studies report that mitochondrial dynamic inhibitors reduced mitochondrial metabolism as well as the activity of the ETC, which leads to neurodegeneration in AD [100, 105-108]. The Mfn-mediated fusion is an important marker to maintain proper ETC function and maintain OXPHOS. In fact, a deficiency in mitochondrial fusion has been revealed to result in mitochondrial dysfunction [109]. For example, double knock out Mfn1/2 mice show reduced number of mtDNA, OCR, and complex II activity [110]. Our current study reports that nilotinib increased the level of mitochondrial fusion marker (Mfn1), which would ascribe specific features to mitochondria for various cellular metabolic procedures, such as up regulation of mitochondrial bioenergetics (OCR, ATP) and OXPHOS.
Mitochondrial fusion has been proposed to provide a new strategy for treating mitochondria-related disorders, such as neurodegenerative disorders [111]. Even mutant-DNA mitochondria can fuse with healthy mitochondria of the same cell, which allows WT-DNA mitochondria to compensate for defects in mutant-DNA mitochondria by sharing RNA and/or protein components [112, 113]. Additionally, mitochondrial fusion can rescue two mutant mitochondria by cross-complementation to one another [114]. It is also suggested that mitochondrial fusion functions as a protective response against stress conditions, carried out by the main participants in the mitochondrial fusion. Disruption in mitochondrial fusion markers (Mfn1/Mfn2) caused severe reduction of mitochondrial DNA (mtDNA) levels [110]. Additionally, mitochondrial fusion disorder significantly decreases mitochondrial function and ultimately causes death in a mouse model with high levels of mtDNA mutations, proposing a protective role for Mfn1/2 against mtDNA mutations [110]. Overall, mitochondrial fusion can lead to compensation of damage, thus rescuing damaged organelles and failure of mitochondrial fusion may lead to neurodegenerative diseases. The current study shows that nilotinib increased the expression of mitochondrial fusion. It is suggested that mitochondrial fusion acts as a compensatory mechanism to preserve energy by balancing proteins from one healthy mitochondrion to another, which might be damaged. Therefore, mitochondrial fusion may rescue damaged organelles and create a pool of healthy mitochondria by increasing fusion regularity without compromising the mitophagy of dysfunctional mitochondria [115]. The current study suggests that Mfn1 is compensating for an increase in mitochondrial biogenesis. These findings propose that regulation of mitochondrial dynamics and biogenesis signaling may indicate a new mitochondria-targeted therapeutic strategy for AD.
Emerging evidence has also revealed that mitochondrial dysfunction triggers inflammation [116]. Among a number of different transcription factors that are involved in the inflammation NF-κB is a key inflammatory regulator with activation in glia following brain injury or infection [117-119]. Several studies reported that NF-κB controls inflammation in astroglia [120-122]. Additionally, the activity of NF-κB has been reported in glia isolated from AD rodent models, a Huntington’s disease mouse model, and the spinal cord of patients with amyotrophic lateral sclerosis (ALS) [123-125]. Also, glial NF-κB is associated with the pathology of neurodegenerative disorders such as AD [126]. The NF-κB pathway was activated in glia cultures in response to Aβ treatment, which leads to increased expression of inflammatory cytokines such as interleukin-6 and interleukin-1 beta, and tumor necrosis factor α (TNFα) [84, 127-129]. Jiang and Cadenas 2014 found that increased inflammation triggers mitochondrial OXPHOS in astroglia [84]. These findings are consistent with our study, while nilotinib increases the expression of inflammatory marker (NF-κB) and OXPHOS. These data suggest NF-κB might play a compensatory role and can have beneficial effects in preserving brain health through an immune response. It is also important to consider NF-κB is not only involved in inflammation, but also plays a critical role in learning and memory formation [130-132]. Therefore, our finding suggests that up regulation of NF-κB via nilotinib might lead to memory improvement. Further research examining the effects of nilotinib in improving memory in AD mice is required.
Both mitochondrial dysfunction and neuroinflammation play critical roles in AD progression; However, molecular mechanisms underpinning enhanced mitochondrial function with nilotinib in astroglia are lacking. NF-κB regulates energy metabolism and metabolic activity by upregulating mitochondrial respiration in cancer cells [53]. Nevertheless, how NF-κB controls mitochondrial function in AD brain cells is not understood [57]. To assess whether energy metabolism is enhanced by nilotinib through NF-κB associated pathways, 3xTg astroglia were treated with nilotinib alone or with Bay11-7082, an NF-κB inhibitor. Mitochondrial OCR levels were reduced in the presence of Bay11-7082; however, co-treatment of Bay11-7082 and nilotinib did not compensate for any mitochondrial deficits. These findings suggest that nilotinib could trigger mitochondrial function through an NF-κB-dependent signaling pathway.
Furthermore, immunofluorescence confirmed that the NF-κB p50 subunit is translocated from the cytoplasm to the nucleus in nilotinib-treated 3xTg astroglia, showing that nilotinib initiates the transcriptional induction of NF-κB-inflammatory signaling. Moreover, immunoblotting and EMSA demonstrated that nilotinib increased transcriptional activity of NF-κB in 3xTg astroglia, which might be explained by increased NF-κB nuclear translocation under nilotinib treatment. These findings indicate that the mechanism underlying the effect of nilotinib on cellular respiration involves an NF-κB-dependent pathway.
Several studies show that NF-κB has been specially linked to CaMKII activation [133, 134]. CaMKII plays critical roles in regulating inflammation as well as various mitochondrial functions, such as mitochondrial biogenesis [92, 135]. An increase in CaMKII expression, in the presence of increased NF-κB transcriptional activity, as found here with nilotinib in 3xTg astroglia, is also consistent with literature indicating that CaMKII (upstream target) is required for NF-κB activation [136]. PGC-1α, a downstream target of CaMKII that has been implicated in mitochondrial biogenesis, was significantly increased as a function of nilotinib. The PGC-1α is involved in AD pathogenesis and upregulation of PGC-1α improves mitochondrial biogenesis through induction of transcription factor Nrf2, which is linked to NF-κB in pathological conditions [137-139]. The current study found upregulation of PGC-1α, in nilotinib-treated 3xTg astroglia, consistent with increased Nrf2 (downstream target of PGC-1α), which is a key modulator of mitochondrial metabolism and biogenesis [140-142]. These findings are consistent with previous studies indicating a role of Nrf2 in inflammation, mitochondrial biogenesis and bioenergetics and its interaction with PGC-1α in the context of neurodegeneration [64, 143]. Overall, our study suggests that nilotinib may increase the number of mitochondria through a CaMKII- PGC-1α-Nrf2 pathway, which is influenced by NF-κB dynamics in 3xTg astroglia.
Conclusions
Emerging evidence suggests that defects in mitochondrial function are associated with bioenergetic deficiency and neuroinflammation, and could serve as early biomarkers of AD. The present results have important implications for treatment of disorders with mitochondrial dysfunction including AD. The major finding of this study is that nilotinib treatment stimulated oxidative phosphorylation in 3xTg astroglia with upregulation of mitochondrial complex proteins subunits and COX activity. Additionally, nilotinib increases mitochondrial function and dynamics through altered CaMKII-PGC-1α-Nrf2 pathway activity that are involved in the development of neurodegeneration. Nilotinib also increased mitochondrial function via the NF-κB signaling pathway. Collectively, these results contribute to our understanding of the effects of nilotinib on regulating brain mitochondrial bioenergetics and biogenesis (Fig. 13).
Figure 13.

This putative pathway suggests that nilotinib improves mitochondrial function through nuclear factor κ B (NF-κB)-dependent signaling in 3xTg-AD astroglia. It is hypothesized that nilotinib activates CAMKII as well as a signal cascade resulting in degradation of the NF-κB subunit IκB. The NF-κB dimer then translocates from the cytoplasm to the nucleus where it binds to a DNA consensus sequence of target genes. It is also hypothesized that nilotinib triggers the NF-κB complex (downstream target of CAMKII) to move into the mitochondrion, where is thought to interact with OXPHOS genes that leads to bioenergetic improvement (ATP, oxygen consumption rate (OCR)), as well as an increase in the expression of mitochondrial complex proteins subunits (Complexes I, III, IV, and V), cytochrome c oxidase (COX), mitochondrial biogenesis (PGC1-α and Nrf2), and mitochondrial dynamics (Mfn1). Findings from our current study importantly show that mitochondrial OCR and ATP levels were significantly reduced in the presence of the IKK inhibitor (Bay11-7082). This figure was developed using the BioRender online software tool.
Supplementary Materials
The Supplemenantry data can be found online at: www.aginganddisease.org/EN/10.14336/AD.2020.0910.
Acknowledgments
We gratefully acknowledge the Research Manitoba Fellowship to Dr. Adlimoghaddam (Grant No: 1913), and support from the St. Boniface Hospital Research Foundation (Grant Nos.1406-3216 and 1410-3216), the Canadian Institute of Health Research (CIHR; Grant No. PJT-162144), the Alzheimer’s Society of Manitoba, the Honourable Douglas and Patricia Everett, Royal Canadian Properties Limited Endowment Fund (Grant No. 1403-3131) to Dr. Albensi. Dr. Albensi also holds the Honourable Douglas and Patricia Everett and Royal Canadian Properties Limited Endowment Fund Chair and the Manitoba Dementia Research Chair (funded by the Alzheimer’s Soc. of Manitoba & Research Manitoba). He is also a Research Affiliate of the Centre on Aging, and a scientist at the Children’s Hospital Research Inst. of Manitoba (CHRIM) at the University of Manitoba. Special thanks are given to Mr. Kyle M Fontaine for reviewing our manuscript. Thanks to Dr. Klonisch for interpreting the TEM data. Thanks to Ms. Marina Mostafizar and Ms. Claudia Perez for helping with the euthanization of mice. Thanks to Dr. Paul Fernyhough for allowing us to access and use his equipment (Seahorse XF24 Analyzer and fluorescence microscope).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].McKeith I, Cummings J (2005). Behavioural changes and psychological symptoms in dementia disorders. Lancet Neurol, 4:735-742. [DOI] [PubMed] [Google Scholar]
- [2].Glabe CC (2005). Amyloid accumulation and pathogensis of Alzheimer's disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem, 38:167-177. [DOI] [PubMed] [Google Scholar]
- [3].Hardy J, Selkoe DJ (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science, 297:353-356. [DOI] [PubMed] [Google Scholar]
- [4].Cardoso S, Seica RM, Moreira PI (2017). Mitochondria as a target for neuroprotection: implications for Alzheimer s disease. Expert Rev Neurother, 17:77-91. [DOI] [PubMed] [Google Scholar]
- [5].Cadonic C, Sabbir MG, Albensi BC (2016). Mechanisms of Mitochondrial Dysfunction in Alzheimer's Disease. Mol Neurobiol, 53:6078-6090. [DOI] [PubMed] [Google Scholar]
- [6].Correia SC, Santos RX, Cardoso S, Carvalho C, Candeias E, Duarte AI, et al. (2012). Alzheimer disease as a vascular disorder: where do mitochondria fit? Exp Gerontol, 47:878-886. [DOI] [PubMed] [Google Scholar]
- [7].Aliev G, Palacios HH, Walrafen B, Lipsitt AE, Obrenovich ME, Morales L (2009). Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int J Biochem Cell Biol, 41:1989-2004. [DOI] [PubMed] [Google Scholar]
- [8].Adlimoghaddam A, Snow WM, Stortz G, Perez C, Djordjevic J, Goertzen AL, et al. (2019). Regional hypometabolism in the 3xTg mouse model of Alzheimer's disease. Neurobiol Dis, 127:264-277. [DOI] [PubMed] [Google Scholar]
- [9].Hedskog L, Zhang S, Ankarcrona M (2012). Strategic role for mitochondria in Alzheimer's disease and cancer. Antioxid Redox Signal, 16:1476-1491. [DOI] [PubMed] [Google Scholar]
- [10].Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, et al. (2016). Nilotinib Effects in Parkinson's disease and Dementia with Lewy bodies. J Parkinsons Dis, 6:503-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lonskaya I, Desforges NM, Hebron ML, Moussa CE (2013). Ubiquitination increases parkin activity to promote autophagic alpha-synuclein clearance. PLoS One, 8:e83914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [12].Deremer DL, Ustun C, Natarajan K (2008). Nilotinib: a second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin Ther, 30:1956-1975. [DOI] [PubMed] [Google Scholar]
- [13].Slupianek A, Poplawski T, Jozwiakowski SK, Cramer K, Pytel D, Stoczynska E, et al. (2011). BCR/ABL stimulates WRN to promote survival and genomic instability. Cancer Res, 71:842-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hebron ML, Lonskaya I, Moussa CE (2013). Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum Mol Genet, 22:3315-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hebron ML, Lonskaya I, Olopade P, Selby ST, Pagan F, Moussa CE (2014). Tyrosine Kinase Inhibition Regulates Early Systemic Immune Changes and Modulates the Neuroimmune Response in alpha-Synucleinopathy. J Clin Cell Immunol, 5:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hebron ML, Lonskaya I, Moussa CE (2013). Tyrosine kinase inhibition facilitates autophagic SNCA/alpha-synuclein clearance. Autophagy, 9:1249-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Soverini S, Martinelli G, Rosti G, Iacobucci I, Baccarani M (2012). Advances in treatment of chronic myeloid leukemia with tyrosine kinase inhibitors: the evolving role of Bcr-Abl mutations and mutational analysis. Pharmacogenomics, 13:1271-1284. [DOI] [PubMed] [Google Scholar]
- [18].Turner RS, Hebron ML, Lawler A, Mundel EE, Yusuf N, Starr JN, et al. (2020). Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer's Disease. Ann Neurol, 88:183-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tremblay MA, Acker CM, Davies P (2010). Tau phosphorylated at tyrosine 394 is found in Alzheimer's disease tangles and can be a product of the Abl-related kinase, Arg. J Alzheimers Dis, 19:721-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schlatterer SD, Acker CM, Davies P (2011). c-Abl in neurodegenerative disease. J Mol Neurosci, 45:445-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Imam SZ, Zhou Q, Yamamoto A, Valente AJ, Ali SF, Bains M, et al. (2011). Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson's disease. J Neurosci, 31:157-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, et al. (2010). Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin's ubiquitination and protective function. Proc Natl Acad Sci U S A, 107:16691-16696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schlatterer SD, Tremblay MA, Acker CM, Davies P (2011). Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J Alzheimers Dis, 25:119-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lawana V, Singh N, Sarkar S, Charli A, Jin H, Anantharam V, et al. (2017). Involvement of c-Abl Kinase in Microglial Activation of NLRP3 Inflammasome and Impairment in Autolysosomal System. J Neuroimmune Pharmacol, 12:624-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee SJ, Seo BR, Choi EJ, Koh JY (2014). The role of reciprocal activation of cAbl and Mst1 in the oxidative death of cultured astrocytes. Glia, 62:639-648. [DOI] [PubMed] [Google Scholar]
- [26].Song GJ, Rahman MH, Jha MK, Gupta DP, Park SH, Kim JH, et al. (2019). A Bcr-Abl Inhibitor GNF-2 Attenuates Inflammatory Activation of Glia and Chronic Pain. Front Pharmacol, 10:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH (2005). Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci, 25:9275-9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fakhoury M (2018). Microglia and Astrocytes in Alzheimer's Disease: Implications for Therapy. Curr Neuropharmacol, 16:508-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH (2003). Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry, 74:788-789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Meraz-Rios MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernandez J, Campos-Pena V (2013). Inflammatory process in Alzheimer's Disease. Front Integr Neurosci, 7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Iglesias J, Morales L, Barreto GE (2017). Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol Neurobiol, 54:2518-2538. [DOI] [PubMed] [Google Scholar]
- [32].Ksiezakowska-Lakoma K, Zyla M, Wilczynski JR (2014). Mitochondrial dysfunction in cancer. Prz Menopauzalny, 13:136-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hsu CC, Tseng LM, Lee HC (2016). Role of mitochondrial dysfunction in cancer progression. Exp Biol Med (Maywood), 241:1281-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Albensi BC (2019). Dysfunction of mitochondria: Implications for Alzheimer's disease. Int Rev Neurobiol, 145:13-27. [DOI] [PubMed] [Google Scholar]
- [35].Obulesu M, Jhansilakshmi M (2014). Neuroinflammation in Alzheimer's disease: an understanding of physiology and pathology. Int J Neurosci, 124:227-235. [DOI] [PubMed] [Google Scholar]
- [36].Obulesu M, Lakshmi MJ (2014). Apoptosis in Alzheimer's disease: an understanding of the physiology, pathology and therapeutic avenues. Neurochem Res, 39:2301-2312. [DOI] [PubMed] [Google Scholar]
- [37].Silva DF, Selfridge JE, Lu J, E L, Roy N, Hutfles L, et al. (2013). Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet, 22:3931-3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Scatena R (2012). Mitochondria and cancer: a growing role in apoptosis, cancer cell metabolism and dedifferentiation. Adv Exp Med Biol, 942:287-308. [DOI] [PubMed] [Google Scholar]
- [39].Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L (2018). Mitochondrial metabolism and cancer. Cell Res, 28:265-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, et al. (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging, 30:1574-1586. [DOI] [PubMed] [Google Scholar]
- [41].Merlo E, Freudenthal R, Maldonado H, Romano A (2005). Activation of the transcription factor NF-kappaB by retrieval is required for long-term memory reconsolidation. Learn Mem, 12:23-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH (2006). Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet, 15:1437-1449. [DOI] [PubMed] [Google Scholar]
- [43].Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD (2011). NF-kappaB in Aging and Disease. Aging Dis, 2:449-465. [PMC free article] [PubMed] [Google Scholar]
- [44].Adlimoghaddam A, Sabbir MG, Albensi BC (2016). Ammonia as a Potential Neurotoxic Factor in Alzheimer's Disease. Front Mol Neurosci, 9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Amsalem Z, Arif T, Shteinfer-Kuzmine A, Chalifa-Caspi V, Shoshan-Barmatz V (2020). The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers(Basel), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chua LM, Lim ML, Wong BS (2013). The Kunitz-protease inhibitor domain in amyloid precursor protein reduces cellular mitochondrial enzymes expression and function. Biochem Biophys Res Commun, 437:642-647. [DOI] [PubMed] [Google Scholar]
- [47].Reddy PH, Manczak M, Yin X (2017). Mitochondria-Division Inhibitor 1 Protects Against Amyloid-beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer's Disease. J Alzheimers Dis, 58:147-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang X, Su B, Fujioka H, Zhu X (2008). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol, 173:470-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci, 29:9090-9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oliver D, Reddy PH (2019). Dynamics of Dynamin-Related Protein 1 in Alzheimer's Disease and Other Neurodegenerative Diseases. Cells, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].DiDonato JA, Mercurio F, Karin M (2012). NF-kappaB and the link between inflammation and cancer. Immunol Rev, 246:379-400. [DOI] [PubMed] [Google Scholar]
- [52].Karin M (2009). NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol, 1:a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, et al. (2011). NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol, 13:1272-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shi C, Zhu X, Wang J, Long D (2014). Intromitochondrial IkappaB/NF-kappaB signaling pathway is involved in amyloid beta peptide-induced mitochondrial dysfunction. J Bioenerg Biomembr, 46:371-376. [DOI] [PubMed] [Google Scholar]
- [55].Laforge M, Rodrigues V, Silvestre R, Gautier C, Weil R, Corti O, et al. (2016). NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ, 23:89-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, et al. (2003). NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem, 278:2963-2968. [DOI] [PubMed] [Google Scholar]
- [57].Albensi BC (2019). What Is Nuclear Factor Kappa B (NF-kappaB) Doing in and to the Mitochondrion? Front Cell Dev Biol, 7:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Moretti M, Bennett J, Tornatore L, Thotakura AK, Franzoso G (2012). Cancer: NF-kappaB regulates energy metabolism. Int J Biochem Cell Biol, 44:2238-2243. [DOI] [PubMed] [Google Scholar]
- [59].Lamas O, Moreno-Aliaga MJ, Martinez JA, Marti A (2003). NF-kappa B-binding activity in an animal diet-induced overweightness model and the impact of subsequent energy restriction. Biochem Biophys Res Commun, 311:533-539. [DOI] [PubMed] [Google Scholar]
- [60].Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. (2016). NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell, 164:896-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Canto C, Auwerx J (2010). AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci, 67:3407-3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, et al. (2002). Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science, 296:349-352. [DOI] [PubMed] [Google Scholar]
- [63].Lin J, Handschin C, Spiegelman BM (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab, 1:361-370. [DOI] [PubMed] [Google Scholar]
- [64].Gureev AP, Shaforostova EA, Popov VN (2019). Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front Genet, 10:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ventura-Clapier R, Garnier A, Veksler V (2008). Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res, 79:208-217. [DOI] [PubMed] [Google Scholar]
- [66].Schildge S, Bohrer C, Beck K, Schachtrup C (2013). Isolation and culture of mouse cortical astrocytes. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Marcus DL, Strafaci JA, Freedman ML (2006). Differential neuronal expression of manganese superoxide dismutase in Alzheimer's disease. Med Sci Monit, 12:BR8-14. [PubMed] [Google Scholar]
- [68].Acevedo G, Padala NK, Ni L, Jonakait GM (2013). Astrocytes inhibit microglial surface expression of dendritic cell-related co-stimulatory molecules through a contact-mediated process. J Neurochem, 125:575-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Guttenplan KA, Liddelow SA (2019). Astrocytes and microglia: Models and tools. J Exp Med, 216:71-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lee J, Rhee MH, Kim E, Cho JY (2012). BAY 11-7082 is a broad-spectrum inhibitor with anti-inflammatory activity against multiple targets. Mediators Inflamm, 2012:416036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wan F, Lenardo MJ (2010). The nuclear signaling of NF-kappaB: current knowledge, new insights, and future perspectives. Cell Res, 20:24-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Silveira E, Cavalcante IP, Kremer JL, de Mendonca POR, Lotfi CFP (2018). The tyrosine kinase inhibitor nilotinib is more efficient than mitotane in decreasing cell viability in spheroids prepared from adrenocortical carcinoma cells. Cancer Cell Int, 18:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Weigel MT, Rath K, Alkatout I, Wenners AS, Schem C, Maass N, et al. (2014). Nilotinib in combination with carboplatin and paclitaxel is a candidate for ovarian cancer treatment. Oncology, 87:232-245. [DOI] [PubMed] [Google Scholar]
- [74].Chen TC, Yu MC, Chien CC, Wu MS, Lee YC, Chen YC (2016). Nilotinib reduced the viability of human ovarian cancer cells via mitochondria-dependent apoptosis, independent of JNK activation. Toxicol In Vitro, 31:1-11. [DOI] [PubMed] [Google Scholar]
- [75].Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, et al. (2005). Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell, 7:129-141. [DOI] [PubMed] [Google Scholar]
- [76].Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol, 57:695-703. [DOI] [PubMed] [Google Scholar]
- [77].Cabezas-Opazo FA, Vergara-Pulgar K, Perez MJ, Jara C, Osorio-Fuentealba C, Quintanilla RA (2015). Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer's Disease. Oxid Med Cell Longev, 2015:509654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Fecher C, Trovo L, Muller SA, Snaidero N, Wettmarshausen J, Heink S, et al. (2019). Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat Neurosci, 22:1731-1742. [DOI] [PubMed] [Google Scholar]
- [79].Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, et al. (2019). Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell, 177:1522-1535 e1514. [DOI] [PubMed] [Google Scholar]
- [80].Fernandez-Fernandez S, Almeida A, Bolanos JP (2012). Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem J, 443:3-11. [DOI] [PubMed] [Google Scholar]
- [81].Rose J, Brian C, Woods J, Pappa A, Panayiotidis MI, Powers R, et al. (2017). Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology, 391:109-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Park J, Choi H, Min JS, Park SJ, Kim JH, Park HJ, et al. (2013). Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J Neurochem, 127:221-232. [DOI] [PubMed] [Google Scholar]
- [83].Orihuela R, McPherson CA, Harry GJ (2016). Microglial M1/M2 polarization and metabolic states. Br J Pharmacol, 173:649-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jiang T, Cadenas E (2014). Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell, 13:1059-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Campuzano O, Castillo-Ruiz MM, Acarin L, Castellano B, Gonzalez B (2009). Increased levels of proinflammatory cytokines in the aged rat brain attenuate injury-induced cytokine response after excitotoxic damage. J Neurosci Res, 87:2484-2497. [DOI] [PubMed] [Google Scholar]
- [86].Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, et al. (2013). Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature, 497:211-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ojeda DS, Grasso D, Urquiza J, Till A, Vaccaro MI, Quarleri J (2018). Cell Death Is Counteracted by Mitophagy in HIV-Productively Infected Astrocytes but Is Promoted by Inflammasome Activation Among Non-productively Infected Cells. Front Immunol, 9:2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Lonskaya I, Hebron ML, Desforges NM, Schachter JB, Moussa CE (2014). Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med (Berl), 92:373-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Telford JE, Kilbride SM, Davey GP (2009). Complex I is rate-limiting for oxygen consumption in the nerve terminal. J Biol Chem, 284:9109-9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Davey GP, Peuchen S, Clark JB (1998). Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem, 273:12753-12757. [DOI] [PubMed] [Google Scholar]
- [91].Djordjevic J, Roy Chowdhury S, Snow WM, Perez C, Cadonic C, Fernyhough P, et al. (2020). Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer's Mice. Cells, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Snow WM, Cadonic C, Cortes-Perez C, Roy Chowdhury SK, Djordjevic J, Thomson E, et al. (2018). Chronic dietary creatine enhances hippocampal-dependent spatial memory, bioenergetics, and levels of plasticity-related proteins associated with NF-kappaB. Learn Mem, 25:54-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Soto IC, Fontanesi F, Liu J, Barrientos A (2012). Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim Biophys Acta, 1817:883-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bazil JN, Buzzard GT, Rundell AE (2010). Modeling mitochondrial bioenergetics with integrated volume dynamics. PLoS Comput Biol, 6:e1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Leipnitz G, Mohsen AW, Karunanidhi A, Seminotti B, Roginskaya VY, Markantone DM, et al. (2018). Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci Rep, 8:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Rossignol O, Dodson JJ, Guderley H (2011). Relationship between metabolism, sex and reproductive tactics in young Atlantic salmon (Salmo salar L.). Comp Biochem Physiol A Mol Integr Physiol, 159:82-91. [DOI] [PubMed] [Google Scholar]
- [97].Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, Maechler P, et al. (2008). Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One, 3:e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Westermann B (2012). Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta, 1817:1833-1838. [DOI] [PubMed] [Google Scholar]
- [99].Figge MT, Reichert AS, Meyer-Hermann M, Osiewacz HD (2012). Deceleration of fusion-fission cycles improves mitochondrial quality control during aging. PLoS Comput Biol, 8:e1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, et al. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J Neurochem, 120:419-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Trimmer PA, Borland MK (2005). Differentiated Alzheimer's disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid Redox Signal, 7:1101-1109. [DOI] [PubMed] [Google Scholar]
- [102].Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X (2009). The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem, 109 Suppl 1:153-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Manczak M, Calkins MJ, Reddy PH (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet, 20:2495-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Joshi AU, Ebert AE, Haileselassie B, Mochly-Rosen D (2019). Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington's disease. J Mol Cell Cardiol, 127:125-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chen H, Chomyn A, Chan DC (2005). Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem, 280:26185-26192. [DOI] [PubMed] [Google Scholar]
- [106].Reddy PH (2014). Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J Alzheimers Dis, 40:245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Rappold PM, Cui M, Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L, et al. (2014). Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat Commun, 5:5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, et al. (2005). The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet, 14:1405-1415. [DOI] [PubMed] [Google Scholar]
- [109].Srinivasan S, Guha M, Kashina A, Avadhani NG (2017). Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim Biophys Acta Bioenerg, 1858:602-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell, 141:280-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Szabo A, Sumegi K, Fekete K, Hocsak E, Debreceni B, Setalo G Jr, et al. (2018). Activation of mitochondrial fusion provides a new treatment for mitochondria-related diseases. Biochem Pharmacol, 150:86-96. [DOI] [PubMed] [Google Scholar]
- [112].Yoneda M, Miyatake T, Attardi G (1994). Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol Cell Biol, 14:2699-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Schon EA, Gilkerson RW (2010). Functional complementation of mitochondrial DNAs: mobilizing mitochondrial genetics against dysfunction. Biochim Biophys Acta, 1800:245-249. [DOI] [PubMed] [Google Scholar]
- [114].Yang L, Long Q, Liu J, Tang H, Li Y, Bao F, et al. (2015). Mitochondrial fusion provides an 'initial metabolic complementation' controlled by mtDNA. Cell Mol Life Sci, 72:2585-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Twig G, Shirihai OS (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal, 14:1939-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].van Horssen J, van Schaik P, Witte M (2019). Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci Lett, 710:132931. [DOI] [PubMed] [Google Scholar]
- [117].Gupta AS, Waters MR, Biswas DD, Brown LN, Surace MJ, Floros C, et al. (2019). RelB controls adaptive responses of astrocytes during sterile inflammation. Glia, 67:1449-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Simmons LJ, Surles-Zeigler MC, Li Y, Ford GD, Newman GD, Ford BD (2016). Regulation of inflammatory responses by neuregulin-1 in brain ischemia and microglial cells in vitro involves the NF-kappa B pathway. J Neuroinflammation, 13:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kiebala M, Polesskaya O, Yao Z, Perry SW, Maggirwar SB (2010). Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production. PLoS One, 5:e11875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kaltschmidt B, Kaltschmidt C (2009). NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol, 1:a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Mattson MP, Meffert MK (2006). Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ, 13:852-860. [DOI] [PubMed] [Google Scholar]
- [122].Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y (2013). A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington's disease. Hum Mol Genet, 22:1826-1842. [DOI] [PubMed] [Google Scholar]
- [123].Carrero I, Gonzalo MR, Martin B, Sanz-Anquela JM, Arevalo-Serrano J, Gonzalo-Ruiz A (2012). Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp Neurol, 236:215-227. [DOI] [PubMed] [Google Scholar]
- [124].Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, et al. (2015). NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron, 85:101-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Migheli A, Piva R, Atzori C, Troost D, Schiffer D (1997). c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol, 56:1314-1322. [DOI] [PubMed] [Google Scholar]
- [126].Gilmore TD (2006). Introduction to NF-kappaB: players, pathways, perspectives. Oncogene, 25:6680-6684. [DOI] [PubMed] [Google Scholar]
- [127].Aisen PS, Davis KL (1994). Inflammatory mechanisms in Alzheimer's disease: implications for therapy. Am J Psychiatry, 151:1105-1113. [DOI] [PubMed] [Google Scholar]
- [128].Block ML, Zecca L, Hong JS (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci, 8:57-69. [DOI] [PubMed] [Google Scholar]
- [129].Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541:481-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Albensi BC, Mattson MP (2000). Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse, 35:151-159. [DOI] [PubMed] [Google Scholar]
- [131].Engelmann C, Haenold R (2016). Transcriptional Control of Synaptic Plasticity by Transcription Factor NF-kappaB. Neural Plast, 2016:7027949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Salles A, Romano A, Freudenthal R (2014). Synaptic NF-kappa B pathway in neuronal plasticity and memory. J Physiol Paris, 108:256-262. [DOI] [PubMed] [Google Scholar]
- [133].Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (2003). NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci, 6:1072-1078. [DOI] [PubMed] [Google Scholar]
- [134].Dresselhaus EC, Meffert MK (2019). Cellular Specificity of NF-kappaB Function in the Nervous System. Front Immunol, 10:1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Chin ER (2004). The role of calcium and calcium/calmodulin-dependent kinases in skeletal muscle plasticity and mitochondrial biogenesis. Proc Nutr Soc, 63:279-286. [DOI] [PubMed] [Google Scholar]
- [136].Ishiguro K, Green T, Rapley J, Wachtel H, Giallourakis C, Landry A, et al. (2006). Ca2+/calmodulin-dependent protein kinase II is a modulator of CARMA1-mediated NF-kappaB activation. Mol Cell Biol, 26:5497-5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Sweeney G, Song J (2016). The association between PGC-1alpha and Alzheimer's disease. Anat Cell Biol, 49:1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jornayvaz FR, Shulman GI (2010). Regulation of mitochondrial biogenesis. Essays Biochem, 47:69-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell, 98:115-124. [DOI] [PubMed] [Google Scholar]
- [140].Feige JN, Auwerx J (2007). Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol, 17:292-301. [DOI] [PubMed] [Google Scholar]
- [141].Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P (2008). Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett, 582:46-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Jager S, Handschin C, St-Pierre J, Spiegelman BM (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A, 104:12017-12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Tufekci KU, Civi Bayin E, Genc S, Genc K (2011). The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson's Disease. Parkinsons Dis, 2011:314082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.