

Abstract
TsrM methylates C2 of the indole ring of L-tryptophan (Trp) during the biosynthesis of the quinaldic acid moiety of thiostrepton. It is annotated as a cobalamin-dependent radical S-adenosylmethionine (SAM) methylase; however, TsrM does not reductively cleave SAM to the universal 5ʹ-deoxyadenosyl 5ʹ-radical intermediate, a hallmark of radical-SAM (RS) enzymes. Herein, we report structures of TsrM from Kitasatospora setae, the first of a cobalamin-dependent radical SAM methylase. Unexpectedly, the structures show an essential arginine residue that resides in the proximal coordination sphere of the cobalamin cofactor and a [4Fe–4S] cluster that is ligated by a glutamyl residue and three cysteines in a canonical CxxxCxxC RS motif. Structures in the presence of substrates suggest a substrate-assisted mechanism of catalysis, wherein the carboxylate group of SAM serves as a general base to deprotonate N1 of the tryptophan substrate, facilitating formation of a C2 carbanion.
One sentence summary:
The first crystal structures of a cobalamin-dependent radical SAM methylase reveal an unexpected mode of methylation.
Introduction:
Thiostrepton is a bicyclic thiazolyl peptide (thiopeptide) natural product that exhibits strong potency against many Gram-positive pathogens through its inhibition of protein translation (1, 2). It has also been noted to target breast cancer cells by inhibiting forkhead box M1 expression (3, 4). Currently it is used primarily as a topical treatment in veterinary medicine, being ineffective for human use because of its poor aqueous solubility and gastric absorption (1). Thiostrepton is a ribosomally synthesized and post-translationally modified peptide composed of 17 amino acids that are extensively processed to contain multiple thiazoles, dehydrated alanyl residues and a quinaldic acid moiety that is essential for its antibiotic properties (5) (Figure 1).
Figure 1. Structure of thiostrepton A and details of the TsrM reaction.
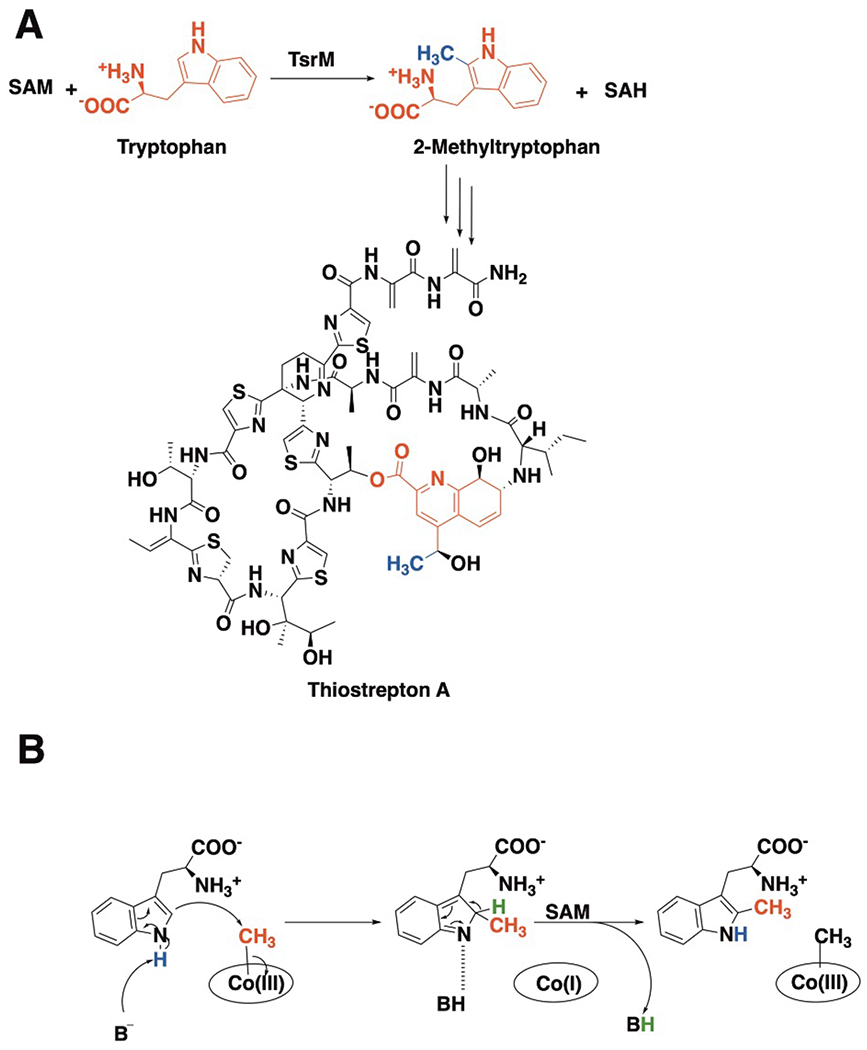
(a) TsrM uses one molecule of SAM to methylate C2 of tryptophan, forming 2-methyltryptophan and SAH. 2-methyltryptophan is an intermediate in the formation of the quinaldic acid moiety (red) of thiostrepton A, with the appended methyl group shown in blue. (b) Proposed TsrM mechanism of catalysis.
The first committed step in the biosynthesis of the quinaldic acid moiety, the methylation of C2 of the indole ring of L-tryptophan (Trp), is catalyzed by TsrM, an enzyme annotated to be a member of the cobalamin-containing subfamily of radical S-adenosylmethionine (SAM) enzymes (Figure 1a) (6, 7). In fact, like all radical-SAM (RS) enzymes, of which now there are more than 115,000 unique full-length sequences (8), TsrM binds a [4Fe-4S] cluster using three cysteines found in an almost universal CxxxCxxC motif. In almost all RS enzymes, this cluster participates intimately in a reductive cleavage of SAM to generate a 5’-deoxyadenosyl 5’-radical (5’-dA•), a species that serves as a key intermediate. The lone exception to date is TsrM, which does not generate a 5’-dA• during catalysis (7, 9, 10). Strikingly, it catalyzes a reaction that does not involve organic radical intermediates at all and, in fact, is now believed to take place via two polar nucleophilic displacements: cob(I)alamin, a supernucleophile, attacks SAM to generate methylcobalamin (MeCbl); and a C2 carbanion on Trp attacks MeCbl to generate methyltryptophan (MeTrp) upon loss of the C2 proton (10) (Figure 1b). Understanding the structural basis of catalysis will provide insight into the expansiveness and mechanistic diversity of the RS superfamily and how Nature can co-opt the RS fold to catalyze non-canonical reactions. This mechanism for catalysis by TsrM elicits at least two major questions: how is it that the weakly nucleophilic C2 carbon of Trp displaces cob(I)alamin, the strongest nucleophile in biology; and why does TsrM use a RS scaffold to catalyze its reaction?
Herein, we report several crystal structures of TsrM from Kitasatospora setae (KsTsrM), which shed light on these two questions and facilitate a deeper understanding of the enzyme’s catalytic mechanism. In particular, we show that a conserved arginyl amino acid occupies the proximal face of the cobalamin cofactor but is not in coordinating distance, which is predicted to influence the cofactor’s reactivity. Moreover, unlike all other RS enzymes characterized to date, in which SAM coordinates to the unique iron of the [4Fe-4S] cluster using its amino and carboxylate functional groups, we show that the [4Fe-4S] cluster on TsrM is fully ligated, with a glutamate amino acid side chain providing the fourth ligand.
RESULTS
KsTsrM domain architecture
KsTsrM was crystallized under anoxic conditions, and its structure was solved to 1.67 Å resolution with two monomers in the asymmetric unit (Supplementary Table 1). Each monomer is composed of three domains from N- to C- terminus: a Rossmann-fold domain (residues 1–205), which binds the Cbl cofactor; an RS domain (residues 206–487) containing a shortened (βα)6 triosephosphate isomerase (TIM) barrel that binds a [4Fe–4S] cluster; and a C-terminal domain (residues 488–567) composed of helices and mobile loops (Figure 2a and Extended Data Figure 1) (11–13). The overall structure of KsTsrM shows marked similarities to the recently determined structure of OxsB, the only other structurally characterized cobalamin-binding RS enzyme to date (Extended Data Figure 2). Although OxsB does not catalyze a methylation reaction and exhibits only 16.5% sequence identity with TsrM, TsrM shares three of the four domains found in OxsB. The N-terminal domain of OxsB, whose function is currently unknown, is not present in TsrM (12).
Figure 2. The X-ray crystal structure of native KsTsrM with both cofactors (cobalamin and a [4Fe–4S] cluster) bound.
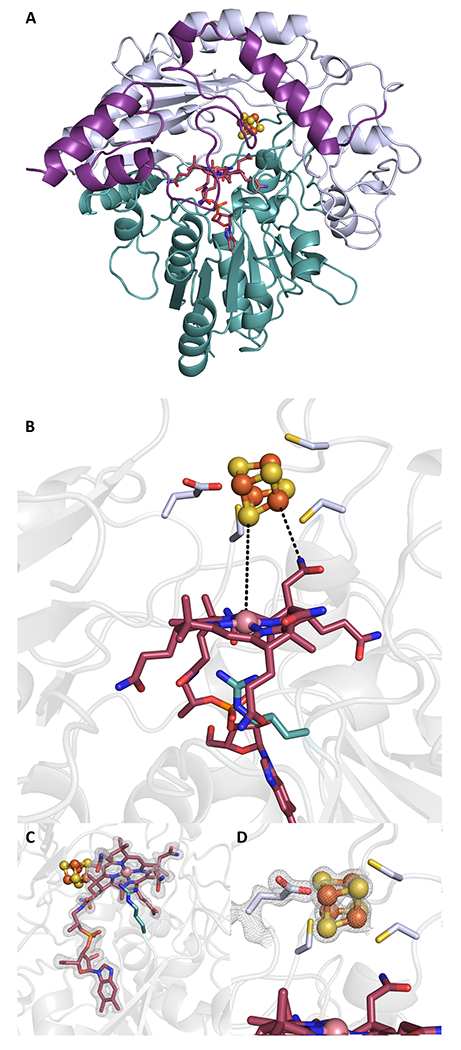
(a) KsTsrM (molecule A) shown as a ribbon diagram, colored by domain (I, II, or III). The cobalamin-binding (Cbl) Rossmann fold (domain I) is shown in teal with the cobalamin cofactor in red sticks. The shortened TIM barrel (βα)6 is shown in light blue (domain II), with the bound [4Fe–4S] cluster shown in orange and yellow spheres. The final domain is a C-terminal domain (domain III), in purple. (b) Close-up of active site of KsTsrM showing the Cbl and [4Fe–4S] cluster. The Cbl has the dimethylbenzimidazole base (DMB) displaced from the bottom axial position and instead has a conserved Arg residue occupying that position. (c) The composite omit map is shown for the Cbl (contoured at 1.5 σ). (d) Close up of the [4Fe–4S] cluster. The three Cys residues of the CxxxCxxC motif ligate three of the irons, while Glu273 ligates the remaining iron of the cluster. The composite omit map (contoured at 2.0 σ) is shown for Glu273 and the cluster, with the electron density indicating a covalent interaction.
The cobalamin-binding domain of KsTsrM
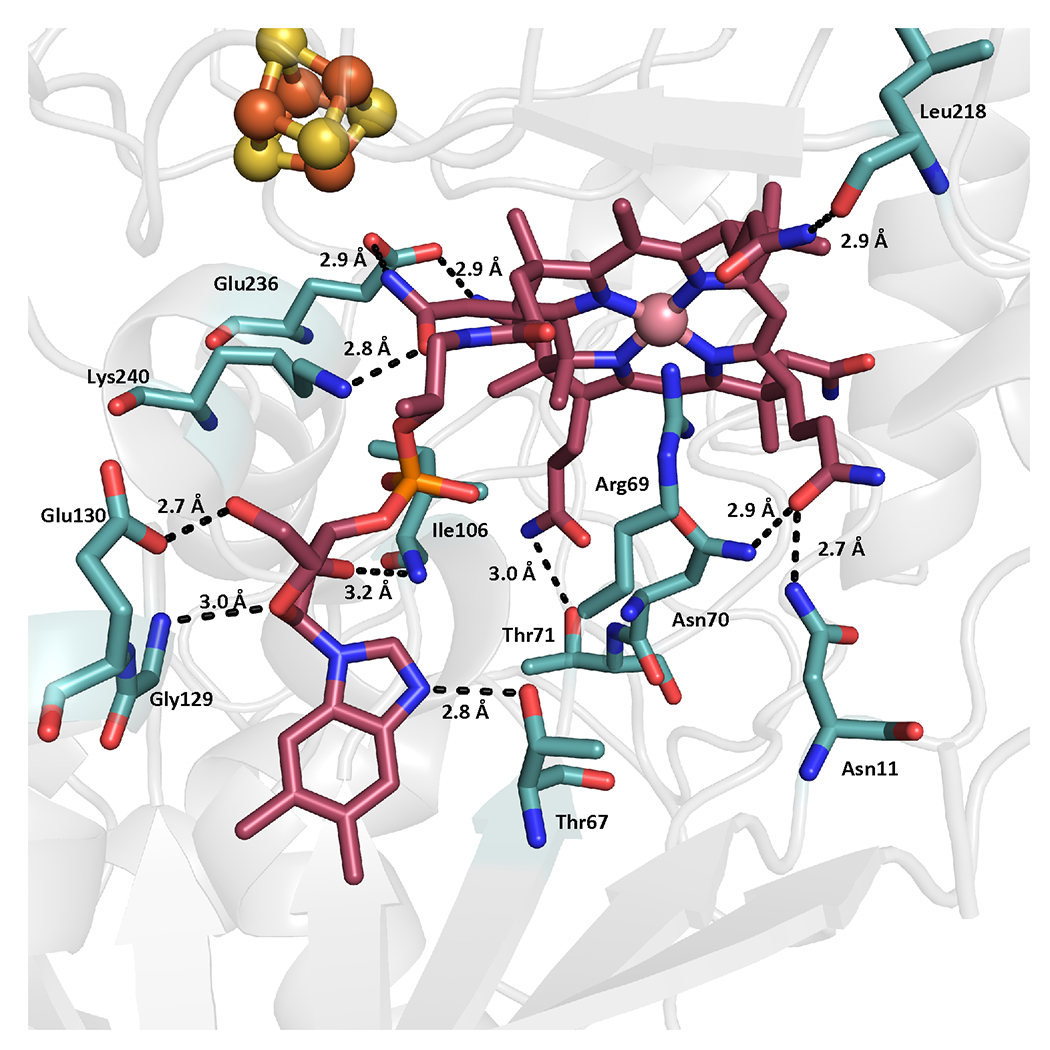
Cobalamin, the key catalytic cofactor in TsrM, is bound to the Rossmann-fold domain with its dimethylbenzimidazole base (DMB) displaced ~13 Å from the Co ion (base-off). Additionally, the cofactor interacts with conserved residues Asn11, Asn70, Gly129, Glu130, Glu236, and Lys240, and exhibits various water–backbone interactions (Extended Data Figure 3). The base-off binding mode is similar to that of the cobalamin-binding domain of methionine synthase (MetH) (r.m.s.d. = 3.04 Å, 100 residues over Cαs) — which was the first structurally characterized Cbl-dependent protein with Cbl in the base-off conformation — and OxsB (r.m.s.d. = 2.59 Å, 261 residues over Cαs) (11–13). Nevertheless, the amino acid sequence of KsTsrM, like that of OxsB, does not contain the canonical His-on cobalamin-binding motif (Asp-X-His-X-X-Gly) observed in MetH, where the His residue (on the loop following β1 of the Rossman fold) replaces DMB and acts as the lower axial ligand to the Co ion (Extended Data Figure 4) (13). Instead, Arg69, which resides on the loop following β3 (i.e. Thr67–Phe82) and is strictly conserved among TsrM orthologs, occupies the lower axial position, but is not in coordinating distance (Figure 2d and Extended Data Figure 4a). The nitrogen atoms of Arg69 are 3.4 and 3.6 Å from the Co, which is too far for a typical Co–N bond (2.5 Å) (14). This observation is consistent with previous electron paramagnetic resonance (EPR) spectroscopic studies, wherein nitrogen superhyperfine splittings were not observed in the paramagnetic cob(II)alamin form (9).
In the likely case that the Arg69 side chain is charged (pKa = 12.5), the interaction between Arg69 and Co would likely stabilize the cob(I)alamin state of the enzyme and destabilize the methylcob(III)alamin state, which would favor the displacement of the strong cob(I)alamin nucleophile by the weakly nucleophilic C2 atom of Trp. In addition, Arg69 blocks water from accessing the lower axial face of Cbl, preventing water from converting the labile pentacoordinated methylcob(III)alamin species to the more stabilized hexacoordinated species (15). Substituting Arg69 in TsrM with Lys results in almost complete loss of activity, but the protein can still methylate cob(I)alamin to form MeCbl, supporting the role of Arg69 mainly in the transfer of the methyl group from MeCbl to Trp (Extended Data Figure 5). An Arg occupying the lower axial face of Cbl has been observed previously in QueG, an epoxyqueuosine reductase that contains Cbl and two [4Fe–4S] clusters (16). However, in QueG, Arg141 is situated 5.0 and 5.6 Å away from the Co and acts to block water or other small ligands from coordinating to the Co. The electrostatic influence of Arg141 in QueG is expected to be less than that of Arg69 in TsrM, given the longer distance (Extended Data Figure 6).
The radical-SAM domain of KsTsrM
Adjacent to the Rossmann-fold domain both in primary and in tertiary structure is the RS domain, containing a (βα)6 truncated TIM barrel that houses a [4Fe–4S] cluster (Figure 2a and Extended Data Figure 1). The (βα)6 barrel is slightly splayed in comparison to the classic RS core fold observed in E. coli pyruvate formate-lyase activating enzyme (Supplementary Figure 1) (17). The difference may result from TsrM having to accommodate the [4Fe–4S] cluster, the Cbl, and the two substrates. In KsTsrM, the [4Fe–4S] cluster is ligated by three Cys residues (Cys227, Cys231, and Cys234) that occur in the canonical CxxxCxxC RS motif (Figure 2d) (18). Notably, Glu273, a completely conserved residue within TsrM proteins, coordinates the unique Fe of the [4Fe–4S] cluster with its carboxylate sidechain as a monodentate ligand (Figure 2d). This Fe is typically coordinated in a bidentate fashion by the carboxylate and amino groups of SAM in RS enzymes (Supplementary Figure 2). Our finding of a fully ligated [4Fe–4S] cluster supports results from previous Mössbauer and EPR spectroscopic studies that suggested that SAM does not bind directly to the cluster, and that the cluster in the as-isolated protein has a site-differentiated Fe ion (9). The observation of an RS enzyme with a fully ligated cluster is unprecedented; however, it is consistent with the finding that TsrM does not reductively cleave SAM to generate a 5ʹ-dA•, as SAM cannot associate directly with the cluster (19). Recent studies indicate that the rate of TsrM turnover is greatest in the presence of a flavodoxin–flavodoxin reductase reducing system, which is hypothesized to be important in priming the enzyme for turnover and maintaining it in an active state, given that cob(I)alamin is easily oxidized to cob(II)alamin and OHCbl (20). At present, we suggest that the FeS cluster is reduced directly by flavodoxin, as is believed for all other RS enzymes that use this physiological reductant. In turn, the reduced cluster can reduce the Cbl cofactor, which is 6 Å from the nearest part of the corrin ring to the nearest ion of the cluster and 11.3 Å from the Co ion to the nearest ion of the cluster (Figure 2b).
The positioning of SAM allows for non-radical chemistry
The C-terminal domain of TsrM is composed of helices and loops. Though the function of this domain is unclear at first glance, corresponding domains in MetH and OxsB have been proposed to be critical for substrate binding. One caveat associated with determining structures of TsrM with bound substrates is that, unlike other RS enzymes, which require that the [4Fe–4S] cluster cofactor be reduced to the +1 core oxidation state for catalysis, TsrM can undergo turnover when all substrates are present, even in the absence of an external reductant (10, 20). Therefore, aza-SAM (S-5Ȳ-azamethionine-5-deoxyadenosine, Supplementary Figure 3) — an unreactive SAM analog in which the sulfonium sulfur atom is replaced with a nitrogen atom (21) — was used in subsequent crystallization experiments.
The structure of TsrM containing both Trp and aza-SAM was solved to 2.2-Å resolution (Figure 3). The overall fold of the protein does not change upon substrate binding (r.m.s.d = 0.88 Å over 539 residues) (Figure 3a); however, a loop in the C-terminal domain of the protein (residues 525–536) moves ~16.8 Å into the active site, forming stabilizing interactions with Trp through Asn527 and Tyr529, which results in capping of the Cbl cofactor and trapping of substrates within the active site (Figure 3b). The structure also reveals how SAM might access the active site. Aza-SAM binds near the top of the β-barrel-like sheet where the residues in its binding site originate from various loops within the TIM barrel (Figure 3d). In determined structures of other RS enzymes, the adenine and ribose portions of SAM are bonded to protein backbone and carboxylate groups from residues on the TIM barrel, and the amino and carboxyl groups coordinate to the unique Fe and also bind the previously identified GGE motif through hydrogen bonds (22). For TsrM, this interaction mode is conserved for adenine and ribose binding; however, the GGE motif in TsrM is actually Asp271–Ser272–Glu273 (i.e. DSE), with Glu273 coordinating the unique Fe, blocking SAM from coordinating to this site. The altered protein environment allows the DSE motif to still bind to SAM, but through hydrogen bonds between SAM’s amino group (Figure 3d) and the protein backbone of Ser272 as well as the side chain of Glu236. The newly discovered binding mode of SAM in TsrM orients its carboxyl group toward Cbl and Trp, which forms hydrogen bonds with a water molecule next to His233 and a water molecule located above the Cbl (Figures 3d and 4a).
Figure 3. The X-ray crystal structure of KsTsrM with bound Trp (teal) and aza-SAM (green) along with both cofactors.
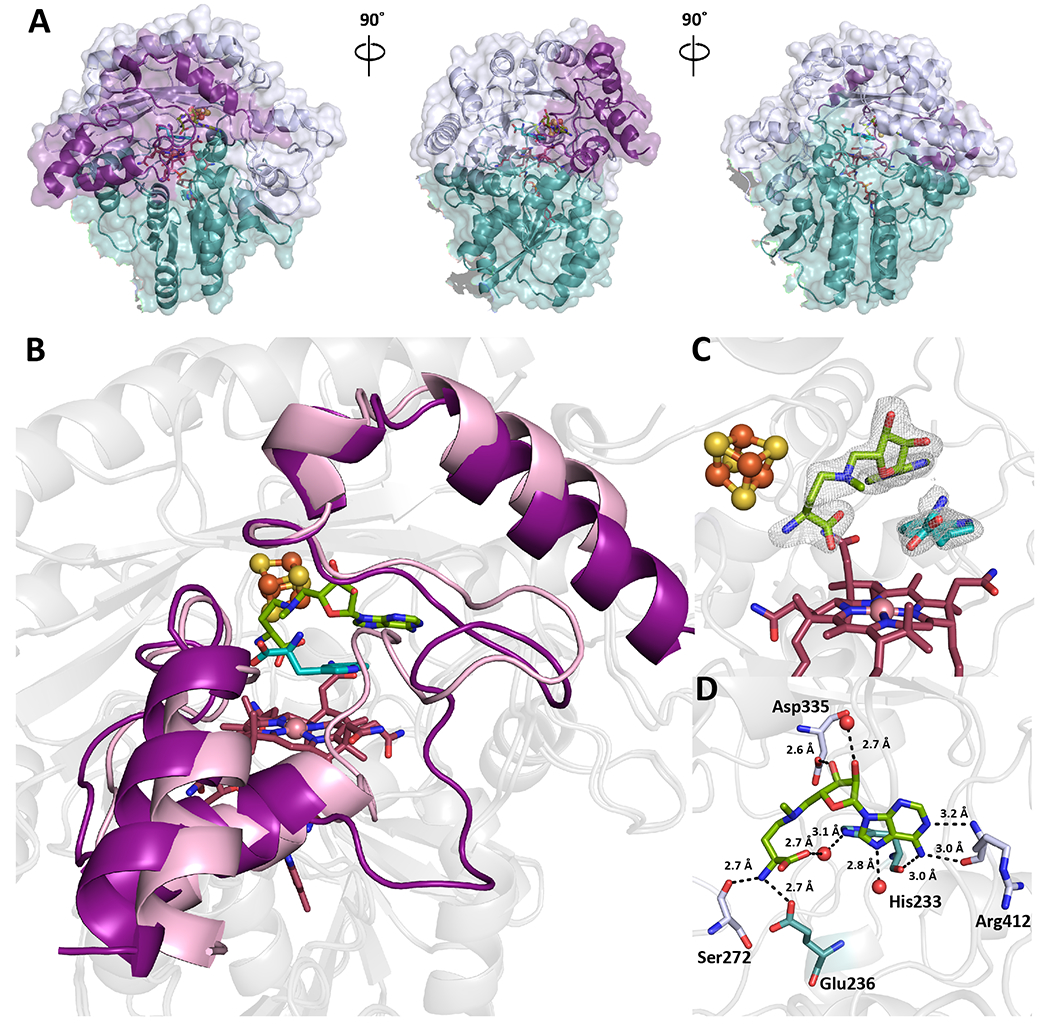
(a) Surface view of KsTsrM with substrates bound. The overall folds do not change substantially, but the C-terminal domain or mobile domain moves into the active site. (b) Ribbon diagram of native KsTsrM (purple) and substrate-bound (pink) KsTsrM overlaid. The Rossmann folds and TIM barrels (shown in light grey) overlay without substantial changes. However, the C-terminal domain undergoes an important change. In the substrate-bound structure, a loop moves into the active site, where it makes hydrogen-bonding contacts with Trp. (c) Close up of active site of substrate-bound KsTsrM. Aza-SAM, a SAM analog that allows for methyl transfer from MeCbl to Trp, and Trp are bound. The composite omit map is shown (contoured at 1.5 σ). SAM is observed to bind close to, but not in contact with, the cluster. (d) The binding of SAM in the active site. Six key hydrogen bonds to amino acid residues are observed, along with two water-to-protein backbone hydrogen bonds. Notably, there are no hydrogen bonds to the carboxylate of SAM; however, a key hydrogen bond between the amino portion of SAM and Ser272, a conserved residue, is present.
The unique binding mode of SAM positions it such that its carboxyl group is 4.0 and 4.8 Å from the nearest portion of the indole ring of Trp (Figure 3d). Trp binds partly over the Cbl (Figure 4a); however, it should be noted that the C2 position of the indole moiety of Trp is 7.0 Å away from the Co ion, which would be too far from the methyl group of MeCbl and not in the proper geometry for methyl transfer. We posit that Trp is not bound in the correct conformation for methyl transfer from MeCbl, because OHCbl is present on the protein (Figure 4a). We were unable to generate a structure with bound MeCbl, most likely because this five-coordinate species is too unstable, especially when exposed to X-rays.
Figure 4. Substrate-assisted catalysis by TsrM.
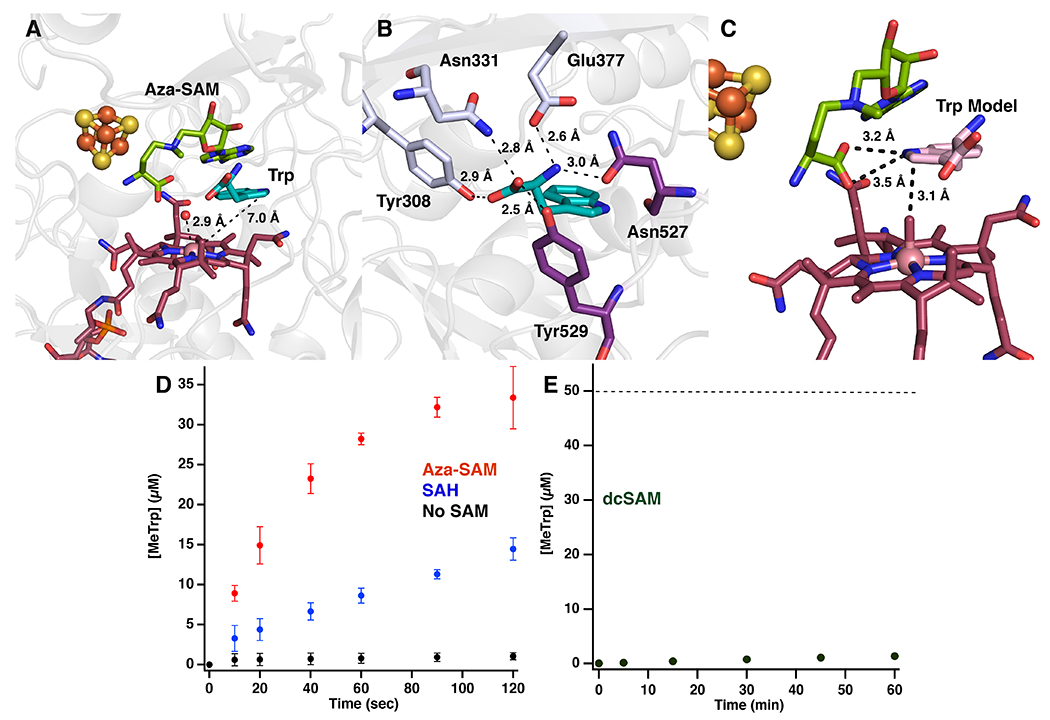
(a) Active site of KsTsrM with bound aza-SAM (green) and Trp (teal). A water molecule is bound above the Cbl 2.9 Å away from the Co ion. C2 of Trp is 7.0 Å away from the Co ion of Cbl. (b) The binding of Trp in the active site. Five hydrogen bonds are made to the carboxylate and amino groups of Trp to four strictly conserved residues located on the TIM barrel (light blue) and the C-terminal domain (purple). There is an additional possible hydrogen bond to the N1 position of Trp from the backbone of Asn527. (c) Pose 2 of the computational docking model of Trp (pink) bound to TsrM, placing the C2 position 3.1 Å above the methyl group of MeCbl and the N1 position 3.2 Å from the carboxylate of aza-SAM. (d) Time-dependent formation of MeTrp under single-turnover conditions. Reactions contained premethylated TsrM (60 μM), Tyr (0.1 mM), Trp (1 mM), and no SAM (black dots), SAH (1 mM) (blue), or azaSAM (1 mM) (red). (e) Time-dependent formation of MeTrp by TsrM (50 μM) in the presence of Trp (1 mM) and dcSAM (1 mM). Dotted line indicates amount of enzyme. All error bars represent the standard deviation of the assays conducted in triplicate, the central point representing the average.
In the Trp-bound structure, there are five hydrogen bonds to the carboxylate and the amine of Trp, which are contributed by Asn527, Glu377, Asn331, Tyr308 and Tyr529 (Figure 4b). We noticed that if we maintained those hydrogen bonds fixed and flipped the indole ring of Trp ~180° manually, C2 of Trp would be located almost directly over the Co. To evaluate this possibility, we performed computational docking of Trp to the aza-SAM+Trp-bound structure of TsrM — in which cobalamin was replaced manually with MeCbl — using the program Glide from the Schrödinger/2016.4 software package (www.schrödinger.com). The best results were obtained with an active site containing Glu377 in its ionized state and three water molecules (HOH 1005, 1062, 1124) bound within 4 Å of Trp in the X-ray crystal structure of TsrM. The two best-scoring docking poses, which are close in energy, are shown in Extended Data Figure 7a. Pose 1 (GScore, −8.9) shows Trp in the flipped conformation, which is virtually identical to its conformation in the structure obtained by X-ray crystallography (Extended Data Figure 7b), confirming the robustness of our docking procedure. Pose 2 (GScore, −8.6) has Trp in what we term the “active” conformation, or how we believe it should exist for catalysis. In pose 2, C2 of Trp is almost directly overhead and 3.1 Å away from the methyl group of MeCbl (Figure 4c). In addition, the carboxylate group of aza-SAM is within 3.2 Å of N1 of Trp, suggesting that it forms a hydrogen bond with this atom (Figure 4c and Extended Data Figure 7c,d). This model also agrees with studies using C6-substituted Trp analogs, all of which showed substantially decreased activity compared to analogs containing the same substitutions on other positions of the ring (10). Substituents at C6 would be expected to clash with the backbone carbonyl (3.1 Å) and R-group (3.8 Å) of Val74.
Based on these crystal structures, as well as a number of biochemical experiments performed with SlTsrM (vide infra), we propose that SAM deprotonates N1 of Trp in the transition state of the reaction, enhancing the nucleophilicity of C2 in order to attack the methyl group of MeCbl and displace cob(I)alamin. Alternatively, this step might be driven by an ionic interaction between the carboxylate of SAM and a positively charged N1 of Trp in the transition state of the reaction. Tyr308, a completely conserved residue in TsrM proteins, undergoes a dramatic flip when substrates are bound (Extended Data Figure 8). Tyr308 is within hydrogen-bonding distance of the carboxylate of Trp; however, in the docking model, the hydroxyl group of Tyr is also 3.7 Å from C2, suggesting that it might be the base that removes the C2 proton in the last step of the reaction (Extended Data Figure 8b). Given that the structure suggests that the methyl group is added to the si face of C2 of Trp, Tyr308 would need to move a short distance to play this role, and/or Trp would need to rotate down toward cobalamin. No other amino acids capable of acting as a general base are found in the vicinity of Trp in either the flipped structure or the active structure.
Considering that TsrM does not catalyze a reductive cleavage of SAM, as well as our previous findings that suggest that the reaction proceeds via polar SN2 chemistry rather than through radical intermediates (10), we expected that the reaction would be ping-pong in nature. In the “ping” step, SAM would bind first to the enzyme and donate its methyl group to Cbl to afford MeCbl. Upon dissociation of SAH, Trp would bind and obtain the methyl group from MeCbl, which corresponds to the “pong” step. However, our X-ray crystal structures show clearly that both SAM and Trp can bind simultaneously to the enzyme, which is consistent with a sequential mechanism. Moreover, when we generated MeCbl on TsrM — by incubating the enzyme with methyliodide or SAM in the presence of reductant and then removing small molecules by gel-filtration chromatography — we observed very little turnover when we reacted this form of the enzyme with Trp in the absence of SAM (Figure 4d). To address the kinetic mechanism of TsrM, a steady-state kinetic analysis of the enzyme was conducted by varying concentrations of SAM at fixed concentrations of Trp. Double-reciprocal plots of the initial velocities as a function of substrate concentrations (Supplementary Figure 4) display clear intersecting lines to the left of the 1/V0 axis, which is indicative of a sequential mechanism (23).
A substrate-assisted mode of catalysis
To understand why SAM is required for Trp methylation, we examined the reaction under single-turnover conditions, monitoring the rate of MeTrp formation using premethylated TsrM. In the absence of SAM, premethylated TsrM catalyzes MeTrp formation at a rate of 0.37 μM min−1, which is more than 1,500-fold slower than the rate (~600 μM min−1) in an identical reaction containing excess SAM (Figure 4d) (24). When similar experiments were conducted with SAH or aza-SAM, analogs of SAM that cannot transfer a methyl group, the initial rates of MeTrp formation were 6.9 and 52.5 μM min−1, respectively, which are substantially higher than the rate in the absence of SAM (Figure 4d). The higher rate with aza-SAM suggests that the partial positive charge of the tertiary nitrogen (pKa =7.08), which mimics the positive charge on SAM, may influence the rate of the reaction. These data suggest that, in addition to transferring its methyl group to cobalamin, SAM plays an important role in the methyl transfer from MeCbl to Trp.
In the crystal structure of TsrM with Trp modeled in the “active” state (Figure 4c), the carboxyl group of SAM is positioned within hydrogen-bonding (3.2 Å) distance of N1 of Trp, which might allow it to remove the N1 proton in the transition state of the reaction. To examine the role of SAM’s carboxyl group, we monitored the activity of TsrM in the presence of S-5′-adenosyl-3-methylthiopropylamine (dcSAM), an analog of SAM that lacks the carboxyl group. When 50 μM TsrM is incubated with saturating dcSAM and Trp, formation of MeTrp occurs with a V•[ET]−1 of 4.5 × 10−4 min−1, with less than 1 μM product formed in 1 h (Figure 4e), while V•[ET]−1 is ~10-20 min−1 in the presence of SAM (24). We also monitored the rate of a single-turnover reaction in the presence of dcSAM using premethylated TsrM. As shown in Supplementary Figure 5, dcSAM only minimally enhances the rate of MeTrp formation under these conditions. To verify that dcSAM can bind to TsrM, its ability to inhibit turnover was assessed. As seen in the Dixon plots in Supplementary Figure 6, dcSAM acts as a competitive inhibitor with respect to SAM, exhibiting a Ki value of 2.45 μM.
To determine which step of the reaction is not supported by dcSAM, we monitored formation of TsrM-bound MeCbl in the presence of dcSAM (Extended Data Figure 9). MeCbl forms (Extended Data Figure 9a) at an initial rate of 4.4 μM min−1 and, after 10 min, the cobalamin cofactor is nearly completely converted to its MeCbl form (Extended Data Figure 9b). Although dcSAM cannot methylate the Cbl cofactor at the same rate as can SAM (~100 μM min−1), the rate of cobalamin methylation is substantially greater than the rate of MeTrp formation in the presence of dcSAM (4.4 μM min−1 versus 0.02 μM min−1), which further suggests that the carboxyl group of SAM plays a key role in Trp methylation. These results suggest a dual role for SAM in the TsrM reaction, as a methyl donor and as a key general base. Supplementary Figure 7 depicts the current hypothesis for the kinetic mechanism of the TsrM reaction, showing the dual role of SAM in the reaction. In the priming step, SAM and a reducing agent are used to convert OHCbl or cob(II)alamin to MeCbl. Upon release of SAH, a second molecule of SAM binds, which is followed by Trp binding. Methyl transfer from MeCbl to Trp takes place with the assistance of SAM. MeTrp leaves and then SAM remethylates cob(I)alamin. This sequence of events, wherein cob(I)alamin is generated only when SAM (the methylating agent) is present, would discourage the oxidation of cob(I)alamin to the inactive cob(II)alamin by rapidly converting it to MeCbl.
Discussion
The structure of TsrM provides exciting insight into the mechanism of cobalamin-dependent RS methyltransferases. It would seem that this enzyme evolved directly from a class B RS methyltransferase that most likely methylates an sp3-hybridized carbon center via a radical mechanism. In this instance, both SAM and the substrate undergoing methylation are required to be bound simultaneously, because the 5’-dA• generated from the reductive cleavage of SAM would need to abstract a hydrogen atom directly from the substrate. This substrate radical would then attack the methyl moiety of MeCbl, yielding the methylated product and cob(II)alamin (25). In the TsrM structure, SAM binds near the FeS cluster, but does not ligate to it. Instead, a glutamate residue coordinates to the unique iron of the FeS cluster, which blocks SAM ligation and, therefore, its reductive fragmentation (19). The lack of coordination of SAM to the FeS cluster frees its carboxylate group, allowing it to participate as a general base in the reaction.
Another unexpected finding is the positioning of Arg69, which is on the proximal face of the corrin ring, with its guanidino nitrogens ~3.5 Å away from the Co ion. We posit that this placement of Arg69 is critical for methylation of Trp by MeCbl, because C2 of Trp is weakly nucleophilic, but still can displace cob(I)alamin, the strongest nucleophile in nature. Alkylcobalamins strongly prefer to be hexacoordinate, while pentacoordinated alkylcobalamins are strongly destabilized toward heterolysis of the carbon-cobalt bond (15). Arg69 prevents MeCbl from becoming hexacoordinate by blocking water or any other potential ligand. In addition, the positive charge on Arg69 would serve to destabilize the cob(III)alamin form of the cofactor and stabilize cob(I)alamin, making it a better leaving group. TsrM is a clear outlier in cobalamin-dependent RS methyltransferases as well as in RS enzymes in general, but its structure details how RS architecture and machinery can be co-opted to catalyze non-radical reactions. Given that there are more than 115,000 unique sequences of RS enzymes, most of which are unannotated, additional surprises are surely on the horizon.
Online Methods
Protein purification and crystallization.
Eight homologs of SlTsrM (Uniprot ID: C0JRZ9) were identified by BLAST search and aligned (Supplementary Figure 8). KsTsrM (UniProt ID: E4N8S5) was overexpressed in Escherichia coli as a SUMO fusion construct in the presence of pBAD42-BtuCEDFB and pDB1281 plasmids and then purified and processed to remove the SUMO tag as described previously (24, 26). All manipulations were carried out in a Coy (Grass Lake, MI) anaerobic chamber at room temperature. Size-exclusion chromatography was conducted on a HiPrep 16/60 S200 column using an ÅKTA fast protein liquid chromatography (FPLC) system housed in the anaerobic chamber (GE Biosciences). The protein solutions were prepared from stock protein solutions (>40 mg/mL) that were diluted in water to a concentration of 8 mg/mL KsTsrM, 13.3 mM HEPES pH 7.5, 53.2 mM KCl, 2.6% (w/v) glycerol, and 0.2 mM DTT. All crystals, with or without substrates, were flat brown plates that were obtained within 24 h using the hanging-drop vapor-diffusion method. Native crystals formed in 0.1 M KCl, 12% (w/v) PEG 8000, and 5% (w/v) glycerol as the precipitating reagent by mixing 1 μl of protein with 1 μl precipitating solution in the hanging drop and equilibrating against a 0.5 M LiCl well solution. Crystals were briefly dipped in cryoprotectant (50% (v/v) precipitating reagent and 50% (v/v) ethylene glycol), mounted on nylon loops, and flash-cooled in liquid nitrogen. The high-resolution native dataset was obtained from crystals that formed overnight using a 0.2 M sodium acetate, 0.1 M MES, pH 6.5, and 15% (w/v) PEG 8000 solution as the precipitating reagent, and adding 1 μl of protein with 1 μl precipitating agent and then equilibrating against a 0.5 M LiCl well solution. Crystals were briefly dipped in cryoprotectant (80% (v/v) precipitating reagent and 20% (v/v) ethylene glycol). For substrate-containing structures, KsTsrM was diluted as above, after which aza-SAM and Trp were added to 2 mM and 4 mM final concentrations, respectively. Protein was incubated for 15 mins with substrate before setting up crystal screens. Substrate-containing crystals formed using 0.1 M tri-sodium citrate, pH 5.6, 20% (v/v) isopropanol, 20% (w/v) PEG 4000, and 0.05 M nickel (II) dichloride hexahydrate as the precipitating reagent, and adding 1 μl of protein with 1 μl precipitating solution and equilibrating against either a 0.5 M LiCl or a 0.5 M calcium acetate well solution. The crystals were briefly dipped in cryoprotectant (perfluoropolyether cryo oil from Hampton Research), mounted on nylon loops, and flash-cooled in liquid nitrogen.
Crystallographic structure determinations
X-ray diffraction datasets were collected at the General Medical Sciences and Cancer Institutes Collaborative Access Team (GM/CA-CAT) and the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source, Argonne National Laboratory, and at the Berkeley Center for Structural Biology (BCSB) beamlines at the Advanced Light Source at Lawrence Berkeley National Laboratory. Anomalous and native KsTsrM datasets were processed using the HKL2000 or HKL3000 package. High-resolution (HR) native KsTsrM and KsTsrM +azaSAM +Trp datasets were processed using XDS (27, 28). Because the HR native KsTsrM dataset was collected on non-merohedrally twinned crystals, reflections from each lattice were split from one another during the COLSPOT stage of XDS. These lattices were indexed, integrated, and scaled separately, and the reflections from only the cleanest lattice were used for subsequent analysis. For both the HR native KsTsrM and KsTsrM +azaSAM +Trp datasets, all reflections between a resolution range of 3.65 to 3.68 Å were excluded due to a prevalent ice ring. Structures were determined by single anomalous dispersion (SAD) phasing using autosol/HySS or by molecular replacement using the program PHASER (29–32). Model building and refinement were performed with Coot and phenix.refine (29, 33). Data collection and refinement statistics are shown in Supplementary Table 1. Ramachandran plots were calculated by Phenix and figures were prepared using PyMOL (34, 35).
Diffraction data for SAD phasing were collected at the iron absorption peak (λ =1.7433 Å) with 360° of data measured using a 0.2° oscillation range to 2.50 Å resolution. Additionally, a 2.00-Å resolution native data set was collected at λ =1.0332 Å (Supplementary Table 1). The peak dataset was subjected to Phenix Autosol, which uses HySS to identify heavy atom sites (29). HySS identified eleven heavy atom sites in total, which is close to the expected ten sites for two molecules of TsrM in the asymmetric unit, each containing four Fe ions in an [4Fe-4S] cluster and one Co ion in a cobalamin molecule. The eleventh site identified was an artifact. The initial overall figure-of-merit (FOM) for refinement of these eight Fe sites and two Co sites was 0.271 to 2.50 Å resolution after solvent flattening in RESOLVE (29). Despite the modest FOM, the density modified experimental maps were of sufficient quality for Phenix Autobuild to construct an initial model (residues 1-170, 172-526, 532-567 in chain A, residues 1-165, 179-482, 490-519, 533-565 in chain B). This model was subjected to one cycle of rigid body refinement before being used to obtain phase information for the 2.00-Å resolution native dataset by molecular replacement using Phenix Phase-MR (29). Rfree flags were maintained so that the same 5% of the reflections were used as the test set. Coot was used to add the [4Fe-4S] cluster and Cbl in the early stages of refinement of the model against the native data whereas water and small molecules were added in the later stages. The structure was refined using iterative rounds of positional refinement and individual B-factor refinement, performed in Phenix, and model building, model adjustment, and addition of water molecules were performed in Coot (33). Non-crystallographic symmetry (NCS) restraints were used throughout refinement.
A high-resolution (HR, 1.67 Å) native structure was collected at λ = 0.97872 Å. Using phenix.refine with rigid body refinement, the 2.00-Å model was refined into the HR map data using the same Rfree test sets. The structure was refined using iterative rounds of positional refinement and individual B-factor refinement, which were performed in Phenix, and model building and model adjustment were performed in Coot. In the later stages of refinement for the HR native structure, Translation/Libration/Screw (TLS) parameters were additionally used. The final HR model consists of residues 1-567 (of 575 residues), one [4Fe-4S] cluster, and one cobalamin cofactor in Chain A; residues 1-165, 177-482, 495-519, 532-565 (of 575 residues), one [4Fe-4S] cluster, and one cobalamin cofactor in Chain B. The final structure also contains 18 ethylene glycol molecules and 718 water molecules. Analysis of the Ramachandran statistics showed that 97.52%, 2.30%, and 0.18% of residues are in favored, allowed, and disallowed regions respectively (34). Data collection and refinement statistics are shown in Supplementary Table 1.
Structures containing substrates were processed the same as above, using the same Rfree test set, and the HR model refined into the map. The structures were then manually adjusted with Coot, fitting in substrates if appropriate and building in loops. The final model of TsrM with bound aza-SAM and Trp consists of residues 2-165, 178-483, 495-566 (of 575 residues), one [4Fe-4S] cluster, one cobalamin cofactor, one aza-SAM, and one Trp in Chain A; residues 2-165, 180-483, 493-563 (of 575 residues), one [4Fe-4S] cluster, one cobalamin cofactor, one aza-SAM, and one Trp in Chain B. The final structure also contains 377 waters. Analysis of the Ramachandran statistics showed that 97.57%, 2.43%, and 0.00% of residues are in favored, allowed, and disallowed regions respectively (34).
Oliver Smart at Global Phasing generously provided refinement restraints for cobalamin. Restraints for [4Fe-4S] clusters were based on M. thermoacetica carbon monoxide dehydrogenase/acetyl-CoA synthase (PDB ID: 3I01) (36). Restraints for aza-SAM were from Grade Web Server (Global Phasing). The distance between the unique iron of the [4Fe-4S] cluster and the carboxylate side chain of Glu273 was restrained at 2.0 Å based on Fo-Fc electron density map and previously reported values (37). Composite-omit electron density maps calculated by Phenix Composite_omit_map were used to verify all models (29). Data collection and refinement statistics are shown in Supplementary Table 1.
Synthesis of SAM and SAM analogs
SAM was synthesized from ATP and methionine and purified as described previously (38). Aza-SAM was synthesized as previously described (39). S-5′-adenosyl-3-methylthiopropylamine (dcSAM) was synthesized enzymatically using SAM decarboxylase, a generous gift from Dr. Megan Phillips at UT Southwestern. SAM decarboxylase was overproduced in E. coli BL21 cells grown in 2× YT media and purified by immobilized metal-affinity chromatography using previously described methods (40). The synthesis of dcSAM was derived from an assay previously reported by Xiong et al. (40). Briefly, SAM decarboxylase was added to a reaction containing DTT, SAM, and putrescine, which was incubated for 60 min at 37 °C (40). After quenching in cold water, the reaction mixture was loaded onto a CM cellulose column (5 × 15 cm) to separate dcSAM from SAM. After eluting with a linear gradient of 0 – 0.5 M NaCl in 10 mM ammonium formate buffer, pH 4.5, fractions were analyzed by HPLC with detection by UV-vis spectroscopy at 260 nm. The pooled fractions of dcSAM were then purified on a P2 gel-filtration column (2.5 × 105 cm).
General methods and instruments
All UV-vis spectra were measured in an anaerobic cuvette and recorded on a Varian Cary 50 spectrometer (Agilent, Santa Clara, CA) using the WinUV software. E. coli flavodoxin (flv) and flavodoxin reductase (flr) were also purified as described previously (41). Ultra-performance liquid chromatography with detection by tandem mass spectrometry (LC-MS/MS) was conducted on an Agilent Technologies system coupled to a 6460 QQQ mass spectrometer. The system was operated with the associated MassHunter software package, which was also used for data collection and analysis. Product quantification by LC-MS/MS was done as previously described (9). Graphical figures were made using IgorPro v. 8.0 unless otherwise noted.
Overproduction and Purification of SlTsrM
pRham-SUMO-TsrM was overproduced in the ΔbtuR strain of E. coli in the presence of plasmids pDB1282 and pBAD42-BtuCEDFB as previously described (9, 24). Cells were cultured in LB media supplemented with hydroxocobalamin. At an O.D.600 of 0.3, expression of genes encoded on pDB1282 and pBAD42-BtuCEDFB was induced with 0.2% (w/v) of l-arabinose, while adding FeCl3 and l-cysteine to final concentrations of 25 μM and 75 μM, respectively. Once the cells reached an O.D.600, the flasks were placed on ice for 20 min prior to induction with 0.2% (w/v) l-rhamnose and supplementation with FeCl3 and l-cysteine at final concentrations of 75 μM and 150 μM, respectively. Cultures were incubated at 18 °C for 16 h prior to harvesting. The purification of TsrM and all subsequent experiments were conducted in a Coy anaerobic chamber. A SUMO-TsrM fusion protein was purified by immobilized metal-affinity chromatography, and as described previously, native TsrM was subsequently generated by incubating the fusion protein with the SUMO express protease (Lucigen) (9, 24). The concentration of TsrM was determined by the Bradford assay, as has been previously described (9, 24).
Determination of TsrM Activity
LC-MS/MS was used to quantify product formation using methods previously described (26, 42). All reactions monitoring the activity of TsrM contained the following: SAM or SAM analog (1 mM), Trp (1 mM), 0.1 mM tyrosine (Tyr) (internal standard (IS)), and TsrM (0.1 – 50 μM). Reactions were quenched in 100 mM H2SO4 at designated time points. Standard curves for product quantification by LC-MS/MS contained 0.1 mM Tyr and varying concentrations of MeTrp and SAH (0.25 – 250 μM).
Quenched reaction mixtures were injected onto an Agilent Zorbax Eclipse-C18 RRHD column (2.1 mm × 50 mm, 1.8 μm particle size) equilibrated in solvent A (0.1% formic acid, pH 2.6) and 9.5% solvent B (methanol). A gradient of 9.5-9.7% solvent B was applied from 0.1 to 0.6 min, which was followed by a gradient of 9.7 – 35% solvent B from 0.6 to 0.65 min. Subsequently, a gradient of 35 – 45% solvent B was applied from 0.65 to 1.1 min, which was held constant for 0.4 min before being returned to 9.5% solvent B. The column was re-equilibrated for 0.6 min before subsequent sample injections. Products were detected using Agilent Jet Stream electrospray ionization in positive mode with multiple reaction monitoring.
Kinetic analysis of TsrM reaction
All reaction mixtures contained the following: 50 mM HEPES, pH 7.5, 200 mM KCl, 5% glycerol, 0.1 mM Tyr, 0.15 μM TsrM, 20 μM flv, 10 μM flr, 1 mM NADPH, 55 – 250 μM SAM, and 30, 60, or 90 μM Trp. All reactions were initiated with Trp. LC-MS/MS was used for product quantification. Data analysis was conducted using GraFit (Erithacus Software) and fitting the data globally to Equation 1.
| Equation 1 |
Single turnover assays
All reactions containing premethylated TsrM were conducted in dim lighting in an anaerobic chamber to prevent photolysis of MeCbl. Premethylated TsrM was generated by incubating 400 μM TsrM with 1.5 mM SAM, 40 μM flv, 20 μM flr, and 1 mM NADPH for 45 min at room temperature. The reaction was exchanged into assay buffer (50 mM HEPES, pH 7.5, 200 mM KCl, 5% glycerol, and 0.5 mM DTT) using a NICK column. After premethylated TsrM was collected, it was immediately added to reactions. Assays contained premethylated TsrM (60 μM), Tyr (0.1 mM), Trp (1 mM) and no SAM, or SAM (1 mM), SAH (1 mM), azaSAM (1 mM), or dcSAM (1 mM). Reactions were initiated with premethylated TsrM and quenched in acid at various time points from 0 – 2 min or 0 – 60 min. Quantification of MeTrp by LC-MS/MS was conducted as described above.
Determination of Ki value for dcSAM
All assays contained 0.1 mM Tyr (IS), 0.1 μM TsrM, 1 mM Trp, 40 μM flv, 20 μM flr, 1 mM NADPH, and 55 – 250 μM SAM. The concentrations of dcSAM were 0, 5, 10, and 25 μM. All reactions were initiated with Trp. LC-MS/MS was used for product quantification. Data analysis was conducted using GraFit, fitting the data globally to Equation 2.
| Equation 2 |
Quantification of cobalamin by LC-MS/MS
Reactions were quenched in 100 mM H2SO4 and injected onto an Agilent Zorbax Extend-C18 RRHD column (2.1 mm × 50 mm, 1.8 μm particle size) equilibrated in 95% solvent A (0.1 % formic acid, pH 2.6) and 5% solvent B (acetonitrile). A gradient of 5 - 10% acetonitrile was applied from 0 to 0.4 min, followed by the following gradients: 10 – 40% acetonitrile from 0.4 to 0.8 min, 40 – 50% acetonitrile from 0.8 to 1.6 min, 50 – 5% acetonitrile from 1.6 to 1.9 min, and re-equilibration in 5% acetonitrile from 1.9 to 2.4 min. Products were detected using Agilent Jet Stream electrospray ionization in positive mode with multiple reaction monitoring. The concentrations of AdoCbl, OHCbl, and MeCbl were determine by UV-Vis spectroscopy after treatment with KCN to form the dicyano adduct as previously described (9), and a standard curve of all three molecules (62.5 nM – 64 μM) as well as tyrosine (internal standard) was prepared for analysis by LC-MS/MS. AdoCbl and MeCbl were detected in their +2 charge states, exhibiting m/z values of 790.6 and 673.0, respectively. OHCbl was detected in its +2 charge state after the loss of its hydroxyl group (m/z = 664.9).
Docking analysis of TsrM
The crystal structure of TsrM with bound aza-SAM and Trp was used for docking using the Schrödinger/2016.4 software package (www.schrödinger.com). Protein and ligand were prepared at pH 7.0 ± 2.0 using the Protein Preparation Wizard and LigPrep software, respectively, which are part of the Schrödinger package (43, 44). Protein preparation was performed on chain A in the presence of only three active-site water molecules (HOH 1005, 1062, 1124) located within 4 Å of the bound Trp ligand. Other than the 3 water molecules, bound Trp, cobalamin, aza-SAM and the Fe/S cluster were also kept in crystal structure during protein preparation. The protein preparation includes the following steps; assigning bond orders, adding hydrogen atom, hydrogen bonding optimization and restrained minimization at 0.3 root-mean-square deviation of atomic positions (RMSD). During hydrogen bond optimization step the side chain of active site Glu377 and His268 were kept in charged and neutral form respectively. After preparation, Trp was deleted from the protein structure, and the resulting structure was used to generate the receptor grid using the coordinates from the centroid of bound Trp. The dimensions of the inner and outer box of the receptor grid were set at X=9 Å; Y=10 Å; Z=9 Å (inner box), and X=Z=19.7 Å; Y=20.7 Å (outer box). The 2D structure file (SDF) of L-Trp was downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov). LigPrep was used to determine all possible tautomers, protonation states (with Epik) and stereoisomers of L-Trp at pH 7.0 ± 2.0. Only one structure was reported in the LigPrep output file when the stereochemistry of C-2 was fixed as S. Trp docking was performed using Glide (Schrödinger package) under standard precision and flexible ligand sampling conditions (45–47). The post docking minimization was performed for first 10 poses. The most favorable pose (or poses) was determined both on the basis of docking score (DScore) and glide score (GScore). All the energy minimization related to the entire procedure were performed using OPLS3 forcefield.
Data Availability:
Atomic coordinates and structure factors for the reported crystal structures in this work have been deposited to the Protein Data Bank (PDB) under accession numbers 6WTE (native KsTsrM) and 6WTF (aza-SAM and Trp bound KsTsrM).
Extended Data
Extended Data Fig. 1. Topology diagram of KsTsrM depicting the different domains.
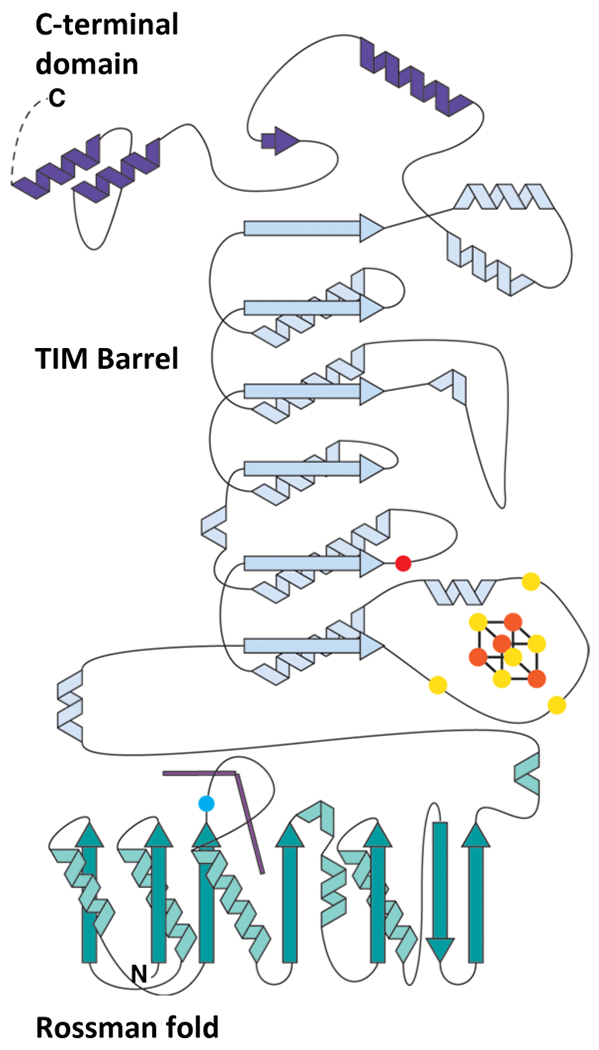
The blue dot represents the conserved Arg residue that occupies the lower axial position of the Cbl, which is shown as two purple lines. The three yellow spheres on the loop after α1 of the TIM barrel represent the three conserved cysteines that coordinate the [4Fe-4S] cluster. The orange sphere on the loop following β2 of the TIM barrel is a conserved Glu residue that coordinates the unique Fe of the cluster.
Extended Data Fig. 2. Structural comparison of TsrM and OxsB.
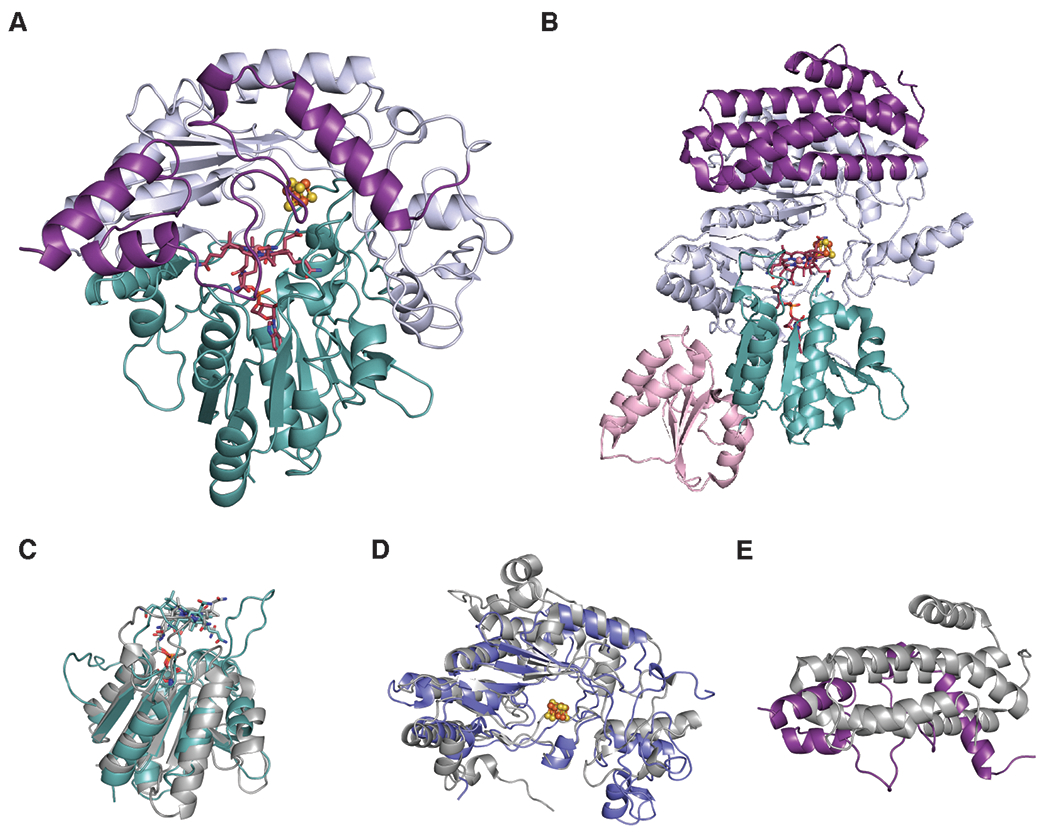
Ribbon diagrams of (A) KsTsrM and (B) OxsB with domains colored to highlight structural similarities: Rossman fold (teal), TIM barrel (light blue), and C-terminal domain (purple). The N-terminal domain of OxsB is colored pink. Overlays of the Rossmann fold (C), TIM barrel (D) and C-terminal domain (E) of KsTsrM (teal, light blue, and purple respectively) with OxsB (grey).
Extended Data Fig. 3. Binding of cobalamin in the Rossmann fold of TsrM.
Conserved stabilizing residues among TsrMs are depicted.
Extended Data Fig. 4. Comparison of lower axial ligands of cobalamin.
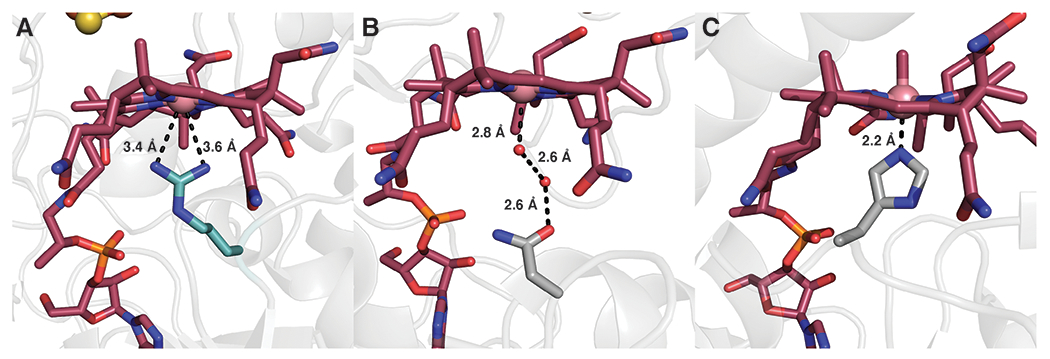
The different axial ligands of (A) KsTsrM, (B) OxsB, and (C) MetH.
Extended Data Fig. 5. Time-dependent production of MeTrp by wild-type KsTsrM and the Arg69Lys variant.
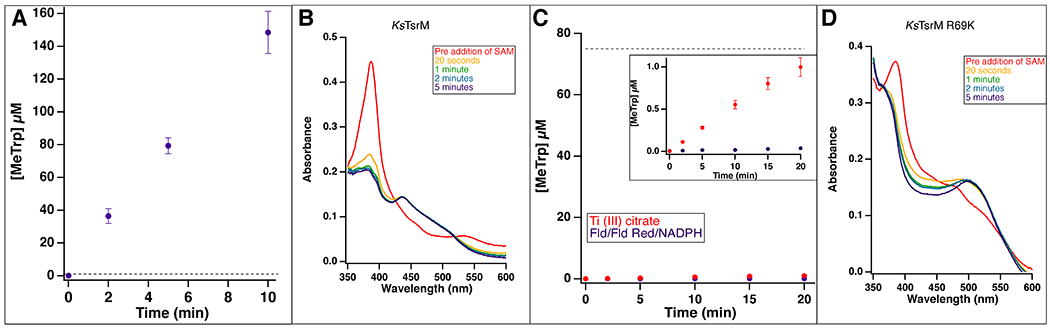
(A) Time-dependent production of MeTrp by KsTsrM (1 μM) using the flavodoxin/flavodoxin reductase/NADPH reducing system. kcat is 14 min−1. (B) UV-vis analysis of KsTsrM. KsTsrM was reduced with Ti (III) citrate to form cob(I)alamin. Upon addition of SAM to the reaction, the peak at 390 nm, indicative of cob(I)alamin, decays, while a peak at 520 nm, indicative of MeCbl, grows in. (C) Time-dependent production of MeTrp by the KsTsrM R69K variant using either Ti (III) citrate (red) or Fld/Fld Red/NADPH (purple) as reductant. Despite using 75 μM enzyme (indicated by dotted line), not even a full turnover is observed over 20 min by KsTsrM R69K with either reducing system. The inset displays a reduced Y-axis range to allow the small amount of activity in the presence of Ti (III) citrate to be observed. (D) UV-vis analysis of KsTsrM R69K. As in (B), KsTsrM R69K was reduced with Ti (III) citrate to generate cob(I)alamin. When SAM is added, cob(I)alamin decays and MeCbl grows in, indicating that MeCbl formation is not significantly diminished with this variant. Error bars represent the standard deviation of reactions conducted in triplicate, with the center point representing the average.
Extended Data Fig. 6. Different roles of Arg residues that occupy the proximal face of cobalamin.
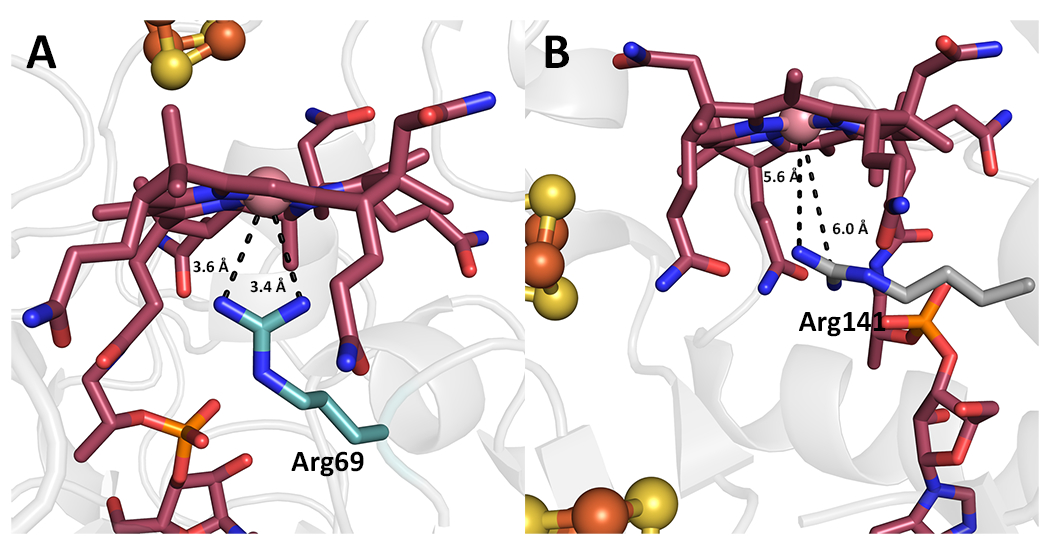
Comparison of the lower proximal face of Cbl in (A) KsTsrM and (B) QueG (PDB: 5D08).
Extended Data Fig. 7. Computational docking studies of Trp positioning in active site.
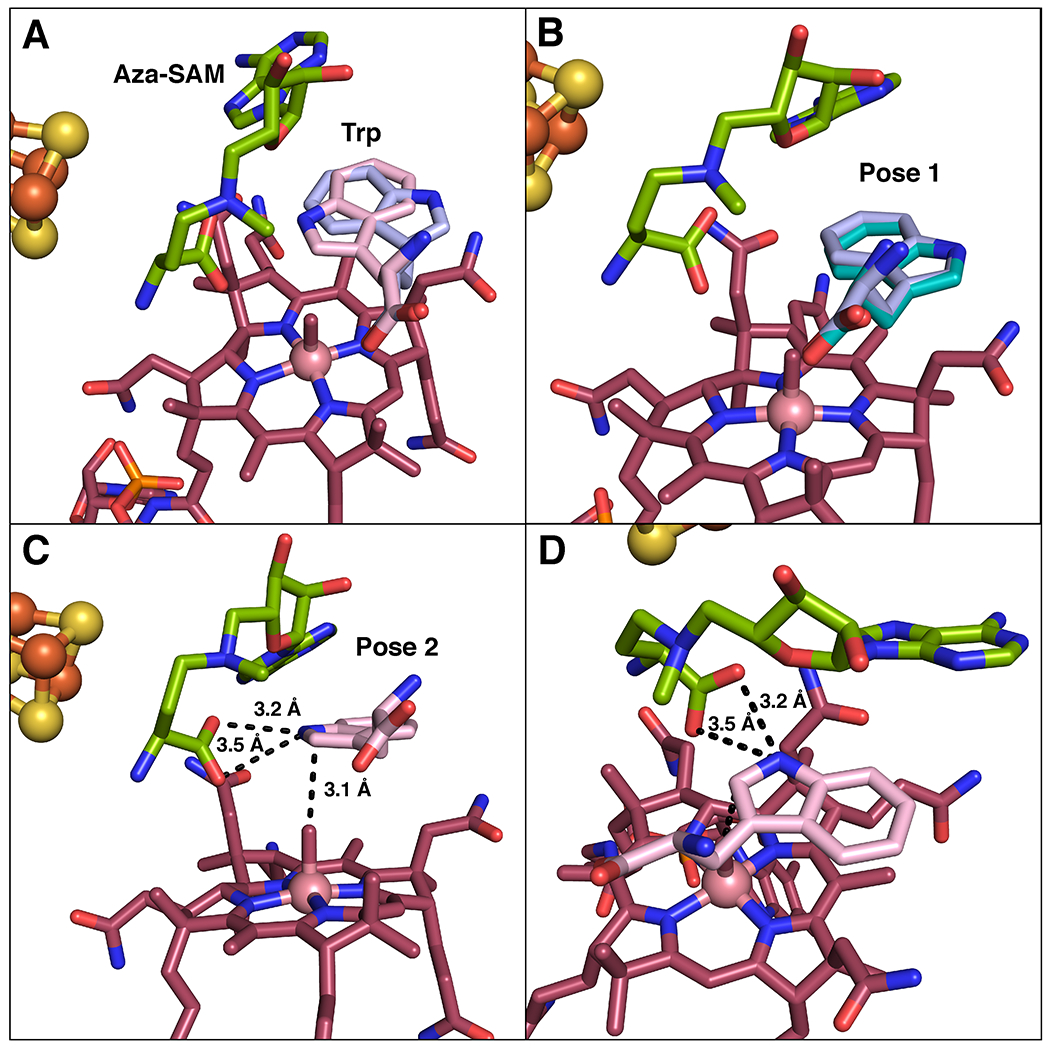
(A) Overlay of the two most likely poses for Trp in the computational docking model: pose 1 (lavender) and pose 2 (pink). (B) Comparison of computational docking model pose 1 (lavender) to flipped Trp observed in crystal structure (teal). (C) Associated distances between carboxylate of aza-SAM to Trp in docking model pose 2. (D) Alternative view of pose 2, demonstrating distances from aza-SAM.
Extended Data Fig. 8. Comparison of Tyr308 in native and substrate-bound structures.
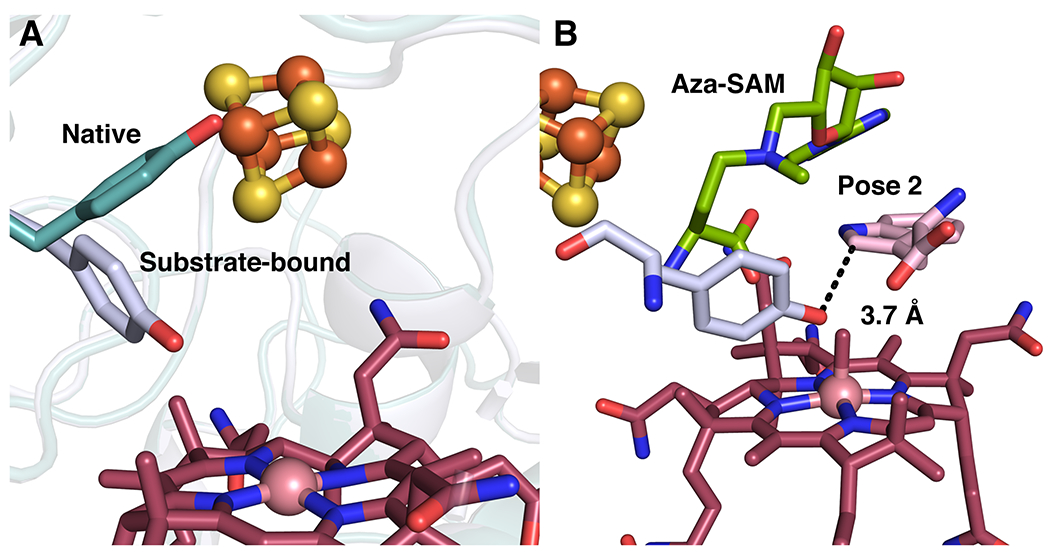
(A) Overlay of KsTsrM native structure (teal) with substrate bound structure (light blue), showing change in conformation of Tyr308. (B) Positioning of Tyr308 in docking model with pose 2, showing proximity to C2 position of Trp.
Extended Data Fig. 9. Analysis of MeCbl formation using dcSAM.
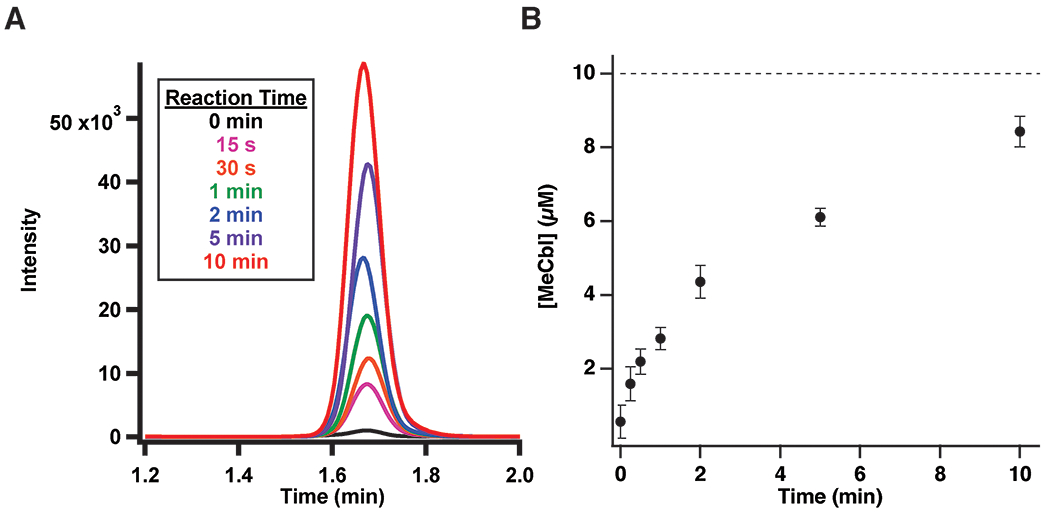
(A) LC-MS/MS analysis of MeCbl formation in a reaction containing TsrM (10 μM), flavodoxin/flavodoxin reductase/NADPH, and dcSAM (1.5 mM). (B) Time-dependent formation of MeCbl. Error bars represent the standard deviation of triplicate determinations. The dashed line indicates the concentration of enzyme in the reaction, with the central point representing the average.
Supplementary Material
ACKNOWLEDGMENTS
Funding Sources
This work was supported by NIH (GM-122595 to S.J.B. and GM-126982 to C.L.D.), and the Eberly Family Distinguished Chair in Science (S.J.B.). S.J.B. and C.L.D. are investigators of the Howard Hughes Medical Institute. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector at GM/CA-XSD was funded by NIH grant S10 OD012289. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). This research also used the resources of the Berkeley Center for Structural Biology supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231. The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169.
Footnotes
The authors declare no competing financial interests
References
- 1.Kelly WL, Pan L, Li CX, Thiostrepton Biosynthesis: Prototype for a New Family of Bacteriocins. J Am Chem Soc 131, 4327–4334 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Bagley MC, Dale JW, Merritt EA, Xiong X, Thiopeptide antibiotics. Chem. Rev 105, 685–714 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Millour J, Lam EW, FOXM1 is a transcriptional target of ERa and has a critical role in breast cancer endocrine sensitivity and resistance. Breast Cancer Res 12, S3–S3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegde NS, Sanders DA, Rodriguez R, Balasubramanian S, The transcription factor FOXM1 is a cellular target of the natural product thiostrepton. Nat Chem 3, 725–731 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Liao RJ et al. , Thiopeptide Biosynthesis Featuring Ribosomally Synthesized Precursor Peptides and Conserved Posttranslational Modifications. Chem Biol 16, 141–147 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frenzel T, Zhou P, Floss HG, Formation of 2-Methyltryptophan in the Biosynthesis of Thiostrepton - Isolation of S-Adenosylmethionine - Tryptophan 2-Methyltransferase. Arch Biochem Biophys 278, 35–40 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Pierre S et al. , Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes. Nat. Chem. Biol 8, 957–959 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Holliday GL et al. , Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a “Plug and Play” Domain. Methods Enzymol 606, 1–71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaszczyk AJ et al. , Spectroscopic and Electrochemical Characterization of the Iron-Sulfur and Cobalamin Cofactors of TsrM, an Unusual Radical S-Adenosylmethionine Methylase. J Am Chem Soc 138, 3416–3426 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Blaszczyk AJ, Wang B, Silakov A, Ho JV, Booker SJ, Efficient methylation of C2 in L-tryptophan by the cobalamin-dependent radical S-adenosylmethionine methylase TsrM requires an unmodified N1 amine. J. Biol. Chem 292, 15456–15467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridwell-Rabb J, Drennan CL, Vitamin B-12 in the spotlight again. Curr Opin Chem Biol 37, 63–70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bridwell-Rabb J, Zhong AS, Sun HG, Drennan CL, Liu HW, A B-12-dependent radical SAM enzyme involved in oxetanocin A biosynthesis. Nature 544, 322–U321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drennan CL, Huang S, Drummond JT, Matthews RG, Ludwig ML, How a Protein Binds B-12 - a 3.0-Angstrom X-Ray Structure of B-12-Binding Domains of Methionine Synthase. Science 266, 1669–1674 (1994). [DOI] [PubMed] [Google Scholar]
- 14.Kime NE, Ibers JA, Co(3)-N Bond-Length in Relation to Co(2)-N Bond-Length . Crystal Structure of Hexaamminecobalt(3) Iodide [Co(Nh3)6]I3. Acta Crystall B-Stru B 25, 168-& (1969). [Google Scholar]
- 15.Kräutler B, Thermodynamic trans-effects of the nucleotide base in the B12 coenzymes. Helv Chim Acta 70, 1268–1278 (1987). [Google Scholar]
- 16.Dowling DP et al. , Molecular basis of cobalamin-dependent RNA modification. Nucleic Acids Res 44, 9965–9976 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vey JL et al. , Structural basis for glycyl radical formation by pruvate formate–lyase activating enzyme. Proc. Natl. Acad. Sci. U S A 105, 16137–16141 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broderick JB, Duffus BR, Duschene KS, Shepard EM, Radical S-Adenosylmethionine Enzymes. Chem Rev 114, 4229–4317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horitani M et al. , Radical SAM catalysis via an organometallic intermediate with an Fe-[5′-C]-deoxyadenosyl bond. Science 352, 822–825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaszczyk AJ, Knox HL, Booker SJ, Understanding the role of electron donors in the reaction catalyzed by Tsrm, a cobalamin-dependent radical S-adenosylmethionine methylase. J Biol Inorg Chem 24, 831–839 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinckley GT, Ruzicka FJ, Thompson MJ, Blackburn GM, Frey PA, Adenosyl coenzyme and pH dependence of the [4Fe–4S]2+/1+ transition in lysine 2,3-aminomutase. Arch. Biochem. Biophys 414, 34–39 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Vey JL, Drennan CL, Structural Insights into Radical Generation by the Radical SAM Superfamily. Chem Rev 111, 2487–2506 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cleland WW, in The Enzymes, Boyer PD, Ed. Academic Press, Ed. (1970), vol. 2, pp. 1–65. [Google Scholar]
- 24.Lanz ND et al. , Enhanced Solubilization of Class B Radical S-Adenosylmethionine Methylases by Improved Cobalamin Uptake in Escherichia coli. Biochemistry 57, 1475–1490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang B et al. , Stereochemical and Mechanistic Investigation of the Reaction Catalyzed by Fom3 from Streptomyces fradiae, a Cobalamin-Dependent Radical S-Adenosylmethionine Methylase. Biochemistry 57, 4972–4984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
References (methods only)
- 26.Blaszczyk AJ, Wang RX, Booker SJ, TsrM as a Model for Purifying and Characterizing Cobalamin-Dependent Radical S-Adenosylmethionine Methylases. Method Enzymol 595, 303–329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabsch W, Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W, Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133–144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams PD et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bunkoczi G et al. , Phaser.MRage: automated molecular replacement. Acta Crystallogr D 69, 2276–2286 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M, HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr D 62, 859–866 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W, Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen VB et al. , MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D-Structural Biology 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The PyMOL Molecular Graphics Systems, Version 2.0 Schrödinger, LLC. [Google Scholar]
- 36.Kung Y, Doukov TI, Seravalli J, Ragsdale SW, Drennan CL, Crystallographic Snapshots of Cyanide- and Water-Bound C-Clusters from Bifunctional Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase. Biochemistry 48, 7432–7440 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding MM, Nowicki MW, Walkinshaw MD, Metals in protein structures: a review of their principal features. Crystallography Reviews 16, 247–302 (2010). [Google Scholar]
- 38.Iwig DF, Booker SJ, Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-L-methionine. Biochemistry-Us 43, 13496–13509 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Joce C, Caryl J, Stockley PG, Warriner S, Nelson A, Identification of stable S-adenosylmethionine (SAM) analogues derivatised with bioorthogonal tags: effect of ligands on the affinity of the E. coli methionine repressor, MetJ, for its operator DNA. Org Biomol Chem 7, 635–638 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiong HS, Stanley BA, Pegg AE, Role of cysteine-82 in the catalytic mechanism of human S-adenosylmethionine decarboxylase. Biochemistry-Us 38, 2462–2470 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Lanz ND et al. , RImN and AtsB as Models for the Overproduction and Characterization Radical SAM Proteins. Natural Product Biosynthesis by Microorganisms and Plants, Pt B 516, 125–152 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Blaszczyk AJ, Wang B, Silakov A, Ho JV, Booker SJ, Efficient methylation of C2 in L-tryptophan by the cobalamin-dependent radical S-adenosylmethionine methylase TsrM requires an unmodified N1 amine. J Biol Chem 292, 15456–15467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olsson MH, Sondergaard CR, Rostkowski M, Jensen JH, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput 7, 525–537 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27, 221–234 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Friesner RA et al. , Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49, 6177–6196 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Friesner RA et al. , Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47, 1739–1749 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Halgren TA et al. , Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47, 1750–1759 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures in this work have been deposited to the Protein Data Bank (PDB) under accession numbers 6WTE (native KsTsrM) and 6WTF (aza-SAM and Trp bound KsTsrM).