

Abstract
Cancer patients have an increased risk of both arterial and venous thrombotic events compared to the general population. Both the site and stage of cancer are known to contribute to the increased risk of thrombotic events. In addition, several treatment related factors enhance the risk of thrombosis, including hospitalization, surgery, central venous catheters, radiation, and anti-cancer agents. Chemotherapy serves as a mainstay treatment for a broad range of malignancies. Chemotherapeutic agents typically exert anti-neoplastic effects through either direct cytotoxicity or inhibition of cellular processes necessary for the proliferation of malignant cells. Unfortunately, in addition to targeting malignant cells chemotherapeutic agents are also cytotoxic to non-malignant cells. More recently, targeted agents have been developed that offer improved selectivity towards malignant cells. However, some of these agents are also associated with an increased risk of both arterial and venous thrombosis. Here, we review the association between specific anti-cancer agents and thrombotic events in cancer patients. Despite an established association in most cases the mechanism by which these agents increase thrombosis is incompletely understood. Anti-cancer agents may damage the endothelium, decrease anticoagulants and/or increase procoagulants leading to activation of coagulation, and/or activate platelets. An improved understanding of the mechanisms that drive the increased risk of thrombotic events associated with anti-cancer agents will enable treatment strategies that mitigate this risk.
Keywords: Cancer, Chemotherapy, Coagulation, Thrombosis
Introduction
Cancer is a leading cause of mortality worldwide resulting in approximately 9.6 million deaths per annum 1. Cancer patients have a higher rate of thrombosis compared with the general population that is an important cause of morbidity and mortality 2–5. The incidence rate of arterial thromboembolism (ATE) and venous thromboembolism (VTE) in cancer patients is estimated to be 2–5% and 4–20%, respectively 2, 6–8. Both the site and stage of cancer have been found to contribute to the risk of thrombotic disease 6, 7, 9, 10. For instance, cancers of the pancreas, brain and stomach are associated with a relatively high risk of VTE, whereas cancers of the breast and prostate are associated with a relatively low risk of VTE (Table 1) 2, 11, 12. In addition, patients with advanced stage cancers, particularly those with metastatic disease, have a relatively high risk of VTE compared to those with early stage localized disease 2, 11, 12.
Table 1:
Incidence of VT by cancer site
| Cancer Site | 1 year cumulative incidence VTE (%) | |
|---|---|---|
| Brain | 6.9 | |
| Pancreas | 5.3 | High |
| Stomach | 4.5 | |
| AML | 3.7 | |
| Esophagus | 3.6 | |
| Renal cell | 3.5 | |
| Ovary | 3.3 | |
| Lymphoma | 2.8 | Medium |
| CLL | 2.7 | |
| ALL | 2.6 | |
| Lung | 2.4 | |
| Colon | 2.3 | |
| Liver | 1.7 | |
| Uterus | 1.6 | |
| CML | 1.5 | |
| Bladder | 1.5 | Low |
| Prostate | 0.9 | |
| Breast | 0.9 | |
| Melanoma | 0.5 |
ALL, acute lymphocytic leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia. Adapted from 11
Treatment related factors also contribute to the increased risk of thrombosis in patients with cancer. These treatment related factors include hospitalization, radiation, surgery, central venous catheters and anti-cancer agents 13–16. Anti-cancer agents, which includes both chemotherapies and targeted agents, complement other therapeutic strategies, such as radiotherapy and surgery. To date, more than a hundred chemotherapy and targeted therapy drugs have been approved for the treatment of a variety of malignancies 17. The growth and spread of a tumor depends on the proliferative and invasive capacity of malignant cells as well as the metastatic potential 18. Chemotherapeutic agents typically exert anti-neoplastic effects through either cytotoxic or cytostatic mechanisms, owing to their ability to kill malignant cells or inhibit their division 19. However, although chemotherapeutic agents are often highly effective in impairing the replicative capacity of malignant cells they also exert cytotoxicity towards non-malignant cells, which can lead to adverse off-target toxicities 20. It was envisaged that the development of a new generation of targeted therapeutics, that more selectively target malignant cells, would reduce the degree of off-target toxicity; however, a number of these agents have significant toxicity to non-malignant cells 21.
A variety of anti-cancer agents have been shown to increase the risk of thrombotic events (TE) in cancer patients (Table 2) 15, 16, 22, 23. However, it has been suggested that there is a significant underestimation of the burden of VTE associated with certain types of chemotherapy regimens due to the method of reporting adverse events in clinical trials in oncology 24. In addition, clinical trials often have strict criteria for the exclusion of patients with a recent history of ATE or VTE. In contrast, there has been concern that a failure to adjust for time-on-treatment may over-estimate the relative risk of thrombosis associated with new therapies because patients receiving these agents may live longer than those in the control arm.
Table 2:
Anti-cancer agents and thrombosis
| Agent | VT | AT |
|---|---|---|
| Platinum-based agents | ||
| Cisplatin | ++ | − |
| Carboplatin | ++ | − |
| Oxaliplatin | + | − |
| Anthracyclines | ||
| Doxorubicin | + | NR |
| Daunorubicin | NR | NR |
| Epirubicin | NR | NR |
| Pyrimidine antagonists | ||
| 5-Fluorouracil | − | − |
| Gemcitabine | − | − |
| L-asparaginase | NR | NR |
| Tamoxifen | + | + |
| Immunomodulatory agents | ||
| Thalidomide | ++ | NR |
| Lenalidomide | ++ | NR |
| Pomalidomide | + | NR |
| Anti-EGFR antibodies | ||
| Cetuximab | + | − |
| Panitumumab | + | − |
| Necitumumab | + | − |
| VEGF targeted molecules | ||
| Bevacizumab | − | ++ |
| Aflibercept | − | NR |
| VEGFR RTKI | ||
| Sunitinib | − | ++ |
| Sorafenib | − | ++ |
| Axitinib | − | ++ |
| Pazopanib | − | + |
| Vandetinib | − | + |
| Lenvatinib | NR | NR |
| Cabozantinib | NR | NR |
| BCR-ABL RTKI | ||
| Imatinib | − | − |
| Dasatinib | ++ | ++ |
| Nilotinib | ++ | ++ |
| Ponatinib | ++ | ++ |
| Bosatinib | NR | NR |
| CDK inhibitors | ||
| Palbociclib | + | NR |
| Abemaciclib | ++ | NR |
| Ribociclib | + | NR |
+, associated; −, not associated; ?, unclear; NR, not reported
The mechanisms that drive cancer therapy associated thrombosis are not fully understood. A primary mechanism may be the activation or disruption of the endothelium by the anti-cancer agents (Figure 1). In addition, these agents may decrease anticoagulants and increase procoagulants, such as tissue factor, leading to activation of coagulation (Figure 1). Finally, anti-cancer drugs may directly or indirectly activate platelets. (Figure 1).
Figure 1: Potential mechanisms of cancer-therapy associated thrombosis.
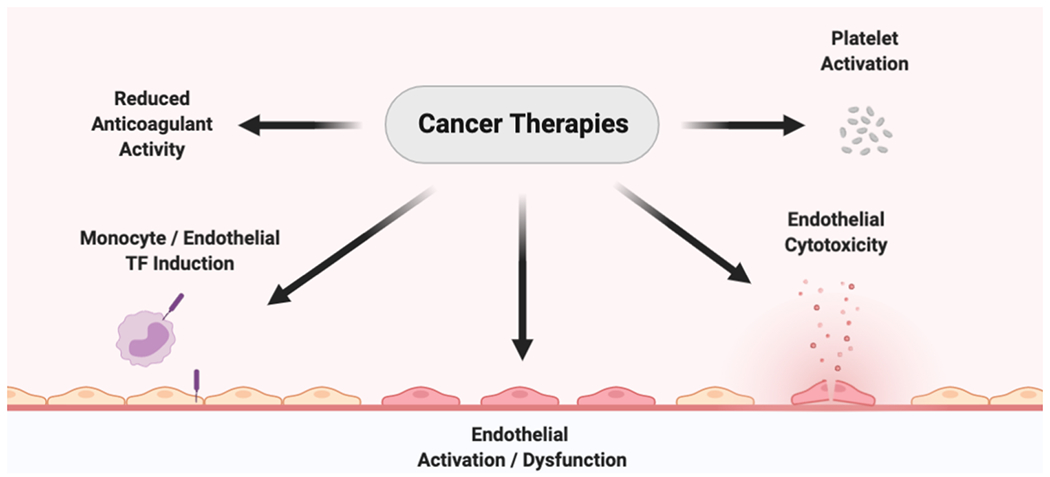
Anti-cancer agents may induce a prothrombotic state through a number of potential mechanisms including reduced levels of endogenous anticoagulants, induction of tissue factor procoagulant activity, activation of the endothelial, endothelial cytotoxicity and activation of platelets. Created with BioRender.com
It is important to note that it is difficult to separate the prothrombotic activity of the tumor itself from the prothrombotic activity of different anti-cancer treatments and their effect on non-malignant cells. Moreover, cancer patients typically receive multiple treatments, including combinations of cytotoxic drugs. This can make it challenging to determine the relative contribution of each agent to the thrombotic risk. In other cases, an anti-cancer drug, such as thalidomide, may not be prothrombotic when administered alone but can be prothrombotic when combined with other drugs, such as dexamethasone, as has been observed in patients with multiple myeloma (MM).
Here, we review the clinical evidence of an association between anti-cancer agents and the risk of ATE and VTE in cancer patients. In addition, we discuss potential mechanisms for thrombosis if known with the different anti-cancer agents.
Platinum-based agents
Platinum-based agents are a class of cytotoxic drugs that induce formation of toxic platinum-DNA adducts that can induce tumor cell apoptosis 25. Platinum-based agents have proven to be effective in the treatment of a wide range of solid tumors, including testicular, ovarian, bladder, stomach and prostate cancer 25. The first-generation agent, cisplatin, and to a lesser extent next generation agents, such as carboplatin and oxaliplatin, have been associated with a range of dose limiting off-target toxicities 25.
In a study of patients with germ cell cancer treatment with a cisplatin-containing combination regimen was associated with a significantly increased incidence of VTE compared to a regimen without cisplatin (8.4% versus 3.4%) 26. A similar finding was made in a complementary study 27. A meta-analysis showed that cisplatin-based therapy had a significantly increased risk of VTE compared with patients receiving non-cisplatin-based therapy, 1.92% versus 0.79% (relative risk [RR] 1.67) 28. In a complementary meta-analysis derived from the same series of trials no significant difference in the relative risk of ATE was observed between cisplatin and non-cisplatin-based therapies 29. Together, this suggests that cisplatin containing regimens increase the risk of VTE but not ATE.
Interestingly, carboplatin does not offer an improved toxicity profile with respect to TE when compared to cisplatin. In patients with lung cancer treated with either cisplatin or carboplatin no significant difference in the incidence of TE was observed between the two agents 30, 31. Consistent with these findings another study of patients with urothelial cell carcinoma found no significant difference in the incidence of TE between carboplatin and cisplatin treatment 32. In contrast, oxaliplatin-based regimens appear to be associated with a lower incidence of TE compared to cisplatin-based regimens. In a phase III trial of patients with metastatic gastroesophageal adenocarcinoma treatment with a combination therapy (5-Fluorouracil (5-FU), leucovorin), the arm receiving oxaliplatin was found to have a markedly lower incidence of TE than the cisplatin arm, 0.9% versus 7.9%, respectively 33. In another phase III trial patients receiving a cisplatin-based regimen with epirubicin and 5-FU or capecitabine exhibited an incidence of VTE of 15.1% whereas patients receiving an oxaliplatin-based regimen with epirubicin and 5-FU or capecitabine had an incidence of VTE of 7.6% 34.
The primary mechanism by which platinum-based agents induce a prothrombotic state has yet to be determined. In vitro studies showed that cisplatin increased tissue factor activity of mouse peritoneal exudate cells treated with LPS and induced apoptosis of endothelial cells and release of extracellular vesicles 35,36. In addition, cancer cells exposed to cisplatin or carboplatin released increased numbers of extracellular vesicles 37, 38. Surprisingly, levels of extracellular vesicle tissue factor activity were either unchanged after treatment of patients with metastatic testicular cancer with cytolytic chemotherapy or decreased in MM patients or pancreatic cancer patients treated with chemotherapy 39–41. At present, the role of extracellular vesicles in the prothrombotic effect of platinum-based agents is unclear.
In a small study of cancer patients, cisplatin-based combination chemotherapy increased circulating von Willebrand Factor in a subset of patients 42. Another study found that administration of cisplatin and gemcitabine to patients with non-small cell lung cancer activated the coagulation system, measured by increased levels of thrombin-antithrombin complexes and D-dimer, increased levels of platelet-derived extracellular vesicles and activated platelets 43.
Platinum-based agents are associated with a significantly increased risk of VTE. However, the absolute risk of VTE associated with this class of agent remains low. The cytotoxic effects of these drugs lead to endothelial damage, activation of coagulation and platelets that all likely contribute to thrombosis (Figure 2).
Figure 2: Prothrombotic mechanisms of specific anti-cancer agents.
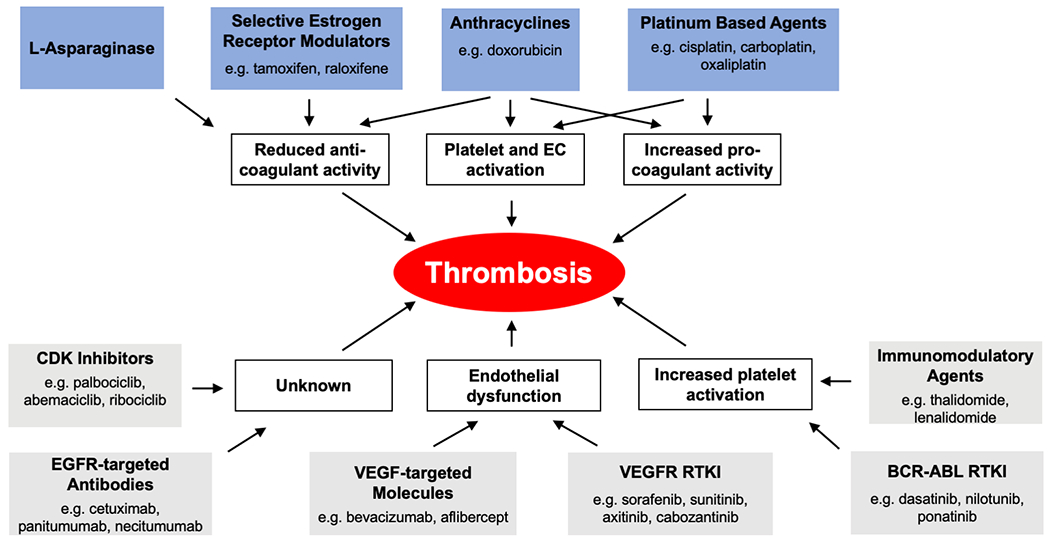
A number of mechanisms have been identified that may contribute to the observed association between both conventional chemotherapies (blue boxes) or targeted therapies (grey boxes) and an increased risk of thrombotic events. CDK, cyclin dependent kinase; EGFR, epidermal growth factor receptor, RTKI, receptor tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Anthracyclines
Anthracyclines, including doxorubicin (also called adriamycin), daunorubicin, and epirubicin, are a family of cytotoxic chemotherapy agents that interrupt topoisomerase mediated ligation of DNA during replication and induce apoptotic cell death. The use of the topoisomerase II inhibitor doxorubicin is associated with both early and late cardiomyopathy 44. Doxorubicin is also associated with an increased risk of VTE. In patients with non-Hodgkin lymphoma doxorubicin treatment was associated with a significantly increased risk of VTE (hazard ratio [HR] 1.96) 45. A prospective phase III trial of MM patients compared VTE rates in two cohorts that received a combination chemotherapy and thalidomide that differed only by the inclusion of doxorubicin 46. The regimen with doxorubicin was associated with a significantly increased risk of VTE (16.0% versus 2.5%) 46. There have been relatively few studies investigating the effect of other topoisomerase inhibitors on VTE risk.
Doxorubicin and epirubicin have been shown to increase the activity of pre-existing tissue factor on cultured endothelial cells by inducing the surface exposure of phosphatidylserine 47. Similarly, doxorubicin and daunorubicin have been found to increase tissue factor activity in a monocytic leukemia cell line (THP-1) 48. Doxorubicin has also been found to induce release of extracellular vesicles that may support activation of coagulation 49, 50. In addition, doxorubicin disrupted the anticoagulant protein C pathway in cultured endothelial cells by reducing levels of endothelial protein C receptor 51.
Doxorubicin and cyclophosphamide have been found to increase plasma thrombin-antithrombin complexes and D-dimer in a small study of breast and lung cancer patients 52. Another study with breast cancer patients treated with epirubicin or doxorubicin showed modestly increased levels of thrombin-antithrombin complexes, protein C and activated protein C, but not other markers, including extracellular vesicle tissue factor activity 53. Doxorubicin has also been found to induce endothelial injury in pediatric patients 54.
Preclinical studies demonstrate that doxorubicin is prothrombotic. Administration of doxorubicin to mice led to platelet activation and enhanced platelet binding to the endothelium 55, 56. Further, doxorubicin and epirubicin administration increased plasma thrombin-antithrombin complexes in mice 57. Doxorubicin also increased thrombosis in a rat aortic loop model 58. In a rat inferior vena cava (IVC) stasis model doxorubicin significantly increased thrombus weight 59. Interestingly, treatment with the platelet inhibitors aspirin and clopidogrel and the thrombin inhibitor hirudin, but not the anticoagulant heparin, significantly reduced the doxorubicin dependent increase in thrombus weight in this model 59. Similarly, in a rat IVC ferric chloride injury model of venous thrombosis doxorubicin dose-dependently increased thrombus burden 60.
Anthracyclines, such as doxorubicin, appear to induce a prothrombotic state by inducing endothelial injury, reducing the protein C pathway, increasing tissue factor procoagulant activity and activating platelets (Figure 2).
Pyrimidine antagonists
Pyrimidine antagonists are a class of anti-metabolic chemotherapy agents that disrupt synthesis of pyrimidine containing nucleotides cytosine, uracil and thymine. Pyrimidine antagonists are all prodrugs that require metabolism by endogenous pyrimidine biosynthetic pathways. Metabolism of pyrimidine antagonist prodrugs leads to the generation of cytotoxic nucleosides and nucleotides. Pyrimidine antagonists, such as 5-FU and gemcitabine, are used to treat a wide range of malignancies, including colorectal, pancreatic, breast, head and neck, and non-small cell lung cancers.
5-FU was developed in the late 1950s and as a result there is a relatively limited amount of controlled clinical data regarding the effect of this agent on the risk of TEs. This is further complicated by the frequent use of 5-FU as part of combination chemotherapy regimens. In a phase I trial of patients with metastatic gastrointestinal cancer treated with 5-FU and leucovorin the rate of VTE was 17% 61. In a phase I trial of metastatic renal cell carcinoma treated with a combination of gemcitabine and 5-FU the rate of VTE was 43% 62. However, in a meta-analysis there was a non-significant increase in VTE and ATE for gemcitabine containing regimens compared to non-gemcitabine containing regimens 62. Similarly, a recent study showed that gemcitabine treatment was not associated with a significant increase in VTE 27. Gemcitabine-associated thrombotic microangiopathy has been reported but this is rare 63.
Administration of 5-FU to rabbits caused disruption of the endothelial monolayer, exposure of the subendothelial matrix and accumulation of platelet aggregates 64, 65. Exposure of cultured endothelial cells to 5-FU resulted in increased reactive oxygen species generation, reduced prostacyclin release, and reduced expression of endothelial nitric oxide synthetase 66–68. 5-FU has also been associated with coronary vasospasm 69. Administration of 5-FU to mice increased levels of thrombin-antithrombin complexes 57. The effect of 5-FU and other pyrimidine antagonists has yet to be assessed in preclinical models of thrombosis.
Even though pyrimidine antagonists are cytotoxic to the endothelium in preclinical models, there is no clear association between pyrimidine antagonist treatment and an increased risk of TEs in patients with cancer.
L- Asparaginase
L- Asparaginase is an enzyme that catalyzes the hydrolysis of the amino acid asparagine and is used as a chemotherapeutic agent in the treatment of acute lymphocytic leukemia (ALL) 70. The anti-neoplastic potential of L-asparaginase in ALL stems from the dependence of leukemic lymphoblasts on asparagine present in the blood 70. Loss of the ability to synthesize asparagine is a hallmark of leukemic lymphoblasts 70.
A challenge in determining the thrombogenic potential of L-asparaginase is that the rate of thrombosis in pediatric ALL patients without L-asparaginase treatment is not clear. A study of pediatric ALL patients observed a TE rate of 1–2%, most of which were intracranial thrombi 71. In a meta-analysis of prospective studies, pediatric ALL patients receiving L-asparaginase had a TE rate of 5.2% 72. In one study of adult ALL patients the rate of TE acutely after L-asparaginase and combination chemotherapy treatment was 4.2% 73. In a multicenter study, the incidence of VTE was 10% for adult ALL patients treated with L-asparaginase 74. In a recent large prospective study the incidence of VTE in adult ALL patients treated with L-asparaginase was 16% despite antithrombin supplementation in the majority of the patients 75. In another study of adult acute leukemia patients a 4.9 fold increase in TE was observed in patients treated with L-asparaginase compared to those not receiving L-asparaginase (11.1% versus 2.0%) 76. Interestingly, the type of VTE associated with L-asparaginase treatment is atypical, frequently involving cerebral venous thrombosis in both pediatric and adult ALL patients 77, 78.
L-asparaginase treatment is associated with the induction of a procoagulant state 79. Treatment of ALL patients with L-asparaginase leads to decreased plasma levels of several coagulation proteins, including both procoagulants (FIX, FX and prothrombin) and anticoagulants (protein C, protein S, and antithrombin), as well as acute hypofibrinogenemia 80–85. The initial reduction in levels of protein C, protein S and antithrombin is thought to induce a temporal procoagulant state 82, 83. Interestingly, levels of procoagulant and anticoagulants decreased in proportion to their half-lives suggesting that L-asparaginase decreased synthesis of these proteins 82. L-asparaginase did not affect vitamin K-dependent carboxylation of the Gla domains because there was no change in the antigen/activity ratio of the clotting proteins 82. In addition, L-asparaginase induced tissue factor expression in endothelial cells but not monocytes, and one small study showed that L-asparaginase treatment was associated with increased tissue factor activity in plasma 86. Consistent with activation of coagulation, L-asparaginase treatment increased levels of thrombin-antithrombin complexes and D-dimer in patients with ALL and acute lymphoblastic lymphoma 87.
Attempts to reduce VTE in ALL patient treated with L-asparaginase have used various strategies with inconsistent results. Prophylactic fresh frozen plasma reduced the rate of VTE from 19% to 6% in adult ALL patients treated with L-asparaginase74. However, a more recent study found that antithrombin supplementation or fresh frozen plasma did not affect the rate of VTE whereas administration of fibrinogen concentrates increased the rate of VTE 75. Overall, it appears that L-asparaginase treatment causes a prothrombotic state in ALL patients by decreasing levels of anticoagulants and possibly by increasing TF expression (Figure 2).
Tamoxifen
The estrogen receptor antagonist tamoxifen is used in the treatment of estrogen receptor-positive breast cancer patients 88. Early studies with tamoxifen showed that it increased the risk of TE 89, 90. The increased risk of VTE associated with tamoxifen treatment is particularly apparent in older women consistent with age being a contributing factor to the risk of tamoxifen-associated VTE 91, 92. A randomized clinical trial showed more TE in patients receiving tamoxifen compared with those receiving placebo (0.9% versus 0.2%) 93. Another study found that the 5-year risk of VTE was 1.2% in women receiving tamoxifen and 0.5% for women not taking tamoxifen 91. In addition, in a large clinical trial more TE were observed with tamoxifen compared to anastrozole, which inhibits the synthesis of estrogen 90. Finally, ATE and VTE were evaluated in breast cancer patients treated with chemotherapy with or without tamoxifen 94. Adjunct therapy with tamoxifen was associated with a significant increase in TE compared with patients on observation (5.4% versus 1.6%) 94. Tamoxifen significantly increased the risk of both ATE (1.6% versus 0.0%) and VTE (2.8% versus 0.8%) when administered with chemotherapy in premenopausal patients 94. In a population-based study a significant temporal increase in the incidence of VTE was observed for breast cancer patients treated with tamoxifen compared to those not treated with tamoxifen in the first two years (0.45% versus 0.12%, RR 3.5) 91.
It was hypothesized that tamoxifen may decrease levels of anticoagulants. Interestingly, different results have been observed. An early study found no differences in plasma levels of antithrombin, protein C or protein S 95. However, another study found that tamoxifen reduced levels of antithrombin and protein S, but not protein C, compared to levels before treatment 96. More recently, it was reported that tamoxifen decreased levels of antithrombin, protein S and protein C 97. Tamoxifen has also been found to induce activated protein C resistance 98, 99. Finally, tamoxifen has been shown to increase levels of FVIII, FIX and von Willebrand Factor 97.
The effect of tamoxifen on platelets is less clear. Tamoxifen metabolites have been shown to increase production of reactive oxygen species by platelets and potentiate agonist-induced platelet aggregation 100, 101. Conversely, tamoxifen treatment has also been found to inhibit platelet adhesion and thrombin or collagen induced platelet aggregation 102, 103. In a preclinical study, tamoxifen was found to inhibit thrombus formation in a mouse mesenteric arteriole ferric chloride model of thrombosis 103. The effect of tamoxifen in preclinical models of venous thrombosis has yet to be determined.
Tamoxifen is associated with a significantly increased risk of both ATE and VTE. However, the absolute risk of VTE in the patient population receiving tamoxifen is very low. Mechanistic studies suggest that tamoxifen decreases levels of anticoagulants and increases levels of procoagulants (Figure 2).
Immunomodulatory agents
Immunomodulatory drugs, such as thalidomide, have found considerable utility as treatments for patients with MM. The dose-limiting toxicity observed with thalidomide treatment in patients with MM led to the development of several analogs, including lenalidomide and pomalidomide. The therapeutic effect of lenolidamide and pomalidamide requires the presence of cereblon, an E3 ubiquitin ligase 114. Indeed, the gene expression changes induced by lenalidomide were reduced in the absence of cereblon. Downstream targets of cereblon include several transcription factors, such as interferon regulatory factor 4. More recently, it was shown that lenalidomide provides anti-cancer activity by inducing selective degradation of the B cell specific transcription factors Ikaros family members 1 and 3 115, 116.
Several clinical studies have demonstrated that patients with MM treated with thalidomide are at an increased risk of VTE. MM patients have an increased risk of VTE (5–10%) compared to the general population 104. The incidence of VTE associated with thalidomide alone is relatively low (~3%) 105. However, the addition thalidomide to a multiagent chemotherapy regimen, including dexamethasone, has been found to increase the rate of deep vein thrombosis from 3–4% to 20–28% in MM patients 106, 107. In a meta-analysis, thalidomide in combination with dexamethasone significantly increased the risk of VTE in MM patients compared to dexamethasone alone (12.5% versus 6.5%, odds ratio [OR] 2.6) 108. At present, it is unclear why combined therapy with thalidomide and dexamethasone confers an increased risk of VTE when neither agent alone is associated with increased risk.
The second-generation thalidomide analogue lenalidomide is also associated with an increased risk of VTE in MM patients. Like results seen with thalidomide, two clinical trials showed that there was a higher rate of VTE on patients receiving lenalidomide together with dexamethasone compared with dexamethasone alone (14.7% versus 3.4%, and 11.4% versus 4.6%) 109, 110. Another study showed a higher rate of VTE in patients receiving lenalidomide together with high-dose dexamethasone compared with patients receiving lenalidomide together with low-dose dexamethasone (26% versus 12%)111. In a meta-analysis of randomized controlled trials lenalidomide treatment was associated with a significantly increased risk of VTE compared to non-lenalidomide treated controls, 5.8% versus 2.2% (RR 2.52) 112. In a phase III trial of pomalidomide plus high-dose dexamethasone versus low-dose dexamethasone in MM patients the frequency of VTE events was 2.7% vs 0%, respectively, despite pomalidomide treated patients receiving thromboprophylaxis 113.
The mechanism by which thalidomide and its analogs combine with dexamethasone to increase thrombosis has not been identified. However, several studies have demonstrated platelet hyperactivation in response to thalidomide treatment (Figure 2). Patients treated with thalidomide were found to have increased platelet P-selectin exposure and an increased abundance of platelet-monocyte aggregates 117. Additionally, platelet function assessed in response to collagen and ADP stimulation was enhanced by thalidomide and dexamethasone treatment 118. Limited evidence also indicates that thalidomide exposure increased apoptosis and reduced cell viability in cultured endothelial cells 119. Further mechanistic studies are warranted to elucidate the prothrombotic potential of thalidomide in combination with dexamethasone.
Due to the increased risk of VTE in MM patients treated with thalidomide and its analogs, patients receive various thromboprophylaxis agents (aspirin, low molecular weight heparin or warfarin) 120. The beneficial effects of aspirin are consistent with studies showing that thalidomide activates platelets. A meta-analysis of MM patients with low TE risk found that a prophylactic dose of low molecular weight heparin or therapeutic dose of warfarin was most effective in the reduction of VTE risk associated with thalidomide and lenalidomide 108, 121. However, one review concluded that the type of thromboprophylaxis strategy to recommend is still unclear 121. Interestingly, in a phase III trial of MM patients receiving thalidomide no significant difference in the risk of VTE was observed between groups receiving aspirin, low molecular weight heparin and warfarin thrombophrophylaxis 122.
EGFR targeted antibodies
Epidermal growth factor (EGF) is a protein that signals through the epidermal growth factor receptor (EGFR) promoting cell growth and differentiation. Mutations in EGFR that lead to overexpression of this receptor, constitutive receptor activation, and excessive EGF mediated cell growth are common in patients with a variety of malignancies, including lung, colorectal and brain cancer 123, 124. This has led to the development of several antibody-based therapies that inhibit EGFR signaling, such as cetuximab, panitumumab, necitumumab. Treatment with EGFR targeted antibodies has been associated with an increased risk of VTE. In a retrospective study of 4 trials of non-small cell lung cancer patients, treatment with necitumumab significantly increased the risk of VTE compared to the control arm (9.9% versus 6.6%) (RR 1.58 95%) 125. Two meta-analyses of EGFR targeted antibody treatment, that included cetuximab and panitumumab, showed that treatment was associated with a significantly increased risk of VTE (RR 1.32 and 1.46) 126, 127. Interestingly, no significant association between anti-EGFR antibody therapies and ATE has been observed 126.
Clinical data indicates that patients treated with EGFR targeted antibodies are at a moderately increased risk of VTE. Mechanistic studies are required to provide insights into how inhibition of EGFR mediated-signaling may contribute to the observed increased risk of VTEs (Figure 2).
VEGF targeted therapies
Vascular endothelial growth factor (VEGF) signaling through VEGF receptors (VEGFRs) on endothelial cells is essential to tumor vessel angiogenesis. This led to the development of several agents that target this axis. The first of these agents to be used clinically was bevacizumab, a humanized anti-VEGF-A antibody. Bevacizumab is used in the treatment of colorectal, lung, brain, kidney, cervical, and ovarian cancer. In addition, an inhibitory VEGFR1 and VEGR2 fusion protein called aflibercept has been developed and is used in the treatment of colorectal cancer.
Meta-analyses have demonstrated a significant association between bevacizumab treatment and risk of ATE in cancer patients 128–130. In the first of these meta-analyses, the incidence of ATE was 3.8% for bevacizumab treated patients versus 1.7% for control (HR 2.0) 128. Two other meta-analyses found that bevacizumab treatment increased the RR of ATE 1.46 and 2.08 129, 130. Although it is possible that differences in time on treatment affected the observed association, it was reported that most ATE events occurred acutely after study initiation, potentially limiting the impact of this confounder 128. There have also been a number of reports indicating that bevacizumab causes renal thrombotic microangiopathy 131.
The clinical association between bevacizumab treatment and VTE remains contentious. In an early meta-analysis, treatment of cancer patients with bevacizumab was found to be associated with a significantly increased risk of VTE compared to patients not receiving bevacizumab, 12.8% versus 9.8% (RR 1.33) 132. However, there was concern that the observed increased risk may have been attributed to an increased time on treatment owing to prolonged progression free survival in the bevacizumab treatment arm. Two subsequent meta-analyses found no significant association between bevacizumab treatment and VTE risk when adjusted for time on treatment 128, 133. Similarly, a meta-analysis found no association between aflibercept treatment and risk of VTE 134.
Preclinical studies support the notion that VEGF targeted antibody therapy is prothrombotic in venous thrombosis models. In a mouse model of lung cancer bevacizumab treatment increased venous thrombus formation in the inferior vena cava stenosis and saphenous vein ferric chloride models 135. Treatment with bevacizumab enhanced expression of the anti-fibrinolytic protein plasminogen activator inhibitor 1 that directly contributed to the prothrombotic phenotype associated with bevacizumab 135. Deletion of the gene encoding VEGF-A in kidney podocytes was found to induce a thrombotic microangiopathy-like phenotype in mice like that observed in bevacizumab treated patients 136. This suggests that VEGF-A expressed by podocytes is critical for endothelial homeostasis in the kidney and that disruption of VEGF signaling in this organ can explain the thrombotic microangiopathy observed with bevacizumab 136.
The VEGF targeted antibody bevacizumab is associated with a moderate increased risk ATE and renal thrombotic microangiopathy but not VTE. The mechanism is likely to be due to effects on the endothelium (Figure 2).
VEGFR targeted receptor tyrosine kinase inhibitors
Small molecule inhibitors called receptor tyrosine kinases inhibitors (RTKIs) have been developed that block signaling downstream of the different VEGFR receptors (VEGFR1 -3). Inhibitors in this class have potent anti-angiogenic activity. The number of approved VEGFR-targeted RTKIs has expanded rapidly and includes sorafenib, sunitinib, axitinib, pazopanib, cabozantinib, lenvatinib and vandetanib. It is important to note that these VEGFR targeted RTKIs vary significantly in their selectivity and specificity for various VEGFRs and other cell surface RTKs. This class of agent is primarily used in the treatment of metastatic renal cell carcinoma. Endogenous VEGFR signaling plays an important role in endothelial cell homeostasis with VEGFR targeted RTKIs associated with a number of vascular toxicities 137.
A large body of data from placebo-controlled phase III trials exists for VEGFR targeted RTKIs. Meta-analysis of trials showed a significantly increased risk of ATE with sorafenib and sunitinib compared to controls (2.2% versus 0.7%, RR 3.03) 138. A meta-analysis that included a broader range of VEGFR targeted RTKIs (sunitinib, sorafenib, pazopanib, axitinib, vandetanib) showed a significant risk of increased ATE (1.4% versus 0.5%, OR 2.26) 139. The most common events were myocardial ischemia/infarction 139. Some caution is warranted given that the studies do not adjust for time on treatment that is significantly longer in arms receiving VEGFR targeted RTKIs 140. In contrast to ATE, meta-analyses indicate that RTKIs did not increased the risk of VTE 141–144. A challenge in interpreting the contribution of specific VEGFR targeted RTKIs to thrombotic risk is that numerous agents with varying specificities are combined for the meta-analysis. This approach may mask the effect of specific agents on thrombotic risk.
Despite the available clinical evidence indicating that VEGFR targeted RTKIs do not increase the risk of VTE in patients, preclinical studies have shown that defective angiogenesis delays venous thrombus resolution. For instance, deletion of VEGFR2 in endothelial cells was found to decrease venous thrombus resolution in a murine model of IVC stenosis 145. Impaired venous thrombus resolution was also observed in mice treated with axitinib and subjected to an IVC stenosis model of thrombosis 146.
The mechanisms driving the observed increased risk of ATE in patients treated with VEGFR targeted RTKIs have yet to be determined but likely involves some form of disruption of the endothelium (Figure 2).
BCR-ABL targeted receptor tyrosine kinase inhibitors
Several BCR-ABL targeted RTKIs have been developed as anti-cancer agents and are used in the treatment of chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia. Imatinib was the first of this family of agents to be used in the treatment of CML. However, the presence of specific point mutations in ABL can lead to imatinib resistance in some CML patients. This led to the development of a second generation of BCR-ABL inhibitors that includes dasatinib, nilotunib, bosutinib and ponatinib. Ponatinib has the widest inhibitory spectrum of CML tyrosine kinases. The first-generation inhibitor, imatinib, has not been associated with an increased risk of TEs. However, second-generation inhibitors have been associated with an increased risk of TEs.
Meta-analysis of trials demonstrated that treatment with second generation BCR-ABL RTKIs was associated with a significantly increased risk of TEs compared to imatinib treatment (5.88% for second generation RTKIs versus 1.04% for imatinib, OR 3.45) 147. Similar results were observed with an independent meta-analysis where second generation agents were associated with a significantly increased risk of ATE when compared to imatinib (4.78% versus 0.96%, OR 3.26) 148. The prothrombotic potential of ponatinib has been specifically highlighted in the literature 149. In a phase II trial of ponatinib in CML patients the frequency of adverse vascular events was 8.9% and 55.9% at 11 and 60 months, respectively 150, 151. The high frequency of adverse vascular events appears to be primarily driven by ATE 151. Indeed, a phase III trial of ponatinib in patients with CML was terminated prematurely citing concerns over the frequency of adverse vascular events 152. Interestingly, the increased frequency of ATE associated with ponatinib was equally distributed between cardiovascular, cerebrovascular and peripheral vascular events 152. The second generation RTKIs dasatinib, nilotinib or ponatinib were also individually associated with a significant increased risk of VTE compared to imatinib with OR of 3.86, 3.42 and 3.47, respectively 147.
Preclinical studies have been used to explore the prothrombotic potential of BCR-ABL RTKIs in models of arterial thrombosis. Administration of nilotinib and ponatinib to mice increased thrombus formation in the mesenteric arteriole and carotid artery models of thrombosis whereas imatinib had no effect on thrombosis 153–155. Treatment of mice with ponatinib also increased aortic endothelial-associated von Willebrand Factor expression and platelet binding 5 to 6-fold compared with controls as well as global microvascular angiopathy indicating endothelial activation 156. Surprisingly, despite the increased risk of TEs in dasatinib treated patients in preclinical studies, dasatinib reduced thrombus formation in the mesenteric arteriole and carotid artery ferric chloride mouse models 153. Platelet activation appears to be main mechanism of ponatinib associated arterial thrombosis 154, 155. Ponatinib was found to induce platelet hyperactivation in response to the glycoprotein VI agonist collagen related peptide155. Consistent with this glycoprotein VI -mediated mechanism ponatinib induced increased platelet adhesion to collagen 154.
Second-generation BCR-ABL RTKIs, particularly ponatinib, are strongly associated with an increased risk of both ATE and VTE. Preclinical studies support the prothrombotic potential of these agents and suggest that ATE is primarily caused by activation of both the endothelium and platelets (Figure 2).
Cyclin dependent kinase inhibitors
Cyclin dependent kinase (CDK) inhibitors have emerged as an important therapy for the treatment of advanced estrogen receptor-positive and HER2-negative breast cancer. This class of agent includes the CDK 4/6 inhibitors palbociclib, abemaciclib and ribociclib. Initial clinical trials with these agents indicated that they increased the risk of VTE 157, 158. In a retrospective study, a VTE incidence of 6.3% was observed in metastatic breast cancer patients receiving CDK 4/6 inhibitors 159. In a meta-analysis of breast cancer patients receiving endocrine therapy, CDK 4/6 inhibitor treatment was associated with a significantly increased risk of VTE compared to control, 2% versus 0.6% (RR 2.62) 160. Subgroup analysis indicated that this increased VTE risk was primarily driven by abemaciclib containing regimens 160. The cellular and molecular mechanism causing the increased risk of VTE in patients treated CDK 4/6 inhibitors has yet to be determined (Figure 2).
Immune checkpoint inhibitors
Immune check point inhibitors (ICIs) impair tumor escape mechanisms by targeting programmed cell death 1, its ligands (PD-1/PD-L1) or cytotoxic T-lymphocyte-associated protein 4161. There are six approved ICIs (atezolizumab, avelumab, ipilimumab, nivolumab, pembrolizumab and atezolizumab). ICIs are associated with various immune-related adverse effects that differ from those associated with chemotherapeutic agents 162. A recent retrospective, single institution study reported the cumulative incidence of VTE and ATE of 12.9% and 0.6% for patients treated with ICIs over a median 8.5 months follow-up 163. However, the study did not have a control group and it is unclear if ICIs contributed to the TE in the cancer patients. A meta-analysis found that the overall rate of VTE and ATE in patients treated with ICIs was 2.7% and 1.1% respectively. In randomized trials, ICIs did not increase the RR of VTE compared with non-ICI containing arms 164. This suggests that ICIs may not significantly increase the risk of TEs.
Thromboprophylaxis
The American Society of Clinical Oncology has recently updated its guidelines for VTE prophylaxis and treatment in patients with cancer 165. Routine thromboprophylaxis is not recommended for all outpatients with cancer. However, thromboprophylaxis may be offered to high-risk patients. The only recommendation for thromboprophylaxis associated with a treatment is for MM patients receiving thalidomide- or lenalidomide-based regimes with chemotherapy and/or dexamethasone. It is recommended that lower-risk MM patients receive either aspirin or low molecular weight heparin whereas high-risk MM patients receive low molecular weight heparin.
Future Investigations
There are a number of areas in this field that warrant further investigation. In the majority of cases the underlying mechanism by which these agents drive thrombosis has yet to be elucidated. Further, the prothrombotic mechanisms associated with specific combinations of therapies, such as thalidomide and dexamethasone, requires additional investigation. The arsenal of therapeutics approved for use in the treatment of patients with cancer continues to expand. Evaluation of the vascular toxicity of agents currently undergoing development may help to identify those candidates with an increased prothrombotic potential prior to extensive clinical study. Critically, through a better understanding of the mechanisms that drive cancer therapy associated thrombosis it may be possible to develop optimal anticoagulant regimens that minimize thrombotic risk. In the clinical setting systematic reviews and metanalyses of new agents using data derived from prospective randomized controlled trials is warranted. Single-agent analyses or sub-analyses would also enable the identification of specific agents in a class that might otherwise be masked. Systematic review-based approaches are preferable to the increasing number of retrospective uncontrolled studies being published that do not establish the risk of TE associated with a given agent.
Conclusions
Although cancer type and stage remain the leading drivers of thrombosis in patients with cancer, anti-cancer agents also increase the risk of TE in cancer patients. In general, anti-cancer agents appear to be associated with a more pronounced effect of the incidence of VTE than ATE, likely attributed to the lower absolute risk for ATE in cancer patients. The relative increased risk of TEs varies markedly between agents suggesting that a therapy specific approach to anticoagulation may be required. Further, TEs appear to be particularly prevalent in patients treated with multi-agent regimens where they may be synergistic effects at play. Interestingly, newer more specific targeted agents do not appear to be associated with a reduced association with TEs compared with conventional anti-cancer agents. It is possible that TEs associated with targeted therapies represent pathological “on-target” effects as opposed to “off-target” effects attributed to conventional chemotherapy. The molecular and cellular mechanisms that underpin anti-cancer agent associated thrombosis remain incompletely understood and further investigations are warranted. Critically, with the increasing number of anti-cancer agents, new issues involving adverse TEs are likely to occur.
Highlights.
Specific cancer therapies are associated with a significantly increased risk of thrombosis
Cancer therapies can induce a procoagulant state through a number of molecular and cellular mechanisms
For numerous cancer therapies, however, the mechanism by which they induce thrombotic complications remain incompletely understood
Acknowledgments
a) Acknowledgements: None
b) ources of funding: This work was supported by an AHA postdoctoral fellowship 19POST34370026 (S.P.G), the NIH NHLBI R01HL147149 (N.M) and the John C. Parker professorship (N.M).
c) Disclosures: The authors have no conflicts of interest to disclose.
Abbreviations
- ALL
Acute lymphocytic leukemia
- ATE
Arterial thromboembolism
- CDK
Cyclin dependent kinase
- CML
Chronic myeloid leukemia
- EGF
Epidermal growth factor
- F
Factor
- HR
Hazard ratio
- ICI
Immune checkpoint inhibitor
- MM
Multiple myeloma
- OR
Odds ratio
- RR
Relative risk
- RTKI
Receptor tyrosine kinase inhibitor
- TE
Thrombotic event
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
- VTE
Venous thromboembolism
- 5-FU
5-Fluorouracil
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424 [DOI] [PubMed] [Google Scholar]
- 2.Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122:1712–1723 [DOI] [PubMed] [Google Scholar]
- 3.Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293:715–722 [DOI] [PubMed] [Google Scholar]
- 4.Khorana AA, Dalal M, Lin J, Connolly GC. Incidence and predictors of venous thromboembolism (vte) among ambulatory high-risk cancer patients undergoing chemotherapy in the united states. Cancer. 2013;119:648–655 [DOI] [PubMed] [Google Scholar]
- 5.Heit JA. Epidemiology of venous thromboembolism. Nat Rev Cardiol. 2015;12:464–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hisada Y, Mackman N. Cancer-associated pathways and biomarkers of venous thrombosis. Blood. 2017;130:1499–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horowitz NA, Brenner B. Thrombosis in hematological malignancies: Mechanisms and implications. Thromb Res. 2020;191 Suppl 1:S58–S62 [DOI] [PubMed] [Google Scholar]
- 8.Grilz E, Königsbrügge O, Posch F, Schmidinger M, Pirker R, Lang IM, Pabinger I, Ay C. Frequency, risk factors, and impact on mortality of arterial thromboembolism in patients with cancer. Haematologica. 2018;103:1549–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falanga A, Russo L, Milesi V, Vignoli A. Mechanisms and risk factors of thrombosis in cancer. Crit Rev Oncol Hematol. 2017;118:79–83 [DOI] [PubMed] [Google Scholar]
- 10.Khorana AA, Francis CW, Culakova E, Fisher RI, Kuderer NM, Lyman GH. Thromboembolism in hospitalized neutropenic cancer patients. J Clin Oncol. 2006;24:484–490 [DOI] [PubMed] [Google Scholar]
- 11.Chew HK, Wun T, Harvey D, Zhou H, White RH. Incidence of venous thromboembolism and its effect on survival among patients with common cancers. Arch Intern Med. 2006;166:458–464 [DOI] [PubMed] [Google Scholar]
- 12.Paneesha S, McManus A, Arya R, Scriven N, Farren T, Nokes T, Bacon S, Nieland A, Cooper D, Smith H, O’Shaughnessy D, Rose P, Investigators V. Frequency, demographics and risk (according to tumour type or site) of cancer-associated thrombosis among patients seen at outpatient dvt clinics. Thromb Haemost. 2010;103:338–343 [DOI] [PubMed] [Google Scholar]
- 13.Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer. 2007;110:2339–2346 [DOI] [PubMed] [Google Scholar]
- 14.Kuter DJ. Thrombotic complications of central venous catheters in cancer patients. Oncologist. 2004;9:207–216 [DOI] [PubMed] [Google Scholar]
- 15.Oppelt P, Betbadal A, Nayak L. Approach to chemotherapy-associated thrombosis. Vasc Med. 2015;20:153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ. Risk factors for deep vein thrombosis and pulmonary embolism: A population-based case-control study. Arch Intern Med. 2000;160:809–815 [DOI] [PubMed] [Google Scholar]
- 17.DeVita VT, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653 [DOI] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70 [DOI] [PubMed] [Google Scholar]
- 19.Malhotra V, Perry MC. Classical chemotherapy: Mechanisms, toxicities and the therapeutic window. Cancer Biol Ther. 2003;2:S2–4 [PubMed] [Google Scholar]
- 20.Nurgali K, Jagoe RT, Abalo R. Adverse effects of cancer chemotherapy: Anything new to improve tolerance and reduce sequelae? Front Pharmacol. 2018;9:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niraula S, Seruga B, Ocana A, Shao T, Goldstein R, Tannock IF, Amir E. The price we pay for progress: A meta-analysis of harms of newly approved anticancer drugs. J Clin Oncol. 2012;30:3012–3019 [DOI] [PubMed] [Google Scholar]
- 22.Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost. 2007;5:632–634 [DOI] [PubMed] [Google Scholar]
- 23.Canale ML, Bisceglia I, Lestuzzi C, Parrini I, Force AC- OT. Arterial thrombosis in cancer: Spotlight on the neglected vessels. Anticancer Res. 2019;39:4619–4625 [DOI] [PubMed] [Google Scholar]
- 24.Carrier M, Khorana AA, Zwicker JI, Lyman GH, Le Gal G, Lee AY, ISTH soHaMftSot. Venous thromboembolism in cancer clinical trials: Recommendation for standardized reporting and analysis. J Thromb Haemost. 2012;10:2599–2601 [DOI] [PubMed] [Google Scholar]
- 25.Kelland L The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584 [DOI] [PubMed] [Google Scholar]
- 26.Weijl NI, Rutten MF, Zwinderman AH, Keizer HJ, Nooy MA, Rosendaal FR, Cleton FJ, Osanto S. Thromboembolic events during chemotherapy for germ cell cancer: A cohort study and review of the literature. J Clin Oncol. 2000;18:2169–2178 [DOI] [PubMed] [Google Scholar]
- 27.Moik F, van Es N, Posch F, Di Nisio M, Fuereder T, Preusser M, Pabinger I, Ay C. Gemcitabine and platinum-based agents for the prediction of cancer-associated venous thromboembolism: Results from the vienna cancer and thrombosis study. Cancers (Basel). 2020;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seng S, Liu Z, Chiu SK, Proverbs-Singh T, Sonpavde G, Choueiri TK, Tsao CK, Yu M, Hahn NM, Oh WK, Galsky MD. Risk of venous thromboembolism in patients with cancer treated with cisplatin: A systematic review and meta-analysis. J Clin Oncol. 2012;30:4416–4426 [DOI] [PubMed] [Google Scholar]
- 29.Proverbs-Singh T, Chiu SK, Liu Z, Seng S, Sonpavde G, Choueiri TK, Tsao CK, Yu M, Hahn NM, Oh WK, Galsky MD. Arterial thromboembolism in cancer patients treated with cisplatin: A systematic review and meta-analysis. J Natl Cancer Inst. 2012;104:1837–1840 [DOI] [PubMed] [Google Scholar]
- 30.Mellema WW, van der Hoek D, Postmus PE, Smit EF. Retrospective evaluation of thromboembolic events in patients with non-small cell lung cancer treated with platinum-based chemotherapy. Lung Cancer. 2014;86:73–77 [DOI] [PubMed] [Google Scholar]
- 31.Kim ES, Baran AM, Mondo EL, Rodgers TD, Nielsen GC, Dougherty DW, Pandya KJ, Rich DQ, van Wijngaarden E. Risk of thromboembolism in cisplatin versus carboplatin-treated patients with lung cancer. PLoS One. 2017;12:e0189410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tully CM, Apolo AB, Zabor EC, Regazzi AM, Ostrovnaya I, Furberg HF, Rosenberg JE, Bajorin DF. The high incidence of vascular thromboembolic events in patients with metastatic or unresectable urothelial cancer treated with platinum chemotherapy agents. Cancer. 2016;122:712–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Batran SE, Hartmann JT, Probst S, Schmalenberg H, Hollerbach S, Hofheinz R, Rethwisch V, Seipelt G, Homann N, Wilhelm G, Schuch G, Stoehlmacher J, Derigs HG, Hegewisch-Becker S, Grossmann J, Pauligk C, Atmaca A, Bokemeyer C, Knuth A, Jäger E, Onkologie AI. Phase iii trial in metastatic gastroesophageal adenocarcinoma with fluorouracil, leucovorin plus either oxaliplatin or cisplatin: A study of the arbeitsgemeinschaft internistische onkologie. J Clin Oncol. 2008;26:1435–1442 [DOI] [PubMed] [Google Scholar]
- 34.Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, Middleton G, Daniel F, Oates J, Norman AR, Kingdom UGCSGotNCRIotU. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358:36–46 [DOI] [PubMed] [Google Scholar]
- 35.Wheeler HR, Geczy CL. Induction of macrophage procoagulant expression by cisplatin, daunorubicin and doxorubicin. Int J Cancer. 1990;46:626–632 [DOI] [PubMed] [Google Scholar]
- 36.Lechner D, Kollars M, Gleiss A, Kyrle PA, Weltermann A. Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J Thromb Haemost. 2007;5:2445–2452 [DOI] [PubMed] [Google Scholar]
- 37.Lv LH, Wan YL, Lin Y, Zhang W, Yang M, Li GL, Lin HM, Shang CZ, Chen YJ, Min J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J Biol Chem. 2012;287:15874–15885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao X, Yu S, Li S, Wu J, Ma R, Cao H, Zhu Y, Feng J. Exosomes: Decreased sensitivity of lung cancer a549 cells to cisplatin. PLoS One. 2014;9:e89534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kasthuri RS, Hisada Y, Ilich A, Key NS, Mackman N. Effect of chemotherapy and longitudinal analysis of circulating extracellular vesicle tissue factor activity in patients with pancreatic and colorectal cancer. Res Pract Thromb Haemost. 2020;4:636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Hengel LG, van Steijn-van Tol AQ, Bertina RM, Versteeg HH, Osanto S. Microparticle-associated tissue factor activity in plasma is unaffected by cytolytic chemotherapy treatment in metastatic testicular cancer patients. Thromb Res. 2013;131:187–189 [DOI] [PubMed] [Google Scholar]
- 41.Auwerda JJ, Yuana Y, Osanto S, de Maat MP, Sonneveld P, Bertina RM, Leebeek FW. Microparticle-associated tissue factor activity and venous thrombosis in multiple myeloma. Thromb Haemost. 2011;105:14–20 [DOI] [PubMed] [Google Scholar]
- 42.Licciardello JT, Moake JL, Rudy CK, Karp DD, Hong WK. Elevated plasma von willebrand factor levels and arterial occlusive complications associated with cisplatin-based chemotherapy. Oncology. 1985;42:296–300 [DOI] [PubMed] [Google Scholar]
- 43.Ma R, Bi Y, Kou J, Zhou J, Shi J. Enhanced procoagulant activity of platelets after chemotherapy in non-small cell lung cancer. Cancer Biol Ther. 2017;18:627–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wenningmann N, Knapp M, Ande A, Vaidya TR, Ait-Oudhia S. Insights into doxorubicin-induced cardiotoxicity: Molecular mechanisms, preventive strategies, and early monitoring. Mol Pharmacol. 2019;96:219–232 [DOI] [PubMed] [Google Scholar]
- 45.Sanfilippo KM, Wang TF, Gage BF, Luo S, Riedell P, Carson KR. Incidence of venous thromboembolism in patients with non-hodgkin lymphoma. Thromb Res. 2016;143:86–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zangari M, Siegel E, Barlogie B, Anaissie E, Saghafifar F, Fassas A, Morris C, Fink L, Tricot G. Thrombogenic activity of doxorubicin in myeloma patients receiving thalidomide: Implications for therapy. Blood. 2002;100:1168–1171 [DOI] [PubMed] [Google Scholar]
- 47.Swystun LL, Shin LY, Beaudin S, Liaw PC. Chemotherapeutic agents doxorubicin and epirubicin induce a procoagulant phenotype on endothelial cells and blood monocytes. J Thromb Haemost. 2009;7:619–626 [DOI] [PubMed] [Google Scholar]
- 48.Boles JC, Williams JC, Hollingsworth RM, Wang JG, Glover SL, Owens AP, Barcel DA, Kasthuri RS, Key NS, Mackman N. Anthracycline treatment of the human monocytic leukemia cell line thp-1 increases phosphatidylserine exposure and tissue factor activity. Thromb Res. 2012;129:197–203 [DOI] [PubMed] [Google Scholar]
- 49.Aubertin K, Silva AK, Luciani N, Espinosa A, Djemat A, Charue D, Gallet F, Blanc-Brude O, Wilhelm C. Massive release of extracellular vesicles from cancer cells after photodynamic treatment or chemotherapy. Sci Rep. 2016;6:35376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yarana C, Carroll D, Chen J, Chaiswing L, Zhao Y, Noel T, Alstott M, Bae Y, Dressler EV, Moscow JA, Butterfield DA, Zhu H, St Clair DK. Extracellular vesicles released by cardiomyocytes in a doxorubicin-induced cardiac injury mouse model contain protein biomarkers of early cardiac injury. Clin Cancer Res. 2018;24:1644–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woodley-Cook J, Shin LY, Swystun L, Caruso S, Beaudin S, Liaw PC. Effects of the chemotherapeutic agent doxorubicin on the protein c anticoagulant pathway. Mol Cancer Ther. 2006;5:3303–3311 [DOI] [PubMed] [Google Scholar]
- 52.Weitz IC, Israel VK, Waisman JR, Presant CA, Rochanda L, Liebman HA. Chemotherapy-induced activation of hemostasis: Effect of a low molecular weight heparin (dalteparin sodium) on plasma markers of hemostatic activation. Thromb Haemost. 2002;88:213–220 [PubMed] [Google Scholar]
- 53.Mukherjee SD, Swystun LL, Mackman N, Wang JG, Pond G, Levine MN, Liaw PC. Impact of chemotherapy on thrombin generation and on the protein c pathway in breast cancer patients. Pathophysiol Haemost Thromb. 2010;37:88–97 [DOI] [PubMed] [Google Scholar]
- 54.Chow AY, Chin C, Dahl G, Rosenthal DN. Anthracyclines cause endothelial injury in pediatric cancer patients: A pilot study. J Clin Oncol. 2006;24:925–928 [DOI] [PubMed] [Google Scholar]
- 55.Ben Aharon I, Bar Joseph H, Tzabari M, Shenkman B, Farzam N, Levi M, Shalgi R, Stemmer SM, Savion N. Doxorubicin-induced vascular toxicity--targeting potential pathways may reduce procoagulant activity. PLoS One. 2013;8:e75157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lv H, Tan R, Liao J, Hao Z, Yang X, Liu Y, Xia Y. Doxorubicin contributes to thrombus formation and vascular injury by interfering with platelet function. Am J Physiol Heart Circ Physiol. 2020;319:H133–H143 [DOI] [PubMed] [Google Scholar]
- 57.Swystun LL, Mukherjee S, Liaw PC. Breast cancer chemotherapy induces the release of cell-free dna, a novel procoagulant stimulus. J Thromb Haemost. 2011;9:2313–2321 [DOI] [PubMed] [Google Scholar]
- 58.Poggi A, Kornblihtt L, Delaini F, Colombo T, Mussoni L, Reyers I, Donati MB. Delayed hypercoagulability after a single dose of adriamycin to normal rats. Thromb Res. 1979;16:639–650 [DOI] [PubMed] [Google Scholar]
- 59.Bernat A, Herbert JM. Effect of various drugs on adriamycin-enhanced venous thrombosis in the rat: Importance of paf. Thromb Res. 1994;75:91–97 [DOI] [PubMed] [Google Scholar]
- 60.Kim SH, Lim KM, Noh JY, Kim K, Kang S, Chang YK, Shin S, Chung JH. Doxorubicin-induced platelet procoagulant activities: An important clue for chemotherapy-associated thrombosis. Toxicol Sci. 2011;124:215–224 [DOI] [PubMed] [Google Scholar]
- 61.Grem JL, McAtee N, Murphy RF, Hamilton JM, Balis F, Steinberg S, Arbuck SG, Setser A, Jordan E, Chen A. Phase i and pharmacokinetic study of recombinant human granulocyte-macrophage colony-stimulating factor given in combination with fluorouracil plus calcium leucovorin in metastatic gastrointestinal adenocarcinoma. J Clin Oncol. 1994;12:560–568 [DOI] [PubMed] [Google Scholar]
- 62.Qi WX, Lin F, Sun YJ, Tang LN, Shen Z, Yao Y. Risk of venous and arterial thromboembolic events in cancer patients treated with gemcitabine: A systematic review and meta-analysis. Br J Clin Pharmacol. 2013;76:338–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Humphreys BD, Sharman JP, Henderson JM, Clark JW, Marks PW, Rennke HG, Zhu AX, Magee CC. Gemcitabine-associated thrombotic microangiopathy. Cancer. 2004;100:2664–2670 [DOI] [PubMed] [Google Scholar]
- 64.Cwikiel M, Zhang B, Eskilsson J, Wieslander JB, Albertsson M. The influence of 5-fluorouracil on the endothelium in small arteries. An electron microscopic study in rabbits. Scanning Microsc. 1995;9:561–576 [PubMed] [Google Scholar]
- 65.Kinhult S, Albertsson M, Eskilsson J, Cwikiel M. Antithrombotic treatment in protection against thrombogenic effects of 5-fluorouracil on vascular endothelium: A scanning microscopy evaluation. Scanning. 2001;23:1–8 [DOI] [PubMed] [Google Scholar]
- 66.Focaccetti C, Bruno A, Magnani E, Bartolini D, Principi E, Dallaglio K, Bucci EO, Finzi G, Sessa F, Noonan DM, Albini A. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ros production in endothelial cells and cardiomyocytes. PLoS One. 2015;10:e0115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cwikiel M, Eskilsson J, Albertsson M, Stavenow L. The influence of 5-fluorouracil and methotrexate on vascular endothelium. An experimental study using endothelial cells in the culture. Ann Oncol 1996;7:731–737 [DOI] [PubMed] [Google Scholar]
- 68.Altieri P, Murialdo R, Barisione C, Lazzarini E, Garibaldi S, Fabbi P, Ruggeri C, Borile S, Carbone F, Armirotti A, Canepa M, Ballestrero A, Brunelli C, Montecucco F, Ameri P, Spallarossa P. 5-fluorouracil causes endothelial cell senescence: Potential protective role of glucagon-like peptide 1. Br J Pharmacol. 2017;174:3713–3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sara JD, Kaur J, Khodadadi R, Rehman M, Lobo R, Chakrabarti S, Herrmann J, Lerman A, Grothey A. 5-fluorouracil and cardiotoxicity: A review. Ther Adv Med Oncol. 2018;10:1758835918780140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Narta UK, Kanwar SS, Azmi W. Pharmacological and clinical evaluation of l-asparaginase in the treatment of leukemia. Crit Rev Oncol Hematol. 2007;61:208–221 [DOI] [PubMed] [Google Scholar]
- 71.Priest JR, Ramsay NK, Steinherz PG, Tubergen DG, Cairo MS, Sitarz AL, Bishop AJ, White L, Trigg ME, Levitt CJ, Cich JA, Coccia PF. A syndrome of thrombosis and hemorrhage complicating l-asparaginase therapy for childhood acute lymphoblastic leukemia. J Pediatr. 1982;100:984–989 [DOI] [PubMed] [Google Scholar]
- 72.Caruso V, Iacoviello L, Di Castelnuovo A, Storti S, Mariani G, de Gaetano G, Donati MB. Thrombotic complications in childhood acute lymphoblastic leukemia: A meta-analysis of 17 prospective studies comprising 1752 pediatric patients. Blood. 2006;108:2216–2222 [DOI] [PubMed] [Google Scholar]
- 73.Gugliotta L, Mazzucconi MG, Leone G, Mattioli-Belmonte M, Defazio D, Annino L, Tura S, Mandelli F. Incidence of thrombotic complications in adult patients with acute lymphoblastic leukaemia receiving l-asparaginase during induction therapy: A retrospective study. The gimema group. Eur J Haematol. 1992;49:63–66 [DOI] [PubMed] [Google Scholar]
- 74.Lauw MN, Van der Holt B, Middeldorp S, Meijers JC, Cornelissen JJ, Biemond BJ. Venous thromboembolism in adults treated for acute lymphoblastic leukaemia: Effect of fresh frozen plasma supplementation. Thromb Haemost. 2013;109:633–642 [DOI] [PubMed] [Google Scholar]
- 75.Orvain C, Balsat M, Tavernier E, Marolleau JP, Pabst T, Chevallier P, de Gunzburg N, Cacheux V, Huguet F, Chantepie S, Caillot D, Chalandon Y, Frayfer J, Bonmati C, Lheritier V, Ifrah N, Dombret H, Boissel N, Hunault-Berger M. Thromboembolism prophylaxis in adult patients with acute lymphoblastic leukemia treated in the graall-2005 study. Blood. 2020;136:328–338 [DOI] [PubMed] [Google Scholar]
- 76.De Stefano V, Sorà F, Rossi E, Chiusolo P, Laurenti L, Fianchi L, Zini G, Pagano L, Sica S, Leone G. The risk of thrombosis in patients with acute leukemia: Occurrence of thrombosis at diagnosis and during treatment. J Thromb Haemost. 2005;3:1985–1992 [DOI] [PubMed] [Google Scholar]
- 77.Couturier MA, Huguet F, Chevallier P, Suarez F, Thomas X, Escoffre-Barbe M, Cacheux V, Pignon JM, Bonmati C, Sanhes L, Bories P, Daguindau E, Dorvaux V, Reman O, Frayfer J, Orvain C, Lhéritier V, Ifrah N, Dombret H, Hunault-Berger M, Tanguy-Schmidt A. Cerebral venous thrombosis in adult patients with acute lymphoblastic leukemia or lymphoblastic lymphoma during induction chemotherapy with l-asparaginase: The graall experience. Am J Hematol. 2015;90:986–991 [DOI] [PubMed] [Google Scholar]
- 78.Klaassen ILM, Lauw MN, Fiocco M, van der Sluis IM, Pieters R, Middeldorp S, van de Wetering MD, de Groot-Kruseman HA, van Ommen CH. Venous thromboembolism in a large cohort of children with acute lymphoblastic leukemia: Risk factors and effect on prognosis. Res Pract Thromb Haemost. 2019;3:234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Truelove E, Fielding AK, Hunt BJ. The coagulopathy and thrombotic risk associated with l-asparaginase treatment in adults with acute lymphoblastic leukaemia. Leukemia. 2013;27:553–559 [DOI] [PubMed] [Google Scholar]
- 80.Conard J, Cazenave B, Maury J, Horellou MH, Samama M. L-asparaginase, antithrombin iii, and thrombosis. Lancet. 1980;1:1091. [DOI] [PubMed] [Google Scholar]
- 81.Conard J, Horellou MH, Van Dreden P, Potevin F, Zittoun R, Samama M. Decrease in protein c in l-asparaginase-treated patients. Br J Haematol. 1985;59:725–727 [DOI] [PubMed] [Google Scholar]
- 82.Vigano’D’Angelo S, Gugliotta L, Mattioli Belmonte M, Cascione ML, Pattarini E, D’Angelo A. L-asparaginase treatment reduces the anticoagulant potential of the protein c system without affecting vitamin k-dependent carboxylation. Thromb Res. 1990;59:985–994 [DOI] [PubMed] [Google Scholar]
- 83.Bezeaud A, Drouet L, Leverger G, Griffin JH, Guillin MC. Effect of l-asparaginase therapy for acute lymphoblastic leukemia on plasma vitamin k-dependent coagulation factors and inhibitors. J Pediatr. 1986;108:698–701 [DOI] [PubMed] [Google Scholar]
- 84.Hernández-Espinosa D, Miñano A, Martínez C, Pérez-Ceballos E, Heras I, Fuster JL, Vicente V, Corral J. L-asparaginase-induced antithrombin type i deficiency: Implications for conformational diseases. Am J Pathol. 2006;169:142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hunault-Berger M, Chevallier P, Delain M, Bulabois CE, Bologna S, Bernard M, Lafon I, Cornillon J, Maakaroun A, Tizon A, Padrazzi B, Ifrah N, Gruel Y, Sang) GGO-EdLAeMd. Changes in antithrombin and fibrinogen levels during induction chemotherapy with l-asparaginase in adult patients with acute lymphoblastic leukemia or lymphoblastic lymphoma. Use of supportive coagulation therapy and clinical outcome: The capelal study. Haematologica. 2008;93:1488–1494 [DOI] [PubMed] [Google Scholar]
- 86.Schneider P, Van Dreden P, Rousseau A, Kassim Y, Legrand E, Vannier JP, Vasse M. Increased levels of tissue factor activity and procoagulant phospholipids during treatment of children with acute lymphoblastic leukaemia. Br J Haematol. 2010;148:582–592 [DOI] [PubMed] [Google Scholar]
- 87.Gugliotta L, D’Angelo A, Mattioli Belmonte M, Viganò-D’Angelo S, Colombo G, Catani L, Gianni L, Lauria F, Tura S. Hypercoagulability during l-asparaginase treatment: The effect of antithrombin iii supplementation in vivo. Br J Haematol. 1990;74:465–470 [DOI] [PubMed] [Google Scholar]
- 88.Deitcher SR, Gomes MP. The risk of venous thromboembolic disease associated with adjuvant hormone therapy for breast carcinoma: A systematic review. Cancer. 2004;101:439–449 [DOI] [PubMed] [Google Scholar]
- 89.Nevasaari K, Heikkinen M, Taskinen PJ. Tamoxifen and thrombosis. Lancet. 1978;2:946–947 [DOI] [PubMed] [Google Scholar]
- 90.Meier CR, Jick H. Tamoxifen and risk of idiopathic venous thromboembolism. Br J Clin Pharmacol. 1998;45:608–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hernandez RK, Sørensen HT, Pedersen L, Jacobsen J, Lash TL. Tamoxifen treatment and risk of deep venous thrombosis and pulmonary embolism: A danish population-based cohort study. Cancer. 2009;115:4442–4449 [DOI] [PubMed] [Google Scholar]
- 92.Lin HF, Liao KF, Chang CM, Lin CL, Lai SW, Hsu CY. Correlation of the tamoxifen use with the increased risk of deep vein thrombosis and pulmonary embolism in elderly women with breast cancer: A case-control study. Medicine (Baltimore). 2018;97:e12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fisher B, Costantino J, Redmond C, Poisson R, Bowman D, Couture J, Dimitrov NV, Wolmark N, Wickerham DL, Fisher ER. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N Engl J Med. 1989;320:479–484 [DOI] [PubMed] [Google Scholar]
- 94.Saphner T, Tormey DC, Gray R. Venous and arterial thrombosis in patients who received adjuvant therapy for breast cancer. J Clin Oncol. 1991;9:286–294 [DOI] [PubMed] [Google Scholar]
- 95.Pemberton KD, Melissari E, Kakkar VV. The influence of tamoxifen in vivo on the main natural anticoagulants and fibrinolysis. Blood Coagul Fibrinolysis. 1993;4:935–942 [PubMed] [Google Scholar]
- 96.Cushman M, Costantino JP, Bovill EG, Wickerham DL, Buckley L, Roberts JD, Krag DN. Effect of tamoxifen on venous thrombosis risk factors in women without cancer: The breast cancer prevention trial. Br J Haematol. 2003;120:109–116 [DOI] [PubMed] [Google Scholar]
- 97.Cosman F, Baz-Hecht M, Cushman M, Vardy MD, Cruz JD, Nieves JW, Zion M, Lindsay R. Short-term effects of estrogen, tamoxifen and raloxifene on hemostasis: A randomized-controlled study and review of the literature. Thromb Res. 2005;116:1–13 [DOI] [PubMed] [Google Scholar]
- 98.Eilertsen AL, Liestøl S, Mowinckel MC, Hemker HC, Sandset PM. Differential impact of conventional and low-dose oral hormone therapy (ht), tibolone and raloxifene on functionality of the activated protein c system. Thromb Haemost. 2007;97:938–943 [PubMed] [Google Scholar]
- 99.Rühl H, Schröder L, Müller J, Fimmers R, Sukhitashvili S, Welz J, Kuhn WC, Oldenburg J, Rudlowski C, Pötzsch B. Tamoxifen induces resistance to activated protein c. Thromb Res. 2014;133:886–891 [DOI] [PubMed] [Google Scholar]
- 100.Vitseva O, Flockhart DA, Jin Y, Varghese S, Freedman JE. The effects of tamoxifen and its metabolites on platelet function and release of reactive oxygen intermediates. J Pharmacol Exp Ther. 2005;312:1144–1150 [DOI] [PubMed] [Google Scholar]
- 101.Shah VP, Chegini HA, Vishneski SR, Weatherman RV, Blackmore PF, Dobrydneva Y. Tamoxifen promotes superoxide production in platelets by activation of pi3-kinase and nadph oxidase pathways. Thromb Res. 2012;129:36–42 [DOI] [PubMed] [Google Scholar]
- 102.Chang Y, Lee JJ, Chen WF, Chou DS, Huang SY, Sheu JR. A novel role for tamoxifen in the inhibition of human platelets. Transl Res. 2011;157:81–91 [DOI] [PubMed] [Google Scholar]
- 103.Nayak MK, Singh SK, Roy A, Prakash V, Kumar A, Dash D. Anti-thrombotic effects of selective estrogen receptor modulator tamoxifen. Thromb Haemost. 2011;106:624–635 [DOI] [PubMed] [Google Scholar]
- 104.Srkalovic G, Cameron MG, Rybicki L, Deitcher SR, Kattke-Marchant K, Hussein MA. Monoclonal gammopathy of undetermined significance and multiple myeloma are associated with an increased incidence of venothromboembolic disease. Cancer. 2004;101:558–566 [DOI] [PubMed] [Google Scholar]
- 105.Glasmacher A, Hahn C, Hoffmann F, Naumann R, Goldschmidt H, von Lilienfeld-Toal M, Orlopp K, Schmidt-Wolf I, Gorschlüter M. A systematic review of phase-ii trials of thalidomide monotherapy in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2006;132:584–593 [DOI] [PubMed] [Google Scholar]
- 106.Zangari M, Anaissie E, Barlogie B, Badros A, Desikan R, Gopal AV, Morris C, Toor A, Siegel E, Fink L, Tricot G. Increased risk of deep-vein thrombosis in patients with multiple myeloma receiving thalidomide and chemotherapy. Blood. 2001;98:1614–1615 [DOI] [PubMed] [Google Scholar]
- 107.Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR, Group ECO. Phase iii clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: A clinical trial coordinated by the eastern cooperative oncology group. J Clin Oncol. 2006;24:431–436 [DOI] [PubMed] [Google Scholar]
- 108.El Accaoui RN, Shamseddeen WA, Taher AT. Thalidomide and thrombosis. A meta-analysis. Thromb Haemost. 2007;97:1031–1036 [PubMed] [Google Scholar]
- 109.Weber DM, Chen C, Niesvizky R, Wang M, Belch A, Stadtmauer EA, Siegel D, Borrello I, Rajkumar SV, Chanan-Khan AA, Lonial S, Yu Z, Patin J, Olesnyckyj M, Zeldis JB, Knight RD, Investigators MMS. Lenalidomide plus dexamethasone for relapsed multiple myeloma in north america. N Engl J Med. 2007;357:2133–2142 [DOI] [PubMed] [Google Scholar]
- 110.Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau JL, Dmoszynska A, San Miguel J, Hellmann A, Facon T, Foà R, Corso A, Masliak Z, Olesnyckyj M, Yu Z, Patin J, Zeldis JB, Knight RD, Investigators MMS. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123–2132 [DOI] [PubMed] [Google Scholar]
- 111.Rajkumar SV, Jacobus S, Callander NS, Fonseca R, Vesole DH, Williams ME, Abonour R, Siegel DS, Katz M, Greipp PR, Group ECO. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: An open-label randomised controlled trial. Lancet Oncol. 2010;11:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang B, Yu RL, Chi XH, Lu XC. Lenalidomide treatment for multiple myeloma: Systematic review and meta-analysis of randomized controlled trials. PLoS One. 2013;8:e64354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miguel JS, Weisel K, Moreau P, Lacy M, Song K, Delforge M, Karlin L, Goldschmidt H, Banos A, Oriol A, Alegre A, Chen C, Cavo M, Garderet L, Ivanova V, Martinez-Lopez J, Belch A, Palumbo A, Schey S, Sonneveld P, Yu X, Sternas L, Jacques C, Zaki M, Dimopoulos M. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (mm-003): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14:1055–1066 [DOI] [PubMed] [Google Scholar]
- 114.Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S, Chang XB, Bjorklund CC, Fonseca R, Bergsagel PL, Orlowski RZ, Stewart AK. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118:4771–4779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, Kaelin WG. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science. 2014;343:305–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, Ebert BL. Lenalidomide causes selective degradation of ikzf1 and ikzf3 in multiple myeloma cells. Science. 2014;343:301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dunkley S, Gaudry L. Thalidomide causes platelet activation, which can be abrogated by aspirin. J Thromb Haemost. 2007;5:1323–1325 [DOI] [PubMed] [Google Scholar]
- 118.Robak M, Treliński J, Chojnowski K. Hemostatic changes after 1 month of thalidomide and dexamethasone therapy in patients with multiple myeloma. Med Oncol. 2012;29:3574–3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Echart CL, Somaini S, Distaso M, Palumbo A, Richardson PG, Fareed J, Iacobelli M. Defibrotide blunts the prothrombotic effect of thalidomide on endothelial cells. Clin Appl Thromb Hemost. 2012;18:79–86 [DOI] [PubMed] [Google Scholar]
- 120.Palumbo A, Rajkumar SV, San Miguel JF, Larocca A, Niesvizky R, Morgan G, Landgren O, Hajek R, Einsele H, Anderson KC, Dimopoulos MA, Richardson PG, Cavo M, Spencer A, Stewart AK, Shimizu K, Lonial S, Sonneveld P, Durie BG, Moreau P, Orlowski RZ. International myeloma working group consensus statement for the management, treatment, and supportive care of patients with myeloma not eligible for standard autologous stem-cell transplantation. J Clin Oncol. 2014;32:587–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carrier M, Le Gal G, Tay J, Wu C, Lee AY. Rates of venous thromboembolism in multiple myeloma patients undergoing immunomodulatory therapy with thalidomide or lenalidomide: A systematic review and meta-analysis. J Thromb Haemost. 2011;9:653–663 [DOI] [PubMed] [Google Scholar]
- 122.Palumbo A, Cavo M, Bringhen S, Zamagni E, Romano A, Patriarca F, Rossi D, Gentilini F, Crippa C, Galli M, Nozzoli C, Ria R, Marasca R, Montefusco V, Baldini L, Elice F, Callea V, Pulini S, Carella AM, Zambello R, Benevolo G, Magarotto V, Tacchetti P, Pescosta N, Cellini C, Polloni C, Evangelista A, Caravita T, Morabito F, Offidani M, Tosi P, Boccadoro M. Aspirin, warfarin, or enoxaparin thromboprophylaxis in patients with multiple myeloma treated with thalidomide: A phase iii, open-label, randomized trial. J Clin Oncol. 2011;29:986–993 [DOI] [PubMed] [Google Scholar]
- 123.da Cunha Santos G, Shepherd FA, Tsao MS. Egfr mutations and lung cancer. Annu Rev Pathol. 2011;6:49–69 [DOI] [PubMed] [Google Scholar]
- 124.Xu H, Zong H, Ma C, Ming X, Shang M, Li K, He X, Du H, Cao L. Epidermal growth factor receptor in glioblastoma. Oncol Lett. 2017;14:512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Young K, Paz-Ares L, Thatcher N, Spigel DR, Shahidi J, Soldatenkova V, Grau G, Kurek R, Shepherd FA. Venous thromboembolism with egfr monoclonal antibody necitumumab in stage iv non-small cell lung cancer: A retrospective cohort analysis. Thromb Res. 2018;167:50–56 [DOI] [PubMed] [Google Scholar]
- 126.Petrelli F, Cabiddu M, Borgonovo K, Barni S. Risk of venous and arterial thromboembolic events associated with anti-egfr agents: A meta-analysis of randomized clinical trials. Ann Oncol. 2012;23:1672–1679 [DOI] [PubMed] [Google Scholar]
- 127.Miroddi M, Sterrantino C, Simmonds M, Caridi L, Calapai G, Phillips RS, Stewart LA. Systematic review and meta-analysis of the risk of severe and life-threatening thromboembolism in cancer patients receiving anti-egfr monoclonal antibodies (cetuximab or panitumumab). Int J Cancer. 2016;139:2370–2380 [DOI] [PubMed] [Google Scholar]
- 128.Scappaticci FA, Skillings JR, Holden SN, Gerber HP, Miller K, Kabbinavar F, Bergsland E, Ngai J, Holmgren E, Wang J, Hurwitz H. Arterial thromboembolic events in patients with metastatic carcinoma treated with chemotherapy and bevacizumab. J Natl Cancer Inst. 2007;99:1232–1239 [DOI] [PubMed] [Google Scholar]
- 129.Schutz FA, Je Y, Azzi GR, Nguyen PL, Choueiri TK. Bevacizumab increases the risk of arterial ischemia: A large study in cancer patients with a focus on different subgroup outcomes. Ann Oncol. 2011;22:1404–1412 [DOI] [PubMed] [Google Scholar]
- 130.Ranpura V, Hapani S, Chuang J, Wu S. Risk of cardiac ischemia and arterial thromboembolic events with the angiogenesis inhibitor bevacizumab in cancer patients: A meta-analysis of randomized controlled trials. Acta Oncol. 2010;49:287–297 [DOI] [PubMed] [Google Scholar]
- 131.Eremina V, Quaggin SE. Biology of anti-angiogenic therapy-induced thrombotic microangiopathy. Semin Nephrol. 2010;30:582–590 [DOI] [PubMed] [Google Scholar]
- 132.Nalluri SR, Chu D, Keresztes R, Zhu X, Wu S. Risk of venous thromboembolism with the angiogenesis inhibitor bevacizumab in cancer patients: A meta-analysis. JAMA. 2008;300:2277–2285 [DOI] [PubMed] [Google Scholar]
- 133.Hurwitz HI, Saltz LB, Van Cutsem E, Cassidy J, Wiedemann J, Sirzén F, Lyman GH, Rohr UP. Venous thromboembolic events with chemotherapy plus bevacizumab: A pooled analysis of patients in randomized phase ii and iii studies. J Clin Oncol. 2011;29:1757–1764 [DOI] [PubMed] [Google Scholar]
- 134.Kanukula R, Ganta S, Sirumalla Y, Salam A, Baddam R, Pasupuleti BC. Risk of venous thromboembolic events in patients with cancer treated with aflibercept: A systematic review and meta-analysis of randomized controlled trials. Am J Ther. 2019;26:e549–e552 [DOI] [PubMed] [Google Scholar]
- 135.Chen N, Ren M, Li R, Deng X, Li Y, Yan K, Xiao L, Yang Y, Wang L, Luo M, Fay WP, Wu J. Bevacizumab promotes venous thromboembolism through the induction of pai-1 in a mouse xenograft model of human lung carcinoma. Mol Cancer. 2015;14:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. Vegf inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with vegf inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens. 2018;12:409–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Choueiri TK, Schutz FA, Je Y, Rosenberg JE, Bellmunt J. Risk of arterial thromboembolic events with sunitinib and sorafenib: A systematic review and meta-analysis of clinical trials. J Clin Oncol. 2010;28:2280–2285 [DOI] [PubMed] [Google Scholar]
- 139.Qi WX, Shen Z, Tang LN, Yao Y. Risk of arterial thromboembolic events with vascular endothelial growth factor receptor tyrosine kinase inhibitors: An up-to-date meta-analysis. Crit Rev Oncol Hematol. 2014;92:71–82 [DOI] [PubMed] [Google Scholar]
- 140.Minor DR. Risk of arterial thrombosis not increased by sorafenib or sunitinib. J Clin Oncol. 2010;28:e619; author reply e620 [DOI] [PubMed] [Google Scholar]
- 141.Qi WX, Min DL, Shen Z, Sun YJ, Lin F, Tang LN, He AN, Yao Y. Risk of venous thromboembolic events associated with vegfr-tkis: A systematic review and meta-analysis. Int J Cancer. 2013;132:2967–2974 [DOI] [PubMed] [Google Scholar]
- 142.Sonpavde G, Je Y, Schutz F, Galsky MD, Paluri R, Rosenberg JE, Bellmunt J, Choueiri TK. Venous thromboembolic events with vascular endothelial growth factor receptor tyrosine kinase inhibitors: A systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. 2013;87:80–89 [DOI] [PubMed] [Google Scholar]
- 143.Zhang D, Zhang X, Zhao C. Risk of venous and arterial thromboembolic events associated with anti-vegf agents in advanced non-small-cell lung cancer: A meta-analysis and systematic review. Onco Targets Ther. 2016;9:3695–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Abdel-Qadir H, Ethier JL, Lee DS, Thavendiranathan P, Amir E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat Rev. 2017;53:120–127 [DOI] [PubMed] [Google Scholar]
- 145.Alias S, Redwan B, Panzenboeck A, Winter MP, Schubert U, Voswinckel R, Frey MK, Jakowitsch J, Alimohammadi A, Hobohm L, Mangold A, Bergmeister H, Sibilia M, Wagner EF, Mayer E, Klepetko W, Hoelzenbein TJ, Preissner KT, Lang IM. Defective angiogenesis delays thrombus resolution: A potential pathogenetic mechanism underlying chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2014;34:810–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Evans CE, Grover SP, Humphries J, Saha P, Patel AP, Patel AS, Lyons OT, Waltham M, Modarai B, Smith A. Antiangiogenic therapy inhibits venous thrombus resolution. Arterioscler Thromb Vasc Biol. 2014;34:565–570 [DOI] [PubMed] [Google Scholar]
- 147.Douxfils J, Haguet H, Mullier F, Chatelain C, Graux C, Dogné JM. Association between bcr-abl tyrosine kinase inhibitors for chronic myeloid leukemia and cardiovascular events, major molecular response, and overall survival: A systematic review and meta-analysis. JAMA Oncol. 2016;2:625–632 [DOI] [PubMed] [Google Scholar]
- 148.Haguet H, Douxfils J, Mullier F, Chatelain C, Graux C, Dogné JM. Risk of arterial and venous occlusive events in chronic myeloid leukemia patients treated with new generation bcr-abl tyrosine kinase inhibitors: A systematic review and meta-analysis. Expert Opin Drug Saf. 2017;16:5–12 [DOI] [PubMed] [Google Scholar]
- 149.Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, le Coutre P. Vascular safety issues in cml patients treated with bcr/abl1 kinase inhibitors. Blood. 2015;125:901–906 [DOI] [PubMed] [Google Scholar]
- 150.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, DiPersio J, DeAngelo DJ, Abruzzese E, Rea D, Baccarani M, Müller MC, Gambacorti-Passerini C, Wong S, Lustgarten S, Rivera VM, Clackson T, Turner CD, Haluska FG, Guilhot F, Deininger MW, Hochhaus A, Hughes T, Goldman JM, Shah NP, Kantarjian H, Investigators P. A phase 2 trial of ponatinib in philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre PD, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, DeAngelo DJ, Abruzzese E, Rea D, Baccarani M, Müller MC, Gambacorti-Passerini C, Lustgarten S, Rivera VM, Haluska FG, Guilhot F, Deininger MW, Hochhaus A, Hughes TP, Shah NP, Kantarjian HM. Ponatinib efficacy and safety in philadelphia chromosome-positive leukemia: Final 5-year results of the phase 2 pace trial. Blood. 2018;132:393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lipton JH, Chuah C, Guerci-Bresler A, Rosti G, Simpson D, Assouline S, Etienne G, Nicolini FE, le Coutre P, Clark RE, Stenke L, Andorsky D, Oehler V, Lustgarten S, Rivera VM, Clackson T, Haluska FG, Baccarani M, Cortes JE, Guilhot F, Hochhaus A, Hughes T, Kantarjian HM, Shah NP, Talpaz M, Deininger MW, investigators E. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17:612–621 [DOI] [PubMed] [Google Scholar]
- 153.Alhawiti N, Burbury KL, Kwa FA, O’Malley CJ, Shuttleworth P, Alzard M, Hamadi A, Grigg AP, Jackson DE. The tyrosine kinase inhibitor, nilotinib potentiates a prothrombotic state. Thromb Res. 2016;145:54–64 [DOI] [PubMed] [Google Scholar]
- 154.Hamadi A, Grigg AP, Dobie G, Burbury KL, Schwarer AP, Kwa FA, Jackson DE. Ponatinib tyrosine kinase inhibitor induces a thromboinflammatory response. Thromb Haemost. 2019;119:1112–1123 [DOI] [PubMed] [Google Scholar]
- 155.Merkulova A, Mitchell SC, Stavrou EX, Forbes GL, Schmaier AH. Ponatinib treatment promotes arterial thrombosis and hyperactive platelets. Blood Adv. 2019;3:2312–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Latifi Y, Moccetti F, Wu M, Xie A, Packwood W, Qi Y, Ozawa K, Shentu W, Brown E, Shirai T, McCarty OJ, Ruggeri Z, Moslehi J, Chen J, Druker BJ, López JA, Lindner JR. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the bcr-abl1 tyrosine kinase inhibitor ponatinib. Blood. 2019;133:1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, Fowst C, Huang X, Kim ST, Randolph S, Slamon DJ. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, her2-negative, advanced breast cancer (paloma-1/trio-18): A randomised phase 2 study. Lancet Oncol. 2015;16:25–35 [DOI] [PubMed] [Google Scholar]
- 158.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, Park IH, Trédan O, Chen SC, Manso L, Freedman OC, Garnica Jaliffe G, Forrester T, Frenzel M, Barriga S, Smith IC, Bourayou N, Di Leo A. Monarch 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35:3638–3646 [DOI] [PubMed] [Google Scholar]
- 159.Gervaso L, Montero AJ, Jia X, Khorana AA. Venous thromboembolism in breast cancer patients receiving cyclin-dependent kinase inhibitors. J Thromb Haemost. 2020;18:162–168 [DOI] [PubMed] [Google Scholar]
- 160.Thein KZ, Htut TW, Ball S, Swarup S, Sultan A, Oo TH. Venous thromboembolism risk in patients with hormone receptor-positive her2-negative metastatic breast cancer treated with combined cdk 4/6 inhibitors plus endocrine therapy versus endocrine therapy alone: A systematic review and meta-analysis of randomized controlled trials. Breast Cancer Res Treat. 2020;183:479–487 [DOI] [PubMed] [Google Scholar]
- 161.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK. Pd-1 and pd-l1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158–168 [DOI] [PubMed] [Google Scholar]
- 163.Moik F, Chan WE, Wiedemann S, Hoeller C, Tuchmann F, Aretin MB, Fuereder T, Zöchbauer-Müller S, Preusser M, Pabinger I, Ay C. Incidence, risk factors and outcomes of venous and arterial thromboembolism in immune checkpoint inhibitor therapy. Blood. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Solinas C, Saba L, Sganzerla P, Petrelli F. Venous and arterial thromboembolic events with immune checkpoint inhibitors: A systematic review. Thromb Res. 2020;196:444–453 [DOI] [PubMed] [Google Scholar]
- 165.Key NS, Khorana AA, Kuderer NM, Bohlke K, Lee AYY, Arcelus JI, Wong SL, Balaban EP, Flowers CR, Francis CW, Gates LE, Kakkar AK, Levine MN, Liebman HA, Tempero MA, Lyman GH, Falanga A. Venous thromboembolism prophylaxis and treatment in patients with cancer: Asco clinical practice guideline update. J Clin Oncol. 2020;38:496–520 [DOI] [PubMed] [Google Scholar]