

Introduction
Growing evidence indicates that most, if not all, chronic diseases and geriatric syndromes share common pathways with aging and that these mechanisms may be modified. This Geroscience Hypothesis arose from the observation that aging is the greatest risk factor for most chronic diseases, such as cardiovascular disease, type II diabetes, cancer and many degenerative and neurodegenerative disorders[1]. The main advantage of looking at aging through the lens of the Geroscience Hypothesis is that, if true, then by targeting these shared aging mechanisms, it may be possible to prevent, delay and perhaps even reverse multiple aging-related conditions and phenotypes simultaneously [2–4] Such interventions would increase the human healthspan (quality, healthy, independent, productive years of life), while decreasing the burden of long disease-ridden years later in life on both caretakers and economy[5, 6]. Several molecular pathways contributing to aging have been identified collectively called “the Hallmarks of Aging” [7].More recently, these have been grouped into 4 main categories, or fundamental aging mechanisms: 1) chronic, low-grade sterile inflammation and fibrosis, 2) Macromolecular/organelle dysfunction, 3) Stem and progenitor cell dysfunction, and 4) cellular senescence[8, 9]. Several studies show that these mechanisms are somewhat intertwined: initiating one can activate others and targeting one usually affects others. Genetically and pharmacologically targeting cellular senescence, in particular, has been the focus of much recent research with very promising pre-clinical results [10–24]. The goal of this review is to describe advances in discovering senolytics, drugs that eliminate senescent cells (SnCs) to delay or alleviate geriatric diseases and syndromes, introduce techniques and models used for their discovery, and where we are in terms of clinical translation.
Cellular Senescence
Like apoptosis and differentiation, cellular senescence is a cellular fate[25]. SnCs are dysfunctional cells that have permanently exited the cell cycle in response to stress or molecular damage. They are resistant to apoptosis, accumulating in many organs 1) with aging, 2) as a result of therapies that cause DNA damage such as chemotherapy or radiation in cancer patients, and 3) at sites of pathology of chronic diseases such as atherosclerosis, obesity, Alzheimer’s and idiopathic pulmonary fibrosis (IPF). Their most detrimental aspect is believed to be their secretome, the senescence-associated secretory phenotype, SASP[25, 26]. The SASP is highly inflammatory, and is composed of many pro-inflammatory cytokines, chemokines and proteases that can be damaging to organs, ultimately resulting in dysfunction. The SASP is so noxious that a small percentage of SnCs might cause substantial tissue damage. This is supported by our studies showing that transplanting just 1 million SnCs (<0.03% of cells throughout the body) intraperitoneally into young, healthy mice, can cause the onset of aging-like symptoms, increase senescence elsewhere and decrease lifespan[27, 10]. Additionally, studies done in transgenic animals, where SnCs (those highly expressing the cyclin-dependent kinase inhibitor p16) were ablated using genetic approaches showed marked improvements in several aging phenotypes[18, 19, 22, 28]. Additionally, senescence in stem cell niches reduces the regenerative capacity of tissues, leading to deterioration and pathology[29, 13].
Cumulatively, pre-clinical data suggest that reducing SnC burden or preventing their accumulation may alleviate or delay the onset of aging-related conditions. Several methods for accomplishing this are being developed and include senolytics (drugs that specifically target and induce the death of SnCs), senomorphics (drugs that dampen or inhibit the SASP), promoting reactivation of proliferation of SnCs (but this may accidentally induce tumorigenesis) and immunotherapy (boosting the immune system to re-instate its ability to combat SnCs as in young organisms)[30].
Senolytics
Ideal senolytics for translation to human use would be drugs that can specifically target and kill harmful SnCs without affecting other cells or harming the individual.[31, 32] The discovery of such senolytics, however, has been made difficult by several factors: 1) Most of our knowledge about SnCs comes from in vitro studies using SnCs induced by irradiation or treatment with DNA-damaging drugs, and although we showed that these in vitro-generated SnCs replicate some of the key altered pathways in naturally occurring SnCs, they may not reflect precisely what occurs in vivo[33]; 2) The percentage of SnCs in various tissues in vivo is relatively small (0.5–20%); 3) There are no sensitive, specific or standard biomarkers for identifying SnCs, and the most routinely used markers such as senescence-associated beta-galactosidase, p16 and p21 are not found in all SnCs and can also be found in non-SnCs (p16 is expressed in many terminally differentiated cell types); 4) SnCs might be as heterogeneous as the cell types they arise from, and may therefore require targeting in terms of matching specific drug combinations with cell type.
Administration of senolytics is envisioned to be intermittent. This proposed “hit-and-run” nature of senolytic treatment reduces the possibility of undesirable off-target effects involved in traditional continuous administration of a drug[34]. Damaged cells take 2–6 weeks to senesce, and a certain threshold of SnCs has to be reached in the body before they can cause damage. Thus, senolytics, designed to kill a large portion of SnCs in a few doses, only needs to be repeated when enough SnCs have returned. The rate of re-accumulation of SnCs is expected to vary among different people, and thus sensitive and reliable biomarkers for SnCs are desired to measure the SnC load in individuals, allowing for more tailored senolytic therapy (precision medicine)[35]. With all of these considerations in mind, we will review most major classes of senolytic drugs developed to date.
Dasatinib and Quercetin
Zhu et al. used a hypothesis-driven approach combined with bioinformatics to identify Dasatinib and Quercetin as the first senolytic agents[12]. They reasoned that SnCs, reported to be resistant to apoptosis, have upregulated pro-survival pathways and down-regulated pro-apoptotic pathways[36]. Comparing proteomic and transcriptomic profiles of senescent and non-SnCs allowed them to identify several Senescent Cell Anti-Apoptotic Pathways (SCAPs). Using RNA interference studies to target these pathways, they identified ones that specifically induced death of SnCs rather than proliferating cells, and realized that different SCAPs need to be targeted to induce apoptosis in different cell types. Dasatinib, a tyrosine kinase inhibitor used in cancer therapy, interferes with members of the ephrin survival-regulating dependence receptors (EFNB) appears to exhibit specificity in terms of killing senescent preadipocytes. In contrast, quercetin, a flavonoid that inhibits kinases (including PI3K), serpines, and certain BCL-2 family members, demonstrates greater specificity for killing senescent endothelial cells. Furthermore, combining the two (D+Q) kills more senescent cell types than either alone by simultaneously targeting multiple SCAPs. The in vivo effects of D+Q senolytic therapy have been tested under dozens of conditions in mouse models including physical dysfunction, osteoporosis, insulin resistance, Alzheimer’s, kidney dysfunction, vasomotor dysfunction, liver steatosis, pulmonary fibrosis, anxiety and lifespan reduction and ex vivo on human tissue with promising results [14, 37–41, 11].
D+Q has moved to human trials of incurable diseases where SnCs have been found to play a causal role such as idiopathic pulmonary fibrosis (IPF), diabetic kidney disease (DKD) and early stage symptomatic tau+ Alzherimer’s disease to determine safety, tolerability and effectiveness (Figure 1) [42, 43, 34, 44, 37, 45, 46]. Early proof of concept human studies reported no serious drug effects, reduced SnC burden, reduction in SASP factors and improved physical function[42, 43, 34]. Follow-up double blinded placebo-controlled trials will be needed before it becomes possible to study the ability of D+Q to prevent or reverse age-related conditions and improve healthspan in older adults.
Figure 1. Senolytics Clinical Trials in Humans.
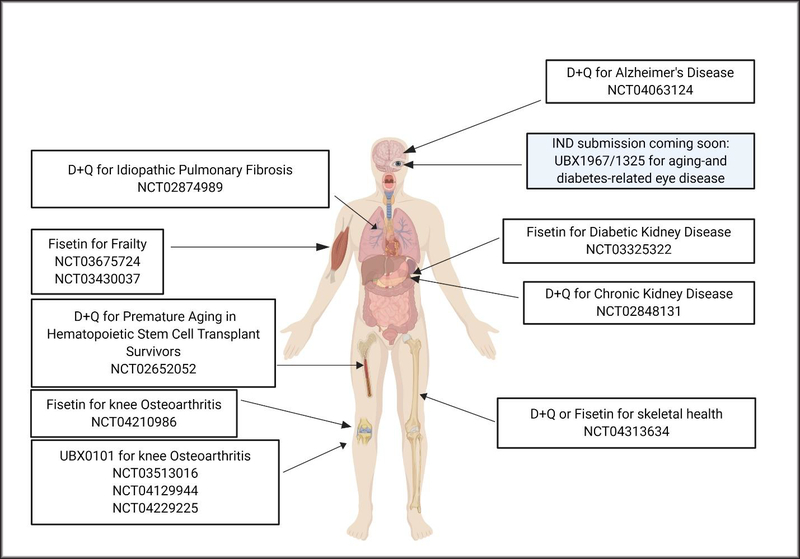
Shown are some completed, recruiting, ongoing and planned trials. Before being tested in healthy adults for delaying or alleviating aging-related diseases and syndromes, senolytics are being tested for safety and efficacy of removing senescent cells from patients with serious conditions involving senescent cell accumulation for which there are currently no other cures. These include idiopathic pulmonary fibrosis (IPF), diabetic/chronic kidney disease, tau+ Alzheimer’s disease, accelerated aging resulting from hematopoietic stem cell transplantation, frailty and osteoarthritis. Some completed Phase I trials have reported no serious adverse effects (eg, D+Q, UBX0101) and are moving on to Phase II trials. (Image created using Biorender.)
Fisetin
Fisetin, a flavonoid very similar in structure to Quercetin, was identified in a screen of flavonoids for more potent senolytics[17, 47]. Fisetin was found to clear SnCs of different lineages and reduce SASP in vivo in several mouse aging models, reduce senescence in human ex vivo tissue explants, and increase lifespan and healthspan. It alleviated frailty in mice even when administered late in life (starting at 20 months, equivalent to approximately 60 human years). Fisetin is attractive for therapy because it is widely available as a supplement with few apparent side effects and better bioavailability, making it eminently translatable. Additionally, it has other desirable effects such as being anti-inflammatory, anti-oxidant, anti-carcinogenic and its targets include PI3 kinase δ, some BCL-2 family members, nuclear factor-kappa B (NF-κB), and mTOR [48–51]. Furthermore, Fisetin was described to be a caloric restriction mimetic, an intervention shown to increase lifespan in mammals[52]. Its well-established safety profile and many benefits have made fisetin one of the quickest senolytics to enter into human clinical trials, with studies currently underway examining its potential benefits in frailty, osteoarthritis and chronic kidney diseases (Figure1).
Targeting the BCL-2 family (Navitoclax (ABT-263), UBX1967/1325, ABT737, EF24)
In a bioinformatics screen for the identification of pathways that may be conferring the resistance of SnCs to apoptosis, Zhu et al. identified upregulation of the BCL-2 pro-survival pathway as one of the SCAPs[12]. This family includes the anti-apoptotic proteins BCL-2, BCL-W, BCL-XL, MCL-1 and A1. Soon thereafter, two reports identified Navitoclax (or ABT-263) as a senolytic[13, 53]. Navitoclax can target several BCL-2 family members (BCL-2, BCL-W and BCL-XL), which are upregulated in senescent HUVECs and IMR-90 cells, but not in primary human preadipocytes. Consequently, Navitoclax was found to be senolytic in HUVECs and IMR-90s but not preadipocytes, with promising in vivo results for rejuvenating stem cells of the hematopoietic and skeletal muscle systems[13]. However, Navitoclax has undesirable effects, mainly toxicity to platelets and neutrophils, making it complicated for translation. Because this undesired off-target toxicity may be caused by specific inhibition of BCL-2, drugs that target other BCL-2 family members, were tested with the hope of less toxicity. A1331852 and A1155463, which specifically target BCL-XL (but not BCL-2) were later found to be senolytic [13, 47, 54, 53]. It is currently unknown whether these two drugs might have less deleterious side effects.
Several in vivo mouse studies have since shown the ability of ABT-263 to kill SnCs and attenuate several diseases or conditions in which SnCs have been shown to accumulate. ABT263 eliminated senescent astrocytes and improved cognitive function in an accelerated brain aging model induced by whole brain irradiation, ameliorated hyperglycemia and improved β-cells in an aging model, high fat diet model and insulin resistance models of Type II Diabetes, cleared senescent cardiomyocytes in hearts of aged mice, alleviated myocardial remodeling, attenuated expression of profibrotic mediators and improved the maintenance of cardiac function following MI, resulting in decreased post-MI mortality, attenuated tau phosphorylation and aggregation in a mutant tau protein model of neurodegeneration, protected lung tissues from chemically induced pulmonary emphysema and improved radiation induced pulmonary fibrosis in mice[55, 21, 23, 56, 57].
ABT-737 is another BCL-2 family inhibitor reported to be senolytic (targets BCL-2, BCL-W and BCL-XL) and to functionally rejuvenate hematopoietic stem cells in naturally aged mice and mice exposed to sublethal radiation[54].
EF24, a curcumin analog screened for its senolytic actions based on curcumin’s reported anti-aging effects in C. elegans and D. melanogaster, was shown to be senolytic in many cell types induced to senesce by different methods[58]. EF24 induced apoptosis of SnCs, and caused a reduction in Bcl-xl and Mcl-1 proteins but not their mRNA, pointing to a possible post-transcriptional mechanism. Proteasome degradation of the BCL-2 family appeared to be the mechanism by which EF24 was senolytic. EF24 combined with ABT-263 had synergistic effects, possibly allowing the lowering of the dose of ABT-263 to decrease its side effects (thrombocytopenia), making it useful for clinical translation.
Some BCL-2 family inhibitors (UBX1967 and UBX1325, Unity Biotechnology) are being prepared for testing in human trials for targeting diseases of the aging eye, such as age-related macular degeneration and glaucoma (Figure1). However, the evidence of efficacy of these drugs in animal models is very limited.
Piperlongumine
Piperlongumine is a natural product made by species from the genus Piper (pepper plants or pepper vines). It was identified as a senolytic in the same screen of a small library of structurally diverse, rationally selected small molecules that target pathways predicted to be important for survival of SnCs that identified ABT-263 by Chang et al[13]. Piperlongumine has low toxicity in mice, kills SnCs by apoptosis, and has significant synergistic senolytic effect when combined with ABT-263, suggesting a different mechanism of action of Piperlongumine from ABT-263[59]. However, the exact mechanism is still unknown for piperlongumine to induce apoptosis in SnCs, and it appears to target several signaling and survival pathways in SnCs.
Targeting p53 (UBX0101, FOXO4 peptide and USP7 inhibitors)
P53 is a transcriptional factor whose levels must be tightly controlled in cells because it regulates various cellular processes including apoptosis, senescence, and proliferation[60]. It can induce apoptosis in a transcription-dependent way (by inducing the expression of pro-apoptotic genes) or transcription-independent mechanism (translocates to the mitochondria and interferes in the interaction between anti-apoptotic BCL-family proteins and pro-apoptotic proteins)[61, 62]. It is also downregulated in many tissues with aging, which may drive higher cancer incidence and SnC accumulation. P53 is upregulated upon activation of the DNA damage response in many cell types, but sometimes goes down in cells that upregulate p16 to maintain their proliferation arrest and senescence, whereas its activity may stay up in cells that do not upregulate p16. Evidence also exists for a role of p53 in SASP suppression by a mechanism involving p38MAPK and NF-κB, and for inducing apoptosis of SnCs, making the activation of p53 an attractive goal for senotherapy[63–65].
Baar et al. also hypothesized that anti-apoptotic pathways must be up-regulated in SnCs, making them resistant to apoptosis and performed RNA sequencing on proliferating and senescent IMR90 human cells[66, 36]. Their transcriptomics results didn’t fully support that hypothesis (eg, senescent IMR-90s had higher PUMA and BIM (pro-apoptotic) and lower BCL-2 (anti-apoptotic) than non-senescent IMR-90s), and so they reasoned that transcription factors may be interfering with the execution of the apoptotic program and focused on FOXO4, a transcription factor linked to apoptosis and target of Insulin/IGF signaling. They found FOXO4 expression to be increased in SnCs and that its inhibition could lead to apoptosis of SnCs. They showed that FOXO4 inhibits apoptosis by binding p53 thus inhibiting p53-mediated apoptosis, causing cells to senesce instead of die in response to DNA damage. They designed a cell permeable peptide to interfere with the FOXO4-p53 interaction and showed that it selectively induced apoptosis in SnCs IMR-90 fibroblasts, by excluding p53 from the nucleus and sending it to the mitochondria, resulting in transcription-independent apoptosis[67, 68]. Using three in vivo senescence models, they show the FOXO4 peptide (FOXO4-DRI) could be clinically useful for use against conditions associated with SnCs (Table 1).
Table 1.
Models Used for Successfully Testing Senolytics in the Different Senolytics Discovery Studies Presented.
| Senolytic | TIS Mouse Model (Radio- or Chemotherapy) | Progeroid Mouse Models | Other Mouse Models | Natural Aging in Mice | Human Samples (ex vivo) | Ref. | |
|---|---|---|---|---|---|---|---|
| Kinase inhibitors | Dasatinib and Quercetin | Irradiated single mouse leg | DNA damage repair deficient Ercc1−/Δ progeroid mouse model (models human XFE progeria) | Improved cardiovascular function, Improved blood albumin, phosphate and amylase levels Improved rotarod activity |
12 | ||
| Fisetin | DNA damage repair deficient Ercc1−/Δ progeroid mouse model (models human XFE progeria) with a p16 reporter | Decreased senescent cells of different lineages and in many tissues Increased healthspan and lifespan in mice, even when started very late in life |
Adipose tissue explants | 17 | |||
| Fibrates | Fenofibrate | Aged and osteoarthritic chondrocytes | 75 | ||||
| BCL-2 family inhibition | ABT-737 | Irradiated mice | p53 activation induced senescence in skin | 54 | |||
| Navitoclax (ABT-263) | Irradiated mice (models premature aging of hematopoietic system) | Cleared senescent cells in bone marrow, lungs and muscle, reducing SASP Improved defects in HSC clonogenicity, long-term repopulating ability and imbalances in multinlineage differentiation (myeloid skewing). Improved muscle stem cell clonogenicity |
13 | ||||
| HSP90 inhibition | 17-DMAG | DNA damage repair deficient Ercc1−/Δ progeroid mouse model (models human XFE progeria) with a p16 reporter | 73 | ||||
| Cardiac Glycosides | Ouabain | 1) Irradiated mice 2) Doxorubicin-treated mice |
Decreased senescent cells in several tissues Improved blood albumin, phosphate and amylase levels Improved rotarod activity Decreased immune infiltration in liver |
76 | |||
| Digoxin | Injected human cancer cells into immunosuppressed mice, administered senescence-inducing cancer drugs combined with Digoxin | Intratracheally administered senescent or proliferating IMR90 fibroblasts into lungs of immunosuppressed mice to model lung fibrosis. | SABG positive osteoarthritic chondrocytes | 77 | |||
| p53 activation | USP7 inhibitors (P5091) | Doxorubicin-treated p16-3MR mice | 71 | ||||
| UBX0101 | Post-traumatic osteoarthritis induced by ACLT surgery in mice (intra-articular injections) | Naturally aged mice with injury-induced OA- reduced SnCs in articular cartilage and synovium, reduced pain, reduced p16, p21 and SASP factors | SABG positive osteoarthritic chondrocytes | 20 | |||
| FOXO4-DRI | Doxorubicin-treated mice | Human progeroid syndrome model (Trichothiodystrophy) | IP injected FOXO4-DRI into naturally aged mice Reduced p16+ve senescent cell burden Restored renal filtering capacity Improved fur density and responsiveness |
66 |
Only in vivo mouse models and human samples are shown in this table, which excludes in vitro cell studies for lack of space. As seen in this table, therapy-induced senescence (TIS) which includes mouse irradiation (usually total body irradiation) with a sublethal radiation dose (6–8 Gray) or treatment with a chemotherapeutic drug (such as doxorubicin), results in DNA damage that causes upregulation of the senescent cell load in several tissues and organs, including the hematopoietic system, and leads to premature aging phenotypes. Accelerated aging models of human diseases (progeroid) where senescent cell accumulation has been established are also used for testing senolytics. Naturally aged mice (19–24 months used in studies described in this review) are the gold standard for testing senolytics in mice, with some of the effects of senolytics described in the studies presented shown. Some groups are coming up with different ways to model human aging phenotypes correlated with senescent cell accumulation to test whether senolysis would attenuate disease (eg, transplanting senescent cells, but not proliferating ones, in mouse lungs intratracheally caused fibrosis, which was reversed by treatment with the senolytic Digoxin, a cardiac glycoside).
Another means of post-transcriptionally activating p53 is by preventing its interaction with the murine double minute 2 (MDM2), a ubiquitin ligase[69]. MDM2 and p53 are connected by a negative-feedback loop, where elevated p53 increases MDM2 expression, which in turn promotes the ubiquitination and proteasome degradation of p53, reducing its activity[70]. He et al. show that small molecule inhibitors of the ubiquitin-proteasome system 7 (UPS7), which deubiquitinates MDM2 preventing its degradation by UPS, are senolytic[71]. USP7 inhibitors activate p53 and cause apoptosis in cancer cells by promoting MDM2 auto-ubiquitination and degradation[72]. This indirect inhibition of MDM2 is well tolerated in mouse studies, compared to direct inhibition of MDM2, making it a more attractive target. In their studies, they find that USP7 inhibitors upregulated p53 and led to apoptosis only partly by MDM2 degradation, indicating other p53 transcription-dependent and independent mechanisms might also contribute. USP inhibitors successfully killed SnCs in a doxorubicin-treated mouse model of senescence and decreased SASP expression in several organs.
UBX0101, a senolytic that completed a Phase 1 human trial for safety and tolerability (Figure 1) to target moderate to severe painful osteoarthritis (OA) of the knee, is stated on Unity Biotechnology’s website (not by peer-reviewed literature) to be a p53/MDM2 interaction inhibitor. Jeon et al showed a causative role of SnCs in trauma-and aging-induced OA, and that eliminating SnCs genetically and pharmacologically with UBX0101 can attenuate OA progression and symptoms[20]. UBX0101 alleviated pain, decreased articular erosion and increased cartilage regeneration in knees of mice that developed post-traumatic OA following anterior cruciate ligament transection (ACLT), while decreasing many senescence markers and SASP molecules in both articular cartilage and synovioum of the knee joint. It is important to note that MDM2 inhibitors are toxic, and may only be useful therapeutically in local (versus systemic) administration as is being tested for OA in humans.
HSP-90 inhibitors
Fuhrmann-Stroissnigg et al. took advantage of the accelerated senescence of DNA damage repair deficient Mouse Embryonic Fibroblasts (MEFs) from Ercc−/− mice (progeroid mouse model) to establish a screen for senotherapeutics[73, 74]. They observed that, when grown at atmospheric oxygen (20%), approximately 50% of these cells became senescent as measured by several senescence markers. They also showed that an SA β-galactosidase (SABG)-based assay using the substrate C12FDG, which fluoresces upon cleavage by β-galactosidase, could accurately determine the proportion of senescent to non-senescent Ercc−/− MEFs. They used both flow cytometry and automated confocal microscopy to quantify senescent and non-SnCs, and performed screens using the microscopy detection method. Because their cultures of Ercc−/− MEFs contained both senescent and non-SnCs, they could determine the effect of different drugs on both cells in the same well, and showed the assay could differentiate between senomorphics (Rapamycin and NDGA, which reduced the number of SnCs without affecting the total number of cells) and senolytics (D+Q and Navitoclax, reduced both the number of SnCs and total cell number, suggesting the SnCs were being killed). They screened a library of 97 autophagy regulators, and identified 13 compounds as senotherapeutics, which can modulate cellular senescence. After excluding compounds that were highly toxic, known to affect lysosomal pH (would yield a false positive in their SABG-based screen), or were senomorphic but not senolytic, they identified two promising candidates, tanespimycin (17AAG) and geldanamycin, both of which are heat shock protein (HSP90) inhibitors. Testing 7 HSP90 inhibitors from different classes showed that all were senolytic, and that they could kill SnCs from different human and mouse cell types induced to senesce with different methods. Specifically, they appeared to induce apoptosis of SnCs by disrupting the stabilization of active, phosphorylated AKT (anti-apoptotic factor upregulated in SnCs with important role in regulating the PI3K/AKT anti-apoptotic pathway) by HSP90. Finally, to establish the in vivo senolytic activity of HSP90 inhibitors, they tested the effect of 17-DMAG (improved, more water soluble, clinically tested geldanamycin derivative) on age-related phenotypes in the Ercc1−/Δ progeroid mouse model, and found a significant decrease in a composite score of aging symptoms as well as a reduction in p16 expression in kidneys (but not liver) of drug-treated mice, signifying a positive effect on healthspan, as previously observed with D+Q (Table1).
Fibrates
Nogueira-Recalde et al used high throughput screening to identify drugs that can simultaneously modulate senescence and autophagy, both shown to play a role in aging-associated articular cartilage degeneration and OA[75]. They used a human chondrocyte-based cell culture imaging assay, where senescence was induced by IL-6 treatment, causing an increase in SABG. After screening 1120 compounds, they found that 279 had senotherapeutic activity. They then tested the effect of these identified senescence modulators on autophagy, and found that 14/279 also increased autophagic flux. They focused on fenofibrate (FN), a peroxisome proliferator-activated receptor alpha (PPARα) agonist and therapeutic target for lipid metabolism dysfunction, because it was previously shown to be important for chondrocyte homeostasis. In addition to being senolytic in human chondrocytes (by inducing apoptosis), they found FN to be senolytic in human IMR90 lung fibroblasts and Ercc1−/− MEFs. Furthermore, they showed that genetic ablation of PPARα induces senescence and SASP, and reduces autophagic flux, confirming its role in senescence and autophagy. They also showed a decrease in PPARα-positive chondrocytes in knee cartilage from surgical OA mice, naturally aged mice, and in samples from OA-patients. Finally, they compared genetically matched subjects from the Osteoarthritis Initiative (OAI) cohort taking fibrates to ones not taking fibrates and found a significant difference in self-reported function, fewer knee replacements and a trend towards less pain in the fibrate-treatment group. Their results suggest fibrates show promise in alleviating OA symptoms, perhaps through their simultaneous effect on senescence and autophagy in chondrocytes.
Interestingly, their primary screen also identified Digitoxigenin, a cardiac glycoside (CG), as a senotherapeutic. Three months later, CGs were reported to be senolytic simultaneously by 2 independent studies[76, 77].
Cardiac glycosides
Guerrero et al. screened the LOPAC 1,280 library of pharmacologically active compounds, in various senescent cells [76]. They found that Ouabain and other CGs could specifically induce apoptosis of SnCs in all conditions tested for inducing senescence in IMR-90 fibroblasts. They showed that Ouabain could be used in vivo as a senolytic using a model of tumor initiation in the liver and a pituitary tumor model, in which SnCs have been shown to accumulate. They also tested the suitability of CGs in the elimination of SnCs produced as a result of treatment with chemotherapy, radiation therapy and targeted anti-cancer drugs and found that, indeed, they sensitized these therapy-induced SnCs to undergo apoptosis, both in in vivo models (looking at SnCs accumulating in the lungs of irradiated mice or mice treated with doxorubicin, Table1) and in several cancer cell culture models. Finally, they saw improvements in metabolic blood markers, markers of physical function, decreased p16 expression in liver, heart and kidneys and decreased immune infiltration in the liver of old Ouabain treated mice relative to controls (Table1).
Triana-Martinez et al screened the Prestwick chemical library in bleomycin-induced senescent cells, and identified the CG Proscillaridin A as a senolytic[77]. As Proscillaridin is not currently being used clinically, they chose to test the CGs Digoxin, which is commonly used for treatment of heart conditions, and Ouabain. They confirmed the senolytic activity of these CGs in multiple senescent cell types (tumor and primary cells) via different induction methods. Screening several additional libraries, they found many more CGs to be senolytic. They also showed Digoxin acted via apoptosis induction (not ferroptosis or necroptosis) for inducing SnC death. To test whether Digoxin could be used for killing cancer-therapy-induced SnCs in vivo, they used mouse models to show that combining CGs with senescence-inducing cancer drugs significantly decreased tumor size. Finally, they found that Digoxin could kill SnCs in a model of lung fibrosis, and alleviate fibrosis in the lungs (Table 1). This finding further supports the use of senolytics in IPF patients (Figure 1)[78–81].
Data from both studies suggest that the senolytic activity of CGs might come from inhibiting the Na+/K+ ATPase. SnCs were more sensitive to changes in membrane potential caused by Digoxin, which causes loss in membrane potential and cellular acidification, because their membranes are slightly depolarized and they are more acidic than proliferating cells at baseline. CGs appear to tip SnCs past a threshold, inducing apoptosis.
Other Methods for killing SnCs in vivo
In addition to senolytic drugs described above, several other methods are currently being developed to kill SnCs in vivo. Muñoz-Espín et al described the use of nanoparticles coated with galacto-oligosaccharides and containing cytotoxic agents to specifically deliver poisons to senescent cells, many of which have a high lysosomal content and lysosomal β-galactosidase, allowing the digestion of the coat and release of the encapsulated drug[82].
Guerrero et al also took advantage of the higher β-galactosidase activity of SnCs to test whether galactose-modified duocarmycin cytotoxic prodrugs would be selectively toxic to SnCs[83]. They showed successful senolysis in vitro and in in vivo mouse models (irradiation model and cancer model).
Another delivery method (Lipid Nanoparticles) taking advantage of a different aspect of the biology of many SnCs (high expression of p16) is being developed by Oisín Biotechnologies. Their SENSOlytics® will deliver an apoptotic gene under control of a p16 promoter. This gene will only be expressed in cells with high p16 expression, and they will undergo cell death.
Although both methodologies have resulted in a marked in vivo decrease in SnC burden in animal models, they both suffer from the shortcoming that the aspect of biology they target (high lysosomal content or high p16 activity) might not be specific to SnCs, and may cause side effects by targeting non-senescent, terminally differentiated cells that express p16 or cells with high lysosomal content as part of their biology such as macrophages. On the other hand, these strategies might have the unique advantage of reducing the potential off-target toxicity once their precise targets have been optimized. Furthermore, these methods could be combined with senolytics to achieve additive benefits.
Conclusions
Although senolytics are being tested for treatment of varied chronic diseases of aging and geriatric syndromes, it may be a while before these compounds can be safely used in healthy individuals for the purpose of prevention, delaying or reversing normal aging phenotypes. Nevertheless, the field of senolytic discovery is moving rapidly through more efficient and specific screens, followed by testing and validation of promising candidates in various preclinical models, and ultimately rigorous testing in clinical trials. Our understanding of the basic biology of SnCs in vivo is still limited, but new techniques such as single cell sequencing are offering new capabilities for better understanding these small populations and then facilitating next-generation senolytic drugs development. Using senolytics for treating diseases where SnCs play a well-established role may soon become a reality, with several clinical trials under way for determining safety and efficacy (Figure 1). In the meantime, these senolytics will certainly improve prognosis for cancer patients who suffer from premature aging likely caused by therapy-induced accumulation of SnCs, providing more evidence for their power against a fundamental aging mechanism.
Acknowledgements
This work was supported by Glenn Foundation for Medical Research and AFAR Grant for Junior Faculty (M.X.), and NIH grants R21AG063528 (G.A.K. and M.X.).
Footnotes
Competing Financial Interests
Dr. Xu has financial interest related to senolytics. Patents on senolytic drugs (including PCT/US2016/041646, filed at the US Patent Office) are held by Mayo Clinic.
Dr. Kuchel has nothing to disclose.
Dr. Al-Naggar has nothing to disclose.
Human and Animal Rights
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Miller RA. Extending life: scientific prospects and political obstacles. Milbank Q. 2002;80(1):155–74. doi: 10.1111/1468-0009.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geroscience Sierra F. and the challenges of aging societies. Aging Med (Milton). 2019;2(3):132–4. doi: 10.1002/agm2.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sierra F The Emergence of Geroscience as an Interdisciplinary Approach to the Enhancement of Health Span and Life Span. Cold Spring Harb Perspect Med. 2016;6(4):a025163. doi: 10.1101/cshperspect.a025163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaeberlein M Translational geroscience: A new paradigm for 21(st) century medicine. Transl Med Aging. 2017;1:1–4. doi: 10.1016/j.tma.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melov S Geroscience approaches to increase healthspan and slow aging. F1000Res. 2016;5. doi: 10.12688/f1000research.7583.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huffman DM, Justice JN, Stout MB, Kirkland JL, Barzilai N, Austad SN. Evaluating Health Span in Preclinical Models of Aging and Disease: Guidelines, Challenges, and Opportunities for Geroscience. J Gerontol A Biol Sci Med Sci. 2016;71(11):1395–406. doi: 10.1093/gerona/glw106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer AK, Gustafson B, Kirkland JL, Smith U. Cellular senescence: at the nexus between ageing and diabetes. Diabetologia. 2019;62(10):1835–41. doi: 10.1007/s00125-019-4934-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirkland JL. Translating the Science of Aging into Therapeutic Interventions. Cold Spring Harb Perspect Med. 2016;6(3):a025908. doi: 10.1101/cshperspect.a025908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246–56. doi: 10.1038/s41591-018-0092-9.•This report shows a causal role for senescent cells in aging-like phenotypes in mice by transplanting senescent or proliferating cells into young and old mice and demonstrating that increased senescent cell load causes physical dysfunction, increased senescence elsewhere and reduced lifespan. Furthermore, they demonstrate that treating transplanted mice with the senolytic cocktail D+Q improves lifespan and healthspan in these mice.
- 11.Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell. 2019;18(3):e12950. doi: 10.1111/acel.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–58. doi: 10.1111/acel.12344.•This is the paper that describes the first senolytics, Dasatinib and Quercetin, and the transcriptomics analysis combined with RNA interference studies that allowed the identification of survival pathways differentially regulated in senescent vs proliferating cells.
- 13.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature medicine. 2016;22(1):78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–7. doi: 10.1111/acel.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moncsek A, Al-Suraih MS, Trussoni CE, O’Hara SP, Splinter PL, Zuber C et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2(−/−)) mice. Hepatology. 2018;67(1):247–59. doi: 10.1002/hep.29464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8:15691. doi: 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018;36:18–28. doi: 10.1016/j.ebiom.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–6. doi: 10.1038/nature10600.•These two Baker et al papers describe the INK-ATTAC mouse model which provided the first genetic evidence supporting a causative role of senescent cells in modulation of aging and aging-related diseases. INK-ATTAC mice carry a drug-inducible caspase driven by part of the p16INK4A promoter, so that when that drug is administered, cells that highly express p16INK4A undergo apoptotic cell death. In addition to alleviation of aging-related diseases such as heart hypertrophy, cataracts, glomerulosclerosis and lipodystrophy, they were able to extend lifespan of progeroid (BubR1) and naturally aged mice.
- 20.Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–81. doi: 10.1038/nm.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562(7728):578–82. doi: 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–33. doi: 10.1016/j.devcel.2014.11.012.•Describes another essential genetic mouse model, the p16–3MR model, essential for establishing the causal role of p16+ve senescent cells in many processes and conditions and providing important proof of concept studies for clearing SnCs for attenuating aging-like phenotypes and conditions.
- 23.Yabluchanskiy A, Tarantini S, Balasubramanian P, Kiss T, Csipo T, Fulop GA et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience. 2020. doi: 10.1007/s11357-020-00154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patil P, Dong Q, Wang D, Chang J, Wiley C, Demaria M et al. Systemic clearance of p16(INK4a) -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell. 2019;18(3):e12927. doi: 10.1111/acel.12927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286(42):36396–403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu M, Bradley EW, Weivoda MM, Hwang SM, Pirtskhalava T, Decklever T et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J Gerontol A Biol Sci Med Sci. 2017;72(6):780–5. doi: 10.1093/gerona/glw154.•This report establishes a causal role of senescent cells in osteoarthritis by showing that injecting senescent mouse ear fibroblasts into knee joints of healthy mice results in OA-like phenotype, whereas a non-senescent cell injection does not.
- 28.Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. 2015;4:e12997. doi: 10.7554/eLife.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506(7488):316–21. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 30.von Kobbe C Targeting senescent cells: approaches, opportunities, challenges. Aging (Albany NY). 2019;11(24):12844–61. doi: 10.18632/aging.102557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL. Discovery, Development, and Future Application of Senolytics: Theories and Predictions. FEBS J. 2020. doi: 10.1111/febs.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Deursen JM. Senolytic therapies for healthy longevity. Science. 2019;364(6441):636–7. doi: 10.1126/science.aaw1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang B, Liu Z, Chen VP, Wang L, Inman CL, Zhou Y et al. Transplanting cells from old but not young donors causes physical dysfunction in older recipients. Aging Cell. 2020:e13106. doi: 10.1111/acel.13106.•This is the first study to use single cell transcriptomics to identify a population of naturally occurring SnCs in aged mice, and showed that these cells resemble some of the key altered pathways in in vitro-generated SnCs, which have been widely used for cellular senescence study.
- 34.Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–56. doi: 10.1016/j.ebiom.2019.08.069.•This human clinical trial reports reduced senescent cell burden in adipose tissue, and decreased circulating SASP factors of patients with diabetic kidney disease (DKD) after receiving a 3 day oral course of D+Q. They report no side effects in this open-label trial which is still ongoing.
- 35.Justice JN, Ferrucci L, Newman AB, Aroda VR, Bahnson JL, Divers J et al. A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: report from the TAME Biomarkers Workgroup. Geroscience. 2018;40(5–6):419–36. doi: 10.1007/s11357-018-0042-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang E Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995;55(11):2284–92. [PubMed] [Google Scholar]
- 37.Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17(6):e12840. doi: 10.1111/acel.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22(5):719–28. doi: 10.1038/s41593-019-0372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cavalcante MB, Saccon TD, Nunes ADC, Kirkland JL, Tchkonia T, Schneider A et al. Dasatinib plus quercetin prevents uterine age-related dysfunction and fibrosis in mice. Aging (Albany NY). 2020;12(3):2711–22. doi: 10.18632/aging.102772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL et al. Targeting cellular senescence prevents age-related bone loss in mice. Nature medicine. 2017;23(9):1072–9. doi: 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. 2019;18(3):e12931. doi: 10.1111/acel.12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554–63. doi: 10.1016/j.ebiom.2018.12.052.••First study of use of senolytics in human patients. 14 patients with IPF received 9 doses orally of the senolytic combination D+Q over a 3 week period in a pilot, open-label clinical trial. Reports safety and tolerability of the drug and showed improved physical function in these patients.
- 43.Kritchevsky SB, Justice JN. Testing the Geroscience Hypothesis: Early Days. J Gerontol A Biol Sci Med Sci. 2020;75(1):99–101. doi: 10.1093/gerona/glz267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendelsohn AR, Larrick JW. Cellular Senescence as the Key Intermediate in Tau-Mediated Neurodegeneration. Rejuvenation Res. 2018;21(6):572–9. doi: 10.1089/rej.2018.2155. [DOI] [PubMed] [Google Scholar]
- 45.Schafer MJ, Haak AJ, Tschumperlin DJ, LeBrasseur NK. Targeting Senescent Cells in Fibrosis: Pathology, Paradox, and Practical Considerations. Curr Rheumatol Rep. 2018;20(1):3. doi: 10.1007/s11926-018-0712-x. [DOI] [PubMed] [Google Scholar]
- 46.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017;9(3):955–63. doi: 10.18632/aging.101202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khan N, Syed DN, Ahmad N, Mukhtar H. Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal. 2013;19(2):151–62. doi: 10.1089/ars.2012.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adhami VM, Syed DN, Khan N, Mukhtar H. Dietary flavonoid fisetin: a novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem Pharmacol. 2012;84(10):1277–81. doi: 10.1016/j.bcp.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Syed DN, Adhami VM, Khan MI, Mukhtar H. Inhibition of Akt/mTOR signaling by the dietary flavonoid fisetin. Anticancer Agents Med Chem. 2013;13(7):995–1001. doi: 10.2174/18715206113139990129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farsad-Naeimi A, Alizadeh M, Esfahani A, Darvish Aminabad E. Effect of fisetin supplementation on inflammatory factors and matrix metalloproteinase enzymes in colorectal cancer patients. Food Funct. 2018;9(4):2025–31. doi: 10.1039/c7fo01898c. [DOI] [PubMed] [Google Scholar]
- 52.Singh S, Singh AK, Garg G, Rizvi SI. Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci. 2018;193:171–9. doi: 10.1016/j.lfs.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15(3):428–35. doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190. doi: 10.1038/ncomms11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M et al. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int J Radiat Oncol Biol Phys. 2017;99(2):353–61. doi: 10.1016/j.ijrobp.2017.02.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walaszczyk A, Dookun E, Redgrave R, Tual-Chalot S, Victorelli S, Spyridopoulos I et al. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell. 2019;18(3):e12945. doi: 10.1111/acel.12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mikawa R, Suzuki Y, Baskoro H, Kanayama K, Sugimoto K, Sato T et al. Elimination of p19(ARF) -expressing cells protects against pulmonary emphysema in mice. Aging Cell. 2018;17(5):e12827. doi: 10.1111/acel.12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W, He Y, Zhang R, Zheng G, Zhou D. The curcumin analog EF24 is a novel senolytic agent. Aging (Albany NY). 2019;11(2):771–82. doi: 10.18632/aging.101787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, Chang J, Liu X, Zhang X, Zhang S, Zhang X et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). 2016;8(11):2915–26. doi: 10.18632/aging.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Levine AJ. Reviewing the future of the P53 field. Cell Death Differ. 2018;25(1):1–2. doi: 10.1038/cdd.2017.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22(56):9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 62.Speidel D Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2010;20(1):14–24. doi: 10.1016/j.tcb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 63.Johmura Y, Nakanishi M. Multiple facets of p53 in senescence induction and maintenance. Cancer Sci. 2016;107(11):1550–5. doi: 10.1111/cas.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johmura Y, Sun J, Kitagawa K, Nakanishi K, Kuno T, Naiki-Ito A et al. SCF(Fbxo22)-KDM4A targets methylated p53 for degradation and regulates senescence. Nat Commun. 2016;7:10574. doi: 10.1038/ncomms10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johmura Y, Yamashita E, Shimada M, Nakanishi K, Nakanishi M. Defective DNA repair increases susceptibility to senescence through extension of Chk1-mediated G2 checkpoint activation. Sci Rep. 2016;6:31194. doi: 10.1038/srep31194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell. 2017;169(1):132–47 e16. doi: 10.1016/j.cell.2017.02.031.•This reports the use of a peptide (FOXO4-DRI) rather than a small molecule, to block the p53-FOXO4 interaction. Once p53 is released, the senescent cell can undergo p53-dependent apoptosis. The peptide was designed as a fusion with HIV-TAT, which is a basic and hydrophilic sequence that allows its entry into cells without energy expenditure through transient pore formation. The FOXO4-DRI had senolytic activity both in vitro and in vivo in several mouse models.
- 67.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11(3):577–90. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 68.Mihara M, Moll UM. Detection of mitochondrial localization of p53. Methods Mol Biol. 2003;234:203–9. doi: 10.1385/1-59259-408-5:203. [DOI] [PubMed] [Google Scholar]
- 69.Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27(4):254–71. doi: 10.7555/JBR.27.20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1(14):1001–8. [PubMed] [Google Scholar]
- 71.He Y, Li W, Lv D, Zhang X, Zhang X, Ortiz YT et al. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell. 2020;19(3):e13117. doi: 10.1111/acel.13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13(6):879–86. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- 73.Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. 2017;8(1):422. doi: 10.1038/s41467-017-00314-z.•Describes a novel, high-throughput platform using DNA repair deficient Ercc−/− MEFs induced to senesce in high oxygen coupled with a senescence-associated β-galactosidase fluorescent substrate for identifying drugs with senotherapeutic activity using a semi-automated confocal microscopy detection system. Their screen of a library of autophagy regulators identified HSP90 inhibitors as a new class of senolytics.
- 74.Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD. Hsp90 inhibitors as senolytic drugs to extend healthy aging. Cell Cycle. 2018;17(9):1048–55. doi: 10.1080/15384101.2018.1475828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nogueira-Recalde U, Lorenzo-Gomez I, Blanco FJ, Loza MI, Grassi D, Shirinsky V et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine. 2019;45:588–605. doi: 10.1016/j.ebiom.2019.06.049.•Report a novel two-step method for screening drugs for their senotherapeutic and autophagy modulating activity simultaneously. Their screen uses the fact that IL-6 treated human chondrocytes senesce and express senescence-associated β-galactosidase combined with the fluorescent substrate for identifying drugs with senotherapeutic activity. In a secondary screen, they test the effect of positive hits on autophagic flux using a chondrocyte cell line transfected with a fluorescent LC3 reporter. This helped them identify fibrates as a previously unknown family of senolytics with pro-autophagic activity.
- 76.Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R et al. Cardiac glycosides are broad-spectrum senolytics. Nat Metab. 2019;1(11):1074–88. doi: 10.1038/s42255-019-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Triana-Martinez F, Picallos-Rabina P, Da Silva-Alvarez S, Pietrocola F, Llanos S, Rodilla V et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun. 2019;10(1):4731. doi: 10.1038/s41467-019-12888-x.•They report a novel high throughput technique for screening for senolytics in a human lung adenocarcinoma cell line. Cells were transfected either with a RFP reporter or GFP reporter using lentiviral transduction. Cells with the RFP reporter were then made to senesce with bleomycin while GFP reporter cells were left untreated. RFP and GFP transduced cells were mixed in a 1:3 ratio (proliferating:senescent) and used to screen for drugs that changed the GFP:RFP cell ratios.
- 78.Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res. 2013;162(3):156–73. doi: 10.1016/j.trsl.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 79.Yanai H, Shteinberg A, Porat Z, Budovsky A, Braiman A, Ziesche R et al. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY). 2015;7(9):664–72. doi: 10.18632/aging.100807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuwano K, Araya J, Hara H, Minagawa S, Takasaka N, Ito S et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig. 2016;54(6):397–406. doi: 10.1016/j.resinv.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 81.Waters DW, Blokland KEC, Pathinayake PS, Burgess JK, Mutsaers SE, Prele CM et al. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2018;315(2):L162–L72. doi: 10.1152/ajplung.00037.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Munoz-Espin D, Rovira M, Galiana I, Gimenez C, Lozano-Torres B, Paez-Ribes M et al. A versatile drug delivery system targeting senescent cells. EMBO Mol Med. 2018;10(9). doi: 10.15252/emmm.201809355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guerrero A, Guiho R, Herranz N, Uren A, Withers DJ, Martinez-Barbera JP et al. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell. 2020:e13133. doi: 10.1111/acel.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]