

Abstract
Melanoma evolution is a complex process. The role epidermal keratinocytes and dermal fibroblasts play in this process and the mechanisms involved in tumor-stroma interactions remain poorly understood. Here, we used a microfluidic platform to evaluate the cross-talk between human primary melanoma cells, keratinocytes and dermal fibroblasts. The microfluidic device included multiple circular chambers separated by a series of narrow connection channels. The microdevice design allowed us to develop a new cell patterning method based on air-walls, removing the need for hydrogel barriers, porous membranes, or external equipment. Using this method, we co-cultured melanoma cells in the presence of keratinocytes and/or dermal fibroblasts. The results demonstrated that the presence of dermal fibroblasts and keratinocytes led to changes in melanoma cell morphology and growth pattern. Molecular analysis revealed changes in the chemokine secretion pattern, identifying multiple secreted factors involved in tumor progression. Finally, optical metabolic imaging showed that melanoma cells, fibroblasts, and keratinocytes exhibited different metabolic features. Additionally, the presence of stromal cells led to a metabolic shift in melanoma cells, highlighting the role the skin microenvironment on melanoma evolution.
Graphical Abstract
Introduction
Melanoma is one of the most common malignancies in the U.S. and western countries1. Additionally, melanoma incidence has more than tripled in the last decades (1975–2020). Estimates from the American Cancer Society indicate that more than one hundred thousand new melanoma cases will be diagnosed, with more than ten thousand deaths during 2020 only in the US1. Melanoma is caused by transformed melanocytes (i.e., pigment-producing cells located in the skin) that start to proliferate out of control2. Once melanoma cells leave the epidermis (i.e. the most superficial layer of the skin) and enter the underlying tissue layers (e.g., the dermis and the submucosa), the tumor has become invasive, which increases the risk of metastasis and worsens the patien’s prognosis3. However, the molecular and environmental factors driving melanoma progression and metastasis are still poorly understood. In this context, the ability of melanoma cells to invade the dermis alone is not a malignant feature by itself, as most “naevi” (e.g., moles or birthmarks) are associated with the presence of melanocytes (i.e., not cancerous cells) in the dermis that do not lead to melanoma4.
Previous studies have shown that melanoma cell invasion is a complex process where the interactions between melanoma and stromal cells (e.g., dermal fibroblasts and keratinocytes) play a critical role5–7. Fibroblasts, keratinocytes, and melanoma cells secrete multiple growth factors and cytokines that affect numerous cellular functions such as cell migration, proliferation, and metabolism6. In addition, tumor-stroma crosstalk has also been shown to modulate treatment response (e.g., stromal cells induce resistance to BRAF inhibitors)8. Thus, understanding the cell cross-talk between melanoma and stromal cells is critical for the identification of efficient predictors of melanoma progression, and for the development of successful therapies.
Microfluidic devices allow the user to control cell location and patterning, offering a versatile tool to study cellular cross-talk 9–13. Additionally, numerous studies have demonstrated that microscale systems favor cellular cross-talk due to signaling molecule (e.g., chemokines) concentration14. In this context, microfluidic systems require significantly lower media volume (in the scale of few μL) compared with traditional assays based on Petri dishes (which require a volume in the mL scale). As a consequence, microfluidic devices are ideally suited to detect cellular cross-talk and cell-cell signaling15. Additionally, microscale systems allow more precise control of cell seeding and culture geometry, which in turn has an impact on cell response (e.g., endothelial cells cultured as lumen behave more similar to their in vivo counterparts compared with traditional 2D monolayers)16. Previous microfluidic devices relied on porous membranes to separate multiple microfluidic chambers and generate patterned co-cultures10, 17–19. Depending on the membrane properties and porous size, these membranes can block the diffusion of cells during the seeding process, while they allow chemokines and other signaling factors to diffuse through the membrane pores. Other microdevices have used hydrogels to generate patterned co-cultures12, 20–22. In these systems, a solution of collagen, Matrigel, fibrinogen, or other extracellular matrix protein, is flown through a microfluidic channel flanked by two or more lateral chambers. The hydrogel provides a physical barrier that allows the user to seed one cell type on each side of the hydrogel barrier. Multiple studies have used this method to study cell cross-talk (e.g., tumor-induced angiogenesis23, chemotaxi12s, and immune infiltration24). However, the presence of the hydrogel pose some limitations: 1) some cells might not be able to migrate through the hydrogel (e.g., fibrin hydrogels are difficult to degrade for multiple cell types); 2) hydrogels can bind secreted proteins such as chemokines and growth factors, hindering or biasing cellular cross-talk; 3) hydrogels are prone to evaporation, limiting their use when the volume of hydrogel is limited. Thus, in this manuscript, we have leveraged a new cell patterning method based on the use of air-walls to study melanoma cross-talk. We fabricated a microfluidic device with a central chamber flanked by two lateral chambers, connected by a series of narrow microchannels. We use air-walls to pattern melanoma cells, dermal fibroblasts, and keratinocytes in the multiple chambers of the microdevice. After removing these air-walls, we studied the effects of fibroblasts and keratinocytes on melanoma cell morphological phenotype, chemokine secretion, and metabolism.
Materials and Methods
Microdevice Fabrication and operation
Details of the microdevice fabrication can be found in25. Two templates, top and bottom, were generated using illustrator and fabricated with SU-8 lithography techniques. Upon completion, this template was used to generate the microdevices. PDMS was poured on both the top and bottom template and a minimum of 30lbs was placed on the templates as it polymerized for 4 hours at 80 °C. The devices were peeled off the wafer and additional polymerized PDMS covering the ports was removed. Then, the PDMS was placed in an isopropyl solution for 10 minutes, after the two sides were placed together creating a cavity. A 340 μm-diameter PDMS rod was inserted. Then the device underwent plasma bonding to a 60 mm glass-bottom Petri dish. Before use in cell culture, the devices were sterilized with 15 minutes of UV exposure. Finally, the microdevices were treated with polyethyleneimine (Sigma-Aldrich, 03880) diluted in water at 2% for 10 minutes and then treated with glutaraldehyde (Sigma-Aldrich, G6257) diluted at 0.4% in water for 30 minutes.
The first step to fabricate any microdevice is to get the SU-8 design of choice on the Silicon wafer. This is done by using a complementary photomask which only exposes the area of choice in the SU-8 and lets that polymerize. Upon the wafer, liquid PDMS is poured and polymerized. Solid PDMS is then peeled off and plasma bonded to a petri dish. The design for this experiment involves a central chamber and 2 lateral chambers, each with a loading port. Our microdevice included a central circular microchamber to culture melanoma cells with two lateral chambers to seed dermal fibroblasts and/or keratinocytes. The central chamber was connected to the lateral chambers by a series of concentric narrow microchannels (i.e., 10μm height). The microdevice was designed to confine different cell populations in their respective chambers (i.e. lateral vs central), allowing cellular crosstalk and migration. Previous studies have relied on hydrogel patterning in other designs including similar connection microchannels to generate physical barriers or walls. These hydrogels allowed researchers to pattern cells in adjacent chambers while allowing cellular crosstalk. Thus, we treated the microdevices with oxygen plasma to render the material hydrophilic, ensuring that the collagen solution flowed through all the chambers and connection microchannels. Next, we aspirated the collagen solution from the central and lateral chambers, leaving the collagen solution contained inside the connection microchannels. However, our results showed that hydrogel patterning may be challenging, or even impossible, in systems where evaporation occurs rapidly due to the small volume contained in such microchannels (Figure 1B and Supporting movie 1). The time-lapse images showed that the 4 mg/ml collagen solution confined in the connection microchannels rapidly evaporated in less than 2 min, making cell patterning impossible (Supporting Figure 1). In the absence of these hydrogel-walls, cells injected in the lateral chambers immediately flowed through the connection microchannels, invading the central chamber and vice versa. Although the microchannel design can be optimized to generate a high fluidic resistance path (e.g., in our design the connection channels had a cross-section 50 times smaller than the central channels, theoretically providing 2500-fold increase in fluidic resistance), cell patterning is extremely challenging without the presence of the physical barrier provided by the hydrogel (Supporting Figure 1). Thus, in our design we explored the use of air-walls to assist cell confinement without the use of hydrogels or other solutions (Figure 1C). In our PDMS-based microdevice aqueous solutions filled first the fluidic path with the highest cross-section (i.e., the central and lateral chambers, as opposed to the connection channels). A cell suspension containing 4 million melanoma cells/ml (labelled in red) was injected through the central inlet. The cell suspension flowed through the central microchamber and reached the central outlet without invading the connection microchannels (Figure 1A–E). Next, a second cell suspension containing 4 million fibroblasts or kertinocytes/ml (labelled in green) was perfused through the lateral chamber, reaching the lateral outlet without invading the connection channels. This approach allowed us to perfectly confine the multiple cell types, leveraging the air-walls trapped in the connection channel. After seeding the cells, the microdevices were placed in the incubator (i.e., 37 °C and 5% CO2) overnight, allowing the air-walls to dissolve in a few hours. Once the air-wall is completely dissolved, the liquid bridges the lateral and central chambers, allowing cellular cross-talk and fibroblast and melanoma cell migration, whereas most keratinocytes remained forming cell clusters in their respective chamber (Figure 1F). The time required for the air-walls to dissolve depended on the air-wall volume, allowing the user to design air-walls with a different volume to control the dynamics of the process (Figure 1G–I).
Figure 1. Microdevice design and air-wall confinement.

A) Scheme depicting the PDMS-based microfluidic device containing the central chamber and two lateral chambers on the sides, connected by a series of concentric narrow microchannels. B) Brightfield time-lapse images showed the evaporation kinetics of liquid-walls. Microdevices were treated with oxygen plasma to render them hydrophilic and ensure the collagen solution flowed through all the chambers and connection microchannels. Next, the collagen solution was aspirated from the lateral and central chambers, leaving the collagen solution confined inside the connection microchannels (upper panel). Given the small volume contained inside the microchannels, the hydrogel solution rapidly evaporated in 2 min (mid and lower panel). C and D) Time-lapse images showing the dynamics of the air-wall dissolving. Microdevices were not treated with plasma, to ensure the liquid injected in the lateral and central chambers did not invade the connection microchannels, generating an air-wall inside these microchannels. The air-wall took several hours to completely dissolve in 10μm-height microchannels, providing the cells enough time to attach to the bottom of the chambers before physically connecting them. E) Melanoma cells and dermal fibroblasts (stained in red and green) were confined in the central and lateral chambers respectively using air-walls. F) After 24 hours, the air-wall was completely dissolved, and fibroblasts and melanoma cells started to migrate through the connection microchannels. G) Scheme of the microdevice design. H) Microdevices designed with 10μm and 40 μm-height connection microchannels. Images demonstrate air-walls dissolve at different speeds depending on their volume, showing the 40 μm-height air-wall disappear slower than the 10 μm-height.I) Bar graph showed the percentage of air-wall dissolved at different time-points in the 10 and 40μm-height air-wall microdevices. J) Fluorescence microscopy images showed the confinement achieved by the air-wall method. Melanoma cells, dermal fibroblasts, and keratinocytes were labelled in red, green, and infra-red (shown in yellow) respectively.
Cell isolation
Primary dermal fibroblasts and keratinocytes were isolated from fresh neonatal foreskin specimens obtained from a local birthing unit (Meriter Hospital, Madison, WI.). Specimens are collected in 10mL DMEM (Gibco) with 1% antibiotics/antimycotics (Gibco) and 2% FBS (Corning) and stored at 4C until processing begins (within 24 hours of collection). After collecting the samples, tissue specimens were placed in 10cm-dishes and washed three times with 4°C HBSS (Hyclone) supplemented with 1% antibiotics and antimycotics to remove residual blood from the circumcision process. Excess fat and connective tissue were removed with curved surgical scissors, placed in a new 10cm-dish, and rinsed five times with 4°C antibiotic-supplemented HBSS. After the 5th wash, the specimen was cut into 4×4mm pieces with a scalpel blade and stored in a 10cm-dish containing 0.5% trypsin-EDTA 10X (Fisher Scientific) with the most superficial layer of skin, or epidermis, facing up for 16–24h at 4 °C. After trypsin digestion, the small pieces of tissue were transferred into a 6cm-dish containing supplemented HBSS with 10% FBS to inactivate trypsin. The epidermis containing keratinocytes was then removed from the dermis using forceps and remained in supplemented HBSS solution. Keratinocytes were released from the epidermal sheet by vigorous pipetting. The epidermal sheet and cell suspension were filtered through a 70m cell strainer to remove tissue debris and washed with 5mL supplemented HBSS solution and collected in a 50mL conical tube. The volume of the collected cell suspension was pipetted into a 15mL conical tube and centrifuged at 1200rpm for 5 minutes to pellet the cells. After supernatant is discarded, keratinocyte specific media was used to resuspend the cells (i.e., 154 media (Gibco) containing Human Keratinocyte Growth Medium (Gibco) and 1% penicillin/streptomycin (Gibco), and the cell suspension was plated in a 6 well plate and incubated at 37C and 5% CO2. Media is changed 24 hours after initial plating and every other day until cultures reach 70% confluency. Fibroblasts were dissociated from the dermal tissue by placing the dermis pieces in a 50mL conical tube containing dispase and collagenase solution (1mg/mL) and incubated at 37C and 5% CO2 for two hours. DMEM containing 10% FBS was then added to inactivate the enzyme solution. Dermal tissue and cell suspension were vigorously pipetted to release fibroblasts, filtered through a 70 μm cell strainer and collected in a 50mL conical tube. The cell strainer was washed with 4°C antibiotic-supplemented HBSS solution. The cell suspension was centrifuged at 1200rpm for 5 minutes and the cell pellet was resuspended in DMEM +10% FBS +1% pen/strep, plated in a 6 well plate and incubated at 37C and 5% CO2. Media was changed 24 hours after initial plating and every other day until cultures reached 70% confluency.
Cell Culture and microdevice operation
The melanoma cell line used was WM-115, a primary melanoma with competence for metastasis, obtained from Rockland Immunochemicals (Limerick, PA). The cell line consists of the BRAFV600D mutation and the PTEN loss of function due to hemizygous deletion and was established from 55-year old female patient. Melanoma cells were cultured in Tumor Specialized Media containing 10 % FBS and Pen/Strep. Keratinocytes were cultured in Gibco 154 medium supplemented with Human Keratinocyte Growth Serum and 1% Pen/Strep. Fibroblasts were cultured in Gibco DMEM with 10% FBS and 1% Pen/Strep.
Cell staining and fluorescence microscopy
Melanoma cells, keratinocytes, and dermal fibroblasts were stained in red with 10 μM cell tracker red (C34552); green with 10 μM calcein AM (C1430); and infra-red with 20 μM Vybrant (DiD V22887, Thermo Fisher) to monitor cell confinement and migration in the microdevice. Cells were trypsinized and stained in the desired staining for 5 min. Next, cells were centrifuged and washed twice with 15 ml of PBS to remove the excess of staining. After seeding the cells in the microdevice, cell confinement, migration, and morphology were analyzed using a Nikon TiE microscope with temperature and CO2 control system set at 37 °C and 5% respectively.
Multiplexed assay for measurement of cytokine secretions
To measure cytokines secreted in the media of monoculture and co-culture systems, a multiplexed bead-based assay was used. Media was collected after 48 and 72 hours in culture and samples were prepared and analyzed using the Inflammation 20-Plex Human ProcartaPlex™ Panel (EPX200–12185-901, Thermo Fisher) following the manufacturer’s guidelines. Briefly, cytokines were detected by sequentially incubating with antibody bead cocktail solution, detection antibody and streptavidin-R-phycoerythrin. Magnetic beads were washed using a magnetic plate washer prior to mixing with samples and after each incubation. Samples were read on MAGPIX Luminex Xmap system (Luminex Corporation, Austin, Texas) using Luminex xPonent software. The results were expressed as mean fluorescence intensity (MFI) for each analyte in each sample. Average MFI values from standards were converted to concentrations (pg/mL) using cytokine-specific standard curve data.
Optical metabolic imaging
A custom-built inverted multiphoton microscope (Bruker Fluorescence Microscopy, Middleton, WI), was used to acquire fluorescence intensity and lifetime images. The equipment consists of an ultrafast laser (Spectra Physics, Insight DSDual), an inverted microscope (Nikon, Eclipse Ti), and a 40× water immersion (1.15NA, Nikon) objective. Next, NAD(P)H and FAD images were obtained for the same field of view. FAD fluorescence was isolated using an emission bandpass filter of 550/100 nm and excitation wavelength of 890 nm. NAD(P)H fluorescence was isolated using an emission bandpass filter of 440/80 nm and an excitation wavelength of 750 nm. Subsequently, fluorescence lifetime images were collected using time-correlated single-photon counting electronics (SPC-150, Becker and Hickl) and a GaAsP photomultiplier tube (H7422P-40, Hamamatsu). 512-pixel images were obtained using a pixel dwell time of 4.8 μs over 60s total integration time. To guarantee adequate photon observations for lifetime decay fits and no photobleaching, the photon count rates were maintained at 1–2 × 105 photons/s. The instrument response function was calculated from the second harmonic generation of urea crystals excited at 900 nm, and the full width at half maximum (FWHM) was measured to be 244 ps. A Fluoresbrite YG microsphere (Polysciences Inc.) was imaged as a daily standard for fluorescence lifetime. The lifetime decay curves were fit to a single exponential decay and the fluorescence lifetime was measured to be 2.1 ns (n=7), which is consistent with published values.
Image and analysis
Redox ratio values for all conditions were normalized to the melanoma only condition (i.e., melanoma alone mean value = 1). NAD(P)H and FAD intensity and lifetime images were analyzed using SPCImage software (Becker & Hickl) as described previously26. The fluorescence lifetime decay curve was deconvolved with the instrument response function and fit to a two-component exponential decay model at each pixel, I(t)=α1*e(−t/τ1)+α2*e(-t/τ2) + C, where I(t) represents the fluorescence intensity at time t after the laser excitation pulse, α accounts for the fractional contribution from each component, C represents the background light, and τ is the fluorescence lifetime of each component. Since both NAD(P)H and FAD can exist in two conformational states, bound, or unbound to enzymes, a two-component model was used. The short and long lifetime components reflect the bound and unbound conformations, respectively for FAD. While the opposite is true for NAD(P)H, the short and long lifetime components correspond with the unbound and bound conformations, respectively. The mean lifetime (τm) was calculated using, τm = α1τ1+ α2τ2 for both NAD(P)H and FAD. The optical redox ratio was determined from the NAD(P)H and FAD lifetime data by integrating the photons detected at each pixel in the image to calculate the total intensity. For each pixel, the intensity of NAD(P)H was then divided by the intensity of FAD. Using Cell Profiler, an automated cell segmentation pipeline was created27. This system identified pixels belonging to nuclear regions by using a customized threshold code. Cells were recognized by propagating out from the nuclei within the image. To refine the propagation and to prevent it from continuing into background pixels, an Otsu Global threshold was used. The cell cytoplasm was defined as the cell borders minus the nucleus. Values for NAD(P)H τm, FAD τm, NAD(P)H intensity, FAD intensity, and the optical redox ratio (NAD(P)H/FAD intensity) were averaged for all pixels within each cell cytoplasm. At least 100 cells per sample were analyzed, and every experiment was repeated at least three times.
Confocal microscopy images were analyzed by FIJI. To examine molecule diffusion and cell viability, a rectangle-shape region was drawn, and the intensity profile was calculated using FIJI software.
Statistical analysis
All experiments were repeated at least three independent times. Replicates were consistent and showed similar trends. The normal distribution assumption for statistical tests was confirmed by the Kolmogorov-Smirnov test. Statistical significance was set at p < 0.05. For nonparametric comparisons, a Kruskal-Wallis test was performed followed by the Mann-Whitney U test. Analysis was performed in GraphPad Prism 9 (https://www.graphpad.com/scientific-software/prism/).
Results and Discussion
Microfluidic model and cell culture
Melanoma progression is a complex process and how dermal fibroblasts, and neighboring epidermal keratinocytes affect melanoma cell behavior is not well understood. Here, we fabricated a microfluidic device to evaluate the influence of keratinocytes and stromal fibroblasts on primary melanoma cells (Figure 1A). We injected melanoma cells, dermal fibroblasts, and keratinocytes in the central, left, and right microchamber, respectively. Using the air-walls described in the material section, we ensured these cell types remained confined in their respective chambers after the cell seeding. The microdevices were placed in the incubator and after a few hours, the cells attached to the bottom and the air-walls dissolved, allowing cell cross-talk and migration. In this context, after 24 hours, fibroblasts and melanoma cells migrated into the connection channels, reaching the cells located in the adjacent chamber. The use of these air-walls allowed us to pattern melanoma cells, dermal fibroblasts, and keratinocytes in the central and lateral chambers (Figure 1G). Fluorescence microscopy revealed cell remained viable (>90% cell viability) after 3 days and they were able to proliferate (Supporting Figure 3). The analysis revealed melanoma cells exhibited the fastest proliferation rate, probably as a consequence of their tumorigenic nature, whereas primary keratinocytes displayed the slowest proliferation rate. . We injected melanoma cells, dermal fibroblasts, and keratinocytes in the central, left, and right microchamber, respectively. Using the air-walls described in the material section, we ensured these cell types remained confined in their respective chambers after the seeding. The microdevices were placed in the incubator and after a few hours, the cells attached to the bottom and the air-walls dissolved, allowing cell cross-talk and migration. In this context, after 24 hours, fibroblasts and melanoma cells migrated into the connection channels, reaching the cells located in the adjacent chamber. The use of these air-walls allowed us to pattern melanoma cells, dermal fibroblasts, and keratinocytes in the central and lateral chambers (Figure 1G). Fluorescence microscopy revealed cell remained viable (>90% cell viability) after 3 days and they were able to proliferate (Supporting Figure 3). The analysis revealed melanoma cells exhibited the fastest proliferation rate whereas primary keratinocytes displayed the slowest proliferation rate.
Stromal cells induced changes in melanoma cell morphology
Epidermal keratinocytes and stromal cells such as dermal fibroblasts surround primary melanoma cells and have the potential to modify tumor evolution and progression. Thus, we co-cultured primary melanoma cells in the presence of dermal fibroblasts and keratinocytes leveraging the air-wall confinement method. Melanoma cells were stained with a red-fluorescent cell tracker and after 3 days in the microdevices, cells were visualized by fluorescence microscopy (Figure 2A). To avoid any potential bias due to different media composition, we used the same media in all the mono-culture and co-culture experiments performed in the microdevice (i.e., a 1:1:1 mixture of melanoma cells, fibroblast, and keratinocyte media). The microscopy images revealed that when cultured in monoculture, melanoma cells formed multicellular clusters with multiple cells grouped together (Figure 2B). On the other hand, when keratinocytes or dermal fibroblasts were present, melanoma cells changed their morphology and transition to a single cell phenotype. The morphological analysis demonstrated that in monoculture, melanoma cells exhibited a circular-like shape (i.e., low aspect ratio), whereas in the presence of dermal fibroblasts or keratinocytes they became more elongated, exhibit a spindle-like morphology (Figure 2C). To ensure we had a homogenous sample, different fields of view were imaged in all the different conditions (i.e., next to the diffusion ports and center of the chambers), observing no significant differences between the different locations. We also repeated these experiments embedding the multiple cell populations on a 3D collagen hydrogel to explore this concept and demonstrate the capability of air-wall to operate in 3D assays. The results demonstrated the profound effects collagen density and collagen polymerization temperature exert on collagen structure (Supporting Figure 4A–C). Next, we observed how different cell types modify the collagen extracellular matrix. Dermal fibroblasts exhibited spatial orientation, contracting and aligning the collagen fibers in the same direction, whereas melanoma cells led to no marked changes in the collagen architecture (Supporting Figure 4D and E). Regarding changes in cell morphology in 3D environments, we observed similar changes as observed in 2D, namely, the co-culture with dermal fibroblasts, keratinocytes, or both, led to a significant melanoma cell elongation (Supporting Figure 5). Future studies could use this approach to explore whether the presence of both cell types (i.e., fibroblasts and keratinocytes) has an additive effect on melanoma cell morphology, or one of them is enough to induce the morphological switch.
Figure 2. Morphological analysis.
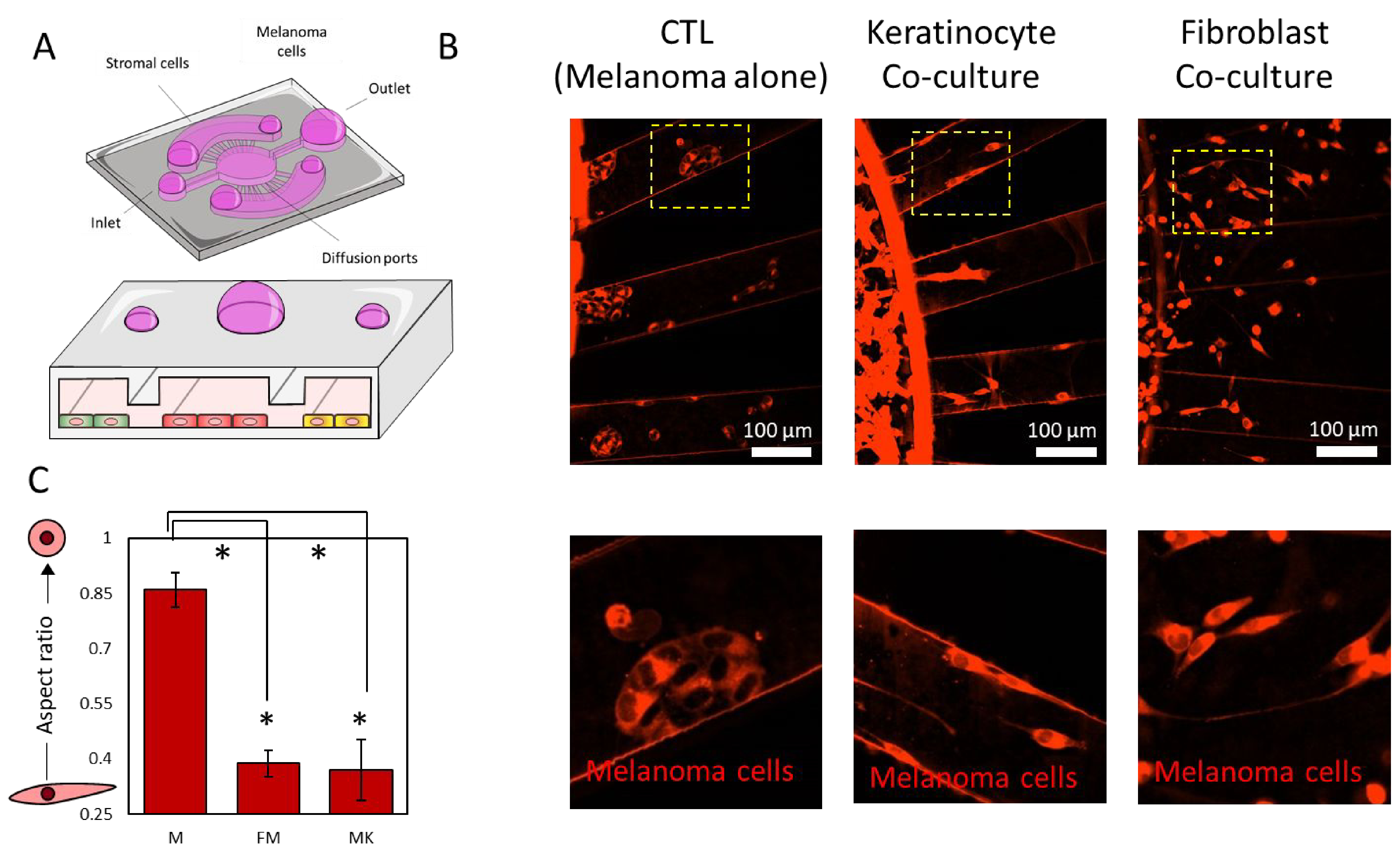
A) Scheme illustrating the experimental protocol: melanoma cells were confined in the central chamber using the air-wall method with/without dermal fibroblasts/keratinocytes in the lateral chambers. After 3 days in culture, the microdevices were imaged to visualize cell migration and morphology. B) Fluorescence microscopy images showed melanoma cells stained in red after 3 days in culture in monoculture (control condition) and in the presence of fibroblasts, or keratinocytes, or both in the lateral chambers. When cultured alone, melanoma cells formed multicellular clusters. When cultured in the presence of fibroblasts, keratinocytes, or both, melanoma cells spread and appeared as isolated cells. C) Graph showed the melanoma cell aspect ratio in monoculture and in the presence of dermal fibroblasts and keratinocytes. Asterisks denote p-value<0.05. Experiments were repeated at least three independent times with replicates showing showed similar trends.
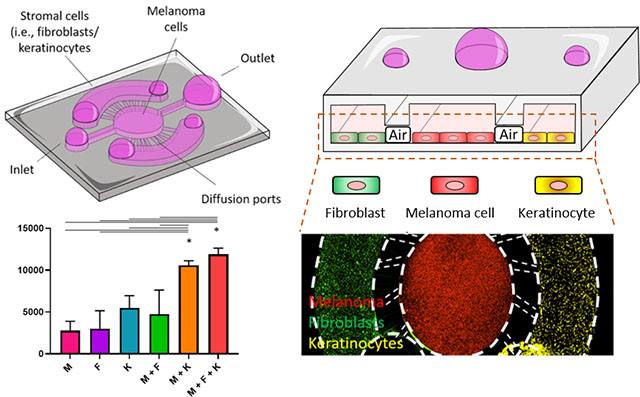
After analyzing the morphological changes induced by the presence of skin cells, we set out to evaluate the molecular cross-talk between dermal fibroblasts, keratinocytes, and melanoma cells. We confined the three cell populations in the microdevice and after 3 days in culture, we analyzed secretion of multiple chemokines and growth factors (Figure 3A). The results demonstrated that the presence of fibroblasts, keratinocytes, or both, led to significant changes in chemokine secretion (Figure 3B). More specifically, keratinocyte monoculture secreted higher levels of IL-6, which were further increased in the triple co-culture with fibroblasts and melanoma cells. IL-6 is a pleiotropic chemokine produced by multiple cell types including melanoma cells and keratinocytes and dermal fibroblasts in response to inflammation or UV exposure. In melanoma, IL-6 was shown to decrease tumor apoptosis and increase tumor cell proliferation in advanced tumor stages28, 29. The analysis revealed that the three cell types secreted IL-8, which has been also associated with increased melanoma invasion, and metastatic potential. Additionally, IL-8 also regulates tumor-induced angiogenesis30, 31. Exposure to UV light leads to overexpression of IL-8 in melanoma cells and is known to accelerate tumor growth. In the co-culture conditions, we observed higher amounts of IL-8, but the levels were not significantly higher than the sum of the fibroblasts, keratinocyte, and melanoma mono-culture conditions combined. However, the observations that keratinocytes and dermal fibroblasts secreted IL-8 suggested their tumor-promoting role in melanoma evolution. IL-1β is an immunomodulatory chemokine secreted by multiple immune cells in response to inflammatory signals32. In normal conditions, IL-1β contributes to resolve the last steps of the inflammatory response. However, during the chronic inflammation response generated inside the tumor, IL-1β promotes tumor growth and metastasis by multiple mechanisms (e.g., NF-κB, MAPK, AKT, and WNT pathways)32. Additionally, IL-1β induces the secretion of other chemokines including the IL-1 family which plays a critical role in tumor progression. In this context, we also observed that keratinocytes secreted high levels of IL-1α. Secretion of IL-1α represses the expression of the transcription factor MITF-M, which controls melanocyte differentiation. As a consequence, IL-1α contributes to maintaining an undifferentiated state in melanoma cells, which contributes to tumor growth. Additionally, IL-1α decreases the generation of immunogenic antigens, contributing to an immunosuppressive microenvironment within the tumor33. Additionally, IL-1α decreases the redox state of melanoma cells and induces the generation of reactive oxygen species (ROS), which in turn also contribute to maintaining a tumor-promoting inflammatory response32. These results suggested dermal fibroblasts, and especially keratinocytes, may be playing a tumor-promoting role. However, keratinocytes also secreted IP-10 and MCP-1, which may help to control tumor growth. IP-10 is an angiostatic that has shown a potent antiangiogenic effect34. Previous animal studies demonstrated that melanoma models overexpressing IP-10 were associated with lower microvascular density, slower tumor proliferation, and increased apoptosis. However, the specific molecular mechanisms controlling this response remain poorly understood. Recent studies suggested that IP-10-induced angiostatic effects are mediated through the CXCR3 pathway, which has been associated with lymph node metastasis in melanoma35. On the other hand, our experiments also revealed that fibroblasts, melanoma cells, and keratinocytes secreted MCP-1, which has been associated with immune regulation36. The co-culture of melanoma cells with fibroblasts and/or keratinocytes showed an additive effect rather than synergistic regarding MCP-1 secretion. MCP-1 is a chemoattractant protein that primarily recruits macrophages to the tumor microenvironment37. In melanoma, macrophages can play a dual role, promoting or inhibiting tumor growth depending on other co-stimulatory factors present in the surrounding microenvironment. Interestingly, studies have shown that macrophages attracted by MCP-1 can lead to tumor formation in non-tumorigenic lesions. Altogether, these results highlight the complex interactions between melanoma cells and other surrounding stromal cells such as dermal fibroblasts and keratinocytes, which in turn, can control melanoma transition from non-tumorigenic stage to an invasive tumor and metastatic disease. Whereas multiple studies have focused on the role of fibroblasts in melanoma progression, the effects of keratinocytes on primary melanoma have not been studied. In this context, our results suggest that keratinocytes may also play an important role in melanoma evolution by secreting multiple tumor-promoting chemokines. One limitation of the techniques used in this study (i.e., MagPIX analysis) is the fact that the magnetic beads used for analysis only bind to chemokines dissolved in the media, preventing us from identifying the cell type(s) responsible for secreting these chemokines. Additionally, in this study we only analyzed chemokine secretion whereas cellular cross-talk can involve multiple mechanisms such as exosomes, RNA, or even mechanical forces. Screening studies could explore the role of these factors individually to try to pinpoint the specific factors driving melanoma progression. Finally, identification of the key pathways controlling melanoma progression might lead to new successful therapies to prevent and treat melanoma.
Figure 3. Cellular cross-talk.
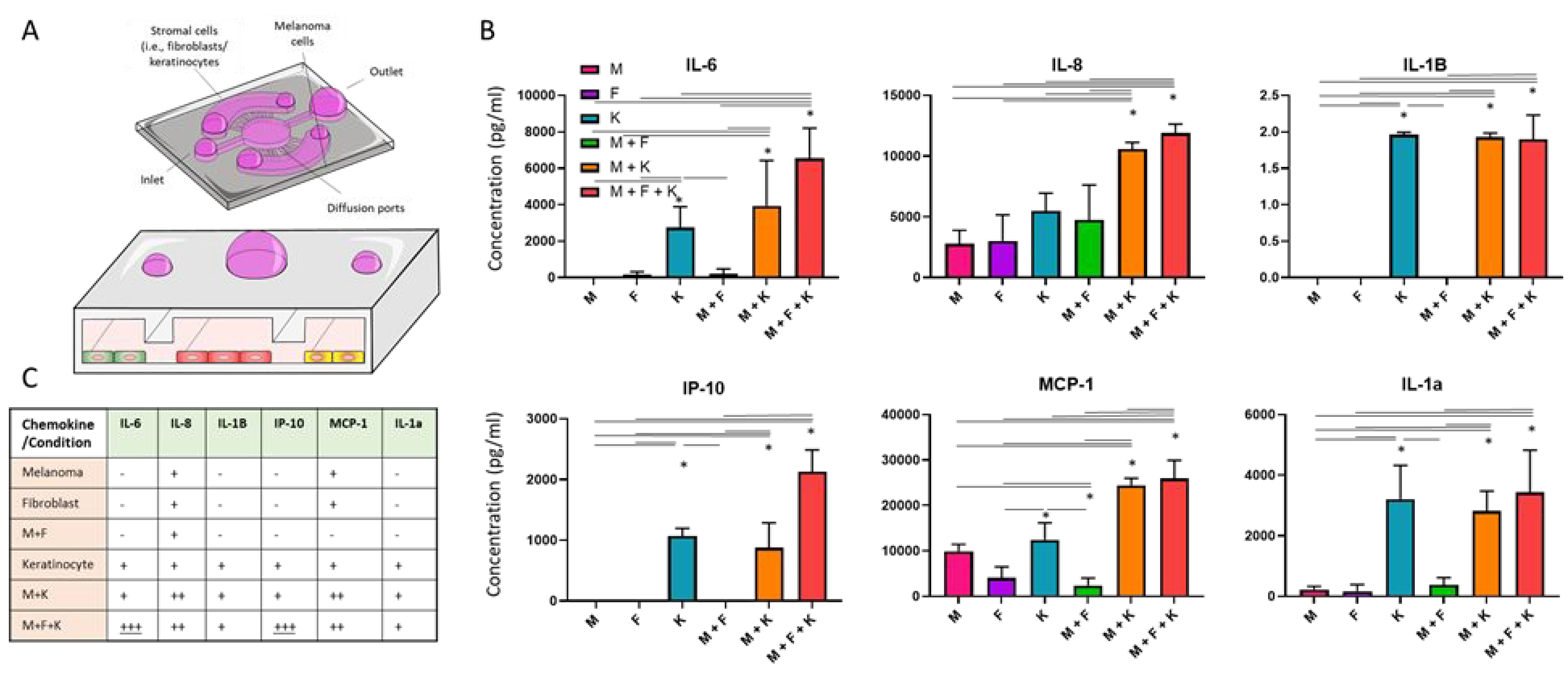
A) Melanoma cells were seeded in the central chamber alone or with dermal fibroblasts and/or keratinocytes on the lateral chambers. After 3days, chemokine secretion was analyzed by MAGPIX.B) Bar graphs showed the chemokine secretion in melanoma cells alone (M), dermal fibroblasts alone (F), keratinocytes alone (K), melanoma with dermal fibroblasts (M + F), melanoma cells and keratinocytes (M + K), and the triple co-culture (M + F + K). C) Table summarizes the trends observed. “–“ denotes low/no expression; “+” denotes expression detected; “++” denotes higher expression compared with “+” but it is compatible with an additive increase; “+++” denotes higher expression than additive increase. Asterisks denote p-value<0.05. Experiments were repeated at least three independent times with replicates showing showed similar trends.
Cell metabolism
Tumor cells exhibit higher proliferation rates compared with normal cells. Increase prolfieration forces them to adapt their metabolism to satisfy the high demand of polysaccharides, proteins, and fatty acids38. As a consequence, tumor metabolism offers a promising opportunity to selectively destroy cancer cells without damaging the normal tissue39. Thus, we compared cell metabolism in melanoma cells, dermal fibroblasts, and keratinocytes using optical metabolic imaging (OMI). OMI relies on the use of multi-photon microscopy to visualize the autofluorescence of NAD(P)H and FAD, molecules deeply involved in cell metabolism and cell redox balance (Figure 4A)40. Rapidly proliferating cells, such as cancer cells, commonly rely on glycolysis to generate macromolecules to support cell proliferation, generating an excess of NAD(P)H as a by-product of their anaerobic metabolism38. Therefore, we quantified NAD(P)H autofluorescence intensity divided by FAD autofluorescence intensity to calculate the optical redox ratio (Figure 4B and C). The NAD(P)H/FAD ratio has been used in previous studies to monitor cellular redox potential in live cells without any external labeling agent26, 40, 41. When cultured alone, melanoma cells exhibited a higher redox ratio compared with dermal fibroblasts and keratinocytes (i.e., normal cells). Interestingly, the co-culture with keratinocytes, dermal fibroblasts, or both, led to a significant change in melanoma cell redox ratio, suggesting a metabolic shift in melanoma cells. When we repeated the experiments in a 3D environment, we observed similar metabolic patterns as in 2D (Supporting Figure 6). First, melanoma cells exhibited a higher redox ratio compared with keratinocytes and fibroblasts, which had the lowest redox ratio. The co-culture with fibroblasts and keratinocytes led to a reduction of melanoma cell redox ratio in 3D environments. However, the difference between the melanoma monoculture and the co-culture with fibroblasts or keratinocytes did not reach statistical significance.
Figure 4. Optical metabolic imaging.
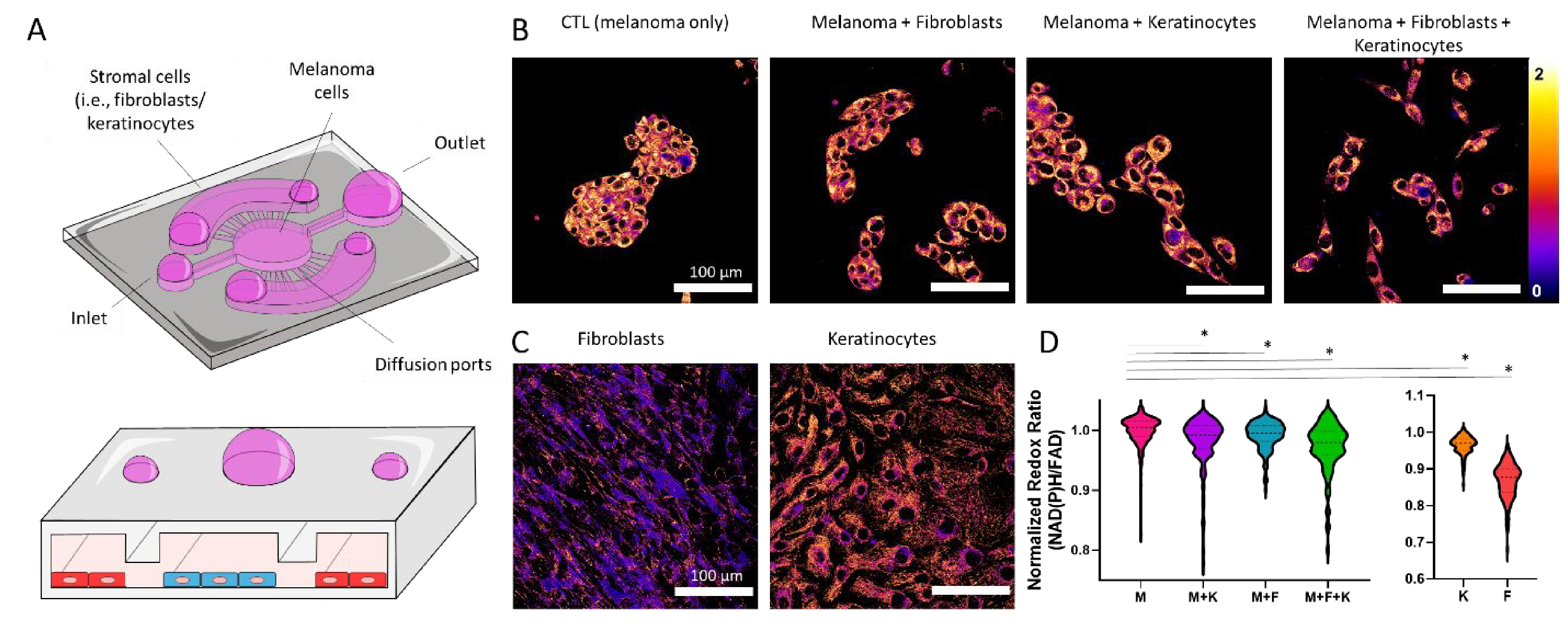
A) Schematic representation of the triple co-culture inside the microdevice, including melanoma cells, dermal fibroblasts, and keratinocytes. B) Melanoma cells were imaged by multi-photon microscopy. Images show ratio of NAD(P)H autofluorescence divided by FAD autofluorescence, defined as the optical redox ratio. Melanoma cells were imaged alone and in the presence of dermal fibroblasts and/or keratinocytes. Images of fibroblasts and keratinocytes alone are also shown. C-D) Left graph shows the quantification of the optical redox ratio of melanoma cells alone and in the co-culture conditions. Right graph shows the optical redox ratio of dermal fibroblasts and keratinocytes alone. Melanoma cells alone (M), dermal fibroblasts alone (F), keratinocytes alone (K), melanoma with dermal fibroblasts (M + F), melanoma cells and keratinocytes (M + K), and the triple co-culture (M + F + K). Asterisk denotes a p-value<0.05. Experiments were repeated at least three independent times with replicates showing showed similar trends.
Additionally, we analyzed NAD(P)H fluorescence lifetime (NAD(P)H τm) in melanoma cells, dermal fibroblasts, keratinocytes, and multiple combinations (Figure 5). NAD(P)H τm is modulated by the ratio of NAD(P)H free in the cytoplasm to the amount bound to enzymes (e.g., mitochondrial respiratory complexes)42. Thus, an increase in NAD(P)H τm is associated with an increase in the ratio NAD(P)H-bound vs NAD(P)H-free, which happens during oxidative phosphorylation42, 43. Our results demonstrated that melanoma cells had a higher NAD(P)H τm compared with normal cells such as dermal fibroblasts and keratinocytes, demonstrating again the different metabolic phenotype exhibited by melanoma cells. Interestingly, when co-cultured with dermal fibroblasts, keratinocytes, or both, melanoma cells showed a decrease in NAD(P)H τm in agreement with an increase in the amount of NAD(P)H free in the cytoplasm. Next, we repeated these experiments embedding the cells in a 3D collagen hydrogel (Supporting Figure 7). In 3D, fibroblasts had a lower NAD(P)H τm compared with keratinocytes, suggesting a metabolic shift compared with 2D environments. On the other hand, the presence of fibroblasts, keratinocytes, or both, led to similar changes in melanoma cell NAD(P)H τm. These results agree with the chemokine analysis (i.e., secretion of tumor-promoting chemokines), suggesting that dermal fibroblasts and keratinocytes play a tumor-supporting role. Enabled by this platform, in-depth proteomics, and metabolomics analysis (e.g., mass spectrometry) could pinpoint the specific molecular alterations driving tumor-stroma cross-talk and melanoma progression.
Figure 5. NAD(P)H Fluorescence Lifetime Microscopy.
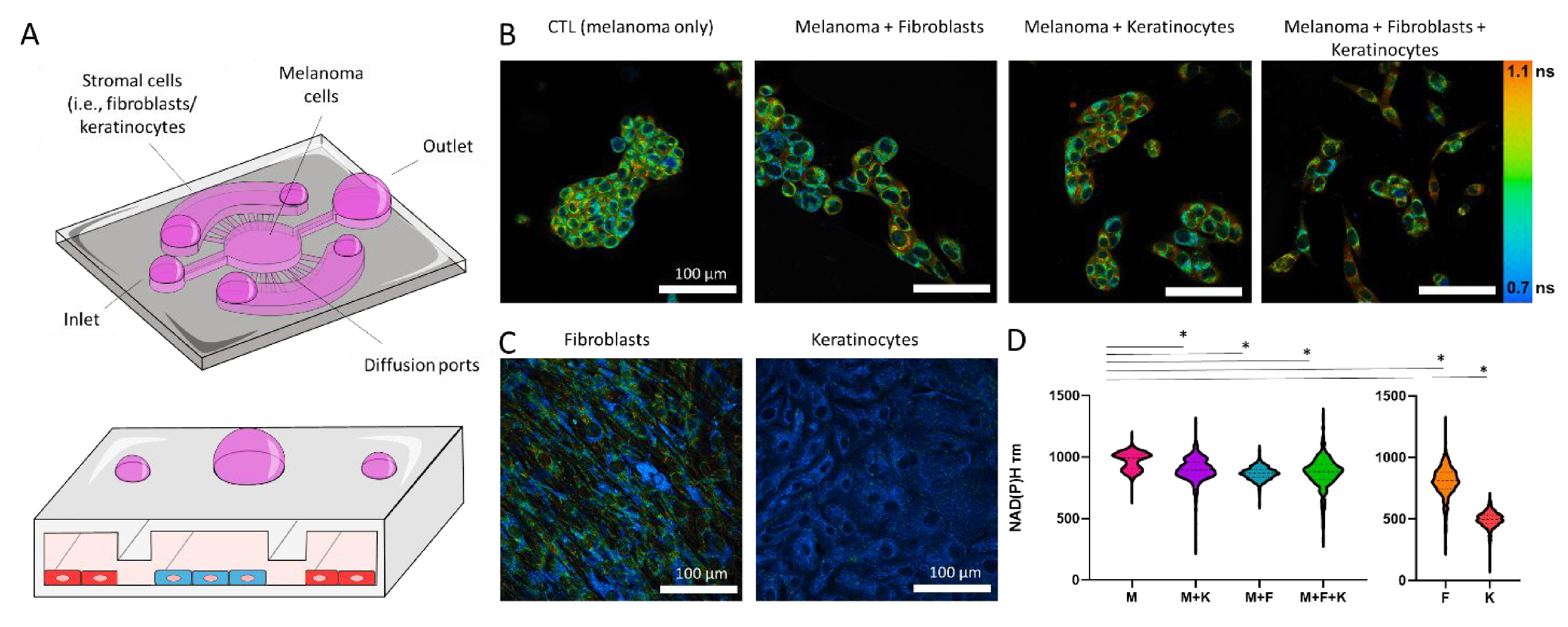
A) Schematic representation of the triple co-culture inside the microdevice, including melanoma cells, dermal fibroblasts, and keratinocytes. B) Melanoma cells were imaged by multi-photon fluorescence lifetime microscopy and images show NAD(P)H mean fluorescence lifetimes (τm). Melanoma cells were imaged alone and in the presence of dermal fibroblasts and/or keratinocytes. Images of NAD(P)H tm for fibroblasts and keratinocytes alone are also shown. C-D) Left graph shows the quantification of melanoma cell NAD(P)H τm alone and in the co-culture conditions. Right graph shows keratinocyte and dermal fibroblast NAD(PH) τm. Melanoma cells alone (M), dermal fibroblasts alone (F), keratinocytes alone (K), melanoma with dermal fibroblasts (M + F), melanoma cells and keratinocytes (M + K), and the triple co-culture (M + F + K). Asterisk denotes a p-value<0.05. Experiments were repeated at least three independent times with replicates showing showed similar trends.
Conclusion
Melanoma progression is a complex process where multiple cell types such as melanoma cells, dermal fibroblasts, and keratinocytes are involved. Fibroblasts and keratinocytes may play a dual role, supporting or inhibiting tumor growth depending on the specific mutational and environmental landscape (e.g., BRAF mutations, hypoxia). Therefore, in vitro tools that allow researchers to evaluate the cross-talk between these cell types might accelerate the identification of successful therapies against melanoma. Here, we presented a methodology to control cell patterning in microfluidic in vitro platforms without the need for external equipment, porous membranes, or hydrogels. Our protocol relied on the use of air-walls to confine the different cell populations in adjacent microfluidic chambers. Once the multiple cell populations were attached, these air-walls spontaneously dissolved in a predictable fashion, allowing cellular cross-talk and migration. The time needed to dissolve these air-walls was controlled by adjusting the air-wall volume, giving researchers more precise control over their co-culture conditions. This methodology allowed us to study the effects of dermal fibroblasts and keratinocytes on melanoma cells. Our results demonstrated that the co-culture with fibroblasts, keratinocytes, or both, led to significant changes in melanoma cell morphology, secretion pattern, and metabolic phenotype. Thus, the use of air-walls could provide a versatile tool to accelerate the identification of new therapies targeting tumor cross-talk.
Supplementary Material
Acknowledgments
University of Wisconsin Carbone Cancer Center (AAB7173). Morgridge Research Institute. NIH grants R01 CA164492, R01 CA185747, R01 CA205101, and NSF grant CBET-1642287. Department of Defense grant W81XWH-18-PRCRP-IASF (CA181014).
Footnotes
Conflict of interests
David J. Beebe holds equity in Bellbrook Labs LLC, Tasso Inc., Turba LLC, Salus Discovery LLC, Stacks to the Future LLC, Lynx Biosciences, Inc. and Onexio Biosystems LLC.
References
- 1.Anonymous, https://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html#:~:text=How%20common%20is%20melanoma%3F,4%2C610%20men%20and%202%2C240%20women). (accessed 2020–09-03).
- 2.Vultur A and Herlyn M, Cancer cell, 2013, 23, 706–706 e701. [DOI] [PubMed] [Google Scholar]
- 3.Gaggioli C and Sahai E, Pigment cell research, 2007, 20, 161–172. [DOI] [PubMed] [Google Scholar]
- 4.Damsky WE and Bosenberg M, Oncogene, 2017, 36, 5771–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou L, Yang K, Andl T, Wickett RR and Zhang Y, Journal of Cancer, 2015, 6, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang JX, Fukunaga-Kalabis M and Herlyn M, Journal of cell communication and signaling, 2016, 10, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Kilsdonk JW, Bergers M, VanKempen LC, Schalkwijk J and Swart GW, Melanoma research, 2010, 20, 372–380. [DOI] [PubMed] [Google Scholar]
- 8.Flach EH, Rebecca VW, Herlyn M, Smalley KS and Anderson AR, Mol Pharm, 2011, 8, 2039–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayuso JM, Virumbrales-Munoz M, McMinn PH, Rehman S, Gomez I, Karim MR, Trusttchel R, Wisinski KB, Beebe DJ and Skala MC, Lab on a chip, 2019, 19, 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee HH, Alves SE, Salmon M, Ferrante TC, Weaver JC, Bahinski A, Hamilton GA and Ingber DE, Nature methods, 2016, 13, 151–157. [DOI] [PubMed] [Google Scholar]
- 11.Phan DTT, Wang X, Craver BM, Sobrino A, Zhao D, Chen JC, Lee LYN, George SC, Lee AP and Hughes CCW, Lab on a chip, 2017, 17, 511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayuso JM, Basheer HA, Monge R, Sanchez-Alvarez P, Doblare M, Shnyder SD, Vinader V, Afarinkia K, Fernandez LJ and Ochoa I, Plos One, 2015, 10, e0139515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adriani G, Ma D, Pavesi A, Goh ELKand Kamm RD, Ieee Eng Med Bio, 2015, 338–341. [DOI] [PubMed] [Google Scholar]
- 14.Bischel LL, Sung KE, Jimenez-Torres JA, Mader B, Keely PJ and Beebe DJ, Faseb J, 2014, 28, 4583–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sackmann EK, Fulton AL and Beebe DJ, Nature, 2014, 507, 181–189. [DOI] [PubMed] [Google Scholar]
- 16.Virumbrales-Munoz M, Ayuso JM, Gong MM, Humayun M, Livingston MK, Lugochannels Cintron KM, McMinn P, Alvarez-Garcia YR and Beebe DJ, Chemical Society reviews, 2020, 49, 6402–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang KJ, Mehr AP, Hamilton GA, McPartlin LA, Chung S, Suh KY and Ingber DE, Integrative biology: quantitative biosciences from nano to macro, 2013, 5, 1119–1129. [DOI] [PubMed] [Google Scholar]
- 18.Deng J, Wei W, Chen Z, Lin B, Zhao W, Luo Y and Zhang X, Micromachines, 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jalili-Firoozinezhad S, Gazzaniga FS, Calamari EL, Camacho DM, Fadel CW, Bein A, Swenor B, Nestor B, Cronce MJ, Tovaglieri A, Levy O, Gregory KE, Breault DT, Cabral JMS, Kasper DL, Novak R and Ingber DE, Nature biomedical engineering, 2019, 3, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M and Kamm RD, Proceedings of the National Academy of Sciences of the United States of America, 2015, 112, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shirure VS, Bi Y, Curtis MB, Lezia A, Goedegebuure MM, Goedegebuure SP, Aft R, Fields RC and George SC, Lab on a chip, 2018, 18, 3687–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Duinen V, van den Heuvel A, Trietsch SJ, Lanz HL, van Gils JM, van Zonneveld AJ, Vulto P and Hankemeier T, Sci Rep, 2017, 7, 18071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim C, Kasuya J, Jeon J, Chung S and Kamm RD, Lab on a chip, 2015, 15, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, Miccolis C, Ceccarello E, Adriani G, Raimondi MT, Kamm RD and Bertoletti A, Jci Insight, 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pak C, Callander NS, Young EW, Titz B, Kim K, Saha S, Chng K, Asimakopoulos F, Beebe DJ and Miyamoto S, Integrative biology: quantitative biosciences from nano to macro, 2015, 7, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayuso JM, Gillette A, Lugo-Cintron K, Acevedo-Acevedo S, Gomez I, Morgan M, Heaster T, Wisinski KB, Palecek SP, Skala MC and Beebe DJ, EBioMedicine, 2018, 37, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walsh A and Skala M, An automated image processing routine for segmentation of cell cytoplasms in high-resolution autofluorescence images, SPIE, 2014. [Google Scholar]
- 28.Hoejberg L, Bastholt L and Schmidt H, Melanoma research, 2012, 22, 327–333. [DOI] [PubMed] [Google Scholar]
- 29.Kucera R, Topolcan O, Treskova I, Kinkorova J, Windrichova J, Fuchsova R, Svobodova S, Treska V, Babuska V, Novak J and Smejkal J, Anticancer research, 2015, 35, 3537–3541. [PubMed] [Google Scholar]
- 30.Norrby K, Cell proliferation, 1996, 29, 315–323. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava SK, Bhardwaj A, Arora S, Tyagi N, Singh AP, Carter JE, Scammell JG, Fodstad O and Singh S, British journal of cancer, 2015, 112, 1772–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bent R, Moll L, Grabbe S and Bros M, International journal of molecular sciences, 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kholmanskikh O, van Baren N, Brasseur F, Ottaviani S, Vanacker J, Arts N, van der Bruggen P, Coulie P and De Plaen E, Int J Cancer, 2010, 127, 1625–1636. [DOI] [PubMed] [Google Scholar]
- 34.Yang J and Richmond A, Mol Ther, 2004, 9, 846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawada K, Sonoshita M, Sakashita H, Takabayashi A, Yamaoka Y, Manabe T, Inaba K, Minato N, Oshima M and Taketo MM, Cancer Res, 2004, 64, 4010–4017. [DOI] [PubMed] [Google Scholar]
- 36.Yoshimura T, Cellular & molecular immunology, 2018, 15, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakasone Y, Fujimoto M, Matsushita T, Hamaguchi Y, Huu DL, Yanaba M, Sato S, Takehara K and Hasegawa M, The American journal of pathology, 2012, 180, 365–374. [DOI] [PubMed] [Google Scholar]
- 38.Keibler MA, Wasylenko TM, Kelleher JK, Iliopoulos O, Vander Heiden MG and Stephanopoulos G, Cancer & metabolism, 2016, 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nat Rev Drug Discov, 2010, 9, 503–504. [DOI] [PubMed] [Google Scholar]
- 40.Walsh AJ, Cook RS, Sanders ME, Aurisicchio L, Ciliberto G, Arteaga CL and Skala MC, Cancer Res, 2014, 74, 5184–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walsh AJ, Cook RS, Manning HC, Hicks DJ, Lafontant A, Arteaga CL and Skala MC, Cancer Res, 2013, 73, 6164–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lakowicz JR, Szmacinski H, Nowaczyk K and Johnson ML, Proceedings of the National Academy of Sciences of the United States of America, 1992, 89, 1271–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blacker TS and Duchen MR, Free radical biology & medicine, 2016, 100, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.