

Abstract
Acne inversa is a chronic inflammatory follicular disease with autosomal dominant inheritance. In recent years, many functional mutations in the NCSTN genes have been identified as the cause of familial acne inversa. Herein, we recruited four patients and seven unaffected individuals from a Chinese family and performed Sanger sequencing of the NCSTN gene. One novel frameshift mutation, c.450_459del (p.Ser 151GlnfsX48), was identified in exon 5 of the NCSTN gene. Three normal-looking children carrying the mutation were proven to be patients. We also presented a literature review from previous studies of acne inversa, suggesting that NCSTN is a hotspot gene for acne inversa. Most affected individuals experienced onset in adolescence. We confirmed the diagnosis in this family based on the mutation. This finding will help expound the relationship between the NCSTN gene and the pathogenesis of acne inversa and emphasize the value of genetic diagnosis in monogenic disorder.
Keywords: Chinese patients, Hidradenitis suppurativa, Mutation analysis, NCSTN gene
INTRODUCTION
Acne inversa (AI; Online Mendelian Inheritance in Man [OMIM] #142690), also called hidradenitis suppurativa (HS), remains a challenging disease for both patients and clinicians. AI is an autosomal dominant monogenic disorder with genetic heterogeneity, usually presenting after puberty on the apocrine gland-bearing areas of the body1. The average onset of AI is in the early 20s, and substantial data suggest a female-predominance with a 3:1 sex ratio2. AI commonly occurs in the armpit and inguinal folds as well as near the genitalia, the inframammary skin and buttocks3. Early skin lesions take the form of acne, papules, and nodules, followed by the formation of cysts, abscesses, and sinuses, causing abnormal pus to flow out and scars to form. The pathogenesis of AI is not fully understood, although it is a multifaceted disease triggered by the follicular occlusions, hereditary components, immune dysregulation and other factors3,4. AI patients are at significantly increased risk for various autoimmune diseases and metabolic syndromes, consisting of rheumatoid arthritis, inflammatory bowel disease, psoriasis, systemic lupus erythematous, diabetes, hypertension, and pilonidal cysts5,6. Depending upon the severity of the disease, AI patients are treated with different methods, such as antibiotics, immunosuppressives, biological agents, antidiabetics, glucocorticoids, retinoids, laser therapy and surgery.
So far, three different genes have been identified in AI patients, including NCSTN, PSENEN, and PSEN17. Genetic inactivation of these genes in mouse skin produces epidermal and follicular abnormalities that are histopathologically similar to those observed in human AI8. After this discovery, loss-of-function mutations in these genes were identified in AI families and AI sporadic individuals, and the most common of these is mutation of the NCSTN gene9. The diagnosis of AI is mainly based on the clinical features. However, with the development of the sequencing technology, sequencing has been used as a high-efficiency tool to make an early diagnosis due to the particular advantage in finding mutations of rare diseases. To confirm the value of genetic diagnosis and further investigate the genetic mechanisms underlying AI, we conducted a mutation analysis of NCSTN in a Chinese family with AI.
CASE REPORT
Patients and controls
A family from Guangdong Province of China were recruited (Fig. 1A). The proband was a 27-year-old male who presented inflammatory papules, painful nodules, sinus tracts and atrophic scars especially on his armpit, back and buttocks over 11 years (Fig. 1B, C). Histopathologic results of the proband showed hyperkeratinization of hair follicles and structural destruction of hair follicles, extensive infiltration of neutrophils and lymphoid cells, which further confirmed the reliability of the diagnosis (Fig. 2A, B). His father was a 64-year-old male who had exhibited similar skin lesions on his back and buttocks for more than 50 years, showing widespread skin abscesses, disfiguring scars and post-inflammatory hyperpigmentation (Fig. 1D, E). The age of onset of 4 patients was 14 years old (I1), 15 years old (II6), 12 years old (II8), and 16 years old (II10). Thus far, the third generation (III1, III2, III3, III4, III5) in this family has no clinical phenotype. There was no autoimmune or metabolic disease associated with this AI family. Written informed consents were obtained from 11 members of this family. This study was approved by the ethics committee of Anhui Medical University (IRB No. 20150051) and was managed in accordance with Declaration of Helsinki principles. We received the patient's consent form about publishing all photographic materials.
Fig. 1. (A) Genealogical tree of the acne inversa family. The arrow in the pedigree refers to the proband. (B, C) Inflammatory papules, painful nodules, sinus tracts and atrophic scarring are distributed on the back and buttocks of the proband. (D, E) The father of the proband showed widespread sinus tracts, inflamed cysts, skin abscesses, disfiguring scars and post-inflammatory hyperpigmentation on his back and buttocks.
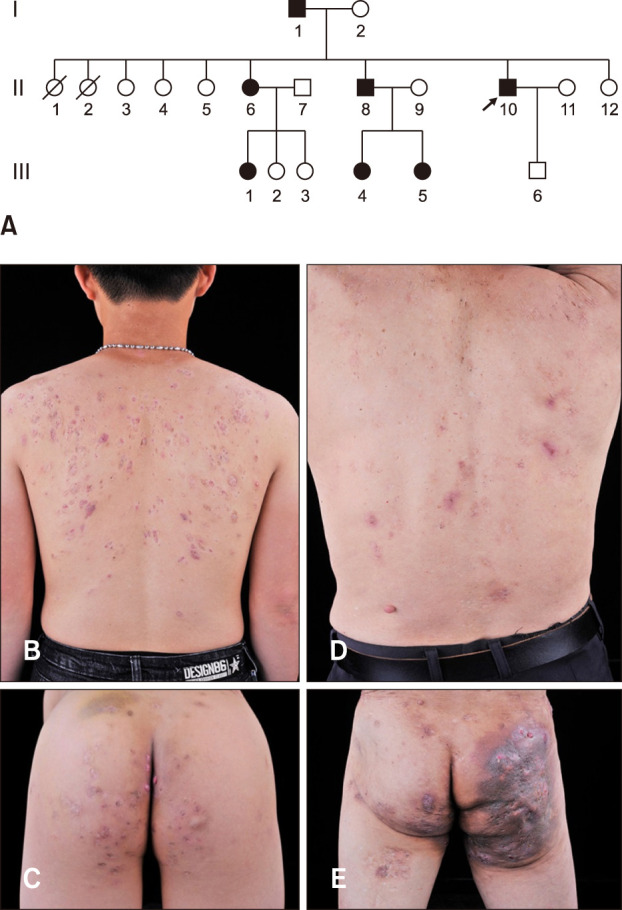
Fig. 2. (A, B) Skin biopsy from the proband showed hyperkeratinization and structural destruction of hair follicles, extensive infiltration of neutrophils and lymphoid cells (A, H&E, ×40; B, H&E, ×100). (C, D) Sanger sequencing confirmed a novel frameshift mutation in the NCSTN gene (C, Control; D, Patient, c.450_459del and p.Ser151GlnfsX48).

Mutation sequencing and analysis
DNA was extracted from 2 ml venous blood using the QIAamp DNA blood mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA samples were amplified using a polymerase chain reaction (PCR) with specially designed primer pairs covering all exons and exon-introns of NCSTN (Table 1). The DNA was amplified by cycling at 95℃ for 15 minutes; 11 cycles of 94℃ for 15 seconds, 62℃ to 0.5℃ per cycle for 40 seconds, 72℃ for 1 minute; 24 cycles of 94℃ for 15 seconds, 57℃ for 30 seconds, 72℃ for 1 minute; 72℃ for 2 minutes. Primers were designed using primer3 (http://bioinfo.ut.ee/primer3-0.4.0/; HHMI, San Francisco, CA, USA). Direct DNA sequencing of NCSTN was performed using an ABI3730XL genetic analyzer (Thermo Fisher Scientific, Waltham, MA, USA) and the results were analyzed by the Polyphred software (UW, Seattle, WA, USA) and revised manually.
Table 1. NCSTN gene exon polymerase chain reaction amplification primer sequence information.
| Exon | Forward primer | Reverse primer | Product length |
|---|---|---|---|
| 1 | GCGCTCTTGGTCTCGAATTT | CTTTGCCTGGACCTGAAGGT | 516 |
| 2–3 | TGATCACCTGTGCAACCA | GTCCTGCTTAGGAGATTTGACA | 520 |
| 4 | TTGTGACATCTTTAGGGGATAAAA | CTGGTCTGGTCAGGGGAGAG | 340 |
| 5 | CCTCCCTGGGGTCCTTACTC | TTCTTTGAGTCACCACCTCCT | 653 |
| 6 | TCACCACCTCCTTTTGAACTCTT | CTTTCCCCACATTTTGCTCC | 611 |
| 7 | AGTCTGCAACCCTTTGTAACTC | TTTCCAATGTTGCCTTTATAGC | 636 |
| 8–9 | GGCAGCTTGTTCCTAAAGTGG | TGACCTAAGTTGTTAACCAGCACA | 795 |
| 10–11 | AGCTGATGTTCATCTTAGACCTTT | GGTGTTGGCCAAAGGATGA | 752 |
| 12 | TAGGGTAGCTCCCCAAGCAG | ACAAGCATGGGAAGGATGGT | 620 |
| 13–14 | CACCCCTTTCTTCTGATGCT | CTCATGCCCCAGAGAGCTCT | 564 |
| 15–16 | TCTATCTGGCCAGTCTGGTTC | GAAAGTTGGAGGTTCTTCTAGACCT | 678 |
| 17 | GGAAAGTTGGAGGTTCTTCTAGACC | TAGCCCTTTCCATCTCCCAT | 540 |
| 18_1 | CAGCTGGGATGAGTCAGTCT | CCTGTTTAACTCCCTAGTTACCCA | 749 |
| 18_2 | CCTGGGCCTGTCTCAGATT | GAGGACCCAAGGAGTAAGGC | 703 |
NCSTN mutation identification and analysis
Four patients (I1, II6, II8, II10), two controls (I2, II9), five children without clinical phenotype (III1, III2, III3, III4, III5) were sequenced using Sanger sequencing (Applied Biosystems, Foster City, CA, USA) (Fig. 2C, D). A novel frameshift mutation, c.450_459del (p.Ser151GlnfsX48), in exon 5 of NCSTN (NM_015331.2) existed in all the affected individuals (I1, II6, II8, II10) and three children with normal phenotype (III1, III4, III5). Notably, this mutation was absent in other two unaffected adults (I2, II9) and two children without a clinical phenotype (III2, III3). According to the result of the function annotation, the mutation found in this family is properly disease-causing.
DISCUSSION
AI is a chronic, debilitating, painful inflammatory disease for which inherent unpredictability poses a major challenge for patients in terms of disease progression and response to treatment.
NCSTN exerts anti-proliferation and differentiation-promoting effects in human keratinocytes, and functional deletion mutation of NCSTN in familial AI promotes proliferation and inhibits differentiation of keratinocytes mainly through Notch and PI3K-AKT signaling pathways10. Moreover, epidermal and follicular hyperkeratosis and epidermal cyst formation are associated with disruption of Notch signaling pathways, leading to subsequent blockage, rupture and infection, a process strikingly similar to the pathogenesis of familial AI11. In addition, the NCSTN/miR-30a-3p/RAB31 contributed to the impaired activation of epidermal growth factor receptor signaling pathway which was followed by dysregulated keratinocyte differentiation in AI progression with the NCSTN mutation12. These findings confirmed the association of NCSTN with proliferation, cell cycle control and differentiation of keratinocytes, which is closely related to the pathogenetic mechanism of AI.
We searched the literature of AI or HS cases in PubMed. A total of 37 unique mutations have been identified in familial or sporadic AI patients with a diversity of mutation types in Chinese, Caucasian, Japanese, British, Germany, French, Indian, or African ethnic origin (Table 2). Twenty-seven located in NCSTN (73.0%, 27/37), six located in PSENEN, two located in PSEN1 and two located in POFUT1. Of which eight resulted in frameshifts and nine resulted in splice sites, seven were missense mutations and thirteen were nonsense mutations (Table 2). Obesity, smoking, hormones, bacterial infections, mechanical friction, and diabetes can aggravate AI symptoms. Four AI patients with NCSTN mutation were combined with cutaneous squamous cell carcinoma, three AI patients with PSENEN mutation and two AI patients with POFUT1 mutation were combined with Dowling-Degos disease. These observations have enriched our understanding of AI and highlighted that NCSTN may play a pivotal role in AI. Moreover, most affected individuals who have been reported experience onset in adolescence with lesions often occurring in the neck, back, armpits, breast, buttocks and groin areas. These previous studies have helped us to rationally select hotspot mutation genes for sequencing when encounter patients with AI.
Table 2. All mutations identified in NCSTN, PSEN1, PSENEN, and POGLUT1 for acne inversa patients to date.
| No. | Familial/sporadic | Gene | Exon | Mutation type | Nucleotide mutation | Protein alteration | Origin |
|---|---|---|---|---|---|---|---|
| 1 | Familial | NCSTN | Ex3 | Frameshift | c.210-211delAG | p.Thr70fs18X | Chinese |
| 2 | Familial | NCSTN | Ex3 | Missense | c.223G>A | p.Val75lle | Chinese |
| 3 | Familial | NCSTN | Ex4 | Nonsense | c.218delC | p.P73Lfs*15 | Chinese |
| 4 | Familial | NCSTN | Ex4 | Nonsense | c.349C>T | p.Arg117X | Chinese African |
| 5 | Case | NCSTN | Ex5 | Missense | c.553G>A | p.Asp185Asn | British |
| 6 | Familial | NCSTN | Int5 | Splice site | c.582+1delG | p. F145fs_X54 | Japanese |
| 7 | Familial | NCSTN | Ex5 | Frameshift | c.487delC | p.Gln163SerfsX39 | French |
| 8 | Familial | NCSTN | Ex5 | Nonsense | c.477C>A | p.C159X | Chinese |
| 9 | Familial | NCSTN | Ex6 | Nonsense | c.617C>A | p.S206X | Chinese |
| 10 | Case | NCSTN | Ex6 | Missense | c.632C>G | p.P211R | Chinese |
| 11 | Familial | NCSTN | Ex6 | Missense | c.647A>C | p.Q216P | Chinese |
| 12 | Familial | NCSTN | Ex6 | Frameshift | c.687insCC | p.Cys230ProfsX31 | Indian |
| 13 | Case | NCSTN | Int8 | Splice site | c.996+7G>A | p.L282_G332del | British |
| 14 | Familial | NCSTN | Ex8 | Missense | c.944C>T | p.Ala315Val | Chinese |
| 15 | Familial | NCSTN | Int9 | Splice site | c.1101+1G>A | p.E333_Q367del | British |
| 16 | Case | NCSTN | Int9 | Splice site | c.1101+10A>G | p.E333_Q367del | African |
| 17 | Familial | NCSTN | Int11 | Splice site | c.1352+1G>A | p.Q393fs_X9 | Chinese |
| 18 | Familial | NCSTN | Ex11 | Nonsense | c.1300C>T | p.Arg434X | French |
| 19 | Familial | NCSTN | Ex11 | Nonsense | c.1258C>T | p.Q420X | Chinese |
| 20 | Familial | NCSTN | Int11 | Splice site | c.1180-5C>G | British | |
| 21 | Familial | NCSTN | Int13 | Splice site | c.1551+1G>A | p.A486-T517del | Chinese |
| 22 | Familial | NCSTN | Ex15 | Frameshift | c.1752delG | p.E584DfsX44 | Chinese |
| 23 | Familial | NCSTN | Ex15 | Nonsense | c.1635C>G | p.Tyr545 | Iranian |
| 24 | Familial | NCSTN | Ex15 | Nonsense | c.1695T>G | p.Y565X | Chinese |
| 25 | Familial | NCSTN | Ex15 | Missense | c.1768A>G | p.Ser590AlafsX3 | French |
| 26 | Familial | NCSTN | Ex15 | Nonsense | c.1702C>T | p.Gln568Term | Japanese |
| 27 | Familial | NCSTN | Ex16 | Nonsense | c.1799delTG | p.Leu600X | Indian |
| 1 | Familial | PSENEN | Ex3 | Frameshift | c.66delG | p.F23LfsX46 | Chinese |
| 2 | Familial | PSENEN | Ex3 | Frameshift | c.279delC | p.F94SfsX51 | Chinese |
| 3 | Familial | PSENEN | Ex3 | Frameshift | c.66-67insG | p.F23VfsX98 | British |
| 4 | Familial | PSENEN | Ex3 | Splice site | c.167-2A>G | p.G55-101Pdel | Chinese |
| 5 | Familial | PSENEN | Ex3 | Missense | c.194T>G | p.L65R | Chinese |
| 6 | Case | PSENEN | Ex3 | Nonsense | c.168T>G | p.Y56X | Germany |
| 1 | Familial | PSEN1 | Ex7 | Frameshift | c.725delC | p.P242LfsX11 | Chinese |
| 2 | Familial | PSEN1 | Ex9 | Nonsense | c.953A> G | p.Glu318Gly | British |
| 1 | Case | POFUT1 | Ex9 | Nonsense | c.814C>T | p.R272* | Caucasian |
| 2 | Case | POFUT1 | Ex4 | Splice site | c.430-1G>A | p.K246_392Ldel | Caucasian |
Int: intron, Ex: exon.
Furthermore, three mutations located in exon 5 of NCSTN causing AI have been reported so far. A nonsense mutation, c.477C>A (p.C159X), in exon 5 of the NCSTN gene was detected that causes messenger RNA and protein expression evident reduction in the lesion10. A frameshift mutation, c.487delC, resulted in early termination of the codon (p.Gln163SerfsX39) and caused haploinsufficiency and severe reduction of NCSTN transcript levels in AI patients13. Finally, the missense variant in exon 5 of NCSTN (c.553G>A, p.Asp185Asn) was identified in a 45-year old female who presented inflammatory nodules, abscesses, and scarring in the breast area. It was predicted that this mutation caused the evolutionary conserved aspartic acid residue to be replaced by an asparagine residue14. In addition, we identified another novel frameshift mutation, c.450_459del (p.Ser151GlnfsX48) in exon 5 of NCSTN in this study, which has not been reported in the National Center for Biotechnology Information (NCBI) or OMIM database.
From the current physical examination, 7 individuals had no lesions. However, the results of the genetic test proven that 3 children of them were actual patients. According to the age in the onset of AI lesions in this family, these three children were too young to develop the skin rash. As summarized above in the literature, AI is not a disease with clinical manifestations at birth. From this point, the mutation analysis is a reliable supplement, suggesting the value of genetic diagnosis. Considering that all four patients in this family have a clinical phenotype presenting after the age of 12, these 3 children are likely to develop the disease in future, suggesting the significance of a timely and regular follow-up.
In conclusion, patients with AI experience chronic pain and have substantial physical, emotional and psychological effects. Our research reveals the cause of this Chinese family and extends the gene database of AI in China, which helps us to conduct clinical diagnosis and early intervention. Moreover, the ongoing recognition of unique mutations may allow people to understand the mechanisms that are still unknown in the development of AI and provide valuable information for further studies of treatment programs.
ACKNOWLEDGMENT
We gratefully appreciate the all the family members for participating in this study. This study was supported by National Natural Science Foundation of China (81573065).
Footnotes
CONFLICTS OF INTEREST: The authors have nothing to disclose.
References
- 1.Kiss N, Plázár D, Lőrincz K, Bánvölgyi A, Valent S, Wikonkál N. [Gynecological aspects of hidradenitis suppurativa] Orv Hetil. 2019;160:291–299. doi: 10.1556/650.2019.31319. Hungarian. [DOI] [PubMed] [Google Scholar]
- 2.Vinkel C, Thomsen SF. Hidradenitis suppurativa: causes, features, and current treatments. J Clin Aesthet Dermatol. 2018;11:17–23. [PMC free article] [PubMed] [Google Scholar]
- 3.Negus D, Ahn C, Huang W. An update on the pathogenesis of hidradenitis suppurativa: implications for therapy. Expert Rev Clin Immunol. 2018;14:275–283. doi: 10.1080/1744666X.2018.1449647. [DOI] [PubMed] [Google Scholar]
- 4.Theut Riis P, Thorlacius LR, Jemec GB. Investigational drugs in clinical trials for hidradenitis suppurativa. Expert Opin Investig Drugs. 2018;27:43–53. doi: 10.1080/13543784.2018.1412430. [DOI] [PubMed] [Google Scholar]
- 5.Lee JH, Kwon HS, Jung HM, Kim GM, Bae JM. Prevalence and comorbidities associated with hidradenitis suppurativa in Korea: a nationwide population-based study. J Eur Acad Dermatol Venereol. 2018;32:1784–1790. doi: 10.1111/jdv.15071. [DOI] [PubMed] [Google Scholar]
- 6.Ergun T. Hidradenitis suppurativa and the metabolic syndrome. Clin Dermatol. 2018;36:41–47. doi: 10.1016/j.clindermatol.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Wang B, Yang W, Wen W, Sun J, Su B, Liu B, et al. Gamma-secretase gene mutations in familial acne inversa. Science. 2010;330:1065. doi: 10.1126/science.1196284. [DOI] [PubMed] [Google Scholar]
- 8.Danby FW, Margesson LJ. Hidradenitis suppurativa. Dermatol Clin. 2010;28:779–793. doi: 10.1016/j.det.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Ratnamala U, Jhala D, Jain NK, Saiyed NM, Raveendrababu M, Rao MV, et al. Expanding the spectrum of γ-secretase gene mutation-associated phenotypes: two novel mutations segregating with familial hidradenitis suppurativa (acne inversa) and acne conglobata. Exp Dermatol. 2016;25:314–316. doi: 10.1111/exd.12911. [DOI] [PubMed] [Google Scholar]
- 10.Xiao X, He Y, Li C, Zhang X, Xu H, Wang B. Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3-kinase/AKT signalling pathways. Br J Dermatol. 2016;174:522–532. doi: 10.1111/bjd.14223. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto N, Tanigaki K, Han H, Hiai H, Honjo T. Notch/RBP-J signaling regulates epidermis/hair fate determination of hair follicular stem cells. Curr Biol. 2003;13:333–338. doi: 10.1016/s0960-9822(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 12.He Y, Xu H, Li C, Zhang X, Zhou P, Xiao X, et al. Nicastrin/miR-30a-3p/RAB31 axis regulates keratinocyte differentiation by impairing EGFR signaling in familial acne inversa. J Invest Dermatol. 2019;139:124–134. doi: 10.1016/j.jid.2018.07.020. [DOI] [PubMed] [Google Scholar]
- 13.Miskinyte S, Nassif A, Merabtene F, Ungeheuer MN, Join-Lambert O, Jais JP, et al. Nicastrin mutations in French families with hidradenitis suppurativa. J Invest Dermatol. 2012;132:1728–1730. doi: 10.1038/jid.2012.23. [DOI] [PubMed] [Google Scholar]
- 14.Pink AE, Simpson MA, Desai N, Dafou D, Hills A, Mortimer P, et al. Mutations in the γ-secretase genes NCSTN, PSENEN, and PSEN1 underlie rare forms of hidradenitis suppurativa (acne inversa) J Invest Dermatol. 2012;132:2459–2461. doi: 10.1038/jid.2012.162. [DOI] [PubMed] [Google Scholar]