

Abstract
A serious adverse effect of prescription opioid analgesics is addiction, both to these analgesics and to illicit drugs like heroin that also activate the mu opioid receptor (MOR). Opioid Use Disorder (OUD) and opioid overdose deaths represent a current American health crisis, and the prescription of opioid analgesics has contributed significantly to this crisis. While prescription opioids are highly effective analgesics, there currently exists no facile way to use them for extended periods without the risk of addiction. If addiction caused by MOR-targeting analgesics could be blocked by blending in a new “anti-addiction” ingredient that does not diminish analgesia and does not introduce its own therapeutically limiting side effects, then continued clinical use of prescription opioids for treating pain could be maintained (or even enhanced) instead of curtailed. In this narrative review, we contextualize this hypothesis, first with a brief overview of the current American opioid addiction crisis. The neurobiology of two key receptors in OUD development, MOR and the kappa opioid receptor (KOR), is then discussed to highlight the neuroanatomical features and circuitry in which signal transduction from these receptors lie in opposition – creating opportunities for pharmacological intervention in curtailing the addictive potential of MOR agonism. Prior findings with mixed MOR/KOR agonists are considered, before exploring new potential avenues such as biased KOR agonists. New pre-clinical data are highlighted demonstrating that the G protein-biased KOR agonist nalfurafine reduces the rewarding properties of MOR-targeting analgesics and enhances MOR-targeting analgesic-induced anti-nociception. Finally, we discuss the recent discovery that a Regulator of G protein Signaling (namely, RGS12) is a key component of signaling bias at KOR, presenting another drug discovery target towards identifying a single agent, or adjuvant to be added to traditional opioid analgesics, that could reduce or eliminate the addictive potential of the latter drug.
Brief History of the United States Opioid Epidemic
Opium, a powerful analgesic producing euphoria, has been used medicinally and recreationally since its first recorded harvest by Sumerians in 3400-BCE.1 In the 1800’s, Serturner isolated morphine, one of opium’s active ingredients.2 Morphine use increased with advent of the hypodermic needle3 in 1853 and was extensive during the Civil-War, with subsequent addiction developing consistently enough to earn the name “the Army-Disease”.4 The addictive potential of rapid, systemic morphine administration via hypodermic needle, while overlooked by the medical community at the time, is demonstrated in the evidence of needle sharing3 as early as 1914. Not long after morphine’s medicalization began pursuit of a non-addictive substitute, including Wright’s synthesis of diacetylmorphine.2 Today, diacetylmorphine (heroin) is used illicitly, with lifetime use prevalence among Americans predicted to be >13%.5 Without a non-addictive replacement, opioid analgesics were largely avoided outside of acute and cancer-related pain until claims in the 1980s that there was little risk of addiction when opioids were prescribed to patients without drug abuse histories.2,6 Prescribing loosened thereafter, and pain was soon seen as “the fifth vital sign,” with chronic-pain management becoming part of hospital accreditation.7 Extended-release oxycodone constituted a major portion of the resultant increase in opioid prescriptions and was a central player in the rise in opioid use disorder (OUD); the extended release formulation was easily circumvented, allowing for large, instant oxycodone release.8 In 2017, U.S. opioid prescriptions numbered 200-million, while OUD diagnoses exceeded 2.1-million9.
OUD is defined10 as “…compulsive, prolonged self-administration…” of opioids without legitimate medical purpose, or at higher doses than therapeutically required, despite “…clinically significant impairment or distress.” While OUD is commonly thought to involve heroin, 1.7-million of the 2.1-million-Americans diagnosed with OUD in 2017 were using prescription opioids.9 While opioids for acute and cancer-related pain are regarded as standard and necessary, new persistent opioid use has been seen in up to 6.5% of post-surgical patients in recent studies.11,12 The use of opioids for chronic-pain is contentious.1 A recent meta-analysis found only a small reduction in pain and a small increase in functioning with opioid analgesics prescribed for chronic pain.13 Long-term outpatient opioid prescriptions for chronic-pain constituted the majority of the increase in opioid prescriptions and carried with them increased abuse potential.14 8–12% of patients on long-term opioids for chronic-pain will develop OUD,15,16 while 25% will engage in “aberrant medication-taking behaviors” such as opioid diversion.15,16 Addiction to, and misuse of, prescription opioids are also linked to heroin use, with 80% of new heroin addicts reporting first abusing prescription opioids.17 With strong correlation of the rise of OUD to prescription-rates, it is hoped that alterations in prescribing practices will help curb this rise.16
MOR, KOR, and Addiction
The opioid family of neuron signaling-inhibitory G protein-coupled receptors (GPCRs) not only includes “classical” mu and kappa opioid receptors (MOR and KOR), but also the delta opioid receptor (DOR) and the “non-classical” nociceptin-receptor.18 While all modulate aspects of opioid-induced behaviors in rodents, MOR and KOR exhibit the strongest influence on the development of addiction.19–22 Agonism of MOR, the primary target of opioid analgesics, produces both analgesia and euphoria.19,20 KOR agonism produces analgesia as well (albeit more weakly), but also evokes clinically limiting effects like dysphoria and psychotomimesis.23 Conversely, KOR antagonism is important to the multifactorial pharmacology of the partial MOR agonist and OUD therapeutic medication buprenorphine; while buprenorphine is considered an effective “anti-addiction” analgesic to prescribe to those in pain with a history of OUD,24 it has also become a street drug in its own right.25 Better understanding of the neurobiology and pharmacology of the two opioid receptors MOR and KOR, as they relate to analgesia as well as addiction development and maintenance, is warranted if KOR, in particular, is to be exploited in developing future anti-addiction pharmacotherapy.
Analgesia
Clinical success with MOR-targeting analgesics arises from activity at both spinal and supraspinal sites. MOR localizes to presynaptic terminals of primary nociceptive afferents transmitting pain signals into the spinal cord,26 as well as post-synaptic soma of secondary nociceptive efferents transmitting pain signals to higher brain centers.26 MOR activation also modulates pain signals through activity in the periaqueductal gray (PAG),27–29 a midbrain region that controls numerous functions, from fear and anxiety responses to blood pressure.27–30
The PAG modulates pain by controlling projections from midbrain nuclei to the dorsal horn of the spinal cord, a descending pathway that inhibits nociception.27–30 This pathway begins in the PAG, where glutamatergic neurons projecting to serotonergic and noradrenergic cell bodies in the rostral-ventromedial-medulla (RVM) remain under tonic inhibition by a local GABAergic circuit.27–30 Activation of MORs located on PAG GABAergic neurons disinhibits the glutamatergic projection neurons, in turn stimulating serotonergic and noradrenergic neurons of the RVM.27 These RVM neurons then project to the dorsal horn, wherein release of serotonin and norepinephrine inhibits transmission of nociceptive signals.27 In this way, MOR-targeting analgesics prevent nociceptive transmission through actions at receptors in the dorsal horn and indirectly through modulation of regulatory projections.
The role of KOR in spinal analgesia is similar to MOR activation outcomes, as the distribution of KOR resembles that of MOR26 and KOR activation inhibits transmission of nociceptive signals through the dorsal horn.26,31 However, the role of KOR remains unclear in the PAG and descending pain-inhibitory circuit. For example, microinjection of the KOR-agonist pentazocine into the PAG produces analgesia against noxious heat but not against pressure, while fentanyl, a MOR-selective-agonist, produced analgesia against both pain modalities.32 A second group, however, used a thermal stimulus to show that morphine (a MOR-selective-agonist), but not U50,488 (a KOR-selective-agonist), produced analgesia when microinjected directly into the PAG.33 In addition to being a KOR-agonist, pentazocine also acts as a partial-agonist of MOR, while U50,488 acts more selectively at KOR and not MOR.34,35 This difference likely explains the dissimilarity in these two findings.
As a further complication, the distribution of MOR and KOR in these descending circuits remains unclear. While overlap may exist, as reported in some histological studies,36 others report that MOR and KOR act in specific RVM neuron subpopulations,37 and produce opposing analgesic effects37 (as discussed below). While less potent for somatic pain, KOR-agonists display greater potency for visceral pain, in both pre-clinical studies38 (Table 1) and in a clinical trial using a peripherally restricted KOR-agonist39. As the underlying mechanisms and circuitry of somatic and visceral pain differ,40 it is possible that differences in MOR and KOR distribution play a role in their differing effects.
Table 1.
Pre-clinical Effects of MOR and KOR Agonism
| Type of Pain: | Effect of MOR Agonism | Effect of KOR Agonism | Relative Efficacy | References |
|---|---|---|---|---|
| Spinal | analgesia | Analgesia | MOR ≈ KOR | 142 |
| Supraspinal | analgesia; euphoria | analgesia; dysphoria | MOR ⋙ KOR | 142 |
| Inflammatory | analgesia | Analgesia | MOR ≈ KOR | 142–146 |
| Visceral | analgesia | Analgesia | MOR < KOR | 72,147 |
| Somatic | analgesia | Analgesia | MOR > KOR | 142–144 |
| Neuropathic | can worsen | Mixed | - | 142–146 |
While both MOR and KOR produce analgesia through activities in the spinal cord and descending pain-modulatory pathways, MOR-agonists provide greater analgesia than KOR-agonists given functional divergence in higher brain centers. Compared with KOR-agonists, MOR-targeting analgesics retain broad applicability to nearly all types of pain, in part because of their ability to produce euphoria, thereby reducing the affective component of pain and preventing “agony” even when the conscious perception of a nociceptive stimulus remains.41 This ability, however, also leads to the dramatic abuse potential of MOR-targeting analgesics. In contrast, KOR agonism produces anxiety and dysphoria, limiting the clinical utility of KOR-agonists and playing a major role in the cycle of addiction through its role in producing negative affect.
Euphoria and Dysphoria
Both MOR and KOR play central roles in the cycle of addiction. While MOR activation drives initial development of drug preference through its euphorigenic properties,19,20 KOR activation reinforces habitual drug-taking through its dysphoric properties, producing negative affect and “emotional pain”.19,42,43 In the CNS, MOR and KOR normally function in opposition to produce an affective homeostasis. When perturbed, however, this system may spiral to lower and lower set-points of negative affect, leading to the destructive cycle of drug-seeking known as addiction.
(i). Euphoria, reward, and MOR
While MOR is distributed widely throughout the brain, its highest levels are in regions involved in pain processing (e.g., thalamus, PAG) and in motivated behavior (mesolimbic system),44 with the latter playing the strongest role in addiction development. Endomorphins are considered to be the natural, MOR-binding opioids central to pain relief. Another class of endogenous MOR-ligand, β-endorphin, is produced in the hypothalamus as well as the nucleus tractus solitarius,45 and is expressed throughout the brain as a homeostatic mechanism of reducing stress and producing reward in response to novel stimuli, stress, and exercise.46 Elevations in β–endorphin increase mesolimbic dopamine release47, produce place preference48, and augment the reinforcing properties of xenobiotic chemicals (e.g., drugs of abuse)49,50 and natural rewards (e.g., sex, food).46,51 Conversely, β-endorphin ablation or deletion of Oprm1 (MOR gene) in mice increases anxiety52, attenuates motivational drive to acquire natural rewards53, and reduces sensitivity to reward-predictive cues or stimuli.54 Ultimately, MOR signaling largely produces positive affect and motivated behavior through effects on the mesolimbic system.
The mesolimbic pathway comprises dopaminergic projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAcc), and plays key roles in reward prediction and encoding the incentive salience of naturally rewarding stimuli.55 When encountering a rewarding stimulus, ranging from basic rewards (e.g., food) to complex social rewards (e.g., praise), VTA burst firing releases dopamine in the NAcc and produces positive reinforcement of the behavioral pattern that produced the rewarding stimulus.56 VTA dopamine neurons remain under tonic inhibition by local GABAergic interneurons that express MOR.57 Endogenous opioid release in this area serves to inhibit these GABAergic interneurons, leading to disinhibition of VTA dopaminergic projections to the NAcc, producing positive reinforcement.57 This action on VTA MORs is the primary mechanism of opioid analgesic-induced reward.55,57 Intra-VTA application of the MOR agonist DAMGO stimulates local dopamine release in wildtype mice, but not MOR knockout mice58; moreover, systemic administration of morphine increases dopamine release in the NAcc, but this effect is markedly reduced in MOR knockout mice59. Accordingly, morphine or heroin-induced locomotor activation59,60, conditioned place preference60,61, intravenous self-administration62, and opioid dependence and withdrawal63 are all abolished in MOR knockout mice. Pharmacological studies show that place preference for MOR-targeting analgesics is blocked in rats by administering a MOR antagonist directly into the VTA;64 conversely, MOR-agonists are self-administered by rats when applied directly to the VTA.65 While animals will self-administer MOR-agonists when applied to several other brain regions as well66, their actions in the VTA are necessary and sufficient for the reinforcing properties of opioid analgesics.55 Furthermore, disinhibition of dopamine release likely plays a role in the ability of MOR-targeting opioid analgesics to reduce the affective experience of pain, as decreased function of midbrain dopaminergic neurons leads to increased pain,67 while dopaminergic agents can produce analgesia themselves (e.g., ref.68). Mice lacking tyrosine hydroxylase (i.e., dopamine-deficient mice) show reduced thermal anti-nociception in response to morphine,69 supporting a role for dopaminergic signaling in the analgesic response to traditional opioid analgesics.
This model above, however, is not without complications. Destroying dopaminergic projections from the VTA to the anterior cingulate cortex, for example, blocks acquisition of place preference to morphine administered to rodents either systemically or directly into the VTA, implicating mesocortical circuitry in opioid reinforcement.70 In addition, while dopamine antagonists block acquisition of morphine-induced place preference, the potency of this effect varies with antagonist specificity across the various dopamine receptor subtypes.71 More confounding still are findings that dopamine-deficient mice69 still display place preference for morphine, implicating factors beyond dopaminergic signaling in producing opioid reward.
While the intricacies of dopamine signaling in addiction require further inquiry, the initial phase of opioid addiction development -- acute reward and euphoria -- remain best explained by disinhibition of mesolimbic circuitry. The potent ability of opioids to disinhibit this circuitry underpins both their profound analgesic effects and their devastating propensity to incentivize compulsive use. Given its power, the brain employs potent means to regulate this mesolimbic circuitry, none linked more tightly to the cycle of addiction than KOR.
(ii). Dysphoria, altered set-points, and KOR
While KOR distribution mirrors that of MOR in many regions, oppositional localization in specific regions produces a profound difference in the ultimate effects of KOR stimulation vs MOR stimulation (Table 1).44 MOR agonist-induced reward and analgesia are maintained in KOR knockout mice63,72 and prodynorphin knockout mice73,74; conversely, KOR agonist-induced G protein coupling, analgesia, and aversion are unaffected by MOR ablation75,76, supporting that the physiological effects of MOR and KOR are distinct and separable. While MOR stimulation produces euphoria, KOR activation produces dysphoria, a negative affective state that includes components of anxiety, anhedonia, and depression.22,77 Dynorphin, the endogenous KOR-ligand neuropeptide cleaved to six different lengths from the precursor prodynorphin, is produced in the hypothalamus,45 the amygdala,78 and direct pathway GABAergic medium spiny neurons of the striatum79 (among other regions), and is released in response to stress.77 Unlike β-endorphin, dynorphin plays the role of a negative homeostatic regulator, producing the negative experiences of stress80 and modulating the central effects of corticotropin releasing factor (CRF), a major hormonal regulator of the stress response.80 The areas most heavily implicated in producing this negative state include the VTA-to-NAcc circuit, along with the amygdala and dorsal raphe nucleus (DRN), the latter being the brain’s major serotonergic nucleus.81 In each of these regions, KOR activation produces effects largely in opposition to those of MOR, culminating in the overall negative affective state of dysphoria.
In the mesolimbic system, KOR activation reduces dopamine release, with KOR expressed on both the cell bodies of dopaminergic VTA neurons (Figure 1), as well as the presynaptic terminals of these neurons within the NAcc of mice.82,83 This distribution is different in the rat brain, with KORs located on the terminals of NAcc projections, but only on the bodies of VTA neurons projecting to the medial prefrontal cortex and amygdala,84 and this distribution may vary within the human brain as well. In both the NAcc and VTA, KOR activation acts in negative feedback regulation of mesolimbic dopamine release, restoring homeostatic function within this circuit. When dopamine D1 receptor-expressing neurons within the NAcc are stimulated by dopamine, they release both dynorphin and GABA from axonal projections terminating in the VTA to reduce the firing rate of the dopaminergic neurons located therein.85,86 Disinhibition of this circuitry by MOR-agonists produces reward, whereas inhibition by KOR-agonists produces the opposite (i.e., aversion, as measured in rodents using conditioned place aversion [CPA]82,83); as a result, KOR-agonists do not support self-administration in rats or rhesus monkeys87,88. Prior studies82,83 strongly link these aversive properties of KOR-agonists to actions on mesolimbic dopamine neurons. For example, systemic administration of a KOR agonist produces place aversion in wildtype mice, but not constitutive or dopamine neuron-specific KOR knockout mice.83 More recent work demonstrates that dynorphin/KOR microcircuits within the NAcc shell mediate negative affect engendered by inflammatory pain89, and that discrete subpopulations of dynorphin/KOR-expressing neurons in the ventral and dorsal NAcc shell differentially drive KOR-mediated aversion and ‘paradoxical’ reward, respectively.90
Figure 1. Schematic of relative locations of KOR and MOR receptors in the mesolimbic pathway of the human (A) and mouse (B) brains.
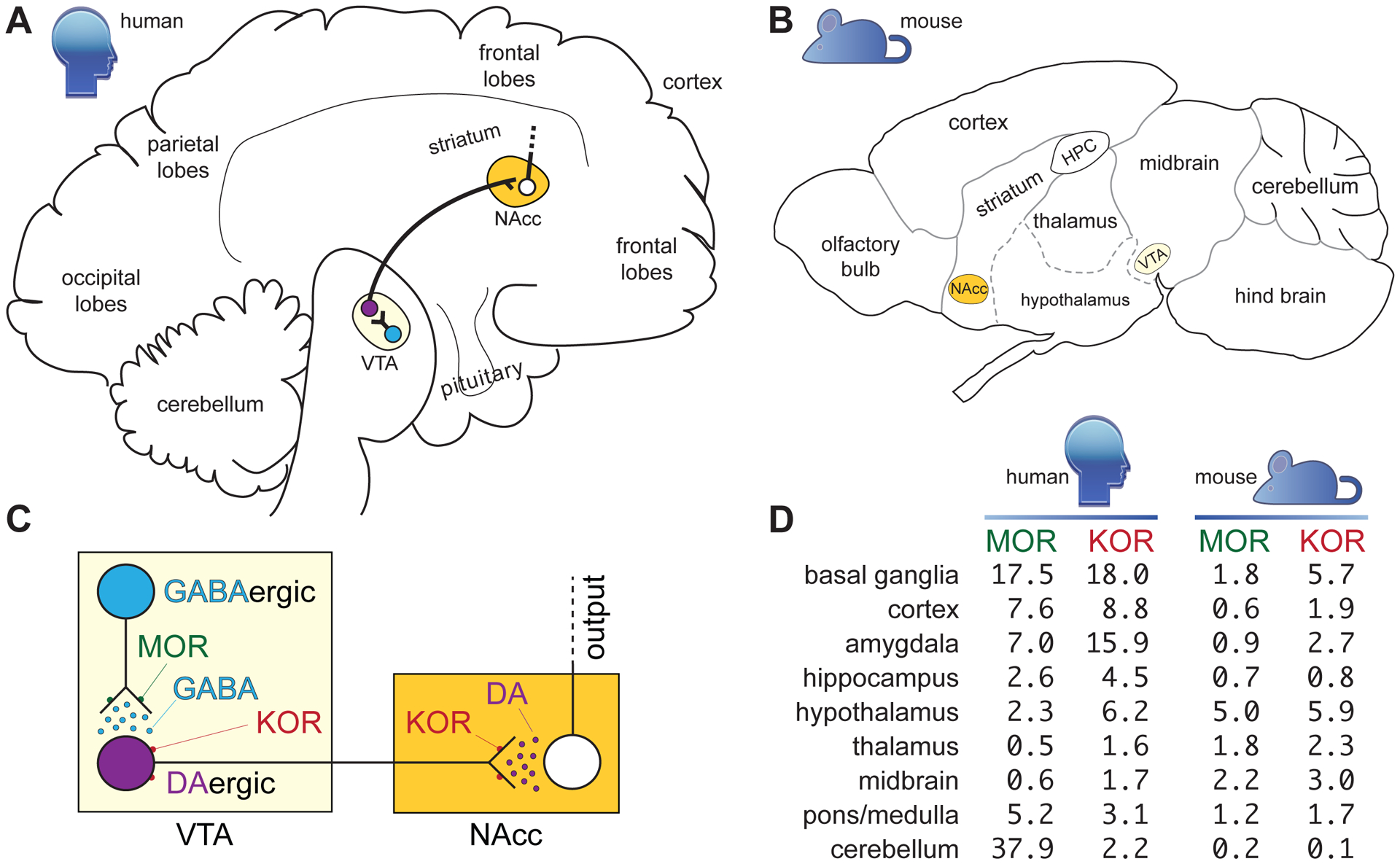
Panel C depicts activation of MORs on GABAergic interneurons of the VTA disinhibiting dopamine release and producing reward. Activation of KORs on dopaminergic neurons inhibits dopamine release, producing dysphoria. Panel D lists normalized mRNA expression levels for the transcripts encoding MOR and KOR in indicated regions of the human and mouse brains. Consensus Normalized eXpression (NX) values were generated as previously described by the Human Protein Atlas (https://www.proteinatlas.org/about/assays+annotation#normalization_rna).
KOR actions in the amygdala appear to modulate fear and anxiety related behaviors, possibly through modulation of local GABAergic neurotransmission and long-term potentiation.91 Fear conditioning in rats is reported to increase Oprk1 mRNA in this region, and local KOR inhibition reduces this response.92 In addition, KOR antagonism in the amygdala blocks the anxiogenic effects of a natural stressor (repeated forced swimming) or of intra-amygdalar CRF injection.93 The DRN also plays a central role in mediating the affective response to KOR-agonists. KOR stimulation in this region reduces extracellular serotonin,94 likely through increasing serotonin transporter levels via a p38α mitogen-activated protein kinase (MAPK)-dependent mechanism,95 an effect seen in mice after both local KOR-agonist administration and social defeat stress95. Furthermore, while the KOR-agonist U50,488H did not produce place aversion in mice lacking KOR, viral re-expression of KORs on DRN neurons projecting to the NAcc restored U50,488H CPA in these KOR-deficient mice96.
The effects of KOR activation within these brain regions suggests that the dynorphin/KOR system is central in mediating the aversive effects of stress.21,43,96 Dynorphin levels increase during stress97, and blockade of KOR or genetic deletion of Oprk1 in mice prevents the pro-depressive and anxiogenic effects of forced swim, social defeat, and foot-shock stressors.80,93,95 Systemic KOR-agonist administration produces place aversion83 and depressive-like phenotypes in forced swim and intra-cranial self-stimulation (ICSS) assays.98 By producing these negative affective states, the dynorphin/KOR system also figures heavily into the addictive cycle. Stressful stimuli reinstate extinguished drug-seeking behaviors in both place preference and self-administration paradigms.99 Pre-treating mice with the KOR-antagonist nor-binaltorphimine (norBNI), or genetic deletion of either Oprk1 or Pdyn, prevents reinstatement of conditioned place preference (CPP) for cocaine by foot shock or forced swim stressors, as well as to a single administration of U50,488H100. In addition, CPP for cocaine increased in mice exposed to forced swim stress prior to conditioning, an effect blocked by norBNI pre-treatment.101
In light of its role in regulating limbic function and mediating stress, KOR should be viewed as central to the “Allostatic Model”102 that contextualizes addiction as a pathologically altered homeostatic set point, or “allostasis”.103 The dynorphin/KOR system contributes to allostasis largely through modulating dopamine tone within the mesolimbic system.42,43 As described above, MOR-targeting analgesics increase dopaminergic tone in the NAcc, a property common to nearly all drugs-of-abuse20. Repeated use increases production of cAMP response element binding protein within the NAcc85 and subsequent upregulation of dynorphin.43,85,86 With cessation of opioid use, this elevated dynorphin tone leads to dysphoria, increasing the hedonic valence of the drug and increasing susceptibility to relapse in an attempt to attain the now pathologically altered homeostatic set point.42,43,102
In summary, while activation of MOR or KOR produces analgesia in response to stressful stimuli, the psychological functions of these receptors diverge dramatically and represent the opposing faces of opioid addiction. The drive to alleviate pain placed the profound effects of MOR stimulation at the forefront of medicine in the early 2000s. Now, because of the resultant OUD crisis spawned by these powerful MOR-targeting analgesics, KOR antagonists sit among NIDA’s ten most wanted drugs for the treatment of OUD,104 specifically with the thought that KOR antagonism during OUD might reduce endogenous dynorphin signaling, and its contribution of stress, to drug-seeking behaviors and addiction reinstatement. In a separate means of exploiting pharmacological manipulation of KOR signaling towards a clinical benefit, KOR agonism has the potential to create blended KOR-agonist/MOR-agonist “combination” analgesics that diminish or eliminate the risk of acquiring OUD while on analgesics in the first place.
New approaches for improving opioid analgesics
Searches for a non-addictive analgesic began nearly the moment Serturner first isolated morphine in the 1800’s.2 Many opioids are sold today as combinations – also containing non-steroidal anti-inflammatories to reduce the dose of opioid necessary to treat pain, otherwise known as “dose sparing”.105 Other combination therapies, such as Suboxone (buprenorphine/naloxone), focus on preventing improper opioid use by compounding them with opioid antagonists active only when taken through improper routes.24 While many approaches are currently being investigated, from targeting little-understood MOR splice-variants106 to designing opioids that function only in the presence of inflammation,107 the following sections focus on two widely studied and promising avenues: combination therapies activating multiple opioid receptors, and the utilization of “biased agonism.”
Combining MOR and KOR activation
In the 1990s, researchers began utilizing the analgesic and non-reinforcing properties of KOR-agonists to augment traditional MOR-agonists. While KOR-agonists alone proved less potent in producing analgesia,108 combining fentanyl (MOR-agonist) with U69,593 (KOR-agonist; Table 2) showed additive analgesic effects in rhesus monkeys while also reducing self-administration.109 Furthermore, combining MOR and KOR-agonists prevented liabilities of opioid tolerance, hyperalgesia, and respiratory depression.110,111 While pre-clinical experiments demonstrated utility of combined MOR and KOR agonism, enthusiasm waned after early clinical experiments utilizing mixed MOR/KOR-agonists. While nalbuphine (KOR-agonist and MOR partial agonist) demonstrated efficacy in reducing CPP to morphine in pre-clinical studies, suggesting a benefit against addiction induction,112 human studies demonstrated dose-dependent increases in dysphoria, curtailing clinical utility.113 Pentazocine (similar to nalbuphine) and butorphanol (KOR-agonist yet MOR-antagonist) also demonstrated classic dysphoric effects of traditional KOR-agonists in humans at higher doses,23,113–116 limiting therapeutic potential.
Table 2.
| Drug / Compound name: | Selectivity (estimated Ki [nM]) at the human receptor* | Approved for use? | ||
|---|---|---|---|---|
| MOR | KOR | DOR | ||
| β-Endorphin | 1.18 | 31.7 | 1 to 5.4 | endogenous |
| Dynorphin A | 32 | 0.5 | 1,000 | endogenous |
| Enkephalin | 1.77 | 506.17 | 200 to 1,430 | endogenous |
| Morphine | 2.16 | 50 to >2,000 | 1,545 to 2,500 | USA (FDA) for analgesia |
| Oxycodone | 780 | >2,000 | >10,000 | USA (FDA) for analgesia |
| Fentanyl | 1 | 169.9 | 152 to >1,000 | USA (FDA) for analgesia |
| Pentazocine | 5.4 | 2.6 | 114.5 | USA (FDA) for analgesia |
| Butorphanol | 0.12 | 0.22 | 12 | USA (FDA) for analgesia |
| Buprenorphine | 0.85 (partial agonism) | 0.71 (antagonism) | 3.7 (antagonism) | USA (FDA) for OUD |
| Nalfurafine* (TRK-820) | 3.2 to 5.2 | 0.025 to 0.075 | 161 to 289 | Japan (MHLW) for pruritus |
| EOM Salvinorin B | >1 | 0.32 | >1 | no |
| ICI 199,441 | 54 | 0.04 | 24 | no |
| U50,488 | >1,000 | 0.16 | 645 | no |
| U69,593 | >1,000 | 1.53 | >1,000 | no |
| RB-64 | >10,000 | 0.59 | >10,000 | no |
| Salvinorin A | >10,000 | 1.82 | >1,000 | no |
| Triazole 1.1 | >10,000 | 0.25 | >2,000 | no |
Estimated nalfurafine Ki (nM) values are presented at the rat MOR, human KOR, and mouse DOR, respectively.
Of note, however, is one report117 that co–administration of the MOR and KOR-antagonist naltrexone with pentazocine in human subjects “unmasks” the KOR-related dysphoric effects of pentazocine at lower doses, but blocks both MOR-dependent and KOR-dependent effects at higher doses. As the antagonist naltrexone possesses greater affinity for MOR than KOR, these results support the premise that MOR agonism can mitigate KOR agonism-related dysphoria to some degree. While a single compound (like a mixed MOR/KOR-agonist) is generally considered a more attractive drug candidate than a dual-drug formulation, this study117 suggests that relative activity at each opioid receptor plays a critical role in the interaction between these two receptor systems. Studies in patients receiving methadone maintenance therapy demonstrate that administering nalbuphine, pentazocine, or butorphanol can precipitate withdrawal,118–120 indicating that none of these three, mixed-action drugs achieves activity at MOR comparable to methadone, perhaps due to incomplete agonism. While new mixed MOR/KOR drugs under development may possess more favorable activity profiles, such as the 6β-naltrexamines and orvinols,121 a co-administration paradigm may allow for greater control over relative activity at each receptor.
The recent rise of OUD has reinvigorated interest in KOR-agonists. A peripherally restricted KOR-agonist -- CR845/difelikefalin -- is currently in Phase III trials.122 In addition to peripheral restriction of KOR agonism (i.e., to provide analgesia without concomitant CNS-centered dysphoria), pre-clinical research is also focused on utilizing a newer concept in pharmacology: “biased agonism.” This approach utilizes variations in the efficacy of different compounds to engage various aspects of intracellular machinery downstream of receptor stimulation.
Biased Agonism: All the good and none of the bad?
G protein-coupled receptors regulate diverse physiological processes and represent the target of greater than a third of all FDA approved drugs.123 GPCRs contain seven transmembrane domains that couple to an intracellular, heterotrimeric complex of Gα, Gβ and Gγ subunits.124 The Gα subunit associates with guanine nucleotide diphosphate (GDP) in the resting (inactive) conformation. When bound by agonist, the activated GPCR catalyzes exchange of GDP for guanine nucleotide triphosphate (GTP), at which point the Gα·GTP subunit dissociates from the Gβγ heterodimer.124 Both the Gα GTP and Gβγ complexes initiate distinct signaling cascades that vary depending upon the receptor and the cellular context124; all opioid receptors couple to the inhibitory Gi/o class of Gα proteins.31 The Gi/o heterotrimer, when activated, generally functions to inhibit cAMP production, as well as increase potassium conductance (hyperpolarizing the cell) and inhibit calcium conductance (preventing vesicular release of neurotransmitters).27 Opioid receptor activation inhibits neuronal function through these G protein-dependent mechanisms.
In addition to G protein-dependent signaling, GPCRs also initiate signaling through a different class of proteins -- the β-arrestins.125 Activated GPCRs often recruit G protein-receptor kinases (GRKs),124 which phosphorylate the GPCR, subsequently leading to β-arrestin recruitment.124,125 As their name implies, β-arrestin proteins arrest further G protein activation, and also can lead to subsequent receptor internalization. The internalized β-arrestin/GPCR complex then activates signaling pathways distinct from those activated by G proteins.125,126 When a particular GPCR agonist activates either the G protein- or β-arrestin-dependent pathway to a greater extent than the other, this agonist is considered “biased” towards that pathway.125 Physiological and behavioral effects classically associated with activation of specific GPCRs may occur to a greater or lesser extent upon application of biased agonists.82,126 The possibility of discovering novel KOR agonists that minimize the adverse effects currently preventing their utility as adjuvants to conventional opioid analgesics has led to a recent surge in research focused on biased KOR compounds and biased KOR signal transduction.
(i). G protein-biased KOR agonists
G protein-dependent signaling downstream of KOR activation produces analgesia via inhibiting pain transmission through the dorsal horn of the spinal cord.26,31 Conversely, β-arrestin2 signaling is implicated in the therapeutically limiting dysphoric effects of KOR agonists.82,127,128 As discussed above, natural stressors (such as forced swim) activate KOR in mice; the dysphoric component of this stress can be prevented by pharmacological inhibition of p38 MAPK downstream of KOR-initiated β–arrestin2 signaling,128 or by preventing β-arrestin2 recruitment to activated KOR through deletion of Adrbk2 (encoding the upstream G protein-receptor kinase βARK2/GRK3).127 Furthermore, deletion of Mapk14 (encoding p38α-MAPK) from dopamine neurons prevents conditioned place aversion (CPA) to the unbiased KOR agonist U50,488.82 These results and others have spurred investigations into developing a “non-dysphoric” replacement of traditional MOR-targeting opioid analgesics with a single KOR agonist that is biased toward G protein signal transduction.
A G protein-biased KOR-agonist named triazole 1.1 was found to have reduced dysphoric liabilities in pre-clinical tests:98 triazole 1.1 did not suppress ICSS thresholds, reduce NAcc dopamine release, nor inhibit novelty-induced locomotion at doses producing analgesia equivalent to U50,488. These results support the hypothesis that a G protein-biased KOR agonist will not produce the sedative and dysphoric effects that have limited the clinical utility of traditional, unbiased KOR agonists. However, another G protein-biased KOR-agonist, RB-64, was observed to produce CPA in both wild-type and β-arrestin2-deficient mice without inhibiting novelty-induced locomotion in either genotype.129
Because newer, G protein-biased KOR-agonists are thought to bias against eliciting dysphoric effects, we recently re-tested the hypothesis that co-administration of a G protein-biased KOR activator with a traditional opioid analgesic reduces the rewarding property of the latter drug without reducing its desired analgesic effect and without eliciting untoward dysphoria. We first profiled several G protein-biased KOR agonists,130 including triazole 1.1 and nalfurafine, against an unbiased KOR agonist (U50,488). Based on our recent analyses130 (summarized in Fig. 2), we agree that triazole 1.1 is G protein-biased, but also highlight that other G protein-biased compounds (e.g., nalfurafine, EOM Sal B) have even greater G protein-bias in vitro and, therefore, decreased potential to elicit dysphoria in clinical usage. In particular, nalfurafine has high translational potential as an “anti-addiction” adjuvant for existing MOR-targeting analgesics, given nalfurafine’s safe usage in Japan since 2009 to treat uremic pruritis131,132 without producing psychotomimesis or undue sleep disturbances (either insomnia or somnolence).132 Most importantly, nalfurafine is also known to be free of abuse potential.131
Figure 2. Triazole 1.1, nalfurafine, and EOM Sal B are G protein-biased KOR agonists.
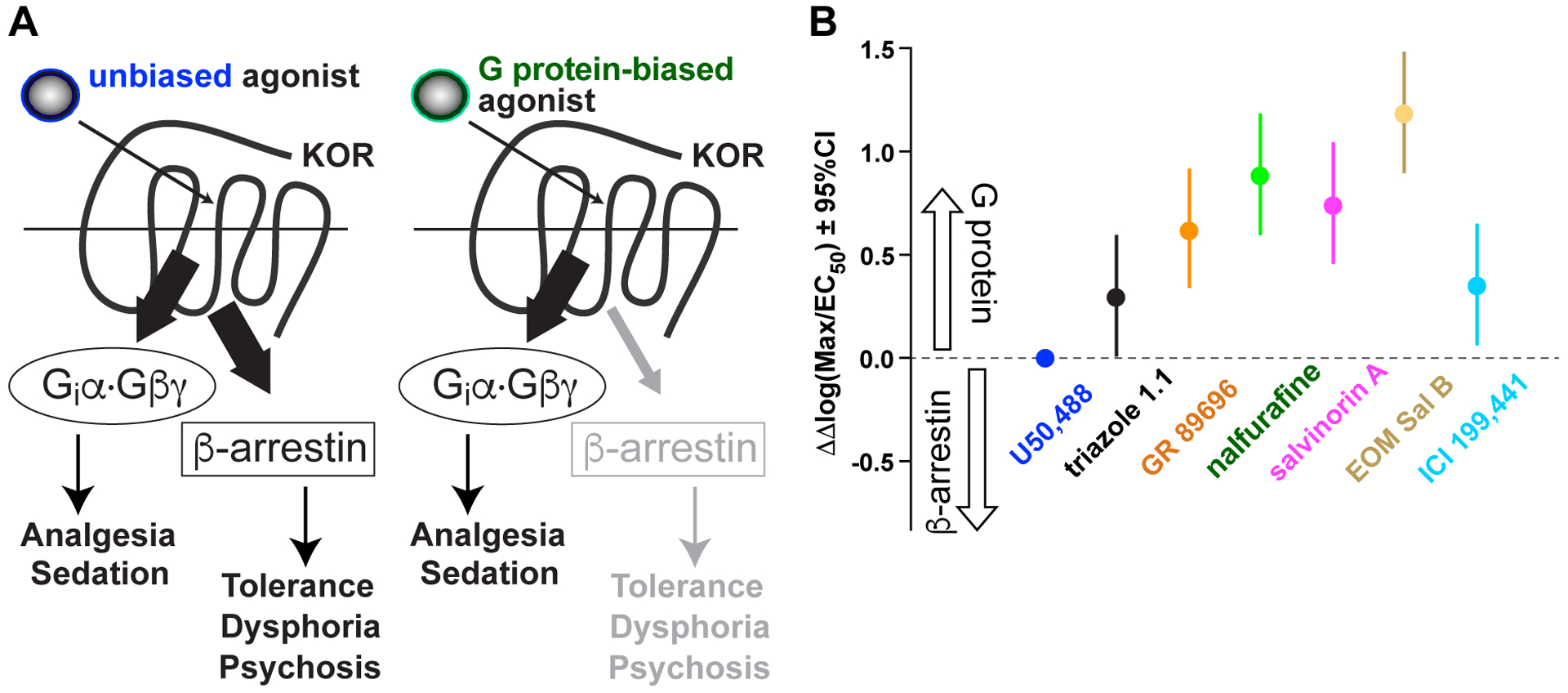
(A) Unlike unbiased KOR agonists (e.g., (±)U50,488), G protein-biased agonists have diminished potency in signaling to β–arrestin recruitment. (B) Summary of bias factors for each compound as obtained from maximal efficacy (Emax or “Max”) and potency (EC50) values. ΔΔlog(Max/EC50) values, with 95% confidence intervals (CIs), are plotted for each compound to indicate relative bias towards G protein signaling (versus U50,488). Bias factors in the plot of panel B are derived from ref.130.
Using co-administration in C57BL/6J mice, nalfurafine was observed to reduce CPP for morphine, yet enhance morphine-induced supraspinal analgesia, the latter suggesting that nalfurafine addition to an opioid analgesic could also lead to a reduction of dose of the latter.130 Testing in male rhesus monkeys has further demonstrated that nalfurafine elicits a reduced degree of sedative-like and motor-impairing effects compared to typical KOR-agonists,133 and furthermore reduces self-administration of oxycodone, in rhesus monkeys, in mice, and in rats134–136 – hopeful signs that nalfurafine could be used as an “anti-addiction” adjuvant to traditional opioid analgesics. Newer KOR agonists with G protein signaling bias are also being created with this anti-addiction application in mind (e.g., USPTO-application US20200131162A1 “Synthesis of 20-nor-salvinorin-A” published 2020-04-30).
(ii). RGS12 inhibitors?
β-arrestin proteins, as their name suggests, arrest G protein-mediated signaling from activated GPCRs. Another protein family – the ‘Regulators of G-protein Signaling’ (or RGS proteins) – also serve to extinguish GPCR signaling, but via a different mechanism: namely, acceleration of GTP hydrolysis by the Gα subunit to return the G proteins to their inactive, heterotrimeric state.124 One particular member, RGS12, is enriched within the ventral striatum (vSTR); genetic ablation of Rgs12 increases dopamine transporter (DAT) expression and dopamine uptake within the vSTR of mice.137,138 (Similar CNS effects of RGS12 loss are also seen in changes to serotonin transporter expression and serotonin uptake139). The most likely direct targets for RGS12’s inhibitory action are striatal presynaptic KORs, activation of which is known to attenuate striatal dopaminergic tone.140 Given that Oprk1 (KOR) and Rgs12 mRNAs overlap in their CNS expression, and that KOR and RGS12 proteins co-immunoprecipitate as a complex from vSTR extracts,137 it is likely that the increased DAT expression / function exhibited by RGS12-null mice is caused by removing a critical negative influence on signaling downstream of KOR activation. RGS12 overexpression markedly and selectively blunts G protein-dependent cAMP inhibition by KOR activation but greatly enhances β-arrestin recruitment to activated KOR.137 (This same report137 demonstrated that RGS12 does not interact with MOR in the same fashion: no co-immunoprecipitation was observed, nor the same degree of reducing MOR-agonist potency upon RGS12 overexpression). Consistent with the in vitro results suggesting an important role for RGS12 in blunting cAMP inhibition yet enhancing β-arrestin recruitment to activated KOR, RGS12 loss in mice enhances KOR-induced analgesia and attenuates KOR-induced CPA – G protein- and β–arrestin-dependent behaviors, respectively.82,96,129 Given these data, we hypothesize that RGS12 is a hitherto unappreciated key regulator of KOR signaling, acting on both G protein- and β–arrestin-dependent signals to modulate the output of dynorphin/KOR signaling to dopamine reuptake and to the behavioral responses of both analgesia and aversion (Fig. 3). An inhibitor of RGS12, if discovered via compound screening (e.g., ref.141), could therefore also have utility as an adjuvant to either a KOR agonist or traditional MOR-targeting analgesic, leading to a reduction in either KOR agonist-induced or dynorphin-induced dysphoria, respectively.
Figure 3. Model of RGS12 action in the differential behavioral outputs of kappa opioid receptor (KOR) signaling, as derived from recent published work using Rgs12-deficient mouse strains.
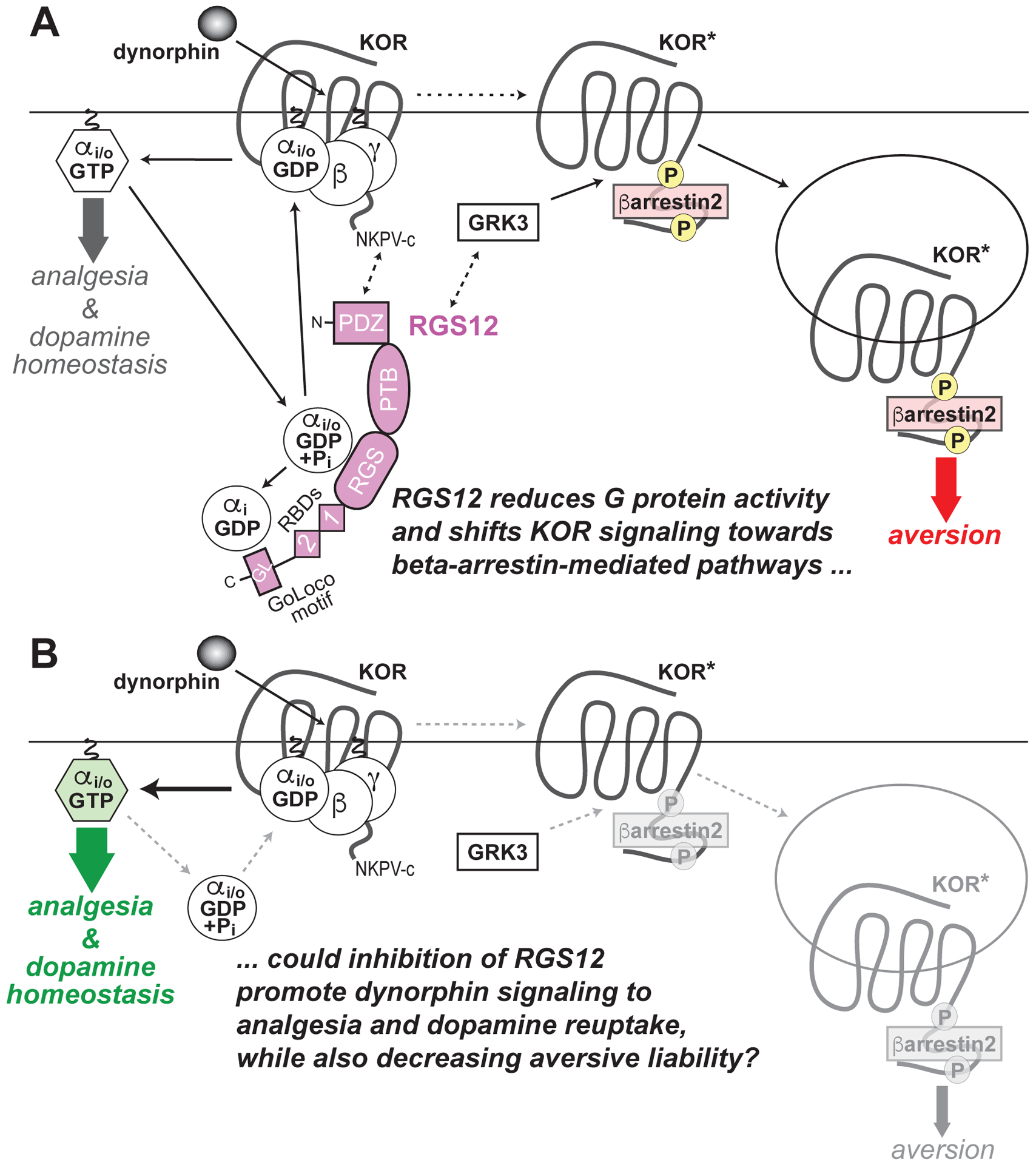
137,138 (A) Dynorphin-induced activation of KOR, as occurs during stress, dissociates Gi/o-coupled heterotrimers into free G protein subunits, evokes G protein-dependent signal transduction (thick grey arrow) and, ultimately, produces analgesia and also regulates dopamine homeostasis by the pre-synaptic dopamine transporter (DAT). RGS12 expression is known to reduce KOR-mediated G protein signaling via its central RGS domain (by accelerating GTP hydrolysis of Gαi/o·GTP) and its C–terminal GoLoco motif (by trapping Gαi·GDP). RGS12 also contains an N-terminal PDZ domain that likely interacts with the C-terminal PDZ domain docking site of KOR (‘NKPV-c’). RGS12 also contains tandem Ras-binding domains (RBDs) that we previously established interact with activated H-Ras and Raf to enable MAPK/ERK scaffolding properties150. Agonist-stimulated KOR activation also drives GRK3-dependent βarrestin2 recruitment, resulting in receptor internalization, βarrestin-dependent signal transduction (thick red arrow) and, ultimately, aversive behavior (in parallel to decreased extracellular dopamine). RGS12 expression augments KOR agonist-stimulated βarrestin recruitment independent of its GAP and GDI activity137, suggesting that RGS12 modulates GRK3 activity on activated KOR (KOR*) and/or enhances βarrestin2 recruitment and/or function by as yet undetermined molecular mechanisms. (B) Loss of RGS12 in mice results in concomitant increases in KOR-mediated G protein signaling and decreases in βarrestin2 recruitment, thus resulting in enhanced KOR agonist-stimulated analgesia and enhanced DAT function causing enhanced dopamine reuptake (thicker green arrow), as well as an attenuation of KOR-stimulated aversion (thinner grey arrow), respectively.
These recent studies have revealed the complexities, and potential promise, of signaling pathway-selective KOR-agonists and RGS12 inhibitors in reducing the deadly addiction liability of traditional MOR-targeting opioid analgesics. Specific G protein-biased KOR-agonists, such as triazole 1.1 and nalfurafine, may produce analgesia on their own without producing untoward dysphoria, and/or serve as additions to traditional opioid analgesics to lower their dose and reduce their addictive potential. A great deal more research into this field of behavioral pharmacology is required to disentangle the complexities of such biased KOR-agonists and their potential utility as replacements for, or adjuvants to, traditional MOR-targeting analgesics. This further research on the pharmacotherapeutic utility of biased KOR agonism is especially important to transact in light of the recent challenges to the existence of (and utility of) biased agonism at MOR.151
Funding:
NIH grants F30 DA044711 (to S.W.K.), F32 DA051139 (to J.D.G.); R36 DA051703 (to A.N.W.), and R01 DA048153 (to D.P.S.)
Glossary
- Agonist
compound which activates receptor upon binding
- Antagonist
compound which prevents receptor activation upon binding
- Adrbk2
gene encoding GRK3
- Arrb2
gene encoding β-arrestin2
- BCE
before the common era
- cAMP
cyclic adenosine monophosphate
- CPA
conditioned place aversion
- CPP
conditioned place preference
- CRF
corticotropin releasing factor
- DAT
dopamine transporter
- DRN
dorsal raphe nucleus
- EOM Sal B
an ethoxymethyl ether derivative of salvinorin A
- GABA
the inhibitory neurotransmitter gamma-aminobutyric acid
- GABAergic
pertaining to, or elaborating, the neurotransmitter GABA
- GPCR
G protein-coupled receptor
- G protein
guanine nucleotide-binding protein
- GDP
guanosine diphosphate
- GRK
G protein-coupled receptor kinase
- GTP
guanosine triphosphate
- ICSS
intra-cranial self-stimulation
- KOR
kappa opioid receptor
- MAPK
mitogen-activated protein kinase
- MOR
mu opioid receptor
- NAcc
nucleus accumbens
- norBNI
the KOR antagonist nor-binaltorphimine
- OPRK1
gene encoding KOR protein
- OPRM1
gene encoding MOR protein
- OUD
opioid use disorder
- PAG
periaqueductal gray
- PDYN
gene encoding dynorphin
- RGS
regulator of G protein signaling
- RGS12
regulator of G protein signaling type 12
- RVM
rostral ventromedial medulla
- SUD
substance use disorder
- vSTR
ventral striatum
- VTA
ventral tegmental area
Footnotes
Conflicts of Interest: None declared.
References:
- 1.Rosenblum A, Marsch LA, Joseph H, Portenoy RK. Opioids and the treatment of chronic pain: controversies, current status, and future directions. Experimental and clinical psychopharmacology. 2008;16(5):405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilkerson RG, Kim HK, Windsor TA, Mareiniss DP. The Opioid Epidemic in the United States. Emerg Med Clin North Am. 2016;34(2):e1–e23. [DOI] [PubMed] [Google Scholar]
- 3.Kotwal A. Innovation, diffusion and safety of a medical technology: a review of the literature on injection practices. Social Science & Medicine. 2005;60(5):1133–1147. [DOI] [PubMed] [Google Scholar]
- 4.Courtwright DT. Opiate Addiction as a Consequence of the Civil War. Civil War History. 1978;24(2):101–111. [DOI] [PubMed] [Google Scholar]
- 5.Schuler MS, Dick AW, Stein BD. Heterogeneity in Prescription Opioid Pain Reliever Misuse Across Age Groups: 2015–2017 National Survey on Drug Use and Health. J Gen Intern Med. 2020;35(3):792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Portenoy RK, Foley KM. Chronic use of opioid analgesics in non-malignant pain: Report of 38 cases. Pain. 1986;25(2):171–186. [DOI] [PubMed] [Google Scholar]
- 7.Phillips DM. JCAHO Pain Management Standards Are Unveiled. JAMA. 2000;284(4):428–429. [DOI] [PubMed] [Google Scholar]
- 8.Remillard D, Kaye AD, McAnally H. Oxycodone’s Unparalleled Addictive Potential: Is it Time for a Moratorium? Current Pain and Headache Reports. 2019;23(2). [DOI] [PubMed] [Google Scholar]
- 9.McCance-Katz EF. SAMHSA/HHS: An Update on the Opioid Crisis. In: SAMHSA; 2018. [Google Scholar]
- 10.American_Psychiatric_Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed 2013. [Google Scholar]
- 11.Clarke H, Soneji N, Ko DT, Yun L, Wijeysundera DN. Rates and risk factors for prolonged opioid use after major surgery: population based cohort study. BMJ. 2014;348:g1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brummett CM, Waljee JF, Goesling J, et al. New Persistent Opioid Use After Minor and Major Surgical Procedures in US Adults. JAMA Surg. 2017;152(6):e170504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busse JW, Wang L, Kamaleldin M, et al. Opioids for Chronic Noncancer Pain: A Systematic Review and Meta-analysis. JAMA. 2018;320(23):2448–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah A, Hayes CJ, Martin BC. Factors Influencing Long-Term Opioid Use Among Opioid Naive Patients: An Examination of Initial Prescription Characteristics and Pain Etiologies. The Journal of Pain. 2017;18(11):1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vowles KE, McEntee ML, Julnes PS, Frohe T, Ney JP, van der Goes DN. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain. 2015;156(4):569–576. [DOI] [PubMed] [Google Scholar]
- 16.Volkow ND, McLellan AT. Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. N Engl J Med. 2016;374(13):1253–1263. [DOI] [PubMed] [Google Scholar]
- 17.Jones CM. Heroin use and heroin use risk behaviors among nonmedical users of prescription opioid pain relievers – United States, 2002–2004 and 2008–2010. Drug and Alcohol Dependence. 2013;132(1):95–100. [DOI] [PubMed] [Google Scholar]
- 18.Mollereau C, Parmentier M, Mailleux P, et al. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994;341(1):33–38. [DOI] [PubMed] [Google Scholar]
- 19.Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278(5335):52–58. [DOI] [PubMed] [Google Scholar]
- 20.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chavkin C, Koob GF. Dynorphin, Dysphoria, and Dependence: the Stress of Addiction. Neuropsychopharmacology. 2016;41(1):373–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crowley NA, Kash TL. Kappa opioid receptor signaling in the brain: Circuitry and implications for treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2015;62:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenwald MK, Stitzer ML. Butorphanol agonist effects and acute physical dependence in opioid abusers: comparison with morphine. Drug Alcohol Depend. 1998;53(1):17–30. [DOI] [PubMed] [Google Scholar]
- 24.Chen KY, Chen L, Mao J. Buprenorphine–Naloxone Therapy in Pain Management. 2014;120(5):1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hswen Y, Zhang A, Brownstein JS. Leveraging black-market street buprenorphine pricing to increase capacity to treat opioid addiction, 2010–2018. Prev Med. 2020;137:106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11(12):823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghelardini C, Di Cesare Mannelli L, Bianchi E. The pharmacological basis of opioids. Clin Cases Miner Bone Metab. 2015;12(3):219–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yaksh TL, Jessell TM, Gamse R, Mudge AW, Leeman SE. Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature. 1980;286(5769):155–157. [DOI] [PubMed] [Google Scholar]
- 29.Basbaum AI, Fields HL. Endogenous pain control mechanisms: review and hypothesis. Ann Neurol. 1978;4(5):451–462. [DOI] [PubMed] [Google Scholar]
- 30.Behbehani MM. Functional characteristics of the midbrain periaqueductal gray. Prog Neurobiol. 1995;46(6):575–605. [DOI] [PubMed] [Google Scholar]
- 31.Stein C. Opioid Receptors. Annual Review of Medicine. 2016;67(1):433–451. [DOI] [PubMed] [Google Scholar]
- 32.Llewelyn MB, Azami J, Gibbs M, Roberts MH. A comparison of the sites at which pentazocine and morphine act to produce analgesia. Pain. 1983;16(4):313–331. [DOI] [PubMed] [Google Scholar]
- 33.Smith DJ, Perrotti JM, Crisp T, Cabral MEY, Long JT, Scalzitti JM. The μ opiate receptor is responsible for descending pain inhibition originating in the periaqueductal gray region of the rat brain. 1988;156(1):47–54. [DOI] [PubMed] [Google Scholar]
- 34.Hoskin PJ, Hanks GW. Opioid agonist-antagonist drugs in acute and chronic pain states. Drugs. 1991;41(3):326–344. [DOI] [PubMed] [Google Scholar]
- 35.Clark JA, Pasternak GW. U50,488: a kappa-selective agent with poor affinity for mu1 opiate binding sites. Neuropharmacology. 1988;27(3):331–332. [DOI] [PubMed] [Google Scholar]
- 36.Gutstein HB, Mansour A, Watson SJ, Akil H, Fields HL. Mu and kappa opioid receptors in periaqueductal gray and rostral ventromedial medulla. Neuroreport. 1998;9(8):1777–1781. [DOI] [PubMed] [Google Scholar]
- 37.Pan ZZ, Tershner SA, Fields HL. Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature. 1997;389(6649):382–385. [DOI] [PubMed] [Google Scholar]
- 38.Kamp EH, Jones RC 3rd, Tillman SR, Gebhart GF. Quantitative assessment and characterization of visceral nociception and hyperalgesia in mice. Am J Physiol Gastrointest Liver Physiol. 2003;284(3):G434–444. [DOI] [PubMed] [Google Scholar]
- 39.Arendt-Nielsen L, Olesen AE, Staahl C, et al. Analgesic efficacy of peripheral kappa-opioid receptor agonist CR665 compared to oxycodone in a multi-modal, multi-tissue experimental human pain model: selective effect on visceral pain. Anesthesiology. 2009;111(3):616–624. [DOI] [PubMed] [Google Scholar]
- 40.Cervero F, Laird JM. Visceral pain. Lancet. 1999;353(9170):2145–2148. [DOI] [PubMed] [Google Scholar]
- 41.Porreca F, Navratilova E. Reward, motivation, and emotion of pain and its relief. Pain. 2017;158 Suppl 1:S43–S49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koob GF. Addiction is a Reward Deficit and Stress Surfeit Disorder. Front Psychiatry. 2013;4:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Merrer J, Becker JA, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev. 2009;89(4):1379–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Froehlich JC. Opioid peptides. Alcohol Health Res World. 1997;21(2):132–136. [PMC free article] [PubMed] [Google Scholar]
- 46.Veening JG, Barendregt HP. The effects of beta-endorphin: state change modification. Fluids Barriers CNS. 2015;12:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spanagel R, Herz A, Bals-Kubik R, Shippenberg TS. Beta-endorphin-induced locomotor stimulation and reinforcement are associated with an increase in dopamine release in the nucleus accumbens. Psychopharmacology (Berl). 1991;104(1):51–56. [DOI] [PubMed] [Google Scholar]
- 48.Amalric M, Cline EJ, Martinez JL Jr., Bloom FE, Koob GF. Rewarding properties of beta-endorphin as measured by conditioned place preference. Psychopharmacology (Berl). 1987;91(1):14–19. [DOI] [PubMed] [Google Scholar]
- 49.Roth-Deri I, Schindler CJ, Yadid G. A critical role for beta-endorphin in cocaine-seeking behavior. Neuroreport. 2004;15(3):519–521. [DOI] [PubMed] [Google Scholar]
- 50.Trigo JM, Zimmer A, Maldonado R. Nicotine anxiogenic and rewarding effects are decreased in mice lacking beta-endorphin. Neuropharmacology. 2009;56(8):1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dutia R, Meece K, Dighe S, Kim AJ, Wardlaw SL. beta-Endorphin antagonizes the effects of alpha-MSH on food intake and body weight. Endocrinology. 2012;153(9):4246–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grisel JE, Bartels JL, Allen SA, Turgeon VL. Influence of beta-Endorphin on anxious behavior in mice: interaction with EtOH. Psychopharmacology (Berl). 2008;200(1):105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Papaleo F, Kieffer BL, Tabarin A, Contarino A. Decreased motivation to eat in mu-opioid receptor-deficient mice. Eur J Neurosci. 2007;25(11):3398–3405. [DOI] [PubMed] [Google Scholar]
- 54.Kas MJ, van den Bos R, Baars AM, et al. Mu-opioid receptor knockout mice show diminished food-anticipatory activity. Eur J Neurosci. 2004;20(6):1624–1632. [DOI] [PubMed] [Google Scholar]
- 55.Fields HL, Margolis EB. Understanding opioid reward. Trends Neurosci. 2015;38(4):217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012;76(3):470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leone P, Pocock D, Wise RA. Morphine-dopamine interaction: ventral tegmental morphine increases nucleus accumbens dopamine release. Pharmacol Biochem Behav. 1991;39(2):469–472. [DOI] [PubMed] [Google Scholar]
- 58.Chefer VI, Denoroy L, Zapata A, Shippenberg TS. Mu opioid receptor modulation of somatodendritic dopamine overflow: GABAergic and glutamatergic mechanisms. Eur J Neurosci. 2009;30(2):272–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chefer VI, Kieffer BL, Shippenberg TS. Basal and morphine-evoked dopaminergic neurotransmission in the nucleus accumbens of MOR- and DOR-knockout mice. Eur J Neurosci. 2003;18(7):1915–1922. [DOI] [PubMed] [Google Scholar]
- 60.Contarino A, Picetti R, Matthes HW, Koob GF, Kieffer BL, Gold LH. Lack of reward and locomotor stimulation induced by heroin in mu-opioid receptor-deficient mice. Eur J Pharmacol. 2002;446(1–3):103–109. [DOI] [PubMed] [Google Scholar]
- 61.Hall FS, Li XF, Goeb M, et al. Congenic C57BL/6 mu opiate receptor (MOR) knockout mice: baseline and opiate effects. Genes Brain Behav. 2003;2(2):114–121. [DOI] [PubMed] [Google Scholar]
- 62.Becker A, Grecksch G, Brodemann R, et al. Morphine self-administration in mu-opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2000;361(6):584–589. [DOI] [PubMed] [Google Scholar]
- 63.Matthes HW, Maldonado R, Simonin F, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383(6603):819–823. [DOI] [PubMed] [Google Scholar]
- 64.Olmstead MC, Franklin KB. The development of a conditioned place preference to morphine: effects of microinjections into various CNS sites. Behav Neurosci. 1997;111(6):1324–1334. [DOI] [PubMed] [Google Scholar]
- 65.Zangen A, Ikemoto S, Zadina JE, Wise RA. Rewarding and psychomotor stimulant effects of endomorphin-1: anteroposterior differences within the ventral tegmental area and lack of effect in nucleus accumbens. J Neurosci. 2002;22(16):7225–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.David V, Cazala P. Differentiation of intracranial morphine self-administration behavior among five brain regions in mice. Pharmacol Biochem Behav. 1994;48(3):625–633. [DOI] [PubMed] [Google Scholar]
- 67.Sophie M, Ford B. Management of Pain in Parkinson’s Disease. CNS Drugs. 2012;26(11):937–948. [DOI] [PubMed] [Google Scholar]
- 68.Cobacho N, de la Calle JL, Paíno CL. Dopaminergic modulation of neuropathic pain: Analgesia in rats by a D2-type receptor agonist. Brain Research Bulletin. 2014;106:62–71. [DOI] [PubMed] [Google Scholar]
- 69.Hnasko TS, Sotak BN, Palmiter RD. Morphine reward in dopamine-deficient mice. Nature. 2005;438(7069):854–857. [DOI] [PubMed] [Google Scholar]
- 70.Narita M, Matsushima Y, Niikura K, et al. Implication of dopaminergic projection from the ventral tegmental area to the anterior cingulate cortex in μ-opioid-induced place preference. Addiction Biology. 2010;15(4):434–447. [DOI] [PubMed] [Google Scholar]
- 71.Manzanedo C, Aguilar MA, Rodriguez-Arias M, Minarro J. Effects of dopamine antagonists with different receptor blockade profiles on morphine-induced place preference in male mice. Behav Brain Res. 2001;121(1–2):189–197. [DOI] [PubMed] [Google Scholar]
- 72.Simonin F, Valverde O, Smadja C, et al. Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J. 1998;17(4):886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zimmer A, Valjent E, Konig M, et al. Absence of delta −9-tetrahydrocannabinol dysphoric effects in dynorphin-deficient mice. J Neurosci. 2001;21(23):9499–9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mizoguchi H, Watanabe C, Osada S, et al. Lack of a rewarding effect and a locomotor-enhancing effect of the selective mu-opioid receptor agonist amidino-TAPA. Psychopharmacology (Berl). 2010;212(2):215–225. [DOI] [PubMed] [Google Scholar]
- 75.Matthes HW, Smadja C, Valverde O, et al. Activity of the delta-opioid receptor is partially reduced, whereas activity of the kappa-receptor is maintained in mice lacking the mu-receptor. J Neurosci. 1998;18(18):7285–7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Skoubis PD, Matthes HW, Walwyn WM, Kieffer BL, Maidment NT. Naloxone fails to produce conditioned place aversion in mu-opioid receptor knock-out mice. Neuroscience. 2001;106(4):757–763. [DOI] [PubMed] [Google Scholar]
- 77.Knoll AT, Carlezon WA Jr. Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marchant NJ, Densmore VS, Osborne PB. Coexpression of prodynorphin and corticotrophin-releasing hormone in the rat central amygdala: evidence of two distinct endogenous opioid systems in the lateral division. J Comp Neurol. 2007;504(6):702–715. [DOI] [PubMed] [Google Scholar]
- 79.Steiner H, Gerfen CR. Role of dynorphin and enkephalin in the regulation of striatal output pathways and behavior. Exp Brain Res. 1998;123(1–2):60–76. [DOI] [PubMed] [Google Scholar]
- 80.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28(2):407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luo M, Zhou J, Liu Z. Reward processing by the dorsal raphe nucleus: 5-HT and beyond. Learn Mem. 2015;22(9):452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ehrich JM, Messinger DI, Knakal CR, et al. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons. J Neurosci. 2015;35(37):12917–12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chefer VI, Backman CM, Gigante ED, Shippenberg TS. Kappa opioid receptors on dopaminergic neurons are necessary for kappa-mediated place aversion. Neuropsychopharmacology. 2013;38(13):2623–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Margolis EB, Mitchell JM, Ishikawa J, Hjelmstad GO, Fields HL. Midbrain Dopamine Neurons: Projection Target Determines Action Potential Duration and Dopamine D2 Receptor Inhibition. 2008;28(36):8908–8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nestler EJ, Carlezon WA Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59(12):1151–1159. [DOI] [PubMed] [Google Scholar]
- 86.Mysels D, Sullivan MA. The kappa-opiate receptor impacts the pathophysiology and behavior of substance use. Am J Addict. 2009;18(4):272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Serra V, Fattore L, Scherma M, et al. Behavioural and neurochemical assessment of salvinorin A abuse potential in the rat. Psychopharmacology (Berl). 2015;232(1):91–100. [DOI] [PubMed] [Google Scholar]
- 88.Ruan X, Hall SM, Kaye AD. Nalfurafine hydrochloride, a selective kappa opioid receptor agonist, has no reinforcing effect on intravenous self-administration in rhesus monkeys. J Pharmacol Sci. 2016;132(1):113–114. [DOI] [PubMed] [Google Scholar]
- 89.Massaly N, Copits BA, Wilson-Poe AR, et al. Pain-Induced Negative Affect Is Mediated via Recruitment of The Nucleus Accumbens Kappa Opioid System. Neuron. 2019;102(3):564–573 e566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Al-Hasani R, McCall JG, Shin G, et al. Distinct Subpopulations of Nucleus Accumbens Dynorphin Neurons Drive Aversion and Reward. Neuron. 2015;87(5):1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gilpin NW, Roberto M, Koob GF, Schweitzer P. Kappa opioid receptor activation decreases inhibitory transmission and antagonizes alcohol effects in rat central amygdala. Neuropharmacology. 2014;77:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Knoll AT, Muschamp JW, Sillivan SE, et al. Kappa opioid receptor signaling in the basolateral amygdala regulates conditioned fear and anxiety in rats. Biol Psychiatry. 2011;70(5):425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bruchas MR, Land BB, Lemos JC, Chavkin C. CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PLoS One. 2009;4(12):e8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tao R, Auerbach SB. mu-Opioids disinhibit and kappa-opioids inhibit serotonin efflux in the dorsal raphe nucleus. Brain Res. 2005;1049(1):70–79. [DOI] [PubMed] [Google Scholar]
- 95.Bruchas MR, Schindler AG, Shankar H, et al. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71(3):498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Land BB, Bruchas MR, Schattauer S, et al. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106(45):19168–19173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nabeshima T, Katoh A, Wada M, Kameyama T. Stress-induced changes in brain Met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci. 1992;51(3):211–217. [DOI] [PubMed] [Google Scholar]
- 98.Brust TF, Morgenweck J, Kim SA, et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal. 2016;9(456):ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mantsch JR, Baker DA, Funk D, Le AD, Shaham Y. Stress-Induced Reinstatement of Drug Seeking: 20 Years of Progress. Neuropsychopharmacology. 2016;41(1):335–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl). 2008;200(1):59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23(13):5674–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koob GF, Schulkin J. Addiction and stress: An allostatic view. Neurosci Biobehav Rev. 2018. [DOI] [PubMed] [Google Scholar]
- 103.Koob GF. Hedonic Homeostatic Dysregulation as a Driver of Drug-Seeking Behavior. Drug Discov Today Dis Models. 2008;5(4):207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rasmussen K, White DA, Acri JB. NIDA’s medication development priorities in response to the Opioid Crisis: ten most wanted. Neuropsychopharmacology. 2019;44(4):657–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maund E, McDaid C, Rice S, Wright K, Jenkins B, Woolacott N. Paracetamol and selective and non-selective non-steroidal anti-inflammatory drugs for the reduction in morphine-related side-effects after major surgery: a systematic review. 2011;106(3):292–297. [DOI] [PubMed] [Google Scholar]
- 106.Xu J, Lu Z, Narayan A, et al. Alternatively spliced mu opioid receptor C termini impact the diverse actions of morphine. Journal of Clinical Investigation. 2017;127(4):1561–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Spahn V, Del Vecchio G, Labuz D, et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science. 2017;355(6328):966–969. [DOI] [PubMed] [Google Scholar]
- 108.Briggs SL, Rech RH, Sawyer DC. Kappa Antinociceptive Activity of Spiradoline in the Cold-Water Tail-Flick Assay in Rats. Pharmacology Biochemistry and Behavior. 1998;60(2):467–472. [DOI] [PubMed] [Google Scholar]
- 109.Negus SS, Schrode K, Stevenson GW. Micro/kappa opioid interactions in rhesus monkeys: implications for analgesia and abuse liability. Experimental and clinical psychopharmacology. 2008;16(5):386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Khotib J, Narita M, Suzuki M, Yajima Y, Suzuki T. Functional interaction among opioid receptor types: up-regulation of mu- and delta-opioid receptor functions after repeated stimulation of kappa-opioid receptors. Neuropharmacology. 2004;46(4):531–540. [DOI] [PubMed] [Google Scholar]
- 111.Field MJ, Carnell AJ, Gonzalez MI, et al. Enadoline, a selective κ-opioid receptor agonist shows potent antihyperalgesic and antiallodynic actions in a rat model of surgical pain. Pain. 1999;80(1):383–389. [DOI] [PubMed] [Google Scholar]
- 112.Tao PL, Liang KW, Sung WY, Wu YT, Huang EY. Nalbuphine is effective in decreasing the rewarding effect induced by morphine in rats. Drug Alcohol Depend. 2006;84(2):175–181. [DOI] [PubMed] [Google Scholar]
- 113.Walsh SL, Babalonis S. The Abuse Potential of Prescription Opioids in Humans-Closing in on the First Century of Research. Curr Top Behav Neurosci. 2017;34:33–58. [DOI] [PubMed] [Google Scholar]
- 114.Preston KL, Bigelow GE, Liebson IA. Comparative evaluation of morphine, pentazocine and ciramadol in postaddicts. J Pharmacol Exp Ther. 1987;240(3):900–910. [PubMed] [Google Scholar]
- 115.Zacny JP, Lichtor JL, Thapar P, Coalson DW, Flemming D, Thompson WK. Comparing the subjective, psychomotor and physiological effects of intravenous butorphanol and morphine in healthy volunteers. J Pharmacol Exp Ther. 1994;270(2):579–588. [PubMed] [Google Scholar]
- 116.Walsh SL, Chausmer AE, Strain EC, Bigelow GE. Evaluation of the mu and kappa opioid actions of butorphanol in humans through differential naltrexone blockade. Psychopharmacology (Berl). 2008;196(1):143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Preston KL, Bigelow GE. Differential naltrexone antagonism of hydromorphone and pentazocine effects in human volunteers. J Pharmacol Exp Ther. 1993;264(2):813–823. [PubMed] [Google Scholar]
- 118.Preston KL, Bigelow GE, Liebson IA. Butorphanol-precipitated withdrawal in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1988;246(2):441–448. [PubMed] [Google Scholar]
- 119.Preston KL, Bigelow GE, Liebson IA. Antagonist effects of nalbuphine in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1989;248(3):929–937. [PubMed] [Google Scholar]
- 120.Strain EC, Preston KL, Liebson IA, Bigelow GE. Precipitated withdrawal by pentazocine in methadone-maintained volunteers. J Pharmacol Exp Ther. 1993;267(2):624–634. [PubMed] [Google Scholar]
- 121.Greedy BM, Bradbury F, Thomas MP, et al. Orvinols with mixed kappa/mu opioid receptor agonist activity. J Med Chem. 2013;56(8):3207–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cara_Therapeutics_Inc. A Study to Evaluate the Safety and Efficacy of CR845 in Hemodialysis Patients With Moderate-to-Severe Pruritus. https://ClinicalTrials.gov/show/NCT03422653. Published 2018. Accessed.
- 123.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nature Reviews Drug Discovery. 2017;16(12):829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G-protein signaling: back to the future. Cellular and molecular life sciences : CMLS. 2005;62(5):551–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wisler JW, Xiao K, Thomsen ARB, Lefkowitz RJ. Recent developments in biased agonism. Current opinion in cell biology. 2014;27:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Raehal KM. Morphine Side Effects in -Arrestin 2 Knockout Mice. 2005;314(3):1195–1201. [DOI] [PubMed] [Google Scholar]
- 127.Bruchas MR, Land BB, Aita M, et al. Stress-Induced p38 Mitogen-Activated Protein Kinase Activation Mediates -Opioid-Dependent Dysphoria. 2007;27(43):11614–11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. The Journal of biological chemistry. 2006;281(26):18081–18089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.White KL, Robinson JE, Zhu H, et al. The G Protein-Biased -Opioid Receptor Agonist RB-64 Is Analgesic with a Unique Spectrum of Activities In Vivo. Journal of Pharmacology and Experimental Therapeutics. 2014;352(1):98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kaski SW, White AN, Gross JD, et al. Preclinical Testing of Nalfurafine as an Opioid-sparing Adjuvant that Potentiates Analgesia by the Mu Opioid Receptor-targeting Agonist Morphine. J Pharmacol Exp Ther. 2019;371(2):487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ueno Y, Mori A, Yanagita T. One year long-term study on abuse liability of nalfurafine in hemodialysis patients. Int J Clin Pharmacol Ther. 2013;51(11):823–831. [DOI] [PubMed] [Google Scholar]
- 132.Inui S. Nalfurafine hydrochloride to treat pruritus: a review. Clin Cosmet Investig Dermatol. 2015;8:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huskinson SL, Platt DM, Brasfield M, et al. Quantification of observable behaviors induced by typical and atypical kappa-opioid receptor agonists in male rhesus monkeys. Psychopharmacology (Berl). 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zamarripa CA, Naylor JE, Huskinson SL, Townsend EA, Prisinzano TE, Freeman KB. Kappa opioid agonists reduce oxycodone self-administration in male rhesus monkeys. Psychopharmacology (Berl). 2020;237(5):1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zamarripa CA, Patel TR, Williams BC, et al. The kappa-opioid receptor agonist, nalfurafine, blocks acquisition of oxycodone self-administration and oxycodone’s conditioned rewarding effects in male rats. Behav Pharmacol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang Y, Kreek MJ. Nalfurafine modulates the reinforcing effects of oxycodone in male and female adolescent C57BL/6J mice. Neuropharmacology. 2020;176:108244. [DOI] [PubMed] [Google Scholar]
- 137.Gross JD, Kaski SW, Schmidt KT, et al. Role of RGS12 in the differential regulation of kappa opioid receptor-dependent signaling and behavior. Neuropsychopharmacology. 2019;44(10):1728–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gross JD, Kaski SW, Schroer AB, Wix KA, Siderovski DP, Setola V. Regulator of G protein signaling-12 modulates the dopamine transporter in ventral striatum and locomotor responses to psychostimulants. J Psychopharmacol. 2018;32(2):191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.White AN, Gross JD, Kaski SW, et al. Genetic deletion of Rgs12 in mice affects serotonin transporter expression and function in vivo and ex vivo. J Psychopharmacol. 2020:269881120944160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kivell B, Uzelac Z, Sundaramurthy S, et al. Salvinorin A regulates dopamine transporter function via a kappa opioid receptor and ERK1/2-dependent mechanism. Neuropharmacology. 2014;86:228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kimple AJ, Yasgar A, Hughes M, et al. A high throughput fluorescence polarization assay for inhibitors of the GoLoco motif/G-alpha interaction. Comb Chem High Throughput Screen. 2008;11(5):396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cesselin F, Benoliel J-J, Bourgoin S, Collin E, Pohl M, Hamon M. Spinal Mechanisms of Opioid Analgesia. In: Stein C, ed. Opioids in Pain Control: Basic and Clinical Aspects. New York, NY: Cambridge University Press; 1999. [Google Scholar]
- 143.Pizziketti RJ, Pressman NS, Geller EB, Cowan A, Adler MW. Rat cold water tail-flick: A novel analgesic test that distinguishes opioid agonists from mixed agonist-antagonists. European Journal of Pharmacology. 1985;119(1):23–29. [DOI] [PubMed] [Google Scholar]
- 144.Auh QS, Ro JY. Effects of peripheral kappa opioid receptor activation on inflammatory mechanical hyperalgesia in male and female rats. Neurosci Lett. 2012;524(2):111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Garcia MM, Goicoechea C, Avellanal M, Traseira S, Martin MI, Sanchez-Robles EM. Comparison of the antinociceptive profiles of morphine and oxycodone in two models of inflammatory and osteoarthritic pain in rat. Eur J Pharmacol. 2019;854:109–118. [DOI] [PubMed] [Google Scholar]
- 146.Millan MJ, Colpaert FC. Opioid systems in the response to inflammatory pain: sustained blockade suggests role of kappa- but not mu-opioid receptors in the modulation of nociception, behaviour and pathology. Neuroscience. 1991;42(2):541–553. [DOI] [PubMed] [Google Scholar]
- 147.Black D, Trevethick M. The kappa opioid receptor is associated with the perception of visceral pain. Gut. 1998;43(3):312–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Munro TA, Duncan KK, Xu W, et al. Standard protecting groups create potent and selective kappa opioids: salvinorin B alkoxymethyl ethers. Bioorg Med Chem. 2008;16(3):1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhou L, Lovell KM, Frankowski KJ, et al. Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem. 2013;288(51):36703–36716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Willard MD, Willard FS, Li X, Cappell SD, Snider WD, Siderovski DP. Selective role for RGS12 as a Ras/Raf/MEK scaffold in nerve growth factor-mediated differentiation. EMBO J. 2007;26(8):2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gillis A, Kliewer A, Kelly E, et al. Critical assessment of G protein-biased agonism at the mu-opioid receptor. Trends Pharmacol Sci. 2020; doi: 10.1016/j.tips.2020.09.009. [DOI] [PubMed] [Google Scholar]