

SUMMARY
Regulatory T (Treg) cells, vital for maintaining immune homeostasis, also represent a major barrier to cancer immunity, as the tumor microenvironment (TME) promotes Treg cell recruitment, differentiation, and activity1,2. Tumor cells have deregulated metabolism leading to a metabolite-depleted, hypoxic, and acidic TME3, placing infiltrating effector T cells in competition with tumors for metabolites, impairing their function4–6. Conversely, Treg cells maintain high suppressive function within the TME7,8. As previous studies suggested Treg cells possess a distinct metabolic profile from effector T cells9–11, we hypothesized the altered metabolic landscape of the TME and increased activity of intratumoral Treg cells are linked. Here we show Treg cells display broad heterogeneity in utilization of glucose metabolism within normal and transformed tissues and can engage an alternative metabolic pathway to maintain suppressive function and proliferation. Glucose uptake correlated with poorer suppressive function and long-term instability, and high glucose culture impaired Treg cell function and stability. Treg cells rather upregulate pathways in metabolism of the glycolytic byproduct lactic acid. Treg cells withstood high lactate conditions, and lactate treatment prevented the destabilizing effects of high glucose, generating intermediates necessary for proliferation. Treg cell-restricted deletion of MCT1, a lactate transporter, revealed lactate uptake is dispensable for peripheral Treg cell function but required intratumorally, resulting in slowed tumor growth and increased response to immunotherapy. Thus Treg cells are metabolically flexible: they can utilize ‘alternative’ metabolites in the TME to maintain suppressive identity. Further, our studies suggest tumors avoid destruction by not only depriving effector T cells of nutrients, but also metabolically supporting regulatory populations.
Treg cells thrive under glucose restriction
Having previously shown heightened tumor cell metabolism correlates with decreased effector T cell function5, we sought to determine the relationship of heighted tumor metabolism with Treg cell function. We measured the suppressor activity of Treg cells isolated from Foxp3 reporter mice12,13 bearing tumors of increasing glycolytic capacity, revealing glycolysis of tumor cells correlated with suppressive function of intratumoral Treg cells (Fig. 1a). Expectedly, Treg cells were enriched within tumors (Fig. 1b, Extended Data Fig. 1a,b) and were not metabolically suppressed but actively proliferating (Fig. 1c). Given the relationship between tumor glycolysis and Treg cell function, we determined the glycolytic capacity of Treg cells directly ex vivo and immediately after activation, a reprogramming event occurring downstream of the TCR in T cells14. TCR triggering induced less glycolysis in Treg cells compared to naïve or antigen-experienced conventional T cells (Fig. 1d). This resistance to glycolysis persisted under strong activating stimuli (anti-CD3, CD28, and IL-2 for 48 h), and tumor-derived Treg cells displayed even lower glycolytic activity (Extended Fig. 1c). Oligomycin treatment showed Treg cells are resistant to engaging glycolysis even when oxidative metabolism is inhibited, suggesting glucose transport may be limiting in Treg cells (Extended Data Fig. 1c). Indeed, Treg cells had significant reductions in glucose uptake based on the fluorescent glucose tracer 2NBDG compared to Tconv (CD4+ Foxp3−) cells directly ex vivo (Fig. 1e, Extended Data Fig. 1d), regardless of CD44/CD62L expression (notably, the largest difference was observed between CD62Lhi populations) (Extended Data Fig. 1e). To directly compare activation-induced Treg and Tconv glucose uptake, we adoptively transferred congenically marked OT-II CD4+ T cells from Foxp3RFP mice into VacciniaOVA-infected hosts. OT-II Treg cells took up significantly less 2NBDG than their Foxp3− counterparts, despite sharing the same TCR and undergoing robust activation (Fig. 1f). While not selected on the OT-II TCR, activation by the same antigen in vivo under the same inflammatory context results in disparate metabolic phenotypes if Foxp3 is expressed.
Figure 1. Regulatory T cells possess a distinct metabolic profile from conventional T cells in normal and transformed tissues.
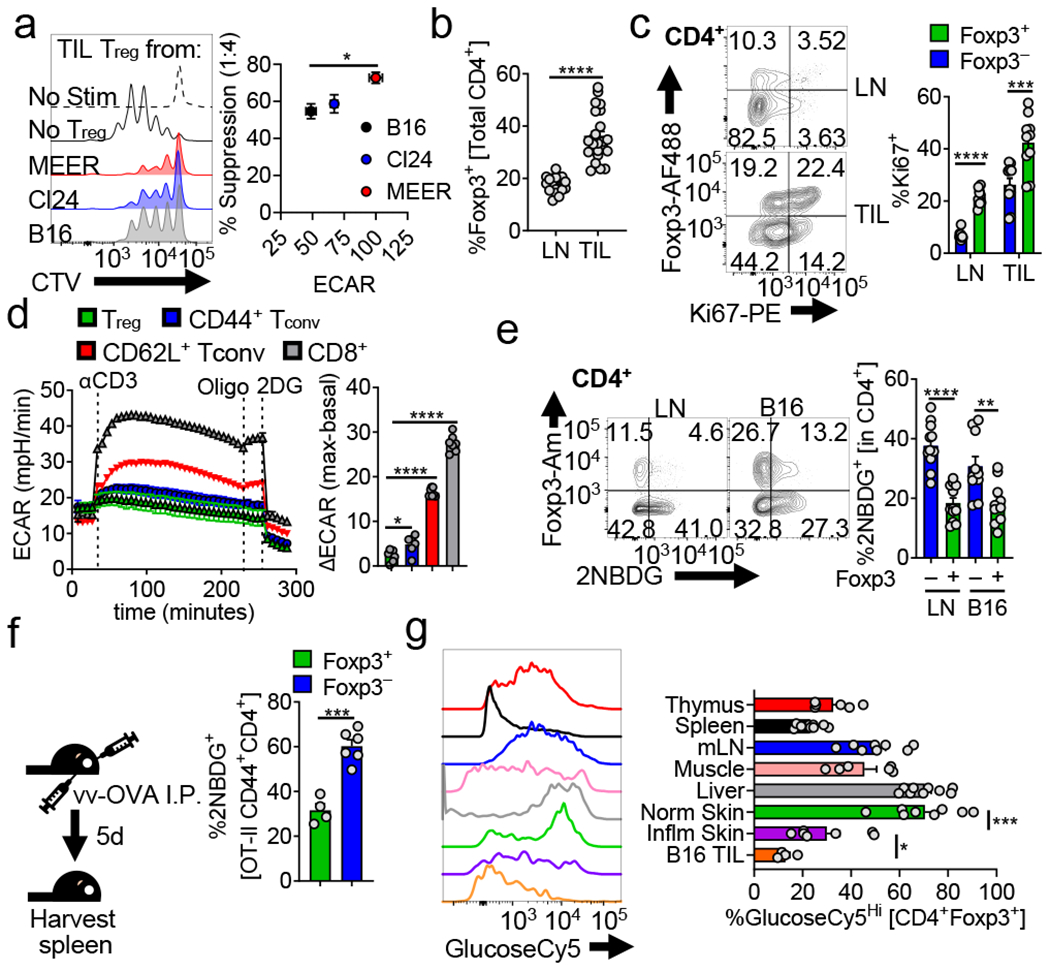
(a) (left) Representative histogram of Tconv responder cell proliferation dye dilution after 72hrs of co-culture with Treg cells isolated from B16, Cl24 (melanoma), or MEER (HPV+ HNSCC) tumors. (right) Capacity of Treg cells to suppress the proliferation of CellTrace Violet (CTV) labelled Tconv cells as a function of the glycolytic extracellular acidification rate (ECAR) of the tumors from which they were isolated (*p=0.047). (b) Percent Treg cells (CD4+ Foxp3+) of total CD4+ T cells from lymph node (LN) and tumor infiltrating lymphocyte (TIL) preparations from C57BL/6 mice bearing B16 melanoma tumors (14 d post intradermal injection) (c) Percent proliferating (Ki67+) Treg and Tconv cells (CD4+ Foxp3-) from mice as in (b). (d) ECAR of in-Seahorse activated Treg, CD8+, CD44+CD62L− and CD44-CD62L+ Tconv cells sorted from LN and spleen of Foxp3 reporter mice. Oligo = oligomycin, 2-DG = 2-deoxy-D-glucose, ΔECAR = max reading after αCD3 minus basal ECAR. (*p=0.022) (e) Flow cytogram depicting ex vivo 2NBDG uptake by Treg and Tconv cells from the LN and B16 TIL of Foxp3 reporter mice. Representative plots gated on CD4+ cells. (**p=0.006) (f) Diagram of experimental procedure and quantification of ex vivo 2NBDG uptake by CD44+ OT-II Treg and Tconv cells isolated 5 days after transfer into congenically mismatched hosts infection with Vaccinia-OVA. (g) Representative histogram and quantification of ex vivo GlucoseCy5 uptake by Treg cells isolated from various tissues (*p=0.047). Results are representative of five (b), three (a,c,e, and g) or two (d,f) independent experiments. (*p< 0.05, **p <0.01, ***p < 0.001 ****p<0.0001) by unpaired two-tailed t test (a,b,d,f,g) or two-way ANOVA with Sidak’s multiple comparisons test (c,e). Data are mean values of biological replicates ±SEM.
To determine if low glucose uptake was a universal phenotype of Treg cells, we interrogated the infiltrate of various tissues at normal and inflammatory states . 2NBDG is not only incompatible with YFP and GFP employed in Foxp3-driven Cre lines, but may have limited utility in some cells as a glucose tracer16. Thus, we synthesized a novel fluorescent glucose tracer (Cy5-linked 1amino-glucose; GlucoseCy517), recapitulating our findings with 2NBDG while demonstrating superior specificity and sensitivity (Extended Data Fig. 1f–h). Treg cell glucose uptake was quite heterogeneous in tissues of Foxp3YFP-iCre mice, but notably low in B16 and imiquimod-inflamed skin (Fig.1g), suggesting inflammation may drive lower glucose uptake in Treg cells. Conventional T cells possessed far less heterogeneity in glucose avidity when assayed from the same environments (Extended Data Fig. 1i).
Glucose avidity predicts Treg cell function
We next purified Treg subsets of low and high glucose avidity and measured their function. Regardless of tissue, 2NBDG avid Treg cells displayed significantly reduced suppressive capacity compared to 2NBDGlo Treg cells (Fig. 2a, Extended Data Fig 2a), not attributable to viability, as 2NBDGhi Treg cells were rather more viable after the assay (Extended Data Fig 2b,c), and retained their initial proclivities for glucose (Extended Data Figure 2b,d). Thus, within a tissue, avidity for glucose predicts poorly suppressive Treg cells. Sequencing 2NBDG low and high LN and B16-infiltrating Treg cells revealed reduced expression of Foxp3, Ikzf2 (Helios), Il2ra (CD25), Nrp1, and other Treg signature genes in 2NBDGhi Treg cells, further confirmed by flow cytometry (Fig. 2b, Extended Data Fig. 2e). These data suggested while glucose avid Treg cells are still Foxp3+, they harbor a ‘weaker’ Treg cell signature. As 2NBDGhi Treg cells harbored some suppressor function, we measured cytokine production after a brief restimulation. 2NBDGhi Treg cells produced significantly more IL-10, suggesting distinct suppressive mechanisms between 2NBDG Treg states, especially intriguing given the differences in mRNA (Fig. 2b,c). These differences may be driven by glycolysis which is tied to cytokine translation in conventional T cells18. To assess long-term stability and function of 2NBDG Treg cell subsets under inflammation, Thy1.2+ Foxp3+ Treg cells of high or low glucose avidity were transferred into Rag1-deficient animals, along with Thy1.1+ Foxp3− Tconv cells to induce colitis. After 12 weeks, mice receiving glucose avid Treg cells began to lose weight due to increased colitis compared to those receiving 2NBDGlo Treg cells, and transferred 2NBDGhi Treg cells lost Foxp3 expression (Fig. 2d,e, Extended Data Fig. 2f). We also investigated whether increased glucose uptake could be observed in Treg cells from an unmanipulated autoimmune setting. Treg cells isolated from pancreatic islets (known to be dysfunctional19) and draining lymph node of pre-diabetic NOD.Foxp3GFP mice took up significantly more glucose than those from non-draining lymph node (ndLN) (Fig. 2f). Thus, glucose avidity in Treg cells correlates with poor suppressive capacity, ultimately leading to instability.
Figure 2. Glucose avidity is associated with reduced Treg cell functional identity.
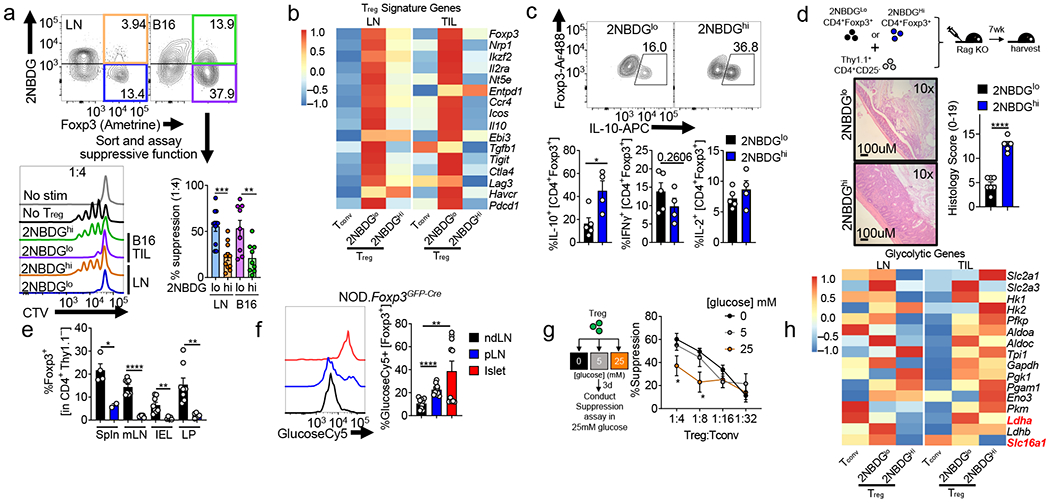
(a) Flow cytogram of CD4+ T cells from B16-bearing Foxp3Ametrine mice depicting sorting strategies for Treg cells based on their glucose uptake. Treg cells sorted based on 2NBDG uptake were assayed for their ability to suppress the proliferation of CellTrace Violet (CTV) labeled Tconv cells. Representative histogram shows 1:4 ratio of Treg:Tconv. (**p=0.0056). (b) Treg cell signature gene expression in RNAseq analysis of 2NBDGhi or 2NBDGlo Treg cells sorted from lymph node (LN) or tumor (TIL) of B16 melanoma-bearing Foxp3 reporter mice as in (a). (c) Production of IL-10, IFN-γ, and IL-2 by sorted 2NBDGlo and 2NBDGhi Treg cells stimulated overnight with PMA/ionomycin and stained intracellularly (*p=0.02). (d) Diagram and colon histology Rag1−/− mice that received either 2NBDGlo or 2NBDGhi Thy1.2+ Treg cells plus Thy1.1+ Tconv cells I.V and followed for 7 wks. (e) Percent of transferred 2NBDGlo or 2NBDGhi Treg cells as in (d) expressing Foxp3 within the spleen (Spln), mesenteric lymph node (mLN), intraepithelial layer (IEL), and lamina propria (LP) (*p=0.02, **p=0.004). (f) Representative histogram and tabulation of GlucoseCy5 uptake by Treg cells isolated from the non-draining lymph node (ndLN), pancreatic lymph node (pLN), and the islet of 10-12-week-old NOD.Foxp3GFP-Cre mice (**p=0.005). (g) (left) Diagram of experimental procedure and (right) capacity of Treg cells conditioned in 0,5, or 25mM glucose media to suppress the proliferation of CellTrace Violet (CTV) labelled Tconv cells. (1:4 *p=0.031, 1:8 *p=0.012 between 0 and 25 mM). (h) Glycolytic pathway gene expression as in (b). Results are representative of three (a,b,c,g,h), or two (d,e,f) independent experiments. Significance (*p< 0.05, **p <0.01, ***p < 0.001, ****p<0.0001) was determined by unpaired two-tailed t test (a,c,d,e,f), or two-way ANOVA with Tukey’s multiple comparison test (g). Data are mean values of biological replicates ±SEM.
We next asked whether exposure to a high glucose environment would drive a less suppressive phenotype, regardless of glucose uptake status. Remarkably, conditioning Treg cells in high glucose dampened suppressive function when later assayed in isoglycemic conditions (Fig. 2g, Extended Data Fig. 2g). We further interrogated the transcriptome of 2NBDGlo Treg cells to identify metabolic pathways that may support their function. 2NBDGlo Treg cells did not possess a transcriptome suggesting preferential lipid metabolism; rather, lipid metabolism genes were enriched in the 2NBDGhi subset (Extended Data Fig. 2h). Both subsets expressed upstream enzymes in glycolysis, suggesting proclivity for glucose was not regulated at the level of transporter or enzyme expression (Fig. 2h). The 2NBDGlo subpopulation, however, had enriched expression for the terminal steps of glycolysis: specifically lactate dehydrogenase (Ldha), and the monocarboxylate transporter MCT1 (encoded by Slc16a1) (Fig. 2h), confirmed by qPCR (Extended Data Fig 2i). While lactate is the end-product of glycolysis, it represents a significant fuel source for many cells20. Lactate uptake is mediated through MCT1, where it is converted by LDH to pyruvate21. Treg cells (but not Tconv cells) conditioned in low or glucose-deficient media increased expression of Ldha and Slc16a1 (Extended Data Fig. 2j). Thus, Treg cells upregulate genes involved in lactate metabolism.
Treg cells can metabolize lactic acid
Lactic acid is highly enriched in the TME22 (Fig. 3a) and known to be immunosuppressive23,24, and, indeed, it curbed function of conventional T cells in vitro (Extended Data Fig. 3a,b). Conversely, both Treg cell suppressor function and proliferation were resistant to tumor-equivalent concentrations of lactic acid (Extended Data Fig. 3a,c). Lactate can stimulate cells via GPR8125 and enter as a metabolite via MCT1, so we asked if Treg cells could consume lactic acid by measuring pH changes26. Lymphocytes from Foxp3YFP-Cre reporter mice were loaded with intracellular pH dye and incubated with lactic acid. Treg cells, but not Tconv cells, took up lactic acid, evidenced by increased fluorescence (Fig. 3b). This assay revealed lactic acid uptake was also heterogeneous within various tissue-derived Treg cells, akin to analysis of glucose uptake, notably low in tissues where Treg cells were glucose avid, such as the liver (Fig. 1g, 3c). Indeed, most dye+ Treg cells did not take up glucose (Extended Data Fig. 3d). In autoimmune NOD mice, islet Treg cells took up less lactic acid than those from the ndLN or pLN (Fig. 3d). pH dye+ Treg cells from LN and B16 TIL possessed increased CD44 and Nrp1 expression, while maintaining similar Foxp3 expression to dye negative cells (Fig. 3e). Thus Treg cells are not only resistant to lactic acid but can take up this metabolite, and those that do display an activated Treg cell signature and low glucose uptake.
Figure 3. Treg cells metabolize lactic acid to support their proliferation and suppressor function.
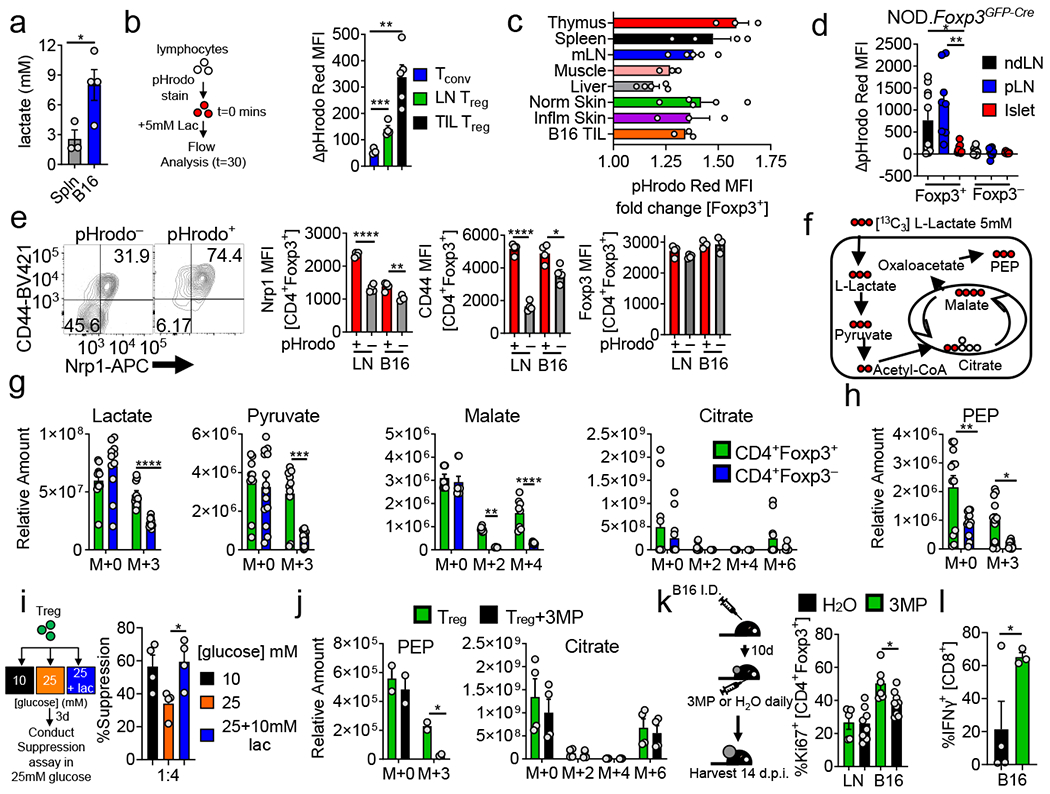
(a) Lactate concentration in spleen or B16 melanoma interstitial fluid (*p=0.041). (b) Lymphocytes from Foxp3Cre mice were loaded with pHrodo Red (intracellular pH dye) and pulsed with 5mM lactic acid. Results are change of MFI from t=0 (**p=0.0012). (c) Lactic acid uptake determined as in (b) by Treg cells infiltrating various tissues. (d) Lactic acid uptake from Treg and Tconv cells isolated from islets, non-draining (ndLN), and pancreatic lymph node (pLN) of 10-12-week-old NOD.Foxp3GFP-Cre mice (*p=0.017, **p=0.001). (e) Flow cytogram and tabulation depicting Nrp1 and CD44 expression in Treg cells based on lactate-elicited pH change (*p=0.018, **p=0.0097). (f) Diagram showing incorporation of 13C derived from lactate into downstream metabolites. (g) Relative abundance determined by mass spectrometry of intracellular lactate, pyruvate, malate, and citrate in Treg and Tconv cells activated overnight then pulsed with uniformly labeled 13C-lactate (m+n equal to the number of incorporated heavy carbons) (**p=0.0025). (h) Relative abundance of PEP derived from 13C-lactate as in (g) (*p=0.036, **p=0.0011). (i) Capacity of Treg cells conditioned for 3 days in 25mM glucose media ±10mM lactic acid to suppress the proliferation of CellTrace Violet (CTV) labelled Tconv cells (*p=0.021). (j) As in (h) with the addition of PEPCK inhibitor 3MP (*p=0.018). (k) Ki67 expression in intratumoral Treg cells of B16 tumor-bearing mice treated with 3MP or water for 3 days (*p=0.015). (l) IFN-γ expression by B16-infiltrating CD8+ T cells from mice treated as in (k) for 5 days. Results are representative of three (a,c,e,g,h,k), or two (b,d,i,j,l) independent experiments. Significance (*p< 0.05, **p <0.01, ***p < 0.001, ****p<0.0001) determined by unpaired two-tailed t test (a,b,d,e,i,j,k,l) or unpaired one-tail t test (l) or two-way ANOVA with Sidak's multiple comparisons test (g,h). Data are mean values of biological replicates ±SEM.
To identify how Treg cells were utilizing lactic acid, we performed high resolution mass spectrometry on activated Treg and Tconv cells pulsed with [U13C]-L-lactate (at pH 6.9) (Fig. 3f). We used LN-derived Treg cells (which do readily take up lactate, Fig. 3b) to facilitate cellular input needed for MS analysis, confirming Treg cells took up significantly more lactate than Tconv cells (Fig. 3g). Treg cells converted 13C-lactate into pyruvate and subsequently into citrate and malate indicating mitochondrial import and entry to the TCA cycle (Fig. 3g). Further analysis of revealed Treg cells also incorporated lactate-derived carbon into phosphoenolpyruvate (PEP) (Fig 3h), formed by phosphoenolpyruvate carboxykinase (PEPCK) when malate leaves the mitochondria and is converted to oxaloacetate. PEP can contribute to upstream glycolytic intermediates essential for proliferation, suggesting lactate may serve as a gluconeogenic fuel source, decreasing a Treg cell’s need for glucose. As high glucose can inhibit Treg cell suppressor function (Fig. 2g), we asked whether lactic acid could mitigate the deleterious effects of glucose. Indeed, conditioning Treg cells in high glucose plus tumor-equivalent concentrations of lactic acid maintained suppressor function (Fig 3i).
We then used the PEPCK inhibitor 3-mercaptopicolinic acid (3MP)27 to dissect the individual contributions of lactate uptake and oxidation from upstream gluconeogenic reactions dependent on lactate-derived PEP. 3MP treatment of 13C-lactate pulsed Treg cells reduced PEP accumulation without impacting incorporation into the TCA cycle (Fig 3j). In vitro 3MP treatment of Treg cells in lactic acid significantly reduced their proliferation (Extended Data Fig. 3i). However, 3MP did not impact suppressive capacity of Treg cells in vitro (Extended Data Fig. 3j). Treating tumor-bearing mice with 3MP decreased proliferation of tumor-resident, but not LN, Treg cells, and increased intratumoral CD8 IFN-γ production (Fig. 3k,l). However, it did not overtly affect the intratumoral percentage of Foxp3+ cells, Tconv/CD8:Treg cells ratios, overt tumor growth, or suppressor function when interrogated ex vivo (Extended Data Fig. 3f–h). Thus Treg cells utilize lactic acid to not only feed the TCA cycle, but to generate PEP, critical for fueling Treg cell proliferation within the tumor.
Lactic acid supports intratumoral Treg cells
To determine the significance of lactic acid utilization by Treg cells in vivo, we generated a constitutive, Treg-specific deletion of MCT1 (Slc16a1f/fFoxp3Cre). qPCR confirmed Slc16a1 deletion, lack of compensation by Slc16a7/MCT2 or Slc16a3/MCT4 upregulation, and no deletion in CD4+Foxp3− cells (Extended Data Fig. 4a,b). Expectedly, deletion of Slc16a1 resulted in loss of lactic acid uptake (Fig. 4a). These mice did not show any overt signs of autoimmunity or lymphoproliferation (Extended Data Fig. 4c,d). Rather, Treg cells from the LN of Slc16a1f/fFoxp3YFPCre mice displayed similar suppressive capacity, proliferation, and expression of Treg signature genes when assayed ex vivo (Extended Data Fig. 4e–h). MCT1 deficiency in Treg cells revealed no significant differences in percentage of Foxp3+ cells infiltrating various tissues, although several compartments showed increases in glucose uptake at the steady-state (Extended Data Fig. 4i,j). Imiquimod treatment resulted in no increased inflammatory ear thickness, nor loss of Treg cell markers in Slc16a1f/fFoxp3Cre mice (Extended Data Fig. 4k,l). We also determined long-term function and stability of MCT1-deficient Treg cells in transfer colitis. Unlike 2NBDGhi Treg cells, MCT1-deficient Treg cells did not show reduced stability (Extended Data Fig. 4m). However, they were not completely functional: at 7 weeks, mice receiving MCT1-deficient Treg cells had more histologically severe colitis, although less severe than mice receiving 2NBDGhi Treg cells (Extended Data Fig. 4n,o). Thus, MCT1 deficiency appeared dispensable for Treg cell survival and function, although under highly inflammatory conditions they may have decreased activity.
Figure 4. Tumor-infiltrating Treg cells require lactate uptake to maintain their high suppressive function.
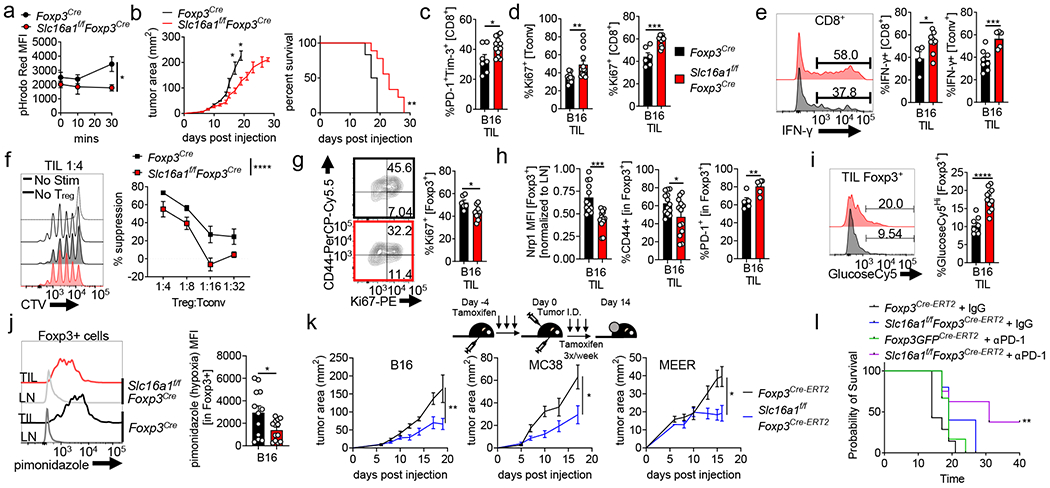
(a) Lactate-elicited pHrodo fluorescence in Foxp3Cre or Slc16a1f/fFoxp3Cre Treg cells (*p=0.041). (b) B16 tumor growth and survival of Foxp3Cre and Slc16a1f/fFoxp3Cre mice (*p=0.0235) (c) Percent of B16-infiltrating PD-1+Tim-3+ CD8+ T cells day 14 post tumor injection in mice as in (a) (*p=0.017). (d) Proliferation of tumor infiltrating Tconv and CD8+ T cells as in (c) (**p=0.009). (e) Tumor-infiltrating CD8+ and Tconv cells restimulated with PMA/ionomycin and stained for IFN-γ (*p=0.039). (f) Capacity of WT or Slc16a1-deficient tumor-infiltrating Treg cells to suppress proliferation of CTV labeled Tconv cells. (g) Percent proliferating tumor infiltrating Treg cells from mice as in (c). (*p=0.011). (h) Percent or MFI of WT and Slc16a1-deficient Treg cells expressing Nrp1, PD-1, CD44 from B16 tumors (TIL) as in (c) (*p=0.029, **p=0.009). (i) GlucoseCy5 uptake by B16 tumor-infiltrating Treg cells as in (c) (j) Pimonidazole staining of WT and Slc16a1-deficient Treg cells from B16 tumors as in (c) (*p=0.029). (k) Tumor growth of B16 (melanoma, **p=0.0011) MC38 (adenocarcinoma, *p=0.023), and MEER (HNSCC, *p=0.0146) in tamoxifen treated Foxp3Cre-ERT2 and Slc16a1f/fFoxp3Cre-ERT2 mice. Mice were given tamoxifen 5 consecutive days prior to tumor injection then 3x per week. (l) Survival of tamoxifen treated Foxp3Cre-ERT2 and Slc16a1f/fFoxp3Cre-ERT2 mice injected with B16 and treated with IgG or anti-PD-1 antibodies 3x weekly (**p=0.002). Results are representative of four (b,c,d,f,g,h(Nrp1, CD44),i,j), three (a, k(B16)), or two (h(PD-1), k(MC38, MEER),l) independent experiments. Significance (*p< 0.05, **p <0.01, ***p < 0.001, ****p<0.0001) determined by the log-rank test for survival curves (b,l) or unpaired two-tailed t test (a,c,d,e,g,h,i,j) or two-way ANOVA (b,f,k). Data are mean values of biological replicates ±SEM.
However, we reasoned MCT1-mediated lactate uptake would be most important within lactate-rich tumor tissue. Inoculating Slc16a1f/fFoxp3Cre mice with B16 melanoma resulted in slowed tumor growth and prolonged survival (Fig. 4b). Characterizing the infiltrate when both genotypes harbored tumors of similar sizes (day 14) showed while CD8+ T cells in Slc16a1f/fFoxp3Cre mice exhibited higher coinhibitory markers PD-1 and Tim-3, indicative of terminal differentiation (Fig. 4c), CD8+ and Tconv cells were more proliferative and competent to produce IFN-γ, suggesting decreased suppressive function by MCT1-deficient Treg cells (Fig. 4d, e). Indeed, intratumoral MCT1-deficient Treg cells had reduced suppressive function ex vivo (Fig. 4f) and were less proliferative, resembling intratumoral Treg cells from 3MP-treated mice (Fig. 4g, 3k). Analysis of 3MP-treated Slc16a1f/fFoxp3Cre mice suggested 3MP’s effects on Treg cells was mediated through MCT1 (Extended Data Fig 3k,l). Characterization of intratumoral MCT1-deficient Treg cells revealed decreases in CD44 and Nrp1 staining, concomitant with elevated PD-1 staining, potentially indicating dysfunctional Treg cells28 (Fig. 4h). Metabolically, MCT1-deficient, tumor-resident Treg cells compensated by becoming glucose avid (Fig. 4i). We found, as a consequence, that MCT1-deficient Treg cells would no longer persist within the lactate rich hypoxic areas of tumors, as measured by pimonidazole staining, despite overall tumor hypoxia remaining similar (Fig. 4j, Extended Data Fig. 4p).
To remove any unforeseen effects of MCT1 deletion on Treg cell development, we interrupted MCT1 expression just prior to B16 melanoma injection using a tamoxifen-inducible, Treg cell-specific model (Slc16a1f/fFoxp3Cre.ERT2) and showed similar immunologic and survival outcomes, including the development of a ‘fragile’29 Treg cell phenotype in which Foxp3+ cells begin expressing IFN-γ (Extended Data Fig. 5). Induced Treg deletion of MCT1 also slowed tumor growth of MC38 adenocarcinoma and MEER HNSCC (Fig. 4k, Extended Data Fig. 5g,h). We thus reasoned loss of lactate uptake in Treg cells produced an environment conducive to immunotherapy. Triggering deletion of MCT1 in Treg cells synergized with anti-PD-1 therapy, resulting in complete regressions in 37.5% of B16-bearing mice (typically insensitive to anti-PD-1) (Fig. 4l). Deletion of MCT1 from Tconv cells using Slc16a1f/fCd4Cre mice did not impact their capacity to produce IL-2 and IFN-γ, suggesting MCT1 was not generally required for T cell function (Extended Data Fig 4q). Thus, MCT1 expression and consequent lactic acid uptake is dispensable for most tissue derived Treg cells but required to maintain high suppressor activity in the TME.
Our study highlights the antagonistic effects of glucose on Treg cell function and identity. While previous data support this9,10,23,30, some studies using transcriptomic and proteomic analysis suggest Treg cells are highly glycolytic31–33. We demonstrate Treg glucose consumption is certainly heterogeneous between tissues (Fig. 1g) and within the Treg population (Fig 2a); however, glucose uptake characterizes poorly suppressive Treg cells, or at least those suppressing via mechanisms not potent in vitro coculture (Fig. 2a,c Extended Data Fig. 2a). Importantly, Treg cells consuming low amounts of glucose still express glycolytic pathway genes (Fig. 2h). As most reactions in glycolysis are reversible, Treg cells likely build higher-order intermediates from carbon sources like lactate (Fig. 3).
Lactate is often thought of as a waste product of glycolytic metabolism, but it represents an important metabolite for cellular function and a likely fuel source for tissues20. Our data suggest Treg cells do not require lactic acid for survival, but are endowed with metabolic flexibility to use this carbon source, both as fuel and as a means to protect their high suppressive capacity from negative effects of glucose (Fig 3i). Consequently, cells do not possess both high lactic acid and glucose uptake (Fig 1g, 2f, 3c,d, Extended Data Figure 3d). Thus, we propose a glucose:lactate axis exists to fine-tune Treg cell functioning depending on the nutrient milieu (Fig 2g, 3i). Lactate not only fuels the TCA cycle but is also exported from the mitochondria, contributing to higher glycolytic pathways via PEP. This gluconeogenic component of lactate metabolism supports Treg cell proliferation, critical in a cell type that does not readily take up glucose. However, the contribution of PEPCK-independent lactate metabolism appears also to be at the level of Treg cell suppression (Fig. 4f), suggesting lactate plays a multi-pronged role in Treg cell biology.
It is unlikely regulatory T cells evolved to thrive in tumors; rather cancers exploit this predilection for alternative substrates to maintain an immunosuppressive environment. Lactate is not only enriched in the tumor but is elevated in immunologically distinct tissue environments such as muscle, adipose, and nervous system34,35. We also suspect lactate is one of a host of alternative metabolites Treg cells can utilize. MCT1 itself can also transport acetate, succinate, propionate, and butyrate, which have been shown to play roles in Treg cells21,36,37. Indeed, our findings using transfer colitis suggest MCT1 deficiency may impact Treg cell function in the gut. Notably, however, we did not observe overt gut inflammation at the steady state in Slc16a1f/fFoxp3Cre mice, suggesting both inflammation and MCT1 substrate availability play roles in maintaining Treg cellsin that environment..
Our study suggests that, as Treg cells must be active when immune activity is at its lowest, they exist metabolically ‘out-of-sync’ with their conventional counterparts. This requires utilizing fuels prevalent in tissue environments to maintain suppressor function when their conventional T cell targets may be competing for nutrients. As lactate can promote suppressive functions of other tolerogenic cell types, like tumor-associated macrophages38, cell types sharing lactate metabolism may share suppressor activity. While this is exploited in the tumor microenvironment, causing suppressive cells to thrive, it is actionable: Treg cell-specific deletion of the lactate transporter not only results in decreased tumor growth but synergy with checkpoint blockade immunotherapy. MCT1 inhibition to directly target lactate metabolism or inhibition of tumor acidity may break this metabolic symbiosis and lower the regulatory T cell barrier to cancer immunity.
MATERIALS AND METHODS
In Vitro Tissue Culture
B16-F10 were obtained from ATCC. MC38 cells were obtained from Dario Vignali (commercially available from Kerafast). Clone24 was originally produced by single cell sorting melanoma tumors arising from Ptenf/fBrafLSL-V600ETyrCreER mice5. MEER cells were obtained from Robert Ferris (originally from 39). B16 cell line was authenticated in 2018 by independent sequencing. MC38 cells were authenticated in 2016 by independent sequencing. Clone24 was authenticated via metabolic profiling and tumor growth compared to Melan-A (normal melanocytes) in 20195. MEER cells were authenticated in 2013 via immunoblot to show E6/E7 and Ras overexpression in MTEC cells. MC38 and MEER were confirmed mycoplasma free in 2016, B16 were confirmed in 2018, Clone24 in 2019. Both primary and immortalized cell lines were maintained in lab-made R10 media [RPMI1640, 10%FBS, 2mM L-glut, PenStrep, NEAA, 1mM Sodium pyruvate, 5mM HEPES, β-ME]. Cultures were incubated in temperature and partial pressure-stable conditions at 37°C and 5% CO2. For in vitro conditioning experiments, isolated Treg or Tconv cells were activated in complete R10 media with 0.1 ug/mL Phorbol 12-myristate 13-acetate (PMA) and 1ug/mL ionomycin (Sigma-Aldrich) with 1000U/mL IL-2 for ~20 hours at 37°C then placed in the conditions indicated in the figure with IL-2. For in vitro 3-mercaptopicolinic acid (3MP, Cayman Chemical) experiments a 250 μM concentration was used.
In Vivo Mice Studies
Sample sizes were not statistically predetermined but were chosen based on previous work in Treg cell functional assays8,29 and tumor immunology4,5,8,14,29. These sample sizes are sufficient because they allowed for the determination of statistical significance between groups and minimized the number of animals or replicates needed for each experiment. Mice were placed into experimental group by nature of their genotype and/or if receiving treatment were randomized within a genotype. For experiments not involving mice, cells were randomized into experimental groups. Blinding was not possible as most of the data acquisition and analysis was done by a single person; two individuals performing every experiment was not feasible during the course of our study. C57BL/6J-Foxp3YFPiCre, -Foxp3GFP.Cre.ERT2, -Rag1−/−, and -Thy1a (Thy1.1 congenic) were purchased from The Jackson Laboratory. Slc16a1f/f mice were a gift from Jeffrey Rothstein and Brett Morrison40 used to generate Slc16a1f/fFoxp3GFP.Cre.ERT2 (termed Slc16a1f/fFoxp3Cre-ERT2) and Slc16a1f/fFoxp3YFP.Cre (termed Slc16a1f/fFoxp3Cre) mice. Foxp3FlpO Ametrine mice were a gift from Dario AA Vignali. Spleens of OT-II Foxp3RFP Thy1.1+ mice were a gift from Geoffrey Camirand. Animal work was done in accordance with the Institutional Animal Care and Use Committee of the University of Pittsburgh (protocol #17071235). All mice were housed in specific pathogen free conditions at an ambient temperature 20-26°C and humidity of 30-70% with a 12:12 hour light:dark cycle prior to use. The maximal tumor size of 15 mm in any direction was not exceed in any experiment. Both male and female mice were used in studies, from five (5) to ten (10) weeks of age.
Flow Sorting and Cytometric Analysis
Single-cell suspensions from murine tumors (day 14 post tumor inoculation), lymph node, liver, thymus, or spleen were derived through mechanical separation and 70 μM filter (Fisherbrand) passage. For hypoxia detection, mice were injected intravenously with pimonidazole (80mg/kg, Hypoxyprobe) in PBS 1.0 hour before sacrifice. Tumors dissolution was aided through lab-made tumor lysis buffer [ 2mg/mL Collagenase Type IV (Gibco), 2U/mL Dispase in Hanks’ Balanced Salt Solution (Stemcell Technologies), 10U/mL Deoxyribonuclease I (Sigma), serum free RPMI medium] injected with a 20G needle and incubated for 30 minutes at 37°C. Prior to sort tumor suspensions were purified by negative selection with MojoSort magnetic bead separation (BioLegend) and biotin-conjugated anti-mouse antibodies to CD105 (MJ7/18, Cat# 120403, Lot# B266720, BioLegend).The skin (ears of mice were used), were finely minced using scissors and resuspended in RPMI1640 (Gibco, Grand Island, NY) with 2.5 mg/mL collagenase XI (Sigma-Aldrich), 0.25 mg/mL hyaluronidase (Sigma-Aldrich), 0.1 mg/mL DNase (Sigma-Aldrich), 0.01 M HEPES (Sigma-Aldrich), and 10% FBS followed by incubation in a shaking incubator for 1 h at 37C at 250 rpm. Following incubation skin was subjected to mechanical separation and passed through a 70 μM filter. Similarly, muscles (quadriceps were used) were finely minced using scissors then resuspended in 0.05% type II collagenase (ThermoFisher) in HBSS for 30 min at 37 °C under constant agitation. Islets were isolated as previously described41. Pimonidazole was visualized using anti-pimonidazole antibodies (Hypoxyprobe, Cat# HP7-100, Lot# 5914) after 5 minutes of 4% PFA fixation followed by the FoxP3 Fix/perm kit (Biolegend). Staining for expression markers was performed with anti-mouse specific antibodies obtaining from the following companies; Biolegend: anti-CD4 (GK1.5, Cat# 100412, Lot# B184560, dil 1:1000), CD8 (53-6.7, Cat# 100707, Lot# B171971, dil 1:1000), CD39 (Duha59, Cat# 143806, Lot# B186014, dil 1:500), CD44 (IM7, Cat# 103032, Lot# B267976, dil 1:500), CD73 (TY/11.8, Cat# 127215, Lot# B260585, dil 1:500), CD279 (PD-1, 29F.1A12, Cat# 135221, Lot# B194160, dil 1:250), HAVcr-2 (TIM-3, RMT3-23, Cat# 119705, Lot# B224472, dil 1:250), Helios (22F6, Cat# 137220, Lot# B155089, dil 1:250), IFNγ (XMG1.2, Cat# 505842, Lot# B270630, dil 1:250), Ki67 (16A8, Cat# 652403, Lot# B208445, dil 1:250), CD304 (Nrp-1, 3E12, Cat# 145212, Lot# B175814 dil 1:200), TNFα (MP6-XT22, Cat# 506322, Lot# B218553, dil 1:500). BD Horizon: CD62L (MEL-14, Cat# 564109, Lot# 7341887, dil 1:500). eBioscience: FoxP3 (FJK-16s, Cat# 53-5773-82, Lot#2011698, dil 1:250). Intracellular staining was performed using FoxP3 Fix/Perm buffer set (BioLegend) as per manufacturer’s protocol and staining was performed on overnight at 4°C. Stained cells were analyzed on a LSRFortessa (BD). Cell doublets were excluded by comparison of side-scatter and forward-scatter width to area. Flow cytometry data and proliferation modeling were analyzed with FlowJo v10 software (Tree Star) and figures were produced in Prism v8 (GraphPad).
Glucose Uptake Assay
Single cell suspensions of tumor and lymph node and other tissues of ~1 million cells/mL were placed in serum free RPMI containing 40 μM 2NBDG or 0.4 μM Glucose-Cy5 for 25 minutes at 37°C. Glucose-Cy5, a Cy5-linked 1-amino-glucose tracer was synthesized in collaboration with Dr. Marcel Bruchez17. For OT-II experiments, CD4+ T cells were isolated via negative selection using magnetic bead separation as previously described4 then injected retroorbital into C57BL/6J mice. Simultaneously, recipient mice were injected I.P. with 1 x 106 vaccinia-OVA viral particles and harvested 5 days later.
Microsuppression & Proliferation Assays
In vitro microsuppression assays were performed as previously described29. Briefly Treg cells (CD4+FoxP3+) populations were isolated via flow assisted sorting from tumor or lymph node from C57BL/6J-Foxp3GFP.Cre.ERT2, -Foxp3YFP.Cre mice or mice crossed to these strains. Responder cells (CD4+Foxp3− Tconv) and antigen presenting cells (CD4−CD8− APCs) were isolated via flow assisted sorting from the spleen of a C57BL/6J-Thy1a mouse. Treg cells were co-cultured for 72 hours at 37°C with APCs and CellTrace Violet (CTV, ThermoFisher Scientific) labeled responder cells at ratios from 1:2 (Treg:responder) to 1:32 in complete RPMI media with 1ug/mL anti-CD3. Proliferation assays were performed by labeling isolated Treg, Tconv, and CD8 T cells with CTV then activating at 1 million cells/mL using anti-CD3/CD28 Dynabeads according to the manufacture’s protocol (ThermoFisher Scientific) and IL-2 in complete RPMI media containing 0 to 10mM lactic acid (Fisher Bioreagents). Cell proliferation of responding cell populations was modeled using FlowJo v10 software (Tree Star).
RT-qPCR
Total RNA was extracted using Trizol reagent (Invitrogen) and cDNA was transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), according to manufacturer protocol. Transcript levels were measured with SybrGreen (ThermoFisher Scientific) and using primers specific to genes of interest, including; Slc16a1: FWD 5’-GCAGTGTTAGTCGGAGCC-3’ REV 5’-GCGATCATTACTGGACGGC-3’, Ldha: FWD 5’-AGCTTCCATTTAAGGCCCCG-3’ REV 5’-TCTTTTGAGACCGCTAGTGC-3’ cDNA concentration was normalized per samples relative to β-actin. All experiments were performed in technical triplicates.
Cytokine Production Assay
Cell suspensions were stimulated in complete R10 media with 0.1 ug/mL Phorbol 12-myristate 13-acetate (PMA) and 1ug/mL ionomycin (Sigma-Aldrich) for ~20 hours at 37°C. Five hours prior to antibody staining, Golgi-Plug (BD Biosciences) was added to the samples. Cells were surfaced stained and then intracellular stained using FoxP3 Fix/Perm buffer set (Invitrogen) as per manufacturer’s protocol. Unstimulated controls were used to determine gating strategies.
Interstitial Lactate Quantification
Tumor and spleen were harvested and placed in empty 15mL conical tubes. Tissues were cut up with scissors then wrapped with a 5-micron Nylon filter paper (Sterlitech) and stuffed filter down into a 1.5 mL conical tube making sure the tissue did not touch the bottom. Tissues were centrifuged at 4000 rpm for 2 hours. Interstitial fluid was assayed for L-lactate concentration using a colorimetric detection kit according to manufacturer’s protocol (abcam).
Isotopic Flux Analysis
Cell suspensions at 1 million cells/mL in complete RPMI media were stimulated for 24 hours at 37°C with plate-bound anti-mouse CD3ε (10 μg for Treg, 3 μg for Tconv, clone 145-2C11, Cat#100239, Lot# B306297, Biolegend) and soluble anti-mouse CD28 (2 μg/mL, clone 37.51, Cat#102115, Lot#B283231, Biolegend) and IL-2 (1000U/mL Treg 50U/mL Tconv). After activation media was exchanged for complete RPMI with 5mM uniformly labeled 13C-L-Lactate (Sigma Aldrich) and IL-2. For some experiments PEPCK inhibitor, 3-mercaptopicolinic acid (3MP, Cayman Chemical) was added at 250 μM. Media was then titrated to pH 6.88 with 1 N HCl. Suspensions were pulsed for 25 hours then washed 2x with room temperature PBS then resuspended in ice cold 80% methanol.
Metabolic quenching and polar metabolite pool extraction was performed using ice cold 80% methanol in water with 0.1% formic acid at a ratio of 400μL per 100μL. Deuterated (D4)-taurine and (D3)-lactate (Sigma-Aldrich) was added to the sample lysates as an internal standard for a final concentration of 100μM. The supernatant was cleared of protein by centrifugation at 16,000xg. 5μL of cleared supernatant was subjected to online LC-MS analysis.
Analyses were performed by untargeted LC-HRMS. Briefly, Samples were injected via a Thermo Vanquish UHPLC and separated over a reversed phase Phenomenex Kinetex C18+ column (2.1×100mm, 1.7μm particle size) maintained at 40°C. For the 20 minute LC gradient, the mobile phase consisted of the following: solvent A (1.5mM ammonium fluoride) and solvent B (100% acetonitrile). The gradient was the following: 0-12.0 min 5% B, to 1000% B, 12.0-15.0 min hold at 100% B, 15.0-15.1100% to 5% B, 15.1-20.0 min 5%B. The Q Exactive mass spectrometer was operated in polarity switching mode, using both positive and negative ion mode, scanning in Full MS mode (2 μscans) from 66.7 to 1000 m/z at 70,000 resolution with an AGC target of 3e6. Source ionization settings were 4.5/3.0 kV spray voltage respectively for positive and negative mode. Source gas parameters were 20 sheath gas, 10 auxiliary gas at 250°C, and 4 sweep gas. Calibration was performed prior to analysis using the PierceTM Positive and Negative Ion Calibration Solutions (Thermo Fisher Scientific). Integrated peak areas were then extracted manually using Quan Browser (Thermo Fisher Xcalibur ver. 2.7). Fully untargeted analysis was completed by using Thermo Compound Discoverer 3.0 software suite to extract and align molecular features. Graphs and statistical analyses (either t-test or ANOVA) were prepared with GraphPad Prism 7.0 (GraphPad Software, Inc., La Jolla, CA, USA).
In Vivo Tumor Growth and Therapy
Slc16a1f/fFoxp3Cre or Foxp3Cre mice were inoculated with 1.5 x 105 B16-F10 cells in complete RPMI media. To monitor tumor growth, tumors area was determined 3x weekly using digital calipers and stopped once tumors reached 15mm in any direction. For 3MP treated mice, Foxp3Cre or Foxp3Cre.ERT2 mice were inoculated with 1.5 x 105 B16-F10 cells. Starting on day 10 or 12 post tumor inoculation mice were treated daily either with water or 25 mg/kg 3MP (Caymen Chemical) via intraperitoneal (I.P.) injection. On day 14 one hour prior to sacrifice, mice were given a final dose of 25 mg/kg 3MP or water. For 3MP tumor growth, treatment started day 7 post tumor injection and continued daily with 25 mg/kg 3MP or H2O I.P. until tumors reached 15mm in any direction.
For the inducible deletion of MCT1 (Slc16a1) Slc16a1f/fFoxp3GFP.Cre.ERT2 or Foxp3GFP.Cre.ERT2 mice were treated I.P. or P.O. with 1 mg of tamoxifen (T5648, Sigma) in corn oil (C8267, Sigma) from day −4 to 0. On day 0, mice were inoculated intradermally with 2.5x105 B16-F10, MC38, or MEER cells in complete RPMI media and given a dose of 1mg Tamoxifen. Tamoxifen treatment continued 3x weekly until the conclusion of the experiment. On day 7, when tumors reached 1-10 mm2 mice were treated with 0.2mg anti–PD-1 or hamster IgG isotype control (Bio X Cell) 3x weekly until the conclusion of the experiment.
pHrodo Red Lactic Acid Uptake Assay
Cell suspensions from lymph nodes and day 14 B16-F10 tumors were loaded with pHrodo Red AM (ThermoFisher Scientific) according to manufacturer’s protocol in a 20mM HEPES in PBS solution. Cells were surfaced stained for multicolor flow cytometry following the normal protocol in 20mM HEPES/PBS buffer. At the flow cytometer, lactic acid was spiked into each sample at a final concentration of 5mM pH ~6.7. Samples were read at 0, 10, and 30 mins after addition of lactic acid.
Extracellular flux analysis
Isolated Treg and Tconv cells were plated on Cell-Tak coated Seahorse culture plates (250,000/well) in media consisting of minimal, unbuffered DMEM supplemented with 2mM glutamine. Basal rates were taken for 30 min, and then streptavidin-complexed anti-CD3biotin at 3 μg/mL was injected and readings continued for 3 hrs. 2μM oligomycin and 10mM 2-DG were injected to obtain maximal and minimal ECAR values, respectively. Similarly, isolated Treg and Tconv cells were activated using plate bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (2 μg/mL) with 250 U/mL IL-2 for 48hrs then plated on Cell-Tak coated Seahorse culture plates (100,000/well) in media consisting of minimal, unbuffered DMEM supplemented with 2mM glutamine. Basal extracellular acidification rates were taken for 30 mins. Cells were stimulated with 10mM Glucose, 2μM oligomycin, 10mM 2-DG.
Transfer Colitis
The transfer colitis model was performed as previously described42. Briefly, 2NBDGhi and 2NBDGlo Treg cells from Foxp3-Ametrine reporter mice or Treg cells from Slc16a1f/fFoxp3Cre or Foxp3Cre were isolated via flow assisted sorting (CD4-APC+ Foxp3-reporter+). Tconv cells (CD4+ CD25−) from C57BL/6J-thy1a were isolated via flow assisted sorting. Rag1−/− mice were simultaneously given 2 x 104 sorted Treg cells and 1 x 105 thy1.1+ Tconv cells via retroorbital injection. Mice were weighed once a week for 7 weeks (experiment end). Sections of colons were fixed in 10% formalin and paraffin embedded. Slides were stained with hematoxylin and eosin by the University of Pittsburgh Pathology Department, and histology scores were blindly determined using criteria previously described42.
Tumor Histology
After pimonidazole pulsing, tumors were dissected and frozen at -80 deg C in Optimal Cutting Temperature Compound (OCT) (Tissue-Tek) and sectioned (Cryostat microtome). Tissue was fixed in histology-grade acetone (Fisher) at −20 deg C, then rehydrated in staining buffer, stained with hypoxyprobe (Hypoxyprobe, catalog# HP7-100Kit), and DAPI (Life Technologies), and mounted with ProLong Diamond AntifadeMountant (Life Technologies). Sections were imaged with an Olympus IX83 microscope and analyzed with ImageJ and NIS-Elements Imaging Software.
RNA Sequencing
1000 low glucose and high glucose consuming Treg cells were sorted directly into lysis buffer in a 96-well plate. Immediately after sorting cDNA was generated using SMART-seq HT kit (Cat # 634456 Clontech) using 1000 cells. cDNA product was checked by Tape Station D5000 from Agilent technologies 2200 to make sure cDNA was successfully generated. Library construction was done using Nextrera XT kit # 15031942 from Illumina. 1ng cDNA was used in total volume 5ul. Sequencing was done using NextSeq 500 System. High Output 75 Cycles kit with run Parameter Paired Read 150 cycles (2X75).
Sequencing reads were trimmed for adapters using Cutadapt43 prior to being aligned to Mus musculus reference genome (mm10) using the RNA-seq aligner HISAT2. Subread’s featureCounts function was used for gene level quantification and results were normalized to Transcripts Per Kilobase Million (TPM). Using the raw quantification, differential genes were found with the R package DESeq2 and statistical cutoffs of q-value <0.05 and |log2foldchange|>1.5.
Statistical Analysis
The data presented in the figures are mean ± standard error of the mean (S.E.M.). Multiple group comparison in in vivo and ex vivo assays was accomplished with one- or two-way analysis of variance (ANOVA). For single comparison, unpaired two-tailed Student’s t test was used, unless otherwise denoted. In cases of non-Gaussian distribution, nonparametric t-test was used. Survival and tumor growth in in vivo implantable tumor models are presented as Kaplan–Meier survival curves and plotted tumor area with respect to time and were statistically analyzed using log rank test. All analysis was completed with on Prism v5 software (GraphPad). A value of P < 0.05 is statistically significant. In the figures, standard designations of significance were given; *p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001. The specific analysis used per figure in the manuscript can be found within the legends.
Data availability
RNA sequencing data that support the findings of this study (Fig. 2b, h) have been deposited in GEO with the GSE158801 accession code. The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files.
Extended Data
Extended Data Figure 1. 1-amino-Cy5-glucose (GlucoseCy5) can act as a surrogate for 2NBDG in GFP/YFP Treg reporter mice.
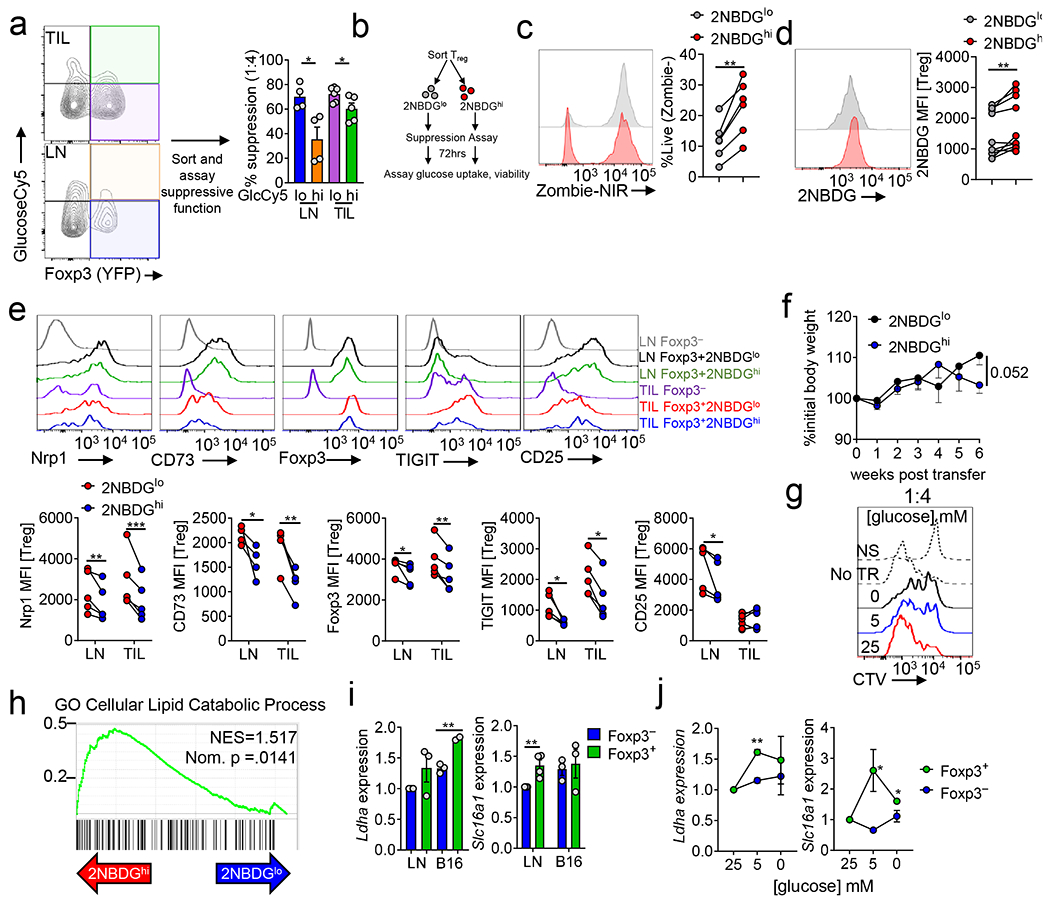
(a) Gating strategy for Treg and Tconv cells (b) Representative plot of percent Treg (CD4+Foxp3+) in the lymph node (LN) and tumor (TIL) of B16 bearing C57BL/6 mice day 14 post tumor inoculation. Representative plots gated on total CD4+ cells. (c) Glycolytic extracellular acidification rates (ECAR) of Treg and Tconv cells sorted from LN and B16 TIL preparations as in (b) 48hrs after activation with αCD3/CD28 and IL-2. Oligo = oligomycin, 2-DG = 2-deoxy-D-glucose, ΔECAR = max reading after glucose minus basal ECAR. Max ECAR = max reading at after oligo minus basal ECAR. (d) Ex vivo 2NBDG uptake mean fluorescence intensity (MFI) by Treg, Tconv, and CD8+ T cells from the LN and B16 TIL (**p=0.0045). (e) Ex vivo 2NBDG uptake by CD44+CD62L− and CD44−CD62L+ Treg and Tconv cells isolated from the LN of Foxp3-Ametrine reporter mice. (f) Lymphocytes from Foxp3-Ametrine reporter mice were simultaneously pulsed with 2NBDG and GlucoseCy5. Representative plot is gated CD4+ Foxp3+ with tabulation of percent GlucoseCy5+ of 2NBDG+ Treg. (g) Ex vivo GlucoseCy5 uptake by CD44+ Treg and Tconv cells from the LN and B16 TIL. Representative plots gated on CD44+ CD4+ cells (**p=0.0025). (h) GlucoseCy5 positivity in Nrp1 negative and positive Treg cells. (i) Ex vivo GlucoseCy5 uptake by Tconv cells isolated from various tissues. Results are representative of three (a-d,f-i) or two (e) independent experiments. Significance (*p< 0.05, **p <0.01, ***p < 0.001, ****p<0.0001) was determined by paired two-tailed t test (e), unpaired two-tailed t test (d,f,g,h) or two-way ANOVA with Sidak's multiple comparisons test (c). Data are mean values of biological replicates ±SEM.
Extended Data Figure 2. Glucose avid Treg cells harbor a weaker Treg cell signature but retain viability and some suppressor activity.
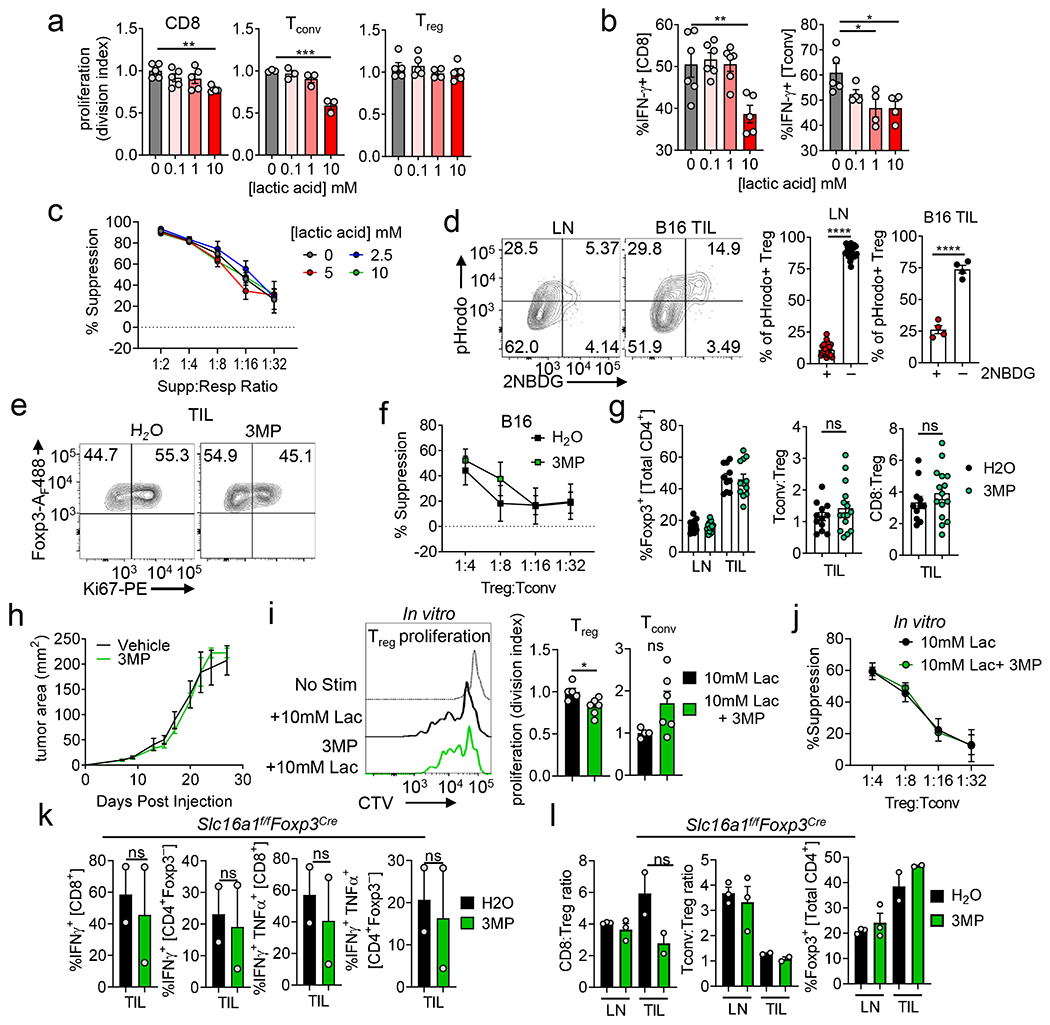
(a) Treg cells were sorted based on GlucoseCy5 uptake and assayed for their ability to suppress the proliferation of CellTrace Violet (CTV) labeled Tconv cells at 1:4 (Treg:Tconv) (LN*p=0.02, TIL *p=0.041). (b) Experimental diagram for (c, d). (c) Representative histogram and quantification of viability of sorted 2NBDGhi and 2NBDGlo Treg cells after 72hrs in a suppression assay. (d) 2NBDG uptake by 2NBDGhi or 2NBDGlo Treg as in (b). (e) Representative histograms and tabulation of Treg signature gene expression between 2NBDG low and high Treg cell subsets (Nrp1 **p=0.0016, CD73 *p=0.029 **p=0.0076, TIGIT LN*p=0.04 TIL*p=0.016, CD25 *p=0.037). (f) Weights of Rag1−/− mice that received an adoptive transfer of either 2NBDGlo or 2NBDGhi Treg cells plus Thy1.1+ Tconv cells I.V. (g) Representative histogram of Tconv responder cell proliferation after 72hrs of co-culture with Treg cells (1:4) conditioned in 0, 5, or 25mM glucose for 3 days. The suppression assay occurred in 25 mM glucose conditions. (h) Geneset enrichment plot of cellular lipid catabolic process from TIL 2NBDGhi vs 2NBDGlo Treg cells. (i) mRNA expression of Slc16a1 and Ldha in LN- and B16-infiltrating Treg and Tconv cells by qPCR (Ldha **p=0.004, Slc16a1 **p=0.009). (j) Slc16a1 and Ldha mRNA expression in Treg or Tconv cells activated overnight and conditioned in the glucose concentration indicated for 3 days (normalized to 25mM glucose, significance between Treg and Tconv) (Ldha **p=0.002 Slc16a1 5 *p=0.030, 0 *p=0.046). Results are representative of three (a,e,h,i,j), or two (c,d,f) independent experiments. Significance (*p< 0.05, **p <0.01, ***p < 0.001) determined by unpaired two-tailed t test (a,f,i,j) or paired two-tailed t test (c,d) or two-way ANOVA with Sidak’s multiple comparisons test (e). Data are mean values of biological replicates ±SEM.
Extended Data Figure 3. Treg cells are resistant to lactic acid and use PEPCK-mediated metabolic pathways to support their proliferation.
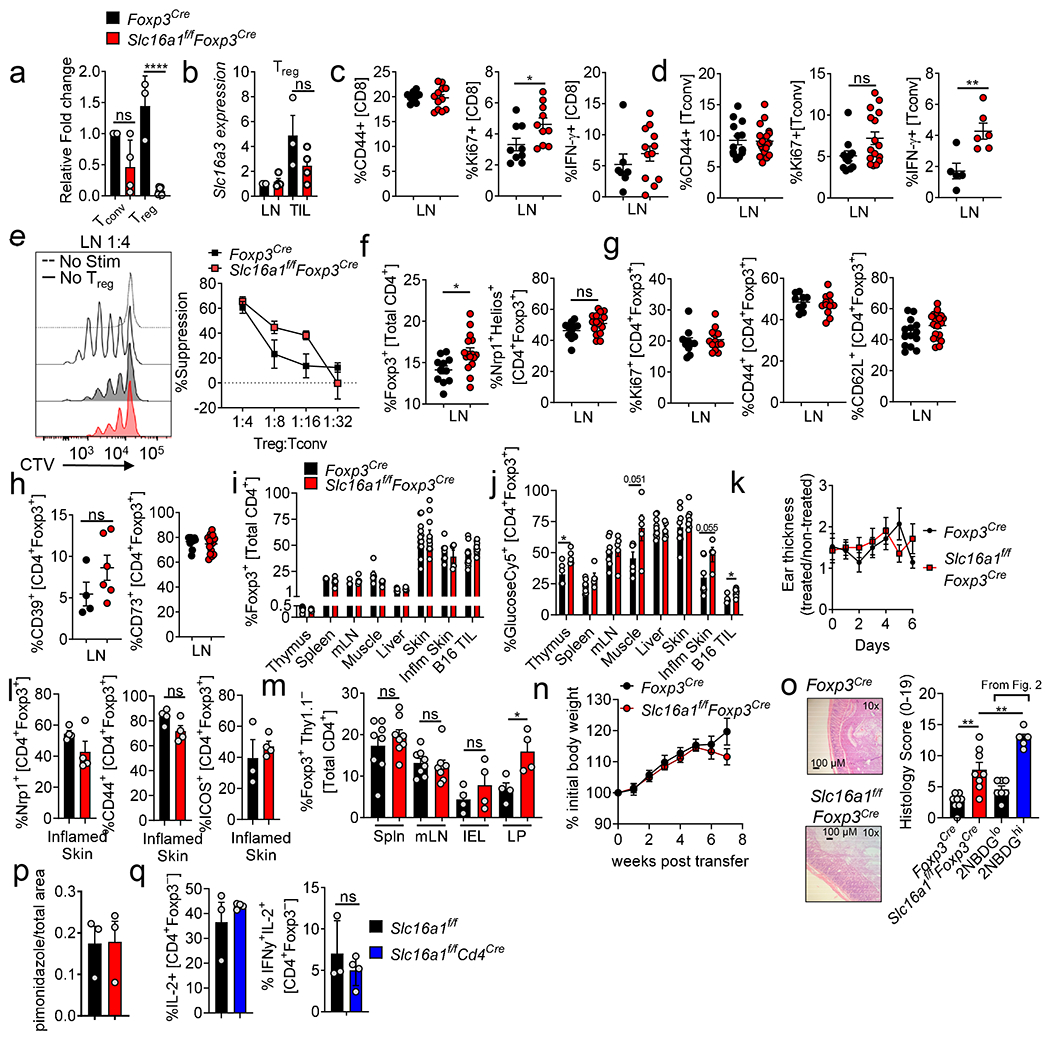
(a) Proliferation of CD8+, Tconv, and Treg cells labeled with CellTrace Violet (CTV) activated in media with lactic acid for 3 days (**p=0.0058). (b) IFN-γ production of CD8 and Tconv cells conditioned as in (a) then restimulated overnight with PMA/ionomycin (CD8 **p=0.0042, Tconv *p=0.02). (c) Suppression assay using Treg cells conditioned as in (a) performed in absence of additional lactic acid. (d) Representative histogram and quantification of pHrodo+ Treg cells taking up 2NBDG. (e) Representative flow plot of Ki67 expression by B16-infiltrating Treg cells from mice treated ±3MP for 3 days. (f) Suppressive capacity of Treg cells, isolated from mice as in Fig.3k. (g) Percent Foxp3+ cells, Tconv:Treg ratio, and CD8:Treg ratio within the TIL of mice treated as in (f). (h) Tumor growth curve of B16 in C57BL/6 mice treated with H2O or 3MP. (i) Proliferation of Treg and Tconv cells activated and cultured in 10mM lactic acid media ±3MP for 3 days (*p=0.016). (j) Capacity of activated Treg cells conditioned in 10mM lactic acid media ± 250μM 3MP for 3 days to suppress the proliferation of CTV labelled Tconv cells. (k) IFN-γ expression by CD8 and Tconv cells in Foxp3Cre or Slc16a1f/fFoxp3Cre mice treated as in (f). (l) Percent Foxp3+ cells, Tconv to Treg ratio, and CD8 to Treg ratio within LN and tumor of Slc16a1f/fFoxp3Cre mice treated as in (f). Results are representative of four (d), three (a,b,c,f,g,i), or two (h,j,k,l) independent experiments. Significance (*p< 0.05, **p <0.01, ***p < 0.001, ****p<0.0001) determined by unpaired two-tailed t test (d,e,g,i,k,l) or one-way ANOVA with Dunnett’s multiple comparisons test (a,b) or two-way ANOVA with Sidak's multiple comparisons test (c,f,h,j). Data are mean values of biological replicates ±SEM.
Extended Data Figure 4. MCT1 is efficiently deleted in Treg cells of Slc16a1f/fFoxp3YFP-Cre mice and is not required for peripheral Treg cell function.
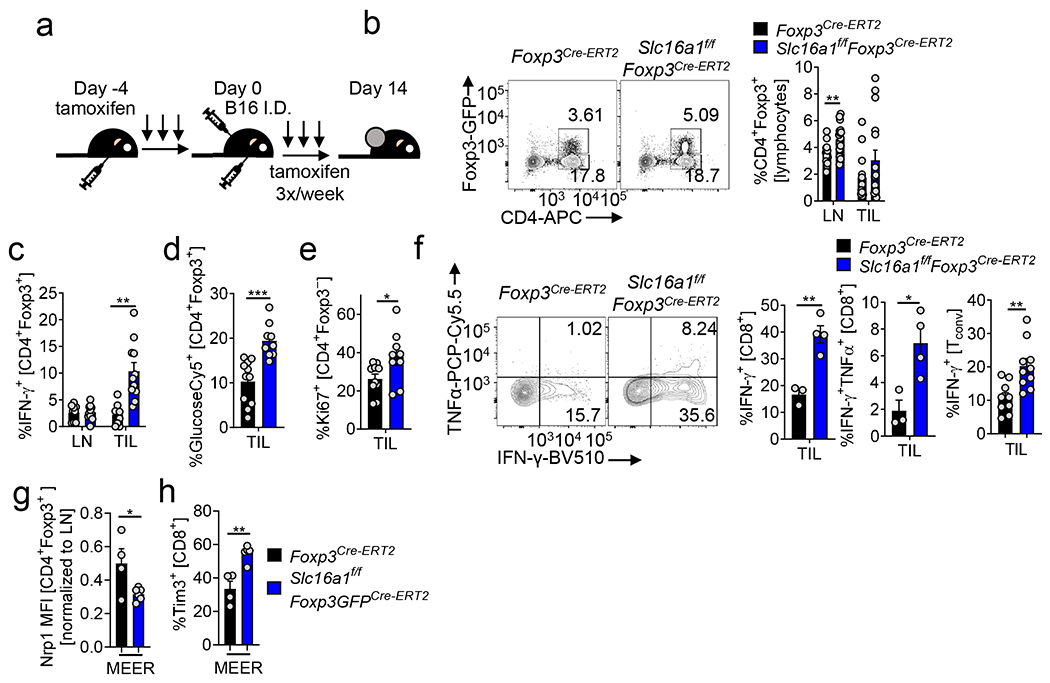
(a) Slc16a1 expression in Tconv and Treg cells from the lymph nodes (LN) of Foxp3Cre or Slc16a1f/fFoxp3Cre mice. (b) Treg cell mRNA expression of Slc16a3 (MCT4) from mice as in (a). CD44, Ki67, and IFN-γ production by LN CD8+ T cells (c, *p=0.039) or Tconv cells (d, **p=0.007) from mice as in (a). (e) Capacity of LN-derived Treg cells as in (a) to suppress proliferation of CellTrace Violet (CTV) labelled Tconv cells. (f) Percent Foxp3+ and Nrp1+Helios+ LN Treg cells from mice as in (a) (*p=0.013). (g) CD44, Ki67, and CD62L expression by LN Treg cells as in (a). (h) CD39 and CD73 expression by LN Treg cells as in (a). Percent of Foxp3+ cells (i) or GlucoseCy5+ Treg cells (j) from various tissues of mice as in (a) (thymus *p=0.014, B16 *p=0.019). (k) Normalized ear thickness of imiquimod treated mice as in (a). (l) Percent marker positive Treg cells from mice treated as in (k). (m) Foxp3 expression in transferred wild type or Slc16a1-deficient Treg infused with Thy1.1+ Tconv cells into Rag1−/− mice iv. (*p=0.02) (n) Weight of Rag1−/− mice over time from (m). (o) Representative sections of the colon and quantified histology scores 7 weeks post transfer from mice as in (m) and Fig.2d (**p=0.001, **p=0.009). (p) Anti-pimonidazole area over tumor area (B16) calculated from immunofluorescence from Foxp3Cre and Slc16a1f/fFoxp3Cre mice. (q) IL-2 and IFN-γ expression by Slc16a1-deficient Tconv cells stimulated overnight with PMA/ionomycin. Results are representative of four (c,d,f,g), three (a,b,e,h,j,p,q), or two (k,l,m,i,o) independent experiments. Significance (*p< 0.05, **p <0.01, ****p < 0.0001) determined by unpaired two-tailed t test (a-d,f-q) or two-way ANOVA (e). Data presented as mean values of biological replicates ±SEM.
Extended Data Figure 5. Acute deletion of MCT1 results in similar immunologic phenotypes in B16 melanoma and predilection towards a fragile Treg cell phenotype.
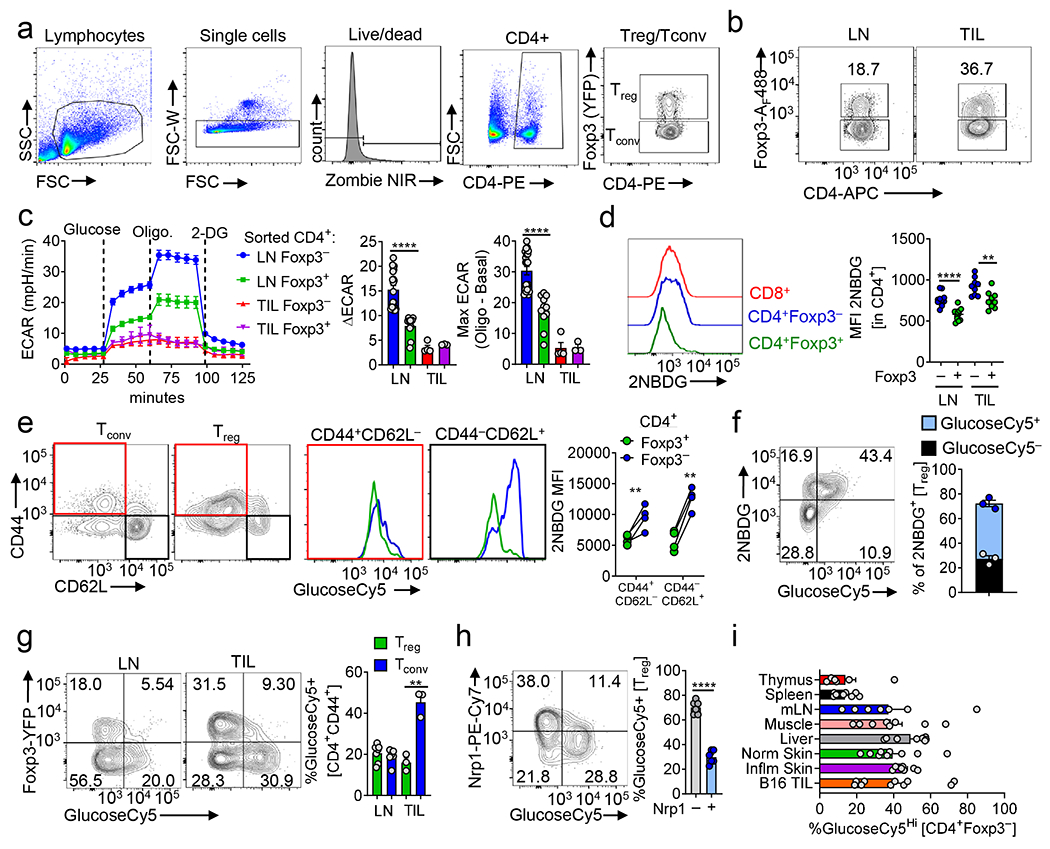
(a) Foxp3Cre-ERT2 and Slc16a1f/fFoxp3Cre-ERT2 mice were treated 5 consecutive days with tamoxifen I.P. prior to inoculation with 1.5 x 105 B16 tumor cells. Following inoculation, tamoxifen was administered I.P. 3 times a week until sacrifice at day 14. (b) Percent Foxp3+ CD4+ cells in the lymph node (LN) and tumor (TIL) as in (a) (**p=0.005). (c) Tabulation of IFN-γ production by Treg cells from the LN and TIL of mice as in (a) (**p=0.002). (d) Glucose consumption by Treg cells from the TIL of mice as in (a). (e) Percent proliferating Tconv cells from the TIL of mice as in (a) (*p=0.037). (f) Representative flow plot and tabulation of IFN-γ and TNFα production by CD8 T cells from the TIL of mice as in (a) (**p=0.003, *p=0.02). Tabulation of IFN-γ production by Tconv cells from the TIL of mice as in (a) (**p=0.003). (g) Nrp1 mean fluorescence intensity on MEER intratumoral Treg cells derived from Foxp3Cre-ERT2 and Slc16a1f/fFoxp3Cre-ERT2 mice treated as in (a) (*p=0.034). (h) Tim-3 expression by MEER derived CD8 T cells from mice as in (g) (**p=0.003). Results are representative of four (b,c,d,e), or three (f), or two (g,h) independent experiments. Significance (*p< 0.05, **p <0.01) determined by unpaired two-tailed t test (b-h). Data presented as mean values of biological replicates ±SEM.
Acknowledgements
The authors wish to thank Amanda Burton and Creg Workman for the generation and gift of the Foxp3FlpO-Ametrine mice and Geoffrey Camirand for the gift of OT-II Foxp3RFP Thy1.1+ spleens. This work was supported by the Sidney Kimmel Foundation, an NIH Director’s New Innovator Award (DP2AI136598), the Hillman Fellows for Innovative Cancer Research Program, a Stand Up to Cancer-American Association for Cancer Research Innovative Research Grant (SU2C-AACR-IRG-04-16), the Alliance for Cancer Gene Therapy, the UPMC Hillman Cancer Center Skin Cancer and Head and Neck Cancer SPOREs (P50CA121973 and P50CA097190), the Mark Foundation for Cancer Research’s Emerging Leader Award, a Cancer Research Institute’s Lloyd J. Old STAR Award, and the Sy Holzer Endowed Immunotherapy Fund (all to G.M.D.). D.A.A.V is supported by R01DK089125, R01CA203689 and P01AI108545. Trainees on this manuscript were supported by T32CA082084 (to M.J.W., P.D.A.V., A.E.O.D., and K.D.), F31AI149971 (to M.J.W.), F30CA247034 (to P.D.A.V.), (F31CA247129 to K.D.), T32AI089443 (to R.M.P and S.G.), F31AI147638 (to S.G.), and a Damon Runyon Cancer Research Fellowship (A.E.O.D.). Mass spectrometry was supported by S10OD023402 (to S.G.W.), and floxed animal generation was supported by R01NS099320 (to J.D.R., and B.M.M). as well as R01 NS086818 (to B.M.M.). Synthesis of Glucose-Cy5 was supported by R21AI135367 (to G.M.D.). Sequencing was supported by the Samuel and Emma Winters Foundation and a Grand Prize Award Grant (2017) from the Immuno-Oncology Young Investigators’ Forum (both to G.M.D.) and was performed at the University of Pittsburgh Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh. RNA sequencing analysis was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided. This work utilized the UPMC Hillman Cancer Center Flow Cytometry and Animal Facilities, supported in part by P30CA047904.
Footnotes
The authors declare no competing interests.
Bibliography
- 1.Wang H, Franco F & Ho P-C Metabolic regulation of tregs in cancer: opportunities for immunotherapy. Trends Cancer 3, 583–592 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S, Yamaguchi T, Nomura T & Ono M Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Scharping NE et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Najjar YG et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho P-C et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 23, 3262–3274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delgoffe GM et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature 501, 252–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michalek RD et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerriets VA et al. Foxp3 and Toll-like receptor signaling balance T reg cell anabolic metabolism for suppression. Nat. Immunol 17, 1459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinberg SE et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubtsov YP et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Rubtsov YP et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menk AV et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 22, 1509–1521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lunt SY & Vander Heiden MG Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol 27, 441–464 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Single cell glucose uptake assays: A cautionary tale. Immunometabolism (2020). doi: 10.20900/immunometab20200029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu H et al. Cyanine-based 1-amino-1-deoxyglucose as fluorescent probes for glucose transporter mediated bioimaging. Biochem. Biophys. Res. Commun 474, 240–246 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Chang C-H et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Alise AM, Ergun A, Hill JA, Mathis D & Benoist C A cluster of coregulated genes determines TGF-beta-induced regulatory T-cell (Treg) dysfunction in NOD mice. Proc. Natl. Acad. Sci. USA 108, 8737–8742 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hui S et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halestrap AP & Wilson MC The monocarboxylate transporter family--role and regulation. IUBMB Life 64, 109–119 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Romero-Garcia S, Moreno-Altamirano MMB, Prado-Garcia H & Sánchez-García FJ Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol 7, 52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angelin A et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 25, 1282–1293.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer K et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Liu C et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem 284, 2811–2822 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Jackson VN & Halestrap AP The kinetics, substrate, and inhibitor specificity of the monocarboxylate (lactate) transporter of rat liver cells determined using the fluorescent intracellular pH indicator, 2’,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein. J. Biol. Chem 271, 861–868 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Robinson BH & Oei J 3-Mercaptopicolinic acid, a preferential inhibitor of the cytosolic phosphoenolpyruvate carboxykinase. FEBS Lett. 58, 12–15 (1975). [DOI] [PubMed] [Google Scholar]
- 28.Lowther DE et al. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Overacre-Delgoffe AE et al. Interferon-γ Drives Treg Fragility to Promote Anti-tumor Immunity. Cell 169, 1130–1141.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macintyre AN et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab. 29, 103–123.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Procaccini C et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity 44, 406–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Priyadharshini B et al. Cutting Edge: TGF-β and Phosphatidylinositol 3-Kinase Signals Modulate Distinct Metabolism of Regulatory T Cell Subsets. J. Immunol 201, 2215–2219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consoli A, Nurjhan N, Reilly JJ, Bier DM & Gerich JE Contribution of liver and skeletal muscle to alanine and lactate metabolism in humans. Am. J. Physiol 259, E677–84 (1990). [DOI] [PubMed] [Google Scholar]
- 35.Proia P, Di Liegro CM, Schiera G, Fricano A & Di Liegro I Lactate as a metabolite and a regulator in the central nervous system. Int. J. Mol. Sci 17, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arpaia N et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colegio OR et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung Y-S et al. CD200: association with cancer stem cell features and response to chemoradiation in head and neck squamous cell carcinoma. Head Neck 37, 327–335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jha MK et al. Monocarboxylate transporter 1 in Schwann cells is critical for maintenance of sensory nerve myelination during aging. BioRxiv (2019). doi: 10.1101/686832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lennon GP et al. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity 31, 643–653 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ostanin DV et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am. J. Physiol. Gastrointest. Liver Physiol 296, G135–46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA sequencing data that support the findings of this study (Fig. 2b, h) have been deposited in GEO with the GSE158801 accession code. The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files.