

Abstract

The geometrical characteristic and the degree of CO2 activation of the CO2-coordinated Ni(0) complexes were investigated computationally by quantum chemical means for bidentate and tridentate ligands of PP, PPMeP, and PNP, and sometimes with co-complexing Fe(II) to differently coordinate CO2. We show that the coordination geometry of the central metal is determined by the ligand geometry. The charge and the energy decomposition analyses show that the charge transfer energy through orbital mixing has a strong correlation with CO2 net charge, while the binding energy cannot due to the lack of the coordination number and the deformation energy of the ligand. Among the examined ligands, PNP with negatively charged secondary amine makes Ni(0) an electron-rich atom, which results in an ∼20% higher CO2 activation than those of PP and PPMeP. In particular, Fe(II)-PNP in the CO2-bridged diatomic complex enhances CO2 activation by another ∼20%, partly through the inductive effect of Fe(II), which pulls electron density from Ni-PNP across the CO2-bridge and partly by the backward donation from Fe(II)-PNP. Therefore, the present study encourages us to design a strongly electron-donating ligand and a CO2-bridged diatomic complex to develop more efficient homogeneous catalyst.
I. Introduction
As human civilization has continued to develop, energy consumption has increased exponentially, especially after the industrial revolution. Because this energy demand has mostly been met by burning carbonaceous fossil fuels such as oil, coal, and natural gas, the liberated carbon has increased the CO2 level in the air, which is believed to be responsible for global warming and the ongoing climate disaster on this planet.1 As a result, developing renewable energies and related technologies for environmental protection has become an urgent and imminent issue. Although there are diverse approaches to mitigate the CO2 level in the atmosphere, the most promising strategy is to reduce CO2 to any form of a valuable material because carbon itself is also a necessary resource for energy circulation and sustaining the framework of organic substances.2 By chemical reduction, depending on the reduction pathway and the degree of reduction, CO2 is converted into an energy resource such as CO, methanol, or methane. In the full pathway of CO2 reduction, the first reduction step of CO2 + e– → CO2– shows the highest activation barrier of 1.9 eV.3 Inevitably, this higher activation barrier necessitates the assistance of the catalyst toward decreasing the energy cost in the CO2 reduction.
There have been many developments in finding catalysts for CO2 activation in the areas of both homogeneous4 and heterogeneous catalysis.5 Regardless of the area, to understand the nature of CO2 in the activated form with the characteristics of the catalysts themselves will be important in developing new strategies of activating CO2.6 In the homogeneous area, the molecular catalyst is solvated in a solution, normally in water, and it takes part in a catalyzing reaction in free solution or by being bound on the electrode surface. These catalysts are conventionally composed of a central metal (M), normally a transition metal (TM), and surrounding ligands. Carbon dioxide then adds to a vacancy in a metal atom or replaces a loosely bound ligand for further reaction. Many combinations of diverse metals and ligands have been studied in detail.7 Among them, Ni-cyclam8−12 (cyclam = 1,4,8,11-tetraazacyclotetradecan) and Fe- and Co-porphyrin families7 have shown interesting behaviors in terms of their performance and chemical mechanisms. Particularly in the cyclam series, [Ni(II)(cyclam)]Cl2 showed superior performance to those of Co2+ and Fe2+ with a low bias of −1.05 V and higher CO selectivity.9,10 A mechanistic study by Song et al.8 revealed that Ni(II)(cyclam) is first bound to the electrode surface and reduced by one electron with −1.07 V (experimentally −1.23 V),12 Ni(I)(cyclam). This step is followed by thermoneutral CO2 binding where CO2 is reduced by one electron and Ni(I) is oxidized to Ni(II). In the whole catalytic reaction, the role of Ni(I) is crucial, as CO2 is attached to Ni(cyclam)+. Also, the amount of charge moving from Ni(I) to CO2 in this step would control the feasibility of the whole reduction mechanism.
The effect of the ligand was further studied experimentally by substituting H with a methyl group in the secondary amine position of the cyclam. The substitution lowered the overpotential compared to the pristine Ni(cyclam)2+, but it enhanced the Gibbs energy of CO2 binding, which overall decreased the catalyst performance.8 The performance deterioration was ascribed to the thermodynamic stability of the CO2 adduct and its steric hindrance. Even though important insights have been drawn from these studies, many questions remain with regard to the characteristics of ligands (e.g., steric and/or inductive effect, roles of anchor atoms such as N, P, and S) and the central metals (e.g., the shape of d- and f-orbitals, HOMO-LUMO energy gap, and the oxidation state). In the case of polydentate ligands, the coordination bond between the central metal and the coordinating atoms can be strained and affect the catalytic role.
Because the state of the central metal, Ni(I) in cyclam rather than Ni(II), is crucial for CO2 reduction, as stated above,8 the study for the low-valent metal-CO2 adducts has attracted much interest. After the characterization of (PCy3)2Ni(η2-CO2)13 (Cy = cyclohexyl) with zero-valent Ni, referred to as Ni0, many Ni-CO2 adducts with low valency were compounded.14,15 When the metal makes a coordination bond to C of CO2, Ni0 with large electron density can act as a strong nucleophile. Therefore, it is expected that the lower valent Ni will reduce CO2 much more and make it more feasible to reduce CO2 to other materials.
Recently, Lee’s group synthesized a series of CO2-coordinated Ni0 complexes where each ligand has a denticity of two or three for each Ni0 atom.16−19 Therefore, it is very heuristic to investigate the nature of Ni0-CO2 interaction systematically for this series. The models to be studied are represented in Scheme 1. The first model is (dtbpe)Ni-(η2-CO2) (dtbpe = 1,2-bis(di-tert-butylphosphino)ethane)14 denoted by 1-PP, where dtbpe is a bidentate neutral ligand with two P’s as anchor atoms. The second one is (PPMeP)Ni-(η2-CO2)16 denoted by 2-PPMeP, where PPMeP (PMe[2-PiPr2-C6H4]2, PiPr2 = di-iso-propylphosphino-) is the modification of PPP anion of bis(2-di-iso-propylphosphinophenyl)phosphide20 as a tridentate ligand. Note that all P’s are tertiary phosphine excluding coordination to Ni. The third one is (PNP)Ni-η1-CO2-κC (PNP = N[2-PiPr2–4-Me-C6H3]2–)19,21 denoted by 3-PNP, where PNP is an anionic ligand of the charge of −1. Note that the middle N in PNP is a secondary amine with two lone pairs, which results in a formal charge of −1. The fourth one is (PNP)Ni-μ-CO2–κC:κ2O,O′-Fe(PNP)18 as denoted by 4-Ni-CO2-Fe, where two metal centers are bridged by CO2. This is a biomimetic complex of carbon monoxide dehydrogenase (CODH), which converts CO2 and CO reversibly.22,23 In the active site of CODH, CO2 is bridged between two metal centers of Ni and Fe (Ni-μ-CO2–κC:κO-Fe), as shown in Figure S1. Some calculations have been performed for a hypothetical model, as shown in Scheme S1, which replaces the middle P in 2-PPMeP with the tertiary N, referred to as 2′-PNMeP, to understand the difference of the effects of the anchor atom species and its valence electrons. 2′-PNMeP resembles 2-PPMeP with respect to the shape of valence electrons while it has the same anchor atom species as 3-PNP. We try to analyze the structural characteristics and the degree of CO2 activation for these models based on DFT calculations. For the estimation of the degree of CO2 activation, the electron charge density of CO2 is analyzed because the amount of electron charges on a CO2 molecule would facilitate the CO2 reduction reaction.
Scheme 1. Schematic Presentation of Four Models.

The metallic oxidation number and total charge are denoted. R, R2, and R3 represent the methyl, isopropyl, and isobutyl groups, respectively.
II. Computational Results and Discussion
For theoretical investigation, the hybrid density functional of B3LYP/6-31G(d) with dispersion correction24 equipped in Q-Chem25 was applied for geometry optimization, and the triple zeta basis set of G3Large was used for energy calculation. Each model was fully optimized, and the natural bond orbital (NBO)26 analysis was applied for the charge of metals (M = Ni, Fe) and CO2. The energy decomposition analysis (EDA)27 was done between the fragments of CO2 and the metal complex for each model. The charges and spin multiplicities used in the calculation are 0, 0 for Ni(0) and +2, 5 for Fe(II), and the overall charge of ligand PNP is −1, whereas all the other ligands are neutral.
a. Analysis of Geometry
The optimized structures are shown in Figure 1, and some geometrical parameters are summarized in Table 1. In Figure 1, H was removed, and C of ligands is drawn as a point for clear view. The full geometrical presentations including H are presented in Figure S2 in the Supporting Information.
Figure 1.
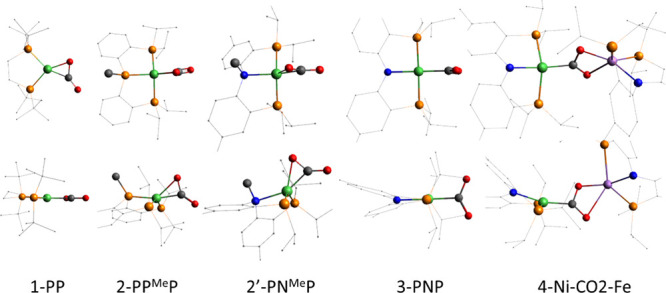
The optimized structures of five models in Scheme 1 and Scheme S1 are drawn with the top view (top) and side view (bottom) using B3LYP/6-31G(d) + D. H’s are removed and some C’s in ligands are drawn as points for clarity. Color scheme: Ni, green; P, brown; C, black; O, red; N, blue; Fe, violet.
Table 1. Geometry Parameters for Models in Scheme 1 Using B3LYP/6-31G(d) + Da.
| parameters | 1-PP | 2-PPMeP | 3-PNP | 4-Ni-CO2-Fe |
|---|---|---|---|---|
| chg, mul | 0, 1 | 0, 1 | –1, 1 | 0, 5 |
| geometry Ni | Pl (Pl)14 | Td (Td)16 | Pl (Pl)19 | Pl (Pl)18 |
| d(C–O) (Å) | 1.289, 1.212 (1.266,1.200) | 1.252, 1.224 (1.252, 1.217) | 1.258, 1.257 (1.248, 1.247) | 1.318, 1.255 (1.289, 1.269) |
| d(Ni–C) | 1.811 (1.868) | 1.875 (1.904) | 1.880 (1.911) | 1.844 (1.831) |
| d(Ni–O) | 1.877 (1.904) | 2.234 (2.191) | 2.664 (2.614) | 2.856, 2.575 (2.705, 2.772) |
| ∠O–C–O(°) | 137.8 (138.0) | 133.4 (135.1) | 129.6 (128.5) | 118.4 (116.5) |
| d(Fe–O) | 1.946, 2.503 (2.008, 2.289) | |||
| ∠O–Fe–O(°) | 58.0 (61.0) |
The values in parenthesis are given from crystallography. Pl and Td stand for planar and tetrahedral structures, respectively.
Because there are so many CO2 adsorption modes for a single-atomic and diatomic catalyst,4 all the modes addressed in this work are presented in Figure S3. It is known that η2-C,O is the most stable for Ni0 without any ligands.28 The ground state of Ni0 with the coordination of a ligand is a singlet state of d10 (Table S1). As presented in the previous work,281-PP and 2-PPMeP have η2-C,O mode as shown in Figure 1. While many CO2-adducts appear as η2-mode, it is known that some CO2-adducts have η1-C mode when a metal is in a relatively low oxidation state so that the energy of the d-orbital is relatively high, as well as when a central metal has a square planar or square pyramidal structure.4 Then, in 3-PNP, Ni0 has square planar-like structure without CO2, which makes it prefer η1-C mode, as shown in Figure 1. As for Fe(II) in model 4-Ni-CO2-Fe, it has κ2O,O′ mode with respect to CO2. Note that the charge of PNP-Fe is +1 as it is composed of Cl– in synthesis.17 Hereafter, Ni(0)-PNP and Fe(II)-PNP without the coordination to CO2 are simply denoted as Ni-PNP and Fe-PNP, respectively, without any complexity in notation. The calculation shows that Fe2+ has a multiplicity of quintets with high spin in PNP-Fe, as shown in Table S2. All the charges and multiplicities of models in calculation are listed in Table 1.
The values with parentheses in Table 1 come from X-ray crystal structures. In this calculation regime, the coordination geometries of all the models are well reproduced except for 4-Ni-CO2-Fe, where two bulky PNP ligands are closely located and the calculation overestimates the dispersion correction, causing the ligands to become slightly twisted, as shown in Figure S4.
From 1-PP to 3-PNP, there is a systematic change of ligands. As for Ni0 with two coordination to P’s, there is an in-depth theoretical study with minimal monodentate ligands of two PH3, where two ligands have no geometrical constraint.29 In the 1-PP model, although two ligands are connected by the ethyl group, the geometry with Ni and CO2 is not much different from that with the two monodentate ligands. All the atoms of CO2 lie in the molecular plane, as shown in Figure 1, as is the case with Ni(PH3)2.29
Moving to 2-PPMeP, the coordination number is increased to three with the tridentate ligand of PPMeP, where Ni has a Td-like structure with three P’s and CO2 in η2-C,O mode. The CO2 plane is perpendicular to the ligand plane, which is composed of the three anchor atoms of the ligand. To understand the effect of the tridentate ligands on coordination geometries, three tridentate ligands are shown in Figure 2. The optimized structure of PPMeP is shown in Figure 2 a, where some valence orbitals are drawn with parity. Those orbitals mainly correspond to the lone pair electrons of the anchor atoms and are directed to above the ligand plane displayed with green triangle. Each MO is separately drawn in Figure S5. The directions of the lone pairs are ascribed to the sp3 structure of the tertiary P as shown with the black arrow. Therefore, Ni can be located on top of the ligand plane, which results in the tetrahedral structure of Ni, as shown in Figure 1.
Figure 2.

The optimized ligand structure of (a) PPMeP, (b) PNMeP, and (c) PNP, and (d) structure of [Ni(0)(PCH3)2NH2]−1-CO2 calculated by B3LYP/6-31G(d) + D. In the ligand structures (a–c), H was removed for clarity. The black arrows denote the direction of the occupied molecular orbitals (MO) near the HOMO level, and the green triangle shows the ligand plane. The MOs in ligand structures are overlaid as much as the number of lone pairs. Colors scheme: Ni, green; C, gray; P, orange; N, blue; H, white; Fe, brown (drawn using Jmol).
The effect of the substitution of the tertiary N for the middle PMe in PPMeP, referred to as PNMeP, is shown in Figure 2b, where MOs are almost identical to PPMeP in Figure 2a. Therefore, the geometry of the hypothetical model of 2′-PNMeP remains almost the same as 2-PPMeP, but the bond length of N–Ni and the angle of Me-N-Ni were decreased by about 0.05 Å and 25°, respectively, compared to 2-PPMeP due to the size and the electronegativity of N.
In 3-PNP, Ni-PNP makes a planar structure with η1-C for CO2, where all the anchor atoms and Ni locate in the same plane together with C of CO2, as shown in Figure 1. The structure of ligand PNP and the four uppermost occupied MOs are shown in Figure 2c, where MOs are located in the ligand plane. The two lone pairs of N are completely symmetric with respect to the ligand plane as shown in Figure S6, and the ligand shows the complete C2 symmetry, as shown in Figure S7a. Therefore, Ni0 as well as Fe(II) is located in the ligand plane, making the Pl structure of Ni-PNP and Fe-PNP. From the ligand field theory, Ni0 with a strongly donating ligand makes the electronic configuration of d10,10 which leaves sp3 orbitals for M-L (metal–ligand) coordination, resulting in a Td structure. Therefore, if the PNP ligand is replaced by three monodentate ligands such as two P(CH3)3 and one [NH2]−1, it surely goes to a Td structure with η2-C,O mode for CO2 coordination, as shown in Figure 2d. Therefore, although Ni0 prefers a Td structure in its low oxidation state, when it is enforced to have a Pl structure, for example, due to the constraint of the chelating PNP ligand, it favors the η1-C mode in CO2 binding.
In 4-Ni-CO2-Fe, CO2 is bridged by Ni-PNP and Fe-PNP in the type of μ-CO2-κC:κ2O,O′. This molecule is prepared by two different metal complexes of Ni-PNP and Fe-PNP. Note that although the coordination structure of Fe-PNP is Pl in its optimized structure, it becomes Td in 4-Ni-CO2-Fe.
b. Analysis of Electronic Structure
To study the electronic properties of CO2-coordinated Ni0 complexes, the energy levels and the atomic basis coefficients of molecular orbitals (MOs) were analyzed. The MO energy levels of 1-PP are shown in Figure 3a with its two fragments of CO2 (right side) and the Ni complex (left side) referred to as 1-PP-A in their fixed geometries. When a free CO2 is bent, the doubly degenerate HOMO (πgn, nonbonding orbital with 1 node) and LUMO (πu*, antibonding orbital with 2 nodes) levels are broken and the HOMO-LUMO gap is decreased as the HOMO level is increased and LUMO level is lowered, as shown in Figure S8. In the broken degeneracy, the MOs in the CO2 plane, also called in-plane mode, take up LUMO (πip*) and HOMO (πipn), while the MOs perpendicular to them, also called out-of-plane mode, are located at LUMO+1 (πoop*) and HOMO-1 (πoopn). In Figure 3a, the red lines denote the occupied orbital levels, and the blue ones denote the unoccupied orbital levels. In Figure 3a, only the selected MO levels are drawn, which are related to the atomic orbitals of Ni and CO2 for clarity. The green dashes denote the corresponding MOs between before and after CO2 coordination, which can be just the energy shift or an orbital interaction. All the levels and the relaxed structures of both fragments are shown in Figure S9. From the relaxed geometry to the fixed geometry (1st → 2nd and 5th → 4th column in Figure S9), the occupied levels are raised and the unoccupied levels are lowered around HOMO and LUMO levels, which explains the energetically unstable structure due to geometrical deformation. In the binding of 1-PP-A and CO2 in Figure 3a, the MO levels of Ni complex decrease while those of CO2 increase. This large shift of the energy levels is ascribed to the charge transfer from Ni to CO2, where the electron–electron repulsion energy is raised in CO2 and lowered in Ni. The backward donation normally occurs through the orbital interactions between LUMO of the bent CO2 (πip*) and any occupied orbitals of M complex, whereas the forward donation occurs through the orbital interactions between HOMO of the bent CO2 (πipn) and any unoccupied orbitals of M complex if it exists. In 1-PP, HOMO of 1-PP-A interacts with πip* of CO2, which means that the backward donation occurs dominantly and is responsible for the CO2 activation.
Figure 3.

(a) MO levels of 1-PP and its fragments of 1-PP-A and CO2 in fixed geometries. Only selected MOs are drawn, which are related to Ni and CO2. Representation of MOs of (b) HOMO of 1-PP-A (top view from z axis), (c) HOMO of 1-PP (side view from y axis), (d) LUMO+1 of 1-PP (slanted to view dxy-Ni), and (e) HOMO-7 of 1-PP. Colors scheme: Ni, green; C, gray; P, orange; N, blue; H, white (drawn using Jmol).
The five occupied orbitals below HOMO in 1-PP-A mainly belong to Ni-3d orbitals but also include 4s, 4p orbitals due to the pre-existing coordination bond to P’s. HOMO of 1-PP-A is mainly composed of dxy-Ni, as shown in Figure 3b. Note that the molecular ring is located on the xz plane. In Figure 3c, πip*-CO2 is well developed in HOMO of 1-PP, which comes from HOMO of 1-PP-A. Therefore, this demonstrates that dxy-Ni charges are moved to the empty CO2 orbital, which confirms the backward donation of CO2. The correspondent antibonding appears in LUMO+1 as shown in Figure 3d, where the parity of CO2 is changed compared to Figure 3c, while the parity of dxy-Ni remains the same. HOMO and HOMO-1 of the bent CO2 are located at HOMO-5 and -6 in 1-PP, respectively, which are just the shift of MO levels primarily due to the electrostatic effect ascribed to the increased charge density. Some complicated interactions appear as shown in Figure 3d and Figure S10; however, no trace of the forward donation was detected through MO analysis, so the binding of CO2 on Ni(0) occurs mainly through the backward donation.
The MO levels of 2-PPMeP and its fragments of CO2 and Ni complex (Ni-PPMeP-A) in fixed geometries are shown in Figure 4a, and there is not much difference from those of 1-PP. The overall molecular orbital levels including the relaxed geometries are also presented in Figure S11. In Figure 4a, there appears backward donation of CO2 with dx2–y2 and dxy in HOMO and HOMO-2 of 2-PPMeP-A, respectively. Then, the bonding and antibonding orbitals are shown in Figure 4c,d, and another backward donation at HOMO-2 in 2-PPMeP is detected Figure 4e.
Figure 4.

(a) MO levels of 2-PPMeP and its fragments of 2-PPMeP-A and CO2 in fixed geometries. Only selected MOs are drawn, which are related to Ni, and C and O in CO2. Representation of MOs of (b) HOMO of 2-PPMeP-A, (c) HOMO of 2-PPMeP, (d) LUMO+5 of 2-PPMeP, and (e) HOMO-2 of 2-PPMeP. Colors scheme: Ni, green; C, gray; P, orange; N, blue; H, white (drawn using Jmol).
The substitution of the tertiary P with the tertiary N for 2′-PNMeP does not make a significant difference, as shown in Figure S12, as expected through the similar valence electron structure. However, the secondary N-substitution effect on MO levels in 3-PNP is significant as shown in Figure 5a. The HOMO level of 3-PNP-A is much higher than the level of πip*-CO2. This is ascribed to the raised HOMO level of 3-PNP-A due to the negatively charged N, while the level of πip*-CO2 stays at a similar energy level. The MO level difference between HOMO of 3-PNP-A and LUMO of CO2 reaches 0.198 Hartree, which is much larger than those of 0.025–0.043 Hartree for the previous models. Therefore, this large energy difference can be a driving force for charge transfer from Ni complex to CO2. Due to the high energy of HOMO, PNP itself has a strong tendency to donate electrons when combined with any other molecule. Therefore, any M-PNP complex is expected to have a strong tendency to donate electrons when any molecule is coordinated to the central metal. Figure 5b shows HOMO of dx2–y2 of 3-PNP-A, which interacts with πip*-CO2. The bonding and antibonding orbitals are drawn in Figure 5c,d, and another backward donation appear, as shown in Figure 5e.
Figure 5.

(a) MO levels of 3-PNP and its fragments of 3-PNP-A and CO2 in fixed geometries. Only selected MOs are drawn, which are related to Ni and CO2. Representation of MOs of (b) HOMO of 3-PNP-A, (c) HOMO of 3-PNP, (d) LUMO+5 of 3-PNP, and (e) HOMO-2 of 3-PNP. Colors scheme: Ni, green; C, gray; P, orange; N, blue; H, white (drawn using Jmol).
Lastly, MO levels of the CO2-bridged bimetal complex of 4-Ni-CO2-Fe are presented in Figure 6a with those of its fragments in the fixed geometries. The green lines represent only the backward donations between metals and CO2. As for 4-Ni-CO2-Fe-A, two separate molecules of Ni-PNP and Fe-PNP exist, so they were calculated together as a supramolecule, as shown in Figure S14. The MO splitting in Figure 6a shows that both α- and β-spin contribute to the backward donation to CO2. In the bonding orbital of α-HOMO-11, two MOs from 4-Ni-CO2-Fe-A contribute, one from Ni (HOMO-3) and the other from Fe (HOME-5). The bonding and antibonding orbitals for α-spin are drawn in Figure 6b,c. In the α-bonding orbital, the C-pz orbital of πip*-CO2 bonds with Ni-dx2–y2 while one of two O-pz’s of πip*-CO2 interacts with the Fe-dxz orbital. Considering the parity of the Fe-dxz orbital and πip*-CO2, only one O1-pz orbital (blue color) can overlap with the d-orbital of Fe (blue color) because the other O2-pz (blue color) has the opposite parity to Fe (red color), as shown in Figure 6b. Therefore, we can say that some portion of the backward donation can occur through the Fe-PNP complex when co-complexing CO2 in κ2O,O′mode. Note that the α-spin is fully occupied in high spin of Fe2+, even though the d-orbital is not fully occupied. As for β-spin, the considerable backward donation appears only through Ni, as shown in Figure 6d. Because Fe2+ has only one electron of β-spin in high spin configuration, no meaningful backward donation occurs from Fe2+. Resultantly, Fe(II)-PNP partly contributes to the backward donation to CO2 in a concerted mode with Ni0 in its high spin configuration.
Figure 6.

(a) MO levels of 4-Ni-CO2-Fe and its fragments of 4-Ni-CO2-Fe-A and CO2 in fixed geometries. Only selected MOs are drawn, which are related to Ni, Fe, and C and O in CO2. Representation of MOs of (b) α-HOMO-11, (c) α-HOMO-2, (d) β-HOMO-6, and (e) β-LUMO+10 of 4-Ni-CO2-Fe. Colors scheme: Ni, green; Fe, brown; C, gray; P, orange; N, blue; H, white (drawn using Jmol).
c. Charge and Energy Decomposition Analysis
Energy decomposition analysis (EDA)27 was done and displayed in Figure 7a for the five models in Scheme 1 and Scheme S1. In Figure 7a, FRZ of EDA components stands for the electrostatic repulsion energy attributed to the frozen electron density when two fragments of CO2 and the Ni complex are combined in the fixed geometries. POL is the polarization energy through intrafragment relaxation from the frozen density, and CT is due to the interfragment electronic relaxation of MOs. Note that FRZ makes the system unstable while POL and CT make the system stable, so FRZ has an opposite sign. In Figure 7a, the sign of FRZ was reversed for comparison. The total SCF energy, referred to as SCF_total in Figure 7a, is given by CT + POL – FRZ. FRZ (orange line) is increasing from 1-PP to 4-Ni-CO2-Fe, which means that the overlap interaction between two fragments is increasing. The POLs (violet line) of 1-PP and 4-Ni-CO2-Fe are higher than those of 2-PPMeP and 3-PNP, but their differences are relatively small compared to FRZ and CT. The large differences in the total SCF (blue line) mainly come from CT (green line) so both energies show a similar trend in Figure 7a. Roughly estimated, it looks like the relative energy differences between models in FRZ and POL counterbalance each other; thus, SCF resembles CT.
Figure 7.

(a) Energy decomposition analysis (EDA). FRZ denotes frozen electron density, POL, polarization, CT, charge transfer, SCF_TOTAL, the sum of CT, POL, and -FRZ. (b) Comparison between CO2 charges from the natural atomic orbital and CT energy from EDA.
Therefore, the SCF energy is largely affected by the CT energy, which comes from the charge delocalization. Because the CO2 binding occurs mainly through the backward donation, CT and BE are expected to be aligned. However, by definition, BE includes the deformation energy of each fragment with reference to the fully relaxed structures of each fragment, which is different from SCF_TOTAL in EDA. BE in Figure S15a increases from 2-PPMeP to 4-Ni-CO2-Fe smoothly. However, the abnormally high BE of 1-PP is ascribed to the lack of coordination bonds to Ni, which increased POL in Figure 7a. The difference between SCF and BE is much more for 4-Ni-CO2-Fe, where the coordination geometry of Fe-PNP changes greatly from Pl to Td. This deformation energy abruptly reduced the BE of 4-Ni-CO2-Fe, which induces the inconsistency between BE and total SCF in this model series.
The natural atomic orbital (NAO) charge in natural bond orbital (NBO)26 package was applied to measure the amount of charges on CO2, which can directly determine the degrees of CO2 activation. The charges on CO2 of NAO and CT of EDA show strong correlation in Figure 7b. The NAO charges on CO2 are similar from 1-PP to 2-PPMeP but largely increase via 3-PNP to 4-Ni-CO2-Fe. This trend is similar to CT, which is represented by the green line in Figure 7b.
To quantitatively investigate the change of charges in the model series, NAO charges are analyzed as presented in Table 2, and all the atomic charges are in Table S3. The charge of M is the partial charge of M in each model. ΔeM and Δe(M,A’s) stand for the charge difference of M alone and the sum of M and its anchor atoms, respectively, between the cases before and after CO2-coordination. Some reference charges before CO2 coordination are presented in Figure 8a,c,e and Table S3. The amount of the negative charge of CO2 is around −0.80e in 1-PP and 2-PPMeP but increased to −1.04e in 3-PNP and −1.21e in 4-Ni-CO2-Fe. Note that as shown in Figure S16, the charge of CO2 cannot be correlated with that of the central metal alone, but when it is summed with the charges of the anchor atoms, some correlation appears. Therefore, from 1-PP to 2-PPMeP, CO2 gets a little less charged, although Ni0 becomes more positive. This is ascribed to the additional anchor atom, P, which is more electronegative (EN = 2.19) than Ni (EN = 1.91). The additional electronegative anchor atom would make the central atom less active for CO2 activation. As for the PNP ligand, N has formal charge of −1 with two lone pair electrons that can donate charges to the next metal Ni(0) and Fe(II). As shown in Figure 8c, Ni in 3-PNP is less positive compared to 2-PPMeP in Figure 8a. Then, after CO2 is coordinated, more electron charges on Ni can move to CO2 by the amount of about 0.31e (3-PNP), compared to 0.21e in 2-PPMeP, which results in CO2 activation of about −1.04e. As for the CO2-bridged diatomic complex of 4-Ni-CO2-Fe, the charge of CO2 is increased to about −1.21e. First, from the Ni-PNP side, there is a positive charge increase, which means some electron charges are donated to CO2. Before adding Fe-PNP, the remaining part of 4-Ni-CO2-Fe is exactly the same as 3-PNP. Therefore, by coordinating Fe-PNP to 3-PNP as κ2O,O′ mode, Δe(Ni0,A’s) in 4-Ni-CO2-Fe is raised by 0.15e compared to that of 3-PNP, as shown in Table 2. It is very interesting that Fe2+, a strong Lewis acid can draw electron density from Ni-PNP across the CO2 bridge, which means Fe2+ strengthens the electron donation of Ni-PNP to CO2. On the other side, ΔeFe2+ of 4-Ni-CO2-Fe shows an increased charge of about 0.15e, as shown in Table 2, compared to the charge of Fe-PNP shown in Table S3. Considering that the sum of the charges of the anchor atoms is almost not changed, it can be interpreted like this. Fe2+ in Fe-PNP has relatively lower partial charge due to the PNP ligand, compared to Fe2+ with neutral ligands. Therefore, some extra electron charges of Fe2+ in Fe-PNP can be donated to CO2 through the backward donation as was examined in MO analysis previously. Therefore, though Fe2+ is Lewis acidic, when it is combined with an electron-rich ligand such as PNP(−1), the backward donation from Fe2+ to CO2 can occur. Accordingly, Fe-PNP coordinated to CO2 with κ2O,O′ mode can activate CO2 through two methods: (1) by inductively attracting the electrons from the Ni-PNP side and (2) by directly donating the electron charges from itself.
Table 2. Partial Charges from Natural Atomic Orbital Analysisa.
| Ni0 |
Fe2+ |
||||||
|---|---|---|---|---|---|---|---|
| models | CO2 | Ni0 | ΔeNi0 | Δe(Ni0,A’s) | Fe2+ | ΔeFe2+ | Δe(Fe2+,A’s) |
| 1-PP | –0.812 | 0.804 | 0.334 | 0.588 | N/A | ||
| 2-PPMeP | –0.793 | 0.862 | 0.215 | 0.556 | N/A | ||
| 3-PNP | –1.044 | 0.792 | 0.318 | 0.729 | N/A | ||
| 4-Ni-CO2-Fe | –1.214 | 0.908 | 0.434 | 0.884 | 1.424 | 0.147 | 0.143 |
ΔeM and Δe(M,A’s) stand for the difference of atomic charges after CO2 coordination for M and the sum of M and the anchor atoms (A’s) of the ligand, respectively. The reference charges are 0.474 for Ni0 and 1.277 for Fe2+ in M complex without CO2.
Figure 8.

Atomic charges from natural atomic orbitals for (a) 2-PPMeP-A, (b) 2-PPMeP, (c) 3-PNP-A, (d) 3-PNP, (e) Fe-PNP, and (f) 4-Ni-CO2-Fe. Colors scheme: Ni, green; C, gray; P, orange; N, blue; Fe, brown.
III. Conclusions
We investigated the nature of CO2 binding on four Ni(0) complexes: 1-PP (η2-CO2), 2-PPMeP (η2-CO2), 3-PNP (η1-CO2), and 4-Ni-CO2-Fe (μ-CO2-κC:κ2O,O′). The DFT calculations in the regime of B3LYP/6-31G(d) + D accurately reproduced the coordination geometries of the central metals, where PPMeP ligand induces the tetrahedral structure (Td) of Ni and PNP ligand induces planar structure (Pl). Although the ligand field theory predicts that Ni0 prefers Td, the geometry of the chelating ligand enforces the coordination geometry of metal, where PPMeP leads to Td and PNP leads to Pl. As for CO2 binding mode, normally CO2 is strongly bound to the central metal in η2-mode, but planar structure of Ni0-PNP complex prefers η1-C mode.
Based on the analysis of molecular orbitals, the backward donation to LUMO of the bent CO2 is mainly responsible for the CO2 binding, which is also responsible for the degree of CO2 activation. However, the binding energy (BE), although it comes from backward donation, cannot precisely explain the degree of CO2 activation because the coordination number and the deformation of ligand do affect BE. Otherwise, the charges of CO2 show strong correlation with the charge transfer (CT) energy from the energy decomposition analysis (EDA). Compared to 1-PP and 2-PPMeP, 3-PNP shows a largely increased CO2 charge of about 0.2e due to the negatively charged [PNP]− ligand. Ni0 with the coordination to PNP is already in an electron-rich state, so it can activate any adsorbate such as CO2. In the diatomic complex of 4-Ni-CO2-Fe, an additional charge transfer of about 0.2e occurs. Interestingly, after the coordination of Fe-PNP to 3-PNP with κ2O,O′ mode, Fe(II) pulls the electron density from Ni-PNP across the CO2 bridge, which enhances the backward donation from Ni-PNP to CO2. Furthermore, Fe(II) also donates some charge from itself to CO2 through the backward donation mechanism. So, the resultant increased charge on CO2 comes partly from the inductive effect of Fe(II) and partly from the backward donation ascribed to PNP in Fe-PNP complex. Therefore, this finding gives insight to designing a new catalyst and explains why we should aim at the diatomic catalyst for CO2 reduction.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1B07049044 and 2019R1A6A1A10073887). The generous supercomputing time provided by KISTI is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c06257.
The details of the geometry of models, MO levels of Ni(0) atom, ligands, and models, multiplicity of Ni0 and Fe2+ and their complexes, BE and SCF of EDA, and NBO analysis (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- https://www.climate.gov/ (12, 01, 2020).
- Liu Q.; Wu L.; Jackstell R.; Beller M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933–5933. 10.1038/ncomms6933. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H.; Rush J. D. Reduction potential of the carbon dioxide/carbon dioxide radical anion: a comparison with other C1 radicals. J. Phys. Chem. 1987, 91, 4429–4430. 10.1021/j100300a045. [DOI] [Google Scholar]
- Gibson D. H. The Organometallic Chemistry of Carbon Dioxide. Chem. Rev. 1996, 96, 2063–2096. 10.1021/cr940212c. [DOI] [PubMed] [Google Scholar]
- Liu J. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34–59. 10.1021/acscatal.6b01534. [DOI] [Google Scholar]
- Álvarez A.; Borges M.; Corral-Pérez J. J.; Olcina J. G.; Hu L.; Cornu D.; Huang R.; Stoian D.; Urakawa A. CO2 Activation over Catalytic Surfaces. ChemPhysChem 2017, 18, 3135–3141. 10.1002/cphc.201700782. [DOI] [PubMed] [Google Scholar]
- Bonin J.; Maurin A.; Robert M. Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Fe and Co metal based complexes. Recent advances. Coord. Chem. Rev. 2017, 334, 184–198. 10.1016/j.ccr.2016.09.005. [DOI] [Google Scholar]
- Song J.; Klein E. L.; Neese F.; Ye S. The Mechanism of Homogeneous CO2 Reduction by Ni(cyclam): Product Selectivity, Concerted Proton–Electron Transfer and C–O Bond Cleavage. Inorg. Chem. 2014, 53, 7500–7507. 10.1021/ic500829p. [DOI] [PubMed] [Google Scholar]
- Beley M.; Collin J. P.; Ruppert R.; Sauvage J. P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461–7467. 10.1021/ja00284a003. [DOI] [PubMed] [Google Scholar]
- Beley M.; Collin J.-P.; Ruppert R.; Sauvage J.-P. Nickel(II)-cyclam: an extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc., Chem. Commun. 1984, 0, 1315–1315. 10.1039/C39840001315. [DOI] [Google Scholar]
- Craig C. A.; Spreer L. O.; Otvos J. W.; Calvin M. Photochemical reduction of carbon dioxide using nickel tetraazamacrocycles. J. Phys. Chem. 1990, 94, 7957–7960. 10.1021/j100383a038. [DOI] [Google Scholar]
- Froehlich J. D.; Kubiak C. P. Homogeneous CO2 Reduction by Ni(cyclam) at a Glassy Carbon Electrode. Inorg. Chem. 2012, 51, 3932–3934. 10.1021/ic3001619. [DOI] [PubMed] [Google Scholar]
- Aresta M.; Nobile C. F.; Albano V. G.; Forni E.; Manassero M. New nickel–carbon dioxide complex: synthesis, properties, and crystallographic characterization of (carbon dioxide)-bis(tricyclohexylphosphine)nickel. J. Chem. Soc., Chem. Commun. 1975, 0, 636–637. 10.1039/C39750000636. [DOI] [Google Scholar]
- Anderson J. S.; Iluc V. M.; Hillhouse G. L. Reactions of CO2 and CS2 with 1,2-Bis(di- tert -butylphosphino)ethane Complexes of Nickel(0) and Nickel(I). Inorg. Chem. 2010, 49, 10203–10207. 10.1021/ic101652e. [DOI] [PubMed] [Google Scholar]
- Bianchini C.; Mealli C.; Meli A.; Sabat M. Metal-promoted transformation of carbon dioxide into carbon monoxide. X-ray crystal structure of the nickel-carbonate complex [O=PPh2CH2C(CH3)(CH2PPh2)2]Ni(O2CO).0.5H2O.C6H6. Inorg. Chem. 1984, 23, 2731–2732. 10.1021/ic00186a002. [DOI] [Google Scholar]
- Kim Y.-E.; Kim J.; Lee Y. Formation of a nickel carbon dioxide adduct and its transformation mediated by a Lewis acid. Chem. Commun. (Cambridge, U. K.) 2014, 50, 11458–11461. 10.1039/C4CC04800H. [DOI] [PubMed] [Google Scholar]
- Yoo C.; Lee Y. Formation of a tetranickel octacarbonyl cluster from the CO2 reaction of a zero-valent nickel monocarbonyl species. Inorg. Chem. Front. 2016, 3, 849–855. 10.1039/C6QI00011H. [DOI] [Google Scholar]
- Yoo C.; Lee Y. Carbon dioxide binding at a Ni/Fe center: synthesis and characterization of Ni(η1-CO2-κC) and Ni-μ-CO2-κC:κ2O,O′-Fe. Chem. Sci. 2017, 8, 600–605. 10.1039/C6SC03450K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo C.; Oh S.; Kim J.; Lee Y. Transmethylation of a four-coordinate nickel(i) monocarbonyl species with methyl iodide. Chem. Sci. 2014, 5, 3853–3858. 10.1039/C4SC01089B. [DOI] [Google Scholar]
- Mankad N. P.; Rivard E.; Harkins S. B.; Peters J. C. Structural Snapshots of a Flexible Cu2P2 Core that Accommodates the Oxidation States CuICuI, Cu1.5Cu1.5, and CuIICuII. J. Am. Chem. Soc. 2005, 127, 16032–16033. 10.1021/ja056071l. [DOI] [PubMed] [Google Scholar]
- Adhikari D.; Mossin S.; Basuli F.; Dible B. R.; Chipara M.; Fan H.; Huffman J. C.; Meyer K.; Mindiola D. J. A Dinuclear Ni(I) System Having a Diradical Ni2N2 Diamond Core Resting State: Synthetic, Structural, Spectroscopic Elucidation, and Reductive Bond Splitting Reactions. Inorg. Chem. 2008, 47, 10479–10490. 10.1021/ic801137p. [DOI] [PubMed] [Google Scholar]
- Ribbe M. W. Insights into the Mechanism of Carbon Monoxide Dehydrogenase at Atomic Resolution. Angew. Chem., Int. Ed. 2015, 54, 8337–8339. 10.1002/anie.201503979. [DOI] [PubMed] [Google Scholar]
- Can M.; Armstrong F. A.; Ragsdale S. W. Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev. 2014, 114, 4149–4174. 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao K. U.; Herbert J. M. Accurate and Efficient Quantum Chemistry Calculations for Noncovalent Interactions in Many-Body Systems: The XSAPT Family of Methods. J. Phys. Chem. A 2015, 119, 235–252. 10.1021/jp5098603. [DOI] [PubMed] [Google Scholar]
- Shao Y.; Gan Z.; Epifanovsky E.; Gilbert A. T. B.; Wormit M.; Kussmann J.; Lange A. W.; Behn A.; Deng J.; Feng X.; Ghosh D.; Goldey M.; Horn P. R.; Jacobson L. D.; Kaliman I.; Khaliullin R. Z.; Kuś T.; Landau A.; Liu J.; Proynov E. I.; Rhee Y. M.; Richard R. M.; Rohrdanz M. A.; Steele R. P.; Sundstrom E. J.; Woodcock H. L.; Zimmerman P. M.; Zuev D.; Albrecht B.; Alguire E.; Austin B.; Beran G. J. O.; Bernard Y. A.; Berquist E.; Brandhorst K.; Bravaya K. B.; Brown S. T.; Casanova D.; Chang C.-M.; Chen Y.; Chien S. H.; Closser K. D.; Crittenden D. L.; Diedenhofen M.; DiStasio R. A.; Do H.; Dutoi A. D.; Edgar R. G.; Fatehi S.; Fusti-Molnar L.; Ghysels A.; Golubeva-Zadorozhnaya A.; Gomes J.; Hanson-Heine M. W. D.; Harbach P. H. P.; Hauser A. W.; Hohenstein E. G.; Holden Z. C.; Jagau T.-C.; Ji H.; Kaduk B.; Khistyaev K.; Kim J.; Kim J.; King R. A.; Klunzinger P.; Kosenkov D.; Kowalczyk T.; Krauter C. M.; Lao K. U.; Laurent A. D.; Lawler K. V.; Levchenko S. V.; Lin C. Y.; Liu F.; Livshits E.; Lochan R. C.; Luenser A.; Manohar P.; Manzer S. F.; Mao S.-P.; Mardirossian N.; Marenich A. V.; Maurer S. A.; Mayhall N. J.; Neuscamman E.; Oana C. M.; Olivares-Amaya R.; O’Neill D. P.; Parkhill J. A.; Perrine T. M.; Peverati R.; Prociuk A.; Rehn D. R.; Rosta E.; Russ N. J.; Sharada S. M.; Sharma S.; Small D. W.; Sodt A.; Stein T.; Stück D.; Su Y.-C.; Thom A. J. W.; Tsuchimochi T.; Vanovschi V.; Vogt L.; Vydrov O.; Wang T.; Watson M. A.; Wenzel J.; White A.; Williams C. F.; Yang J.; Yeganeh S.; Yost S. R.; You Z.-Q.; Zhang I. Y.; Zhang X.; Zhao Y.; Brooks B. R.; Chan G. K. L.; Chipman D. M.; Cramer C. J.; Goddard W. A.; Gordon M. S.; Hehre W. J.; Klamt A.; Schaefer H. F.; Schmidt M. W.; Sherrill C. D.; Truhlar D. G.; Warshel A.; Xu X.; Aspuru-Guzik A.; Baer R.; Bell A. T.; Besley N. A.; Chai J.-D.; Dreuw A.; Dunietz B. D.; Furlani T. R.; Gwaltney S. R.; Hsu C.-P.; Jung Y.; Kong J.; Lambrecht D. S.; Liang W.; Ochsenfeld C.; Rassolov V. A.; Slipchenko L. V.; Subotnik J. E.; van Voorhis T.; Herbert J. M.; Krylov A. I.; Gill P. M. W.; Head-Gordon M. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2014, 113, 184–215. 10.1080/00268976.2014.952696. [DOI] [Google Scholar]
- Glendening E. D.; Badenhoop J. K.; Reed A. E.; Carpenter J. E.; Bohmann J. A.; Morales C. M.; Weinhold F.. NBO 7.0 PROGRAM CITATION; University of Wisconsin, Madison: (2018) http://nbo6.chem.wisc.edu/biblio_css.htm(12, 01, 2020).
- Khaliullin R. Z.; Cobar E. A.; Lochan R. C.; Bell A. T.; Head-Gordon M. Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals. J. Phys. Chem. A 2007, 111, 8753–8765. 10.1021/jp073685z. [DOI] [PubMed] [Google Scholar]
- Galan F.; Fouassier M.; Tranquille M.; Mascetti J.; Pápai I. CO2 Coordination to Nickel Atoms: Matrix Isolation and Density Functional Studies. J. Phys. Chem. A 1997, 101, 2626–2633. 10.1021/jp9701552. [DOI] [Google Scholar]
- Kégl T.; Ponec R.; Kollár L. Theoretical Insights into the Nature of Nickel–Carbon Dioxide Interactions in Ni(PH3)2(η2-CO2). J. Phys. Chem. A 2011, 115, 12463–12473. 10.1021/jp201140h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.