

Abstract

Sickle cell disease (SCD) is a group of common, life-threatening disorders caused by a point mutation in the β globin gene. Early diagnosis through newborn and early childhood screening, parental education, and preventive treatments are known to reduce mortality. However, the cost and complexity of conventional diagnostic methods limit the feasibility of early diagnosis for SCD in resource-limited areas worldwide. Although several point-of-care tests are commercially available, most are antibody-based tests, which cannot be used in patients who have recently received a blood transfusion. Here, we describe the development of a rapid, low-cost nucleic acid test that uses real-time fluorescence to detect the point mutation encoding hemoglobin S (HbS) in one round of isothermal recombinase polymerase amplification (RPA). When tested with a set of clinical samples from SCD patients and healthy volunteers, our assay demonstrated 100% sensitivity for both the βA globin and βS globin alleles and 94.7 and 97.1% specificities for the βA globin allele and βS globin allele, respectively (n = 91). Finally, we demonstrate proof-of-concept sample-to-answer genotyping of genomic DNA from capillary blood using an alkaline lysis procedure and direct input of diluted lysate into RPA. The workflow is performed in <30 min at a cost of <$5 USD on a commercially available benchtop fluorimeter and an open-source miniature fluorimeter. This study demonstrates the potential utility of a rapid, sample-to-answer nucleic acid test for SCD that may be implemented near the point of care and could be adapted to other disease-causing point mutations in genomic DNA.
Introduction
Sickle cell disease (SCD) comprises a group of inherited blood disorders characterized by at least one βS globin allele and a second pathogenic globin variant that results in the predominant formation of hemoglobin S (HbS).1 Abnormal polymerization of HbS distorts red blood cells into a sickle shape, causing anemia, painful vaso-occlusion, and death. SCD disproportionately affects persons living in low- and middle-income countries (LMICs), with 75% of affected individuals residing on the African continent.2 Despite substantial evidence for the efficacy of early screening programs, the majority of children in sub-Saharan Africa are never tested for SCD or receive a diagnosis only after complications arise.3 As a result, an estimated 50–80% of babies born with SCD in Africa die before the age of 5, and the vast majority die without ever having the correct diagnosis.3
Although life-saving preventive care and treatment for SCD are available,4 the complexity of conventional diagnostic tools for SCD is a major impediment to universal newborn screening in LMICs. Most conventional diagnostics for SCD identify the presence of normal (HbA) or the major abnormal (HbS, HbC) hemoglobins resulting from the genotypes AA (wild type), AS (sickle cell trait), SS (sickle cell anemia), and SC (hemoglobin SC disease). These methods include electrophoresis, isoelectric focusing (IEF), and high-performance liquid chromatography (HPLC), all of which require expensive equipment and trained personnel. Sickle cell anemia comprises the vast majority of disease burden, has the worst clinical outcome, and is the focus of most point-of-care diagnostic efforts.5,6
Several point-of-care devices are becoming available to reduce the necessary infrastructure challenges and cost, including tests based on hemoglobin solubility7,8 and detection of hemoglobin A or S.9 However, any test that detects hemoglobin proteins cannot be used in recently transfused patients, as blood transfusions contain globin proteins from the donor.8,10−14 Because many patients with SCD require blood transfusions, often before they are diagnosed, this is a limitation that contributes to delays in diagnosis.12,15 Additionally, some tests have difficulty diagnosing newborns, who have high concentrations of fetal hemoglobin (HbF) and low HbS.4,16 In contrast to protein-based assays, DNA analysis targets the genetic basis of the disease by identifying point mutations in the β globin gene and is therefore unaffected by transfusions or high concentrations of HbF. DNA analysis is the most reliable approach to diagnose hemoglobin disorders and the only reliable method to confirm SCD diagnosis in patients who have recently received blood transfusions.12,15 However, DNA analysis for SCD is not available at the point of care because of its cost and complexity.
Molecular techniques have previously been employed to detect β globin gene mutations using polymerase chain reaction (PCR) in combination with allele-specific amplification using one of several specificity-enhancing strategies, including the amplification refractory mutation system (ARMS)17 and locked nucleic acids (LNAs).18,19 ARMS introduces a deliberate mismatch, chosen according to the strength of the mismatch at the 3′-terminus, to destabilize the primer and enhance the specificity of amplification.20 A locked nucleic acid (LNA) is a nucleic acid analog with a 2′-O, 4′-C methylene bridge that locks the ribose moiety into a C3′-endo conformation, which enhances mismatch discrimination in comparison to DNA-only primers.18 To reduce instrumentation requirements, recent efforts to detect DNA point mutations for other diseases at the point of care have explored the use of isothermal amplification techniques with LNAs, peptide nucleic acids (PNAs), and ARMS.21,22 However, these approaches have required either complex primer design or multiple rounds of amplification, which increases the likelihood of workspace contamination with amplified DNA. Moreover, these approaches have not yet addressed sample preparation, which is a significant challenge for all low-resource nucleic acid tests. Recent advances in DNA extraction methods for nucleic acid tests have used paper extraction matrices and simple buffers to release DNA from crude samples,23−30 but methods for isolation of genomic DNA from whole blood have not been widely reported.
In this paper, we present a strategy to directly detect the point mutation that gives rise to the most common variant form of β globin, βS(Glu6Val) (Figure 1A), in a single round of isothermal recombinase polymerase amplification (RPA) and within 20 minutes. First, we describe an allele-specific amplification primer that incorporates a penultimate LNA to increase its affinity for its complementary sequence. Then, we combine the results of two real-time RPA reactions, one for the βA allele and one for the βS allele, to yield a determination of the patient genotype (Figure 1B). The RPA reactions contain a sequence-specific fluorescent “exo” probe, which is cleaved to produce fluorescence when amplification occurs (Figure 1C).
Figure 1.

Overview of assay design. (A) Variant forms of hemoglobin are produced when a single base substitution changes the sixth amino acid in the β chain of hemoglobin. (B) Amplification pattern produced by each reaction in response to patient genotype (green check: amplification; red X: no amplification). (C) Schematic showing the overall detection scheme of the assay using an allele-specific forward primer with LNA placed at the penultimate nucleotide, a fluorescent probe, and a common reverse primer (tetrahydrofuran (THF); locked nucleic acid (LNA)).
We evaluate the ability of the allele-specific RPA reactions to detect the presence or absence of the βA and βS alleles in a set of 109 extracted genomic DNA samples of clinically relevant genotypes AA, SS, SC, Sβ+, and Sβ0. Results show that the assay identifies patients with the genotypes relevant to the majority of SCD management (AA, AS, and SS) with >95% accuracy, and predicts the correct genotype for SS patients with recent blood transfusion. Next, we show that the volume of the reactions can be reduced by at least half to substantially reduce per-test cost with minimal effect on assay performance, using clinically relevant numbers of gene copies expected from a capillary blood sample. Finally, we demonstrate proof of concept on two low-resource isothermal platforms for the sample-to-answer selective detection of the βA allele from a 50 μL sample of capillary blood using a two-step, room-temperature alkaline lysis method to release genomic DNA from white blood cells.
Experimental Section
Primer Screen
A primer screen was performed in two steps: (1) unmodified forward and reverse primer candidates purchased from Integrated DNA Technologies (Coralville, Iowa) were screened in reactions with a fluorogenic probe designed according to RPA specifications and purchased from Biosearch Technologies (Novato, CA) (Table S1). These primers were screened to identify sequences that amplified the desired section of the β globin gene with the best efficiency (data not shown). (2) Eight versions of the chosen forward primer sequence, containing ARMS and/or up to three LNA modification(s), were designed and purchased from Qiagen (Germantown, MD) or Integrated DNA Technologies. Candidate forward primers were screened for their ability to selectively amplify target DNA containing the βS point mutation (Table S2). Additional details regarding the primer screen can be found in the Supporting Information.
Whole Blood Samples
Study protocols were reviewed and approved by the Institutional Review Boards at the Rice University and Baylor College of Medicine. Venous and capillary blood samples were obtained from healthy volunteers under a protocol approved by the Rice University IRB (2017–303); these samples were assumed to be AA genotype. Volunteers provided written informed consent prior to study participation. Venous blood samples from patients with varying genotypes seen in the Sickle Cell Program at Texas Children’s Hematology Center were collected and deidentified under an exempt protocol approved by the Rice University and Baylor College of Medicine IRBs (H-35374 Version 6.2). Genotypes, as determined by qualitative IEF combined with quantitative HPLC to confirm a diagnosis, were provided with the blood samples. HbF percentages were available for 28 of the 75 samples received from the Sickle Cell Program, and the percentage of HbF in those samples ranged from 0 to 23.5%. Additionally, at least 15 samples were from patients who were on chronic transfusion or had a recent transfusion. Blood samples were collected in ethylenediaminetetraacetic acid (EDTA) anticoagulant tubes and were stored at 4 °C for up to 4 weeks prior to testing. Samples with discordant clinical results were submitted for Sanger sequencing. For each discordant sample, a 262-bp portion of the β globin gene containing the mutation of interest was amplified in-house by PCR and Sanger-sequenced by GENEWIZ (South Plainfield, NJ).
Extraction of Genomic DNA from Clinical Samples
Extraction of genomic DNA from venous and capillary samples was performed using the Qiagen DNA Micro Kit (56304) according to the manufacturer’s instructions. Purified DNA was eluted in either 50 or 100 μL of buffer AE, and DNA concentration was estimated using a NanoDrop (ND-1000). DNA was stored at −20 °C until use and was diluted to working concentrations in nuclease-free water immediately prior to experiments.
Lysis of Blood Cells in Capillary Blood Samples
Sodium hydroxide (NaOH, 10 N) was purchased from Fisher Scientific (SS267) and used to prepare 0.4 N NaOH. To lyse cells and release genomic DNA, 50 μL of whole blood was combined with 50 μL of 0.4 N NaOH and mixed by pipetting. The solution was incubated at room temperature for 5 min. The lysate was diluted 1:100 in nuclease-free water before being added directly to RPA reactions. No-lysis controls (NLCs) were prepared according to the same protocol with nuclease-free water substituted for NaOH.
Real-Time RPA: Assay Setup
TwistAmp exo RPA kits were purchased from TwistDx, Limited (Maidenhead, U.K., TAEXO02KIT). All primers and probes were prepared at 10 μM working concentrations in 1× TE buffer. Fifty-microliter reactions were performed to characterize the assay using representative samples and test clinical samples. For each 50 μL reaction, 37.5 μL of a master mix containing 29.5 μL of rehydration buffer, 1.5 μL of allele-specific forward primer, 1.5 μL of reverse primer, 2.1 μL of probe, and 2.9 μL of water was added to an enzyme pellet within an 8-tube strip provided by the manufacturer and the solution was pipetted to mix. Magnesium acetate (2.5 μL) was added to tube caps as per the manufacturer’s recommendations. Ten microliters of DNA template in nuclease-free water was added, containing 102, 103, 104, or 105 copies of DNA, depending on the experiment. For clinical sample testing, 103 copies of purified genomic DNA were added to each allele-specific reaction.
For reduced-volume (<50 μL) reactions, standard manufacturer-recommended volumes of rehydration buffer, primers, probe, MgOAc, and water were combined in a master mix that was prepared on ice. The appropriate number of enzyme pellets was added to the master mix last, and the solution was gently vortexed to ensure even distribution of enzymes. Master mix, scaled down proportionally to 80% v/v of the total desired reaction volume, was added to each tube, and the appropriate volume of DNA template (20% v/v) was added to the caps. When DNA was added as a target to reduced-volume reactions, the copy number was kept the same (i.e., the concentration increased); target DNA added to reactions was either 10–104 copies of DNA in nuclease-free water or diluted crude blood lysate, depending on the experiment. To begin all reactions, tube strips were vortexed, briefly spun to combine the material in the caps with the volume in the tubes, and vortexed again before incubation in an isothermal fluorimeter. A T8-ISO isothermal fluorimeter (Axxin Pty, Ltd.) was used for real-time detection of amplification in most experiments. Additional details regarding instrumentation are provided in the Supporting Information.
Analysis of Real-Time Data
Samples were classified as positive or negative based on unprocessed fluorescence intensity, reported by the T8-ISO in millivolts (mV) vs time. A sample was considered positive (i.e., containing the allele complementary to the primer) if the fluorescence intensity reached 2500 mV in 20 min and considered negative (i.e., not containing the allele complementary to the primer) otherwise. All runs of the instrument included a positive and negative control. If either control was not valid, the other results from that master mix were to be considered invalid; however, this was not observed during the course of this study. Raw amplification data from all experiments were exported and analyzed in Microsoft Excel, version 16.40, and GraphPad Prism, version 8.4.3. Amplification curves in figures are shown with relative fluorescence units (RFUs) for clarity, where 1 RFU = 1 mV as reported by the T8-ISO.
Results and Discussion
Point Mutation Detection by RPA
We designed and screened versions of an RPA forward primer that had specificity-enhancing modifications to allow the selective amplification of template with single-nucleotide mutations. Allele-specific primers screened were designed with the 3′ end located on the βS globin mutation (Table S2). The primer that showed the best specificity was used in subsequent experiments; this primer was designed with the allele-specific nucleotide at the 3′ end of the primer and a single LNA at the penultimate position relative to the 3′ end (SC_fP4).
To observe the effect of the LNA in the primer, we first amplified DNA that simulated genotypes AA and SS, as described in the Supporting Information, in triplicate with two versions of the primer specific to the βS mutation: one having a normal nucleotide in place of the LNA (Figure 2A; SC_fP-S) and therefore just a single mismatch with the βA template at the 3′ end, and the other with the 3′ mismatch as well as the penultimate LNA as designed from the primer screen (Figure 2B; SC_fP4). As shown in Figure 2A, no selectivity was observed with the allele-specific primer—both targets amplified with roughly equal efficiency, despite the mismatch between the primer and target that was present in the AA samples. In contrast, Figure 2B shows that including the LNA modification at the penultimate nucleotide location resulted in robust selective amplification with only a slight delay in amplification compared to the allele-specific primer. We believe that the exonuclease present in RPA exo reactions cleaves the mismatched nucleotide when it is located at the 3′ end of the primer without an LNA. However, the presence of the LNA at the penultimate position likely generates complete nuclease resistance and prevents the cleaving of the mismatched nucleotide.31
Figure 2.
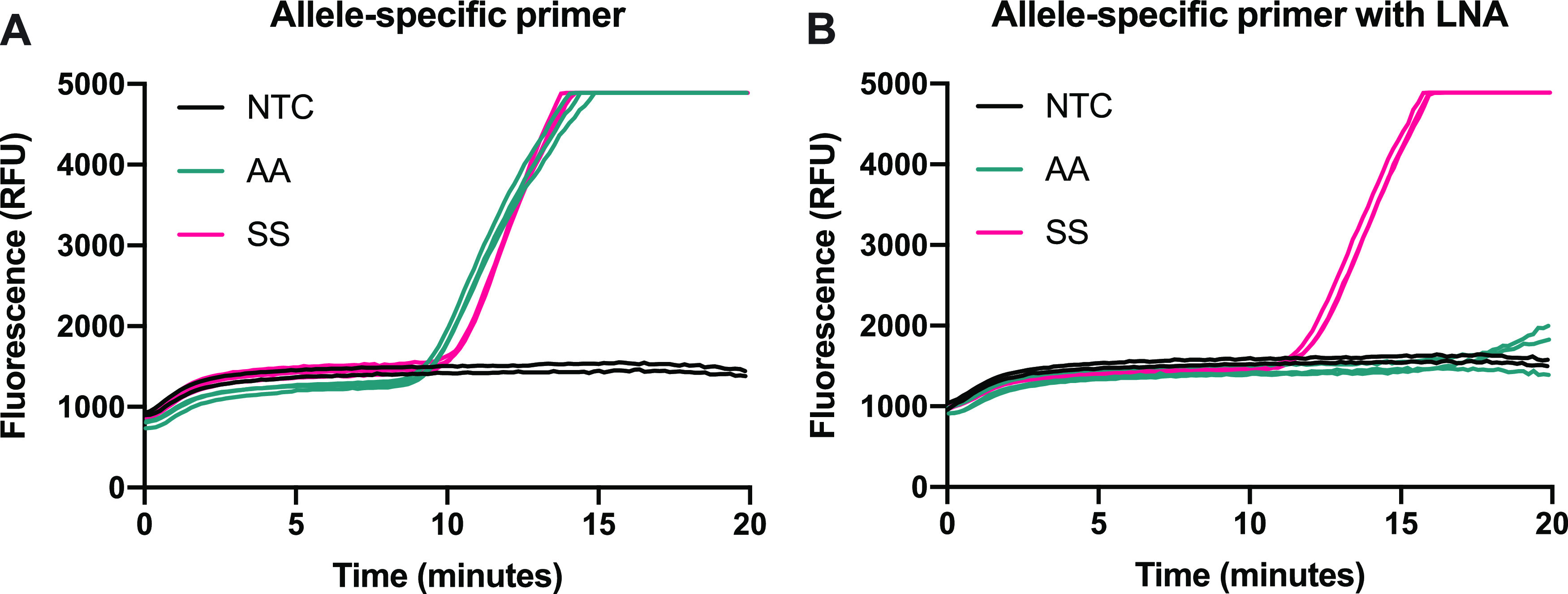
Impact of LNA modification on reaction specificity using 104 input copies of DNA-simulating genotypes AA and SS. Primer screen amplification curves with (A) βS allele-specific primer (no LNA) and (B) βS allele-specific primer with penultimate LNA.
Next, the allele-specific primer with LNA was modified to be complementary to the βA allele, and DNAs of genotypes AA, SS, and AS were tested in duplicate with either the βA or the βS version of the primer to evaluate the feasibility of using two concurrent allele-specific reactions to predict a patient’s genotype. Figure S1 shows the amplification curves with 104 input copies of simulated patient DNA of genotypes AA, AS, and SS. AA DNA produced strong amplification only in the βA allele reaction, SS DNA produced strong amplification only in the βS allele reaction, and AS DNA showed amplification in both reactions. The assay showed robust allele specificity over a dynamic range of 5 orders of magnitude, with a limit of detection of 10–100 copies (data not shown), demonstrating utility beyond the clinically relevant range of genome copies expected from a blood sample.
Assessing Accuracy with Patient Samples
Next, we evaluated the clinical sensitivity and specificity of our assay with DNA extracted from whole blood samples from both sickle cell patients and healthy volunteers. Samples from 34 healthy volunteers and 75 SCD patients (109 total) were tested in the developed assay. Genomic DNA was extracted and purified from 34 AA samples, 59 SS samples, 7 SC samples, 5 Sβ0 samples, and 4 Sβ+ samples; the extracted DNA was added to each of the two RPA reactions of the developed assay at a concentration of 1000 gene copies per reaction. Representative amplification curves of three AA samples and three SS samples are shown in Figure 3. DNA extracted and purified from clinical samples resulted in amplification in 10–15 min.
Figure 3.
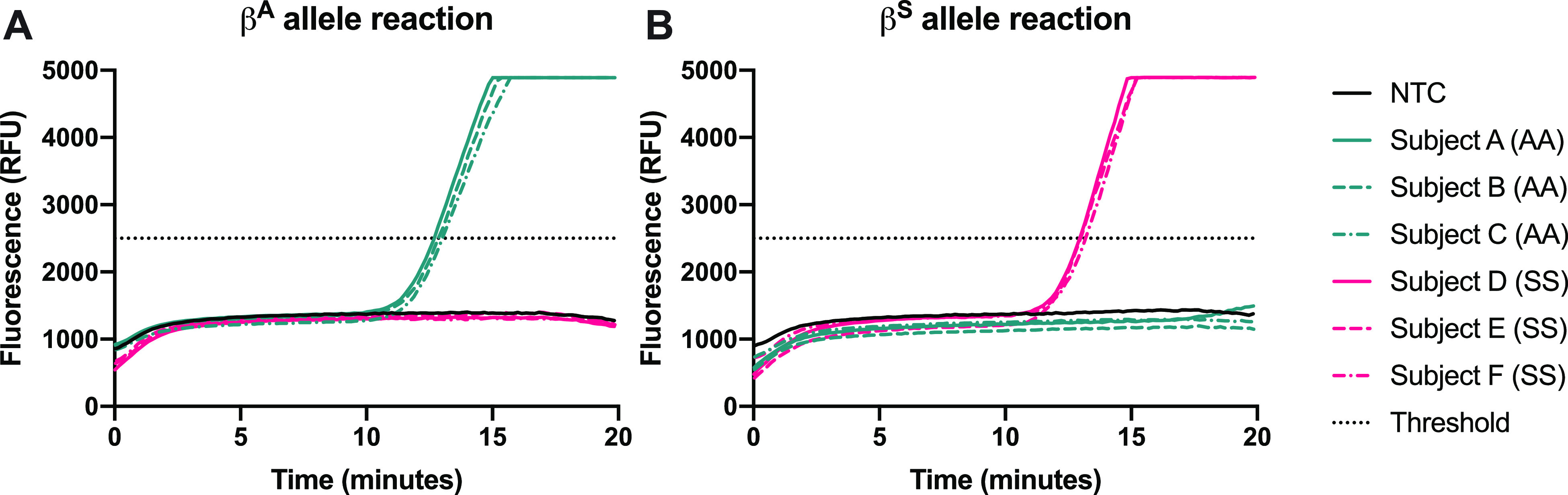
Representative amplification curves of purified genomic DNA from clinical samples with 1000 copies input into the reaction using (A) βA allele primer and (B) βS allele primer. Amplification curves resulting from three samples of each genotype are shown.
Five samples were submitted for Sanger sequencing due to discordant results for allele detection. Results showed that two samples were heterozygous for both the A and S alleles at the mutation site despite an SS diagnosis; these two samples were likely contaminated by amplicons in the laboratory workspace during purification, so they were excluded from further analysis. The sensitivity and specificity of the allele-specific reactions to identify the presence of the βA and βS alleles were calculated (Table 1) using data from the remaining 91 samples that were either AA or SS. Sensitivity was shown to be 100% for both alleles; specificity was calculated to be 94.7 and 97.1% for the βA globin and βS globin alleles, respectively.
Table 1. Sensitivity, Specificity, and Accuracy as Calculated by Allele.
| sensitivity | specificity | accuracy | |
|---|---|---|---|
| βA allele detection | 100.0% (34/34) | 94.7% (54/57) | 96.7% (88/91) |
| βS allele detection | 100.0% (57/57) | 97.1% (33/34) | 98.9% (90/91) |
Table 2A shows a confusion matrix of the genotype predicted by the developed assay (left) compared to the genotypes reported by standard-of-care IEF and HPLC (top). The assay correctly predicted the genotype of 87/91 of the samples with AA and SS genotypes, exhibiting an accuracy of 95.6%. No samples tested returned a negative result in both reactions. The sensitivity and specificity of the assay to predict AA and SS genotypes were evaluated (Table 2B). The assay predicted the AA genotype with 97.1% sensitivity and the SS genotype with 94.7% sensitivity. Specificity was 100% for the prediction of both genotypes.
Table 2. Clinical Results by Genotypea,b.
| |
IEF/HPLC |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| A | AA | AS | SS | total | B | sensitivity | specificity | accuracy | |
| assay predictions | AA | 33 | 0 | 0 | 33 | AA | 97.1% (33/34) | 100.0% (57/57) | 95.6% (87/91) |
| AS | 1 | 0 | 3 | 4 | |||||
| SS | 0 | 0 | 54 | 54 | SS | 94.7% (54/57) | 100.0% (34/34) | ||
| total | 34 | 0 | 57 | 91 | |||||
(A) Confusion matrix of genotypes identified by this assay compared to those obtained by IEF/HPLC gold standard for the 91 AA and SS samples.
(B) Sensitivity, specificity, and overall accuracy by genotype.
Through the evaluation of 16 clinical samples with other hemoglobinopathies, including Sβ0, Sβ+, and SC, we conclude that the RPA assay is unable to detect hemoglobinopathies beyond the βA globin and βS globin alleles for which it was designed (Figure S3 and Table S3). This is a challenge faced by most point-of-care SCD tests as β thalassemia can be caused by hundreds of mutations and deletions.4,32
To directly assess the potential of the RPA assay to be used in cases with recent blood transfusion, blood samples were acquired from three SS patients who had received a blood transfusion within the previous 3 months. The blood samples were tested with two commercially available antibody tests, Sickle SCAN (BioMedomics, Inc.) and HemoTypeSC (Silver Lake Research), according to the manufacturer’s instructions, and DNA extracted from the samples was tested in the RPA assay (Figure S2). Results indicate that both the Sickle SCAN and HemoTypeSC assays misidentified all three samples as AS (Figure S2A,B), while the RPA assay correctly identified all three samples as SS (Figure S2C,D).
Toward a Sample-To-Answer Test
To reduce assay cost and complexity, we reduced the volume of the RPA reactions and utilized a two-step lysis procedure that yielded crude lysate to be directly input to the small volume reactions. Because the RPA reagents make up the vast majority of the overall per-test cost, reducing the reaction volume can reduce the cost of the assay considerably (Figure S4 and Table S4). Recent literature suggests that reaction volume can be reduced to as little as 5 μL without a significant loss of performance.33 We therefore tested a range of reaction volumes from 5 to 50 μL (Figure S5). Amplification was evident at all volumes tested in this range, though the fluorescence vs time plots for volumes below 20 μL exhibited significant noise. The Axxin T8 is specified for use with reaction volumes of 30 μL or greater, and interference at lower volumes is likely due to the scattering of light on the reaction meniscus as well as bubbles caused by ball agitation. Based on these results, a reaction volume of 25 μL was selected for further experiments.
Figure 4 shows the amplification of synthetic templates simulating the genotypes AA and SS in 25 μL of reactions at concentrations ranging from 10 to 104 gene copies per reaction. With a 25 μL reaction volume, the assay has a dynamic range of 3–4 orders of magnitude and maintains single-nucleotide specificity.
Figure 4.
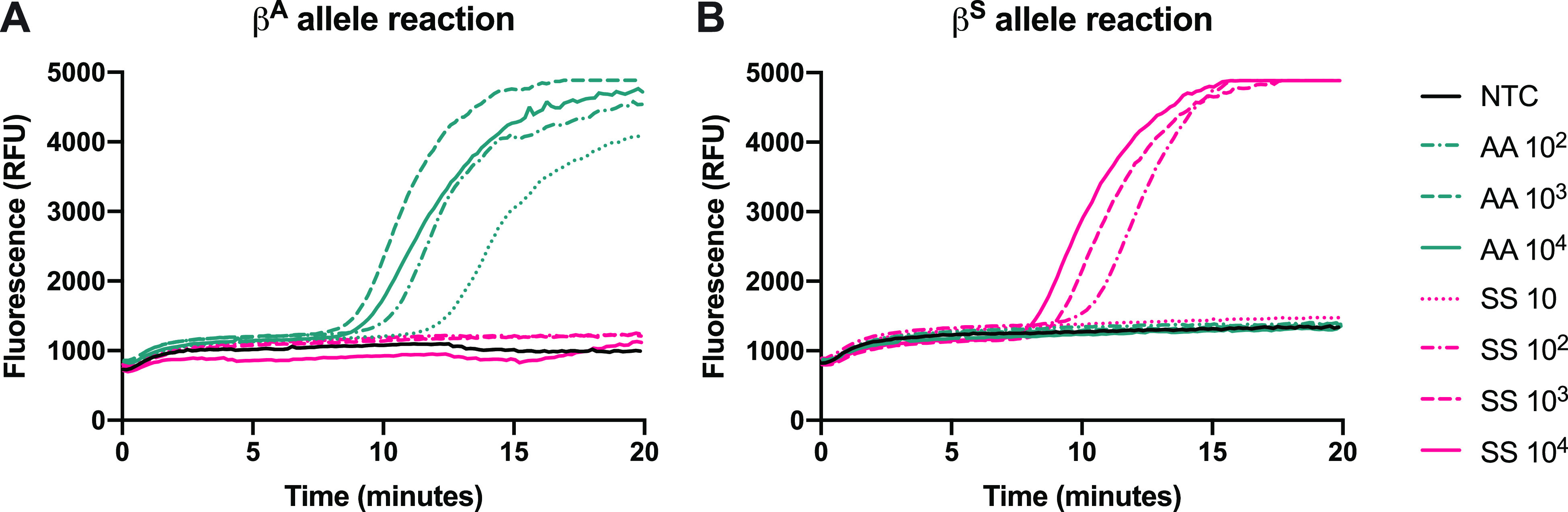
Amplification of synthetic templates of genotypes AA and SS at a range of input concentrations, with 25 μL reaction volumes with (A) βA allele primer and (B) βS allele primer.
Next, we implemented a two-step lysis procedure that is simple to perform and is also compatible with direct amplification in RPA after a dilution step for sample-to-answer testing. The lysis procedure is modified from Rudbeck et al.34 and consists of combining 50 μL of capillary whole blood with 50 μL of 0.4 N NaOH, incubating at room temperature for 5 min, and then diluting the lysate 1:100 in nuclease-free water prior to direct addition to RPA reactions. Similar methods have been used prior to loop-mediated isothermal amplification.35 Incubation at alkaline pH rapidly disrupts cell and nucleus membranes, denatures nucleases, and dissolves DNA. A 1:100 dilution step is necessary following lysis to dilute the components of whole blood that inhibit RPA to levels tolerated by the assay, while still maintaining a high enough concentration of DNA to fall within the dynamic range of the assay.
We experimentally verified the ability to detect the expected number of gene copies within diluted lysate (180–495 gene copies per 25 μL of reaction, as detailed in the Supporting Information). Capillary blood samples were obtained from three healthy volunteers and were subjected to the two-step lysis procedure followed by direct amplification of diluted crude lysate in RPA. This experiment was performed without mixing or with a brief vortex at 4 min, rather than with a mixing ball as in previous experiments, to further reduce the material requirements of the assay. Samples from healthy volunteers with genotype AA positively amplify in the βA allele reaction (Figure 5A,C) but not in the βS allele reaction (Figure 5B,D). This indicates that lysis by NaOH, coupled with the allele-specific amplification assay, provides DNA of sufficient quality and quantity to both amplify in the assay and achieve allelic discrimination. Consistent with others’ observations,33 a mix at 4 min provided a small performance boost in time to amplification and repeatability of technical replicates as compared to a no-mixing condition (Figure 5C).
Figure 5.
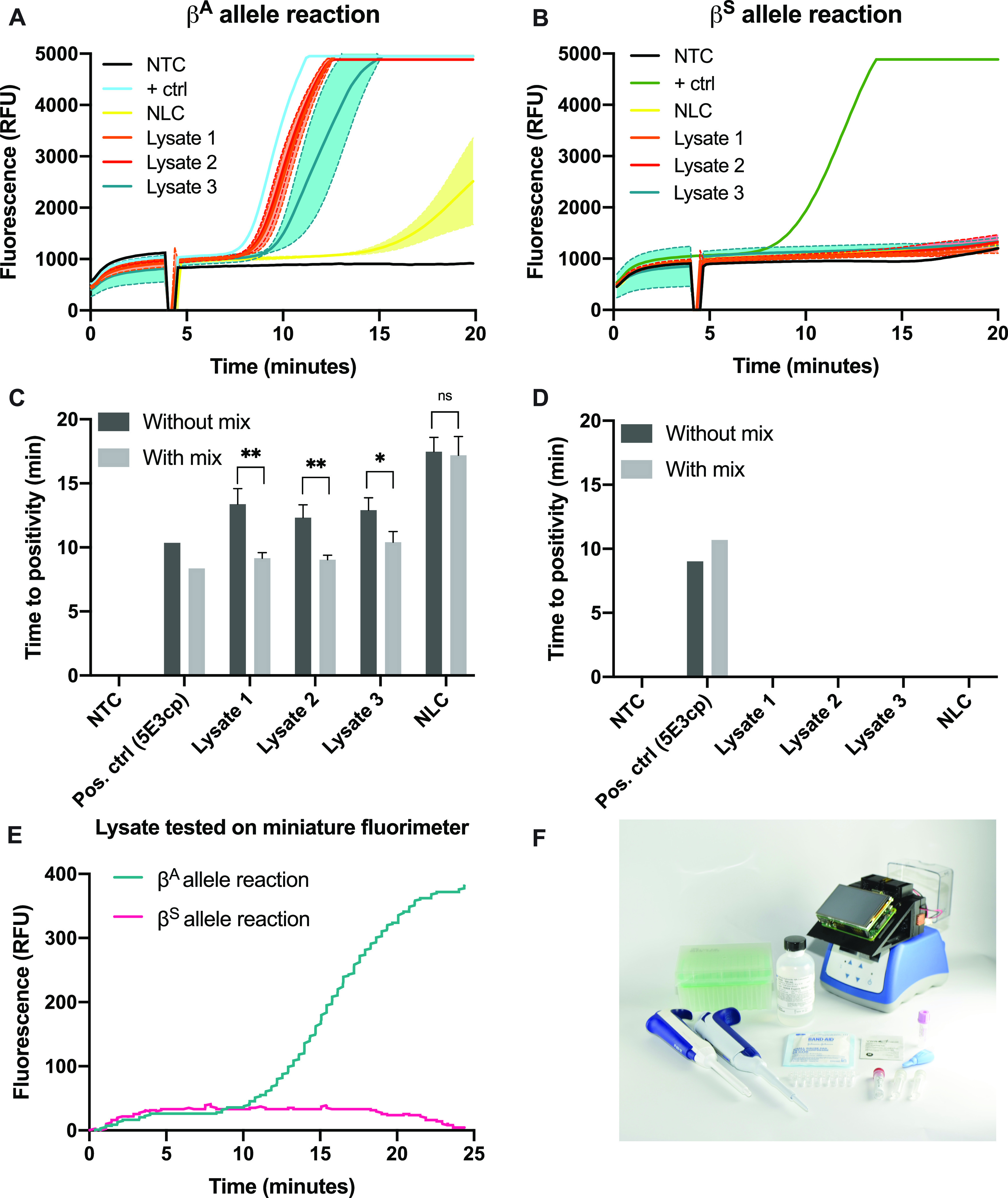
Amplification results of diluted crude lysate from normal volunteers using each allele-specific primer. (A−B) Average fluorescence of three replicates of each lysate sample with (A) the βA allele primer or (B) the βS allele primer; shading denotes 1 standard deviation. The dip in fluorescence at 4 min reflects the removal of the samples to vortex. (C) Time to positivity for the βA allele reaction results in part (A) and results from the same experiment performed without mixing; (D) time to positivity for the βS allele reaction results in part (B) and results from the same experiment performed without mixing. Error bars denote 1 standard deviation. NTC: no-target control; NLC: no-lysis control; *: p < 0.05, **: p < 0.01, ns: not significant. (E) Lysate from a normal volunteer in a 25 μL RPA reaction with each allele-specific primer performed on an open-source miniature fluorimeter. (F) Photo of all materials needed to run the assay. From left to right: P200 pipette; P20 pipette, pipette tips, NaOH, gauze pad, RPA enzyme pellets, miniature fluorimeter, alcohol pad, MgOAc, and RPA rehydration buffer; 2-mm lancet; primer mix; and 1.5-mL microtainer EDTA-coated capillary blood collection tube.
Finally, the diluted lysate from a normal volunteer was tested in 25 μL reactions on a miniature, low-cost, open-source fluorimeter designed to be used near the point of care with any conventional heat block.36Figure 5E shows clear amplification in the βA allele reaction only, correctly genotyping this sample as AA. Figure 5F shows all of the components necessary for the workflow. When evaluated in a laboratory setting using the items pictured in Figure 5F, capillary blood collection required 5 min, lysis required 5 min, and amplification and detection were accomplished in 20 min. The assay was reasonably accomplished in a laboratory setting in 30 min. Compatibility of this assay with a low-cost, open-source fluorimeter overcomes traditional cost barriers associated with fluorimeters while maintaining the advantages of real-time detection. These advantages include the potential for quantitation, the ability to build in an algorithm with result readout, and the ability to discard closed tubes after amplification, limiting the possibility of cross-contamination.
Table S4 details the costs associated with one patient test in the sample-to-answer, reduced-volume format. The total cost of consumables in each complete assay is less than $5 USD, which is comparable to the cost of an immunoassay and considerably cheaper than sequencing. To implement this test with minimal end-user manipulation and meet the World Health Organization’s ASSURED criteria for SCD testing,4 a lysis module could be incorporated. The lysis module could contain prealiquoted lysis buffer and feature a foil pouch that, when punctured, would transfer lysate into tubes containing lyophilized RPA reactions for amplification.
Conclusions
In conclusion, we demonstrated a novel strategy for rapid isothermal detection of the βA and βS alleles that shows >94% sensitivity and specificity with a set of 91 genomic DNA samples from individuals with AA and SS genotypes. Additionally, we have shown that the method is compatible with nucleic acid amplification directly from crude lysate obtained with a commonly available alkaline lysis agent. The sample-to-answer method takes less than 30 min from blood collection to result, costs less than $5 USD with the potential for further cost reduction, and has a low risk of workspace contamination because samples remain enclosed throughout the amplification and detection portion of the test. Additionally, the assay can be run using a T8-ISO, a small, portable benchtop instrument that is easily battery-operated and deployable in the field, as well as a miniature, low-cost, open-source fluorimeter that can be used with any heat block.
This work adds growing evidence to the literature that RPA is in fact suitable for discriminating highly similar DNA sequences. In addition, the demonstration that RPA can be successful following NaOH-based lysis adds to previous findings that very simple lysis methods may be used to obtain genomic DNA from whole blood in preparation for isothermal amplification reactions. Our work also suggests that RPA is tolerant to whole blood lysate when genomic DNA is the target, likely due to the high concentration of genomic DNA present in lysed blood. This finding has important implications for other targets of interest in genomic DNA.
This assay has several limitations that should be addressed in future work. First, two separate reactions are currently required to differentiate A and S templates. Our initial attempts to multiplex were unsuccessful due to a difficult self-dimer region close to the mutation, hindering efforts to design an allele-specific primer in the reverse direction. However, if multiplexing could be accomplished, the cost of the assay would be reduced by almost half. In addition, hemoglobin variants besides βS are not detected with this version. Although the βS allele is responsible for the vast majority of disease burden in resource-limited settings,15,37 the βC allele is the second most common hemoglobin variant and is particularly prevalent in West African populations.38 The assay described here classifies SC genetic material as AS, yet it is important to discriminate between these two genotypes because SC has clinical implications that require treatment, whereas AS does not. Therefore, future work will incorporate the detection of the βC allele and will explore self-contained lateral flow detection, a design that will allow multiplexing.
The nucleic acid test described here can be readily adapted to other point mutations, such as other clinically relevant rare hemoglobin variants that have become geographically concentrated in documented patterns due to global migration patterns (e.g., βD, βE, or βO).39 This test has the potential to streamline diagnosis of SCD patients who have recently undergone a blood transfusion, with a comparable cost to protein-based tests, and could potentially be used to monitor the effectiveness of some emerging gene therapy treatments. Finally, the primer design strategy described could become a platform for other point mutation applications, including other known β thalassemia mutations, drug resistance in highly conserved sequences, and cancer mutations.
Acknowledgments
The authors acknowledge funding received from NIH K23-HL148548-01A1. The authors also acknowledge funding received by our laboratory from the Prevent Cancer Foundation through the 2020 Global Cancer Prevention opportunity and Rice University internal funding, through which other research in the lab has contributed to the findings described in this paper. The authors gratefully acknowledge David Brenes for help with algorithm development and Dr. Meaghan Bond, Alyssa Shapiro, and Norma Estrada for assistance with the clinical study. The authors also thank Dr. Nathaniel Butlin, Dr. Meaghan Bond, and Chelsey Smith for helpful discussions, as well as Alex Kortum, Dr. Yubo Tang, and Imran Vohra for their contributions on the open-source miniature fluorimeter.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c04191.
Description of template DNA used as controls; real-time RPA instrumentation methods; calculation of gene copies expected in a fingerprick blood sample; primer sequences; primer optimization data; assay results with simulated genotypes; assay performance on recently transfused SS patients compared to antibody tests; assay results for β thalassemia and SC genotypes; data from reduced assay volumes from 50 to 5 μL; and discussion of these results (PDF)
The authors declare the following competing financial interest(s): V.N.T. has served as a consultant for Novartis Pharmaceuticals, Global Blood Therapeutics, Forma Therapeutics, and Perkin Elmer. M.N., M.C., K.K., J.C., G.A., and R.R.K. declare no competing interests.
Supplementary Material
References
- Bunn H. F. N. Engl. J. Med. 1997, 337, 762–769. 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- Piel F. B.; Hay S. I.; Gupta S.; Weatherall D. J.; Williams T. N. PLoS Med. 2013, 10, e1001484 10.1371/journal.pmed.1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse S. D.; Odame I.; Atrash H. K.; Amendah D. D.; Piel F. B.; Williams T. N. Am. J. Prev. Med. 2011, 41, S398–S405. 10.1016/j.amepre.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann P. T.; Hoppe C. Blood Cells, Mol., Dis. 2017, 67, 104–113. 10.1016/j.bcmd.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn C. T.; Rogers Z. R.; Buchanan G. R. Blood 2004, 103, 4023–4027. 10.1182/blood-2003-11-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D. C.; Williams T. N.; Gladwin M. T. Lancet 2010, 376, 2018–2031. 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Piety N. Z.; George A.; Serrano S.; Lanzi M. R.; Patel P. R.; Noli M. P.; Kahan S.; Nirenberg D.; Camanda J. F.; Airewele G.; Shevkoplyas S. S. Sci. Rep. 2017, 7, 45488 10.1038/srep45488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter J.; Telen M. J.; Hoppe C.; Roberts C. L.; Kim J. S.; Yang X. BMC Med. 2015, 13, 225 10.1186/s12916-015-0473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele C.; Sinski A.; Asibey J.; Hardy-Dessources M. D.; Elana G.; Brennan C.; Odame I.; Hoppe C.; Geisberg M.; Serrao E.; Quinn C. T. Am. J. Hematol. 2019, 94, 39–45. 10.1002/ajh.25305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed W.; Lane P. A.; Lorey F.; Bojanowski J.; Glass M.; Louie R. R.; Lubin B. H.; Vichinsky E. P. J. Pediatr. 2000, 136, 248–250. 10.1016/S0022-3476(00)70110-7. [DOI] [PubMed] [Google Scholar]
- Michlitsch J.; Azimi M.; Hoppe C.; Walters M. C.; Lubin B.; Lorey F.; Vichinsky E. Pediatr. Blood Cancer 2009, 52, 486–490. 10.1002/pbc.21883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart L. R.; Ambrose E. E.; Raphael K. C.; Hokororo A.; Kamugisha E.; Tyburski E. A.; Lam W. A.; Ware R. E.; McGann P. T. Ann. Hematol. 2018, 97, 239–246. 10.1007/s00277-017-3182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankanja R.; Kadhumbula S.; Tagoola A.; Geisberg M.; Serrao E.; Balyegyusa S. Am. J. Hematol. 2019, 94, E164–E166. 10.1002/ajh.25458. [DOI] [PubMed] [Google Scholar]
- Nnodu O.; Isa H.; Nwegbu M.; Ohiaeri C.; Adegoke S.; Chianumba R.; Ugwu N.; Brown B.; Olaniyi J.; Okocha E.; Lawson J.; Hassan A. A.; Diaku-Akinwumi I.; Madu A.; Ezenwosu O.; Tanko Y.; Kangiwa U.; Girei A.; Israel-Aina Y.; Ladu A.; Egbuzu P.; Abjah U.; Okolo A.; Akbulut-Jeradi N.; Fernandez M.; Piel F. B.; Adekile A. Blood Cells, Mol. Dis. 2019, 78, 22–28. 10.1016/j.bcmd.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimlich J. B.; Chipoka G.; Kamthunzi P.; Krysiak R.; Majawa Y.; Mafunga P.; Fedoriw Y.; Phiri A.; Key N. S.; Ataga K. I.; Gopal S. Br. J. Haematol. 2016, 174, 325–329. 10.1111/bjh.13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piety N. Z.; Yang X.; Kanter J.; Vignes S. M.; George A.; Shevkoplyas S. S. PLoS One 2016, 11, e0144901 10.1371/journal.pone.0144901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra K. K.; Patel P.; Bhukhanvala D. S.; Shah A.; Ghosh K. Anal. Biochem. 2017, 537, 93–98. 10.1016/j.ab.2017.06.014. [DOI] [PubMed] [Google Scholar]
- Latorra D.; Campbell K.; Wolter A.; Hurley J. M. Hum. Mutat. 2003, 22, 79–85. 10.1002/humu.10228. [DOI] [PubMed] [Google Scholar]
- Ugozzoli L. A.; Latorra D.; Pucket R.; Arar K.; Hamby K. Anal. Biochem. 2004, 324, 143–152. 10.1016/j.ab.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Ye S.; Dhillon S.; Ke X.; Collins A. R.; Day I. N. Nucleic Acids Res. 2001, 29, E88 10.1093/nar/29.17.e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itonaga M.; Matsuzaki I.; Warigaya K.; Tamura T.; Shimizu Y.; Fujimoto M.; Kojima F.; Ichinose M.; Murata S. PLoS One 2016, 11, e0151654 10.1371/journal.pone.0151654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng B. Y. C.; Wee E. J. H.; Woods K.; Anderson W.; Antaw F.; Tsang H. Z. H.; West N. P.; Trau M. Anal. Chem. 2017, 89, 9017–9022. 10.1021/acs.analchem.7b01685. [DOI] [PubMed] [Google Scholar]
- Huang S.; Do J.; Mahalanabis M.; Fan A.; Zhao L.; Jepeal L.; Singh S. K.; Klapperich C. M. PLoS One 2013, 8, e47095 10.1371/annotation/45250927-10e7-4545-8fdf-066e688cc125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes S. A.; Bishop J. D.; Lafleur L.; Buser J. R.; Lutz B.; Yager P. Lab Chip 2015, 15, 2647–2659. 10.1039/C5LC00317B. [DOI] [PubMed] [Google Scholar]
- Fan A.; Byrnes S.; Klapperich C. Methods Mol. Biol. 2013, 949, 403–411. 10.1007/978-1-62703-134-9_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.; Hyun J. C.; Yang S. Sci. Rep. 2015, 5, 15167 10.1038/srep15167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan A. V.; Ramachandran S.; Vigil G. D.; Yager P.; Böhringer K. F. Lab Chip 2012, 12, 174–181. 10.1039/C1LC20622B. [DOI] [PubMed] [Google Scholar]
- Rodriguez N. M.; Linnes J. C.; Fan A.; Ellenson C. K.; Pollock N. R.; Klapperich C. M. Anal. Chem. 2015, 87, 7872–7879. 10.1021/acs.analchem.5b01594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauk M.; Liu C.; Song J.; Bau H. Microarrays 2015, 4, 474–489. 10.3390/microarrays4040474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke R. M.; McKenna J. P.; Cox C.; Coyle P. V.; Shields M. D.; Fairley D. J. J. Microbiol. Methods 2016, 129, 103–108. 10.1016/j.mimet.2016.08.016. [DOI] [PubMed] [Google Scholar]
- Di Giusto D. A. Nucleic Acids Res. 2004, 32, e32. 10.1093/nar/gnh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein S. L. Cold Spring Harbor Perspect. Med. 2013, 3, a011700 10.1101/cshperspect.a011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillis L.; Siverson J.; Lee A.; Cantera J.; Parker M.; Piepenburg O.; Lehman D. A.; Boyle D. S. Mol. Cell. Probes 2016, 30, 74–78. 10.1016/j.mcp.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudbeck L.; Dissing J. Biotechniques 1998, 25, 588–592. 10.2144/98254bm09. [DOI] [PubMed] [Google Scholar]
- Soejima M.; Egashira K.; Kawano H.; Kawaguchi A.; Sagawa K.; Koda Y. J. Mol. Diagn. 2011, 13, 334–339. 10.1016/j.jmoldx.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coole J.; Kortum A.; Tang Y.; Vohra I.; Kundrod K.; Natoli M.; Richards-Kortum R.; Maker Y. J. Vis. Exp. 2021, 168, e62148 10.3791/62148. [DOI] [PubMed] [Google Scholar]
- McGann P. T.; Williams A. M.; Ellis G.; McElhinney K. E.; Romano L.; Woodall J.; Howard T. A.; Tegha G.; Krysiak R.; Murray Lark R.; Louise Ander E.; Mapango C.; Ataga K. I.; Gopal S.; Key N. S.; Ware R. E.; Suchdev P. S. Blood Adv. 2018, 2, 3035–3044. 10.1182/bloodadvances.2018023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel R. L.; Fabry M. E.; Steinberg M. H. Blood Rev. 2003, 17, 167–178. 10.1016/S0268-960X(03)00003-1. [DOI] [PubMed] [Google Scholar]
- Mburu J.; Odame I. Int. J. Lab. Hematol. 2019, 41, 82–88. 10.1111/ijlh.13023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.