

Abstract
Early embryos are vulnerable to environmental insults, such as medications taken by the mother. Due to increasing prevalence of hypercholesterolemia, more women of childbearing potential are taking cholesterol-lowering medications called statins. Previously, we showed that inhibition of the mevalonate pathway by statins impaired mouse preimplantation development, by modulating HIPPO signaling, a key regulator for trophectoderm (TE) lineage specification. Here, we further evaluated molecular events that are altered by mevalonate pathway inhibition during the timeframe of morphogenesis and cell lineage specification. Whole transcriptome analysis revealed that statin treatment dysregulated gene expression underlying multiple processes, including cholesterol biosynthesis, HIPPO signaling, cell lineage specification and endoplasmic reticulum (ER) stress response. We explored mechanisms that link the mevalonate pathway to ER stress, because of its potential impact on embryonic health and development. Upregulation of ER stress-responsive genes was inhibited when statin-treated embryos were supplemented with the mevalonate pathway product, geranylgeranyl pyrophosphate (GGPP). Inhibition of geranylgeranylation was sufficient to upregulate ER stress-responsive genes. However, ER stress-responsive genes were not upregulated by inhibition of ras homolog family member A (RHOA), a geranylgeranylation target, although it interfered with TE specification and blastocyst cavity formation. In contrast, inhibition of Rac family small GTPase 1 (RAC1), another geranylgeranylation target, upregulated ER stress-responsive genes, while it did not impair TE specification or cavity formation. Thus, our study suggests that the mevalonate pathway regulates cellular homeostasis (ER stress repression) and differentiation (TE lineage specification) in preimplantation embryos through GGPP-dependent activation of two distinct small GTPases, RAC1 and RHOA, respectively. Translation of the findings to human embryos and clinical settings requires further investigations.
Keywords: blastocyst, unfolded protein response, cell lineage, hypercholesterolemia, geranylgeranylation
Introduction
Many human embryos are lost under natural conditions, particularly during the first weeks after fertilization, which significantly contributes to fertility decline (Macklon et al., 2002; Wilcox et al., 2020). Although genetic abnormality, including aneuploidy, is a known cause for such embryo demise (Nagaoka et al., 2012; Shahbazi, 2020), how environmental factors, such as maternal exposure to pharmacologic agents, contribute to embryo loss, is mostly unclear. Since the first sign of pregnancy occurs as a missed menstruation about 2 weeks after fertilization, women cannot recognize if they have produced an embryo and lost it before implantation due to an environmental insult. Thus, environmental causes of early embryo losses are too elusive to determine by epidemiologic studies in human. To compensate for such difficulties, investigations using the embryos of model animals are invaluable in providing mechanistic insights into how environmental factors may contribute to preimplantation demise.
Preimplantation development culminates in the construction of the blastocyst, the stage that is competent to implant into the uterus to enable the survival and development of the fetus. Two important cell lineages initially emerge at this stage: the extraembryonic tissue called trophectoderm (TE) and the pluripotent tissue called inner cell mass (ICM), which are the precursors of the placenta and fetal body, respectively. Best studied in the mouse model system, the segregation of the two lineages is under the control of the HIPPO signaling pathway (Karasek et al., 2020; Sasaki, 2015). Yes-associated protein (YAP; encoded by Yap1) is the downstream effector of HIPPO signaling, and acts as a co-activator for the TEA domain family (TEAD) of transcription factors. The differential subcellular localization of YAP to the nucleus or cytoplasm leads to the establishment of transcriptional regulatory networks that specify the TE fate in outer cells or ICM fate in inner cells of the embryo, respectively. Environmental factors that disturb these processes would likely interfere with preimplantation development, resulting in early embryo loss.
One type of environmental factors that may impair preimplantation development is statins, a class of medications that are commonly used to treat hypercholesterolemia, which is the condition of having high cholesterol level in the blood. Hypercholesterolemia is a major risk factor for cardiovascular diseases (Stone et al., 2014). The Centers for Disease Control and Prevention estimates that >12% of adults (aged ≥20 years) and 7% of children and adolescents (aged 6–19 years) have high cholesterol in the USA and that about half of these adults are taking cholesterol-lowering medications, such as statins, to prevent cardiovascular diseases (Centers for Disease Control and Prevention [CDC], 2020). Statins inhibit the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) of the mevalonate pathway, a major metabolic process for the synthesis of cholesterol and other bioactive lipids (Buhaescu and Izzedine, 2007) (Fig. 1A). It has been shown previously that mouse preimplantation development is impaired by exposure to statin (Surani et al., 1983), and we demonstrated a potential mechanism connecting the disruption of the mevalonate pathway to the impairment (Alarcón and Marikawa, 2016). We revealed an essential role of a specific lipid product of the mevalonate pathway, geranylgeranyl pyrophosphate (GGPP), in the TE lineage specification. GGPP serves as a substrate for geranylgeranylation, a post-translational modification that modulates the activities of various proteins (Wang and Casey, 2016). GGPP synthesis is independent of cholesterol production, implicating that preimplantation impairment by statin exposure is not due to cholesterol deficiency. Notably, the activity of ras homolog family member A (RHOA) small GTPase, a major target of geranylgeranylation, is required to modulate HIPPO signaling during cell lineage formation (Kono et al., 2014; Shi et al., 2017). Thus, we have provided a mechanistic link between the mevalonate pathway and the TE lineage specification, one of the crucial events in preimplantation development.
Figure 1.

Upregulation of the ER stress-responsive genes in mouse preimplantation embryos by inhibition of the mevalonate pathway. A, a schematic diagram of the mevalonate pathway, highlighting the key intermediates and pharmacological agents that interfere with specific steps. B, bright-field images of E3.5 embryos that have been treated with lovastatin (1 μM) or with lovastatin plus MVA (100 μM). Scale bar, 100 μm. C, relative gene expression levels of the YAP/TEAD targets, lineage specification regulators and ER stress-responsive genes. Means ± standard deviations (n = 3) are shown in bar graphs, in which the means for the control samples (vehicle only) are set as 1. Asterisks indicate significant differences between the two groups marked by horizontal bars.
Nonetheless, the mevalonate pathway produces various bioactive lipids, and is likely to regulate diverse cellular activities essential for preimplantation development in addition to the modulation of HIPPO signaling. Further understanding of the adverse effects of statins is of particular significance in light of the recent prevalence of hypercholesterolemia among younger populations, including women of childbearing potential (Arnett et al., 2005; CDC, 2020). In particular, more women are postponing to conceive until their later years (Heffner, 2004), further increasing the chance of embryonic exposures to medications, such as statins. Because human epidemiologic approaches are ineffective to elucidate the impact of statins during the preimplantation stages, investigations using model animals are crucial to reveal what types of cellular activities are affected in embryos by the inhibition of the mevalonate pathway.
The present study is an extension of our previous study (Alarcón and Marikawa, 2016), in which mouse embryos were treated with statins from the 8-cell stage to early blastocyst stage. This particular window of development is of interest because crucial processes, such as cell lineage specification and morphogenesis (e.g. cavitation), are active to create the blastocyst. To further evaluate the impact of statins, we examined the molecular events that were altered by inhibition of the mevalonate pathway during this developmental window and led to the failure of blastocyst formation. Here, we found that gene expressions linked to endoplasmic reticulum (ER) stress were upregulated by the HMGCR inhibitor lovastatin. The ER is the major site of protein synthesis, folding and trafficking, whose proper regulations are critical for normal cell functions (Oakes and Papa, 2015). When cells encounter exogenous (e.g. environmental insults) and endogenous (e.g. signaling pathways) disturbances, protein folding may be disrupted, leading to the accumulation of proteins that are misfolded or unfolded. As a result, the ER is unable to meet cell demands for proteins that perform essential functions. This condition is known as ER stress, and it initiates a corrective mechanism referred to as the unfolded protein response (UPR) to facilitate adaptation to changes in the cellular environment or elimination of damaged cells. The present study is the first to link the control of ER stress to the mevalonate pathway in the early embryo, revealing how the pathway coordinates repression of ER stress with the TE lineage specification through distinct mechanisms.
Materials and methods
Reagents
All reagents were commercially obtained as follows: lovastatin (#PHR1285; Sigma-Aldrich, St Louis, MO, USA), mevalonic acid (MVA, dl-mevalonic acid lactone, #M4667; Sigma-Aldrich), tunicamycin (#T7765; Sigma-Aldrich), farnesyl pyrophosphate (FPP, #F6892; Sigma-Aldrich), GGPP (#G6025; Sigma-Aldrich), B581 (FTase Inhibitor I, #344510; Calbiochem, La Jolla, CA, USA), GGTI-298 (#G5169; Sigma-Aldrich), RHO inhibitor I (#CT04; Cytoskeleton, Denver, CO, USA) and EHop-016 (#21557; Cayman Chemicals, Ann Arbor, MI, USA). RHO inhibitor I is the exoenzyme C3 transferase that selectively inhibits the activity of RHOA subfamily members (RHOA, RHOB and RHOC), but not other small GTPases, such as Cell division cycle 42 (CDC42) and Rac family small GTPase 1 (RAC1; Clayton et al., 1999; Vogelsgesang et al., 2007; Wilde et al., 2000). EHop-016 is a small molecule that selectively inhibits RAC1 (IC50 of 1.1 μM) but not CDC42 or RHOA (Montalvo-Ortiz et al., 2012). Stock solutions were prepared by dissolving in dimethyl sulfoxide (DMSO) for all reagents, except for MVA, FPP, GGPP (provided by the vendor in a liquid format) and RHO inhibitor I (dissolved in water).
Animals and embryos
The protocol for animal use was approved by the Institutional Animal Care and Use Committee of the University of Hawaii, and is in accordance with the national guidelines for the care and use of animals (National Research Council (US) Committee for the Update of the Guide, 2011). Embryos were obtained from crossings of the F1 (C57BL6 × DBA/2) strain of mice (Charles River Laboratories, Frederick, MD, USA). With superovulation, this hybrid strain produces large numbers of high quality F2 embryos (about 15–30), which develop robustly in vitro. Such high developmental potential not only helps to reduce the number of animals to be sacrificed, but also provides rigorous experimental outcomes to assess biological effects. Female mice were injected with 5 IU of pregnant mare serum gonadotrophin and 5 IU of human chorionic gonadotrophin (hCG) (Millipore, Temecula, CA, USA) at 48 h apart, and mated with males. At about 42 h after the hCG injection, oviducts were dissected from females, from which 2-cell stage (E1.5) embryos were flushed out with the FHM medium (MR-024-D; Millipore). For each experiment, embryos were collected from three to four females and were grouped together as a single batch.
Prior to the embryo collection, the culture dish was prepared and equilibrated in the incubator. This involved dotting a series of 20 μl drops of the KSOM medium (MR-121-D; Millipore) on the bottom of a Petri dish (#351007; Corning, Corning, NY, USA), covering the drops with mineral oil (#M8410; Sigma-Aldrich) and placing the dish in an incubator at 37°C with 5% CO2 humidified air for more than 1 h. The batch of 2-cell-stage embryos were serially transferred through several KSOM drops before being placed in a final drop at a density of up to 100 embryos per drop. The embryos were then cultured in the incubator up to the 8-cell stage (E2.5), and subjected to various experimental treatments. All experimental treatments and embryo cultures were conducted in the atmospheric oxygen level (20%), which is common practice and allows nearly 100% of the F2 embryos to develop into fully expanded blastocysts (under control conditions) that yield fetuses when transferred into pseudopregnant female mice (Alarcón and Marikawa, 2003; Laeno et al., 2013). Bright-field images of embryos were captured, using AxioCam MRm digital camera attached to Axiovert 200 inverted microscope with Hoffman modulation contrast optics (Carl Zeiss, Thornwood, NY, USA).
Pharmacological treatment of embryos in hanging drop culture
Embryos were treated with the pharmacological reagents for 24 h from E2.5 to E3.5 (early blastocyst stage) in hanging drops of the KSOM medium, as previously described (Alarcón and Marikawa, 2016). This was done because most of the reagents are lipophilic and may be absorbed by the mineral oil that is used with the traditional embryo culture method, resulting in the reduction of reagent concentrations in the culture medium. On the other hand, the hanging drop culture method does not employ mineral oil, so that the reagent concentrations are more likely to remain constant. The following reagent concentrations were used for treatments: lovastatin at 1 μM, MVA at 100 μM, tunicamycin at 0.1 μg/ml, FPP at 10 μM, GGPP at 10 μM, B581 at 10 μM, GGTI-298 at 10 μM, RHO inhibitor I at 1 μg/ml and EHop-016 at 0.5 μM. The corresponding controls for lovastatin, tunicamycin, B581, GGTI-298 and EHop-016 treatments contained 0.1% of DMSO in the culture medium. The control for RHO inhibitor I was water added to the culture medium.
Our study focused on the reagent concentrations that exhibit the maximum effect, i.e. complete inhibition of blastocyst formation in all embryos. This is to facilitate analyses of the reagent’s action efficiently at the molecular level, while lower concentrations still have significant but much milder effect. For lovastatin, we previously tested different doses (0.2, 1 and 5 μM) on the development of embryos from the 8-cell stage (E2.5) to the fully expanded blastocyst stage (E4.5) (Alarcón and Marikawa, 2016). The results indicated that 1 μM was the lowest dose that blocked blastocyst formation without arresting cell division, whereas 0.2 μM delayed blastocyst formation. Therefore, 1 μM of lovastatin was used for experimental treatments. The concentration of tunicamycin was determined via pilot studies where several doses were tested, and the results showed that 0.1 μg/ml consistently blocked blastocyst formation. The concentrations of B581 and GGTI-298 were the same as those applied for mouse preimplantation embryos in previous studies (Tao et al., 2012; Alarcón and Marikawa, 2016). The concentration of RHO inhibitor I was the same as the one used in a previous study, and it consistently inhibited blastocyst formation in all embryos, so it was used for experimental treatments in the present study (Kono et al., 2014). For EHop-016, our pilot studies suggested that treatment of E2.5 embryos with 1 μM or higher, but not 0.5 μM, caused cell cycle arrest or cell lysis in a significant number of embryos. Therefore, 0.5 μM of EHop-016 was used in the present study.
RNA-seq
RNA-seq was conducted on embryos that were collected after 24 h of treatment with lovastatin and vehicle control from the 8-cell (E2.5) to early blastocyst (E3.5) stage. RNA extraction, library preparation and sequencing were performed with the assistance of the Genomics and Bioinformatics Shared Resource at the University of Hawaii Cancer Center. A total of four samples, i.e. two independent sets of two samples each (lovastatin-treated and vehicle control embryos), were subjected to RNA-seq analyses. Total RNA was extracted from each sample (about 200 embryos each) using the Direct-zol RNA Microprep Kit (Zymo Research, Irvine, CA, USA), according to the manufacturer’s instructions, including the optional DNase I treatment. About 70 ng of RNA per sample was processed for library preparation using the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA, USA). Libraries were sequenced using the NextSeq 500 System with a Mid Output Flow Cell (130 M reads, 150 cycles; Illumina). Raw sequencing reads were trimmed for both adapters and low-quality bases on the 5′- and 3′-ends (Phred quality score Q < 20), and short reads were filtered (<20 bases), using Cutadapt (Martin, 2011). Trimmed reads were aligned to mouse genome reference build mm10 using STAR (Dobin et al., 2013). Gene expression was quantified using Partek Flow software (Partek, St Louis, MO, USA), and normalized to account for differences in sequencing depth among samples using DESeq2 (Love et al., 2014). An Excel spreadsheet file containing the normalized gene expression data for all four samples is provided in Supplementary Table S2.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from each sample (20–30 embryos), using TRI reagent (Thermo Fisher Scientific, Waltham, MA, USA) and Direct-zol RNA MicroPrep Kit (Zymo Research) and processed for cDNA synthesis using M-MLV Reverse Transcriptase (Promega, Madison, WI, USA) and oligodT (18) primer. Quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed using the CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) as follows: initial denaturation at 94 °C for 5 min, followed by up to 45 cycles of 94 °C for 15 s, 60 °C for 20 s and 72 °C for 40 s. Data files were opened in CFX Manager software (Bio-Rad) and Ct values were transferred to the Excel program for further analyses. The sequences of the primers used are listed in Supplementary Table S1. The expression levels of genes were normalized with Gapdh, and presented as relative expression levels. The relative expression levels in the control samples are set as 1, so that those in the experimental groups represent the fold differences relative to the control. The RNA-seq analysis showed that the relative abundance of Gapdh transcripts was comparable between control and lovastatin-treated samples (Supplementary Table S2). This suggests that Gapdh is suited as a housekeeping gene in the present study, as its expression is apparently unaffected by disturbances in the mevalonate pathway.
Immunofluorescence staining
Fixation, permeabilization and blocking of embryos were performed, as previously described (Laeno et al., 2013). Embryos were incubated in primary antibody at 4 °C overnight and in secondary antibody at room temperature for 2 h. Primary antibodies used were mouse anti-CDX2 (CDX2-88, #ab157524; Abcam, Cambridge, MA, USA) at 1:500 dilution and rabbit anti-SOX2 (#AB5603; Millipore) at 1:200 dilution. Secondary antibodies were goat anti-mouse Alexa 488 at 1:500 dilution and goat anti-rabbit Alexa 546 at 1:1000 dilution (Thermo Fisher Scientific). Stained embryos were mounted in ProLong Gold medium containing 4′,6-diamidino-2-phenyl-indole (DAPI) to stain nuclei (Thermo Fisher Scientific). A total of more than 13 embryos, compiled from two or three independent experiments, were examined for each treatment group.
Confocal microscopy
All embryos from the same experiment were imaged in a single session with FV1000 confocal laser scanning microscope with the Fluoview software (Olympus, Center Valley, PA, USA), using identical configurations, as described previously (Kono et al., 2014). Serial optical sections of entire embryos were obtained at 2 μm intervals under a 40× oil objective lens. Image files were opened with the ImageJ program (http://rsb.infonihgov/ij), and a maximum intensity projection image was generated for each sample.
Statistics
All experiments were conducted at least three times, using different batches of pooled embryos as biological replicates, and compiled data were presented as mean ± standard deviation. When three or more experimental conditions were compared, the one-way analysis of variance was used to determine whether there were any significant differences among their means, which was then followed by post-hoc two-sample t-test to compare between two specific conditions. P < 0.05 was deemed significant in the present study.
Results
Inhibition of the mevalonate pathway upregulates ER stress-responsive genes
The mevalonate pathway controls diverse physiological activities through the production of various bioactive lipids, including cholesterol and isoprenoids. To better understand what aspects of preimplantation development are controlled by the mevalonate pathway, we compared global transcriptomes of lovastatin-treated and vehicle only (control) embryos by RNA-seq (Fig. 1B). Specifically, embryos were exposed to lovastatin or vehicle only from E2.5 (8-cell stage) to E3.5 (early blastocyst stage), because this period entails critical developmental processes, such as cell lineage specification and blastocyst cavity formation. The RNA-seq analysis revealed that various genes were upregulated or downregulated by lovastatin treatment (Supplementary Table S2). In particular, impacts on the following four categories of genes were notable (Table I): (i) Cholesterol biosynthesis enzyme genes (Cyp51, Hmgcr, Lss and Sqle) were upregulated; (ii) Transcriptional targets of YAP/TEAD (Cyr61, Ctgf, Amotl2 and Ajuba) were downregulated; (iii) TE-enriched genes (Cdx2, Gata3 and Id2) were downregulated, whereas the ICM-enriched gene (Sox2) was upregulated; and (iv) ER stress-responsive genes (Trib3, Chac1, Herpud1 and Jun) were upregulated.
Table I.
Results of RNA-seq showing four categories of genes that are affected by lovastatin treatment.
| Gene symbol | Gene name | Fold change (direction)* |
References | |
|---|---|---|---|---|
| Set 1 | Set 2 | |||
| Cholesterol synthesis | ||||
| Cyp51 | Cytochrome P450, family 51 | 1.68 (up) | 1.96 (up) | Gibbons (2002) |
| Hmgcr | 3-hydroxy-3-methylglutaryl-coenzyme A reductase | 1.56 (up) | 1.73 (up) | Sato (2010) |
| Lss | Lanosterol synthase | 1.42 (up) | 1.52 (up) | Hua et al. (2019) |
| Sqle | Squalene epoxidase | 1.36 (up) | 1.58 (up) | Howe et al. (2017) |
| YAP/TEAD target | ||||
| Cyr61** | Cysteine-rich angiogenic inducer 61 | 3.98 (down) | 2.64 (down) | Zhao et al. (2008) |
| Ctgf** | Connective tissue growth factor | 2.47 (down) | 1.82 (down) | Zhao et al. (2008) |
| Amotl2 | Angiomotin-like 2 | 5.81 (down) | 3.72 (down) | Liu et al. (2016) |
| Ajuba | Ajuba LIM protein | 2.48 (down) | 2.13 (down) | Zanconato et al. (2015) |
| Cell lineage-enriched | ||||
| Cdx2 | Caudal type homeobox 2 | 1.96 (down) | 2.00 (down) | Nishioka et al. (2009) |
| Gata3 | GATA binding protein 3 | 1.76 (down) | 1.86 (down) | Home et al. (2009) |
| Id2 | Inhibitor of DNA binding 2 | 2.19 (down) | 2.18 (down) | Guo et al. (2010) |
| Sox2 | SRY (sex determining region Y)-box 2 | 1.83 (up) | 1.75 (up) | Frum et al. (2019) |
| ER stress-related | ||||
| Trib3 | Tribbles pseudokinase 3 | 3.56 (up) | 2.65 (up) | Ohoka et al. (2005) |
| Chac1 | ChaC, cation transport regulator 1 | 7.42 (up) | 3.90 (up) | Galluzzi et al. (2012) |
| Herpud1 | Homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member 1 | 2.52 (up) | 2.10 (up) | Kokame et al. (2000) |
| Jun | Jun proto-oncogene | 1.87 (up) | 1.70 (up) | Fuest et al. (2012) |
Fold difference in normalized transcript levels between control and lovastatin-treated embryos in each set of experiment. Parentheses show direction of change, i.e. (up) indicates upregulation by lovastatin treatment, whereas (down) indicates downregulation by lovastatin treatment.
Cyr61 and Ctgf are also known as Ccn1 (cellular communication network factor 1) and Ccn2 (cellular communication network factor 2), respectively, according to the Mouse Genome Informatics.
Category (i) is in line with a general feedback mechanism observed in various cell types, which is to compensate for the reduced production of cholesterol due to inhibition of the mevalonate pathway (Medina et al., 2012; Sharpe and Brown, 2013). Categories (ii) and (iii) are consistent with our previous study that HIPPO signaling-mediated lineage specification is dependent on the production of isoprenoids, especially GGPP, through the mevalonate pathway (Alarcón and Marikawa, 2016). Category (iv), however, is a novel observation, which implicates that inhibition of the mevalonate pathway induces ER stress in preimplantation embryos.
To validate that the effects on these changes in gene expression are specifically due to HMGCR inhibition, embryos were treated with lovastatin together with MVA, a product of the HMGCR action (Fig. 1A). Co-treatment with lovastatin and MVA permitted blastocyst cavity formation, whereas lovastatin alone inhibited it (Fig. 1B andTable II). Lovastatin-induced alterations in expressions of the YAP/TEAD targets (Amotl2 and Cyr61) and the lineage-specific regulators (Cdx2 and Sox2) were prevented by MVA, such that transcript levels were similar to those in control embryos (Fig. 1C). Notably, lovastatin-induced upregulation of the ER stress-responsive genes (Chac1, Herpud1, Jun and Trib3) was significantly dampened by MVA (Fig. 1C andTable II). This indicates that the changes in gene expressions by lovastatin treatment are due to a reduction in the production of MVA by HMGCR.
Table II.
Summary of the effects of inhibitor treatments on mouse blastocyst formation.
| Reagent | Blastocyst cavity | YAP/TEAD target gene expression | Cell-lineage gene expression |
ER stress-responsive gene expression | |
|---|---|---|---|---|---|
| TE | ICM | ||||
| Lovastatin (HMGCR inhibitor) | Absent | Downregulated 1 | Downregulated 2 | Upregulated | Upregulated |
| Tunicamycin (N-linked glycosylation inhibitor) | Absent |
No change (Amotl2) Upregulated (Cyr61) |
No change | No change | Upregulated |
| C3 exoenzyme (RHOA inhibitor) | Absent | Downregulated 1 | Downregulated 2 | Upregulated | No change |
| EHop-016 (RAC1 inhibitor) | Present |
Downregulated (Amotl2) 1 No change (Cyr61) |
Downregulated 2 | No change | Upregulated |
Amotl2 (angiomotin-like 2) is downregulated by 70% (lovastatin), by 98% (C3 exoenzyme) and by 20% (EHop-016).
Cdx2 (caudal type homeobox 2) is downregulated by 50% (lovastatin), by 95% (C3 exoenzyme) and by 30% (EHop-016).
Abbreviations: Cyr61, cysteine-rich angiogenic inducer 61 (also known as, Ccn1 or cellular communication network factor 1); ER, endoplasmic reticulum; HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; ICM, inner cell mass; RAC1, Rac family small GTPase 1; RHOA, ras homolog family member A; TE, trophectoderm; TEAD, TEA domain family of transcription factors; YAP, Yes-associated protein.
To verify that the genes listed in Category (iv) are indeed responsive to ER stress during preimplantation development, their expression levels were examined in embryos that were treated with a known ER stress inducer, tunicamycin (Latham, 2015), from E2.5 to E3.5. The transcript levels were significantly upregulated in tunicamycin-treated embryos compared to control embryos (Fig. 2 and Table II), confirming that the upregulation of these genes was due to ER stress.
Figure 2.

Upregulation of the ER stress-responsive genes by tunicamycin. A, bright-field images of E3.5 embryos treated with tunicamycin (0.1 μg/ml). Scale bar, 100 μm. B, relative expression levels of the ER stress-responsive genes. Means ± standard deviations (n = 3) are shown in bar graphs, in which the means for the control samples (vehicle only) are set as 1. Asterisks indicate significant differences.
In light of its potential impact on embryonic health and development, we further analyzed how the mevalonate pathway is mechanistically linked to the upregulation of the ER stress-responsive genes in the studies described below.
Insufficient isoprenoid production elevates the expression of ER stress-responsive genes
Isoprenoids FPP and GGPP are lipids produced by the mevalonate pathway downstream of MVA (Fig. 1A). The hydrophobic moieties of FPP and GGPP (i.e. farnesyl and geranylgeranyl groups) are covalently attached to certain proteins to modify their subcellular localization and biological activity (Wang and Casey, 2016). When embryos were co-treated with lovastatin and FPP or lovastatin and GGPP, they developed into blastocysts that were indistinguishable from control blastocysts (Fig. 3A). The expression levels of the YAP/TEAD targets and the lineage-specific regulators were also similar between the lovastatin/FPP- or lovastatin/GGPP-treated embryos and control embryos (Fig. 3B), which is in line with the normal blastocyst morphology. This indicates that the impact of HMGCR inhibition on these genes is mainly due to insufficient production of FPP or GGPP. To test whether the impact on ER stress is also linked to shortage of these isoprenoids, the expression levels of ER stress-responsive genes were compared between embryos treated with lovastatin together with FPP or GGPP. The results showed that elevation of their expressions was alleviated by co-treatment with FPP or GGPP (Fig. 3B). Notably, addition of GGPP was significantly more effective than FPP in reducing the expression of some ER stress-responsive genes, namely Chac1 and Trib3. This suggests that the synthesis of isoprenoids is necessary to repress ER stress.
Figure 3.

Suppression of lovastatin-induced upregulation of the ER stress-responsive genes by the supplementation of FPP and GGPP. A, bright-field images of E3.5 embryos treated with lovastatin (1 μM) plus FPP (10 μM) or GGPP (10 μM). Scale bar, 100 μm. B, relative expression levels of the YAP/TEAD targets, lineage specification regulators and ER stress-responsive genes. Means ± standard deviations (n = 3) are shown in bar graphs, in which the means for the control samples (vehicle only) are set as 1. Asterisks indicate significant differences between the two groups marked by horizontal bars.
Repression of ER stress depends mainly on geranylgeranylation but not farnesylation
The attachment of farnesyl and geranylgeranyl groups, termed farnesylation and geranylgeranylation, respectively, is catalyzed by enzymes farnesyltransferase and geranylgeranyltransferase (Wang and Casey, 2016). To test whether the lack of these lipidation processes is responsible for upregulation of the ER stress-responsive genes, embryos were treated with pharmacological inhibitors of farnesyltransferase (B581; Tao et al., 2012) or geranylgeranyltransferase (GGTI-298; Sorrentino et al., 2014), followed by gene expression analyses. GGTI-298, but not B581, significantly altered the expression of the YAP/TEAD targets and lineage-specific regulators in a manner comparable to lovastatin treatment (Fig. 4A and B). Notably, the expressions of ER stress-responsive genes were also significantly upregulated by GGTI-298 but not by B581 (Fig. 4B). These results suggest that a reduction in protein geranylgeranylation, rather than protein farnesylation, is mostly responsible for the transcriptional impact of the HMGCR inhibition.
Figure 4.

Upregulation of the ER stress-responsive genes by pharmacological inhibition of geranylgeranyltransferase. A, bright-field images of E3.5 embryos treated with B581 (10 μM; farnesyltransferase inhibitor) or GGTI-298 (10 μM; geranylgeranyltransferase inhibitor). Scale bar, 100 μm. B, relative expression levels of the YAP/TEAD targets, lineage specification regulators and ER stress-responsive genes. Means ± standard deviations (n = 3) are shown in bar graphs, in which the means for the control samples (vehicle only) are set as 1. Asterisks indicate significant differences between the two groups marked by horizontal bars.
RHO GTPase family members are differentially required for ER stress repression and TE lineage specification
Major proteins that are modified by geranylgeranylation are members of the RHO family of small GTPases, such as RHOA and Rac family small GTPase 1 (RAC1; Wang and Casey, 2016). Geranylgeranylation is essential for their functions, including reorganization of the actin cytoskeleton, cell polarization and cell differentiation (Phuyal and Farhan, 2019). RHOA regulates HIPPO signaling in a geranylgeranylation-dependent manner in various cancers (Sorrentino et al., 2014; Mi et al., 2015; Kato et al., 2018) and possibly in preimplantation embryos (Alarcón and Marikawa, 2016). Thus, we examined whether interference with RHOA activity is responsible for lovastatin-induced ER stress. Embryos treated with the RHOA inhibitor C3 exoenzyme failed to cavitate and remained a ball of cells (Fig. 5A andTable II). Also, expressions of the YAP/TEAD targets and lineage-specific regulators were significantly altered in both RHOA-inhibited embryos and HMGCR-inhibited embryos, although RHOA inhibition had a more potent effect (Fig. 5B andTable II). These observations were consistent with previous studies (Kono et al., 2014; Shi et al., 2017). However, the expression levels of the ER stress-responsive genes were unchanged in RHOA-inhibited embryos relative to control embryos, which was in stark contrast to the upregulation in HMGCR-inhibited embryos (Fig. 5B andTable II). These results suggest that ER stress is induced by lovastatin through mechanisms that are independent of RHOA inhibition.
Figure 5.

Impact of small GTPase inhibition on embryo morphology and gene expression patterns. A, bright-field images of E3.5 embryos treated with ras homolog family member A inhibitor (RHOi; 1 μg/ml). B, relative expression levels of the YAP/TEAD targets, lineage specification regulators and ER stress-responsive genes. Embryos treated with lovastatin (1 μM) are also examined as a comparison. C, bright-field images of E3.5 embryos treated with EHop-016 (0.5 μM). D, relative expression levels of the YAP/TEAD targets, lineage specification regulators and ER stress-responsive genes. A and C, scale bars, 100 μm. B and D, Means ± standard deviations (n = 3) are shown in bar graphs, in which the means for the control samples (vehicle only) are set as 1. Asterisks indicate significant differences between the two groups marked by horizontal bars.
We then tested whether RAC1 is involved in the induction of ER stress. EHop-016 is a pharmacological inhibitor of RAC1 (IC50 = 1.1 μM), and does not significantly inhibit other related small GTPases, such as RHOA and CDC42, at concentrations less than 5 μM (Montalvo-Ortiz et al., 2012; Dharmawardhane et al., 2013). When treated with EHop-016 at 2 μM, which is higher than the IC50, all embryos died in such a way that cells lysed or failed to divide (data not shown). However, embryos treated with EHop-016 at 0.5 μM developed into blastocysts at an efficiency similar to control embryos (Fig. 5C andTable II). Notably, the expression levels of the ER stress-responsive genes were significantly elevated in EHop-016 (0.5 μM)-treated embryos relative to control embryos (Fig. 5D andTable II). In contrast, the expression levels of Cyr61 (YAP/TEAD target) and Sox2 (ICM-specific regulator) were unaltered by EHop-016 treatment (Fig. 5D). The expression levels of Amotl2 (YAP/TEAD target) and Cdx2 (TE-specific regulator) were reduced by EHop-016 treatment, although the extent of reduction (20 and 30%, respectively) was much smaller than the extent of the induction by the RHOA inhibitor (98 and 95%, respectively; Fig. 5B andTable II). Notably, CDX2 and SOX2 proteins were immunolocalized in the nuclei of outside and inside cells, respectively, in EHop-016-treated embryos in a manner similar to that in control embryos (Fig. 6A). These results suggest that a mild inhibition of RAC1 induces ER stress, while the embryos retain the potential to develop into blastocysts with the correct spatial organization of the two cell lineages.
Figure 6.

Distribution of the cell lineage marker proteins in embryos with upregulated ER stress-responsive genes. A, projected confocal images of embryos at E3.5 that were immunostained for CDX2 (green) and SOX2 (red). Nuclei are stained with DAPI. Images of representative embryos from the control (n = 14), lovastatin-treated (n = 13) and EHop-016-treated (n = 15) groups are shown. B, relative gene expression levels for the YAP/TEAD targets and lineage specification regulators in embryos treated with tunicamycin (0.1 μg/ml). Asterisk indicates significant difference. C, projected confocal images of tunicamycin-treated embryos that were stained for CDX2 (green), SOX2 (red) and nuclei. Images of representative embryos from the control (n = 18) and tunicamycin-treated (n = 18) groups are shown. A and C, scale bars, 50 μm.
Taken together, our findings implicate that two distinct RHO GTPases, namely RAC1 and RHOA, are differentially required to repress ER stress and to specify the TE lineage, respectively, while both of them are under the control of the mevalonate pathway through geranylgeranylation.
Tunicamycin induces cavitation failure and ER stress without inhibiting TE lineage specification
The above experiment with EHop-016 suggests that a mild inhibition of RAC1 causes ER stress without interfering with TE formation (Fig. 5C and D). In contrast, tunicamycin, a well-known inducer of ER stress, prevents the formation of the blastocyst cavity, which is the morphological hallmark of TE formation (Surani et al., 1983; Fig. 2A). This raises the question of whether failure to cavitate is associated with lack of TE specification in the presence of tunicamycin. Tunicamycin treatment elevated the transcript level of YAP/TEAD target Cyr61, but not that of Amotl2, whereas it did not significantly alter the levels of the lineage-specific regulators (Cdx2 and Sox2; Fig. 6B andTable II). Furthermore, CDX2 and SOX2 proteins were immunolocalized in the nuclei of outside and inside cells, respectively, in tunicamycin-treated embryos, suggesting that lineage specification occurred in a cell position-specific manner (Fig. 6C). Thus, the cavitation failure caused by tunicamycin is neither due to lack of TE specification nor due to elevated ER stress.
Discussion
Previously, we showed that metabolic cues from the mevalonate pathway direct the formation of TE, a key feature of the blastocyst, through nuclear enrichment of the transcriptional coactivator YAP of the HIPPO signaling pathway (Alarcón and Marikawa, 2016). Expanding on this finding, our present study demonstrated a novel link between the mevalonate pathway and ER stress. Combined, we propose a model that the mevalonate pathway regulates processes that are essential for normal preimplantation development, namely repression of ER stress and specification of the TE lineage (Fig. 7). These two processes are modulated by distinct members of the RHO GTPase family, RAC1 and RHOA, respectively, both of which require post-translational modification by the mevalonate pathway product GGPP. Notably, regulation of ER stress and of TE specification are separable events, such that ER stress may be provoked without impairing the TE cell fate, and TE specification can be suppressed without inducing ER stress.
Figure 7.
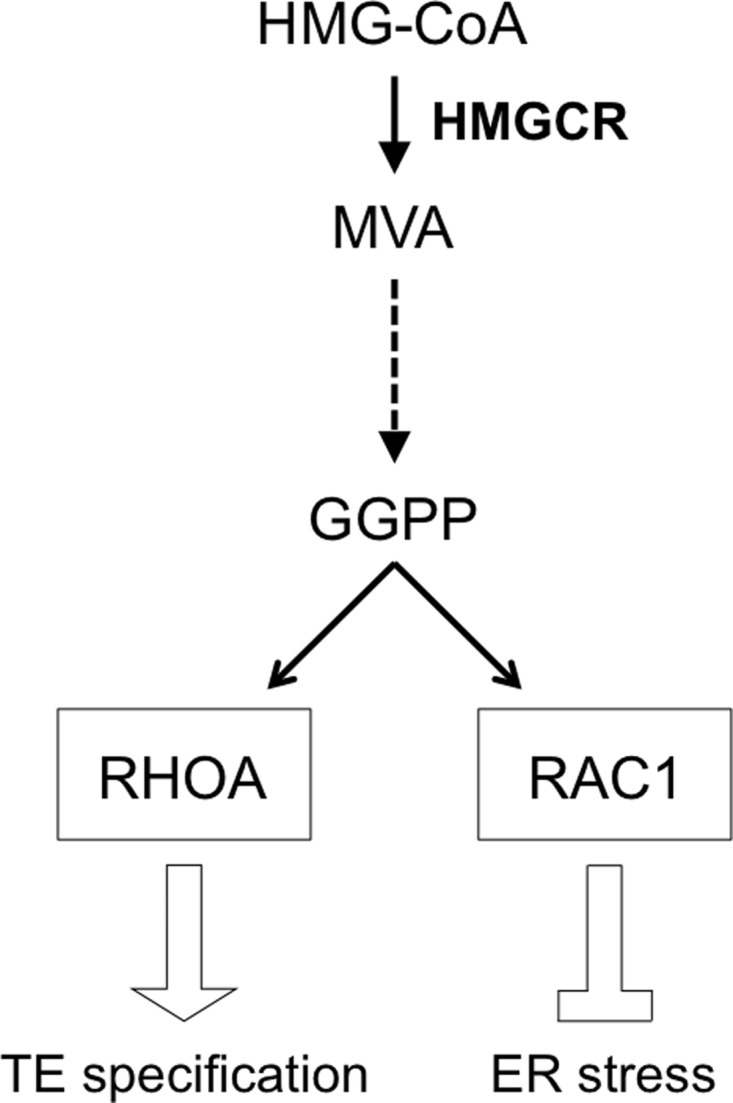
Model of mevalonate pathway regulation of trophectoderm specification and ER stress repression through GGPP-dependent ras homolog family small GTPases in mouse blastocyst formation. See Discussion section for details. Solid arrows, direct relationship; broken arrow, several intervening steps.
Although an elevated expression of the ER stress-responsive genes suggests occurrence of ER stress in lovastatin-treated embryos, the severity and nature of the stress are currently unknown and need to be further investigated. Generally, in stressed cells, incorrectly folded proteins build up, as the protein-folding capacity of the ER is impaired. Consequently, proteins with essential functions become deficient in the cells (Oakes and Papa, 2015). As a reparative measure, ER stress launches the UPR signal transduction in an attempt to adjust protein-folding capacity to meet cellular needs. The molecular mechanisms of the UPR signal transduction have been extensively studied in various adult cells (Hetz and Papa, 2018). ER chaperone 78-kDa glucose-regulated protein (GRP78) plays a major role in protein folding, and it associates with three ER-transmembrane proteins: protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). When GRP78 is transferred to incorrectly folded proteins, each of the three ER-transmembrane proteins transduces distinct downstream signaling events, which reduce new protein synthesis, eliminate abnormally folded proteins and increase protein-folding capacity. How these known mechanisms linked to ER stress are affected by statins in preimplantation embryos is currently unknown. However, the induction of ER stress-related events by statin treatment has been shown in several adult cell types. In mouse macrophages, treatment with lovastatin or fluvastatin increases the levels of GRP78 and spliced X-box binding protein 1 (XBP1; downstream of IRE1 signaling) and confers cytoprotective effects against hypoxia-induced cell death (Chen et al., 2008). In human atrial fibroblast, airway fibroblast and airway smooth muscle cells, simvastatin treatment causes apoptosis while elevating the levels of GRP78, spliced XBP1, activating transcription factor 4 (ATF4; downstream of PERK signaling) and cleaved ATF6 (downstream of ATF6 signaling; Ghavami et al., 2012, 2014). The induction of these events by statins is blunted by co-treatment with MVA, indicating that inhibition of HMGCR is mainly responsible for the ER stress (Chen et al., 2008; Ghavami et al., 2012, 2014). However, the roles of isoprenoid production, namely that of GGPP, were not investigated in these studies. Thus, it is unclear at this point whether these responses in adult cell types are mechanistically comparable to the lovastatin-induced activation of the ER stress-responsive genes in preimplantation embryos.
Another factor that makes comparisons difficult between the situations in adult and embryonic cells is that the above studies with adult cell types were conducted using relatively high concentrations of statins (e.g. 10 μM for lovastatin; Chen et al., 2008) in contrast to the 1 μM used in the present study. Such high concentrations may provoke additional biochemical impacts that are not induced by lower concentrations, as the mevalonate pathway is involved in the production of various lipids with diverse biological functions. Furthermore, disturbance of the mevalonate pathway by statins is likely to result in cellular responses that are dependent on cellular context. Responses to the mevalonate pathway inhibition may be significantly different between embryonic and adult cell types, because the former are dynamically changing in a spatiotemporal manner, whereas the latter are more homeostatic.
Depending on the nature and severity of the stressor, most cells respond to ER stress either by apoptosis or adaptation (Oakes and Papa, 2015). In the latter situation, cells modulate various aspects of the secretory pathway and metabolism to restore protein-folding capacity. An understanding of how these responses are executed in preimplantation embryos is still scarce (Latham, 2015). Embryos exposed to inducers of ER stress, such as tunicamycin and lovastatin, fail to form blastocysts and eventually die by E4.5 (Surani et al., 1983; Alarcón and Marikawa, 2016), which raises the possibility that ER stress induced by these stressors is severe enough to cause cell death. However, the effects of tunicamycin and lovastatin are likely to be pleiotropic, as their pharmacological actions (i.e. inhibitions of N-linked glycosylation and isoprenoid synthesis, respectively) would impair a wide range of biochemical pathways. Thus, death of embryos exposed to these agents may be due to adverse events other than ER stress. Indeed, we showed in the present study that RAC1 inhibitor EHop-016 upregulated the ER stress-responsive genes without blocking blastocyst formation or causing death (Fig. 5C). This suggests that ER stress by itself, at least at this level of severity, is not enough to trigger apoptotic responses in preimplantation embryos. Nonetheless, whether blastocysts exposed to EHop-016 are able to implant and survive after implantation still need to be examined, because detrimental effects of ER stress may manifest several days after the time of exposure to the stressor (Ryoo, 2016).
Thus far, no study has been reported to mechanistically connect RAC1 to ER stress, to the best of our knowledge. However, RAC1 is a known regulator of actin cytoskeleton dynamics (Marei and Malliri, 2017) and F-actin has been linked to IRE1 through interaction with nonmuscle myosin IIB motor protein during ER stress (He et al., 2012). It is possible that alterations in RAC1 activity modulate actin, thereby impacting UPR signaling in the embryo.
The present study was conducted using various pharmacological reagents or small molecule inhibitors, such as lovastatin, GGTI-298 and EHop-016. In principle, any type of small molecule inhibitors can affect more than one intended target depending on the concentrations, environment and cell types. Thus, to validate whether the observed effects of a small molecule inhibitor are specifically caused by the inhibition of the intended target, additional studies may be needed, in which the gene encoding the intended target is knocked out by genetic manipulations, such as the homologous recombination or Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9) technology. However, the application of such genetic approaches to analyze the role of specific targets in preimplantation embryos may also be complicated. This is partly because both maternal gene products (stored in the oocyte) and zygotic gene products regulate various events in preimplantation development, and it may be necessary to knock out both the maternal and zygotic alleles to yield detectable phenotypes. Also, because of functional redundancy, knockout of one gene may be compensated by other genes, which obscures phenotypic outcomes. Regardless, further investigations employing multiple methods, including genetic and pharmacological approaches, should shed light on mechanistic details that connect the mevalonate pathway and ER stress.
The fluid-filled cavity is a distinct feature of the blastocyst, and is a morphological indicator for the presence of the TE (Marikawa and Alarcón, 2012). Nonetheless, our present study revealed that in tunicamycin-treated embryos, TE marker expression was observed in the outer cells in the absence of cavity formation, suggesting that specification of the TE lineage and cavitation are separable processes. It has been hypothesized that mechanical forces, such as stretch and tension, that are associated with cavitation may influence TE cell fate specification (Biggins et al., 2015; Zenker et al., 2018; Chan et al., 2019). In line with this idea, various studies using cultured cells have demonstrated that stretch and tension can modulate HIPPO signaling (Dupont et al., 2011; Wada et al., 2011). Also, a study using mouse preimplantation embryos implicates that mechanical forces can modulate YAP activity to regulate TE differentiation (Maitre et al., 2016). However, the tunicamycin experiment suggests that cavitation or associated mechanical force does not play an essential role in TE specification. It is yet to be investigated whether these non-cavitating embryos with TE gene expressions are competent to implant and generate trophoblasts. Regardless, such a possibility bears relevance to the in vitro production of human embryos in which the quality of blastocysts is assessed mainly based on morphological features, namely robust cavity expansion as an indicator of developmental potential (Gardner and Balaban, 2016). In light of the possibility that TE can form in noncavitating embryos, further characterization of human embryos with deficient cavitation is warranted.
While the present study suggests the occurrence of ER stress in response to the pharmacological inhibition of HMGCR in preimplantation embryos, it is unclear whether such a situation could arise in women who have taken statins before or around the time of conception. In principle, the concentrations of medications, such as statins, dynamically change in the body, as they are influenced by various factors, namely absorption, distribution, metabolism and excretion. The plasma concentrations of statins found in patients are generally much lower than those employed in experimental settings, including the present study (Alarcón and Marikawa, 2016). This is partly because the therapeutic objective of statin use is to reduce, but not to eliminate, the activity of HMGCR. In contrast, the statin use in experimental settings is intended to block the HMGCR action robustly so that outcomes of treatments are more clear-cut for interpretation. Nonetheless, the in vivo data for actual concentrations of statins in the reproductive tract, where preimplantation embryos develop, are essentially absent. In addition, how embryos may be impacted by the various metabolic products of statins, which are generated by the actions of drug-metabolizing enzymes in the liver, is yet to be evaluated. Such additional information is crucial to aptly extrapolate the findings of experimental studies to real-life situations.
Supplementary data
Supplementary data are available at Molecular Human Reproduction online.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgements
We are grateful to Silvia B. Alarcon for her support and encouragement during the course of the writing of this article. We thank the Animal and Veterinary Service staff of the University of Hawaii for providing care for our mice. The authors also thank the Department of Anatomy, Biochemistry and Physiology for the Grant Awards to V.B.A. and Y.M. RNA-seq analysis was performed at the University of Hawaii John A. Burns School of Medicine Bioinformatics Core.
Authors’ roles
Y.M. and V.B.A. designed the study, executed the experiments, analyzed the data and wrote the article. M.M. and Y.D. assisted with the initial analysis of the RNA-seq data. All authors read and approved the final version of the article.
Funding
Hawaii Community Foundation (16ADVC-78882 to V.B.A.); National Institutes of Health (NIH; R03 HD088839 to V.B.A.); NIH Centers of Biomedical Research Excellence Phase 3/Institute for Biogenesis Research (P30 GM131944); NIH (5P30 GM114737, P20 GM103466, 5U54 MD007601, 5P30 CA071789 to Y.D.).
Conflict of interest
The authors declare no conflict of interest.
References
- Alarcón VB, Marikawa Y.. Deviation of the blastocyst axis from the first cleavage plane does not affect the quality of mouse postimplantation development. Biol Reprod 2003;69:1208–1212. [DOI] [PubMed] [Google Scholar]
- Alarcón VB, Marikawa Y.. Statins inhibit blastocyst formation by preventing geranylgeranylation. Mol Hum Reprod 2016;22:350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett DK, Jacobs DR Jr, Luepker RV, Blackburn H, Armstrong C, Claas SA.. Twenty-year trends in serum cholesterol, hypercholesterolemia, and cholesterol medication use: the Minnesota Heart Survey, 1980-1982 to 2000-2002. Circulation 2005;112:3884–3891. [DOI] [PubMed] [Google Scholar]
- Biggins JS, Royer C, Watanabe T, Srinivas S.. Towards understanding the roles of position and geometry on cell fate decisions during preimplantation development. Semin Cell Dev Biol 2015;47–48:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhaescu I, Izzedine H.. Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 2007;40:575–584. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC). High Cholesterol Facts. https://www.cdc.gov/cholesterol/facts.htm(25 August2020, date last accessed).
- Chan CJ, Costanzo M, Ruiz-Herrero T, Monke G, Petrie RJ, Bergert M, Diz-Munoz A, Mahadevan L, Hiiragi T.. Hydraulic control of mammalian embryo size and cell fate. Nature 2019;571:112–116. [DOI] [PubMed] [Google Scholar]
- Chen JC, Wu ML, Huang KC, Lin WW.. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc Res 2008;80:138–150. [DOI] [PubMed] [Google Scholar]
- Clayton L, Hall A, Johnson MH.. A role for Rho-like GTPases in the polarisation of mouse eight-cell blastomeres. Dev Biol 1999;205:322–331. [DOI] [PubMed] [Google Scholar]
- Dharmawardhane S, Hernandez E, Vlaar C.. Development of EHop-016: a small molecule inhibitor of Rac. Enzymes 2013;33:117–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S. et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179–183. [DOI] [PubMed] [Google Scholar]
- Frum T, Watts JL, Ralston A.. TEAD4, YAP1 and WWTR1 prevent the premature onset of pluripotency prior to the 16-cell stage. Development 2019;146:dev179861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuest M, Willim K, MacNelly S, Fellner N, Resch GP, Blum HE, Hasselblatt P.. The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology 2012;55:408–418. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, De Santi M, Crinelli R, De Marco C, Zaffaroni N, Duranti A, Brandi G, Magnani M.. Induction of endoplasmic reticulum stress response by the indole-3-carbinol cyclic tetrameric derivative CTet in human breast cancer cell lines. PLoS One 2012;7:e43249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DK, Balaban B.. Assessment of human embryo development using morphological criteria in an era of time-lapse, algorithms and ‘OMICS’: is looking good still important? Mol Hum Reprod 2016;22:704–718. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Sharma P, Yeganeh B, Ojo OO, Jha A, Mutawe MM, Kashani HH, Los MJ, Klonisch T, Unruh H. et al. Airway mesenchymal cell death by mevalonate cascade inhibition: integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim Biophys Acta 2014;1843:1259–1271. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Yeganeh B, Stelmack GL, Kashani HH, Sharma P, Cunnington R, Rattan S, Bathe K, Klonisch T, Dixon IM. et al. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis 2012;3:e330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons GF. The role of cytochrome P450 in the regulation of cholesterol biosynthesis. Lipids 2002;37:1163–1170. [DOI] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, Robson P.. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell 2010;18:675–685. [DOI] [PubMed] [Google Scholar]
- He Y, Beatty A, Han X, Ji Y, Ma X, Adelstein RS, Yates JR, Kemphues K, Qi L.. Nonmuscle myosin IIB links cytoskeleton to IRE1α signaling during ER stress. Dev Cell 2012;23:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffner LJ. Advanced maternal age—how old is too old? N Engl J Med 2004;351:1927–1929. [DOI] [PubMed] [Google Scholar]
- Hetz C, Papa FR.. The unfolded protein response and cell fate control. Mol Cell 2018;69:169–181. [DOI] [PubMed] [Google Scholar]
- Home P, Ray S, Dutta D, Bronshteyn I, Larson M, Paul S.. GATA3 is selectively expressed in the trophectoderm of peri-implantation embryo and directly regulates Cdx2 gene expression. J Biol Chem 2009;284:28729–28737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe V, Sharpe LJ, Prabhu AV, Brown AJ.. New insights into cellular cholesterol acquisition: promoter analysis of human HMGCR and SQLE, two key control enzymes in cholesterol synthesis. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:647–657. [DOI] [PubMed] [Google Scholar]
- Hua H, Yang T, Huang L, Chen R, Li M, Zou Z, Wang N, Yang D, Liu Y.. Protective effects of lanosterol synthase up-regulation in UV-B-induced oxidative stress. Front Pharmacol 2019;10:947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasek C, Ashry M, Driscoll CS, Knott JG.. A tale of two cell-fates: Role of the Hippo signaling pathway and transcription factors in early lineage formation in mouse preimplantation embryos. Mol Hum Reprod 2020;26:653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Liberona MF, Cerda-Infante J, Sánchez M, Henríquez J, Bizama C, Bravo ML, Gonzalez P, Gejman R, Brañes J. et al. Simvastatin interferes with cancer ‘stem-cell’ plasticity reducing metastasis in ovarian cancer. Endocr Relat Cancer 2018;25:821–836. [DOI] [PubMed] [Google Scholar]
- Kokame K, Agarwala KL, Kato H, Miyata T.. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J Biol Chem 2000;275:32846–32853. [DOI] [PubMed] [Google Scholar]
- Kono K, Tamashiro DA, Alarcon VB.. Inhibition of RHO-ROCK signaling enhances ICM and suppresses TE characteristics through activation of Hippo signaling in the mouse blastocyst. Dev Biol 2014;394:142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laeno AM, Tamashiro DA, Alarcon VB.. Rho-associated kinase activity is required for proper morphogenesis of the inner cell mass in the mouse blastocyst. Biol Reprod 2013;89:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham KE. Endoplasmic reticulum stress signaling in mammalian oocytes and embryos: life in balance. Int Rev Cell Mol Biol 2015;316:227–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park JS. et al. Tead and AP1 coordinate transcription and motility. Cell Rep 2016;14:1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklon NS, Geraedts JP, Fauser BC.. Conception to ongoing pregnancy: the ‘black box’ of early pregnancy loss. Hum Reprod Update 2002;8:333–343. [DOI] [PubMed] [Google Scholar]
- Maitre JL, Turlier H, Illukkumbura R, Eismann B, Niwayama R, Nedelec F, Hiiragi T.. Asymmetric division of contractile domains couples cell positioning and fate specification. Nature 2016;536:344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marei H, Malliri A.. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 2017;8:139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marikawa Y, Alarcón VB.. Creation of trophectoderm, the first epithelium, in mouse preimplantation development. Results Probl Cell Differ 2012;55:165–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 2011;17:10–12. [Google Scholar]
- Medina MW, Theusch E, Naidoo D, Bauzon F, Stevens K, Mangravite LM, Kuang YL, Krauss RM.. RHOA is a modulator of the cholesterol-lowering effects of statin. PLoS Genet 2012;8:e1003058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi W, Lin Q, Childress C, Sudol M, Robishaw J, Berlot CH, Shabahang M, Yang W.. Geranylgeranylation signals to the Hippo pathway for breast cancer cell proliferation and migration. Oncogene 2015;34:3095–3106. [DOI] [PubMed] [Google Scholar]
- Montalvo-Ortiz BL, Castillo-Pichardo L, Hernandez E, Humphries-Bickley T, De la Mota-Peynado A, Cubano LA, Vlaar CP, Dharmawardhane S.. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem 2012;287:13228–13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka SI, Hassold TJ, Hunt PA.. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 2012;13:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (US) Committee for the Update of the Guide. Guide for the Care and Use of Laboratory Animals, 8th edn. Washington DC, USA: National Academies Press, 2011. [Google Scholar]
- Nishioka N, Inoue K, Adachi K, Kiyonari H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N. et al. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell 2009;16:398–410. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Papa FR.. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 2015;10:173–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H.. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. Embo J 2005;24:1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuyal S, Farhan H.. Multifaceted Rho GTPase signaling at the endomembranes. Front Cell Dev Biol 2019;7:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD. Long and short (timeframe) of endoplasmic reticulum stress-induced cell death. Febs J 2016;283:3718–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H. Position- and polarity-dependent Hippo signaling regulates cell fates in preimplantation mouse embryos. Semin Cell Dev Biol 2015;47–48:80–87. [DOI] [PubMed] [Google Scholar]
- Sato R. Sterol metabolism and SREBP activation. Arch Biochem Biophys 2010;501:177–181. [DOI] [PubMed] [Google Scholar]
- Shahbazi MN. Mechanisms of human embryo development: from cell fate to tissue shape and back. Development 2020;147:dev190629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe LJ, Brown AJ.. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem 2013;288:18707–18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Yin Z, Ling B, Wang L, Liu C, Ruan X, Zhang W, Chen L.. Rho differentially regulates the Hippo pathway by modulating the interaction between Amot and Nf2 in the blastocyst. Development 2017;144:3957–3967. [DOI] [PubMed] [Google Scholar]
- Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 2014;16:357–366. [DOI] [PubMed] [Google Scholar]
- Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM. et al. ; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:S1–45. [DOI] [PubMed] [Google Scholar]
- Surani MA, Kimber SJ, Osborn JC.. Mevalonate reverses the developmental arrest of preimplantation mouse embryos by Compactin, an inhibitor of HMG Co A reductase. J Embryol Exp Morphol 1983;75:205–223. [PubMed] [Google Scholar]
- Tao H, Inoue K, Kiyonari H, Bassuk AG, Axelrod JD, Sasaki H, Aizawa S, Ueno N.. Nuclear localization of Prickle2 is required to establish cell polarity during early mouse embryogenesis. Dev Biol 2012;364:138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsgesang M, Pautsch A, Aktories K.. C3 exoenzymes, novel insights into structure and action of Rho-ADP-ribosylating toxins. Naunyn Schmiedebergs Arch Pharmacol 2007;374:347–360. [DOI] [PubMed] [Google Scholar]
- Wada K, Itoga K, Okano T, Yonemura S, Sasaki H.. Hippo pathway regulation by cell morphology and stress fibers. Development 2011;138:3907–3914. [DOI] [PubMed] [Google Scholar]
- Wang M, Casey PJ.. Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol 2016;17:110–122. [DOI] [PubMed] [Google Scholar]
- Wilcox AJ, Harmon Q, Doody K, Wolf DP, Adashi EY.. Preimplantation loss of fertilized human ova: estimating the unobservable. Hum Reprod 2020;35:743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde C, Genth H, Aktories K, Just I.. Recognition of RhoA by Clostridium botulinum C3 exoenzyme. J Biol Chem 2000;275:16478–16483. [DOI] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S.. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 2015;17:1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenker J, White MD, Gasnier M, Alvarez YD, Lim HYG, Bissiere S, Biro M, Plachta N.. Expanding actin rings zipper the mouse embryo for blastocyst formation. Cell 2018;173:776–791.e17. [DOI] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22:1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.