

Abstract
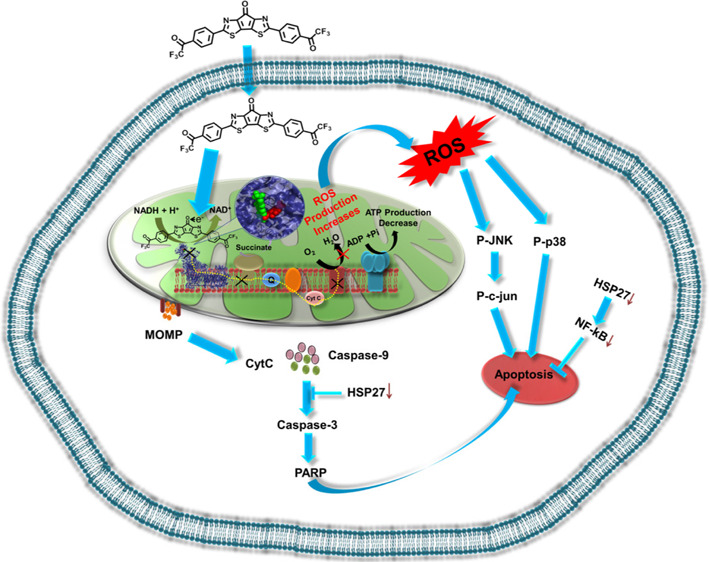
Here, we demonstrate an interesting strategy of modulating mitochondrial reactive oxygen species (ROS) using the organic electron acceptor molecule carbonyl-bridged bithiazole attached with bis-trifluoroacetophenone (BBT). This molecule was found to affect complex I activity. It has the propensity to bind close to the flavin mononucleotide site of complex I of mitochondria where it traps electron released from nicotinamide adenine dinucleotide (NADH) and elevates intracellular ROS, which suggests that the bridged carbonyl in BBT plays a crucial role in the acceptance of electron from NADH. We understand that the potential of the NADH/NAD+ redox couple and low-lying LUMO energy level of BBT are compatible with each other, thus favoring its entrapment of released electrons in complex I. This effect of BBT in ROS generation activates JNK and p38 stress-dependent pathways and resulted in mitochondrial-dependent apoptotic cell death with the reduction in expression of several important cyto-protecting factors (Hsp27 and NFκB), indicating its potential in inhibition of cancer cell relapse. Intriguingly, we found that BBT is not a P-glycoprotein substrate, which further reveals its excellent anticancer potential. This study enlightens us on how the power of electron acceptor ability became an emerging strategy for modulation of intracellular function.
Introduction
Reactive oxygen species (ROS) plays a pivotal role in cancer genesis and progression. Mitochondria are the key metabolically active cellular organelles for the production of intracellular ROS in most mammalian cells.1 In general, complexes I, II, III, and ubisemiquinone of the electron transport chain (ETC) are involved in the production of superoxide (·O2–). NADH is the most important electron donor in eukaryotic cells, and the associated electron transport chain is as follows: NADH → complex I → Q → complex III → cytochrome c → complex IV → O2, where complexes I, III, and IV are proton pumps, while Q and cytochrome c are mobile electron carriers, and the electron acceptor is molecular oxygen. Inhibition of electron transport or inhibition of complex I2 results in the decrease in adenosine triphosphate (ATP) production due to prevention of the conversion of NADH into usable energy such as ATP, which causes the enhancement of ROS generation (free radicals). Other electron carriers such as glutathione (GSH) and CoQH2 (reduced coenzyme Q) carry out regulation of ·O2– in the cell. The extent of ·O2–production solely depends upon NADH/NAD+ and CoQH2/CoQ ratios and the local O2 concentration. For instance, higher production of ·O2–results to higher matrix NADH/NAD+ and reduced FMN (flavin mononucleotide) site in complex I.3 Then, mitochondrial superoxide dismutase (SOD2) can reduce ·O2– to form H2O2. Both ·O2– and H2O2 pass through the membrane into the cytoplasm via a VDAC (voltage-dependent anion channel) and aquaporin.4 Then, cellular ROS can damage cellular proteins, lipids, and DNA and results in mutagenesis.5 ROS regulates the cellular redox signaling6 where excessive ROS induces apoptotic cell death following the JNK/p38 pathway or autophagic pathway.7 Dysregulation of mitochondrial ROS generation in cancer plays important role in tumor growth and malignancy.8 A certain level of ROS is pro-tumorigenic in cancer cells and documented in cancer progression. For instance, mitochondrial ROS promotes mutagenesis of both mitochondrial and nuclear DNA that induces the progression of cancer.9 This ROS-dependent tumorigenesis results in a higher concentration (near to the threshold value) of cancer cells than normal cells. Thus, many chemotherapeutic agents could be used to induce cancer-specific ROS generation leading to cell death.10
The cancer cell death process is highly regulated by different factors.11 Various cytotoxic drugs are involved in inducing necrotic, apoptotic, and autophagic types of cell death processes.11 Cell death through apoptosis can be caspase dependent or independent based on the availability of the required factors.12 ROS inducers show either apoptosis or autophagy or necrosis based on different cell death signaling pathways. Briefly, outer membrane localization of phosphatidylserine, depolarization, or loss of mitochondrial membrane potential symbolizes early events in apoptotic-like cell death. Cleavage-dependent activation of caspases 9 and 3, inactivation of poly(ADP-ribose) polymerase (PARP), and induction of DNA fragmentation are late apoptotic events.13 Autophagy can be related to the localization of LC3 in autophagy bodies.14
Increasing trends in drug resistance to aggressive cancers is the main cause of poor clinical success.15,16 These resistant cancer cells display disease recurrence and malignancy, which ultimately leads to poor prognosis after treatment. Cancer cells render a resistant property by over-expressing multidrug resistant 1 protein (MDR1) also named P-glycoprotein (P-gp), which can modulate many intracellular drug concentrations by its ATP-dependent efflux pump. Due to its broad substrate affinity property, it can pump out various anticancer molecules and is thus considered as an important factor for cancer drug resistance and poor prognosis.17 Further, cancer cells show a higher accumulation of genomic mutations to modulate different internal factors to make the cells survive at extreme conditions like stress (increased oxidative stress) and chemo- or radiation therapy. There are several factors involved in promoting cancer cell resistance, whereas activation of HSP2718 and NFκB19 have an indispensable role in the cytoprotective effect conferring cancer cell resistance to death stimuli. Thus, analysis of the effect of the resistant and cytoprotective factors on the activity of anticancer molecules is essential.
Our molecule BBT contains a carbonyl-bridged bithiazole module for ROS generation (Figure 1A).20 This hybrid molecule is a strong electron acceptor in nature due to the presence of a carbonyl-bridged bithiazole moiety, which initially accepts electrons and then distributes them to two trifluoroacetophenone terminals (Figure 1A).21 We have shown how this organic electron acceptor elicits ROS production both in the mitochondria and cytoplasm of the cancer cells. Herein, we proposed both experimentally and computationally that our molecule inhibits mitochondrial complex I by inhibition of electron transport from NADH, which enhances the ROS. Further, its affinity with P-gp and its role in the regulation of important disease resistance genes have also been investigated. Finally, the tumor inhibitory effect has been analyzed in the more aggressive 3D model of cancer cells, HeLa.
Figure 1.

BBT induces mitochondrial ROS production. Structure of CS, FAP, BBT (ROS-producing structure is denoted as a box), and LBT. (A) Microscopic images represent MitoSOX assay in MCF7 cells after treatment with only 12.5 μM BBT or 12.5 μM BBT along with MitoTempo (10 μM). Scale bars correspond to 20 μm, and the magnification is 40×. (B) Microscopic images represent MitoSOX assay in HeLa cells. Cells were incubated in the absence (control) or presence of only 12.5 μM BBT. Then, cells were subjected to the MitoSOX reagent and visualized under a microscope. Scale bars correspond to 20 μm. (C) Quantification of mitochondrial ROS using MitoSOX in MCF7 and HeLa cells treated with BBT. (D) Data are shown as means ± SD and have been analyzed using unpaired T test. ROS: reactive oxygen species, BBT: carbonyl-bridged bithiazole attached with bis-trifluoroacetophenone, LBT: linear bithiazole, CS: central scaffold, and FAP: trifluoroacetophenone.
Results
BBT as an Electron Acceptor Can Confer ROS Production
To verify the hypothesis of BBT as a ROS inducer, the cellular redox state has been analyzed using various techniques. Again, it is well known that mitochondria serve as an important source of ROS in cells and most importantly in cancer cells.22 It is interesting to note that BBT can form intermediates in the presence of NADH and O2, which can trigger ROS production. Thus, mitochondrial ROS production was initially analyzed after treatment with BBT in both MCF7 and HeLa cancer cells using microscopy and flow cytometric analyses. At first, we examined the change in the mitochondrial redox potential of MCF7 cancer cells by evaluating the changes in the flavoprotein fluorescence (autofluorescence, green) in cells. Interestingly, we observed a good increment in the green fluorescence in 12 h BBT-treated MCF7 cells, which represents the shift of mitochondria toward the more oxidative state (Figure S1). Further, mitochondrial oxidative stress has been analyzed using a MitoSOX/mitochondrial ROS detection probe in both MCF7 and HeLa cancer cells. In each experiment, cells were treated with BBT for 12 h and incubated with the MitoSOX to determine the presence of the superoxide inside the cell. Here, microscopic images show significant intense red fluorescence in MCF7 and HeLa cells treated with BBT than that in untreated controls (Figure 1B–D). This increase in red fluorescence represents the higher production of mitochondrial superoxide due to BBT. Furthermore, inhibition of the mitochondrial ROS generation in MitoTempo treatment along with BBT resulted in a significant decrease in red fluorescence in MCF7 cells (Figure 1B). This is due to the antioxidant property of MitoTempo, which can specifically minimize the ROS production in mitochondria. Therefore, BBT has been found to have a potential role in the enhancement of mitochondrial superoxide generation. Following the microscopic experiment, flow cytometric evaluation of MitoSOX experiments in MCF7 and HeLa cells reveals significantly higher mean red fluorescence intensity in the case of an increasing concentration of BBT compared to that of the control (Figure 1D and Figures S2 and 3). In MCF7 cells, there is a ∼2-fold increase in fluorescence intensity with the increase in concentration of BBT from 6.25 to 12.5 μM. Further, mitochondrial ROS production efficiency of BBT is compared with CS. Here, no significant increment in the fluorescence intensity of cells treated with CS has been observed in comparison with the control. This result indicates that the presence of the trifluoroacetophenone moiety plays a major role in the modification of the superoxide status in cancer cells. Similarly, BBT shows the efficient mitochondrial superoxide formation in HeLa cells (Figure 1C and Figure S3). This can be assumed because of the ∼3-fold increments of red fluorescence in cells treated with BBT than in untreated control cells. Again, the mitochondrial ROS production was found to enhance with the increasing concentration of BBT in HeLa. This is quite predictable from the result showing a maximum ∼25-fold increase in red fluorescence after treatment with 25 μM BBT than the control. Again, to validate the importance of structural moieties of BBT, the effect of LBT on mitochondrial ROS generation was assayed. Here, the flow cytometric result confirms that ROS generation upon treatment with various concentrations of LBT was quite less. This could also be discussed with our previous observation regarding the cyclic voltammetry assay revealing the high electron accepting nature of BBT compared to the LBT.21 These results indicate that the mitochondrial ROS generation requires the availability of both bridged carbonyl and trifluoroacetophenone, which can synergize the ROS production, and thus BBT can enhance the generation of mitochondrial superoxide.
Next, microscopic images of DCFDA analysis after treatment of MCF7 cells with 12.5 μM BBT show intense green fluorescence than control cells (Figure S4A). Similarly, flow cytometric analysis reveals a ∼34-fold increase in fluorescence compared to control cells (Figures S4B and S5). These results denote an accumulation of the higher amount of cytoplasmic ROS in the case of BBT-treated cells. In reduced form, DHE shows blue fluorescence and localizes at the cytosol of the cell.23 Upon oxidation, DHE is converted to 2-hydroxyethidium by cellular superoxide, emits red fluorescence, and gets intercalated into the DNA, which can be visualized using the microscope and quantified by FACS. Microscopic images show significant blue fluorescence in the case of the control, representing a reduced form of DHE and a lower level of ROS species. However, BBT (6.25 μM)-treated MCF7 cells show an accumulation of red fluorescence of DHE, representing higher cellular oxidative stress (Figure S4C). The flow-cytometric analysis represents a dose-dependent increase in fluorescence intensity in BBT-treated MCF7 cells. Even the red fluorescence intensity observed in a lower concentration (3.125 μM) of BBT-treated cells is significantly higher by ∼1.8-fold in comparison to the control cells (Figures S4D and S6). Hence, BBT in contrary to CS and LBT is found to induce mitochondrial ROS, which eventually gets accumulated in the cytoplasm.
Mechanistic Understanding of Mitochondrial ROS Generation by BBT
We know that NADH, being a key molecule, plays a vital role in the redox process in mitochondria and can donate electrons. From this information, we speculated fundamentally that if NADH donates electrons to BBT, then there must be a redox potential compatibility among NADH, BBT, and O2. In this instance the oxidation potential of NADH should be energetically close to the reduction potential of BBT in mitochondria. To find out whether NADH can transfer electrons to BBT, LBT, CS, and FAP, we have compared the electrochemical redox potentials. According to the previous reports, the formal potential (E0′) of the NADH/NAD+ redox couple is −0.560 V vs SCE (pH 7.0 at 25 °C),24 which is highly dependent on pH and temperature,25 whereas E0′ of the O2/O2·– redox couple is −0.73 to −0.82 V vs SCE in aprotic media.26 Previously, we reported21 that the electrochemical reduction potential of BBT in fluorobenzene is −1.16 V vs ferrocene/ferrocenium ion couple, corresponding to a redox potential of −0.74 V vs SCE and the lowest unoccupied molecular orbital (LUMO) energy level of −3.6 eV. These redox potentials are very close, and thus it is considered that NADH reduces BBT in aqueous solution, which can mediate the reduction of O2 to O2·–. Then, the LUMO energy states for BBT, LBT, CS, and FAP have been evaluated using density functional theory (DFT) calculations (Figure 2A). The LUMO energy level of BBT was calculated to be −3.57 eV, which is well consistent with the experimental value (−3.6 eV). In contrary to BBT, CS and FAP molecules have higher LUMO energy (more negative redox potential), thus their reduction by NADH and the mediation of O2 reduction are quite difficult. Hence, NADH can easily reduce BBT, which eventually resulted in BBT radical anion formation. Next, the BBT radical anion caused the formation of superoxide ion, which elevates oxidative stress (Figure 2B). As both NADH and O2 have quite high concentrations in the mitochondria, we assume that the molecules are entering the mitochondria and carried out the change in the redox process of the ETC that generates a large amount of ROS in the cell. Moreover, we assume that BBT binds at complex I in the mitochondrial respiratory chain as we postulate that BBT traps the electron releases from NADH. Thus, we have analyzed the effect of the BBT on complex I activity. Thus, we have checked the activity of complex I in the presence of BBT (Figure 2C). We have used rotenone, a known complex I inhibitor, as a control. Here, we observed significant dose-dependent inhibition of the complex I activity in the presence of BBT. This represents that BBT goes inside the mitochondria and interacts with complex I and inhibits its activity. Like rotenone, BBT affects complex I activity and might be generating ubisemiquinone. This ubisemiquinone consequently donates electrons for the generation of mitochondrial superoxides. To further understand the mechanism of BBT in ROS generation, we have tried to understand the effect of mitochondrial ROS in the presence of a complex I inhibitor, such as rotenone and BBT. This has been conducted using MitoSOX by flow cytometry as discussed above.
Figure 2.

Electron accepting power of BBT modulates the mitochondrial complex I function. (A) Diagrammatic representation of the molecular structure and HOMO/LUMO energy levels of CS, FAP, and BBT. Molecular docking image of FMN (red) in complex I (blue). (B) Diagrammatic illustration of the molecule having 1,4-dihydropyridine-3-carboxamide that can transfer the electron to the BBT to form its intermediate, which reversely transfers the electron to oxygen to form reactive oxygen species. (C) Complex I activity analysis in the presence of rotenone (Rot; 12.5 μM) and BBT (6.25, 12.5, and 25 μM). Data are shown as means ± SD and have been analyzed using one-way ANOVA (***p < 0.0001). (D) Molecular docking of BBT (green) with mitochondrial complex I (blue), where the FMN is represented as red spheres. (E) Molecular interaction of BBT with different amino acids of complex I, which is near to the FMN site. FMN: flavin mononucleotide, HOMO: highest occupied molecular orbital, and LUMO: lowest unoccupied molecular orbital.
Here, HeLa cells were treated with either 40 μM rotenone or 12.5 μM BBT or their combination for 4 h. In flow cytometric analysis, we observed that the use of rotenone increased the mitochondrial ROS, and an increase in mitochondrial ROS was observed when rotenone was applied along with BBT in comparison with the treatment of only BBT (Figure S7). This represents an increase in mitochondrial ROS generation even after a short time of incubation with BBT. Still, a very negligible change in ROS has been noticed between BBT and BBT with rotenone-treated cells. This might be because electrons from NADH get transferred to BBT but not to FMN, which participates in ROS generation (Figure 3A,B). Next, it is worth to understand the binding location of BBT in complex I. For this purpose, we performed blind docking of BBT in complex I (Figure 2D). Interestingly, we found that BBT binds very close to the FMN site of complex I through H bonding of the carbonyl group of one of the trifluoroacetophenone moieties and bridged carbonyl group with ARG-86 and GLY-350 of complex I, respectively, and −N= of the central moiety of BBT with LEU-349 of complex I with an affinity of −9.5 kcal/mol (Figure 2E).
Figure 3.
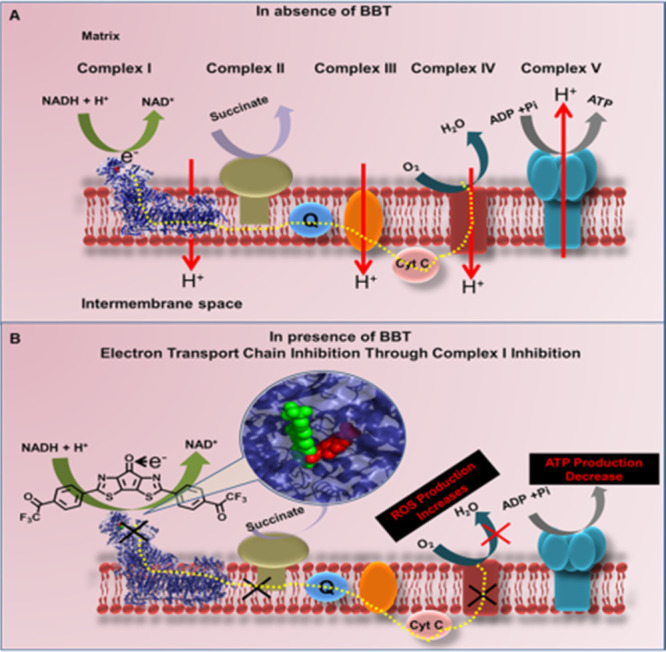
(A) Schematic illustration of mitochondrial active ETC complexes. NADH as a source of electron to start the electron transfer through various complexes to produce ATP in the absence of BBT. (B) Whereas in the presence of BBT (green spheres) that binds close to the FMN site (red spheres), it shows electron acceptance from NADH, thus hindering the overall pathway of electron transfer and ATP generation, resulting to an increase in ROS production. NADH: nicotinamide adenine dinucleotide hydrogen, and ATP: adenosine triphosphate.
BBT Induces Cell Death Predominantly by Apoptosis
Cancer cells display higher accumulation of oxidative stress. Therefore, a small increase in oxidative stress can make the cells vulnerable. As BBT is found to induce oxidative stress, we were interested in verifying its cytotoxic effect as well. Initially, we have checked the cell viability using various cancer cells like cervical cancer (HeLa and SiHa), breast cancer (MCF7), lung adenocarcinoma (A549), and glioblastoma (U87MG) (Figure S8) after treatment with BBT, and the IC50 values for the cells are HeLa = 5.12 μM, A549 = 8.88 μM, U87MG = 4.75 μM, MCF7 = 3.7 μM, and SiHa = 6.6 μM. Here, we have observed significant cytotoxicity of BBT against various types of cancer. However, BBT did not show cytotoxicity in normal lung fibroblast WI-38 cells, proving that it is not cyto-toxic in normal cells. (Figure S9). From this effect of BBT, we moved to understand the effect of BBT on cancer cell cycle progression, and we have performed flow cytometric analysis. Results show a gradual increase in cell population in the sub G0 phase in a dose-dependent manner up to 25 μM concentrations of BBT (Figure 4A and Figure S10). This represents that there is no significant cell cycle arrest at any checkpoint but rather an increased fragmentation of DNA after treatment. To demonstrate the cytotoxic effect of BBT as the type of cell death, we have adopted annexin-PI assay. Here, in HeLa cells, early (71.8%) and late (21.0%) apoptotic cells have been found after BBT treatment, while in the control (untreated cells), only 4.9% cells were found in early apoptosis and 10.7% cells in late apoptosis. (Figure 4B). Next, we studied autophagy, which can be detected by understanding the LC3 status.27,28 Hence, we have used the flow-cytometric method to quantify the autophagic cancer population based on LC3 localization. Histogram representation depicts less increase in LC3 detection in 12.5 μM-treated cells than control cells (Figure S11). This denotes that a small population of cells undergoes autophagic cell death. Overall, the cell death analysis denotes apoptosis as the predominant pathway than autophagy in BBT-treated cancer cells.
Figure 4.

BBT activates JNK-, p38-, and caspase-dependent apoptosis. (A) Cell cycle histograms of Hela cells after treatment with various concentrations of BBT. Analysis of apoptosis using the annexin V and PI method. (B) HeLa cells were incubated in the absence (control) or presence of BBT (12.5 μM). Then, cell death was analyzed using annexin V and PI through flow cytometry. (C) Flow-cytometric analysis of mitochondrial membrane potential (ΔΨm) with the help of JC-1 staining in treated and untreated samples.(D) Immunoblotting analysis of cell lysates collected from HeLa cells after treatment of 12.5 μM BBT. Stress response pathway (p-JNK1/2, p-c-jun, and p-p38) and apoptotic factors (cleaved caspase 9 and 3 and PARP) were seen to be upregulated on treatment with BBT. α-Tubulin was used as a loading control. All experiments have been performed thrice (n = 3). PI: propidium iodide, PARP: poly(ADP-ribose) polymerase, and JNK: Janus kinase.
BBT Induces Loss of Mitochondrial Membrane Potential
Now, we have analyzed the pathway of cell death. ROS generation can modulate the mitochondrial mode of cell death.28 Change in the mitochondrial membrane potential is considered as an early-stage detection of apoptosis.29 Thus, to analyze these events, we initially analyzed the loss of mitochondrial potential using a cell-permeable JC1 molecule. Thus, observation of fluorescence in cells before and after treatment of BBT has been conducted using a flow cytometer. Here, most of the untreated cells depict a lower green/red fluorescence ratio representing intact membrane potential. On the other hand, cells treated with 12.5 μM BBT resulted in the population having an increased green/red fluorescence ratio and emitting mostly at the green fluorescence channel (Figure 4C and Figure S12). This change in the red/green fluorescence ratio from control cells after treatment represents increase in mitochondrial outer membrane permeabilization (MOMP) potential, which affects the JC1 aggregation at mitochondria.
BBT Induces Cyt C Release from Mitochondria
Microscopic images show co-localization of green cyt C with red mitochondria in control cells. This represents occupancy of cyt C at its usual location, i.e., mitochondria. In the case of BBT treated cells, dispersion of the green signal around the cytoplasm has been observed as well as the limited co-localization of the green signal with red mitochondria (Figure S13). This indicates release of cyt C from the mitochondria of HeLa cells after treatment of BBT.
BBT Activates Stress-Related JNK/c-Jun and p38 Pathways
Stress such as oxidative damages activates JNK and p38 signaling pathways.30 Thus, we have analyzed the activation of JNK or stress-activated protein kinases expression after treatment of BBT. Here, we have quantified phosphorylated JNK at Thr183 and Tyr182 in HeLa cells, which indicates the active form of the protein. To further validate the activation of JNK, activation of c-jun, which is its most important downstream protein, has been analyzed. Figure 4D represents that, in contrast to untreated cells, significantly phosphorylated c-jun has been found in BBT-treated cells. Like JNK, we also observed p38 activation as the striking increased in the presence of phospho-p38 protein in BBT-treated HeLa cells than in untreated cells (Figure 4D). Further increasing pieces of evidence show that JNK/c-jun activation can regulate mitochondria-dependent cellular apoptosis. Briefly, JNK activation can trigger the cytoplasmic release of cyt C31 and result in the mitochondrial pathway of apoptosis.
BBT Triggers the Mitochondria-Dependent Apoptotic Pathway
As BBT induces apoptosis, we initially checked the activation of caspase 9 using immunoblot experiments. The result indicates an intense band of the cleaved form of caspase 9 for cells treated with the compound compared to untreated (control) cells (Figure 4D), indicating the activation of caspase 9 by BBT.
Next, we investigated the activation of caspase 3 as downstream of activation of caspase 9. Thus, we initially performed an analysis of its activity based on the colorimetric analysis. In principle, PARP, which is an important substrate of active caspase 3, has a conserved cleaving sequence (DEVD).32 Here, only BBT-treated cells shows an ∼4-fold increase in free pNA absorbance at a 405 nm wavelength compared to control. Additionally, 2 μM doxorubicin (DOX) has been used for the positive control of the experiment and that shows only ∼2-fold increase in comparison to control cells (Figure S14). This indicates the activation of caspase 3 due to BBT treatment, which is higher than the conventional anticancer drugs. Also, caspase 3 activity has been analyzed using the flow cytometer. Here, a significant number of cells with cleaved caspase 3 have been observed against the control, which represents activation of caspase 3 after treatment (Figure S15). Further, the activity of caspase 3 in detecting its cleaved form before and after treatment of BBT has been conducted using immunoblotting experiments (Figure 4D). The result shows activation of caspase 3 after treatment of BBT. Similarly, time-dependent analysis of active caspase 3 has been evaluated after treatment with BBT for different time intervals (0, 4, 6, and 12 h). Here, we have observed time-dependent higher occurrence of the active or cleaved form of caspase 3 (Figure 5D). The above result indicates that BBT activates caspase 3. Here, we tried to understand whether ROS is produced after induction of apoptosis or ROS takes part in the activation of apoptosis. To understand this, caspase 3 activity has been analyzed in BBT-treated cells co-incubated with an ROS inhibitor, N-acetylcysteine (NAC). Here, we observed a negligible change in absorbance in comparison with control cells (Figure S14). This indicates that caspase 3 activation required BBT-induced ROS generation inside the HeLa cells. Again, activation of the caspase-dependent pathway has been verified as PARP cleavage. Thus, we have performed immunoblotting experiments of HeLa cells after treatment with BBT for 12 h. In comparison to the control cells, BBT-treated cells showed an intense band of cleaved PARP that indicates their inactivation (Figure 4D). Hence, BBT induces activation of the JNK/SPAK pathway, mitochondrial permeability transition, the release of cyt C, activation of caspase 9 and 3, and cleavage of PARP.
Figure 5.

Unrestrained action of BBT on various drug-resistant factors. (A) Molecular docking of BBT (green) and LBT (blue) in P-gp receptor protein (brown). Interaction of (B) BBT and (C) LBT with amino acids of P-gp. (D) Western blots showing the expression of cleaved caspase 3, p-HSP27, and NFκB in HeLa cells after treatment with 12.5 μM BBT after various time points of treatment. The α-tubulin expression has been used as a loading control. (E) Cancer cell recurrence efficiency using MTT assay: MTT assay after treatment of BBT for 48 h in one set or 24 h with and without BBT in SiHa cells in another set. All experiments have been performed thrice (n = 3). P-gp: P-glycoprotein, HSP27: heat shock protein 27, and NFκB: nuclear factor kappa B.
Cancer Drug Resistance and Disease Relapse
Many potent anticancer molecules are inefficient against the more aggressive form of cancer.33 The cancer cell shows resistance to various blockbuster anticancer drugs due to higher accumulation of resistance-related factors, which causes poor prognosis and eventually cell death. This is the reason we need to analyze the effect of the molecule on various disease resistant factors, namely, P-gp receptor, Hsp27, and NFκB.
The inevitable role of the P-gp receptor in multidrug resistance (MDR) in cancer cells can be denoted as the first line of the defense system of cancer cells against a various range of anticancer drugs, most importantly doxorubicin, paclitaxel, etoposide, colchicine, vinblastine, and so on.15 With this remarkable feature of interaction with structurally divergent molecules, P-gp plays a pivotal role in chemo-resistance. Most of the cancer cells at the early to the late stage of cancer accumulate P-gp transporter to efflux the drugs out of the cells and eventually reduce their cellular concentration. Further use of P-gp inhibitors in combination with anticancer drugs can lead to toxicity especially to the brain where P-gp is observed to be an important regulator of the blood–brain barrier (BBB). Thus, we have analyzed the P-gp receptor binding efficacy of BBT. This has been performed using two bio-informatic tools. At first, we have analyzed P-gp binding using SwissADME (link: http://www.swissadme.ch/).34 Here, we observed that the molecule is not a P-gp substrate and also could not pass through the BBB. Thus, we next examined different BBT analogues using the same tool to understand the importance of the different moieties (Figure S16). Here, we observed that both CS and FAP do not show P-gp substrate activity. However, LBT is found to be a P-gp substrate. Further, we observed that the replacement of nitrogen from BBT and CS results in a P-gp interaction. However, the presence of nitrogen in BBT renders no P-gp binding. To confirm the results, we have performed docking with the P-gp substrate. Ferreira et al. described the possible binding sites of the molecules in P-gp that are designated as R, M, and H sites.35 To understand the binding of BBT and LBT with P-gp, we performed molecular docking. According to this, we observed no such interaction between BBT and P-gp (Figure 5A,B). However, we observed the location of LBT at the R site of the P-gp (Figure 5A,C). This represents that the presence of the central carboxyl group not only plays an important role in ROS generation but also helps the molecule to be rescued from P-gp-dependent exocytosis. Thus, both the analyses confirm that BBT is not a P-gp substrate and its intracellular transport cannot be affected because of the higher expression of P-gp on cancer cells.
Again, another factor like Hsp27 is the small heat shock protein cytoprotective chaperone that plays a pivotal role in the rescue of the cells from cell death by modulating ROS/glutathione levels36 in the cytochrome C/apoptosome complex37 and activating NFκB that consequently triggers cell survival pathways. Again, various cancer cells display overexpression of Hsp27 than the normal or non-transformed cells. Further, confluent cells of the solid tumor results in maximum activation of Hsp27 than proliferative 2D cells.38 Hsp27 confers to the high resistance of tumor toward various anticancer drugs like dexamethasone, paclitaxel, doxorubicin, gemcitabine, and so on.39 The effect of the BBT on p-Hsp27 (ser82) has been evaluated using immunoblotting assay over different times of treatments (0, 4, 6, 12 h) with 12.5 μM BBT in HeLa cells. Here, we observed a decrease in p-Hsp27 protein after 12 h of treatment (Figure 5D). This result indicates that Hsp27 might have been rendered inactive after time-dependent incubation with BBT. Similarly, NFκB is an important transcription factor having an indispensable role in oncogenesis, cancer progression, and chemo-resistance. Previous reports documented the role of NFκB-dependent suppression of JNK signaling.40 Here, expression of NFκB (p65) has been verified after different time intervals (0, 4, 6, and 12 h) of treatment of BBT in HeLa cells. The immunoblotting experiment demonstrates diminished expression of NFκB after 4 h of expression, which is found in the other two time intervals (6 and 12 h) (Figure 5D).
Relapse or recurrence of tumors after chemotherapy results in a poor prognosis and compromises the effectiveness of the chemotherapeutic agents. This is due to the increment of the drug-resistant cancer cell population. Thus, we have evaluated the effect of BBT on more aggressive cervical cancer (SiHa) cell relapse. Briefly, SiHa cells were divided in two groups. In the first group, cells were treated with BBT at different lower concentrations and incubated up to 48 h. In the next group, having BBT treatment of similar concentrations was removed after 12 h, and cells were incubated with complete media. A less significant change has been observed in cell death in both the groups (Figure 5E). This result indicates that even at lower concentrations, BBT may possess the property to prevent the re-appearance of cancer-initiating cell populations, thereby preventing the phenomenon of cancer recurrence.
BBT Affects Migration and Growth of Tumor-Mimicking Cancer Cell Spheroids
A tumor in vitro spheroid model is closer to in vivo tumor models in both phenotypical and genotypical manners.41 Previously, we have reported various biochemical and drug distribution properties between the tumor spheroid and monolayer culture system.42 This is mostly due to the activation of various resistant proteins in the spheroid culture. Further, the spheroid culture contains cells that can regenerate the complete tumor even after chemotherapy and can result in a high chemo-resistant and malignant tumor.43 Conversely, BBT was found to be highly active against monolayer cancer cells and was not affected by the resistance protein P-gp and various cytoprotective factors of cancer cells. Thus, the understanding of BBT properties against a more complex cancer system like the tumor spheroid model is quite necessary. Initially, we have analyzed the effect of BBT on cancer cell migration from the tumor spheroid because migration of cancer cells in the late phase in cancer leads to poor prognosis and is extremely lethal. Many promising anticancer molecules were unable to affect these heterogeneous cancer populations. Thus, we have verified the effect of BBT on the migratory property of cells using the tumor spheroid. This is because the full-grown spheroid shows the availability of heterogeneous cells than monolayer cells.44 Also, the population of cancer stem cells (CSCs), which drives the migratory signal, is rich in the anchorage-independent culture.45 In brief, full-grown tumor spheroids were transferred to the tissue culture-treated cover glass and incubated for 12 h in serum containing DMEM. Migratory cells can be detected as surface-attached monolayer cells around the spheroid. PI has been used to detect dead cells. To analyze the effect of BBT, spheroids were treated with 6.25 and 12.5 μM BBT for 8 h. There is a significant migratory cell that was observed around the untreated spheroid (control), whereas relatively less migratory population were observed in 6.25 and 12.5 μM BBT-treated spheroids (Figure 6A). Also, PI staining represents an increased in cell death in spheroids with an increasing concentration of BBT. From the above results, it can be concluded that dual target-based activity of BBT can not only trigger monolayer cancer cell death more aggressively but also affect the growth and migration of tumor-mimicking CSC-rich spheroid cells. This can be due to the ROS stress-induced pathways of regulation of migration inhibition and cancer cell death.46−49
Figure 6.

BBT inhibits both migration and growth of tumor spheroid. (A) Microscopic image analysis of cell migration from the HeLa cell spheroid after treatment with 0, 6.25, and 12.5 μM concentrations of BBT. Dead cells were stained with PI. Scale bars correspond to 100 μm. (B) DIC images represent the growth of 3D spheroid HeLa cells incubated with 12.5 μM BBT up to a period of 8 days. Scale bars correspond to 100 μm. (C) Growth kinetics curve of the HeLa spheroid after treatment with 0 (blue) and 12.5 μM (red) BBT for 8 days. Data are shown as means ± SD. All experiments have been performed thrice (n = 3).
Finally, these results motivated us to verify the effect of the molecule on tumor spheroid growth. From the microscopic image and growth kinetic plot of HeLa spheroids, we observed significant inhibition in spheroid growth due to BBT treatment (Figure 6B,C). These results imply the substantial in vitro tumor growth inhibitory role of BBT.
Discussion
In this study, we showed an organic molecular electron trap, which contains structural modules important for the elevation of mitochondrial and cellular ROS. We understood in detail the importance of the BBT molecular structure as its core scaffold (CS) and trifluoroacetophenone molecules are not self-sufficient to generate mitochondrial ROS, indicating the importance of multi-structural modality. So, according to our previous report,21 BBT is a highly electron-accepting organic molecule, where the central core mainly takes part in electron acceptance, and then the two terminal trifluoroacetophenone moieties also act as additional electron acceptor units. In the presence of strong electron donors, BBT can form a radical anion. Then, it can transfer an electron to O2, which results in ·O2– formation and thus can increase the oxidative stress. In cells, the 1,4-dihydropyridine-3-carboxamide moiety in the NADH molecule can transfer electrons or hydride ion to the complex of the ETC in the presence of NADH dehydrogenase. Thus, we postulate that BBT can be reduced in the presence of NADH and can form various free radicals in the presence of O2, which will eventually increase the ROS of the cells. Next, we tried to understand how BBT generates mitochondrial ROS inside the cell.50,51 Because of the high electron accepting nature of BBT, we presumed its various ROS generating intermediates inside the cells where NAD(p)H/NAD+ is higher. To understand in detail how BBT generates ROS in mitochondria, we investigated the redox potentials of both BBT and NADH since NADH is present in high amounts in mitochondria. To our surprise, we found that the potential of the NADH/NAD+ redox couple is close to the reduction potential of BBT,52 which basically makes the situation amicable for donating an electron from NADH to BBT. Molecular docking of BBT with complex I reveals that BBT binds close to the FMN site. We envision that this binding property of BBT inhibits the transfer of electrons into the FMN, which resulted in the inhibition of the electron transport chain of complex I to complex IV. We envision that this is the reason BBT efficiently triggered the ROS generation in mitochondria, subsequently triggering ROS generation in the cytoplasm. Interestingly, from complex I activity assay, we have observed that BBT affects complex I activity. This result indicates the interaction of BBT with complex I of the ETC. Further inhibition of complex I can generate superoxide in mitochondria, where ubisemiquinone plays an important role in donating electrons. However, electron donation from other mitochondrial and cytoplasmic components to BBT is highly possible due to the complex mitochondrial and cytoplasmic environment. Next, we tried to understand the cellular fate due to the high amount of ROS generation by BBT. As cancer cells are already overloaded with these ROS molecules, thus, an increase in the ROS generation can induce stress-dependent cell death pathways. So, we have analyzed the status of ROS in the cytoplasm. This is because mitochondrial ROS can be transferred into the cytoplasm and thus can trigger redox signaling or oxidative stress-dependent cell death. For that purpose, DCFDA and DHE probes have been used to both detect and quantify ROS in the cytoplasm. DCFDA is a cell-permeable probe and cleaved by non-specific cellular esterase after entering into the cell, which now makes a non-permeable byproduct that, when interacting with H2O2 and other peroxides, gets converted to a green fluorescence-releasing molecule, which can be visualized in a fluorescence microscope and quantified using flow cytometry. We observed that BBT gets converted into different ROS producing molecules, which can eventually affect cell survival. Here, we found that BBT is a potent anticancer molecule against various cancer cells.
Intriguingly, BBT shows an electron accepting property and induction of ROS-producing intermediates. It is evident as to how the small molecule can affect cancer cell environment, but the role of a highly electron accepting molecule in cancer cell fate determination is not well revealed before. For the first time, we observed that the electron acceptor BBT can modulate oxidative stress and survival of the cancer cell. Further, we were interested to find out the effect of this molecule on various types of cancer cells. This implies that the cytotoxicity of BBT is not much specific toward the type of cancer cell that has been tested. As we observed the potent cytotoxic effect of BBT against various cancer cells, we are now interested to understand the type of cell death triggered with this molecule. As the cell cycle and cell death are an integral part of each other, we first evaluated any change in the cell cycle after treatment. Further, dysregulation of the cancer cell cycle is the most important event in cancer progression, which leads to a poor prognosis. So, we tried to understand the effect of BBT on cell fate. First, cell cycle assay reveals the absence of any cell cycle arrest due to BBT but with an increase in the cell population at sub G0 that represents the fragmentation of DNA or apoptosis. This indicates that it is a cytotoxic molecule rather than cytostatic, thus having a chance to develop resistance to cancer is reasonably difficult. Moreover, the increase in annexin V and PI positive cell population after treatment with BBT in HeLa and A549 cells notifies apoptotic cell death. The report also shows that ROS generation induces apoptotic and/or autophagy cell deaths.14 Thus, we looked on whether BBT caused autophagic death in cancer cells or not. Autophagy, like apoptosis in the presence of oxidative stress or stimuli, triggers cell death. It starts with the formation of an autophagosome, translocation of LC3 protein, fusion of the lysosome, and finally degradation. Basically, LC3-I resides in the cytoplasm, and LC3-II sequesters at the membrane of the autophagosome. However, flow cytometric analysis of LC3 after treatment represents a minute increase in LC3-II. This indicates that BBT predominates apoptotic death rather than autophagy in cancer cells. Next, we tried to understand the mechanistic pathway governing apoptosis; we examined the role of mitochondria as an ROS-dependent cell death pathway. In the intrinsic pathway of apoptosis, the change in mitochondrial membrane potential results in increased membrane permeabilization and release of cytochrome C (cyt C), which takes part with different cellular factors in the activation of caspase 3. Change in MOMP was studied using the JC1 dye, which is a cationic dye that accumulates in healthy mitochondria with a characteristic emission at red wavelengths (∼590 nm). Loss of mitochondrial membrane potential is the outcome of JC1 release into the cytoplasm with characteristic emission of green fluorescence (∼525 nm). Here, BBT displays a potent inducer of MOMP, which regulates the activation of several apoptosis regulators. Consequently, we have observed the release of cyt C in cancer cells. Further, we observed that stress due to ROS causes activation of JNK1/2 that consequently activates c-jun both in HeLa cells. We found an activation of another stress-related protein, p38, in HeLa cells upon treatment with BBT. We have observed how BBT affects mitochondria to elicit the apoptotic pathway due to different stress-induced factors. So, we were interested to analyze the role of downstream factors for apoptosis due to treatment of BBT. Again, apoptosis can follow two important pathways, such as a caspase-dependent or -independent pathway. In the caspase-dependent pathway, apoptosome complex formation after the release of cyt C from mitochondria due to MOMP is obvious, following the activation of caspase 9 and caspase 3, where the former one is an initiator caspase found to be activated during the JNK/p38 pathway and leads to activation of the latter one, which is an executioner caspase. We observed the activation of caspase 9 and consequently caspase 3 and cleavage of PARP protein, confirming caspase-dependent apoptosis.
Chemo-resistance is the challenging part of anticancer drug discovery. In this direction, we looked at the potential of BBT and its analogues as the P-gp substrate using the SwissADME method. We found that BBT is not a P-gp substrate. BBT and its different moieties display no binding affinity to any of the important binding sites of the P-gp complex. In contrast, LBT appears to be a good P-gp substrate with strong binding with the R site of P-gp. This shows that the presence of a central carbonyl moiety in BBT affects P-gp binding and helps to be rescued from disease resistance through P-gp-dependent efflux of the molecule. Next, we tried to understand whether BBT can modulate the cytoprotective factors in the cancer cells or not. We observed stable caspase 3 activation upon time-dependent treatment of BBT, indicating a less cytoprotective effect. Thus, the functions of Hsp27 as an inhibitor of caspase activation and NFκB as a JNK inhibitor were examined. Time-dependent degradation of Hsp27 and NFκB in BBT-treated HeLa cells indicates a lesser chance of cellular escape from apoptosis. This can be well documented from the relapse experiment showing that even after the withdrawal of the BBT, the cellular survival rate is minimized, and an insignificant difference was observed in the continuous treatment of BBT. These results indicate that the BBT molecule induces apoptotic cell death while restraining cell escape. Finally, we observed that BBT also displays potent inhibition of spheroid growth and metastatic cancer migration.
Conclusions
Altogether, we demonstrated a new concept of using an organic electron acceptor, BBT, to modulate the mitochondrial respiratory chain by inhibiting the activity of complex I. This BBT participates in the reversible electron transfer that exerts the generation of a high amount of mitochondrial and intracellular ROS. Subsequently, mitochondrial outer membrane permeabilization has been increased with an increase in cytoplasmic delocalization of cyt C. Furthermore, treatment of BBT resulted in stress-dependent JNK/p38 and caspase-dependent apoptosis in cancer cells. Conversely, BBT reduces the expression of important cytoprotective or drug resistance factors (Hsp27 and NFκB), indicating its potential inhibition of cancer cell resistance and relapse. Interestingly, we found that BBT is not a P-gp substrate, which reveals its excellent anticancer potential. These properties empower BBT to inhibit the migration and growth of more resistant and complex in vitro tumor spheroids.
Materials and Methods
Materials
The cell lines MCF7 and HeLa have been obtained from NCCS, Pune. Dimethylsulfoxide (DMSO) and methanol (MeOH) were purchased from Spectrochem, Mumbai. TritonX-100 was purchased from SRL, Mumbai. 2-[4-(2-Hydroxyethyl) piperazin-1-yl]ethanesulfonic acid (HEPES) was purchased from Himedia. Ethylenebis(oxyethylenenitrilo)tetraacetic acid (EGTA), 4-piperazinediethanesulfonic acid (PIPES), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), bovine serum albumin (BSA), 5(6)-carboxy fluorescence, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dulbecco’s modified Eagle’s medium (DMEM), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), trypsin–EDTA solution, dimethylsulfoxide for cell culture, formaldehyde solution for molecular biology, propidium iodide (PI), 2′,7′-dichlorofluorescin diacetate (DCFDA) (Sigma D6665), dihydroethidium (DHE) (Sigma D7008), and 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL) were purchased from Sigma Aldrich, Missouri, United States. Fetal bovine serum (FBS) was purchased from Invitrogen, California, United States. Goat polyclonal anti-rabbit IgG H&L (Cy3.5) (ab6941) was purchased from Abcam, Cambridge, United Kingdom. Mouse monoclonal anti-alpha tubulin antibody (T9026) , anti-cyt C primary antibody (05–479), goat anti-mouse IgG-cy3 conjugated secondary antibody (AP124C), anti-mouse IgG (H&L) HRP conjugated (12-509), anti-rabbit IgG (H/L) HRP conjugated antibody (AP187P), RNase A, and FlowCellect Autophagy LC3 antibody-based autophagy detection kit (FCCH100171) were purchased from Millipore, Massachusetts, United States. The annexin V/PI apoptosis detection kit (cat. # 556547), cleaved caspase analysis kit (cat. 559341), and BD MitoScreen (cat. 551302) were purchased from BD New Jersey, United States. Bisbenzimide H 33258 (Hoechst) was purchased from Calbiochem, San Diego, United States.
Cell Culture
MCF7, HeLa, A549, U87MG, WI-38, and SiHa cell lines were brought from National Centre for Cell Science (NCCS), Pune, India. Cells were cultured and maintained under a 5% CO2 humidified incubator atmosphere at 37 °C using Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, penicillin (50 units/mL), streptomycin (50 μg/mL), and kanamycin sulfate (110 mg/L). Trypsin–EDTA solution was used to digest the monolayer cell culture.
Evaluation of ROS Generation
Flow Cytometric Analysis
The ROS studies have been performed following our previously described study.53 Briefly, quantification of fluorescence of MitoSOX was carried out using FACS. Briefly, MCF7 cells (1 × 106 cells/mL) were seeded overnight in six-well plates (5% CO2 and 37 °C). Then, cells were treated with 12.5 and 6.25 μM BBT and central scaffold (CS) for 12 h in a medium with 1% serum. Then, the cells were collected in suspension and incubated with a 5 μM MitoSOX working solution for 10 min. After that, cells were washed with PBS and examined using FACS. Similarly, HeLa cells were treated with an increasing concentration (0, 6.25, 12.5, and 25 μM) of BBT or LBT (bithiazole attached with bis-trifluoroacetophenone) and were examined for mitochondrial ROS generation using 5 μM MitoSOX by a flow cytometer. Further, HeLa cells were treated with 12.5 and 25 μM LBT for 12 h in a medium with 1% serum and examined for fluorescence intensity using FACS. Again, for the rotenone experiment, HeLa cells were treated either with 12.5 μM BBT or 40 μM rotenone or both for 4 h in a medium with 1% serum followed by incubation with a 5 μM MitoSOX working solution for 10 min. Then, cells were washed and analyzed using FACS.
Flow cytometric evaluation of DCFDA was carried out using the abovementioned method. Similarly, flow cytometric analysis was conducted after treatment of MCF7 cells with various concentrations of BBT (0, 3.125, 6.25, 12.5, and 25 μM). After treatment, cells were given a wash with PBS and incubated with DHE as mentioned above. Then again, a wash was given, and fluorescence quantification was conducted using FACS.
Microscopic Evaluation
To determine the mitochondrial ROS, cells (MCF7 and HeLa) were treated with 12.5 μM BBT for 12 h followed by a PBS wash and further incubated with a 5 μM MitoSOX solution for 10 min at 37 °C. To inhibit the mitochondrial ROS generation, cells were co-administrated with 10 μM MitoTEMPO with 12.5 μM BBT. Then, cells were washed with PBS, and images were captured using an Olympus microscope in bright field and 561 nm wavelength filters using a 40× objective, and lamp intensity was 5.40. Cytoplasmic ROS has been determined using DCFDA and DHE staining. Briefly, MCF7 cells were seeded in a cover glass bottom dish. Then, the cells were given a PBS wash and then incubated with 12.5 μM BBT for 12 h. After that, cells were washed and incubated with 20 μM DCFDA for 30 min. Again, a PBS wash was given to remove residual DCFDA followed by observation under the microscope. Here, cells were treated with 25 μM BBT. Similarly, MCF7 cells were examined for DHE fluorescence after treatment with 12.5 μM BBT using a microscope. Here, cells were given a PBS wash and supplemented with 5 μM DHE in serum-free colorless DMEM and incubated for 30 min at 37 °C. Then, cells were again given a wash and fresh DMEM was added. The ROS generation was examined by change in fluorescence of DHE from blue to red fluorescence.
Cell Viability Analysis
Cell viability was performed using MTT assay according to a previous report.54
Molecular Docking Analysis
Molecular docking has been conducted using the previously described technique.54 However, here, we have used ETC complex I [PDB 5XTD] to find the interaction of BBT using a 96 × 114 × 102 affinity grid box. Similarly, the P-gp receptor [PDB 3G5U] has been used to find the binding of BBT and its various analogues. Here, the 66 × 70 × 74 affinity grid box was centered on the substrate binding sites that mostly include R, M, and H sites of P-gp.
Cell Cycle
For asynchronous cell cycle analysis,54 HeLa and MCF7 cells (5–6 × 106 cells/mL) were cultured in a six-well tissue culture dish overnight before treatment with BBT (1.56, 3.125, and 6.25 μM) for 12 h. Then, cells were fixed overnight with 70% ethanol at −20 °C. To perform staining of cellular DNA, cells were incubated with 100 μg/mL PI and 10 μg/mL RNase for 45 min at room temperature. Then, the cell cycle was analyzed using PI channels of a BD LSRFORTESA flow cytometer using emission filters at 610 nm.
Cell Death Analysis
Cell death has been analyzed using annexin V and propidium iodide (BD Biosciences)-based flow-cytometric studies.54 Further, autophagy was analyzed by a FlowCellect Autophagy LC3 antibody-based assay kit (Merck Millipore) using the manufacturer’s protocol. Briefly, cells (5–6 × 106 cells/mL) were harvested overnight in six-well plates followed by treatment with BBT for 12 h. Then, cells were selectively permeabilized to remove cytoplasmic LC3 but maintaining autophagosome-sequestered LC3 protected. Then, LC3 was detected using an anti-LC3 (FITC) antibody by a flow cytometer.
Mitochondrial Membrane Potential
Mitochondrial membrane potential was analyzed using the JC1 kit (BD MitoScreen, BD Biosciences) following the manufacturer’s protocol.
Caspase Activity Assay
Caspase activity was analyzed using Ac-DEVD-pNA that was performed according to a previously described method.53 Flow cytometric analysis of cleaved caspase 3 was conducted using a PE-rabbit anti-cleaved caspase 3 assay kit (BD Biosciences).
Spheroid Culture and Migration Assay
A HeLa cell in vitro tumor spheroid was prepared using an anchorage independent culture method as described previously.54 As spheroids reached a diameter of ≥100 μm, they were grouped as treated and untreated. At day 1, spheroids were treated with 6.25 and 12.5 μM BBT, then the growth of the spheroid was evaluated up to 8 days using bright field under an inverted microscope. Then, the change in the spheroid size is calculated in each day to have a growth kinetic plot. Migration assay was conducted using the fully grown HeLa spheroid.44 Briefly, the non-adherent culture of HeLa cell spheroids was performed as discussed above. Spheroids were transferred to a covered glass dish for attachment. After 5–6 days of spheroid growth, spheroids were collected intact and centrifuged at 200 × g for 3 min. Then, re-suspended spheroids were now transferred to adherent-type cover glasses. PI staining was performed to discriminate between live and dead migratory cells. The radius of the spheroid and migratory cells was defined as r and R, respectively.
DFT
All calculations were conducted using the Gaussian 09 program. The geometry was optimized with the restricted Becke Hybrid (B3LYP) at the 6-31G (d, p) level.
Statistical Analysis
All statistical calculations were done using GraphPad Prism 5.0 by performing one-way and two-way ANOVA. Statistical values of *p < 0.005and ***p < 0.0001 were allocated for different experiments accordingly.
Acknowledgments
Authors wish to thank Dr. Susanta Roychowdhury, Saroj Gupta Cancer Centre & Research Institute, Prof. Samit Chattopadhyay, Dr. Partha Chakrabarti, and Dr. Debabrata Biswas, CSIR-IICB for critically assessing this work and providing invaluable suggestions to improve the manuscript. S.M. thanks UGC, G.D. thanks ICMR, P.M. thanks CSIR, and V.G. thanks DST, India for their fellowships. S.G. kindly acknowledges DBT, India (BT/PR19159/NNT/1043/2016) for providing full financial support to carry out all the biological experiments and CSIR-IICB for the infrastructure. Authors wish to thank Ms. Juhee for critical reading and English editing of the manuscript.
Glossary
Abbreviations
- ROS
reactive oxygen species
- BBT
carbonyl-bridged bithiazole derivative
- LBT
linear bithiazole derivative
- HOMO
highest occupied molecular orbital
- LUMO
lowest unoccupied molecular
- Hsp27
heat shock protein 27
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- LC3
microtubule-associated protein 1A/1B-light chain 3
- pNA
p-nitroaniline
- NADH
nicotinamide adenine dinucleotide
- ETC
electron transport chain
- P-gp
P-glycoprotein
- GSH
glutathione
- FMN
flavin mononucleotide
- VDAC
voltage-dependent anion channel
- SOD2
superoxide dismutase
- ATP
adenosine triphosphate
- PARP
poly(ADP-ribose) polymerase
- MDR1
multidrug resistant 1 protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00308.
Microscopic images show the oxidative state of flavoprotein in green fluorescence, and mitochondria have been stained with MTRC (MitoTracker Red CMXRos) showing red fluorescence; histograms and graph represent MitoSOX assay in MCF7 cells; histograms and graph represent mitochondrial ROS in HeLa cells using MitoSOX; microscopic images of DCFDA assay in MCF7 cells, flow-cytometric quantification of DCFDA assay in cells after treatment of 25 μM BBT, microscopic images representing DHE assay in MCF7, and DHE assay based flow-cytometric quantification of ROS generation in MCF7 after treatment with an increasing concentration of BBT; histogram represents quantification of cytoplasmic ROS using DCFDA in MCF7 cells; histogram represents quantification of cytoplasmic ROS using DHE in MCF7 cells and bar diagram representation of quantification of ROS using DHE in MCF7; analysis of mitochondrial ROS using MitoSOX dye in HeLa cells after treatment of 40 μM Rot (rotenone), 12.5 μM BBT, and Rot with BBT using FACS; cell viability curve of various cancer cells after treatment of various concentrations of BBT; cell viability curve of BBT-treated WI-38 cells; bar diagram represents the proportion of MCF7 cells in each phase on treatment with varying doses of BBT; histogram represents autophagic cell population using anti-LC3 (FITC) antibody by FACS in the absence (control; green histogram) and presence of 12.5 μM BBT; bar diagram representation of the JC-1 assay measured through the green/red ratio of the JC-1 dye; microscopic images show immunolocalization of cytochrome C (cyt C) in HeLa cells; activation of caspase 3 by using DEVD-pNA using a spectrophotometer; cleaved caspase 3 analysis in HeLa cells before (control) and after (12.5 μM BBT) treatment by flow cytometry; and classification of BBT and its analogues as the P-gp substrate or non-P-gp substrate (PDF)
Author Contributions
⊥ S.M., G.D., and V.G. contributed equally.
Author Contributions
S.M. and G.D. performed major experiments, analyzed the presented data, and prepared the figures in this manuscript and helped S.G. in writing the manuscript. V.G. helped S.M. in various cell-based assays and in vivo experiments. P.M. performed FRET and computational experiments. M.N. and Y.I. synthesized, purified, and characterized all the compounds used in this manuscript. S.C. performed the DFT calculations. S.G. conceived the idea, designed, and monitored all the biological experiments. Y.A. supervised the synthesis and wrote the chemistry part as well as DFT part of the manuscript.
S.G. kindly acknowledges DBT, India (BT/PR19159/NNT/1043/2016) for providing full financial support to carry out all the biological experiments and IIT Jodhpur and CSIR-IICB for the infrastructure. S.G. and Y.A. thank DST, India and JSPS, Japan for initiating this project under the Indo-Japan Science Cooperative Program. Y.A. thanks “Dynamic Alliance for Open Innovation Bridging Human, Environmental and Materials” from The Ministry of Education, Culture, Sports, Science and Technology, Japan for supporting the chemistry part of this project.
The authors declare no competing financial interest.
Supplementary Material
References
- Sena L. A.; Chandel N. S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov R. G.; Baradaran R.; Sazanov L. A. The architecture of respiratory complex I. Nature 2010, 465, 441–445. 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- Efremov R. G.; Sazanov L. A. Structure of the membrane domain of respiratory complex I. Nature 2011, 476, 414–420. 10.1038/nature10330. [DOI] [PubMed] [Google Scholar]
- Bienert G. P.; Møller A. L. B.; Kristiansen K. A.; Schulz A.; Møller I. M.; Schjoerring J. K.; Jahn T. P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- Ďuračková Z. Some Current Insights into Oxidative Stress. Physiol. Res 2010, 59, 459–469. 10.33549/physiolres.931844. [DOI] [PubMed] [Google Scholar]
- Balaban R. S.; Nemoto S.; Finkel T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Kamata H.; Honda S.-I.; Maeda S.; Chang L. F.; Hirata H.; Karin M. Reactive oxygen species promote TNF alpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Sullivan L. B.; Chandel N. S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 2049–3002. 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharwal S. S.; Schumacker P. T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel?. Nat. Rev. Cancer 2014, 14, 709–721. 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira V.; Hay N. Molecular Pathways: Reactive Oxygen Species Homeostasis in Cancer Cells and Implications for Cancer Therapy. Clin Cancer Res. 2013, 19, 4309–4314. 10.1158/1078-0432.CCR-12-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H.; Mak T. W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. 10.1038/nrc1412. [DOI] [PubMed] [Google Scholar]
- Kolenko V. M.; Uzzo R. G.; Bukowski R.; Finke J. H. Caspase-dependent and -independent death pathways in cancer therapy. Apoptosis 2000, 5, 17–20. 10.1023/A:1009677307458. [DOI] [PubMed] [Google Scholar]
- Sastry K. S. R.; Al-Muftah M. A.; Li P.; Al-Kowari M. K.; Wang E.; Chouchane A. I.; Kizhakayil D.; Kulik G.; Marincola F. M.; Haoudi A.; Chouchane L. Targeting proapoptotic protein BAD inhibits survival and self-renewal of cancer stem cells. Cell Death Differ. 2014, 21, 1936–1949. 10.1038/cdd.2014.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I.; Ueno T.; Kominami E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Persidis A. Cancer multidrug resistance. Nat. Biotechnol. 1999, 17, 94–95. 10.1038/5289. [DOI] [PubMed] [Google Scholar]
- Ramirez M.; Rajaram S.; Steininger R. J.; Osipchuk D.; Roth M. A.; Morinishi L. S.; Evans L.; Ji W.; Hsu C.-H.; Thurley K.; Wei S.; Zhou A.; Koduru P. R.; Posner B. A.; Wu L. F.; Altschuler S. J. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 1. 10.1038/ncomms10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R. B. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug. Metab. Rev. 2002, 34, 47–54. 10.1081/DMR-120001389. [DOI] [PubMed] [Google Scholar]
- Arrigo A. P.; Virot S.; Chaufour S.; Firdaus W.; Kretz-Remy C.; Diaz-Latoud C. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid. Redox Signaling 2005, 7, 414–422. 10.1089/ars.2005.7.414. [DOI] [PubMed] [Google Scholar]
- De Smaele E.; Zazzeroni F.; Papa S.; Nguyen D. U.; Jin R.; Jones J.; Cong R.; Franzoso G. Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 2001, 414, 308–313. 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- Limnios D.; Kokotos C. G. 2,2,2-Trifluoroacetophenone: An Organocatalyst for an Environmentally Friendly Epoxidation of Alkenes. J. Org. Chem. 2014, 79, 4270–4276. 10.1021/jo5003938. [DOI] [PubMed] [Google Scholar]
- Ie Y.; Nitani M.; Karakawa M.; Tada H.; Aso Y. Air-Stable n-Type Organic Field-Effect Transistors Based on Carbonyl-Bridged Bithiazole Derivatives. Adv. Funct. Mater. 2010, 20, 907–913. 10.1002/adfm.200901803. [DOI] [Google Scholar]
- Ames B. N.; Shigenaga M. K.; Hagen T. M. Oxidants, Antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 7915–7922. 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Zhao J.; Han G.; Liu Z.; Liu C.; Zhang C.; Liu B.; Jiang C.; Liu R.; Zhao T.; Han M.-Y.; Zhang Z. Real-Time Discrimination and Versatile Profiling of Spontaneous Reactive Oxygen Species in Living Organisms with a Single Fluorescent Probe. J. Am. Chem. Soc. 2016, 138, 3769–3778. 10.1021/jacs.5b12848. [DOI] [PubMed] [Google Scholar]
- Tse D. C. S.; Kuwana T. Electrocatalysis of dihydronicotinamide adenosine diphosphate with quinones and modified quinone electrodes. Anal. Chem. 1978, 50, 1315–1318. 10.1021/ac50031a030. [DOI] [Google Scholar]
- Saleh F. S.; Rahman M. R.; Okajima T.; Mao L.; Ohsaka T. Determination of formal potential of NADH/NAD+ redox couple and catalytic oxidation of NADH using poly(phenosafranin)-modified carbon electrodes. Bioelectrochemistry 2011, 80, 121–127. 10.1016/j.bioelechem.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Vasudevan D.; Wendt H. Electroreduction of oxygen in aprotic media. J. Electroanal. Chem. 1995, 392, 69–74. 10.1016/0022-0728(95)04044-O. [DOI] [Google Scholar]
- Tanida I.; Waguri S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 2010, 648, 193–214. 10.1007/978-1-60761-756-3_13. [DOI] [PubMed] [Google Scholar]
- Mitra K.; Kim W.; Daniels J. S.; Gates K. S. Oxidative DNA cleavage by the antitumor antibiotic leinamycin and simple 1,2-dithiolan-3-one 1-oxides: Evidence for thiol-dependent conversion of molecular oxygen to DNA-cleaving oxygen radicals mediated by polysulfides. J. Am. Chem. Soc. 1997, 119, 11691–11692. 10.1021/ja971359z. [DOI] [Google Scholar]
- Gottlieb E.; Armour S. M.; Harris M. H.; Thompson C. B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. 10.1038/sj.cdd.4401231. [DOI] [PubMed] [Google Scholar]
- Dhillon A. S.; Hagan S.; Rath O.; Kolch W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Tournier C.; Hess P.; Yang D. D.; Xu J.; Turner T. K.; Nimnual A.; Bar-Sagi D.; Jones S. N.; Flavell R. A.; Davis R. J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- Le Rhun Y.; Kirkland J. B.; Shah G. M. Cellular responses to DNA damage in the absence of poly(ADP-ribose) polymerase. Biochem. Biophys. Res. Commun. 1998, 245, 1–10. 10.1006/bbrc.1998.8257. [DOI] [PubMed] [Google Scholar]
- Huang C. C.; Chia W. T.; Chung M. F.; Lin K. J.; Hsiao C. W.; Jin C.; Lim W. H.; Chen C. C.; Sung H. W. An Implantable Depot That Can Generate Oxygen in Situ for Overcoming Hypoxia-Induced Resistance to Anticancer Drugs in Chemotherapy. J. Am. Chem. Soc. 2016, 138, 5222–5225. 10.1021/jacs.6b01784. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R. J.; Ferreira M.-J. U.; dos Santos D. J. V. A. Molecular Docking Characterizes Substrate-Binding Sites and Efflux Modulation Mechanisms within P-Glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. 10.1021/ci400195v. [DOI] [PubMed] [Google Scholar]
- Mehlen P.; Kretz-Remy C.; Préville X.; Arrigo A. P. Human hsp27, Drosophila hsp27 and human alpha B-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against TNF alpha-induced cell death. EMBO J. 1996, 15, 2695–2706. 10.1002/j.1460-2075.1996.tb00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido C.; Bruey J.-M.; Fromentin A.; Hammann A.; Arrigo A. P.; Solary E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 1999, 13, 2061–2070. 10.1096/fasebj.13.14.2061. [DOI] [PubMed] [Google Scholar]
- Garrido C.; Ottavi P.; Fromentin A.; Hammann A.; Arrigo A. P.; Chauffert B.; Mehlen P. HSP27 as a mediator of confluence-dependent resistance to cell death induced by anticancer drugs. Cancer Res. 1997, 57, 2661–2667. [PubMed] [Google Scholar]
- Mori-Iwamoto S.; Kuramitsu Y.; Ryozawa S.; Mikuria K.; Fujimoto M.; Maehara S. I.; Maehara Y.; Okita K.; Nakamura K.; Sakaida I. Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2007, 31, 1345–1350. 10.3892/ijo.31.6.1345. [DOI] [PubMed] [Google Scholar]
- Papa S.; Zazzeroni F.; Bubici C.; Jayawardena S.; Alvarez K.; Matsuda S.; Nguyen D. U.; Pham C. G.; Nelsbach A. H.; Melis T.; De Smaele E.; Tang W.-J.; D’Adamio L.; Franzoso G. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2004, 6, 146–153. 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- Dean M.; Fojo T.; Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Mohapatra S.; Nandi S.; Chowdhury R.; Das G.; Ghosh S.; Bhattacharyya K. Spectral mapping of 3D multi-cellular tumor spheroids: time-resolved confocal microscopy. Phys. Chem. Chem. Phys. 2016, 18, 18381–18390. 10.1039/C6CP02748B. [DOI] [PubMed] [Google Scholar]
- Li Y.; Rogoff H. A.; Keates S.; Gao Y.; Murikipudi S.; Mikule K.; Leggett D.; Li W.; Pardee A. B.; Li C. J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 1839–1844. 10.1073/pnas.1424171112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinci M.; Box C.; Zimmermann M.; Eccles S. A. Tumor spheroid-based migration assays for evaluation of therapeutic agents. Methods Mol. Biol. 2013, 986, 253–266. 10.1007/978-1-62703-311-4_16. [DOI] [PubMed] [Google Scholar]
- Ishiguro T.; Ohata H.; Sato A.; Yamawaki K.; Enomoto T.; Okamoto K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. 10.1111/cas.13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu L. M.; Ju R. J.; Liu R.; Bu Y. Z.; Zhang J. Y.; Li X. Q.; Zeng F.; Lu W. L. Dual-functional drug liposomes in treatment of resistant cancers. Adv. Drug Delivery Rev. 2017, 115, 46–56. 10.1016/j.addr.2017.04.006. [DOI] [PubMed] [Google Scholar]
- Bhunia D.; Saha A.; Adak A.; Das G.; Ghosh S. A dual functional liposome specifically targets melanoma cells through integrin and ephrin receptors. RSC Adv. 2016, 6, 113487–113491. 10.1039/C6RA23864E. [DOI] [Google Scholar]
- Martin A.; Byrne A.; Burke C. S.; Forster R. J.; Keyes T. E. Peptide-Bridged Dinuclear Ru(II) Complex for Mitochondrial Targeted Monitoring of Dynamic Changes to Oxygen Concentration and ROS Generation in Live Mammalian Cells. J. Am. Chem. Soc. 2014, 136, 15300–15309. 10.1021/ja508043q. [DOI] [PubMed] [Google Scholar]
- Kim E. J.; Bhuniya S.; Lee H.; Kim H. M.; Cheong C.; Maiti S.; Hong K. S.; Kim J. S. An Activatable Prodrug for the Treatment of Metastatic Tumors. J. Am. Chem. Soc. 2014, 136, 13888–13894. 10.1021/ja5077684. [DOI] [PubMed] [Google Scholar]
- Simon H. U.; Haj-Yehia A.; Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. 10.1023/A:1009616228304. [DOI] [PubMed] [Google Scholar]
- Gupta V.; Yang J.; Liebler D. C.; Carroll K. S. Diverse Redoxome Reactivity Profiles of Carbon Nucleophiles. J. Am. Chem. Soc. 2017, 139, 5588–5595. 10.1021/jacs.7b01791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Hu K.; Yu Y.; Rotenberg S. A.; Amatore C.; Mirkin M. V. Direct Electrochemical Measurements of Reactive Oxygen and Nitrogen Species in Nontransformed and Metastatic Human Breast Cells. J. Am. Chem. Soc. 2017, 139, 13055–13062. 10.1021/jacs.7b06476. [DOI] [PubMed] [Google Scholar]
- Saha A.; Mohapatra S.; Das G.; Jana B.; Ghosh S.; Bhunia D.; Ghosh S. Cancer Cell Specific Delivery of Photosystem I Through Integrin Targeted Liposome Shows Significant Anticancer Activity. ACS Appl. Mater. Interfaces 2017, 9, 176–188. 10.1021/acsami.6b13352. [DOI] [PubMed] [Google Scholar]
- Mohapatra S.; Saha A.; Mondal P.; Jana B.; Ghosh S.; Biswas A.; Ghosh S. Synergistic Anticancer Effect of Peptide-Docetaxel Nanoassembly Targeted to Tubulin: Toward Development of Dual Warhead Containing Nanomedicine. Adv. Healthcare Mater. 2017, 6, 1600718. 10.1002/adhm.201600718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.