

Abstract
Cholesterol serves critical roles in enveloped virus fusion by modulating membrane properties. The glycoprotein (GP) of Ebola virus (EBOV) promotes fusion in the endosome, a process that requires the endosomal cholesterol transporter NPC1. However, the role of cholesterol in EBOV fusion is unclear. Here we show that cholesterol in GP-containing membranes enhances fusion and that the membrane-proximal external region and transmembrane domain (MPER/TM) of GP interacts with cholesterol via several glycines in GP2 TM, notably G660. Compared to wild-type counterparts, a G660L mutation caused a more open angle between MPER and TM in a MPER/TM construct, higher probability of stalling at hemifusion for GP2 proteoliposomes, and lower cell entry of virus-like particles (VLPs). VLPs with depleted cholesterol show reduced cell entry and VLPs produced under cholesterol-lowering statin conditions show less entry than respective controls. We propose that cholesterol-TM interactions affect structural features of GP2 thereby facilitating fusion and cell entry.
Keywords: Ebola virus, cholesterol, protein-cholesterol interaction, statin, membrane fusion
Introduction
Ebola virus (EBOV) is a life-threatening pathogen known to cause hemorrhagic fevers (1, 2). The 2013–2016 epidemic in West Africa caused more than 10,000 casualties, and left survivors suffering with post EBOV disease (EVD) syndromes, such as increased intraocular pressure (3, 4). It is unknown whether the virus was cleared from survivors, or if it persisted in specific tissues (5, 6). While several treatment options are being explored against this deadly pathogen (7), a vaccine (Ervebo by Merck;https://www.fda.gov/news-events/press-announcements/first-fda-approved-vaccine-prevention-ebola-virus-disease-marking-critical-milestone-public-health) and a therapeutic cocktail of three monoclonal antibodies (Inmazeb by Regeneron; https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-ebola-virus) have been approved only recently.
The EBOV glycoprotein (GP) is the sole protein expressed in the viral membrane and is consequently the major target for neutralizing antibodies (8, 9). GP, consisting of GP1 and GP2 subunits, is responsible for binding to cell surface attachment factors and mediating cellular entry through endosomes (10–12). Conformational changes in GP are thought to provide energy to overcome the barrier to membrane fusion (13–16). To do this GP binds to its endosomal receptor, Niemann-Pick C1 (NPC1), which in conjunction with low pH and perhaps other factors (10, 17–19), but independent of its cholesterol transporting activity (19), triggers conformational changes that unclamp the hold that GP1 bears on GP2 and releases the fusion loop (FL) in GP2, so that it can embed in the target membrane. Subsequent folding of GP into a hairpin structure causes the viral and endosomal membranes to merge (18, 20–24) (Fig. 1A).
Figure 1. Effect of cholesterol on EBOV membrane fusion.
A) Schematic of EBOV GP-mediated fusion highlighting the two membrane interacting domains. The fusion loop (FL) (blue) initiates fusion by interacting with the host cell membrane eventually pulling it towards the MPER/TM (red) embedded in the viral membrane. B) The EBOV GP2 sequence is shown using the same color scheme. Heptad repeats 1 and 2 that ultimately form the six-helix bundle extend from residues 560–595 and 615–631, respectively. Residues mutated in constructs used in this study are indicated with boxes. C) The relative extent of lipid mixing of GP2 proteoliposomes with target protein-free liposomes as observed by dequenching of FRET-paired lipid probes in the target liposome membrane. The fluorescent lipids NBD-DOPE and Rh-DOPE were included, each at 1.5 mol% in the liposome membrane to allow lipid mixing to be determined by dequenching of NBD fluorescence. The cholesterol content was varied in either the target or GP2 containing membrane while the cholesterol content in the other membrane was kept constant at 20% cholesterol. Data are mean and s.d. of triplicate measurements of the same reconstituted sample. D) Increasing amounts of VLPs, treated as indicated to modulate cholesterol content, were added to HEK293T cells and entry was monitored. Data are mean and s.d. of entry values from triplicate experiments of the indicated input from the same VLP preparation. Based on cholesterol to protein ratios (see online Methods), the cholesterol content of the VLPs, relative to untreated VLPs, were: cholesterol-depleted VLPs, 1 %; cholesterol-repleted VLPs, 94 %; mock-treated VLPs, 79 %. Data for panels C and D are available as Source Data.
The FL of EBOV GP engages host endosomal membranes that contain NPC1, a cholesterol transporter. And, the transmembrane domain of GP is anchored in the viral membrane, which has a lipid composition reflecting that of the plasma membrane of producer cells and hence, a high concentration of cholesterol (10, 19, 25). Lipid compositions of fusing membranes, including the amount of cholesterol, can strongly influence the energy barrier and thereby the efficiency of fusion (26–30), and cholesterol has been shown to be important for the entry of several viruses (31–37). For examples, for HIV and influenza cholesterol in both viral and target membranes is critical for efficient fusion (29, 32, 34, 36–38). However, a mechanistic understanding of any direct interaction of cholesterol with a viral GP is still lacking, and how cholesterol affects the participating membranes and the energy barrier for EBOV fusion have not been explored.
Despite this limited knowledge about potential cholesterol effects on EBOV fusion, there has been considerable debate about employing statins, cholesterol lowering drugs, to combat EBOV. Since EBOV disease is associated with endothelial barrier loss, Fedson et al. suggested benefits of using statins to suppress morbidity and mortality, due to their ability to improve endothelial integrity (in addition to lowering cholesterol levels) (39). In addition, the statin simvastatin emerged in a screen of FDA-approved drugs for activity against EBOV in cell-based assays. Shrivastava-Ranjan et al. subsequently found that statins block EBOV infection of cells in vitro, but the effects were not in the initial round of virus entry (40). Instead relatively high concentrations (20 and 50 μM) of lovastatin led to the production of EBOV particles defective for infecting a next set of cells (41).
To address the question if cholesterol in the viral or target membrane directly affects membrane fusion and thereby viral entry and if it interacts directly with any portion of EBOV GP, we investigated cholesterol’s role in these processes by setting up reconstituted single particle fusion and cell entry assays. The functional studies were complemented with biophysical assays for cholesterol binding to EBOV GP and measurements of tertiary structural changes in EBOV GP in response to cholesterol binding. Our results show that cholesterol physically interacts with the transmembrane domain of EBOV GP; that cholesterol in the viral membrane promotes membrane fusion and cell entry; and that producing viral-like particles in the presence of a few μM of the statin lovastatin results in particles that are severely compromised in their ability to enter cells.
Results
Cholesterol in viral membrane enhances EBOV membrane fusion
Fusion of the EBOV membrane envelope with an endosomal membrane is essential for release of the viral genetic material into the cell cytoplasm to initiate an infection. As the only protein exposed on the external side of the viral membrane, the EBOV glycoprotein (GP) is solely responsible for receptor binding and membrane fusion. By analogy with other class I fusion proteins, the driving force for fusion is thought to be provided by conformational changes in the GP2 subunit that ultimately generate a six-helical bundle (14) (Fig. 1A and B). In other systems lipid compositions of the participating membranes, including their cholesterol content, have been found to play pivotal roles in membrane fusion (26–29, 31–33, 42–45). We therefore explored the role of cholesterol in viral surrogate and target membranes, for EBOV GP-mediated membrane fusion.
The effect of cholesterol on GP2-mediated EBOV fusion was first investigated by reconstituting full length GP2 into proteoliposomes (46) and assessing lipid mixing using a bulk fusion assay. Note that the endosomal receptor for EBOV, NPC1 (10, 19), is not required in this ‘minimal EBOV fusion system’, as there is no need to release the GP1 clamp to expose GP2; GP2 proteoliposome fusion to liposomes is triggered solely by exposure to low pH (also see below). Förster resonance energy transfer (FRET) between Rhodamine (Rh) and nitrobenzoxadiazole (NBD) labeled lipids in target liposomes provided a measure of GP2-mediated lipid mixing, which was induced by lowering the pH to 5.5. Increasing cholesterol in the GP2 (viral analog) membrane enhanced fusion; increasing cholesterol in the target membrane caused a smaller enhancing effect (Fig 1C). Next, we tested the importance of cholesterol in the virus membrane, in the context of the full GP1/GP2 trimer, using a virus-like particle (VLP) cell entry assay. Depletion of VLP cholesterol with methyl-β-cyclodextran (MβCD) reduced entry efficiency by ~10-fold while cholesterol replenishment reversed this effect by ~50% (Fig 1D).
Cholesterol interacts with the EBOV MPER/TM
NMR spectroscopy was used to probe potential interactions between the membrane proximal extracellular region and transmembrane domain (MPER/TM) of GP2 and cholesterol. To do this an EBOV MPER/TM construct was prepared and incorporated into DMPC/DHPC (1:2 mol/mol, q=0.5) bicelles. Native gel electrophoresis showed that this construct behaves as a monomer in dodecyl-phosphocholine (DPC) micelles, but forms a higher oligomer, presumably a trimer, in DMPC/DHPC bicelles (Extended Data Fig.1). 1H-15N TROSY-HSQC spectra of EBOV MPER/TM in bicelles were then acquired at pH 5.5, in the absence or presence of varying concentrations of up to 20 mol% cholesterol (relative to DMPC) in the bicelles (Fig 2A). Three glycines (G655, G657, G660) and two nearby residues (I652, T659) in the TM domain showed chemical shifts upon adding cholesterol, with G660 (red box in Fig. 1B) showing the most prominent change. This glycine is in a GXXXA motif that was previously suggested to bind cholesterol (47). In order to test the hypothesis that G660 is required for the interaction of cholesterol with the EBOV MPER/TM, we mutated this glycine to leucine, (GXXXA to LXXXA), and performed the same solution NMR experiments. No chemical shift deviations were seen for G660L MPER/TM in the presence of cholesterol (Fig 2B). Fig 2C displays the chemical shift perturbations of WT and G660L induced by cholesterol along the sequence of EBOV MPER/TM.
Figure 2. Cholesterol interaction with the EBOV MPER/TM in DMPC/DHPC bicelles.
NMR spectra of WT (A) and G660L (B) EBOV MPER/TM in q=0.5 DMPC/DHPC bicelles were acquired in 0, 5, 10, 15, and 20 mol% cholesterol relative to DMPC. Red arrows denote peaks with the greatest changes. C) Chemical shift perturbations (Δδcomp= [ΔδHN2+ (ΔδN/6.5)2]1/2) in response to addition of 20 mol% cholesterol. WT (blue); G660L (green). D) Amide proton intensity changes with 5 mol% (relative to DMPC) cholesterol or 5 mol% 3β-doxyl-5α-cholestane added to EBOV MPER/TM in DMPC/DHPC bicelles. Red, intensity ratios between cholesterol and cholesterol-free bicelles. Blue, intensity ratios between doxyl-cholestane and cholesterol bicelles. The error bars in panel D were propagated from S/N of peak pairs and calibrated with duplicate measurements of the bicelle sample without cholesterol (analog).
In order to examine whether there is a direct interaction of cholesterol with EBOV MPER/TM, we employed the spin-labeled cholesterol analog 3β-doxyl-5α-cholestane and measured the attenuation of the amide proton signals along the sequence in comparison with the effects of cholesterol (Fig 2D and Extended Data Fig.2). Backbone amide proton resonances were dramatically reduced by the nitroxide paramagnetic spin probe of doxyl-cholestane in the region from W651 to A664, which comprises the headgroup contacting region of the EBOV MPER/TM structure including the GXXXA motif (residues 660–664) at the beginning of the TM domain. The amide proton signals of the remainder of the TM domain were also attenuated but to a lesser extent. These data further support the notion that cholesterol is in close contact with the TM domain of EBOV MPER/TM.
Secondary structure analyses using chemical shift indices of Cα atoms showed the same basic helix-break-helix structure for the G660L MPER/TM that had been previously described for the WT MPER/TM (46) (Extended Data Fig.3A). Local backbone motions measured by heteronuclear NOEs, and by spin-lattice and spin-spin relaxation showed similar values and trends for G660L and WT (Extended Data Fig.3B–D).
The absence of apparent differences in the secondary structure and backbone dynamics of the G660L compared to the WT MPER/TM does not exclude a potential change in tertiary structure. To test this hypothesis, we performed double electron electron resonance (DEER) experiments to measure distances and thus the angle between the two main segments of the EBOV MPER/TM structure. The MPER/TM construct was double labeled with S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl) methyl methanesulfonothioate (MTSL) at residues 643 and 670 and incorporated into DMPC/DHPC bicelles. The distance between the two probes in the mutant (G660L) MPER/TM was increased compared to the WT (Fig 3A) indicating that the angle between the MPER and TM domains is wider in G660L than that same angle previously determined by DEER in the WT MPER/TM structure (46) (Fig 3B and C). Observing DEER signals of MPER/TM reconstituted into POPC liposomes revealed similar trends (Extended Data Fig.4).
Figure 3. Distance distribution obtained using DEER on double-MTSL-labeled WT and G660L EBOV MPER/TM in DMPC/DHPC bicelles.
A) Cartoon showing the position of the labels (N643C and A670C, red) in the EBOV MPER/TM structure (46). The position of G660 is also indicated (blue). Increasing the distance between the two label positions increases the indicated angle between the MPER and TM helices of MPER/TM. B) Background-corrected DEER data for WT and G660L EBOV MPER/TMs. C) Distance distributions obtained from DEER data of WT (green) and G660L (purple) EBOV MPER/TMs. The G660L mutation results in a shift towards longer distance components consistent with an opening of the MPER/TM angle. [AU: please define what is shown in graph, i.e., what the colored area and lines represent]
Viral membrane cholesterol enhances fusion and cell entry
Imaging single fusion events allows the states of docking and fusion to be distinguished (27, 35–37, 43, 48–51). Previously, influenza hemagglutinin incorporated into a planar supported lipid bilayer (SLB) was shown to fuse with labeled liposomes in a pH dependent manner (52). Similarly, we incorporated EBOV GP2 into SLBs and monitored its interaction with individual liposomes dually-labeled with a membrane (DiD) and a self-quenched content (sulforhodamine B) dye using TIRF microscopy. Three distinct types of events were recorded: (A) docking: liposomes bind to the GP2-SLB but do not fuse (Fig 4A). (B) fusion: liposomes bind and then, after some time, fuse with the GP2-SLB (Fig 4B). (C) stalled hemifusion: liposomes bind and undergo lipid mixing, but no content mixing (Fig. 4C). Stalled hemifusion events have previously been characterized in the context of SNARE-mediated fusion as off pathway events that infrequently proceed to full fusion (27). Similarly, during GP2-mediated fusion further changes in membrane or content dye distribution were seldom observed for the hemifusion events. The characteristic fluorescence traces originating from membrane dyes that distinguish stalled hemifusion from full fusion allow imaging to be done using only a membrane dye, as shown in Fig 4D.
Figure 4. Intensity traces of peak pixel intensity of DiD membrane label (red) and sulforhodamine B content label (green) of single vesicle events.
(A) Binding (docking) of a liposome to a supported lipid bilayer containing EBOV GP2 is marked by the increase in DiD intensity seen when the vesicle enters the TIRF field. The sulforhodamine B label is self-quenched and no intensity change is observed. (B) A fusion event followed by binding of a liposome to the GP2-containing supported bilayer. After ~0.5 s the membrane dye diffuses into the supported bilayer at the onset of fusion. The sulforhodamine B label dequenches at the moment of fusion and diffuses away into the cleft under the supported bilayer. (C) A stalled hemifusion event where the membrane dye in the outer leaflet of the liposome diffused into the supported bilayer (loss of ~55% of the intensity) while no change in the content dye is observed. (D) Full and hemifusion events can be observed using particles labeled with only a membrane dye (as shown for Rh-DOPE) by observing if all or half of the membrane dye diffuses into the supported bilayer. (E) Effects of pH and a double-point mutation in the fusion loop, LIAA (purple); see Figure 1A) on liposome binding to GP2-containing supported lipid bilayers. Experiments were conducted as shown in Extended Data Fig 5 and saturation values were determined by single exponential fits. Data points show mean and s.e.m. of saturation values for triplicate experiments performed on 3 different supported bilayers. (F) Cumulative fusion delay times for liposomes bound to GP2-containing supported bilayers at pH 5.5 or 6.5. Data were collected from 4 different supported bilayers. Data for panels E and F are available as Source Data.
The pH dependence of GP2-mediated fusion was explored utilizing this single particle assay. Injecting liposomes at different pH values and recording the total fluorescence within the evanescence field revealed a requirement for low pH for liposomes to efficiently bind to the GP2-SLB (Fig 4E and Extended Data Fig.5). This lipid binding activity is likely due to pH dependent conformational changes in GP2 including changes in the structure of the fusion loop (53), that lead to a quite deep insertion of the GP2 fusion loop into the liposome membrane (53). In order to test that the fusion loop of GP2 is responsible for the pH dependent docking, we utilized a mutant (LIAA) with two substitutions in the fusion loop (L529A and I544A; boxed in blue in Fig 1B). Indeed, replacing WT GP2 with the LIAA mutant of GP2, in the single liposome fusion assay, abolished liposome binding at pH 5.5, which correlates with the previously shown inability of the LIAA EBOV GP fusion loop to bind tightly to target membranes and to mediate fusion and cell entry (21). Single liposome assays allow the quantification of fusion kinetics by measuring the delay time between docking and the onset of fusion. Cumulative distribution functions of these delay times showed that ~40% of the liposomes that bind, fuse within ~0.2 seconds of docking (Fig 4F). Changing the pH to 6.5 greatly lowered the number of docking events (Fig 4E), but for the liposomes that did bind, the fusion kinetics did not change significantly.
Examining the cholesterol dependence of fusion of liposomes (50 nm diameter) with GP2-containing SLBs revealed that in the absence of cholesterol there were few full fusion events, but many stalled hemifusion events (Fig 5A, B). Increasing cholesterol increased the number of full fusion events and decreased the number of stalled hemifusion events (Fig 5A, B). The distribution of the delay times between docking and fusion revealed biphasic kinetics with initial fast and later slow components. The fast component appeared most sensitive to the presence of cholesterol (Fig 5B). A similar enhancing effect of cholesterol was seen for GP2 proteoliposomes (100 nm diameter) interacting with protein-free SLBs (Fig 5C, D). Cross-linking of GP2 in proteoliposomes showed that GP2 formed trimers in POPC:POPG (85:15) bilayers (Extended Data Fig.6).
Figure 5. The cholesterol dependence of fusion.
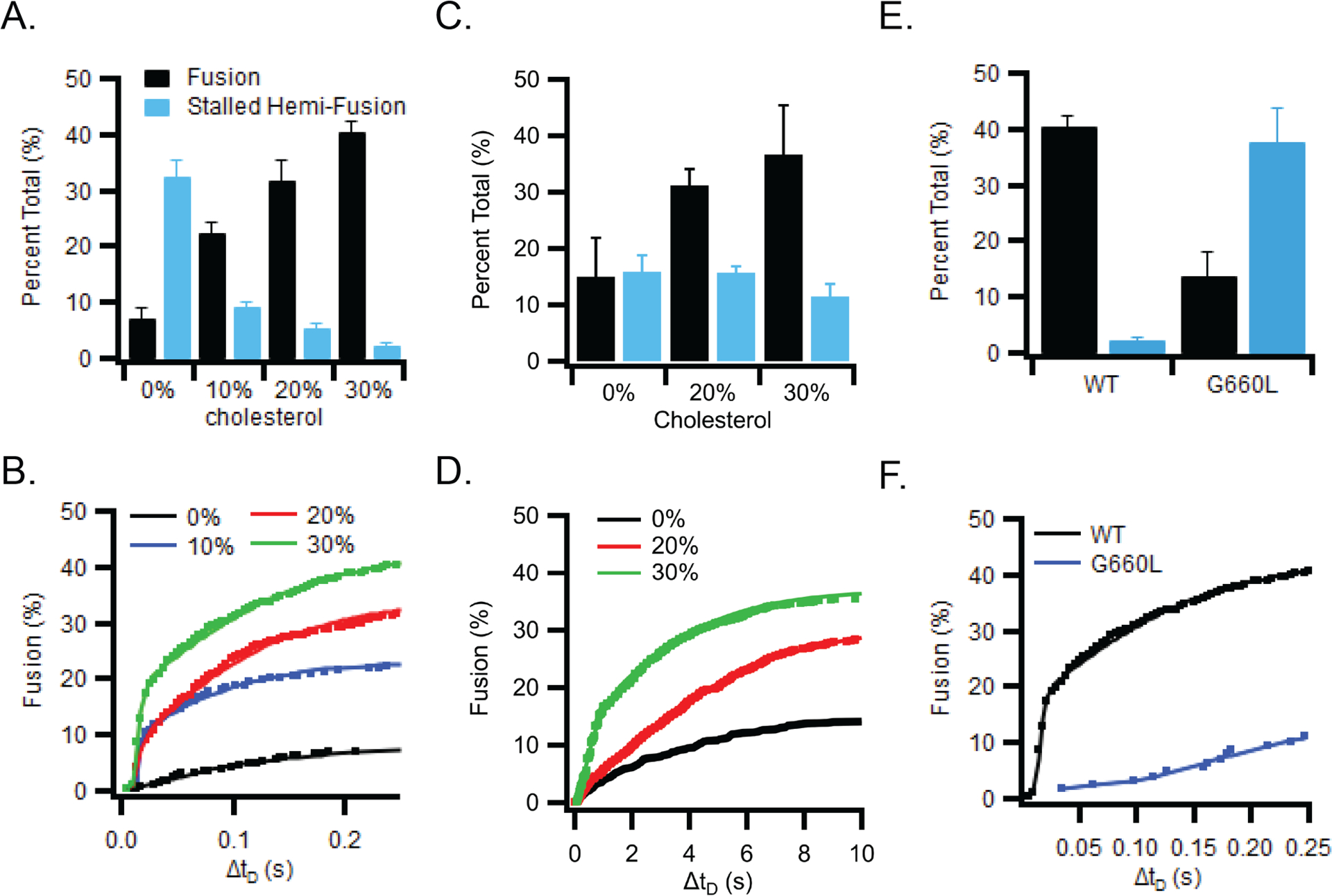
(A) Fusion of liposomes (50 nm diameter) with supported bilayers containing GP2 (lipid:protein ratio of 1000) and increasing amounts of cholesterol. (B) Cumulative distribution of the delay times from the time of binding until the time of fusion of liposomes to GP2-containing supported bilayers with increasing amounts of cholesterol. (C) Fusion of GP2-proteolipsomes (100 nm diameter, POPC plus 1 mol% Rh-DOPE) plus 0, 20, or 30 mol% cholesterol with protein-free supported bilayers (77:20:3 POPC:Chol:DPS). (D) Cumulative distribution of fusion delay times for GP2 proteoliposomes with 0, 20, or 30 mol% cholesterol. (E) Fusion of liposomes to supported bilayers containing wt (black) or G660L (blue) GP2. (F) Cumulative distribution of the delay times from the time of binding until the time of fusion for liposomes to supported bilayers containing wt (black) or G660L (blue) GP2. Bar graphs in panels A, C and E show mean and s.e.m. from 3–6 independent experiments under each condition. Typically, 1000–2000 particles were collectively counted in these experiments. The kinetic fusion data in panels B, D, and F were collected from the same experiments pooling all approximately 1000–2000 events from the 3–6 independent bilayers under each condition. Data including number of repeats and particles measured under each condition are available as Source Data.
The mutant G660L, which showed decreased cholesterol binding to the TM domain (Fig 2B, C) and a wider angle between the MPER and TM domains (Fig 3) behaved differently than WT GP2 when incorporated (as G660L GP2) into an SLB and assayed for fusion with liposomes. With 30% cholesterol in the SLB, G660L GP2 displayed less full fusion and more stalled hemifusion (Fig 5 E,F). The fusion kinetics were also strongly reduced for G660L compared to WT GP2 (Fig 5F). Trimerization of GP2 in proteoliposomes was not impaired by the G660L mutation (Extended Data Fig.6).
We found that the G660L mutation also affects virus entry into HEK293T cells. Filamentous EBOV VLPs were prepared with WT or G660L EBOV GP (trimers of GP1/GP2) and then tested in a VLP entry assay. VLPs with G660L GP were significantly compromised for target cell entry (Fig 6A), despite equivalent incorporation of G660L GP into VLPs (GP/VP40 ratio = 1.36 +/− 0.47 relative to the GP/VP40 ratio for WT GP; based on 3 samples of each prep run on a single SDS gel and blotted for GP and VP40). Over a 5-fold range of VLP inputs, the cell entry efficiency of G660L VLPs was less than 20% that of WT VLPs (Fig 6A). The cholesterol content of G660L VLPs was measured to be 76 % that of WT VLPs. Overall, the results in Fig 5 E,F and Fig 6A verify that residue G660 in the GP2 TM domain is critical for full fusion to occur efficiently.
Figure 6. A mutation (G660L) in the cholesterol binding domain in EBOV GP2 or preparation of EBOV VLPs from statin-treated cells inhibits the entry capacity of EBOV GP VLPs.
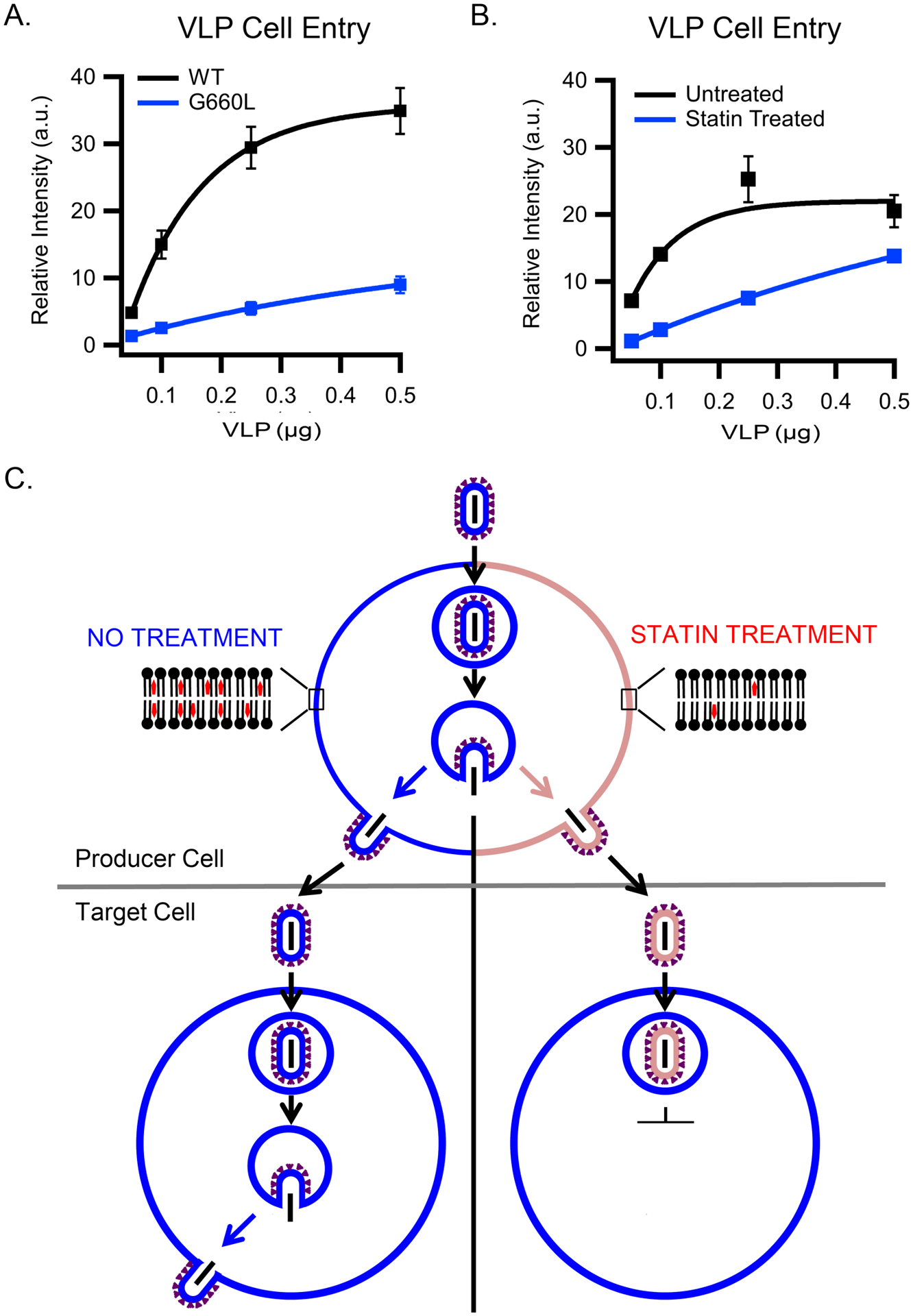
(A) VLPs were prepared with WT or G660L EBOV GP and assayed for VLP cell entry as in the legend to Fig 1D. Data are mean and s.d. of cell entry values from triplicate samples of the indicated input of the indicated VLP preparation. (B) Increasing amounts of VLPs produced from untreated (black) or statin-treated (blue) cells were added to HEK293T cells and assayed (in triplicate) and analyzed for cytoplasmic entry as in (A). The experiment displayed is representative of 2 independent experiments. (C) Schematic depicting (top) VLP production from untreated (left) or statin-treated (right) producer cells and (bottom) their subsequent entry into fresh (untreated) HEK293T cells. Blue denotes VLPs and plasma membranes with normal content of cholesterol; pink depicts VLPs and plasma membranes depleted for cholesterol (by virtue of statin treatment). Data for panels A and B are available as Source Data.
EBOV VLPs from statin-treated cells show reduced entry
Based on the previous results, we hypothesized that VLPs budded from cholesterol-depleted cells would have less cholesterol in their membrane and therefore be impaired in membrane fusion and hence entry capacity. To test this hypothesis, we used a cholesterol-lowering statin to reduce the cholesterol content of the HEK293T cell plasma membrane from which VLPs are produced by budding. Two sets of VLPs were prepared in parallel: one set from mock (DMSO) treated and the second from statin-treated HEK293T cells. VLPs produced from statin treated cells exhibited markedly lower entry efficiency than VLPs produced from mock treated cells (Fig 6B). VLPs prepared from statin-treated cells contained 22% the level of cholesterol and approximately equivalent levels of GP (Extended Data Fig.7) compared to VLPs produced from mock-treated cells.
The findings in Fig 6 are consistent with the demonstration that treatment of VLPs (from non-statin-treated producer cells) with MBCD decreases both their cholesterol content and entry efficiency (Fig 1D). We propose that, because of their lowered cholesterol content, VLPs with WT GP produced from statin treated cells enter target cells less efficiently than VLPs produced from mock treated cells (Fig 6C) because less cholesterol is available to bind to the GP2 TM, and therefore influence the structure of GP for optimal fusion. An inference is that VLPs with G660L GP should show the same (low) entry capacity whether or not depleted of cholesterol or when produced in untreated or statin-treated cells; we have not performed these experiments here because G660L VLP entry is already significantly suppressed compared to WT and a further reduction of entry capacity after cholesterol extraction would be difficult to detect in a conclusive manner. However, such experiments may be interesting to explore in the future.
Discussion
The lipid compositions of the membranes that partner in viral fusion events are important (29), but this aspect has not been explored in detail for EBOV. In this study, we probed the role of cholesterol in EBOV GP-mediated fusion and cell entry. We found that cholesterol, in the membrane of three different viral membrane surrogates, promotes EBOV fusion and entry. NMR experiments revealed that the initiating glycine (G) in a G660XXXA motif in the EBOV GP transmembrane domain promotes an interaction with cholesterol, and DEER experiments indicated that changing G660 to leucine (L) alters the tertiary structure of the membrane proximal external region-transmembrane (MPER/TM) domain of EBOV GP, which correlates with the diminished fusion and entry activity of GP constructs with the G660L mutation. Single particle fusion studies employing supported lipid bilayers (SLBs) and TIRF microscopy further showed that cholesterol in viral membrane surrogates promotes EBOV GP-mediated membrane fusion, and that a mutation (G660L) in the cholesterol binding site greatly reduces membrane fusion. These latter findings correlated with a reduced capacity of EBOV-GP VLPs with either reduced cholesterol content or the G660L mutation to enter cells.
Cholesterol is required in host cell membranes at both the entry and assembly stages of viral lifecycles (54). A cholesterol requirement for virus entry has been demonstrated for HIV (35–37, 55), influenza (31, 32, 56–58), herpes simplex (33), and human parainfluenza 3 (59) viruses. A cholesterol recognition amino acid consensus (CRAC) motif in the MPER of the gp41 subunit has been implicated in HIV’s cholesterol dependence (60, 61), but the mechanism by which this motif promotes fusion has not been elucidated. Influenza also has been identified as having a cholesterol consensus motif (CCM) at the N-terminal end of the TMD of its hemagglutinin protein (62). Hemolysis, lipid mixing, and cell-cell fusion were impaired by disrupting this motif in hemagglutinin (63). While cholesterol has been identified as necessary for entry of other viruses (33, 59), their sites for cholesterol binding and mechanism(s) by which cholesterol enhances their fusion activity remain unknown.
Here, after demonstrating a fusion-promoting role for cholesterol in the membrane of EBOV, we identified a cholesterol binding motif in EBOV GP2, employing NMR chemical shift analysis, with residues I652, G655, G657, T659 and G660, located in the region connecting the MPER and TM domains of MPER/TM, interacting with cholesterol (Fig 2C), with confirmation by PRE data (Fig 2D and Extended Data Fig 2). G660 is part of a GXXXA motif at the beginning of the TM domain of GP2 that was previously mutated to investigate the cell detachment effect caused by ectopic expression of EBOV GP (47); that effect was found to be cholesterol-dependent, and the authors reported greater photoactivatable cholesterol binding to WT (GXXXA) than LXXXL GP. A second study asked whether the GXXXA motif is involved in the ability of EBOV to counter the effect of tetherin, a cellular interferon induced anti-viral protein; that response was found to be cholesterol independent (64), and the authors reported an ~50% reduction in cell entry of pseudoviruses bearing LXXXL GP as well as a delay of infection, and an ~1 log decrease in titer of recombinant VSV bearing LXXXL GP. Our study provides direct biophysical evidence that GXXXA in the TM domain of EBOV GP indeed contributes to cholesterol binding. We also showed that cholesterol in the viral membrane and G660, are critical for optimal EBOV fusion and entry. The GXXXA sequence (G[V/I]IIA) is present in all annotated Ebolavirus GP2 sequences, whereas Marburgvirus GPs have the sequence LSIAV at the equivalent position.
How does cholesterol in the viral membrane enhance Ebola fusion? Cholesterol can affect the function of membrane proteins in cholesterol-containing bilayers by imparting general effects on membrane structure (e.g., curvature, phase behavior, and thickness) and also by binding to specific protein motifs, which include CRACs, CARCs, and CCMs (29). We propose that cholesterol has a direct effect on EBOV GP. Specifically, we suggest that by binding to the GXXXA motif in the EBOV TMD (Fig 2), cholesterol affects the environmental structure of the MPER/TM domain (the angle between the two helical segments is altered; Fig 3), which in turn affects the ability of GP to mediate fusion (Figs 5 and 6). This could be by preferentially positioning the MPER/TM for binding to the fusion loop (46) at the ‘fold-back’ stage of fusion, by altering the structure of the EBOV ectodomain and its consequent fusion ability, and/or by changing the structure or properties of the bilayer surrounding GP. The latter possibilities clearly require future experimentation.
In addition to its role in the viral membrane, cholesterol in the target membrane may also promote fusion. In the case of influenza, cholesterol in the target membrane was shown to promote full fusion and minimize stalled hemifusion reactions (32). For HIV (65) cholesterol is important in the target membrane (66), with fusion occurring preferentially at boundaries between cholesterol-rich ordered (Lo) and cholesterol-poor disordered (Ld) domains within target membranes (35, 37). For EBOV, an early study showed that depletion of cholesterol from target membranes inhibits infection by EBOV GP pseudoviruses (67). And, a 16-residue peptide from within the EBOV fusion loop was found to bind preferentially to cholesterol-rich domains in target membranes (68). However, we observed only a mild enhancing effect of cholesterol in the target membrane (Fig 1C).
Our results also shed new light on how cholesterol lowering statins may reduce the burden of EBOV infections. For example, the statin simvastatin emerged in a screen of FDA-approved drugs for activity against EBOV in cell-based assays (40). We found that VLPs produced from cells treated with 4 μM lovastatin had a reduced cholesterol content and consequently were impaired in their ability to enter cells by membrane fusion. Thus, our findings, using a lower dose (4 μM) of lovastatin, concur with the previous conclusion (41) that treatment of producer cells with lovastatin (albeit higher doses; 20 and 50 μM) generates EBOV particles with reduced entry and infection capacity (Fig 6). Shrivastava-Ranjan et al. (41) attributed the lowered infectivity of EBOV particles produced in the presence of 20–50 μM lovastatin to reduced incorporation of EBOV GP, whereas we posit that a major cause of reduced infectivity in particles produced in the presence of (4 μM) lovastatin is reduced cholesterol binding to the GXXXA motif in the TMD of GP and its consequently reduced fusion activity. The collective results ((39, 41), and this study) suggest that statins warrant consideration as part of a multi-component treatment for patients infected with EBOV.
In conclusion, this study provides direct biophysical evidence for a cholesterol binding (GXXXA) motif in the TM domain of the EBOV GP as well as molecular insight into how cholesterol, via this motif, alters the structure of the MPER/TMD region of GP. This change may affect the structural transformation of the EBOV GP ectodomain (and/or the membrane surrounding GP) that is required to facilitate membrane fusion. Our findings also bear on the mechanism by which statins may result in the production of fusion-inefficient EBOV particles and hence on considerations of whether and how (e.g., as part of a drug cocktail (69)) to use statin drugs to treat EBOV-infected patients.
Online Methods
Expression and purification of MPER/TM domain and GP2 constructs
Expression and purification of EBOV MPER/TM was carried out as described previously with slight modification (46). Briefly, cells were grown to OD600? 0.8 at 37°C and incubated 4 hrs after induction with the final concentration of 1 mM IPTG. MPER/TM was then purified as follows. Cells were harvested and resuspended in 20 mM Tris, 300 mM NaCl, and 10% sucrose, pH 8, sonicated and then centrifuged for 1 hr at 40,000xg at 15°C. The resulting pellets containing inclusion bodies were solubilized with 20 mM Tris, 300 mM NaCl, 8 M urea and 1% Triton X-100, pH 8. Following the same sonication and centrifugation steps, the supernatant was incubated with 5 mL of Ni-NTA beads for 1 hr. The beads were washed with an 8 M to 0 M urea gradient to remove urea and Trition X-100. Thrombin (in 20 mM Tris, 300 mM NaCl, 1% n-octyl-β-D-glucopyranoside [β-OG]) was then added to the Ni-NTA beads and incubated overnight to elute the EBOV MPER/TM.
Preparation of full-length GP2 was carried out as described previously (46) using the previously described pET-24a vector (kanamycin resistant) encoding (N- to C-) a Trp leader protein, a thrombin cleavage site, and Ebola GP2 followed by an N-terminal His-tag; C670 and C672 were mutated to alanine to avoid formation of non-native disulfides (46). One liter of BL21(DE3) cells transformed with the GP2 expression vector was grown to O.D.600 of 0.6–0.8, induced with 1 mM IPTG, and harvested 4 hrs later. The cells were resuspended in 100 mL 20 mM Tris buffer pH 8.0 containing 100 mM NaCl and 10% sucrose, lysed by sonication until homogeneous, and centrifuged for 30 min at 40,000 × g at 10 °C. The pellets, which contained inclusion bodies, were solubilized in 100 mL 20 mM Tris buffer pH 8.0 containing 100 mM NaCl, 8 M urea, and 1% Triton X-100 by sonication and the solution centrifuged as above. The supernatant was collected and incubated with 5 mL of Ni affinity beads for ≥1 hr at 4°C. Finally, a urea gradient (total 500 mL of 8 to 0 M urea in Tris buffer) was applied to the Ni column to remove urea and Triton X-100. The Trp leader protein was then removed by treating resuspended Ni beads with 100 μL of 5 mg/ml thrombin (in 25 mL of DPC buffer: 20 mM Tris buffer pH 8.0 containing 100 mM NaCl and 0.2 % DPC). After 2 hrs (with rotation at RT), the beads were transferred to a column, the GP2 containing eluate was collected, and the beads were washed with another 20 mL of 0.2% DPC buffer thereby collecting all of the GP2 protein. Collected fractions were pooled, concentrated, and run over a Superdex 200 size exclusion column in the final required buffer with 0.2% DPC. For NMR studies, the final buffer was 20 mM Na phosphate pH 7 or pH 5.5 containing 100 mM NaCl and 0.2% DPC. For NMR samples, cells were grown in minimal media containing 15NH4SO4 and 13C-glucose and induction was performed at 25 °C overnight. The mutants of the respective constructs were prepared using the same protocols.
Native gel of MPER/TM
To check the oligomeric state of MPER/TM in DPC micelles and DMPC/DHPC (1:2) bicelles (prepared as for NMR experiments), samples of both were run on a pre-cast 4–16% Bis-Tris native gel (Invitrogen) following the manufacturer’s protocol. Briefly, the gel samples were prepared by adding 0.5 μL of 5% G-250 sample additive (Thermo Fisher), 5 mM DDM and 5 μL of 4X NativePage sample buffer (Invitrogen) to 15 μL of micelle or bicelle samples each containing 15 μg MPER/TM. The samples were then loaded onto the gel and electrophoresis was performed on ice at constant voltage (150 V) for 100 minutes with cold 1X Anode Buffer and 1X Dark Blue Cathode Buffer (Invitrogen).
Cross-linking of GP2 proteoliposomes
POPC:POPG (85:15) proteoliposomes with WT and G660L GP2, with an estimated protein:lipid ratio 1:100, were incubated with 10 mM 3,3’-dithiobis(sulfosuccinimidylpropionate) (DTSSP, Sigma Aldrich) for 0, 15 or 30 mins at room temperature. The reaction was quenched by adding 50 mM Tris pH 7.5. The samples were then boiled in the presence of SDS sample buffer for 5 minutes, and separated by non-reducing 4–20% SDS-PAGE. The gels were stained with silver stain (70).
Incorporation of EBOV MPER/TM into bicelles
The incorporation of EBOV MPER/TM into DMPC/DHPC bicelles was performed as described previously (46). In brief, the EBOV MPER/TM in β-OG buffer (20 mM Tris, 300 mM NaCl, 1% β-OG) was mixed with the appropriate amount of DMPC (16 mg for final 250uL) and dialyzed to remove β-OG. Dialysis was performed 3 days against NMR buffer (25 mM sodium phosphate, 100 mM NaCl) starting at pH 7.5 on the first day, 6.5 on the second day, and 5.5 on the third day at 4°C. Then, EBOV MPER/TM containing liposomes were concentrated and DHPC was added to form bicelles. The q value for all bicelles was 0.5. To prepare bicelles with varying mol% cholesterol or nitroxide cholesterol analog, 3b-doxyl-5a-cholestane (Sigma-Aldrich), preformed bicelles were added to tubes to which specific amounts of cholesterol (or analog) had previously been dried down from their respective stocks in chloroform (under a stream of nitrogen followed by vacuum desiccation). The tubes were then subjected to cycles of freeze-thawing (~5 cycles or until a clear solution was obtained) using liquid N2 and hot (initially ~100 °C) water.
NMR experiments
After obtaining EBOV MPER/TM incorporated bicelles, all NMR spectra of the MPER/TM domain were acquired at 45°C on a Bruker Avance III 800 MHz spectrometer. 1H-15N TROSY based HSQC, 15N-heteronuclear NOEs, 15N T1 and T2 experiments were performed as described (46) and all NMR data were processed using NMRPipe and SPARKY (71, 72). Chemical shift perturbations, defined as Δδcomp= [ΔδHN2+ (ΔδN/6.5)2]1/2 were plotted as a function of residues. The error bars in the intensity ratio plots were propagated from S/N of peak pairs and calibrated with duplicate measurements of the bicelle sample without cholesterol (analog).
EBOV MPER/TM in bicelles and DEER experiments
The EBOV MPER/TM construct was labeled with an MTSL nitroxide probe at both 643 and 670 positions using the method described previously (46). Double MTSL labeled EBOV MPER/TM was incorporated into DMPC/DHPC bicelles following the method described above. Approximately 15 μL of an EBOV MPER/TM bicelle sample with 15% deuterated glycerol was loaded into quartz capillary tubes with an inner diameter of 1.1 mm and outer diameter of 1.6 mm (Vitrocom). Samples were frozen in liquid nitrogen and loaded into a Bruker E580 spectrometer with an EN5107D2 resonator. DEER data were collected at Q-band and 80 K using a dead-time free four-pulse sequence with 16 step phase cycling. Pump and observe pulses were separated by 75 MHz. The program LongDistances by Christian Altenbach (UCLA) was used for the removal of background functions from initial V(t)/V(0) data and the model-free fitting regime was used to extract distance distributions from the resulting F(t)/F(0) (73). The value of the smoothing factor for the fits was 30.
Bulk lipid mixing assay
The fusion assay of full-length GP2 proteoliposomes was carried out as described previously (46). Briefly, POPC/POPG (85/15) liposomes were prepared in HMA buffer (10 mM HEPES/MES/Tris, 100 mM NaCl, pH 7.4) (HEPES: 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid); MES: 2-(N-morpholino)ethanesulfonic acid hydrate, 4-morpholineethanesulfonic acid) by extrusion through 100-nm polycarbonate filters. Then, 20 mM Tris, 100 mM NaCl, pH 8, buffer containing 1% β-OG was added to the extruded liposomes to a final concentration of 0.125% β-OG and then incubated at room temperature for at least 1 hr. Then, EBOV GP2 in DPC was added to give an estimated protein:lipid ratio of 1:100 and incubated for at least 1 hr before dialysis. Extensive dialysis against HMA buffer was performed to remove all detergent and to incorporate EBOV GP2 into the liposomes. Cholesterol concentration was changed from 0 to 30 mol% relative to total lipid concentration while keeping the POPG concentration constant. Labeled liposomes containing 1.5 mol % of both Rh- DOPE and NBD-DOPE were prepared by extrusion as described above.
To measure fusion, fluorescent liposomes and unlabeled proteoliposomes were mixed at a ratio of 1:9 in HMA buffer. Relief of NBD-Rh FRET was recorded at 37°C as a function of time with mixing between each reading. Excitation and emission wavelengths were set at 460 nm and 538 nm, respectively. Percent lipid mixing (fusion) was determined as the fraction of the maximal FRET relief observed after addition of 2% Triton X-100 at the end of each reaction. All mutant lipid mixing data were normalized to the extent of lipid mixing observed with the WT protein.
Preparation of protein-free liposomes
POPC lipids and the desired membrane dye (1 mol% of either rhodamine-DOPE or DiD) were pipetted into a glass tube at the desired concentration from chloroform solutions and then dried down using N2 gas. Lipids were further dried in a vacuum desiccator for >1 hr. Lipids were then suspended in the desired volume of buffer (for content dye experiments, 50 mM sulforhodamine B was included in the buffer that the POPC/DiD liposomes were suspended in). The lipid suspension was freeze-thawed in liquid nitrogen and a water bath 5 times, followed by extrusion through a 100 nm pore polycarbonate filter (Avestin). After extrusion, the membrane labeled (Rhodamine-DOPE) liposomes were used within 36 hrs. The content/membrane labeled liposomes were run over a G-50 superfine Sephadex size exclusion column (GE Healthcare, Piscataway, NJ) to remove the free sulforhodamine B label and the collected liposomes were used within 36 hr.
Preparation of planar supported bilayers containing EBOV GP2
Planar supported bilayers containing EBOV GP2 were prepared by the Langmuir-Blodgett/vesicle fusion technique as described in previous studies (52, 74). Quartz slides were cleaned by dipping in sulfuric acid/hydrogen peroxide (3:1) for 15 mins using a Teflon holder. Slides were then rinsed thoroughly in water. The first leaflet of the bilayer was prepared by Langmuir-Blodgett transfer directly onto the quartz slide using a Nima 611 Langmuir-Blodgett trough by applying POPC and the indicated amount of cholesterol from a chloroform solution to the air-water interface of the trough. Where indicated 3 mol% of 1,2-dimyristoyl-sn-gycero-3-phosphatidylethanolamine-PEG3400-triethoxysilane (DPS) was included in the Langmuir film to produce a polymer-cushioned supported bilayer. After allowing the solvent to evaporate for 10 min, the monolayer was compressed at a rate of 10 cm2/min to reach a surface pressure of 32 mN/m. After equilibration for 5 to 10 min, a clean quartz slide was rapidly (68 mm/min) dipped into the trough and slowly (5 mm/min) withdrawn while a computer maintained a constant surface pressure and monitored the transfer of lipids with head groups down onto the hydrophilic substrate. POPC liposomes with the indicated amount of cholesterol reconstituted with GP2 at a lipid/protein ratio of 1000:1 was incubated with the Langmuir-Blodgett monolayer in a perfusable holding cell to form the outer leaflet of the planar supported bilayer. After incubation of the proteoliposomes for 2 hrs, the excess proteoliposomes were removed by washing with 10 ml of HMA buffer (10 mM Hepes, 10 mM MES, 10 mM NaOAc and 100 mM, NaCl, pH 7.4).
Single particle binding and fusion assay
For planar supported bilayer binding assays and single particle fusion assays, holding cells with the planar supported bilayers were mounted on a Zeiss Axiovert 25 fluorescence microscope (Carl Zeiss, Thornwood, NY) equipped with a 63x water immersion objective (Zeiss; N.A. 0.95) and prism-based TIRF illumination. The light source was an OBIS 532 LS laser from Coherent Inc. (Santa Clara, Ca.). Fluorescence was observed through a 610 nm bandpass filter (D610/60; Chroma, Brattleboro, VT) by an electron multiplying CCD (DU- 860E; Andor Technologies). The EMCCD was cooled to −70°C. The prism-quartz interface was lubricated with glycerol to allow easy translocation of the holding cell on the microscope stage. The beam was totally internally reflected at an angle of 72° from the surface normal, resulting in an evanescent wave that decays exponentially with a characteristic penetration depth of ~100 nm. An elliptical area of 250 × 65 μm was illuminated. The laser intensity, shutter, and camera were controlled by a homemade program written in LabVIEW (National Instruments, Austin, TX).
The binding assay was performed by injecting POPC liposomes (10 μM lipid with 1 mol% rhodamine-DOPE). One image was taken before injecting liposomes, and images were taken every 30 sec thereafter for 5 mins. This was done at the pH values indicated in the text and the total intensity of the field of view was recorded.
Single particle fusion data were obtained by injecting 500 nM lipid of POPC:Chol:Rh-DOPE (79:20:1) containing liposomes. This was first done at pH 7.4 to confirm that no docking or fusion occurred at neutral pH. The planar supported bilayers were then washed and the solution replaced with one containing pH 5.5 (or pH 6.5 as indicated in the text) buffer and a second replacement with 500 nM lipid of liposomes in the same buffer. Five movies were recorded for 1 min for each bilayer at a rate of 4 frames/ms. Single-particle fusion data were analyzed using a program written in LabVIEW (National Instruments). Stacks of images were filtered by a moving average filter. The maximum intensity for each pixel over the whole stack was projected on a single image. Liposomes were located in this image by a single-particle detection algorithm described in Kiessling et al. (75). The peak (central pixel) and mean fluorescence intensities of a 5-pixel × 5-pixel area around each identified center of mass were plotted as a function of time for all particles in the image series. The exact time points of docking and fusion were determined from the central pixel similar to previous work (48, 50). Fusion times of individual events were determined from the time of binding to the time of fusion and cumulative distribution fusion vs. time plots were constructed from typically 200–300 events depending on the condition. Fusion efficiencies were determined from the number of liposomes that underwent fusion compared with the total number of liposomes that bound.
For content and membrane labeled single liposome fusion experiments, planar supported bilayers were mounted on a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss, Thornwood, NY) equipped with a 63x water immersion objective (Zeiss; N.A. 0.95) and a prism-based TIRF illumination system. The beams of a 514 nm line of an argon ion laser (Innova 90C, Coherent, Palo Alto, CA), controlled through an acousto-optic modulator (Isomet, Springfield, VA), and a diode laser (Cube 640, Coherent) emitting light at 640 nm were directed (72° from the normal) into a prism above the quartz slide to illuminate the sample by total internal reflection with a characteristic penetration depth of ~102 and ~130 nm for the 514 and the 640 nm lasers, respectively. The prism-quartz interface was lubricated with glycerol to allow easy translocation of the sample cell on the microscope stage. An OptoSplit (Andor-Technologies, South Windsor, CT) was used to separate the fluorescence from the lipid and soluble dyes. Fluorescence signals were recorded by an electron-multiplying charge-coupled device camera (iXon DV887ESC-BV, Andor, Belfast, UK). The EMCCD camera was cooled to −70°C. The laser intensities, light-blocking shutters, and cameras were controlled by a program written in LabVIEW (National Instruments, Austin, TX). POPC liposomes with a sulforhodamine B content label and a DiD membrane label were injected at a concentration of 0.5 μM lipid. Images were recorded at a frame rate of 20 ms (the slower acquisition rate was in order to observe the content dye). Single fusion events were analyzed as described above. Behavior of the content dye verified that full fusion only occurred when all of the membrane dye diffused away from the fusion site, as seen previously for SNARE-mediated fusion (27). All experimental conditions were repeated at least 4 times with at least 4 independent samples and errors are standard errors of the mean. For each individual experiment, data from on the order of 100 individual particles were collected.
VLP production and cell entry assays
The production of EBOV VLPs and the assay to measure their entry into cells were performed as described previously (46). In brief, HEK293T/17 cells were co-transfected with plasmids encoding VP40, β-lactamase-VP40, mCherry-VP40, and WT or G660L EBOV GP with a C-terminal V5 tag using polyethylenimine. The ratio of respective plasmids was 1:2.25:2.25:1.5. After 48 hr at 37°C, the cell medium was collected, cleared of debris by centrifugation, and the cleared media (containing VLPs) was subjected to ultracentrifugation through a 20% sucrose cushion. VLP pellets were resuspended in HM buffer (20 mM HEPES, 20 mM MES, 130 mM NaCl, pH 7.4) overnight and subsequently repelleted. Final VLP pellets were resuspended in 10% sucrose-HM buffer, aliquoted, and stored at −80 °C.
Methyl-β-cyclodextrin (MβCD) was used to deplete/replenish cholesterol in VLPs after production. For depletion, purified VLPs were incubated with 20 mM MβCD at 37°C for 30 min. After incubation, VLPs were centrifuged at 13,000 rpm (15,700 xg) at 4°C for 2–3 hr. The supernatant was then removed, and the pellet was washed with HM buffer and centrifuged again. After the wash step, VLPs were resuspended in 10% sucrose-HM buffer overnight at 4°C. For replenishment, cholesterol-depleted VLPs were incubated at 37°C for 30 mins with cholesterol saturated MβCD (5 mM final concentration). After incubation, VLPs were subjected to the purification protocol described above.
Production of EBOV VLPs from statin-treated cells was performed as follows. HEK 293T/17 cells were pretreated with DMEM containing 0.01% FBS and 4 μM of lovastatin or DMSO (control) 24 hrs prior to transfection, and the cells were maintained in this media (DMEM with 0.01% FBS and 4 μM of lovastatin or DMSO) throughout VLP production. VLPs were then harvested (after 48 hrs) and purified as described above.
All VLPs were analyzed for total protein concentrations (by BCA assay) and for their content of EBOV GP and VP40 (by Western blotting). Where indicated, the cholesterol content of VLPs was determined using the Amplex cholesterol kit following the manufacturer’s instructions.
To measure cell entry, VLPs were bound to HEK293T/17 cells by centrifugation (250 × g) for 1 hr at 4°C and then allowed to enter for 3 hrs at 37°C in a 5% CO2 incubator. The fluorescent CCF2-AM β-lactamase substrate was then incubated with the cells for 1 hr at RT in the dark. Cells were washed and incubated overnight in the dark at room temperature. Cells were lifted, fixed, and analyzed by flow cytometry on a FACSCalibur flow cytometer. The degree of the shift in fluorescence from green to blue was used to measure entry (67). All data were analyzed using FlowJo software.
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Figure 1. Native PAGE analysis of EBOV MPER/TM in micelles and bicelles.

4–16% polyacrylaminde gel of EBOV MPER/TM in DPC micelle and q=0.5 DMPC/DHPC bicelle, stained with Coomassie Blue.
Extended Data Figure 2. Attenuation of amide 1H NMR peak intensities at increasing concentrations of the nitroxide free-radical cholesterol analog 3β-doxyl-5α-cholestane.
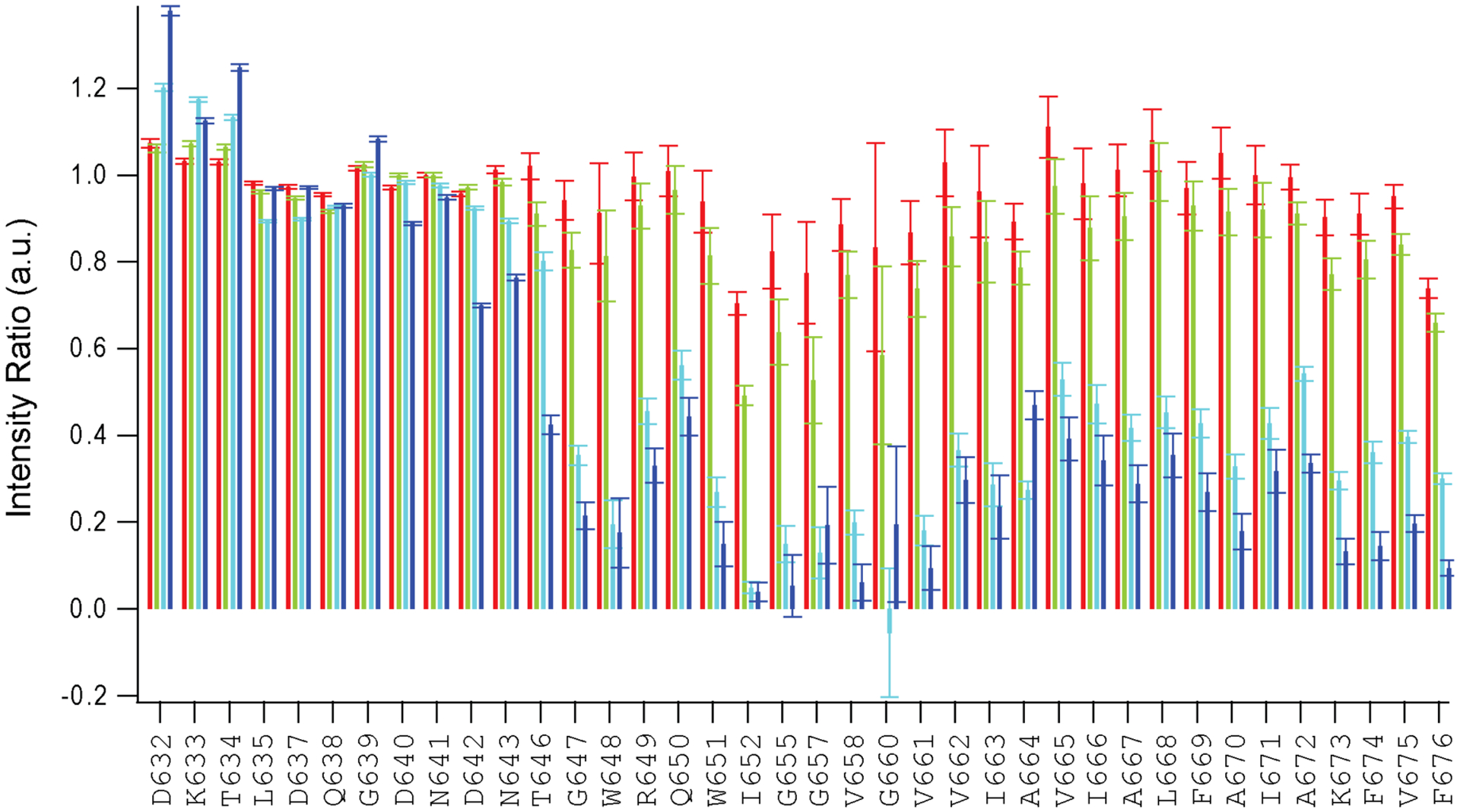
Titration of the paramagnetic cholesterol analog into an EBOV MPER/TM q=0.5 DMPC/DHPC bicelle sample. Amide proton intensity ratios between bicelles with 1 (red), 3 (green), 5 (cyan), 10 (blue) mol% 3β-doxyl-5α-cholestane (relative to DMPC) and cholesterol analog free bicelles are plotted.
Extended Data Figure 3. Secondary structure and polypeptide backbone dynamics of EBOV WT and G660L MPER/TM in DMPC/DHPC bicelles.
A) Cα chemical shift index of WT (green) and G660L mutant (purple). Both show a helix-break-helix motif (see also Fig 3). B-D) Backbone dynamics measurements of WT (green) and G660L mutant (purple) showing that the N-terminus is flexible and the TM domain is rigid in both constructs. B) Heteronuclear 15N-NOEs. C) 15N T1 spin-lattice and D) 15N T2 spin-spin relaxation times. All measurements were carried out at 45°C, in pH 5.5 buffer, and in q=0.5 bicelles.
Extended Data Figure 4. Distance distribution obtained using DEER on double-MTSL-labeled EBOV MPER/TM and G660L in POPC liposomes.
A) Background-corrected DEER data for WT (green) and G660L (purple) EBOV MPER/TM in POPC liposomes. B) Distance distributions obtained by a best fit to the data in (A). As seen with the bicelle data (Fig 3), the addition of the G660L mutation causes a shift towards longer distance elements consistent with an opening of the MPER/TM angle. Measurements were performed at pH 5.5.
Extended Data Figure 5. Binding of protein-free liposomes to GP2 in supported lipid bilayers.
Liposomes (5μM, 79:20:1 POPC:Chol:Rh-DOPE, 50 nm diameter) were added to SLBs (80:20 POPC:Chol) containing GP2 (lipid:protein 1000) at time 0 and the fluorescence within in the TIRF field was recorded. The average fluorescence intensities were determined from initial frames and used to determine the density of liposomes on the SLB. Binding was determined as a function of pH and also assessed for the fusion-deficient LIAA mutant at pH 5.5.
Extended Data Figure 6. SDS-PAGE gels of crosslinked WT and G660L GP2 in POPC:POPG (85:15) proteoliposomes.
Samples of WT and G660L GP2 proteoliposomes (each with ~10 μg GP2) were incubated with 10 mM DTSSP for the indicated times at room temperature. After quenching, the samples were run, at the same time, on parallel SDS-PAGE gels, after which proteins were visualized by silver staining. The positions of the monomeric (M), dimeric (D) and trimeric (T) forms of GP2 are indicated with arrows.
Extended Data Figure 7. Western blot of VLPs produced from untreated HEK293T cells (WT) or HEK293T cells treated with 4 μM lovastatin.
1, 2, and 5 μg of each type of VLP was applied to the gel. After probing for EBOV GP and VP40 (see Methods), the relative amounts of GP to VP40 were calculated for each lane. When normalized to WT VLPs, the ratio of GP:VP40 in Statin VLPs was 1.1 +/− 0.07 that in WT VLPs based on analysis of all lanes. The ratio was 0.86 +/− 0.07 based on analysis of the last 2 lanes of each gel. VLPs produced in cells treated with 20 or 50 μM lovastatin (statin) showed reduced GP incorporation (see ref 41).
Acknowledgments
This work was supported by NIH grants R01 AI030557 (to LKT) and R01 AI114776 (to JMW) and by Human Frontiers Science Program grant RGP0055/2015 (to LKT). D.A.N. was supported by NIH training grant T32 GM080186.
Footnotes
Code Availability
The code of programs used to collect and analyze the single particle fusion data is available from the authors upon request.
Data Availability
NMR chemical shift data for EBOV MPER/TM WT and G660L have been deposited in the Biomolecular Magnetic Resonance Data Bank under accession number 50584 and 50591, respectively.
Competing interest
The authors declare no competing interests.
References
- 1.Feldmann H, Geisbert TW, Ebola haemorrhagic fever. Lancet 377, 849–862 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoenen T, Groseth A, Falzarano D, Feldmann H, Ebola virus: unraveling pathogenesis to combat a deadly disease. Trends Mol Med 12, 206–215 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Carod-Artal FJ, [Illness due the Ebola virus: epidemiology and clinical manifestations within the context of an international public health emergency]. Rev Neurol 60, 267–277 (2015). [PubMed] [Google Scholar]
- 4.Carod-Artal FJ, Post-Ebolavirus disease syndrome: what do we know? Expert Rev Anti Infect Ther 13, 1185–1187 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Scott JT et al. , Post-Ebola Syndrome, Sierra Leone. Emerg Infect Dis 22, 641–646 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burki TK, Post-Ebola syndrome. Lancet Infect Dis 16, 780–781 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Maxmen A, Science under fire: Ebola researchers fight to test drugs and vaccines in a war zone. Nature 572, 16–17 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Ito H, Watanabe S, Takada A, Kawaoka Y, Ebola virus glycoprotein: proteolytic processing, acylation, cell tropism, and detection of neutralizing antibodies. J Virol 75, 1576–1580 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maruyama T et al. , Ebola virus can be effectively neutralized by antibody produced in natural human infection. J Virol 73, 6024–6030 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carette JE et al. , Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondratowicz AS et al. , T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc Natl Acad Sci U S A 108, 8426–8431 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakurai Y et al. , Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995–998 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison SC, Viral membrane fusion. Nat Struct Mol Biol 15, 690–698 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White JM, Delos SE, Brecher M, Schornberg K, Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43, 189–219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malashkevich VN et al. , Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-A resolution. Proc Natl Acad Sci U S A 96, 2662–2667 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC, Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol Cell 2, 605–616 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Feneant L, Szymanska-de Wijs KM, Nelson EA, White JM, An exploration of conditions proposed to trigger the Ebola virus glycoprotein for fusion. PLoS One 14, e0219312 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das DK et al. , Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol 18, e3000626 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cote M et al. , Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477, 344–348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White JM, Whittaker GR, Fusion of Enveloped Viruses in Endosomes. Traffic 17, 593–614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory SM et al. , Ebolavirus entry requires a compact hydrophobic fist at the tip of the fusion loop. J Virol 88, 6636–6649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H et al. , Ebola Viral Glycoprotein Bound to Its Endosomal Receptor Niemann-Pick C1. Cell 164, 258–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fels JM, Spence JS, Bortz RH 3rd, Bornholdt ZA, Chandran K, A Hyperstabilizing Mutation in the Base of the Ebola Virus Glycoprotein Acts at Multiple Steps To Abrogate Viral Entry. mBio 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrison JS, Higgins CD, Chandran K, Lai JR, Designed protein mimics of the Ebola virus glycoprotein GP2 alpha-helical bundle: stability and pH effects. Protein Sci 20, 1587–1596 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adu-Gyamfi E et al. , Host Cell Plasma Membrane Phosphatidylserine Regulates the Assembly and Budding of Ebola Virus. J Virol 89, 9440–9453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Churchward MA, Rogasevskaia T, Hofgen J, Bau J, Coorssen JR, Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J Cell Sci 118, 4833–4848 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Kreutzberger AJ, Kiessling V, Tamm LK, High cholesterol obviates a prolonged hemifusion intermediate in fast SNARE-mediated membrane fusion. Biophys J 109, 319–329 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee DE, Lew MG, Woodbury DJ, Vesicle fusion to planar membranes is enhanced by cholesterol and low temperature. Chem Phys Lipids 166, 45–54 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Yang ST, Kreutzberger AJB, Lee J, Kiessling V, Tamm LK, The role of cholesterol in membrane fusion. Chem Phys Lipids 199, 136–143 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siegel DP, The Gaussian curvature elastic energy of intermediates in membrane fusion. Biophys J 95, 5200–5215 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biswas S, Yin SR, Blank PS, Zimmerberg J, Cholesterol promotes hemifusion and pore widening in membrane fusion induced by influenza hemagglutinin. J Gen Physiol 131, 503–513 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chlanda P et al. , The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat Microbiol 1, 16050 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wudiri GA, Schneider SM, Nicola AV, Herpes Simplex Virus 1 Envelope Cholesterol Facilitates Membrane Fusion. Front Microbiol 8, 2383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Domanska MK et al. , Hemagglutinin Spatial Distribution Shifts in Response to Cholesterol in the Influenza Viral Envelope. Biophys J 109, 1917–1924 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang ST, Kiessling V, Simmons JA, White JM, Tamm LK, HIV gp41-mediated membrane fusion occurs at edges of cholesterol-rich lipid domains. Nat Chem Biol 11, 424–431 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang ST, Kiessling V, Tamm LK, Line tension at lipid phase boundaries as driving force for HIV fusion peptide-mediated fusion. Nat Commun 7, 11401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang ST et al. , HIV virions sense plasma membrane heterogeneity for cell entry. Sci Adv 3, e1700338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun X, Whittaker GR, Role for influenza virus envelope cholesterol in virus entry and infection. J Virol 77, 12543–12551 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fedson DS, Jacobson JR, Rordam OM, Opal SM, Treating the Host Response to Ebola Virus Disease with Generic Statins and Angiotensin Receptor Blockers. MBio 6, e00716 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansen LM et al. , A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci Transl Med 7, 290ra289 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Shrivastava-Ranjan P et al. , Statins Suppress Ebola Virus Infectivity by Interfering with Glycoprotein Processing. MBio 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiessling V et al. , A molecular mechanism for calcium-mediated synaptotagmin-triggered exocytosis. Nat Struct Mol Biol 25, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Domanska MK, Kiessling V, Tamm LK, Docking and fast fusion of synaptobrevin vesicles depends on the lipid compositions of the vesicle and the acceptor SNARE complex-containing target membrane. Biophys J 99, 2936–2946 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreutzberger AJB et al. , Asymmetric Phosphatidylethanolamine Distribution Controls Fusion Pore Lifetime and Probability. Biophys J 113, 1912–1915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu KN, Boxer SG, Target Membrane Cholesterol Modulates Single Influenza Virus Membrane Fusion Efficiency but Not Rate. Biophys J 118, 2426–2433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J et al. , Structure of the Ebola virus envelope protein MPER/TM domain and its interaction with the fusion loop explains their fusion activity. Proc Natl Acad Sci U S A 114, E7987–E7996 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hacke M et al. , Inhibition of Ebola virus glycoprotein-mediated cytotoxicity by targeting its transmembrane domain and cholesterol. Nat Commun 6, 7688 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Domanska MK, Kiessling V, Stein A, Fasshauer D, Tamm LK, Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem 284, 32158–32166 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Floyd DL, Ragains JR, Skehel JJ, Harrison SC, van Oijen AM, Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A 105, 15382–15387 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiessling V, Domanska MK, Tamm LK, Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J 99, 4047–4055 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kreutzberger AJB et al. , Reconstitution of calcium-mediated exocytosis of dense-core vesicles. Sci Adv 3, e1603208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinterdorfer P, Baber G, Tamm LK, Reconstitution of membrane fusion sites. A total internal reflection fluorescence microscopy study of influenza hemagglutinin-mediated membrane fusion. J Biol Chem 269, 20360–20368 (1994). [PubMed] [Google Scholar]
- 53.Gregory SM et al. , Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc Natl Acad Sci U S A 108, 11211–11216 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schroeder C, Cholesterol-binding viral proteins in virus entry and morphogenesis. Subcell Biochem 51, 77–108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lai AL, Moorthy AE, Li Y, Tamm LK, Fusion activity of HIV gp41 fusion domain is related to its secondary structure and depth of membrane insertion in a cholesterol-dependent fashion. J Mol Biol 418, 3–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Domanska MK, Wrona D, Kasson PM, Multiphasic effects of cholesterol on influenza fusion kinetics reflect multiple mechanistic roles. Biophys J 105, 1383–1387 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goronzy IN, Rawle RJ, Boxer SG, Kasson PM, Cholesterol enhances influenza binding avidity by controlling nanoscale receptor clustering. Chem Sci 9, 2340–2347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zawada KE, Wrona D, Rawle RJ, Kasson PM, Influenza viral membrane fusion is sensitive to sterol concentration but surprisingly robust to sterol chemical identity. Sci Rep 6, 29842 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang Q, Liu P, Chen M, Qin Y, Virion-Associated Cholesterol Regulates the Infection of Human Parainfluenza Virus Type 3. Viruses 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vincent N, Genin C, Malvoisin E, Identification of a conserved domain of the HIV-1 transmembrane protein gp41 which interacts with cholesteryl groups. Biochim Biophys Acta 1567, 157–164 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Chen SS et al. , Identification of the LWYIK motif located in the human immunodeficiency virus type 1 transmembrane gp41 protein as a distinct determinant for viral infection. J Virol 83, 870–883 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Vries M, Herrmann A, Veit M, A cholesterol consensus motif is required for efficient intracellular transport and raft association of a group 2 HA from influenza virus. Biochem J 465, 305–314 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Hu B, Hofer CT, Thiele C, Veit M, Cholesterol binding to the transmembrane region of a group 2 HA of Influenza virus is essential for virus replication affecting both virus assembly and HA’s fusion activity. J Virol 10.1128/JVI.00555-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brinkmann C et al. , The Tetherin Antagonism of the Ebola Virus Glycoprotein Requires an Intact Receptor-Binding Domain and Can Be Blocked by GP1-Specific Antibodies. J Virol 90, 11075–11086 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Graham SM, Impact of HIV on childhood respiratory illness: differences between developing and developed countries. Pediatr Pulmonol 36, 462–468 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE, Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res Hum Retroviruses 17, 1009–1019 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Yonezawa A, Cavrois M, Greene WC, Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: involvement of cytoskeletal proteins and enhancement by tumor necrosis factor alpha. J Virol 79, 918–926 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freitas MS et al. , Measuring the strength of interaction between the Ebola fusion peptide and lipid rafts: implications for membrane fusion and virus infection. PLoS One 6, e15756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dyall J et al. , Identification of Combinations of Approved Drugs With Synergistic Activity Against Ebola Virus in Cell Cultures. J Infect Dis 218, S672–S678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Merril CR, Dunau ML, Goldman D, A rapid sensitive silver stain for polypeptides in polyacrylamide gels. Anal Biochem 110, 201–207 (1981). [DOI] [PubMed] [Google Scholar]
- 71.Delaglio F et al. , NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 72.Goddard T, Kneller DG., SPARKY: NMR Assignment and Intergration Software. University of California, San Francisco: Version 3.114. (2008). [Google Scholar]
- 73.Jeschke G, Polyhach Y, Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys Chem Chem Phys 9, 1895–1910 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Kalb E, Frey S, Tamm LK, Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim Biophys Acta 1103, 307–316 (1992). [DOI] [PubMed] [Google Scholar]
- 75.Kiessling V, Crane JM, Tamm LK, Transbilayer effects of raft-like lipid domains in asymmetric planar bilayers measured by single molecule tracking. Biophys J 91, 3313–3326 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]