

Abstract
Alzheimer’s disease (AD) disproportionately affects certain racial and ethnic subgroups, such as African American/Black and Hispanic adults. Genetic, comorbid, and socioeconomic risk factors contribute to this disparity; however, the molecular contributions have been largely unexplored. Herein, we conducted a pilot proteomics study of postmortem brains from African American/Black and non-Hispanic White adults neuropathologically diagnosed with AD compared to closely-matched cognitively normal individuals. Examination of hippocampus, inferior parietal lobule, and globus pallidus regions using quantitative proteomics resulted in 568 differentially-expressed proteins in AD. These proteins were consistent with the literature and included glial fibrillary acidic protein, peroxiredoxin-1, and annexin A5. In addition, 351 novel proteins in AD were identified, which could partially be due to cohort diversity. From linear regression analyses, we identified 185 proteins with significant race x diagnosis interactions across various brain regions. These differences generally were reflective of differential expression of proteins in AD that occurred in only a single racial/ethnic group. Overall, this pilot study suggests that disease understanding can be furthered by including diversity in racial/ethnic groups; however, this must be done on a larger scale.
Keywords: Alzheimer’s disease, Proteomics, Disparities, Brain, African American, Black, Hippocampus, Inferior parietal lobule, Globus pallidus, Proteins
1. Introduction
The Alzheimer’s Association estimates that 5.7 million Americans have Alzheimer’s disease (AD), (Association, A. S, 2018) although different racial and ethnic subgroups of the population are not affected equally (Barnes, 2019; González et al., 2019). African American/Black adults are 2–3 and Hispanic adults are 1.5–2 times more likely to develop AD and related dementias than non-Hispanic White adults (Association, A. S, 2017; Chin et al., 2011). On the other hand, Native American and Asian American adults (i.e., Japanese Americans) have lower prevalence and incidence of AD than non-Hispanic White adults (Association, A. S, 2018; Association, A. S, 2017; Manly and Mayeux, 2004; Matthews et al., 2018; Mayeda et al., 2015) African American/Black and Hispanic minorities will comprise 40% of 65-year and older individuals and AD sufferers by 2050, (Matthews et al., 2018; Barnes and Bennett, 2014; Lines et al., 2014) which underscores the urgency of better understanding disparities in this disease.
Significant differences in postmortem disease hallmarks, such as amyloid beta (Aβ) plaques and hyperphosphorylated tau tangles (neurofibrillary tangles; NFTs), between the brains of African American/Black and non-Hispanic White adults have not been observed (Chin et al., 2011; Barnes et al., 2015; Gottesman et al., 2017; Wilkins et al., 2006). Cerebral amyloid angiopathy, which often coexists with AD, has similar prevalence and histopathological characteristics between African American/Black and non-Hispanic White adults (Kamara et al., 2018). Global gray matter change is the best predictor of cognitive decline in both African American/Black and non-Hispanic White adults, (Gavett et al., 2018) however, African American/Black adults are more likely to present with mixed AD pathologies and other dementias, particularly Lewy body dementia, infarcts, and cerebrovascular disease (Barnes et al., 2015; Graff-Radford et al., 2016; Filshtein et al., 2019).
Socioeconomic factors, genetics, and comorbidities may also have substantial contributions to higher incidence of AD in African American/Black adults, and highlight the importance of carefully designed biological experiments in this context (Wilkins et al., 2020). Socioeconomic factors include education level, healthcare access, and willingness to seek care and treatment (Chin et al., 2011; Barnes and Bennett, 2014; Mehta and Yeo, 2017; Burke et al., 2017; Gilligan et al., 2012) African American/Black adults, in one study, were less likely to seek care for symptoms of mild cognitive impairment (MCI) (Burke et al., 2017) and in other studies, were less likely than non-Hispanic White adults to receive AD pharmacotherapy treatment (e.g., cholinesterase inhibitors or memantine) upon disease diagnosis (Barnes and Bennett, 2014; Gilligan et al., 2012). Genetic risk factors, particularly the apolipoprotein E (APOE) ε4 allele and single nucleotide polymorphisms of the ATP-binding cassette transporter A7 (ABCA7) gene, differ in prevalence and effect size amongst different racial and ethnic groups (Barnes and Bennett, 2014; Graff-Radford et al., 2016; Hohman et al., 2016; Reitz et al., 2013). APOE, ABCA7, and other risk genes impacting African American/Black adults such as apolipoprotein D (APOD), (Desai et al., 2003) sortilin-related receptor 1 (SORL1),(Lee et al., 2007) and sigma non-opioid intracellular receptor 1 (SIGMAR1) (Ghani et al., 2015) are relevant for lipid metabolism and encode proteins involved in lipid transport, homeostasis, regulation, and cholesterol biosynthesis (Reitz et al., 2013; El Gaamouch et al., 2016; Martins et al., 2009; Rogaeva et al., 2007). Lipid metabolism plays an important role in AD pathogenesis (El Gaamouch et al., 2016; Martins et al., 2009; Liu and Zhang, 2014; Gamba et al., 2012; Burns and Duff, 2002; Sato and Morishita, 2015) and in comorbidities that increase AD risk, such as dyslipidemia, type 2 diabetes mellitus (T2DM), cardiovascular disease, and hypertension (Association, A. S, 2017; Manly and Mayeux, 2004; Barnes and Bennett, 2014; Stepler and Robinson, 2019; Chakrabarti et al., 2015; Matsuzaki et al., 2011). These comorbidities are also prevalent in African American/Black adults (Manly and Mayeux, 2004).
Differences in the immune system and inflammatory pathways are noted in African American/Black compared to non-Hispanic White adults (Babulal et al., 2018). For example, a recent study reported higher cerebrospinal fluid (CSF) levels of interleukin-9 (IL-9) in African American/Black adults correlate with AD but this is not the case in non-Hispanic White adults (Wharton et al., 2019). Cognitively normal (CN) middle-aged African American/Black adults also have lower CSF levels of total and phosphorylated tau, biomarkers for AD, (Rosenmann, 2012; Howell et al., 2017; Morris et al., 2019; Blennow et al., 2001; Wallin et al., 2006; Garrett et al., 2019) and IL-9 compared to non-Hispanic White adults. (Wharton et al., 2019). These findings suggest tau-related pathways may contribute to racial disparities in AD; however, large-scale molecular studies of African American/Black adults using biofluids or postmortem brain are necessary to test this hypothesis (Barnes, 2019).
Discovery-based proteomics can be useful for disease understanding and has been employed broadly to analyze AD postmortem brain (Seyfried et al., 2017; Xu et al., 2019; Ping et al., 2018; Manavalan et al., 2013; Zahid et al., 2014; McKetney et al., 2019). Based on an extensive literature search, African American/Black and other underrepresented minorities have been grossly excluded in proteomic studies of AD, especially in brain (Ping et al., 2018; Manavalan et al., 2013). Significant pathways found in brains of non-Hispanic White adults include innate immune response and the citric acid cycle, while neurotransmitter regulation, monosaccharide/glucose metabolism, and apoptosis/cell cycle regulation primarily differ in regions most severely affected by AD pathology (i.e. hippocampus, entorhinal cortex, and cingulate gyrus) (Xu et al., 2019). The hippocampus has a well-established role in the early to late stages of AD and undergoes changes in cytoskeletal, metabolic, synaptic, and signaling pathways (Xu et al., 2019; Manavalan et al., 2013; Zahid et al., 2014; Mu and Gage, 2011; Hondius et al., 2016; Smith, 2002; Scahill et al., 2002; Begcevic et al., 2013; Schrotter et al., 2017). Oxidative posttranslational modifications in the hippocampus and inferior parietal lobule (IPL) increase in amnestic MCI and AD (Reed et al., 2009; Hensley et al., 1995; Dalle-Donne et al., 2003; Sultana et al., 2007a; Newman et al., 2007). IPL has decreased gray matter volume in AD (Wang et al., 2015; Greene and Killiany, 2010) and increased protein phosphorylation (Triplett et al., 2016). Loss of cholinergic neurons occurs in the globus pallidus (GP) in AD, (Lehéricy et al., 1991) which may be due to the presence of Aβ oligomers in this region (Baker-Nigh et al., 2015). However, GP has less AD pathology compared to other brain regions.
Understanding the extent of molecular contributions and/or outcomes of racial and ethnic disparities in AD is very necessary to further overall disease understanding and to inform prevention, therapeutic, and personalized medicine strategies. Herein, we present a pilot spatial proteomics study of postmortem brain tissues (i.e. hippocampus, IPL, GP) from African American/Black and non-Hispanic White adults. This study included participants from the University of Pittsburgh Alzheimer DiseaseResearch Center (ADRC) who were CN or neuropathologically diagnosed with AD at autopsy. Our findings provide new insights regarding the molecular basis of AD and especially highlight the need for more inclusion of racial/ethnic minorities in proteomics studies of AD.
2. Materials and methods
2.1. Sample selection
Postmortem brain tissues were selected from the University of Pittsburgh ADRC brain bank. The University of Pittsburgh ADRC database was surveyed for African American/Black adults with AD between the time of its inception in 1985 and 12/15/2016 (N = 209; 8.2%). Of these, five were deceased and had brain samples available from hippocampus, IPL, and/or GP. We selected all five of these African American/Black AD brains and the four African American/Black CN brains from these regions, and matched brains from non-Hispanic White adults based on age, sex, and diagnosis. Hippocampal (N = 18), IPL (N = 19), and GP (N = 18) tissues were acquired from African American/Black and non-Hispanic White adults who were CN or neuropathologically diagnosed with AD (Table 1). Race was self-reported as Black or African American (referred to throughout as African American/Black) or White (referred to throughout as non-Hispanic White). Braak staging (Braak et al., 2006; Braak and Braak, 1991) was completed for all samples. This study was approved by the University of Pittsburgh Institutional Review Board (IRB) and Committee for Oversight of Research and Clinical Training Involving Decedents (CORID).
Table 1.
Cohort characteristics.a
| Characteristics | NHW CN | NHW AD | AA CN | AA AD | Diagnosis p-valueb | Race p-valueb |
|---|---|---|---|---|---|---|
| Sex | 3 F, 2 M | 4 F, 2 M | 0 F, 4 M | 2 F, 3 M | 0.301 | 0.065 |
| Agec | 65 ± 13 | 83 ± 8 | 69 ± 15 | 80 ± 6 | 0.011 | 0.986 |
| Years of Educationd | 17 | 15 ± 4 | 8 | 13 | 0.572 | 0.050 |
| PMIe | 14 ± 9 | 6 ± 2 | 13 ± 9 | 11 ±7 | 0.142 | 0.479 |
| APOE Genotypef | 4 ε3/ε3, 1 ε2/ε2 | 5 ε3/ε3, 1 ε3/ε4 | 1 ε3/ε3, 3 N/A | 1 ε3/ε3, 2 ε3/ε4, 2 ε4/ε4 | – | – |
| Aβ A4 Positive | 4 N 1 Y, rare |
6 Y | 2 N 1 Y, rare 1 Y |
5 Y | – | – |
| Braak Stageg | 1 (0–2) | 4 (4–5) | 1 (0–2) | 5 (4–5) | < 0.0001 | 0.698 |
Values for each group are given as average ± standard deviation, unless otherwise noted.
p-values were calculated using two-factor ANOVAs with replication; bold indicates p < 0.05.
Age in years.
Years of education was not available for all individuals. Averages and standard deviations were calculated from available samples for each group (NHW CN: N = 2; NHW AD: N = 6; AA CN: N = 1; AA AD: N = 2).
Postmortem interval (PMI), in hours.
N/A indicates genotype was unavailable.
Average Braak stage (range). Abbreviations: NHW, non-Hispanic White; AA, African American/Black.
2.2. Sample preparation
Brain tissue (20 mg) was homogenized in 1 × phosphate-buffered saline (PBS) with 8 M urea. Briefly, tissues were homogenized with Lysing Matrix A at 4.0 m/s for 20 s using a FastPrep-24™ 5G system (MP Biomedicals). After homogenization, 1 × PBS with 8 M urea, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 0.3 μM aprotinin were added. Homogenate was centrifuged at 4 °C, 13,000 rpm for 15 min and supernatant was collected. Protein concentration was determined using bicinchoninic acid (BCA) assay according to the manufacturer’s protocols (Thermo Fisher Scientific). A pooled sample containing equimolar amounts of protein from the 18 (hippocampus/GP) or 19 (IPL) samples was generated and served as a quality control (QC). Samples were randomized into two batches of 10 (hippocampus/GP) or 11 (IPL), including at least one QC per batch, and were processed separately (Supplementary Note). Protein (100 μg) was placed in 50 mM Tris with 8 M urea and was reduced for 30 min using 25 mM dithiothreitol at 37 °C. Protein was subsequently alkylated with 25 mM iodoacetamide for 30 min on ice in the dark and quenched with 25 mM L-cysteine for 30 min with shaking. Samples were diluted to 1 M urea with 20 mM Tris, 10 mM CaCl2 prior to digestion with trypsin/Lys-C mix (Promega) for 6–8 h at 37 °C (1:50 enzyme:protein ratio). Peptides were acidified with formic acid (FA) and desalted with an HLB cartridge (Waters Corporation; 1 cc/10 mg). TMT10-plex (hippocampus/GP) or TMT11-plex (IPL) reagents were used to label 25 μg of each sample. Each batch mixture was desalted and fractionated using a gradient of acetonitrile at pH 10 to generate 12 fractions (2, 4, 6, 8,10, 12, 14,16, 20, 25, 35, and 50%). All fractions were analyzed individually via LC-MS3 on an Orbitrap Fusion Lumos (Thermo Fisher Scientific) with technical duplicates. Fractions were injected in a randomized order.
2.3. LC-MS3 parameters
An UltiMate 3000 RSLCnano system (Thermo) was coupled to an Orbitrap Fusion Lumos mass spectrometer operated in positive mode. Peptides were loaded onto a self-packed C18 trap column (100 μm i.d. x 2.5–2.6 cm, 200 Å, 5 μm; Bruker) prior to separation on an in-house C18 packed column (100 μm i.d. x 20 cm, 100 Å, 2.5 μm; Waters) over the following 100 min gradient: 0–7 min, 10% B; 7–67 min, 10–30% B; 67–75 min, 30–60% B; 75–77 min, 60–90% B; 77–82 min, 90% B; 82–83 min, 90–10% B; 83–100 min, 10% B. Mobile phase A was 0.1% FA and mobile phase B was 0.1% FA in acetonitrile. Full MS spectra were collected in the Orbitrap (375–1500 m/z, 120,000 resolution, automated gain control (AGC) 4.0E5, maximum injection time 50 ms). The instrument was operated in data-dependent acquisition (DDA) mode to acquire the top 7 MS/MS spectra in the ion trap using collision-induced dissociation (CID; normalized collision energy 35%, isolation width 0.7 m/z, AGC 1.0E4) and dynamic exclusion of 20 s. Synchronous precursor selection (SPS) mode was used to select the top 8 most intense ions from each MS/MS spectrum for MS3 in the Orbitrap using higher-energy collisional dissociation (HCD; 100–400 m/z, normalized collision energy 55%, resolution 60,000, AGC 5.0E4, maximum injection time 118 ms, isolation width 2 m/z).
2.4. Data analysis
RAW files were analyzed using Proteome Discoverer software (version 2.2). All technical replicates and fractions for each batch were combined into one result file and searched against the UniProt human reviewed protein database (hippocampus: 03/22/2018, 20,259 sequences; IPL/GP: 06/25/2018, 20,302 sequences) using SEQUEST-HT. The following modifications were included in this search: fixed modification of cysteine carbamidomethylation and variable modifications of methionine oxidation and TMT10-plex (229.163 Da) on lysine residues and peptide N-termini for hippocampus and GP and both TMT10-plex and TMT11-plex (229.169 Da) for IPL. A maximum of two trypsin miscleavages were allowed in the search.
TMT10-plex (hippocampus/GP) or TMT11-plex (IPL) was set as the quantification method in Proteome Discoverer, and reporter ion quantitation was based on intensity with a reporter signal-to-noise threshold of 10. Protein groups identified are referred to as proteins throughout. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/) via the PRIDE (Vizcaíno et al., 2016) partner repository with the dataset identifiers PXD012114 (hippocampus), PXD014372 (IPL), and PXD014371 (GP).
Processing of hippocampus, IPL, and GP data can be found in Supplementary Data 1–3, respectively. Peptides were filtered to only include those identified with high confidence (< 1% false discovery rate, FDR) and their corresponding proteins. This list of proteins was further filtered by requiring two peptide spectral matches (PSMs) for a protein identification. Post-analysis filtering was performed to only include proteins identified in both batches with reporter ion intensities in ≥80% of samples (≥ 16 of 20 TMT channels for 10-plex or ≥ 17 of 22 for 11-plex), which must include all pooled channels and ≥ 3 samples per group. These proteins were considered to be the quantified proteins from each region. TMT reporter ion intensities of quantified proteins were normalized using a modified two-step process involving within-batch and across-batch normalization (Supplementary Figs. 1–2) (Plubell et al., 2017). The within-batch normalization was based on the intensity of the pooled sample instead of the average across TMT channels, and we adapted the across-batch normalization to having one pool instead of two in most batches.
Differentially-expressed proteins were identified separately for each region. Fold-change cutoffs of < 0.81 and > 1.24 between AD and CN groups were established based on technical and biological variation and level of technical and biological replication (Cao et al., 2014). Main effects of diagnosis on protein intensities were assessed using linear regression with models stratified by race. Further, a race x diagnosis interaction term assessed whether race modifies the association between diagnosis and protein intensities. P-values were corrected for the number of proteins tested within each brain region using the FDR procedure. However, use of corrected p-values resulted in no significant proteins (likely due to small sample size); therefore, throughout this manuscript, differentially-expressed proteins refers to those with uncorrected p-values < 0.05. Additionally, unadjusted R2 values were pulled from the main effects models of race, diagnosis, and race + diagnosis covarying for demographic variables (age, sex, and postmortem interval (PMI)), to assess the additional variance explained by these terms above and beyond demographic variables. Within each region, proteins with coefficients of variation (CVs) greater than two standard deviations from the mean were excluded (CV > 0.49, 0.34, 0.61 in hippocampus, IPL, and GP, respectively; Supplementary Fig. 3). Ingenuity Pathway Analysis (IPA) was used to identify significant biological pathways (p < 0.05). TMT reporter ion intensities for differentially-expressed proteins were uploaded into ClustVis to generate heatmaps and cluster data (https://biit.cs.ut.ee/clustvis/) (Metsalu and Vilo, 2015).
2.5. Western blots
Three samples from each of the four sample groups were randomly selected for verification by Western blot. Protein was fractionated by SDS-PAGE (120 V loading, 160 V for ~80 min). Proteins were transferred to a nitrocellulose membrane using a wet transfer at 100 V for 70 min. After incubation with 5% nonfat milk in Tris-buffered saline with Tween-20 (TBST; 50 mM Tris, 150 mM NaCl, pH 7.4, 0.1% Tween-20) for 30–60 min, the membrane was washed 4 × 4 min with TBST and incubated overnight with antibodies against calcium/calmodulin dependent protein kinase IIα (CAMKIIα; Thermo MA1-048; 1:5000), peroxiredoxin-2 (PRDX2; abcam ab109367; 1:10,000), or fatty acid-binding protein, heart isoform (H-FABP; Hycult Biotech HM 2016; 1:1000) at 4 °C. Membrane was washed 4 × 4 min with TBST and incubated with a 1:10,000 dilution of fluorescent-labeled anti-mouse (StarBright Blue 700; Bio-Rad Laboratories) or anti-rabbit (IRDye 800CW; Li-Cor Biosciences) secondary antibodies for 30–60 min. For β-actin blots, membrane was incubated with a rhodamine-conjugated anti-β-actin antibody (Bio-Rad 12,004,164; 1:10,000) overnight (no secondary antibody necessary). Blots were washed 4 × 4 min with TBST prior to imaging using a ChemiDoc MP imaging system (Bio-Rad). ImageLab software (Bio-Rad, version 6.0) was used for band quantification.
3. Results
Postmortem hippocampus, IPL, and GP tissues were obtained from the University of Pittsburgh ADRC from African American/Black and non-Hispanic White, CN and AD individuals (Table 1; see Online Methods). The grouping of CN and AD is consistent with disease diagnosis at autopsy, Braak staging, and Aβ staining. Because we were limited by brains from the African American/Black groups, the non-Hispanic White groups were closely-matched based on age and sex to the African American/Black groups. We note that CN individuals were younger than those with AD.
3.1. Characterization of dataset
Brain samples were analyzed using a discovery-based quantitative proteomics workflow (Fig. 1a). The numbers of proteins (peptides) identified from hippocampus, IPL, and GP based on 1% FDR and ≥ 2 PSMs were 1883 (8764), 2055 (9071), and 1656 (9891), respectively. Overall, 2613 total unique proteins were identified across the regions with 1229 common in all three regions (Fig. 1b). These identifications were then filtered to include proteins observed in both TMT batches and with reporter ion signal in ≥80% of the TMT channels (including all pools and ≥ 3 per sample group). The numbers of quantified proteins identified from hippocampus, IPL, and GP were 1414, 1487, and 1173 quantified proteins, respectively. This gave a total of 1801 quantified proteins, 943 of which were common in all three regions (Fig. 1c). The greatest overlap in total and quantified protein identifications was between hippocampus and IPL (Fig. 1b–c). Trends in protein expression for selected proteins (β-actin, PRDX2, CAMKIIα, H-FABP) were verified by Western blots and generally supported MS data (Supplementary Fig. 4).
Fig. 1.
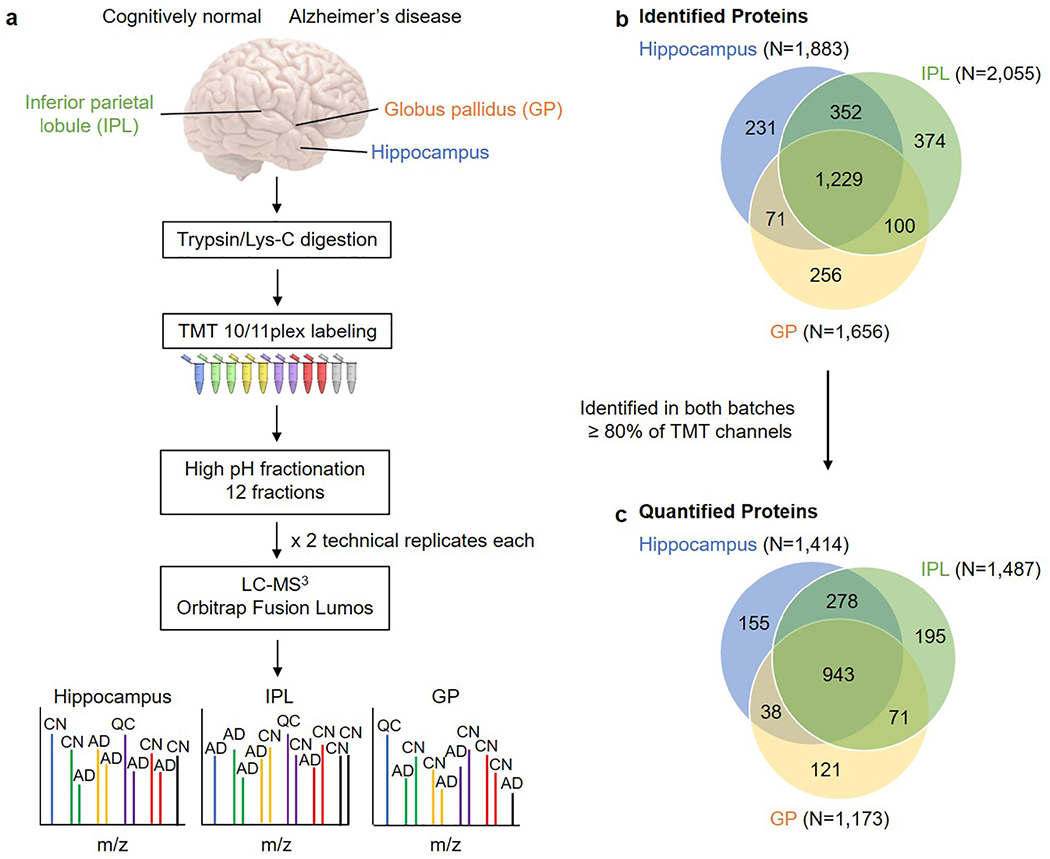
Workflow and summary of identified and quantified proteins by region. a, Experimental workflow used in this study. Samples for each region were randomized into two batches such that each batch contained one pool (QC) and at least one individual from each of the four sample groups. The colored tubes indicate the different TMT11-plex reagents. b, Overlap of identified proteins across the three brain regions. c, Overlap of quantified proteins across the three brain regions. Quantified proteins were identified in both batches with TMT quantification data for ≥ 80% of TMT channels across batches. Brain image modified from “Human brain on white background” by _DJ_ used under CC BY-SA 2.0. Abbreviations: TMT, tandem mass tags; IPL, inferior parietal lobule; GP, globus pallidus; CN, cognitively normal; AD, Alzheimer’s disease; QC, quality control.
3.2. Differentially-expressed proteins by region
Quantified proteins (see Online Methods) were used to assess differences in AD relative to CN individuals within each brain region (implied hereafter). Most differentially-expressed proteins were region-specific, leading to a total of 568 differentially-expressed proteins in AD (Fig. 2a). In these 568 differentially-expressed proteins, covariates (age, sex, PMI, and race) explained 43.42 ± 10.13% of variance in protein intensity, and diagnosis explained an additional 3.24 ± 4.01% of variance in protein intensity above and beyond covariates. In hippocampus, two main clusters of differentially-expressed proteins were observed: a smaller cluster that appears to be mostly increased in AD individuals and a larger cluster decreased in AD individuals (Fig. 3). Individuals cluster into either an AD group or an admixed CN group that also includes a few AD cases. Heatmap analysis showed similar clustering of AD and CN groups in IPL and GP (data not shown). This suggests that the proteomes for some of the AD individuals are more similar to CN individuals. It is important to note that these neuropathologically diagnosed AD individuals that were clustered with the CN group included non-Hispanic White adults and an African American/Black adult, and two of these AD individuals clustered with the CN group in more than one region. Racial subgroups were not clustered within the AD or CN groups from heatmap analysis, likely due to the small sample size.
Fig. 2.
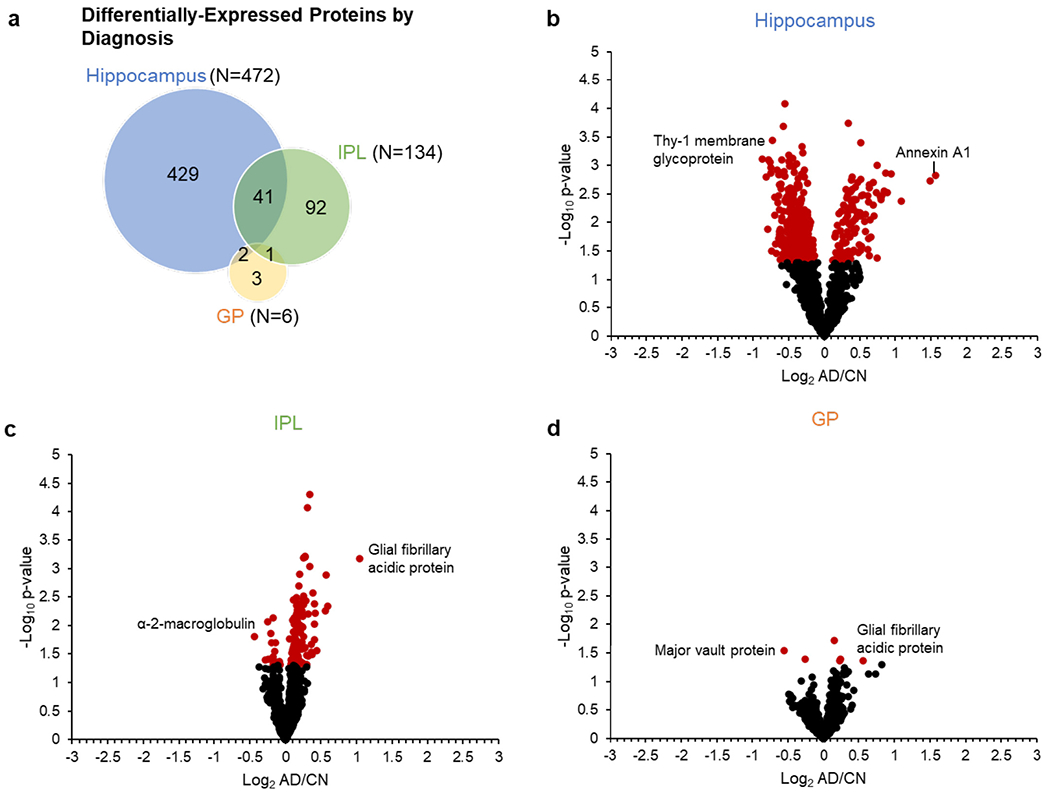
Differentially-expressed proteins by diagnosis in each region. a, Venn diagram showing the regional overlap in differentially-expressed proteins by diagnosis and corresponding volcano plots for b, hippocampus, c, IPL, and d, GP. CV-filtered quantified proteins are shown for each region (hippocampus N = 1338; IPL N = 1407; GP N = 1103). Red data points indicate differentially-expressed proteins with uncorrected p-value < 0.05; black data points indicate quantified proteins with nonsignificant p-values. Selected proteins with significant changes in AD are highlighted in each plot. Abbreviations: IPL, inferior parietal lobule; GP, globus pallidus; AD, Alzheimer’s disease; CN, cognitively normal. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
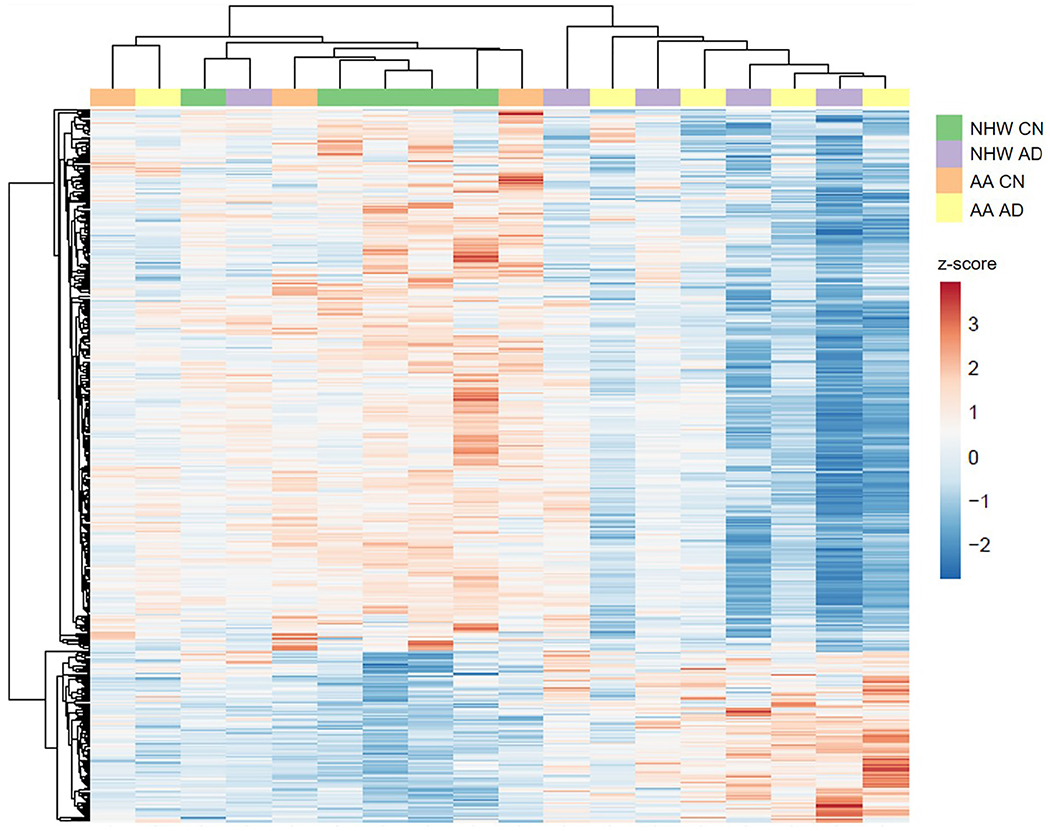
Heatmap with clustering of proteins in hippocampus. ClustVis was used to create a heatmap from the TMT reporter ion intensities for the differentially-expressed proteins in hippocampus (N = 472) across individuals (N = 18; see (Metsalu and Vilo, 2015)). The columns correspond to the individuals while the rows correspond to the proteins. Rows are centered; unit variance scaling is applied to rows. Both rows and columns are clustered using correlation distance and average linkage. The proteins corresponding to the heatmap can be found in Supplementary Data 4. Abbreviations: AA, African American/Black; CN, cognitively normal; NHW, non-Hispanic White; AD, Alzheimer’s disease.
Hippocampus had the most differentially-expressed proteins of the three regions in this study (N = 472; Fig. 2b, Supplementary Table 1), consistent with others,(Xu et al., 2019) with most proteins (N = 359) decreased in AD. Fewer differentially-expressed proteins were observed in IPL (N = 134; Fig. 2c, Supplementary Table 2), most of which (N = 118) were increased in AD, opposite of hippocampus. Only six proteins were differentially expressed in GP, consistent with less noted pathological hallmarks in this region (N = 6; Fig. 2d, Supplementary Table 3). Of the 568 total differentially-expressed proteins, none consistently changed across all regions, though 44 changed in two regions (Fig. 2a). For example, glial fibrillary acidic protein (GFAP) was one of the most robust differentially-expressed proteins across regions and was significantly increased in AD in IPL and GP (Fig 2c–d) as previously reported (Hondius et al., 2016; Begcevic et al., 2013). GFAP was the only differentially-expressed protein shared between IPL and GP, and was significantly increased in hippocampus as well but was filtered out due to a high CV within the AD group. The two proteins that were differentially expressed in both hippocampus and GP were methanethiol oxidase (selenium-binding protein 1), which was increased in AD as in previous work, (Begcevic et al., 2013) and protein FAM49A, which was slightly decreased in AD and has not been reported previously. Forty-one proteins were differentially-expressed in both hippocampus and IPL, including α-2 macroglobulin, glutathione S-trans-ferases Mu 3 and P, peroxiredoxin-1, and annexin A5, which have been reported previously (Zahid et al., 2014; Hondius et al., 2016; Begcevic et al., 2013; Musunuri et al., 2014). The majority of these proteins changed similarly across both regions. However, α-2 macroglobulin was decreased in AD in hippocampus while increased in AD in IPL.
On the other hand, the majority of differentially-expressed proteins differed across regions (Fig. 2a). Hippocampus had the most unique differentially-expressed proteins (N = 429) of the three regions. Example proteins unique to hippocampus include H-FABP, CAMKIIα, PRDX2, annexin A1, thy-1 membrane glycoprotein, α-synuclein, and multiple subunits of hemoglobin, as well as proteins involved in metabolism. IPL also had a significant proportion of unique differentially-expressed proteins (N = 92) including α-enolase, peroxiredoxin-6, acetyl-CoA acetyltransferase, and the brain isoform of fatty acid-binding protein. GP had only three differentially-expressed proteins-elongation factor 2, proteasome subunit α type-3, and major vault protein-none of which have been previously reported in AD.
It is important to note that some of these proteins are blood-derived, including α-2 macroglobulin and the various hemoglobin isoforms. Though it is possible that blood contamination of these brain samples occurred (as is common with human postmortem tissues), several of these proteins have been observed as differentially-expressed in AD brain in prior studies (Zahid et al., 2014; Begcevic et al., 2013). Furthermore, the presence of these blood-derived proteins in the brain could be due to blood-brain barrier leakage and breakdown known to occur with aging and various forms of dementia including AD (Halliday et al., 2016; Nelson et al., 2016).
3.3. Significant pathways in each brain region
The most significant pathways in AD in were mostly region-specific (Supplementary Fig. 5). Of the top 10 most significant pathways shared in multiple regions, 14–3-3-mediated signaling, was the only one in both hippocampus and GP regions. In hippocampus, the most significant pathways include mitochondrial dysfunction, oxidative phosphorylation, synaptogenesis and cell junction signaling (Supplementary Fig. 5; Supplementary Table 7). In IPL, the most significant pathways were related to oxidative stress or metabolism, including gluconeogenesis, glycolysis, glycogen degradation, and xenobiotic metabolism (Supplementary Fig. 5; Supplementary Table 7). In GP, the three differentially-expressed proteins represented diphthamide biosynthesis, 14–3-3-mediated signaling, and p70S6K signaling pathways (Supplementary Fig. 5; Supplementary Table 7).
3.4. Proteins with significant race x diagnosis interactions
Next, we evaluated if self-reported race had an impact on protein changes in AD. We examined the overlap between the differentially-expressed proteins in each region in all AD compared to CN individuals and the differentially-expressed proteins in a race-stratified analysis between only African American/Black AD compared to CN individuals (Supplementary Fig. 6). In hippocampus and IPL, about 20% of differentially-expressed proteins were common between the combined and African American/Black race-stratified comparisons, while in GP, no proteins overlapped between the two comparisons. However, in all regions, there were also proteins (N = 24, 78, 46 in hippocampus, IPL, and GP, respectively) that were differentially-expressed between African American/Black AD and CN groups but were not differentially-expressed in the combined analysis, many of which were decreased in AD in hippocampus and IPL and increased in AD in GP (Supplementary Fig. 6). This suggests that some protein changes in AD would not be detected without examining multiple racial/ethnic groups.
We used linear regression models with a race x diagnosis interaction term to determine whether race modifies the association between diagnosis and protein intensities in each region, which resulted in 185 proteins with significant race x diagnosis interactions (Fig. 4). There were 27, 60, and 105 proteins with significant race x diagnosis interactions in hippocampus, IPL, and GP, respectively (Supplementary Tables 4–6). Seven proteins with significant interactions overlapped in hippocampus and IPL (Fig. 4). Example proteins are highlighted in Fig. 4. In hippocampus, heteronuclear ribonucleoprotein D0 increased in non-Hispanic White adults with AD and had no change in African American/Black adults. In IPL, heteronuclear ribonucleoprotein D0 decreased in African American/Black adults with AD and had no change in non-Hispanic White adults (Fig. 4). Other examples of proteins that differed in the African American/Black and non-Hispanic White AD groups are shown in Fig. 4, for α-2 macroglobulin, α-synuclein, and fructose-bisphosphate aldolase A.
Fig. 4.
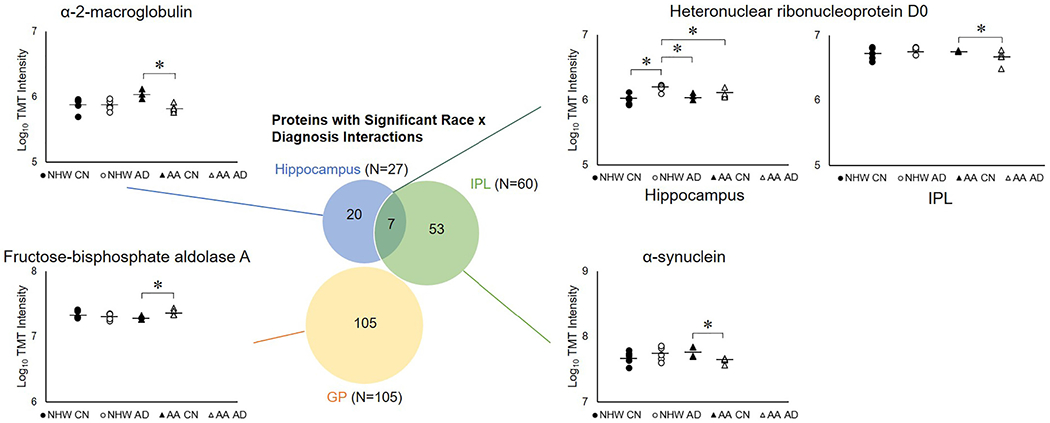
Proteins with significant race x diagnosis interactions in each region. Venn diagram showing the regional overlap in proteins with significant race x diagnosis interactions (p < 0.05), and plots showing examples of these proteins in different regions: heteronuclear ribonucleoprotein D0, α-2-macroglobulin, α-synuclein, and fructose-bisphosphate aldolase A. The plots show log10 TMT intensities plotted for each brain sample in the relevant region. Each data point represents one individual; brains of non-Hispanic White CN adults are filled circles, brains of non-Hispanic White AD adults are open circles, brains of African American/Black CN adults are filled triangles, and brains of African American/Black AD adults are open triangles. Horizontal lines indicate group averages (N = 5 per group, except N = 3 for hippocampus from African American/Black CN adults, N = 4 for IPL and GP African American/Black CN and GP African American/Black AD adults). * indicates p < 0.05. Abbreviations: AA, African American/Black; CN, cognitively normal; NHW, non-Hispanic White; AD, Alzheimer’s disease; IPL, inferior parietal lobule; GP, globus pallidus.
Next, we compared proteins (N = 185) with significant race x diagnosis interactions to those significant from the race-stratified linear regression models for the effect of diagnosis (Table 2). A subset of these proteins were significant in AD in at least one of the race-stratified comparisons: 20 in hippocampus, 39 in IPL, and 40 in GP. Interestingly, most of these proteins changed in AD in one racial/ethnic group and not the other.
Table 2.
Overlap of differentially-expressed proteins in AD with ROS/MAP TMT dataset.
| Brain region | By diagnosis in everyone |
By diagnosis in NHWs |
By diagnosis in AAs |
With Race x diagnosis interaction |
||||
|---|---|---|---|---|---|---|---|---|
| Significant in this study | Overlap with ROS/MAP | Significant in this study | Overlap with ROS/MAP | Significant in this study | Overlap with ROS/MAP | Significant in this study | Overlap with ROS/MAP | |
| Hippocampus | 472 | 155 | 199 | 33 | 114 | 2 | 20 | 0 |
| IPL | 134 | 59 | 86 | 33 | 105 | 3 | 39 | 1 |
| GP | 6 | 3 | 23 | 2 | 45 | 3 | 40 | 3 |
Abbreviations: NHW, non-Hispanic White; AA, African American/Black.
3.5. Comparison to the Religious Orders Study and the Memory and Aging Project (ROS/MAP)
Additionally, we compared the differentially-expressed proteins in our study to a TMT dataset of N = 375 dorsolateral prefrontal cortex samples from the Religious Orders Study and the Memory and Aging Project (ROS/MAP), composed of African American/Black (CN, N = 5 and AD, N = 1) and non-Hispanic White adults (CN, N = 151; MCI N = 90; and AD, N = 120). We performed linear regression analyses and identified 495 significant (corrected p < 0.05) proteins in AD. Comparison of these proteins with differentially-expressed proteins in this study (Fig. 2) resulted in 199 overlapping proteins (Table 2), highlighting the consistency and relevance of our findings. It is apparent that most of our overlap with the ROS/MAP dataset occurred in the non-Hispanic White group even despite different brain regions (i.e., hippocampus, IPL, GP vs. prefrontal cortex). Notably, the published ROS/MAP dataset included only a single African American/Black AD case, further emphasizing the value of the current dataset despite our limited sample size. Given the number of signals identified in our non-Hispanic White-stratified analysis that were confirmed in the larger non-Hispanic White dataset of ROS/MAP, it is likely that many of the novel signals identified in our African American/Black stratified analysis would show similar consistency if a larger African American/Black replication sample were available. Clearly there is a pressing need to increase representation in proteomic analyses of the AD brain.
4. Discussion
Our study identified 2613 total proteins in hippocampus, IPL, and GP which is on par with other proteomics studies of mostly hippocampus and temporal lobe (Xu et al., 2019; Manavalan et al., 2013; McKetney et al., 2019; Hondius et al., 2016; Begcevic et al., 2013; Schrotter et al., 2017; Musunuri et al., 2014; Martins-de-Souza et al., 2014; Xu et al., 2016a; Xu et al., 2016b; Liu et al., 2017; Fernández-Irigoyen et al., 2014). Despite the limited number of brain samples included in this study, 568 total proteins were found to be differentially expressed in AD across hippocampus, IPL, and GP. Hippocampus is the most severely affected brain region in AD and, as such, is previously noted to have substantial protein expression changes (Xu et al., 2019). Our data are consistent with this observation. Furthermore, the 568 differentially-expressed proteins in AD from our study, when compared to 709 differentially- expressed proteins compiled from the literature, (Manavalan et al., 2013; Zahid et al., 2014; Hondius et al., 2016; Begcevic et al., 2013; Musunuri et al., 2014; Qi et al., 2007; Minjarez et al., 2016; Andreev et al., 2012; Sultana et al., 2007b; Butterfield and Lange, 2009; Bai et al., 2020; Wang et al., 2020; Haytural et al., 2020; Vlkolinsky et al., 2001) reveals an overlap of 217 proteins. Most of the differentially-expressed proteins in our study were region-specific with none observed in all regions and only 44 differentially expressed in two regions, most of which were common between hippocampus and IPL (Fig. 2a). Many of these protein changes have been previously reported in AD brain (Zahid et al., 2014; Hondius et al., 2016; Begcevic et al., 2013; Musunuri et al., 2014). Aβ A4 protein was not differentially expressed in any of the regions in our study; it was removed from the analysis in hippocampus and IPL due to high within-group CVs, while it was measured but not differentially expressed in GP. Aβ42 accumulation has been reported in GP neurons, (Baker-Nigh et al., 2015) although Aβ accumulation may not be substantial enough for proteomic differences between AD and CN individuals to be detected. Microtubule-associated protein tau was measured in all three regions, but was not differentially expressed in any region or racial group.
It is important to note that existing human AD brain proteomics literature includes studies with a variety of sample sizes (N = 350 - ≥ 90090) and brain regions such as frontal lobe, (Seyfried et al., 2017; Ping et al., 2018; Zahid et al., 2014; McKetney et al., 2019; Minjarez et al., 2016; Bai et al., 2020; Wang et al., 2020; Johnson et al., 2020; Johnson et al., 2018; Zhang et al., 2018; Mendonça et al., 2019; Adav et al., 2019; Bereczki et al., 2018; Ping et al., 2020) temporal lobe (including hippocampus), (Xu et al., 2019; Manavalan et al., 2013; Zahid et al., 2014; McKetney et al., 2019; Hondius et al., 2016; Begcevic et al., 2013; Musunuri et al., 2014; Andreev et al., 2012; Sultana et al., 2007b; Haytural et al., 2020; Johnson et al., 2020; Mendonça et al., 2019; Xiong et al., 2019) and IPL (McKetney et al., 2019). None included nonpathological regions such as GP. Our study aligns with ~61% of these publications that have had cohorts of N ≤ 20 individuals, (Xu et al., 2019; Manavalan et al., 2013; Zahid et al., 2014; McKetney et al., 2019; Begcevic et al., 2013; Musunuri et al., 2014; Minjarez et al., 2016; Andreev et al., 2012; Sultana et al., 2007b; Wang et al., 2020; Haytural et al., 2020; Adav et al., 2019; Xiong et al., 2019) resulting in group sizes of N = 1–10. Differentially-expressed proteins in our study (N = 197 and 56, respectively) overlapped with studies of both small (N = 4–20 (Manavalan et al., 2013; Zahid et al., 2014; Begcevic et al., 2013; Musunuri et al., 2014; Minjarez et al., 2016; Andreev et al., 2012; Sultana et al., 2007b; Wang et al., 2020; Haytural et al., 2020)) and large (N = 40–201 (Hondius et al., 2016; Bai et al., 2020)) sample sizes. Most of these studies also do not include racial/ethnic diversity of participants. For example, one report exclusively studied Mexican (Minjarez et al., 2016) and another Japanese (Manavalan et al., 2013) adults, while a few included African American/Black, Hispanic, and Native Hawaiian or other Pacific Islander groups representing 11–13% of study participants (Seyfried et al., 2017; Ping et al., 2018; Ping et al., 2020). Therefore, these consistent findings increase the confidence of this study.
However, a majority of differentially-expressed proteins (i.e., 351 proteins) in our study compared to previous reports (Manavalan et al., 2013; Zahid et al., 2014; Hondius et al., 2016; Begcevic et al., 2013; Musunuri et al., 2014; Qi et al., 2007; Minjarez et al., 2016; Andreev et al., 2012; Sultana et al., 2007b; Butterfield and Lange, 2009; Bai et al., 2020; Wang et al., 2020; Haytural et al., 2020; Vlkolinsky et al., 2001) were novel. This is likely due to both the inclusion of a diverse cohort and brain regions: IPL and GP. IPL and GP have been studied in AD, (Reed et al., 2009; Hensley et al., 1995; Dalle-Donne et al., 2003; Newman et al., 2007; Wang et al., 2015; Greene and Killiany, 2010; Triplett et al., 2016; Lehéricy et al., 1991; Castegna et al., 2002; Jacobs et al., 2012; Foerde and Shohamy, 2011; Packard and Knowlton, 2002) but not in the context of global proteomics analyses. Furthermore, proteomic changes from African American/Black adults or other racial/ethnic AD groups in the U.S. are not well-characterized. There were proteins (N = 24, 78, 46 in hippocampus, IPL, and GP, respectively) that were differentially expressed in AD when evaluating only the African American/Black group that were not differentially-expressed in the combined analysis of both racial groups (Supplementary Fig. 6). This underscores the need for racial/ethnic diversity in AD cohorts and ‘omics studies. We also compared the differentially-expressed proteins in our dataset to an existing ROS/MAP TMT dataset from the dorsolateral prefrontal cortex region and identified substantial overlap (Table 2). It is notable that substantial overlap was observed despite differences in brain regions and geography between the two datasets. Also, this overlap was higher in the non-Hispanic White group than in the African American/Black group. Additionally, 52% of proteins with significant race x diagnosis interactions also had significant race-stratified changes in AD in one or both racial groups, the majority of which only had significant changes in AD in one racial group but not the other. This could be because the smaller N is more sensitive to heterogeneous changes in AD that could be neutralized in larger groups. However, it is also possible based on our consistent findings with ROS/MAP TMT data that disease pathogenesis is more heterogeneous at the proteome level, which highlights the need to conduct larger studies that include diverse participants.
4.1. Study strengths and limitations
The most important strength of our study is the inclusion of brain samples from both African American/Black and non-Hispanic White adults. African American/Black adults and adults from other racial/ethnic minorities are highly underrepresented in brain proteomics studies in AD. This is likely due to the need for increased research participation and lower likelihood of some individuals to consent to autopsy to provide brain tissue samples (Bonner et al., 2000; Barnes et al., 2012; Siminoff et al., 2006). Furthermore, African American/Black and Hispanic adults are at increased risk for AD, making molecular understanding of AD pathogenesis in those groups particularly vital. While we did not study Hispanic or Asian American adults, or adults from other racial/ethnic groups here, we suggest that racial/ethnic diversity be included in future ‘omics study designs. However, such studies when including components of race must also consider other social and environmental factors that impact physiological and biological changes (Wilkins et al., 2020). Another strength of our study is that the brain samples from both racial groups and disease states were analyzed within the same experiments. This sample multiplexing minimized error and enabled direct comparison of relative protein abundances across groups. Most brain samples came from the same ADRC, minimizing any potential differences in handling across centers. Although there could be zip code differences, all brains came from the same geographical area.
The inclusion of multiple brain regions in this study is a strength because AD has spatial effects in the brain (Xu et al., 2019; McKetney et al., 2019; Ray and Zhang, 2010). The hippocampus is one of the earliest regions affected in AD (Smith, 2002; Halliday, 2017; West et al., 2000) making it a valuable region to study disease pathogenesis. Furthermore, IPL and GP are not well-studied in AD using proteomics. Since little is known about proteomic changes in these regions, including them in our study is particularly valuable, especially given the potential role of GP in memory. This study adds to the available proteomics literature with new differentially-expressed proteins for disease insight and confirms others from previous studies. Furthermore, where available, the three brain regions were collected from the same individual, which additionally allows regional comparisons within individuals as well as across individuals and groups.
Proteome depth is greatly influenced by front-end LC separations and MS duty cycles. We used a high pH fractionation approach on a solid-phase extraction cartridge to generate 12 fractions and note that this may have limited our total number of proteins identified compared to column high pH separations and the collection of 24–100 fractions. The inclusion of MS3 in our proteomics workflow can be viewed as a limitation because MS3 leads to fewer identified proteins and thus potentially fewer quantified proteins due to longer instrumental duty cycles. However, MS3 also leads to more robust quantitative measurements from TMT reporter ions, which was vital to this pilot study in order to accurately detect differences between disease states and assess impact of covariates (race, diagnosis, age, sex, PMI) (McAlister et al., 2014; Ting et al., 2011).
The main challenge of our study was limited sample availability from African American/Black adults, which in turn limited the statistical power of this study. Postmortem brain samples from African American/Black adults are difficult to obtain, particularly from CN individuals, partially because African American/Black adults are less likely to consent to autopsy than non-Hispanic White adults (Bonner et al., 2000; Barnes et al., 2012; Siminoff et al., 2006) and only few centers have been effective in recruitment to brain autopsy programs (Barnes et al., 2012; Williams et al., 2011). Additional brain samples from African American/Black adults could have been acquired from other ADRCs; however, we thought it detrimental to combine few and unmatched cases and controls from a given center together as it introduces center effects that can impact proteomics results. At the beginning of our study (12/15/2016), African American/Black adults comprised 8.2% of the clinical AD cases in the University of Pittsburgh ADRC (N = 209 African American/Black adults). However, only five African American/Black adults with AD had brain samples available from the selected regions, along with four African American/Black adults that were CN. Thus, we selected all of these brains for our study, and matched brain samples from non-Hispanic White adults based on age, sex, and diagnosis (N = 55).
The limited availability of samples resulted in some differences across the sample groups, specifically in APOE genotype, age, and sex (Table 1). All groups consisted of equal distribution of males and females except the African American/Black CN group, which was all male. There was no significant difference (p = 0.48, race; p = 0.14, diagnosis) in postmortem interval across groups. The APOE genotypes are noted where available and were not used as matching criteria since data was missing in the African American/Black CN group. AD patients were 15 ± 15 years older than CN individuals (p = 0.010). Despite this age difference, diagnosis explained additional variance in protein intensity beyond the variance explained by the covariates of age, sex, PMI, and race. There is significant overlap between changes in the brain during normal aging and AD pathogenesis (Johnson et al., 2020) and due to the age differences across our sample groups, some of the protein expression changes detected in this study could be driven by age-related processes as opposed to disease-related processes. Years of education did not significantly differ between groups (p = 0.050, race; p = 0.572, diagnosis). However, it is important to note that this data was not available for all individuals, so this study cannot adequately address the contribution of education, as measured by number of years, to the proteomic findings. Importantly, in future studies, education, the scope by which it is measured, and quality of education (Sisco et al., 2015) must be included. Furthermore, records of vascular comorbidities were not available for all participants, such that incidence of vascular diseases may have differed between the groups and contributed to the observed differences. Nonetheless, significant differences across racial/ethnic backgrounds remain. In future studies, it will be important to collaborate with other ADRCs and brain banks that have greater brain sample availability from African American/Black adults. Such sample sizes will greatly increase statistical power and better normalize racial groups in consideration of socioeconomic and other demographic factors.
5. Conclusions
African American/Black adults are disproportionately affected by AD in comparison to non-Hispanic White adults. The molecular basis of this disparity is largely unknown. This pilot study aimed to elucidate molecular pathways that can explain these disparities in postmortem brain tissue from non-Hispanic White and African American/Black adults in a pilot cohort using discovery-based quantitative proteomics. Our study identified many differentially-expressed proteins in AD in hippocampus, IPL, and GP that are consistent with prior AD studies. When race was examined as a covariate, we observed proteins that were differentially-expressed in one racial/ethnic group and not in the other. Overall, our preliminary findings strongly highlight the need for diverse groups especially African American/Black adults to be included in proteomics, and likely other ‘omics, studies, to gain a clear picture of disease pathogenesis. The insights gained from this study stress the point that inclusive study designs are necessary in AD research and at a larger scale.
Supplementary Material
Acknowledgements
The authors would like to thank the University of Pittsburgh ADRC Neuropathology Core and Data Management and Statistics Core (Heather Eng, Rocco Mercurio, Shelley Ferson). The authors acknowledge pilot funds from the University of Pittsburgh Alzheimer DiseaseResearch Center funded by the National Institutes of Health and National Institute on Aging (P50-AG005133), the Vanderbilt Interdisciplinary Training Program in Alzheimer’s Disease (T32-AG058524), the Alzheimer’s Association (AARGD-17-533405), the Vanderbilt Institute of Chemical Biology (T32-GM065086), additional grants from the National Institute on Aging (R01-AG064950, K01-AG049164), and Vanderbilt University Start-Up Funds.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.105129.
Declaration of Competing Interest
The authors have declared no conflicts of interest.
References
- Adav SS, Park JE, Sze SK, 2019. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 12, 8. 10.1186/s13041-019-0430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev VP, et al. , 2012. Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res 11, 3053–3067. 10.1021/pr3001546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association AS, 2017. Alzheimer’s disease facts and figures. Alzheimers Dement. 13, 325–373. [Google Scholar]
- Association AS, 2018. Alzheimer’s disease facts and figures. Alzheimers Dement. 14 (367–429), 2018. 10.1016/j.jalz.2018.02.001. [DOI] [Google Scholar]
- Babulal GM, et al. , 2018. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: update and areas of immediate need. Alzheimers Dement. 10.1016/j.jalz.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, et al. , 2020. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron. 10.1016/j.neuron.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Nigh A, et al. , 2015. Neuronal amyloid-β accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 138, 1722–1737. 10.1093/brain/awv024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LL, 2019. Biomarkers for alzheimer dementia in diverse racial and ethnic minorities—a public health priority. JAMA Neurol. 76, 251–253. 10.1001/jamaneurol.2018.3444. [DOI] [PubMed] [Google Scholar]
- Barnes LL, Bennett DA, 2014. Alzheimer’s disease in African Americans: risk factors and challenges for the future. Health Aff. (Millwood) 33, 580–586. 10.1377/hlthaff.2013.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LL, Shah RC, Aggarwal NT, Bennett DA, Schneider JA, 2012. The minority aging research study: ongoing efforts to obtain brain donation from African Americans without dementia. Curr. Alzheimer Res 9, 734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LL, et al. , 2015. Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology 85, 528–534. 10.1212/wnl.0000000000001834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begcevic I, et al. , 2013. Semiquantitative proteomic analysis of human hippocampal tissues from Alzheimer’s disease and age-matched control brains. Clin. Proteomics 10. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3648498/pdf/1559-0275-10-5.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereczki E, et al. , 2018. Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain 141, 582–595. 10.1093/brain/awx352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Vanmechelen E, Hampel HCSF total tau, 2001. Aβ42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol. Neurobiol 24, 87–97. 10.1385/mn:24:1-3:087. [DOI] [PubMed] [Google Scholar]
- Bonner GJ, Darkwa OK, Gorelick PB, 2000. Autopsy recruitment program for African Americans. Alzheimer Dis. Assoc. Disord 14, 202–208. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. 10.1007/bf00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K, 2006. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke SL, Cadet T, Maddux M, 2017. Chronic health illnesses as predictors of mild cognitive impairment among African American older adults. J. Natl. Med. Assoc 110, 314–325. https://www.sciencedirect.com/science/article/pii/S0027968417300883/>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M, Duff K, 2002. Cholesterol in Alzheimer’s disease and tauopathy. Ann. N. Y. Acad. Sci 977, 367–375. 10.1111/j.1749-6632.2002.tb04839.x. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Lange ML, 2009. Multifunctional roles of enolase in Alzheimer’s disease brain: beyond altered glucose metabolism. J. Neurochem 111, 915–933. 10.1111/j.1471-4159.2009.06397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Yende S, Kellum JA, Angus DC, Robinson RAS, 2014. Proteomics reveals age-related differences in the host immune response to sepsis. J. Proteome Res 13, 422–432. 10.1021/pr400814s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castegna A, et al. , 2002. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem 82, 1524–1532. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, et al. , 2015. Metabolic risk factors of sporadic Alzheimer’s disease: implications in the pathology, pathogenesis and treatment. Aging Dis. 6, 282–299. 10.14336/ad.2014.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin AL, Negash S, Hamilton R, 2011. Diversity and disparity in dementia: the impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis. Assoc. Disord 25, 187–195. 10.1097/WAD.0b013e318211c6c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A, 2003. Protein carbonylation in human diseases. Trends Mol. Med 9, 169–176. 10.1016/S1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- Desai PP, et al. , 2003. Genetic variation in apolipoprotein D affects the risk of Alzheimer disease in African-Americans. Am. J. Med. Genet. B Neuropsyehiatr. Genet 98–101. 10.1002/ajmg.b.10798. 116B [DOI] [PubMed] [Google Scholar]
- El Gaamouch F, Jing P, Xia J, Cai D, 2016. Alzheimer’s disease risk genes and lipid regulators. J. Alzheimers Dis 53, 15–29. 10.3233/JAD-160169. [DOI] [PubMed] [Google Scholar]
- Fernández-Irigoyen J, Zelaya MV, Tuñon T, Santamaría E, 2014. Anatomo-proteomic characterization of human basal ganglia: focus on striatum and globus pallidus. Mol. Brain 7. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4236423/>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filshtein TJ, et al. , 2019. Neuropathological diagnoses of demented Hispanic, black, and non-Hispanic white decedents seen at an Alzheimer’s disease center. J. Alzheimers Dis 68, 145–158. 10.3233/JAD-180992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerde K, Shohamy D, 2011. The role of the basal ganglia in learning and memory: insight from Parkinson’s disease. Neurobiol. Learn. Mem 96, 624–636. 10.1016/j.nlm.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba P, et al. , 2012. The link between altered cholesterol metabolism and Alzheimer’s disease. Ann. N. Y. Acad. Sci 1259, 54–64. 10.1111/j.1749-6632.2012.06513.x. [DOI] [PubMed] [Google Scholar]
- Garrett SL, et al. , 2019. Racial disparity in cerebrospinal fluid amyloid and tau biomarkers and associated cutoffs for mild cognitive impairment. JAMA Netw. Open 2. 10.1001/jamanetworkopen.2019.17363>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavett BE, et al. , 2018. Ethnoracial differences in brain structure change and cognitive change. Neuropsychology 32, 529–540. https://psycnet.apa.org/record/2018-15740-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghani M, et al. , 2015. Association of long runs of homozygosity with Alzheimer disease among African American individuals. JAMA Neurol. 72, 1313–1323. 10.1001/jamaneurol.2015.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan AM, Malone DC, Warholak TL, Armstrong EP, 2012. Racial and ethnic disparities in Alzheimer’s disease pharmacotherapy exposure: an analysis across four state Medicaid populations. Am. J. Geriatr. Pharmacother 10, 303–312. 10.1016/j.amjopharm.2012.09.002. [DOI] [PubMed] [Google Scholar]
- González HM, et al. , 2019. A research framework for cognitive aging and Alzheimer’s disease among diverse US Latinos: Design and implementation of the Hispanic Community Health Study/Study of Latinos—Investigation of Neurocognitive Aging (SOL-INCA). Alzheimers Dement. 10.1016/j.jalz.2019.08.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman RF, et al. , 2017. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317, 1443–1450. 10.1001/jama.2017.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford NR, Besser LM, Crook JE, Kukull WA, Dickson DW, 2016. Neuropathological differences by race from the National Alzheimer’s coordinating Center. Alzheimers Dement. 12, 669–677. 10.1016/j.jalz.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene SJ, Killiany RJ, 2010. Subregions of the inferior parietal lobule are affected in the progression to Alzheimer’s disease. Neurobiol. Aging 31, 1304–1311. 10.1016/j.neurobiolaging.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday G, 2017. Pathology and hippocampal atrophy in Alzheimer’s disease. Lancet Neurol. 16, 862–864. 10.1016/S1474-4422(17)30343-5. [DOI] [PubMed] [Google Scholar]
- Halliday MR, et al. , 2016. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab 36, 216–227. 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haytural H, et al. , 2020. The proteome of the dentate terminal zone of the perforant path indicates presynaptic impairment in Alzheimer disease. Mol. Cell. Proteomics 19, 128–141. mcp.RA119.001737. https://www.mcponline.org/content/mcprot/early/2019/11/07/mcp.RA119.001737.full.pdf. https://www.mcponline.org/content/mcprot/19/1/128.full.pdf>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, et al. , 1995. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem 65, 2146–2156. 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- Hohman TJ, et al. , 2016. Global and local ancestry in African-Americans: implications for Alzheimer’s disease risk. Alzheimers Dement. 12, 233–243. 10.1016/j.jalz.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondius DC, et al. , 2016. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimers Dement. 12, 654–668. 10.1016/j.jalz.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Howell JC, et al. , 2017. Race modifies the relationship between cognition and Alzheimer’s disease cerebrospinal fluid biomarkers. Alzheimers Res. Ther 9. 10.1186/s13195-017-0315-1>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs HI, Van Boxtel MP, Jolles J, Verhey FR, Uylings HB, 2012. Parietal cortex matters in Alzheimer’s disease: an overview of structural, functional and metabolic findings. Neurosci. Biobehav. Rev 36, 297–309. 10.1016/j.neubiorev.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Johnson ECB, et al. , 2018. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener 13, 52. 10.1186/s13024-018-0282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, et al. , 2020. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 26, 769–780. https://www.nature.com/articles/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamara DM, et al. , 2018. Cerebral amyloid angiopathy: similarity in African-Americans and Caucasians with Alzheimer’s disease. J. Alzheimers Dis 62, 1815–1826. 10.3233/jad-170954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, et al. , 2007. The association between genetic variants in SORL1 and Alzheimer’s disease in an urban, multiethnic, community-based cohort. Arch. Neurol 64, 501–506. 10.1001/archneur.64.4.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehéricy S, Hirsch EC, Hersh LB, Agid Y, 1991. Cholinergic neuronal loss in the globus pallidus of Alzheimer disease patients. Neurosci. Lett 123, 152–155. 10.1016/0304-3940(91)90918-J. [DOI] [PubMed] [Google Scholar]
- Lines L, Sherif A, Wiener J, 2014. Racial and Ethnic Disparities Among Individuals with Alzheimer’s Disease in the United States: A Literature Review, vol. RR-0024-1412 RTI Press. [Google Scholar]
- Liu Q, Zhang J, 2014. Lipid metabolism in Alzheimer’s disease. Neurosci. Bull 30, 331–345. 10.1007/s12264-013-1410-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Guo Z, Liu W, Sun W, Ma C, 2017. Differential proteome analysis of hippocampus and temporal cortex using label-free based 2D-LC-MS/MS. J. Proteome 165, 26–34. 10.1016/j.jprot.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Manavalan A, et al. , 2013. Brain site-specific proteome changes in aging-related dementia. Exp. Mol. Med 45. 10.1038/emm.2013.76>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly JJ, Mayeux R, 2004. In: Anderson NB, Bulatao RA, Cohen B (Eds.), Critical Perspectives on Racial and Ethnic Differences in Health in Late Life. National Academies Press, Washington D.C. [PubMed] [Google Scholar]
- Martins IJ, et al. , 2009. Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J. Neurochem 11, 1275–1308. 10.1111/j.1471-4159.2009.06408.x. [DOI] [PubMed] [Google Scholar]
- Martins-de-Souza D, et al. , 2014. Deciphering the human brain proteome: characterization of the anterior temporal lobe and corpus callosum as part of the chromosome 15-centric human proteome project. J. Proteome Res 13, 147–157. 10.1021/pr4009157. [DOI] [PubMed] [Google Scholar]
- Matsuzaki T, et al. , 2011. Association of Alzheimer disease pathology with abnormal lipid metabolism: the Hisayama study. Neurology 77, 1068–1075. 10.1212/WNL.0b013e31822e145d. [DOI] [PubMed] [Google Scholar]
- Matthews KA, et al. , 2018. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged >65 years. Alzheimers Dement. 10.1016/j.jalz.2018.06.3063>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeda ER, Glymour MM, Quesenberry CP, Whitmer RA, 2015. Inequalities in dementia incidence between six racial and ethnic groups over 14 years. Alzheimers Dement. 12, 216–224. 10.1016/j.jalz.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, et al. , 2014. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158. 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKetney J, et al. , 2019. Proteomic atlas of the human brain in Alzheimer’s disease. J. Proteome Res 18, 1380–1391. 10.1021/acs.jproteome.9b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta KM, Yeo GW, 2017. Systematic review of dementia prevalence and incidence in United States race/ethnic populations. Alzheimers Dement. 13, 72–83. 10.1016/j.jalz.2016.06.2360. [DOI] [PubMed] [Google Scholar]
- Mendonça CF, et al. , 2019. Proteomic signatures of brain regions affected by tau pathology in early and late stages of Alzheimer’s disease. Neurobiol. Dis 130. https://www.sciencedirect.com/science/article/pii/S096999611930169X. [DOI] [PubMed] [Google Scholar]
- Metsalu T, Vilo J, 2015. ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 43, W566–W570. 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minjarez B, et al. , 2016. Identification of proteins that are differentially expressed in brains with Alzheimer’s disease using iTRAQ labeling and tandem mass spectrometry. J. Proteome 139, 103–121. 10.1016/j.jprot.2016.03.022. [DOI] [PubMed] [Google Scholar]
- Morris JC, et al. , 2019. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 76, 264–273. 10.1001/jamaneurol.2018.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Gage FH, 2011. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener 6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3261815/pdf/1750-1326-6-85.pdf>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuri S, et al. , 2014. Quantification of the brain proteome in Alzheimer’s disease using multiplexed mass spectrometry. J. Proteome Res 13, 2056–2068. 10.1021/pr401202d. [DOI] [PubMed] [Google Scholar]
- Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV, 2016. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 1862, 887–900. 10.1016/j.bbadis.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman SF, et al. , 2007. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res 85, 1506–1514. 10.1002/jnr.21275. [DOI] [PubMed] [Google Scholar]
- Packard MG, Knowlton BJ, 2002. Learning and memory functions of the basal ganglia. Annu. Rev. Neurosci 25, 563–593. 10.1146/annurev.neuro.25.112701.142937. [DOI] [PubMed] [Google Scholar]
- Ping L, et al. , 2018. Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s disease. Sci. Data 5. 10.1038/sdata.2018.36>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping L, et al. , 2020. Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer’s disease. bioRxiv 2020. 10.1101/2020.05.19.105197. 2005.2019.105197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plubell DL, et al. , 2017. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell. Proteomics 16, 873–890. 10.1074/mcp.M116.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi JP, et al. , 2007. Cerebral ischemia and Alzheimer’s disease: the expression of amyloid-beta and apolipoprotein E in human hippocampus. J. Alzheimers Dis 12, 335–341. [DOI] [PubMed] [Google Scholar]
- Ray M, Zhang W, 2010. Analysis of Alzheimer’s disease severity across brain regions by topological analysis of gene co-expression networks. BMC Syst. Biol 4. 10.1186/1752-0509-4-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed TT, Pierce WM Jr., Turner DM, Markesbery WR, Butterfield DA, 2009. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J. Cell. Mol. Med 13, 2019–2029. 10.1111/j.1582-4934.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, et al. , 2013. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ε4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492. 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, et al. , 2007. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer’s disease. Nat. Genet 39, 168–177. 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmann H, 2012. CSF biomarkers for amyloid and tau pathology in Alzheimer’s disease. J. Mol. Neurosci 47, 1–14. 10.1007/s12031-011-9665-5. [DOI] [PubMed] [Google Scholar]
- Sato N, Morishita R, 2015. The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci 7. 10.3389/fnagi.2015.00199>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC, 2002. Mapping the evolution of regional atrophy in Alzheimer’s disease: unbiased analysis of fluid-registered serial MRI. Proc. Natl. Acad. Sci. U. S. A 99, 4703–4707. 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrotter A, et al. , 2017. LMD proteomics provides evidence for hippocampus field-specific motor protein abundance changes with relevance to Alzheimer’s disease. Biochim. Biophys. Acta, Proteins Proteomics 1865, 703–714. 10.1016/j.bbapap.2017.03.013. [DOI] [PubMed] [Google Scholar]
- Seyfried NT, et al. , 2017. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst. 4, 60–72. 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siminoff LA, Burant CJ, Ibrahim SA, 2006. Racial disparities in preferences and perceptions regarding organ donation. J. Gen. Intern. Med 21, 995–1000. 10.1111/j.1525-1497.2006.00516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisco S, et al. , 2015. The role of early-life educational quality and literacy in explaining racial disparities in cognition in late life. J. Gerontol. B-Psychol 70, 557–567. 10.1093/geronb/gbt133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, 2002. Imaging the progression of Alzheimer pathology through the brain. Proc. Natl. Acad. Sci. U. S. A 99, 4135–4137. 10.1073/pnas.082107399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepler KE & Robinson RAS in Reviews on Biomarker Studies in Psychiatric and Neurodegenerative Disorders Vol. 1118 Advances in Experimental Medicine and Biology (ed Guest Paul C.) Ch. 1, 1–28 (Springer International Publishing, 2019).30747415 [Google Scholar]
- Sultana R, et al. , 2007a. Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J. Cell. Mol. Med 11, 839–851. 10.1111/j.1582-4934.2007.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, et al. , 2007b. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J. Alzheimers Dis 11, 153–164. [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W, 2011. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940. 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triplett JC, Swomley AM, Cai J, Klein JB, Butterfield DA, 2016. Quantitative phosphoproteomic analyses of the inferior parietal lobule from three different pathological stages of Alzheimer’s disease. J. Alzheimers Dis 49, 45–62. 10.3233/jad-150417. [DOI] [PubMed] [Google Scholar]
- Vizcaíno JA, et al. , 2016. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 44, D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G, 2001. Decreased brain levels of 2′,3′-cyclic nucleotide-S’-phosphodiesterase in down syndrome and Alzheimer’s disease. Neurobiol. Aging 22, 547–553. [DOI] [PubMed] [Google Scholar]
- Wallin ÅK, Blennow K, Andreasen N, Minthon L, 2006. CSF biomarkers for Alzheimer’s disease: levels of β-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement. Geriatr. Cogn. Disord 21, 131–138. 10.1159/000090631. [DOI] [PubMed] [Google Scholar]
- Wang Z, et al. , 2015. Differentially disrupted functional connectivity of the subregions of the inferior parietal lobule in Alzheimer’s disease. Brain Struct. Funct 220, 745–762. 10.1007/s00429-013-0681-9. [DOI] [PubMed] [Google Scholar]
- Wang Z, et al. , 2020. 27-plex tandem mass tag mass spectrometry for profiling brain proteome in Alzheimer’s disease. Anal. Chem 10.1021/acs.analchem.0c00655>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Kawas CH, Martin LJ, Troncoso JC, 2000. The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann. N. Y. Acad. Sci 908, 255–259. [DOI] [PubMed] [Google Scholar]
- Wharton W, et al. , 2019. Interleukin 9 alterations linked to Alzheimer disease in African Americans. Ann. Neurol 86, 407–418. 10.1002/ana.25543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins CH, Grant EA, Schmitt SE, McKeel DW, Morris JC, 2006. The neuropathology of Alzheimer disease in African American and white individuals. Arch. Neurol 63, 87–90. 10.1001/archneur.63.1.87. [DOI] [PubMed] [Google Scholar]
- Wilkins CH, Schindler SE, Morris JC, 2020. Addressing health disparities among minority populations: why clinical trial recruitment is not enough. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.1614 . https://doi.org/10.1001/jamaneurol.2020.1614https://jamanetwork.com/journals/jamaneurology/articlepdf/2767088/jamaneurology_wilkins_2020_vp_200008.pdf>. https://jamanetwork.com/journals/jamaneurology/articlepdf/2767088/jamaneurology_wilkins_2020_vp_200008.pdf>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MM, Meisel MM, Williams J, Morris JC, 2011. An interdisciplinary out-reach model of African American recruitment for Alzheimer’s disease research. Gerontologist 51, S134–S141. 10.1093/geront/gnq098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong F, Ge W, Ma C, 2019. Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer’s disease. Alzheimers Dement. 15, 429–440. 10.1016/j.jalz.2018.10.006. [DOI] [PubMed] [Google Scholar]
- Xu B, et al. , 2016a. Quantitative protein profiling of hippocampus during human aging. Neurobiol. Aging 39, 46–56. 10.1016/j.neurobiolaging.2015.11.029. [DOI] [PubMed] [Google Scholar]
- Xu B, et al. , 2016b. Temporal lobe in human aging: a quantitative protein profiling study of samples from Chinese human brain Bank. Exp. Gerontol 73, 31–41. 10.1016/j.exger.2015.11.016. [DOI] [PubMed] [Google Scholar]
- Xu J, et al. , 2019. Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun. Biol 2. https://www.nature.com/articles/s42003-018-0254-9.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid S, Oellerieh M, Asif AR, Ahmed N, 2014. Differential expression of proteins in brain regions of Alzheimer’s disease patients. Neurochem. Res 39, 208–215. 10.1007/s11064-013-1210-1. [DOI] [PubMed] [Google Scholar]
- Zhang Q, et al. , 2018. Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer’s disease. Acta Neuropathol. Commun 6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5831854/pdf/40478_2018_Article_524.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.