

Abstract
Background
The spread of a novel coronavirus termed severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) in China and other countries is of great concern worldwide with no effective vaccine. This study aimed to design a novel vaccine construct against SARS-CoV-2 from the spike S protein and orf1ab polyprotein using immunoinformatics tools. The vaccine was designed from conserved epitopes interacted against B and T lymphocytes by the combination of highly immunogenic epitopes with suitable adjuvant and linkers.
Results
The proposed vaccine composed of 526 amino acids and was shown to be antigenic in Vaxigen server (0.6194) and nonallergenic in Allertop server. The physiochemical properties of the vaccine showed isoelectric point of 10.19. The instability index (II) was 31.25 classifying the vaccine as stable. Aliphatic index was 84.39 and the grand average of hydropathicity (GRAVY) was − 0.049 classifying the vaccine as hydrophilic. Vaccine tertiary structure was predicted, refined and validated to assess the stability of the vaccine via Ramachandran plot and ProSA-web servers. Moreover, solubility of the vaccine construct was greater than the average solubility provided by protein sol and SOLpro servers indicating the solubility of the vaccine construct. Disulfide engineering was performed to reduce the high mobile regions in the vaccine to enhance stability. Docking of the vaccine construct with TLR4 demonstrated efficient binding energy with attractive binding energy of − 338.68 kcal/mol and − 346.89 kcal/mol for TLR4 chain A and chain B respectively. Immune simulation significantly provided high levels of immunoglobulins, T-helper cells, T-cytotoxic cells and INF-γ. Upon cloning, the vaccine protein was reverse transcribed into DNA sequence and cloned into pET28a(+) vector to ensure translational potency and microbial expression.
Conclusion
A unique vaccine construct from spike S protein and orf1ab polyprotein against B and T lymphocytes was generated with potential protection against the pandemic. The present study might assist in developing a suitable therapeutics protocol to combat SARSCoV-2 infection.
Keywords: SARS CoV-2, Spike S protein, orf1ab polyprotein, Multiepitopes vaccine, B-lymphocytes, T-lymphocytes
Background
A novel coronavirus termed severe acute respiratory syndrome related coronavirus-2 or SARS-CoV-2 was identified in China in late 2019. The virus is the causative agent of coronavirus disease 2019 (COVID-19) and is contagious through human-to-human transmission [1, 2]. The disease is characterized by severe respiratory illness with symptoms of fever, cough, and shortness of breath and significant mortality, particularly among patients over the 60 years of age and in those suffering from chronic conditions such as diabetes and hypertension [3, 4]. SARS-CoV-2 was first reported in Wuhan, Hubei Province, in China, and swiftly spread all over China and other countries [4]. The causative agent of the outbreak was identified as Betacoronavirus with a genomic sequence closely related to that of the severe acute respiratory syndrome (SARS) coronavirus from 2003, hence the name SARS-CoV-2 [5–8]. The disease had become pandemic and globally spread to many countries and territories, including community transmission in countries like the United States, Germany, France, Spain, Japan, Singapore, South Korea, Iran and Italy with high significant morbidity and mortality rates [9].
SARS-CoV-2 is a positive-strand RNA virus that belongs to the group of Betacoronaviruses. The genome of the virus consists of 29,700 nucleotides with 79.5% sequence similarity to SARS-CoV. The virus encodes multiple structural and non-structural proteins [4, 10]. The orf1ab polyprotein is nonstructural protein at the 5 prime end of the viral genome constitutes two third of the viral proteome and encodes for 15 or 16 non-structural proteins. The 3 prime end of the genome encodes four major structural proteins, including the spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and the envelope (E) protein in addition to nonstructural proteins including orf3a, orf8, orf7a, orf7b, orf6 and orf10 [10, 11].
Like SARS-CoV, SARS-CoV-2 binds to the receptor angiotensin converting enzyme 2 (ACE2) on the host cell via the receptor binding domain (RBD) on the spike S protein of the virus [7, 11]. The spike S protein of SARS-CoV-2 is type I transmembrane glycoprotein with predicted length of 1273 amino acids. Moreover it comprises the major antigenic determinants that induce neutralizing antibodies [12, 13]. SARS-CoV and SARS-CoV-2 demonstrated 89.8% sequence identity in the S2 subunits of their spike (S) protein, which mediate the membrane fusion process. Moreover the S1 subunits of both viruses utilized human angiotensin-converting enzyme 2 (hACE2) as the receptor to infect human cells [7, 14]. Specific amino acids sequence region within the spike S proteins, termed receptor binding domain (RBD), is considered as a functional domain responsible for virus binding to the target cell receptor [15–17]. Most importantly, the RBD present in S1 subunit of spike S protein of SARS-CoV-2 has 10 to 20 fold high affinity to bind to the target cell receptor than that of SARS-CoV. This high affinity may contribute to the higher infectivity and transmissibility of SARS-CoV-2 compared to SARS-CoV [18, 19]. In addition to that the most existing vaccine candidates against SARS CoV were based on the spike S protein and RBD region [12, 13, 15, 20, 21].
The nonstructural orf1ab gene is the largest gene segment of SARS-CoV-2 and it constitutes orf1a and orf1b [2]. The replicase orf1ab is cleaved by papain-like protease (PLpro) and 3C-like protease (3CLpro). Orf1ab is cleaved into many nonstructural proteins (NSP1-NSP16) [2, 22]. Moreover it was shown that proteins or protein domains encoded in orf1ab may serve specific roles in virulence, virus–cell interactions and/or alterations of virus–host response [23]. This indicated that orf1ab polyprotein plays an important role in the virus pathogenesis distinct from or in addition to functions directly involved in viral replication. Recent reverse genetic study confirmed that proteins of orf1ab polyprotein may be involved in cellular signaling and modification of cellular gene expression, as well as virulence. Moreover it has become clear that NSP order, expression level, and proteolytic processing may constitute distinct virulence alleles [23]. Furthermore it was suggested that the orf1ab polyproteins, notably NSP3, may interact with multiple structural and nonstructural proteins, as well as with regulatory sequences in viral RNA [23].
To control SARS-CoV-2 infection, several old drugs such as chloroquine phosphate provided slight positive effect on the treatment of the novel coronavirus pneumonia [24, 25]. Vaccination process is significantly increased to develop a vaccine against pandemic SARS-CoV-2, including the development of several RNA and DNA vaccines, recombinant protein vaccines and cell-culture-based vaccines [9]. The mRNA vaccines are a new type of vaccines to protect against infectious diseases. Recently Food and Drug Administration (FDA) has authorized the emergency use of the Pfizer-BioNTech COVID-19 Vaccine (BNT162b2) to prevent COVID-19 in individuals 16 years of age and older under an emergency use authorization given in two doses 3 weeks apart. However this vaccine showed allergic reactions such as difficulty in breathing, welling of face and throat, fast heartbeat, skin rashes, dizziness and weakness [26, 27]. Another vaccine by ModernaTX, Inc. (mRNA-1273) is recommended for people aged 18 years and older. But the vaccine also showed side effects that usually started within a day or two of getting the vaccine [26, 27].
The advances made in the field of immunoinformatics tools coincided with the knowledge on the host immune response leads to new disciplines in vaccine design against diseases via computer in silico epitope predictions. The epitopes driven vaccine is a new concept that is being successfully applied in multiple studies, particularly to the development of vaccines targeting conserved epitopes in variable or rapidly mutating pathogens [28–30]. Therefore, as the genome and proteome sequences of SARS-CoV-2 is swiftly made available [6–8], this study aimed to use immunoinformatics approach to design multi epitopes vaccine against SARS-CoV-2 infection from the structural spike S protein and the nonstructural orf1ab polyprotein.
Results
Sequences alignment
Sequence alignment of all retrieved strains was performed using ClustalW that presented by Bioedit software. As shown in Fig. 1, the retrieved sequences of the spike S protein and orf1ab polyprotein including those of the new variant strain of Britain (SARS-CoV-2 VUI 202012/01 (MW450666.1) demonstrated high level of epitopes conservancy. The new variant strain was included since it is important to design a vaccine combating the infections from wild-type and mutant forms of SARS-CoV2. The conserved regions from both proteins were recognized by identity of amino acid sequences among the retrieved sequences. All the predicted epitopes that showed 100% conservancy in the tools of B and T lymphocytes were included for further analysis while the non-conserved epitopes were excluded.
Fig. 1.

Multiple sequence alignment (MSA) of the retrieved strains of a spike S protein and b orf1ab polyprotein using Bioedit software and ClustalW. Letters within the rectangular indicated the non-conserved areas and dots indicated the conserve regions
B-cell epitopes prediction
The reference sequences of the spike S protein (YP_009724390.1) and orf1ab polyprotein (YP_009724389.1) were subjected to BepiPred linear epitopes prediction, Emini Surface Accessibility prediction, Kolaskar and Tongaonkar Antigenicity prediction, Karplus and Schulz flexibility and Parker hydrophilicity prediction tools in the IEDB server. The thresholds for each prediction method for each protein were shown in Table 1. The spike S protein and orf1ab polyprotein demonstrated 33 and 178 linear conserved epitopes with different lengths, respectively. When these epitopes further analyzed by the other B cell prediction tools, only one epitope from the spike S protein and four epitopes from orf1ab were passed the B cell tools and were shown to be antigenic, non-allergic and non-toxic. These epitopes, their length and position in each protein were shown in Table 1.
Table 1.
Predicted B cell epitopes, their antigenicity, allergenicity and toxicity from spike S protein and orf1ab polyprotein
| Protein | Epitopea | Start | End | Length | Emini surface accessibility Predictionb |
Kolaskar & Tongaonkar antigenicity Predictionc |
Karplus & Schulz flexibility Predictiond |
Parker hydrophiliciy Predictione |
Vaxijen antigenicityf | Allergenicity | Toxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Spike S protein | VYDPLQPE | 1137 | 1144 | 8 | 1.923 | 1.082 | Flexible | 1.65 | 0.5148 | Nonallergen | Nontoxin |
| orf1ab | SLDTYPSL | 2297 | 2304 | 8 | 1173 | 1066 | Flexible | 1.25 | 0.6746 | Nonallergen | Nontoxin |
| KSVYYTSNP | 1533 | 1541 | 9 | 308 | 1045 | Flexible | 2.833 | 0.6523 | Nonallergen | Nontoxin | |
| DASGKPVPY | 2924 | 2932 | 9 | 1632 | 1046 | Flexible | 3.178 | 0.6980 | Nonallergen | Nontoxin | |
| VKGLQPSVGPKQ | 6600 | 6611 | 12 | 1136 | 1066 | Flexible | 2.408 | 1.0641 | Nonallergen | Nontoxin |
aBepipred linear epitope prediction threshold for both proteins was 0.350
bEmini surface accessibility prediction threshold for both proteins was 1.000
cKolaskar & tongaonkar antigenicity prediction threshold for spike S protein and orf1ab was 1.041 and 1.044, respectively
dKarplus & schulz flexibility prediction threshold for spike S protein and orf1ab was 0.993 and 0.988, respectively
eParker hydrophiliciy prediction threshold for spike S protein and orf1ab was 1.238 and 1.127, respectively
fVaxijen antigenicity threshold for both proteins was 0.4
Cytotoxic T lymphocytes epitopes prediction
The reference sequences of the spike S protein (YP_009724390.1) and orf1ab polyprotein (YP_009724389.1) were analyzed using IEDB MHC-1 binding prediction tools to predict T cell epitopes interacting with MHC Class I alleles. This was performed based on Artificial Neural Network (ANN) with half-maximal inhibitory concentration (IC50) ≤ 100. A total of 218 and 358 epitopes were predicted interacting with different MHC-1 alleles from the spike S protein and orf1ab polyprotein, respectively. The antigenic, nonallergic, nontoxic epitopes that provided high population coverage and high allelic interactions with MHC-1 alleles were elected as vaccine candidates. Accordingly five epitopes from the spike S protein and seven epitopes from the orf1ab were chosen as vaccine candidates. These epitopes, their position and population coverage were provided in Table 2.
Table 2.
The predicted T cytotoxic cells epitopes, their antigenicity, allergenicity, toxicity and the population coverage from spike S protein and orf1ab polyprotein
| Protein | Epitopes | Start | End | Vaxijen antigenicity (0.4) | Allergenicity | Toxicity | Population coverage |
|---|---|---|---|---|---|---|---|
| Spike S protein | FTISVTTEI | 718 | 726 | 0.8535 | Nonallergen | Nontoxin | 52.54% |
| FVFLVLLPL | 2 | 10 | 0.8601 | Nonallergen | Nontoxin | 48.45% | |
| VVFLHVTYV | 1060 | 1068 | 1.5122 | Nonallergen | Nontoxin | 48.45% | |
| VRFPNITNL | 327 | 335 | 1.1141 | Nonallergen | Nontoxin | 41.68% | |
| aFAMQMAYRF | 898 | 906 | 1.0278 | Nonallergen | Nontoxin | 39.96% | |
| orf1ab | aVMYASAVVL | 3683 | 3691 | 0.4778 | Nonallergen | Nontoxin | 57.06% |
| SLIYSTAAL | 2242 | 2250 | 0.452 | Nonallergen | Nontoxin | 51.80% | |
| MMISAGFSL | 6425 | 6433 | 1.0248 | Nonallergen | Nontoxin | 51.47% | |
| FVMMSAPPA | 1804 | 1812 | 0.4871 | Nonallergen | Nontoxin | 48.97% | |
| FLLNKEMYL | 3183 | 3191 | 0.44 | Nonallergen | Nontoxin | 45.42% | |
| aFLLPSLATV | 3639 | 3647 | 0.5954 | Nonallergen | Nontoxin | 40.60% | |
| aSLENVAFNV | 6453 | 6461 | 1.0488 | Nonallergen | Nontoxin | 40.60% |
athe overlapped epitopes between MHCI and MHCII molecules were used once in the vaccine construct as MHCI or MHCII epitopes
Helper T lymphocytes epitopes prediction
The reference sequences of the spike S protein (YP_009724390.1) and orf1ab polyprotein (YP_009724389.1) were analyzed using IEDB MHC-II binding prediction tools to predict T cell epitopes interacting with MHC Class II alleles (HLA-DR, HLA-DQ and HLA-DP). Vast amount of epitopes were predicted interacting with different MHC II alleles from the spike S protein and orf1ab polyprotein. Multiple antigenic, nonallergic and nontoxic epitopes were predicted overlapping between MHC I and MHC II. However, only the MHC II non-overlapping epitopes were considered in this stage. Among them eight epitopes from the spike S protein and ten epitopes from the orf1ab were chosen as vaccine candidates against MHC II based on their high population coverage and high allelic interaction. These epitopes, their position and population coverage were demonstrated in Table 3.
Table 3.
The predicted T helper cells epitopes, their antigenicity, allergenicity, toxicity and the population coverage from spike S protein and orf1ab polyprotein
| Protein | Core peptide (Epitope) | Peptide | Start | End | Vaxijen antigenicity (0.4) | Allergenicity | Toxicity | Population coverage |
|---|---|---|---|---|---|---|---|---|
| Spike S protein | FELLHAPAT | FELLHAPATVCGPKK | 515 | 529 | 0.5409 | Nonallergen | Nontoxin | 98.03% |
| aFAMQMAYRF | FAMQMAYRFNGIGVT | 898 | 912 | 1.0278 | Nonallergen | Nontoxin | 98.93% | |
| FNFSQILPD | FNFSQILPDPSKPSK | 800 | 814 | 0.5831 | Nonallergen | Nontoxin | 99.73% | |
| FGAISSVLN | FGAISSVLNDILSRL | 970 | 984 | 0.5435 | Nonallergen | Nontoxin | 98.66% | |
| FNATRFASV | FNATRFASVYAWNRK | 342 | 356 | 0.5609 | Nonallergen | Nontoxin | 99.23% | |
| LLFNKVTLA | LLFNKVTLADAGFIK | 821 | 835 | 0.615 | Nonallergen | Nontoxin | 99.48% | |
| LLQYGSFCT | LLQYGSFCTQLNRAL | 753 | 767 | 0.8188 | Nonallergen | Nontoxin | 99.21% | |
| NRALTGIAV | NRALTGIAVEQDKNT | 764 | 778 | 0.5302 | Nonallergen | Nontoxin | 99.03% | |
| WTFGAGAAL | WTFGAGAALQIPFAM | 886 | 900 | 0.4918 | Nonallergen | Nontoxin | 99.29% | |
| orf1ab | aVMYASAVVL | VMYASAVVLLILMTA | 3683 | 3697 | 0.4778 | Nonallergen | Nontoxin | 98.28% |
| aFLLPSLATV | FLLPSLATVAYFNMV | 3639 | 3653 | 0.5954 | Nonallergen | Nontoxin | 94.69% | |
| aSLENVAFNV | SLENVAFNVVNKGHF | 6453 | 6467 | 1.0488 | Nonallergen | Nontoxin | 97.84% | |
| FFYVLGLAA | FFYVLGLAAIMQLFF | 2337 | 2351 | 0.8102 | Nonallergen | Nontoxin | 99.72% | |
| YELQTPFEI | YELQTPFEIKLAKKF | 249 | 263 | 0.5468 | Nonallergen | Nontoxin | 98.88% | |
| LIYSTAALG | LIYSTAALGVLMSNL | 2243 | 2257 | 0.5328 | Nonallergen | Nontoxin | 99.77% | |
| LRGTAVMSL | LRGTAVMSLKEGQIN | 7052 | 7066 | 0.8822 | Nonallergen | Nontoxin | 99.82% | |
| LVQMAPISA | LVQMAPISAMVRMYI | 2371 | 2385 | 1.0016 | Nonallergen | Nontoxin | 99.86% | |
| LVQSTQWSL | LVQSTQWSLFFFLYE | 3594 | 3608 | 1.095 | Nonallergen | Nontoxin | 99.22% | |
| PLIVTALRA | PLIVTALRANSAVKL | 4125 | 4139 | 0.9545 | Nonallergen | Nontoxin | 99.70% | |
| VLGLAAIMQ | VLGLAAIMQLFFSYF | 2340 | 2354 | 0.6624 | Nonallergen | Nontoxin | 99.71% | |
| SACVLAAEC | SACVLAAECTIFKDA | 2911 | 2925 | 0.5526 | Nonallergen | Nontoxin | 93.56% | |
| YVLGLAAIM | YVLGLAAIMQLFFSY | 2339 | 2353 | 0.6002 | Nonallergen | Nontoxin | 99.87% |
athe overlapped epitopes between MHCI and MHCII molecules were used once in the vaccine construct as MHCI or MHCII epitopes
The proposed vaccine construct
The total number of proposed epitopes used to built the vaccine construct were five linear B-cell epitopes, 12 T cytotoxic and 18 T helper lymphocytes epitopes from both spike S protin and orf1ab polyprotein. In addition, adjuvants, linkers and His-tag were added to the vaccine construct. Taken together the vaccine construct comprises 526 amino acids (Fig. 2). The vaccine construct was shown to be antigenic in Vaxigen server with score of 0.6194 and nonallergen in the Allertop server.
Fig. 2.

Multi-epitope vaccine design. T helper epitopes (blue colour) and B cell epitopes (red colour) from both spike S protein and orf1ab polyprotein were linked by the short peptide linker KK, while T cytotoxic epitopes (purple color) were linked by GPGPG linker. Human β-defensin-3 (green color) was used as an adjuvant at N and C-terminals and linked by the short peptide EAAAK linkers. C-terminal 6-his was added as his-tag
Physical and chemical properties of the vaccine construct
The Protparam server demonstrated that the molecular weight of the vaccine construct was 56.37327 k dalton with theoretical isoelectric point value (pI) of 10.19. The total number of negatively (Asp+Glu) and positively (Arg + Lys) charged residues was 18 and 84 respectively. The vaccine construct comprises the 12 amino acids entered in the protein biosynthesis or protein structure. The Extinction coefficients (M− 1 cm− 1) at 280 nm measured in water was 40,185 assuming all pairs of Cys residues form cystines. The estimated half-life was 30 h (mammalian reticulocytes, in vitro), > 20 h (yeast, in vivo) and > 10 h (Escherichia coli, in vivo). The instability index (II) was computed to be 31.25. This classifies the protein as stable. Aliphatic index was 84.39 and the grand average of hydropathicity (GRAVY) was − 0.049 that classified the vaccine construct as hydrophilic.
BLAST homology assessment
Homology between the sequence of the vaccine and the host proteome sequence demonstrated that the query coverage of the vaccine protein showed only 17% homology to human proteins. This result showed that the predicted vaccine would not implicate in autoimmunity diseases to the host.
Cluster analysis of the MHC1 restricted alleles
The MHC1 alleles (HLA-A, HLA-B and HLA-C) that interacted with the epitopes from spike S protein and orf1ab polyprotein were clustered by MHCcluster v2.0 server. Sixteen alleles of class I HLA molecules were included in this analysis. Figure 3 showed the cluster analysis of the MHC1 alleles. The figure demonstrated (heatmap) red regions providing strong interaction between the clustering HLA alleles while the yellow regions showed weak allelic interaction between HLA alleles.
Fig. 3.

Cluster analysis of the HLA alleles in heat map representation. The red areas indicated strong interaction between HLA alleles while the yellow areas indicated weak interaction
Secondary structure of the vaccine construct
For the secondary structure prediction and as shown in Fig. 4 the vaccine construct demonstrated 30.8% alpha helix, 5.7% beta turn, 22.24% extended strands and 41.25% random coiled.
Fig. 4.

Secondary structure prediction plot of the vaccine construct. Alpha Helices were shown in blue color, while extended strands and beta turns were shown by red and green colours, respectively. The visualization of the prediction (a) and the score curves for each predicted state (b) were shown
Tertiary structure prediction, refinement and adaptation of the vaccine construct
The 3D structure (PDB file) of the vaccine construct that predicted by I-TASSER sever was submitted to ModRefiner and Galaxyrefiner servers to meliorate the quality of predicted 3D modeled structure (Fig. 5). The PDB file was then evaluated by the Ramachandran plot on Rampage. As shown in Fig. 5 the 3D structure of the vaccine construct predicted by I-TASSER server was further analyzed in Ramachandran plot assessment after refinement. Ramachandran plot showed that the number of residues in favoured region was 91.2% and the number of residues in allowed region was 5.3% with only 3.4% of the residues in the outlier region. Moreover proSA server provided Z-score of − 3.6 representing the good quality of the model.
Fig. 5.

a The 3D model of the vaccine construct obtained after homology modelling on I-TASSER. b The 3D model was refined in modrefiner and galaxyrefiner and c the validated refined model was assessed by Ramachandran plot analysis that demonstrated 91.2%, 5.3% and 3.4% of protein residues in favoured, allowed, and disallowed (outlier) regions respectively. d ProSA-server, giving a Z-score of − 3.6
Solubility and stability (disulfide bonds prediction) of the vaccine construct
Protein-sol server was used to predict the solubility of the vaccine construct. Figure 6 demonstrated that the solubility of the vaccine construct in terms of QuerySol (scaled solubility value) was 0.571. The experimental dataset (PopAvrSol) had a population average of 0.45. Accordingly the solubility of the vaccine construct was larger than 0.45. This result indicated that the vaccine construct is soluble compared to the average solubility of E. coli proteins. The solubility of the vaccine construct was further confirmed by SOLpro server. The vaccine construct showed solubility score of 0.873254 greater than the probability of ≥0.5 of the server. For the stability of the vaccine construct, as shown in Fig. 7, residues in the highly mobile region of the protein sequence were mutated with cysteine to perform disulfide engineering. A total 61 pairs of amino acid residues were shown to be probable forming disulfide bonds. Among them only six regions were evaluated to form disulfide bond based on the chi3 residue screening (between − 87 and + 97), B-factor value (ranged 6.950–17.410) and energy value less than 2.5. These six residues were replaced by cysteine residues. The six residue pairs were LYS204-LEU253; SER297-GLY341; VAL315-ALA329; PRO376-PRO451; PRO427-GLY431 and GLY491-LYS519.
Fig. 6.

Solubility of the vaccine construct as obtained by protein sol server. The solubility of the vaccine construct was shown to be 0.571 compared to 0.45 of the population average solubility of E. coli
Fig. 7.

Stability of the vaccine construct by disulfide bond engineering in a the original form and b the mutant form. Six disulfide bond regions were shown in golden sticky forms indicated by white arrows in the mutant form
Molecular docking of the vaccine construct with TLR4
For the docking analysis, the vaccine construct was docked against TLR4 (PDB1D: 4G8A) alpha and beta chains using the HDOCK server. Figure 8 showed that the vaccine construct bound to the TLR4: chain A with attractive binding energy of − 338.68 kcal/ mol. When the vaccine construct docked with TLR4: chain B the attractive binding energy was − 346.89 kcal/mol. The energy score obtained for both A and B chains were the lowest among all other predicted docked complexes showing highest binding affinity. A low (negative) energy indicated a stable system and thus likely binding interaction.
Fig. 8.

molecular docking between the vaccine construct and the TLR4 chains. The yellow colour represents the vaccine construct while the brown color represents the TLR4 chains. a A cartoon structure of the vaccine construct docked with chain A of TLR4 while b represents the ball structure. c A cartoon structure of the vaccine construct docked with chain B of TLR4 while d represents the ball structure
IFN-γ inducing epitope prediction
Concerning IFN-γ inducing epitope predictions from the vaccine construct, 412 potential epitopes were predicted from the vaccine construct after removal of the adjuvant. This number includes both +ve and –ve prediction scores. A total of 158 epitopes were predicted to be +ve for inducing IFN-γ with higher score ranging from 1to 7 for 28 epitopes. Figure 9 showed the level of IFN-γ induction during the period of the injections compared to the other cytokines. When the prediction was only performed for the adjuvant, 433 overlapped +ve and –ve epitopes were predicted inducing IFN-γ production. Among them 82 epitopes were predicted positive (+ve). However none of the positive epitopes scored greater than 1. Thus they were considered as IFN-γ non-inducing epitopes.
Fig. 9.
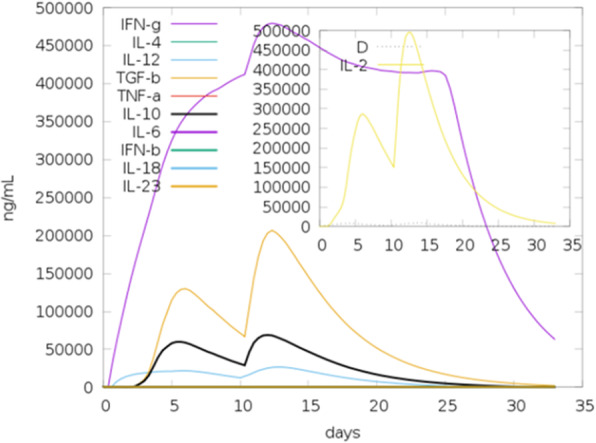
The cytokine levels induced by two injections of the vaccine construct given in interval of 30 days as simulated by C-ImmSim server. The main plot provided the concentration of cytokines and interleukins after the injections. The insert plot showed danger signal together with leukocyte growth factor IL-2 with the Simpson index, D (diversity) shown by the dotted line. The smaller the D value, the lower the diversity
Immune simulation of the vaccine construct
C-ImmSim server was used to mimic the actual immune responses in the body upon exposure to the vaccine construct. Generally the primary immune response occurs as a result of first contact with an antigen and the first antibody produced is mainly IgM, although small amount of IgG are also produced. The amount of antibodies produced depends on nature of antigen and usually produced in low amount. As shown in Fig. 10 the amount of the IgM was markedly started to increase during the first injection of the vaccine construct (antigen) as a primary immune response. Secondary immune response occurs as a result of the second and subsequent exposure to the same antigen and characterized by increased level of IgM and IgG. Also there was marked increased in the level of IgM + IgG and decreased level of the antigen. Moreover there were marked increase in the level of IgM, IgG1 + IgG2, and IgG1 (Fig. 10). This indicated that the antibodies had greater affinity to the vaccine construct (antigen) and would develop immune memory. Consequently, this resulted in increased clearance of the antigen upon subsequent exposures. Concerning the cytotoxic and helper T lymphocytes, high response in the cells populations with corresponding memory development was observed. Most importantly the population of the Helper T lymphocytes remained higher during all exposure time. In the IFN- γ induced epitopes prediction, the results showed that 158 predicted epitopes inducing IFN- γ production without adjuvant. This interpreted the high IFN- γ concentration score compared to the other cytokines. The Simpson index D demonstrated the level of danger when the cytokines level increased that may result in complications during the immune response.
Fig. 10.

The immune simulation with vaccine construct using C-immsim server. a Immunoglobulins production increased in response to exposure to antigen injections with marked decrease in the antigen concentration observed. b Showed the B-cell populations with marked increase in the memory and non-memory immunoglobulins. Figure c and d Showed increased level in the populations of the active T helper and T cytotoxic cells per state after the injections, respectively. The resting state provided cells not exposed to antigen while the anergic state provided tolerance of the T-cells to the antigen exposures
Codon adaptation and in silico cloning
The protein sequence of the vaccine construct was reversed translated into DNA sequence. Codon adaptation index values (CAI-Value) of the improved DNA sequence was 0.9199, indicating the higher proportion of most abundant codons. The GC-content of the improved sequence was 51.58%, indicating favourable GC content. Figure 11, showed that DNA sequence was cloned into pET28a (+) vector typically at the multiple cloning site (MCS) of the vector after linking BamHI and Xho1restriction enzymes cutting sites sequences to the vicinities of the DNA sequence.
Fig. 11.

In silico cloning of the final vaccine construct sequence into the pET30a (+) expression vector. The vector was shown in black color, while the red color provided the gene coding for the vaccine construct protein. The DNA sequence of the vaccine construct was typically cloned in the MCS of the vector between BamH1 and Xho1 cutting sites
Discussion
The availability of a safe and effective vaccine for SARS-CoV-2 is well-recognized as an additional tool to contribute to the control of the pandemic. Furthermore enormous challenges and efforts are needed to rapidly develop, evaluate and produce effective vaccine at large scales. In this regard, the Sinovac Biotech has created a new COVID-19 vaccine by growing the novel coronavirus in the VERO monkey cell line and inactivating it with chemicals [31]. The vaccine has protected the rhesus macaques from infection by the new coronavirus. However the vaccine was an old-fashioned formulation consisting of a chemically inactivated version of the virus. Despite that the vaccine produced no obvious side effects in the monkeys and human trials are under processing, but the number of animals tested was too small to yield statistically significant results. Moreover the vaccine may have caused changes that make it less reflective of the ones that infect humans. Another concern is that monkeys do not develop the most severe symptoms that SARS-CoV-2 causes in humans [31]. Generally such kinds of vaccines may have multiple caveats such as the risk of reversion to a more virulent strain of the virus being vaccinated against. Also they may cause severe complications in immunocompromized individuals. In addition to that they are expensive, time consuming and may include unnecessary proteins particles of the virus that provoke immunity, resulting in allergenic and other deleterious immunological responses [32, 33]. Accordingly, recently the focus has shifted towards the development of subunit vaccines as they are associated with better safety profiles and are logistically more feasible [34]. Beside the Sinovac Biotech vaccine, more than 42 vaccines candidates against the pandemic in the clinical trials phases, and some are currently in phase III trials such as Pfizer-BioNTech COVID-19 Vaccine (BNT162b2), ModernaTX, Inc. (mRNA-1273), Sinopharm, CanSino, AstraZeneca and Novavax vaccines [35].
The restrictions on the use of live or attenuated virus vaccines create the need for a safer and effective vaccine. Epitope-based vaccines demonstrated a novel approach for production of a specific immune response and flee the responses against undesirable epitopes in the antigen [36]. Hence, the spike S protein and orf1ab polyprotein were targeted to generate a vaccine construct against SARS-CoV-2 using reverse vaccinology especially enough data about the genomics and proteomics of SARS-CoV-2 become available.
In the current study, the entire viral proteome of SARS-CoV-2 was retrieved from NCBI database. Each protein in the virus was subjected to protein analysis using protparam analysis tool. Moreover the viral proteins were subjected to Vaxijen server to investigate the antigenicity of each protein. All the viral proteins demonstrated antigenicity (scored more than 0.4). Furthermore the viral proteins were examined for the transmembrane helices (TMHs), where the nonstructural orf1ab polyprotein owned the highest number of TMHs. Also the orf1ab polyprotein is the largest protein with 7096 amino acids [2, 22] and plays vital roles in the viral replication, virulence, virus–cell interactions and/or alterations of virus–host response [23]. In the preclinical studies of vaccines against SARS-CoV and MERS-CoV, the spike S protein is the major antigenic determinants that induce neutralizing antibodies [12, 13, 37, 38] and contains the receptor binding domain (RBD) [15–17]. Moreover the majority of the vaccine candidates against SARS CoV were based on the spike S protein and RBD region [12, 13, 15, 20, 21]. Thus these two proteins were targeted for the generation of the vaccine candidates.
In this study a 100% conserved epitopes amongst the screened sequences of spike S protein and orf1ab polyprotein (including those of the new variant strain of Britain, SARS-CoV-2 VUI 202012/01) that could be recognized by B and T lymphocytes to work as vaccine candidates were proposed. For B cell epitopes prediction, the predicted epitopes were obtained using various tools in the IEDB. The predicted B cell epitopes were tested to be linear, surface accessible, antigenic, flexible and hydrophilic using IEDB prediction tools. Furthermore the resulting epitopes were subjected to antigenicity, allergenicity and toxicity analysis. However, only one epitope from the spike S protein and four epitopes from orf1ab polyprotein successfully passed these criteria (Table 1). Thus were proposed as vaccine candidates against B cells. The scarcity of the number of the predicted B cell epitopes may indicate the nonfavourable interaction between the B cells and the virus. Moreover the humoral response from memory B cells can easily be overcome over time by number of antigens, however, cell mediated immunity often elicits long lasting immunity [39, 40].
For T cells, large numbers of epitopes were shown to interact with MHCI and MHCII alleles from spike S protein and orf1ab polyprotein. Epitopes that shown to be antigenic, nonallergic, nontoxic and with high population coverage were elected as a vaccine candidates (Tables 2 and 3). The epitopes 898FAMQMAYRF906 and 800FNFSQILPD808 were previously proposed as vaccine candidates from spike S protein of SARS CoV [21]. Here in this study, the former epitope was also shown to interact with both MHCI and MHC II alleles, while the later epitope interacted only with MHC II alleles of SARS-CoV-2. In addition to that, the two epitopes were located within S2 region (amino acids from 511 to 1190) of the spike S protein that predicted to interfere with fusion of the viral envelope with the host cell and considered as appropriate target for monoclonal antibody development or as vaccine candidates [15]. This result reflected the importance of these two epitopes in SARS-CoV-2 vaccine construction.
For the vaccine to be considered as a global vaccine, the proposed epitopes that constitute the vaccine should interact with most ethnic polymorphic MHC1 and MHC11alleles with high population coverage scores. In this regard the population coverage of the predicted epitopes interacting with T lymphocytes was investigated. The proposed epitopes demonstrated higher affinity to interact with MHC I and MHC II alleles and bound to different sets of alleles with high population coverage scores (Tables 2 and 3). This result indicated that the proposed epitopes as vaccine candidates could cover large population and effectively interacted with the human common alleles worldwide. This result further strengthens the proposed epitopes to work as vaccine candidates against SARS-CoV-2.
One of the most important features of the vaccine protein is not to provide significant similarity or homology to the host proteins. The high similarity between the vaccine as a protein in nature and the host proteome could guide to autoimmune diseases due to molecular mimicry and the chances of cross reactivity [41–43]. In this study the vaccine construct demonstrated less homology (17%) to the human proteins using BLASTp tool providing the vaccine as an excellent candidate with no autoimmunity. Moreover, MHC superfamilies are considered as an essential player in vaccine construction and development as well as drug development. Thus MHC cluster analysis was also performed to assess the functional relationship between MHC1 clustering variants.
To design a vaccine construct, the elected B and T cells epitopes were fused using appropriate specialized spacer (linkers) sequences in order to generate multi-epitopes peptides [44]. The linkers KK and GPGPG were introduced between the selected B and T cells epitopes to generate a sequence with minimal junctional immunogenicity [45–49]. The EAAAK linker was also added between the adjuvants sequences and the fused epitopes in order to reach a high level of expression and improved bioactivity of the fused epitopes [44, 46]. The adjuvants were previously reported as immunomodulator to ameliorate the activity of multiple vaccines [50, 51]. In this regard the β-defensin adjuvant, experimentally, demonstrated an effective immune-stimulation against different kinds of organisms [52–54]. Thus it was used as an adjuvant in the amino and carboxyl terminals of the vaccine construct in this study. Later the vaccine construct was tested for antigenicity and allergenicity and was shown to be antigenic and nonallergic since vaccines with multiple epitopes are often poorly immunogenic and require coupling to adjuvant [44].
The physical and chemical properties showed that the vaccine construct molecular weight was 56.37 k dalton. The computed instability index (II) classifies the protein as stable. Moreover the aliphatic index showed that the protein contains aliphatic side chains, indicating potential hydrophobicity. Moreover the grand average of hydropathicity (GRAVY) was − 0.049 that classified the vaccine construct as hydrophilic. All these characteristics showed that the vaccine protein is thermally stable and therefore suitable as a vaccine against SARS-CoV-2. Furthermore the secondary and tertiary structures of the vaccine construct were evaluated since they are important in vaccine design [44]. Secondary structure analysis showed that the vaccine construct contains alpha helices, extended strands, beta turns and random coiled structures. The 3D structure of the vaccine construct highly ameliorated by the refined software and demonstrated desirable characteristics on Ramachandran plot predictions. Moreover a major problem in structural biology is the recognition of errors in experimental and theoretical models of protein structures [55]. Thus ProSA program was employed to predict the potential structural and modeling errors in the vaccine. The overall quality score was calculated by ProSA program for a specific input structure. The result was displayed in a plot that showed the scores of all experimentally determined protein chains currently available in the Protein Data Bank (PDB) [55]. In this study the predicted vaccine construct demonstrated a Z-score of − 3.6. This indicated that the quality of the overall model is satisfactory as a vaccine candidate against SARS-CoV-2.
Protein solubility and stability have multiple biologically significant functions. For instance the solubility of the overexpressed recombinant protein in the E. coli host is one of the important requirements of many biochemical and functional analysis [46, 49]. In this study the solubility of the vaccine construct was measured using protein sol and SOLpro servers. The vaccine construct provided solubility indexes greater than the average probabilities of the servers indicating the solubility of the vaccine construct. Disulfide engineering is important for protein folding and stability. Also structural disulfide engineering decreases the possible number of conformations for a given protein, resulting in decreased entropy and increased thermostability [56–58]. Thus the stability of the vaccine construct was indexed if six residues in the vaccine structure mutated to cysteine.
To strengthen the interaction between the vaccine construct and TLR4, molecular protein-protein docking was performed to explore the binding affinity of vaccine construct with TLR4 chain A and chain B. TLR4 is the key receptor for infectious and noninfectious stimuli that induced a proinflammatory response. TLR4 also plays important role as amplifier of the inflammatory response [59]. In this study the attractive binding energy between TLR4 chains and the vaccine construct demonstrated high binding affinity that expressed in negative binding energy values. Thus this interaction with the TLR4 professionally eliciting a potential protective immune response. Furthermore immune simulation was performed to mimic the typical immune responses. Generally there was marked increase in the immunoglobulins coincided with frequent injection of the vaccine construct. This result indicated the development of memory B cells. Also the level of the active T cytotoxic and T helper lymphocytes were significantly increased supporting the enhancement of humoral and adaptive immune responses. The level of the IFN-γ was also observed high at peak level during the injection times.
Most importantly the expression of the vaccine construct in a suitable E. coli expression vector is essential for the production of recombinant proteins [60, 61]. The designed vaccine construct was reverse transcribed and adapted for E. coli strain K12 before cloning into pET28a (+) vector. The codon adaptability index (0.9199) and the GC content (51.58%) provided high-level expression of the protein in bacteria. The vaccine construct gene was typically cloned in the vector in the multiple cloning sites. This result provided the successful cloning of the vaccine protein.
Conclusion
The elimination of the pandemic is coincided with development of novel control measures to combat the infection of SARS-CoV-2. In this study a unique vaccine construct (multiepitopes) was generated from spike S protein and orf1ab polyprotein against B and T lymphocytes via various bioinformatics tools. This proposed vaccine construct could potentially provide protection against the pandemic SARS-CoV-2 and/or used as complementary tool to eliminate the infection. Therefore, the present study might assist in developing a suitable therapeutics protocol to combat SARS-CoV-2 infection.
Methods
The retrieval of the viral whole proteome
The entire viral proteome of SARS-CoV-2 (COVID-19) was retrieved from National Center For Biotechnology Information (NCBI) at (https://www.ncbi.nlm.nih.gov/genome/browse/#!/proteins/86693/757732%7CSevere%20acute%20respiratory%20syndrome%20coronavirus%202/). The virus demonstrated 12 proteins. These 12 proteins accession numbers, lengths and names were shown in Table 4.
Table 4.
Physical and chemical properties, antigenicity and number of the predicted transmembrane helices of SARS CoV-2 proteins
| Viral protein | Accession number | Molecular weight (Dalton) | Instability indexb | Aliphatic index | Theoretical pI | No amino acids | Extinction coefficient | GRAVYc | Vaxijen antigenicityd | TMHs |
|---|---|---|---|---|---|---|---|---|---|---|
| orf1ab polyprotein | YP_009724389.1 | 794,057.79 | 33.31 | 86.87 | 6.32 | 7096 | 942,275 | −0.07 | 0.4624 | 14 |
| orf1a polyprotein | YP_009725295.1 | 489,988.91 | 34.92 | 88.99 | 6.04 | 4405 | 552,175 | −0.023 | 0.4787 | 14 |
| Surface glycoprotein | YP_009724390.1 | 141,178.47 | 33.01 | 84.67 | 6.24 | 1273 | 148,960 | −0.079 | 0.4646 | 1 |
| orf3a protein | YP_009724391.1 | 31,122.94 | 32.96 | 103.42 | 5.55 | 275 | 58,705 | 0.275 | 0.4945 | 3 |
| Envelope protein | YP_009724392.1 | 8365.04 | 38.68 | 144 | 8.57 | 75 | 6085 | 1.128 | 0.6025 | 1 |
| Membrane glycoprotein | YP_009724393.1 | 25,146.62 | 39.14 | 120.86 | 9.51 | 222 | 52,160 | 0.446 | 0.5102 | 3 |
| orf6 protein | YP_009724394.1 | 7272.54 | 31.16 | 130.98 | 4.6 | 61 | 8480 | 0.233 | 0.6131 | 0 |
| aorf7a protein | YP_009724395.1 | 13,744.17 | 48.66 | 100.74 | 8.23 | 121 | 7825 | 0.318 | 0.6441 | 1 |
| orf7b protein | YP_009725296.1 | 5180.27 | 50.96 | 156.51 | 4.17 | 43 | 7115 | 1.449 | 0.6025 | 1 |
| orf8 protein | YP_009724396.1 | 13,831.01 | 45.79 | 97.36 | 5.42 | 121 | 16,305 | 0.219 | 0.6502 | 0 |
| Nucleocapsid phosphoprotein | YP_009724397.2 | 45,625.7 | 55.09 | 52.53 | 10.07 | 419 | 43,890 | −0.971 | 0.5059 | 0 |
| aorf10 protein | YP_009725255.1 | 4449.23 | 16.06 | 107.63 | 7.93 | 38 | 4470 | 0.637 | 0.7185 | 0 |
THMs Transmembrane helices
athe protein contains no tryptophan
binstability index < 40 considered the protein stable
cGRAVY negative sign indicated the protein is hydrophilic
dthe threshold for the Vaxijen antigenicity is 0.4
Physical and chemical properties of the viral proteins, antigenicity and transmembrane topology
ProtParam (http://web.expasy.org/protparam/) is a tool allowed the computation of various physical and chemical parameters for a given protein sequence. Each protein was subjected to Protparam server for the physiochemical properties and the computed parameters covered the molecular weight, theoretical pI, amino acid composition, extinction coefficient, instability index, aliphatic index and grand average of hydropathicity (GRAVY). Moreover the VaxiJen v2.0 server at (http://www.ddg-pharmfac.net/vaxijen/) which based on auto- and cross-covariance transformation of protein sequences into uniform vectors of principal amino acid properties was used to analyze the potent antigenicity of each protein of SARS-CoV-2. The viral proteins were further analyzed for transmembrane topology using TMHMM (http://www.cbs.dtu.dk/services/TMHMM/). Proteins that demonstrated best physiochemical properties, antigenicity and transmembrane topologies were allowed for further analysis. In this essence, as shown in Table 4 only the first three proteins in the table demonstrated best physical and chemical properties despite all the viral proteins were shown to be antigenic by VaxiJen v2.0 passing the threshold of (0.4) and contained varied numbers of TMHs. It is noteworthy that the viral orf1ab polyprotein and orf1a polyprotein upon alignment the later was shown to be partial from the former (orf1ab). Accordingly, the spike S protein and orf1ab polyprotein were targeted for prediction of epitopes as vaccine candidates that could elicit both B and T lymphocytes.
Protein sequences retrieval of spike S proteins and orf1ab polyprotein
A set of available 714 orf1ab polyproteins at (https://www.ncbi.nlm.nih.gov/protein/?term=orf1ab+polyprotein+%5BSevere+acute+respiratory+syndrome+coronavirus+2%5D) and 9 proteins of spike S glycoproteins at (https://www.ncbi.nlm.nih.gov/protein/?term=spike+S+protein+severe+acute+respiratory+syndrome+2+) of SARS-CoV-2 were retrieved from the NCBI. These sequences were retrieved in FASTA format and further used for epitopes conservancy among the retrieved strains. The spike S protein (id= QQL92050.1) and orf1ab protein (id= QQL92048.1) of the new variant strain SARS-CoV-2 VUI 202012/01(MW450666.1) that was recently identified in Britain was also included in the epitopes conservancy analysis.
Sequence alignment and determination of the conserved regions
The retrieved protein sequences of spike S protein and orf1ab polyprotein were further aligned to obtain conserved epitopes using multiple sequence alignment (MSA) tools, Clustal W, embedded in the BioEdit program, version 7.0.9.0 [62]. MSA analysis was performed to analyze 100% conserved epitopes amongst the screened sequences of spike S protein and orf1ab polyprotein.
B-cell epitopes prediction
B-cell epitopes are antigenic determinants recognized by the immune system and represent the specific piece of the antigen to which B lymphocytes bind. These play a vital role in vaccine design. The Immune Epitope Data Base web server (IEDB) at (https://www.iedb.org/) was used for prediction of B cell epitopes from spike S protein and orf1ab polyprotein. A collection of methods to predict B cell epitopes based on sequence characteristics of the antigen using amino acid scales and hidden Markov Models (HMMs) were used. For instance; Linear B-cell epitopes were predicted using BepiPred linear epitopes prediction tool [63–65]. Emini Surface Accessibility prediction method was used to obtain surface epitopes [66]. The antigenicity of the predicted epitopes was performed using Kolaskar and Tongaonkar Antigenicity prediction tools [67]. For prediction of epitopes flexibility and hydrophilicity, the Karplus and Schulz flexibility and Parker hydrophilicity prediction tools were used [68, 69].
Cytotoxic T lymphocytes epitopes prediction
The epitopes binding analysis to Major Histocompatibility Complex class I molecules (MHC class I) from spike S protein and orf1ab polyprotein was performed using IEDB MHC I tools at (http://tools.iedb.org/mhci/). The MHC I epitope molecules that interacted to T lymphocytes was subjected to multiple steps. This prediction method used an amino acid sequence, or set of sequences and determined each subsequence’s ability to bind to a specific MHC class I molecule. The binding of the fragmented peptides to MHC molecules step was predicted by Artificial Neural Network 4.0 (ANN 4.0) method. Prior to the prediction, all lengths of epitope was set as 9mers and all the conserved epitopes that bound to alleles at score of ≤100 half-maximal inhibitory concentration (IC50) were subjected for further analysis [70–73].
Helper T-lymphocytes epitopes prediction
Analysis of peptides binding to MHC II molecules from spike S protein and orf1ab polyprotein was assessed by the IEDB MHC II prediction tool at (http://tools.iedb.org/mhcii/result/). For MHC II binding predication, human allele’s reference sets (human HLA-DR, HLA-DQ, HLA-DP) were used. MHC II groove has the ability to bind different lengths peptides that makes prediction more difficult and less accurate. Thus Neural Network align (NN-align 2.3; Net MHCII 2.3) was used to identify both the binding affinity and MHCII binding core epitopes. Prior to the prediction, the length of peptide was set as 15mers (15 amino acids) and all the conserved epitopes that bound to alleles at score of score of ≤1000 half-maximal inhibitory concentration (IC50) were subjected for further analysis [74].
Antigenicity, allergenicity and toxicity of the predicted epitopes
Analysis of the antigenicity, allergenicity and toxicity of the predicted epitopes from spike S protein and orf1ab polyprotein for B and T lymphocytes, was performed using multiple prediction tools. The predicted epitopes were submitted to the VaxiJen v2.0 server for antigenicity prediction. The threshold of VaxiJen v2.0 server was set to the default threshold (0.4). Epitopes that demonstrated antigenicity were further investigated for their allergenicity using AllerTOP server [75]. Epitopes found to be antigenic and non-allergenic were further assessed for toxicity by ToxinPred server [76].
Population coverage
Epitopes that interacted with MHC I and MHC II from spike S protein and orf1ab polyprotein were subjected to population coverage analysis after they were shown to be antigenic, nonallergic and nontoxic. The population coverage was investigated against the whole world using IEDB population coverage tool at (http://tools.iedb.org/tools/population/iedb_input).
Vaccine construction (multiepitopes vaccine)
Epitopes that passed the criteria of B cell epitopes prediction, epitopes with high allelic interaction and best population coverage scores against cytotoxic and helper T lymphocytes were used to generate the vaccine construct. Epitopes that overlapped in both MHC I and MHC II were used once in the vaccine construct as MHC I or MHC II epitopes. The vaccine construct was generated as previously described [45, 46] with minor modifications. The GPGPG linker was used to fuse the B cell and T helper predicted epitopes. While KK linker was used to link the epitopes of T cytotoxic lymphocytes. EAAAK linker was used to link the epitopes with the human β-defensin (uniprot entry Q5U7J2) that was used as an adjuvant on the amino and carboxyl terminals to ameliorate the immunogenicity of the vaccine construct [77]. β-defensin was shown to induce potential immunogenic responses similar to natural immune responses [77]. Linkers were shown to assist in enhancing expression, stability and folding of the protein by separating the functional domains [43, 78].
Physical and chemical properties of the vaccine construct
The vaccine construct from predicted epitopes was analyzed for the physical and chemical properties using Protparam analysis tool. The computed parameters covered the molecular weight, theoretical pI, amino acid composition, extinction coefficient, estimated half-life, instability index, aliphatic index and Grand average of hydropathicity (GRAVY).
BLAST and assessment of vaccine protein against the human proteins
Protein BLAST was performed to find similarity or homology between the vaccine protein construct with the human proteome via NCBI BLASTp [79, 80]. The aim behind this homology analysis was to avoid autoimmunity that might be caused due the homology between the vaccine and human proteins. The search in Protein BLAST was limited to records that include: Homo sapeins (taxid: 9606). The result of the homology score must be no or least homology (< 40%) to the human proteome [41].
Cluster analysis of the MHC1 restricted alleles
The MHC genomic region in most species is extremely polymorphic. The human MHC genomic region (HLA) is extremely polymorphic comprising several thousand alleles, many encoding a distinct molecule. The potentially unique specificities remain experimentally uncharacterized for the vast majority of HLA molecules [81]. The MHCcluster server is a tool to functionally cluster MHC class I molecules (MHC I) based on their predicted binding specificity was used [81]. The functional relationship between the allelic variants is presented as a phylogenetic tree and/or heat-map between MHC variants [45, 81].
Secondary structure prediction
Self-optimized prediction method (SOPMA) at (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) was used to predict alpha helix, coiled structures and beta sheets in the secondary structure of the vaccine construct [82].
Tertiary structure prediction, refinement and validation
The vaccine construct sequence was submitted to I-TASSER protein folding recognition server [83]. The server is an active development with the goal to provide the most accurate protein structural and functional predictions using state-of-the-art algorithms. The PDB file obtained by I-TASSER was submitted to ModRefiner [84] and GalaxyWEB [85, 86] web servers for protein structure prediction, refinement, and related methods. The refinement was performed to ameliorate the physical quality of the structure. The refined protein structure was further validated through Ramachandran plot assessment at RAMPAGE [87, 88]. Moreover the refined PDB file obtained by I-TASSER server was analyzed by ProSA server for structure potential errors [55]. ProSA-web Z-score is depicted in a plot, which includes the Z-score of experimentally determined structures deposited in PDB.
Solubility and stability (disulfide bonds prediction) of the vaccine construct
Protein-sol (https://protein-sol.manchester.ac.uk/) is a web based suite of theoretical calculations and predictive algorithms for understanding protein solubility [89]. The solubility of the vaccine construct was analyzed compared to solubility in databases. The server predicted the solubility of proteins in terms of QuerySol (scaled solubility value). The experimental dataset (PopAvrSol) had a population average of 0.45. Accordingly the protein solubility scores larger than 0.45 is expected to be soluble compared to the average solubility of E. coli proteins from the experimental solubility dataset and vice versa [45, 90].
The solubility of the vaccine construct was further analyzed by SOLpro server (http://scratch.proteomics.ics.uci.edu/) to predict the solubility upon overexpression.
SOLpro predicts solubility based on the probability of ≥0.5. Thus soluble protein scores ≥0.5 and insoluble protein scores < 0.5. For stability, the disulfide bonds strengthen the geometric conformation of the vaccine construct and provided significant stability. The Disulfide by Design 2.0 (DbD2) is a web-based tool for disulfide engineering in proteins was used to design disulfide bonds in the vaccine construct [91]. For a given protein structural model to predict disulfide bonds, all residue pairs are rapidly assessed for proximity and geometry consistent with disulfide formation, assuming the residues were mutated to cysteine.
Molecular docking of the vaccine construct with TLR4 (protein-protein docking)
Protein–protein and protein–DNA/RNA interactions play a fundamental role in a variety of biological processes. Determining the complex structures of these interactions is valuable, in which molecular docking has played an important role. HDOCK server that used protein-protein and protein-DNA/RNA docking based on a hybrid algorithm of template-based modeling and ab initio free docking was used to dock the vaccine construct with human Toll-Like Receptor4 (TLR4) [92]. The vaccine construct PDB file was submitted to the server with TLR4 (PDB ID: 4G8A) as a receptor for the docking process.
IFN-γ inducing epitope prediction
IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/scan.php) is a module designed for predicting Interferon gamma (IFN-γ) inducing regions in a protein or antigen by generated all possible overlapping peptides (of length or window selected by user) from the protein or antigen. The server identifies best antigenic regions or IFN epitope in a query antigen sequence that can induce IFN-γ. Interferon gamma (IFN-γ), has an impact on the adaptive and innate immune responses, provokes immune system cells and raised response to MHC antigens. The prediction process was performed as previously described [46, 93] with minor modification. The length of the designed peptide was set to15-mers IFN-γ epitopes. The prediction was performed by Supportive Vector Machine approach.
Immune simulation
To further characterize the immunogenicity and immune response profile of the vaccine construct, an in silico immune simulations were conducted using the C-ImmSim server (http://150.146.2.1/C-IMMSIM/index.php) [94]. Two injections with vaccine construct were given at intervals of 30 days. The Simpson index, D (a measure of diversity) was interpreted from the plot.
Codon adaptation and in silico cloning
Codon adaptation and in silico cloning were performed in order to express the final vaccine construct in the E. coli (strain K12) host since codon usage optimization demonstrated differences between human and E. coli strain. The purpose of this cloning was to guarantee the expression of the vaccine construct in the selected host. Java Codon Adaptation Tool (JCAT) server (http://www.prodoric.de/JCat) was firstly used for the reverse translation of the protein sequence of the vaccine construct into DNA sequence. The rho independent transcription termination, prokaryote ribosome binding site and cleavage site of restriction enzyme were avoided [46]. In the JACT, codon adaptation index (CAI) score is 1.0 but > 0.8 is considered a good score [95]. The favourable GC content of a sequence ranged between 30 and 70%. Secondly, BamHI and Xho1restriction enzymes cutting sites sequences were introduced to the DNA sequence obtained by (JCat) server at the N-terminal and C-terminal vicinities, respectively. The SnapGene restriction cloning module [46, 47] was used to insert the DNA sequence into pET28a (+) vector between the BamHI and Xho1.
Acknowledgements
Not applicable
Abbreviations
- SARS-CoV-2
Severe acute respiratory syndrome coronavirus-2
- SARS-CoV
Severe acute respiratory syndrome coronavirus
- COVID-19
Coronavirus disease 2019
- RBD
Receptor binding domain
- ACE2
Angiotensin converting enzyme 2
- PLpro
Papain-like protease
- 3CLpro
3C-like protease
- NSP
Nonstructural proteins
- IEDB
Immune Epitope Data Base web server
- MHC 1
Major Histocompatibility Complex class I
- MHC II
Major Histocompatibility Complex class II
- HLA
Human leucocyte antigen
- ANN
Artificial Neural Network
- NN-align
Neural Network align
- IC50
Half-maximal inhibitory concentration
- pI
Isoelectric point
- GRAVY
Grand average of hydropathicity
- TLR4
Toll like Receptor 4
- BLASTp
Basic local alignment search tool for protein
- 3D structure
Three dimensional structure
- PDBfile
Protein Data Bank file
- QuerySol
Query solubility
- PopAvrSol
Population average solubility
- CAI
Codon adaptation index
- JCAT
Java Codon Adaptation Tool
- MCS
Multiple cloning site
- TMHs
Transmembrane helices
- TMHMM
Transmembrane helices prediction method based on a hidden Markov Model
- NCBI
National Center for Biotechnology Information
- MSA
Multiple sequence alignment
- SOPMA
Self-optimized prediction method
Authors’ contributions
YAA: Conceived the study, designed the study, data analysis, coordinated the study and writing and drafting the manuscript. KA: conceived of the study, designed the study, data analysis and drafting the manuscript. EEME: data analysis coordinated the study and revised the draft manuscript. All authors gave final approval for publication.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Availability of data and materials
The datasets during and/or analyzed during the current study available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yassir A. Almofti and Khoubieb Ali Abd-elrahman contributed equally to this work.
References
- 1.World Health Organization. WHO director-general’s remarks at the media briefing on 2019-nCoV on 11 February 2020. https://www.who.int/dg/speeches/detail/who-director-general-s-remarks-at-the-media-briefing-on-2019-ncov-on-11-february-2020. Accessed 13 Feb 2020.
- 2.Biswas A, Bhattacharjee U, Chakrabarti AK, Tewari DN, Banu H, Dutta S. Emergence of novel coronavirus and COVID-19: whether to stay or die out? Crit Rev Microbiol. 2020. 10.1080/1040841X.2020.1739001. [DOI] [PMC free article] [PubMed]
- 3.Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. 2020. 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed]
- 4.Chen WH, Strych U, Hotez PJ, Bottazzi ME. The SARS-CoV-2 vaccine pipeline: an overview. Curr Trop Med Rep. 2020:1–4. 10.1007/s40475-020-00201-6 Epub ahead of print. PMID: 32219057; PMCID: PMC7094941. [DOI] [PMC free article] [PubMed]
- 5.Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, et al. The species severe acute respiratory syndrome related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, et al. China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amanat F, Krammer F. SARS-CoV-2 vaccines: status report. Immunity. 2020;52(4):583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jinyong Z, Hao Z, Jiang G, Haibo L, Lixin Z, Quanming Z. Progress and prospects on vaccine development against SARS-CoV-2. Vaccines. 2020;8:153. doi: 10.3390/vaccines8020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He Y, Lu H, Siddiqui P, Zhou Y, Jiang S. Receptor-binding domain of severe acute respiratory syndrome coronavirus spike protein contains multiple conformation-dependent epitopes that induce highly potent neutralizing antibodies. J Immunol. 2005;174(8):4908–4915. doi: 10.4049/jimmunol.174.8.4908. [DOI] [PubMed] [Google Scholar]
- 13.Saif LJ. Coronavirus immunogens. Vet Microbiol. 1993;37:285. doi: 10.1016/0378-1135(93)90030-B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong N, Yang X, Ye L, Chen K, Chan EWC, Yang M, Chen S. Genomic and protein structure modelling analysis depicts the origin and infectivity of 2019-nCoV, a new coronavirus which caused a pneumonia outbreak in Wuhan, China. BioRxiv. 2020.
- 15.Babcock GJ, Esshaki DJ, Thomas WD, Jr, Ambrosino DM. Amino acids 270 to 510 of the severe acute respiratory syndrome coronavirus spike protein are required for interaction with receptor. J Virol. 2004;78:4552. doi: 10.1128/JVI.78.9.4552-4560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin converting enzyme 2. J Biol Chem. 2004;279:3197. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Commun. 2003;312:1159. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia S, Liu M, Wang C, et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchholz UJ, Bukreyev A, Yang L, Lamirande EW, Murphy BR, Subbarao K, et al. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc Natl Acad Sci USA. 2004;101(26):9804–9809. doi: 10.1073/pnas.0403492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Almofti YA, Khoubieb AA, Sahar AG, Salih MA. Multi epitopes vaccine prediction against Severe Acute Respiratory Syndrome (SARS) coronavirus using immunoinformatics approaches. AmerJ Microbiol Res. 2018;6(3):94–114. [Google Scholar]
- 22.Rozhgar AK, Muhamad S, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020:100682. [DOI] [PMC free article] [PubMed]
- 23.Graham RL, Sparks JS, Eckerle LD, Sims AC, Denison MR. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008;133:88–100. doi: 10.1016/j.virusres.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, Li Y, Hu Z, Zhong W, Wang M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020;6:16. doi: 10.1038/s41421-020-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N Engl J Med. 2020. 10.1056/NEJMoa2034577https://www.nejm.org/doi/full/10.1056/NEJMoa2034577?query=RP. [DOI] [PMC free article] [PubMed]
- 27.CDC: Centers for disease control and prevention; different COVID-19 vaccines at https://www.cdc.gov/coronavirus/2019-ncov/vaccines/different-vaccines.html. Updated Dec. 28, 2020 [PubMed]
- 28.Gershoni JM, Roitburd A, Siman DD, Tarnovitsk N, Weiss Y. Epitope mapping the first step in developing epitope-based vaccines. Biodrugs. 2007;21:145–156. doi: 10.2165/00063030-200721030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iurescia S, Fioretti D, Fazio VM, Rinaldi M. Epitope-driven DNA vaccine designemploying immunoinformatics against B-cell lymphoma: a biotech’s challenge. Biotechnol Adv. 2012;30:372–383. doi: 10.1016/j.biotechadv.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 30.Abu-haraz AH, Abd-elrahman KA, Ibrahim MS, Hussien WH, Mohammed MS, et al. Multi epitope peptide vaccine prediction against Sudan Ebola virus using immuno-informatics approaches. Adv Tech Biol Med. 2017;5:203. doi: 10.4172/2379-1764.1000203. [DOI] [Google Scholar]
- 31.Cohen J. COVID-19 vaccine protects monkeys from new coronavirus, Chinese biotech reports posted in: health coronavirus. 2020. [Google Scholar]
- 32.Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK. Peptide vaccine: progress and challenges. Vaccines. 2014;2:515–536. doi: 10.3390/vaccines2030515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lo YT, Pai TW, Wu WK, Chang HT. Prediction of conformational epitopes with the use of a knowledge-based energy function and geometrically related neighboring residue characteristics. BMC Bioinformatics. 2013;14(4):S3. doi: 10.1186/1471-2105-14-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vartak A, Sucheck SJ. Recent advances in subunit vaccine carriers. Vaccines (Basel). 2016;4(2). 10.3390/vaccines4020012. [DOI] [PMC free article] [PubMed]
- 35.Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516–527. doi: 10.1038/s41586-020-2798-3. [DOI] [PubMed] [Google Scholar]
- 36.Moise L, Gutierrez A, Kibria F, Martin R, Tassone R, Liu R, Terry F, Martin B, De Groot AS. iVAX: an integrated toolkit for the selection and optimization of antigens and the design of epitope-driven vaccines. Hum Vaccin Immunother. 2015;11(9):2312–2321. doi: 10.1080/21645515.2015.1061159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yong CY, Ong HK, Yeap SK, Ho KL, Tan WS. Recent advances in the vaccine development against Middle East respiratory syndrome-coronavirus. Front Microbiol. 2019;10:1781. doi: 10.3389/fmicb.2019.01781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graham RL, Donaldson EF, Baric RS. A decade after SARS: strategies for controlling emerging coronaviruses. Nat Rev Microbiol. 2013;11:836–848. doi: 10.1038/nrmicro3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bacchetta R, Gregori S, Roncarolo MG. CD4+ regulatory T cells: mechanisms of induction and effector function. Autoimmun Rev. 2005;4:491–496. doi: 10.1016/j.autrev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 40.Igietseme J, Eko F, He Q, Black CM. Antibody regulation of T-cell immunity: implications for vaccine strategies against intracellular pathogens. Expert Rev Vaccines. 2014;3:23–34. doi: 10.1586/14760584.3.1.23. [DOI] [PubMed] [Google Scholar]
- 41.Rojas M, Restrepo-Jimenez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, Leung PSC, Ansari AA, Gershwin ME, Anaya JM. Molecular mimicry and autoimmunity. J Autoimmun. 2018;95:100–123. doi: 10.1016/j.jaut.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 42.Kanduc D. Peptide cross-reactivity: the original sin of vaccines. Front Biosci. 2012;4:1393–1401. doi: 10.2741/s341. [DOI] [PubMed] [Google Scholar]
- 43.Ojha R, Pareek A, Pandey RK, Prusty D, Prajapati VK. Strategic development of a next-generation multi-epitope vaccine to prevent Nipah virus zoonotic infection. ACS Omega. 2019;4:13069–13079. doi: 10.1021/acsomega.9b00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meza B, Ascencio F, Sierra-Beltrán AP, Torres J, Angulo C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: an in silico approach. Infect Genet Evol. 2017;49:309–317. doi: 10.1016/j.meegid.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 45.Hasan M, Ghosh PP, Azim KF, Mukta S, Abir RA, Nahar J, Hasan Khan MM. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb Pathog. 2019;130:19–37. doi: 10.1016/j.micpath.2019.02.023. [DOI] [PubMed] [Google Scholar]
- 46.Shey RA, Ghogomu SM, Esoh KK, Nebangwa ND, Shintouo CM, Nongley NF, Asa BF, Ngale FN, Vanhamme L, Souopgui J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep. 2019;9(1):4409. doi: 10.1038/s41598-019-40833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandey RK, Ojha R, Aathmanathan VS, Krishnan M, Prajapati VK. Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection. Vaccine. 2018;36:2262–2272. doi: 10.1016/j.vaccine.2018.03.042. [DOI] [PubMed] [Google Scholar]
- 48.Ali M, Pandey RK, Khatoon N, et al. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci Rep. 2017;7:9232. doi: 10.1038/s41598-017-09199-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khatoon N, Pandey RK, Prajapati VK. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep. 2017;7:82–85. doi: 10.1038/s41598-017-08842-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohan T, Verma P, Rao DN. Novel adjuvants & delivery vehicles for vaccines development: a road ahead. Indian J Med Res. 2013;138(5):779–795. [PMC free article] [PubMed] [Google Scholar]
- 51.Solanki V, Tiwari V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci Rep. 2018;8(1):9044. doi: 10.1038/s41598-018-26689-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mohan T, Sharma C, Bhat AA, Rao DN. Modulation of HIV peptide antigen specific cellular immune response by synthetic α- and β-defensin peptides. Vaccine. 2013;31:1707–1716. doi: 10.1016/j.vaccine.2013.01.041. [DOI] [PubMed] [Google Scholar]
- 53.Mohan T, Mitra D, Rao DN. Nasal delivery of PLG microparticle encapsulated defensin peptides adjuvanted gp41 antigen confers strong and long-lasting immunoprotective response against HIV-1. Immunol Res. 2013;58:139–153. doi: 10.1007/s12026-013-8428-5. [DOI] [PubMed] [Google Scholar]
- 54.Yang D, Biragyn A, Kwak LW, Oppenheim JJ. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol. 2002;23:291–296. doi: 10.1016/S1471-4906(02)02246-9. [DOI] [PubMed] [Google Scholar]
- 55.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Web Server issue):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berkmen M. Production of disulfide-bonded proteins in Escherichia coli. Protein Expr Purif. 2012;82(1):240–251. doi: 10.1016/j.pep.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 57.Zhang T, Bertelsen E, Alber T. Entropic effects of disulphide bonds on proteinstability. Nat Struct Biol. 1994;1:434–438. doi: 10.1038/nsb0794-434. [DOI] [PubMed] [Google Scholar]
- 58.Creighton TE. Disulfide bonds as probes of protein folding pathways. Methods Enzymol. 1986;131:83–106. doi: 10.1016/0076-6879(86)31036-X. [DOI] [PubMed] [Google Scholar]
- 59.Molteni M, Gemma S, Carlo R. The role of Toll-Like Receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm Vol. 2016:Article ID 6978936, 9 10.1155/2016/6978936. [DOI] [PMC free article] [PubMed]
- 60.Chen R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol Adv. 2012;30:1102–1107. doi: 10.1016/j.biotechadv.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 61.Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:172. doi: 10.3389/fmicb.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hall TA. Nucleic acids symposium series. London: Information Retrieval Ltd; 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT; pp. c1979–c2000. [Google Scholar]
- 63.Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ponomarenko JV, Bourne PE. Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct Biol. 2007;7:64. doi: 10.1186/1472-6807-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haste Andersen P, Nielsen M, Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006;15:2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55:836–839. doi: 10.1128/JVI.55.3.836-839.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276:172–174. doi: 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- 68.Karplus PA, Schulz GE. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72:212–213. doi: 10.1007/BF01195768. [DOI] [Google Scholar]
- 69.Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25:5425–5432. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- 70.Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE, Nielsen M, Peters B. Immune epitope database analysis resource. Nucleic Acids Res. 2012;40(Web Server issue):W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12:1007–1017. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–WW12. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008;4:2. doi: 10.1186/1745-7580-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang P, Sidney J, Dow C, Mothe B, Sette A. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4:e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dimitrov I, Bangov I, Flower DR, Doytchinova IA. AllerTOP v.2- a server for in silico prediction of allergens. J Mol Model. 2013;20:2278. doi: 10.1007/s00894-014-2278-5. [DOI] [PubMed] [Google Scholar]
- 76.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R. Open source drug discovery consortium, Raghava GP in silico approach for predicting toxicity of peptides and proteins. PLoS One. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tani K, Murphy WJ, Chertov O, Salcedo R, Koh CY, Utsunomiya I, Funakoshi S, Asai O, Herrmann SH, Wang JM, Kwak LW, Oppenheim JJ. Defensins act as potent adjuvants that promote cellular and humoral immune responses in mice to a lymphoma idiotype and carrier antigens. Int Immunol. 2000;12(5):691–700. doi: 10.1093/intimm/12.5.691. [DOI] [PubMed] [Google Scholar]
- 78.Shamriz S, Ofoghi H, Moazami N. Effect of linker length and residues on the structure and stability of a fusion protein with malaria vaccine application. Comput Biol Med. 2016;76:24–29. doi: 10.1016/j.compbiomed.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 79.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zheng Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schäffer AA, Yu Y-K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thomsen M, Lundegaard C, Buus S, Rasmussen M, Lund O, Nielsen M. MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics. 2013. [DOI] [PMC free article] [PubMed]
- 82.Combet C, Blanchet C, Geourjon C, Deléage G. NPS@: network protein sequence analysis. TIBS. 2000;25(3[291]):147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 83.Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43:W174–W181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101:2525–2534. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shin WH, Lee GR, Heo L, Lee H, Seok C. Prediction of protein structure and interaction by GALAXY protein modeling programs. Bio Design. 2014;2(1):1–11. [Google Scholar]
- 86.Ko J, Park H, Heo L, Seok C. Galaxy WEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012;40(W1):W294–W297. doi: 10.1093/nar/gks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lovell SC, Davis IW, Arendall WB, Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and C beta deviation. Proteins Struct Funct Genet. 2002;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 88.Al-Hakim M, Hasan R, Ali MF, Joy R, Marufatuzzahan ZF. In-silico characterization and homology modeling of catechol 1,2 dioxygenase involved in processing of catechol- an intermediate of aromatic compound degradation pathway. Glob J Sci Front Res G Bio-Tech Genet. 2015;15:1–13. [Google Scholar]
- 89.Hebditch M, Carballo-Amador MA, Charonis S, Curtis R, Warwicker J. Protein-Sol: a web tool for predicting protein solubility from sequence. Bioinformatics. 2017;33(19):3098–3100. doi: 10.1093/bioinformatics/btx345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Niwa T, Ying BW, Saito K, Jin W, Takada S, Ueda T, Taguchi H. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc Natl Acad Sci Unit States Am. 2009;106:4201–4206. doi: 10.1073/pnas.0811922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Craig DB, Dombkowski AA. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics. 2013;14:346. doi: 10.1186/1471-2105-14-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yan Y, Zhang D, Zhou P, Li B, Huang SY. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017;45(W1):W365–W373. doi: 10.1093/nar/gkx407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. 2013:8–30. [DOI] [PMC free article] [PubMed]
- 94.Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE. 2010;5(4):e9862. doi: 10.1371/journal.pone.0009862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morla S, Makhija A, Kumar S. Synonymous codon usage pattern in glycoprotein gene of rabies virus. Gene. 2016;584:1–6. doi: 10.1016/j.gene.2016.02.047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets during and/or analyzed during the current study available from the corresponding author on reasonable request.