

ABSTRACT
Pancreatic ductal adenocarcinoma (PDAC) has traditionally been thought of as an immunologically quiescent tumor type presumably because of a relatively low tumor mutational burden (TMB) and poor responses to checkpoint blockade therapy. However, many PDAC tumors exhibit T cell inflamed phenotypes. The presence of tertiary lymphoid structures (TLS) has recently been shown to be predictive of checkpoint blockade response in melanomas and sarcomas, and are prognostic for survival in PDAC. In order to more comprehensively understand tumor immunity in PDAC patients with TLS, we performed RNA-seq, single and multiplex IHC, flow cytometry and predictive genomic analysis on treatment naïve, PDAC surgical specimens. Forty-six percent of tumors contained distinct T and B cell aggregates reflective of “early-stage TLS” (ES-TLS), which correlated with longer overall and progression-free survival. These tumors had greater CD8+ T cell infiltration but were not defined by previously published TLS gene-expression signatures. ES-TLS+ tumors were enriched for IgG1 class-switched memory B cells and memory CD4+ T cells, suggesting durable immunological memory persisted in these patients. We also observed the presence of active germinal centers (mature-TLS) in 31% of tumors with lymphocyte clusters, whose patients had long-term survival (median 56 months). M-TLS-positive tumors had equivalent overall T cell infiltration to ES-TLS, but were enriched for activated CD4+ memory cells, naive B cells and NK cells. Finally, using a TCGA-PDAC dataset, ES-TLS+ tumors harbored a decreased TMB, but M-TLS with germinal centers expressed significantly more MHCI-restricted neoantigens as determined by an in silico neoantigen prediction method. Interestingly, M-TLS+ tumors also had evidence of increased rates of B cell somatic hypermutation, suggesting that germinal centers form in the presence of high-quality tumor neoantigens leading to increased humoral immunity that confers improved survival for PDAC patients.
AbbreviationsTLS: tertiary lymphoid structures; GC: germinal center(s); PDAC: pancreatic ductal adenocarcinoma; RNA-seq: RNA sequencing; BCRseq: B cell receptor sequencing; HEV: high endothelial venule; PNAd: peripheral node addressin; TMB: tumor mutational burden; TCGA: the cancer genome atlas; PAAD: pancreatic adenocarcinoma; FFPE: formalin fixed paraffin embedded; TIME: tumor immune microenvironment.
KEYWORDS: TLS, b cells, pancreatic cancer, immunotherapy, t cells, neoantigens
Background
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer with a high mortality rate due largely to lack of early detection and poor response to cytotoxic therapy. While many other cancer types have benefited from immune checkpoint blockade (ICB), PDAC remains largely resistant to these interventional strategies.1–3 One hypothesis for the failure of ICB in PDAC is that it is a poorly immunogenic tumor characterized by a lack of T cell mediated inflammation. However, recent studies have challenged this hypothesis demonstrating that in some PDAC patients, there is a significant intratumoral cytotoxic T cell infiltration and a high Tumor Inflammation Signature.4–6 Further, patients with greater T cell infiltrate have improved survival, suggesting that T cell immunity is active in some PDAC patients.7 Tumor mutational burden (TMB) is prognostic for response to ICB in lung cancer and melanoma,8 whereas in PDAC, the neoantigen “quality,” defined as the molecular homology to microbial pathogens, is predictive of survival, not the quantity of mutations as defined in TMB.9 Together, these data suggest T cell responses in PDAC tumors exist but may be limited by unique microenvironments in the pancreas, warranting further investigation.
One distinctive tumor immune microenvironmental (TIME)10 feature present in some patients’ tumors are tertiary lymphoid structures (TLS). TLS are lymphocyte aggregates with varying levels of organization reminiscent of lymph node follicles. TLS are located in peripheral tissues, often as the result of infection, autoimmunity, or chronic inflammation.11 Bona fide, mature TLS (M-TLS) contains: lymphocyte clusters containing distinct yet adjacent T- and B cell zones, peripheral node addressin-positive (PNAd+) high endothelial venules, activated dendritic cells present within the T cell zone, expression of the chemokines CCL19, CCL21, CXCL13 and often evidence of B cell class switching, plasma cell differentiation and germinal center (GC) reactions in the B cell follicle.12–14 This contrasts with early-TLS (E-TLS) that primarily consist of T and B cells without this higher order architecture. Previous publications propose a timeline for TLS development in which E-TLS precede M-TLS.12,13 TLS can develop in tumors and correlate with improved disease-free and overall survival in many cancers, including PDAC.14–16 The presence of TLS predicts ICB response in melanoma, sarcoma, and urothelial cancer, and ICB treatment can increase TLS density.17–20 In non-small cell lung cancers, further stratification of survival is seen with GC formation in TLS compared to TLS+ tumors without GC.12 Consistent with this, TLS with significant plasma cell infiltration correlated with improved CD8+ T cell infiltration and overall survival in ovarian cancer.13 Together these data indicate TLS contribute to anti-tumor immunity, however, their functional roles have yet to be determined. Furthermore, it remains unknown whether TLS are formed as a consequence of immunogenic antigen expression or as the byproduct of chronic inflammation. In this study, we characterize the cellular and molecular differences of PDAC tumors with and without TLS in order to address these questions.
Materials and methods
Study cohort
Patients providing for consent for this study were recruited under study protocol PHS IRB #06-108. Demographic data for patients recruited at Providence are indicated below.
| Characteristic | IHC/mIF | RNAseq |
|---|---|---|
| Male – no. (%) | 37 (58.7) | 25 (64.1) |
| Female – no. (%) | 26 (41.3) | 14 (35.9) |
| Age – yr. | 66.7 ± 8.9 | 68.5 ± 9.6 |
| % Caucasian | 90.4 | 97.4 |
| Stage I – no. (%) | 12 (19.0) | 4 (10.3) |
| Stage II – no. (%) | 32 (50.8) | 35 (89.7) |
| Stage III – no. (%) | 19 (30.2) | 0 (0) |
| R0 resection – no. (%) | 54 (85.7) | 33 (84.6) |
| R1 resection – no. (%) | 9 (14.3) | 6 (15.4) |
| E-TLS – no. (%) | 29 (46.0) | 14 (35.8) |
| M-TLS – no. (%) | 9 (14.3) | 6 (15.4) |
This retrospective cohort had only one inclusion criteria, to be diagnosed by the overseeing pathologist as having previously untreated pancreatic ductal adenocarcinoma that was surgically resected. Serial sections from the diagnostic surgical tissue block were cut for both IHC and RNAseq analysis. Further information regarding use of patient samples in these analyses outlined below was cross-referenced according to the recommendations for tumor marker prognostic studies (REMARK).21
Immunohistochemistry
Formalin fixed paraffin embedded (FFPE) pathology sections were selected per the determination of a board-certified pathologist (C.B.) determined to contain sufficient and representative cancer tissue within the resection specimen. Tumor H&E’s were scored, and then serial sections (5 μm) were cut for IHC staining and RNA extraction (see below). PDL1 (clone SP263, Ventana), CD3 (clone SP7, Roche), CD20 (clone L26, Abcam), CD8 (clone SP16, Roche) and GZMB (ab4059, Abcam) were stained as previously described22 and developed using monochromatic DAB (Jackson Immunoresearch).
Image analysis
Single stained sections were digitally scanned using a Leica autoscanner at 20x magnification. Positive cells/mm2 were analyzed using ScanScope software (Leica Biosystems), or Qupath.23 Whole tissue regions (1 section/patient) were selected for analysis by identifying tumor cells surrounded by clear desmoplastic stroma in consultation with a pathologist (C.B.) Adjacent normal and adipose tissue on the periphery was excluded from analysis. These analysis regions also included TLS when present. Hi vs. low cutoffs were determined by median cut-points. E-TLS positivity was determined by recognition of two or more lymphoid aggregates observed to contain both CD20+ B cells and CD3+T cells. For all patients, a range of 4–8 slides were examined/tissue block that were directly serial to the original H&E used for diagnosis and the sample extracted for RNA-sequencing. These sections were used for IHC and/or additional multiplex immunofluorescence where indicated. Additionally, 5 TLS− patients were randomly selected to assess for the possible presence of TLS in 3 different tissue blocks with pathologically defined cancer whereby all of the available H&E slides cut from those blocks was examined for the presence or absence of a TLS (8–13 slides/block). None of these five patients was determined to contain a TLS.
Multiplex immunofluorescence
CD3, CD21 (clone EP3093, Abcam), CD103 (EPR4166 (2), Abcam), CD8 (SP16, Roche), CD20, PNAd (MECA-79, Biolegend), BCL6 (EPR11410-43, Abcam) and Ki67 (SP6, Abcam) were serially stained on germinal center positive sections as determined by H&E assessment. Following 1 h-o/n primary staining, anti-mouse, anti-Rabbit or anti-Rat/HRP conjugated polymers were incubated for 15 min. All sections were washed post primary and secondary staining with TBS-tween with shaking. OPAL TSA dyes 520, 540, 590, 620 and 690 (Perkin Elmer) were then applied for 10 min at RT for fluorescent detection of primary staining. Stained sections were then subjected to serial staining by first stripping the previous antibody with citrate buffer for 15 min at 95 C followed by peroxidase quenching for 15 min with 3% hydrogen peroxide (in 1x PBS). Whenever possible, mouse, rat and rabbit primary antibodies were alternated to ensure degradation of the previous marker detection. Multiplex protocols were validated according to current standards as previously published.24 All histologically determined germinal centers were confirmed to contain strictly confined regions of CD21+ follicular dendritic cells in a field of larger BCL6+ and Ki67+ B cells (CD20+) surrounded by PNAd+ vessels and qualified as mature (M)-TLS. As unstained sections were not available for the TCGA cohort, morphological assessment of H&E images was used to score TCGA patients for M-TLS. Lymphocyte aggregates with clearly defined germinal center reactions in the middle akin to those seen in Figure 4 were assumed to be M-TLS. QuPath software was used to quantitate immunofluorescent images.
Figure 4.
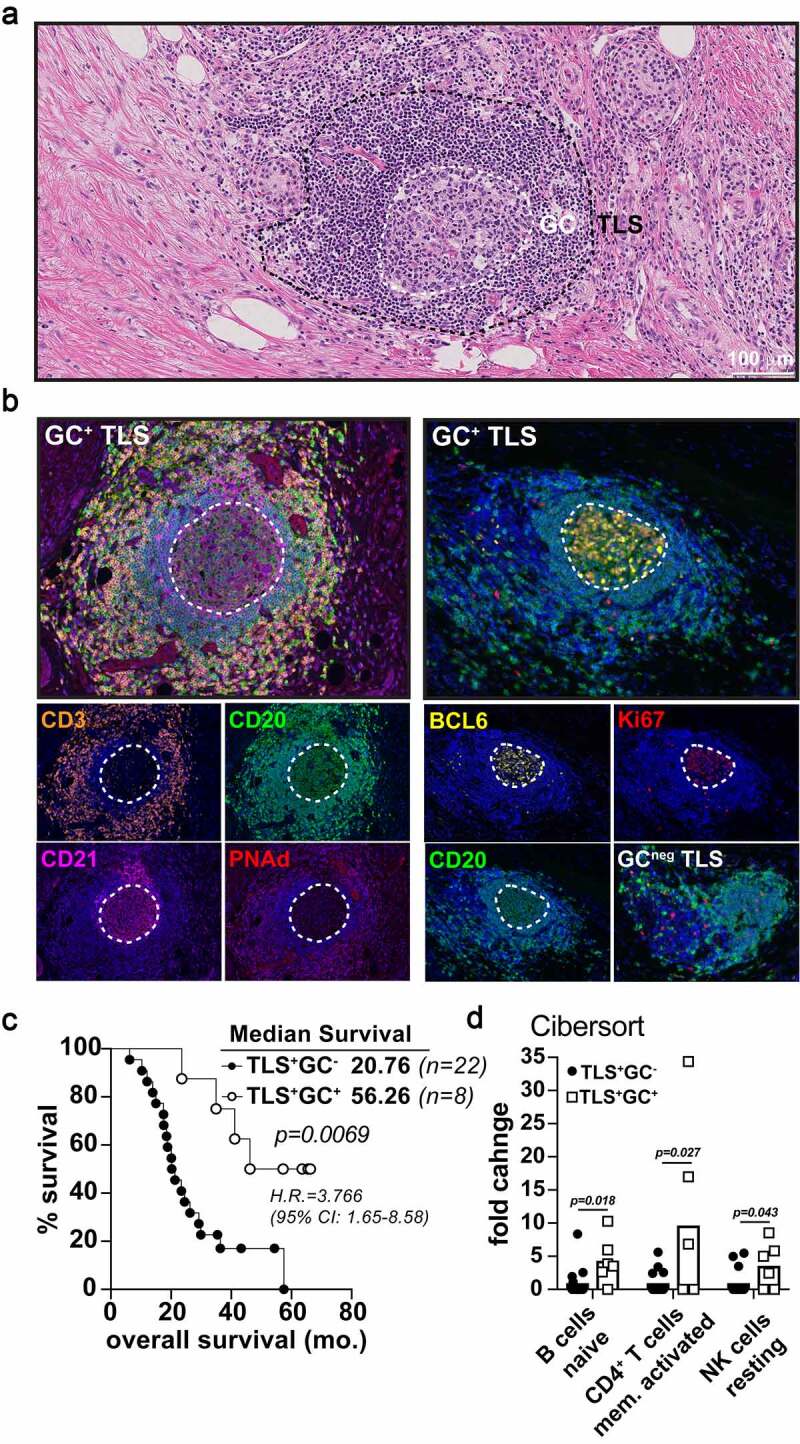
Germinal center reactions identify mature TLS associated long term PDAC survivorship. A-B) Representative 20x H&E micrograph (a) followed by multispectral IHC images (b) demonstrating germinal center marker expression (CD21, BCL6, Ki67) in the midst of CD3 (T cells) peripheral clusters, PNAd+ HEV, and CD20 (B cells) zones. The image on the bottom right in (B) is an example of a GC-negative, early-TLS aggregate lacking BCL6 and Ki67 B cell zones. C) Kaplan-Meier overall survival analysis comparing TLS+GC− patients (n = 20) and TLS+GC+ patients (n = 8) in the Providence upfront resectable cohort. D) CIBERsort analysis on tumor RNA-seq data was performed for both GC+ and GC− groups in the PCI cohort and significant fold enrichment changes are shown for naïve B cells, activated CD4+ T memory cells, and resting NK cells as indicated
IHC and mIF antibodies
| Antigen | Clone | Vendor | Dilution | Species |
|---|---|---|---|---|
| CD8 | SP16 | Abcam | 1/200 | rabbit |
| CD20 | L26 | Abcam | 1/200 | mouse |
| CD3 | SP7 | Abcam | 1/100 | rabbit |
| CD103 | EPR22590 | Abcam | 1/500 | rabbit |
| Ki67 | SP6 | Abcam | 1/200 | rabbit |
| CD21 | EP3093 | Abcam | 1/500 | rabbit |
| PNAd | MECA-79 | Biolegend | 1/500 | rat |
| BCL6 | EPR11410-43 | Abcam | 1/250 | rabbit |
Flow cytometry
Tumor single cell suspensions (TLS− n = 9; TLS+ n = 6) were prepared by enzymatic digestion of tumor samples following surgery as previously described. Briefly, tumors were collected and transported in DMEM media + 1% soybean trypsin inhibitor. Digestion commenced in DMEM + 1 mg/ml collagenase A, 1 mg/ml hyaluronidase, 50 u/ml DNase for 30–45 min. in a 37 C water bath with constant stirring. Digestion was quenched using FCS buffer (1x PBS/1% BSA/2 mM EDTA) and resuspended for FACS staining. Leukocytes were collected from whole blood (TLS− n = 12; TLS+ n = 6) by spinning whole blood collected in EDTA coated tubes (BD Biosciences) at 1500 rpm for 15 min and removing the buffy coat layer. Residual red blood cells were lysed in a hypotonic buffer (Gibco) for 5 min. Antibodies used for FACS staining were incubated with cells for 30 min. at 4 C followed by washing in FCS buffer. Stained cells were acquired on a Cytoflex Flow Cytometer (Beckman Coulter) or BD Fortessa and data was analyzed using FlowJo software.
FACS antibodies
| Antigen | Clone | Fluor | Vendor | Dilution |
|---|---|---|---|---|
| CD45 | HI30 | BV786 | BD Bio | 1/200 |
| CD19 | SJ25 C1 | BUV496 | BD Bio | 1/200 |
| CD20 | 2H7 | BV650 | BD Bio | 1/100 |
| sIgM | G20-127 | BV605 | BD Bio | 1/80 |
| sIgA | 1S11j-8E10 | PerCP-EF710 | Miltenyi | 1/15 |
| sIgG | G18-145 | FITC | BD Bio | 1/20 |
| sIgE | G7-26 | BUV395 | BD Bio | 1/200 |
| sIgD | 1A6-2 | PE-Cy7 | BD Bio | 1/500 |
| CD138 | MI15 | BV510 | BD Bio | 1/30 |
| HLA-DR | G46-6 | PE-CF594 | BD Bio | 1/200 |
| CD40 | 5 C3 | APC-H7 | BD Bio | 1/100 |
| CD27 | M-T271 | BV421 | BD Bio | 1/200 |
| Live/dead 700 | N/A | 700 | BD Bio | 1/10,000 |
RNA-sequencing and analysis
Formalin-fixed, paraffin-embedded (FFPE) serial sections to the H&E sections were deparaffinized, followed by RNA extraction and purification using the Qiagen AllPrep DNA/RNA FFPE kit. Purified RNA was prepared into RNA-seq libraries using RNA Access Library Preparation reagents (Illumina) according to the manufacturer’s instructions. RNA-seq libraries were assessed for quantity and quality by TapeStation (Agilent) and QuBit (Thermo Fisher). Libraries were pooled and sequenced at a depth of 25–50 million reads on a HiSeq 4000 sequencer (Illumina). RNA alignment was performed using STAR alignment to GRCh37(hg19) reference, duplicate reads were marked using Picard’s MarkDuplicate tool, and fragments per kb per million mapped reads (FPKM) values were calculated using cufflinks (Ref GitHub). FPKM levels were used to assess expression of candidate mutations that are identified using whole-exome data. ClustVis was used primarily to create unsupervised clustering heat maps. Details of this method can be found at https://biit.cs.ut.ee/clustvis/and are previously described.25 Volcano plots for differentially expressed genes were created using RStudio. Gene set enrichment analysis was performed GSEA 4.1.0 software from the Broad Institute (UC-San Diego) as previously described.26 Hallmark gene sets used for pathway enrichment analysis were from the Molecular Signatures Database v7.2 (gsea-msigdb.org).
xCell and CIBERsort deconvolution
Two independent publicly available computational methods for immune cell content estimation in tumor samples were utilized: Marker gene-based approach (xCell) and deconvolution-based approach (CIBERsort). Briefly, the analysis was performed as follows:
CIBERsort RNA deconvolution analysis was used to profile tumor immune cells (TILs) using machine learning approach, called support vector regression (SVR) as previously described.27 Bulk RNA from FFPE tissue was used to characterize gene expression profiles based on a validated leukocyte gene signature matrix (LM22) consisting of 22 functionally defined human hematopoietic subsets. The signature matrix file consists of 547 genes that accurately and reproducibly distinguish these 22 hematopoietic populations including seven T cell types, naïve and memory B cells, plasma cells, NK cells and myeloid subsets. To achieve statistical rigor, 100 permutations were performed. Deconvolution results were expressed as relative fractions normalized to 1 (e.g., fractions of total leukocyte content). xCell analysis28 uses the expression values of marker genes (derived from targeted transcriptomics studies characterizing each immune-cell type), where every cell type is quantified independently based on statistical test for enrichment of predefined set of marker genes. As with CIBERSORT, results were normalized to 1 relative to TLS-samples.
Neoantigen prediction pipeline
TCGA-PAAD aligned exome data was downloaded from https://portal.gdc.cancer.gov/. Aligned exome sequences were unaligned from hg38 and realigned to hg19 Human genome reference to be consistent with in-house neoantigen prediction pipeline. Sequence alignment was done using BWA-mem29 followed by GATK best practice to pre-process the aligned reads. Somatic single nucleotide variant (SNV) calling was done using Mutect,30 Somaticsniper,31 Strelka32 and Varscan.33,34 Somatic INDEL calling was done using Strelka and Varscan. Mutations found were merged and further annotated using different annotation sources such as the Catalog Of Somatic Mutations In Cancer (COSMIC), The Exome Aggregation Consortium (ExAC), 1000 Genomes Project, TCGA etc. Any mutation that causes a protein codon change and also observed at mutation allele frequency (MAF)>10% were used to build 25-mer neoantigen peptides. The amino acid change caused by the mutations were placed at 13th amino acid position in the 25-mer neoantigen sequence. Prior to computing HLA-A, B, or C affinity, mutations were further filtered by including only those INDELs that were called by 2 independent programs regardless of MAF. All SNVs were included for further analysis. We used NetMHCpan-4.034 to predict binding affinity of 8–11mer peptides (subset from 25-mer neoantigen peptides) to MHC Class I. Total number of strong (<0.5% rank) and weak binders (0.5–2.0% rank) were considered as total neoantigens predicted. Total strong binding and weak binding neoantigens predicted were correlated with TLS and GC status as described above.
BCR sequencing by TRUST algorithm
B cell receptor sequences were determined from normalized RNAseq data of whole tumor tissue by deployment of the TRUST algorithm as previously described.35,36 Ig heavy chain constant region frequencies were estimated by enumerating the total number of unique B cell clones with a specific Ig isotype sequence and dividing by the total number of unique clones with IGHC predictions. Simpson’s clonality of B cells was determined using the standard equation: Simpson clonality = square root of Simpson’s index where the Simpson’s index = . N = the total number of all B cells and n = the total number of B cells within each clonotype. Somatic hypermutation rate was calculated by adding the total number of somatic hypermutation events (defined by ≤2 amino acid differences/clone) divided by the number of unique clones present in each sample. The percent SHM clonality was determined by adding the number of SHM B cells present in each sample divided by the total number of B cells in each tumor.
TCGA data
The TCGA-PAAD data set (n = 182) was explored and WES and RNAseq data was downloaded via https://portal.gdc.cancer.gov/. Patients that were not ductal adenocarcinoma were removed for a final analysis total of n = 174. The addition of 24 PDAC patients from a previous genomic characterization of this data37 includes 7 previously excluded patients and 17 more samples submitted to TCGA following the 2017 publication. For some exploratory analyses, the cBioPortal for Cancer Genomics was also queried.38 TLS and GC were determined by exploration of digital pathology images of H&E stained sections available for each patient.
Radiomics analysis
Venous phase contrast-enhanced CT scans performed preoperatively were utilized for radiomics analysis. Tumor and ring structures were delineated as previously described by a radiation oncologist using RayDoctor (RaySearch, Stockholm, Sweden). DICOM-RT images and structures were exported into LifeX software for texture analysis as previously described.39
Statistical analysis
Statistical significance was calculated with GraphPad v8.0 software. When comparing two groups in a static timepoint, student’s T tests were used. Three or more groups were analyzed using 1-way Anova with posttest corrections. Only significant p-values are displayed in the figures. The log-rank and Wilcoxon tests were used to compare two groups in a Kaplan-Meier curve. Gene set enrichment analysis is reported as the False Discovery Rate (FDR) as is provided by GSEA 4.1 software (The Broad Institute). Exact p values are only reported for those groups achieving p < .05 unless otherwise indicated. Volcano plot cuts-offs are twofold change and p < .01.
Results
T and B cell aggregation in PDAC tumors correlates with survival
We analyzed a cohort of resectable PDAC patients (N = 63) at our institute who underwent primary tumor surgical resection without prior neoadjuvant therapy. H&E sections of surgical specimens from these patients revealed 29 (46.0%) had at least 2 organized lymphoid aggregates (E-TLS, median 8.5, range 3–39/section) composed of both CD3+ T cells and CD20+ B cells (Figure 1a). Early-TLS were located in various locations throughout the primary tumor, including the margins, tumor center, near adipose tissue and directly adjacent to malignant cells (Figure 1a). CD8+ cells also infiltrated lymphocyte clusters but were primarily positioned in and around the marginal zone away from follicular centers. Patients whose tumors contained putative TLS structures had significantly longer overall survival (Figure 1b, median 26.32 vs. 14.37 months; Log-Rank p = .014, H.R. = 1.96) and disease-free survival (Fig. S1A) compared to patients whose tumors lacked E-TLS, consistent with published data.15 However, in our cohort, the density of TLS did not correlate with overall survival (Fig. S1B) as has been previously reported.15 We and others have previously shown that tumor infiltration of CD3+ T cells and CD8+ T cells is prognostic for outcomes in PDAC patients,7,38 including using this patient cohort.39 In this cohort, CD8+ cell infiltration again was prognostic for overall survival but had a numerically lower median survival than E-TLS+ patients (26.32 vs. 23.43 months) with a noticeable difference in early patient survival following surgery statistically defined the Wilcoxon p-value (Figure 1b-c). Ultimately, however, TLS− and TLS+ patients perished at comparable 5-year survival rates (17% vs. 19% respectively) whereas 20% of CD8hi patients survived past 5 years and 0 CD8low patients survived (Figure 1b-c). Importantly, CD20+ single staining by IHC did not correlate with survival, while patients whose tumors contained high levels of both CD3 and CD20 (CD3hiCD20hi) demonstrated similar survival to patients with high levels of CD3 alone (not significant), suggesting lymphocyte aggregation is distinct from high T and B cell infiltration alone (Figure 1d-e).40 A previously published computed tomography radiomics signature shown to predict tumor T cell infiltrates and ICB response in non-PDAC histologies,39 failed to correlate with CD8 T cell infiltrate or survival in our cohort (Fig. S1C-D), suggesting unique microenvironmental characteristics preside in PDAC tumors compared to other cancer types. The addition of CD8 infiltration to TLS+ patients added no further prognostic value (Fig. S1F). Furthermore, granzyme B+ cells (GZMB) were similar between groups and did not correlate with survival either alone or in combination with CD8+ infiltrate (Fig. S1G, H, I). This suggests E-TLS formation did not influence this aspect of cytotoxic effector function, although this does not exclude alternative methods of tumor cell killing such as expression of cytostatic cytokines.41,42 These data indicate E-TLS are a distinct immunologic predictor of survival in PDAC, unique from assessing T and B cell infiltrates alone.
Figure 1.
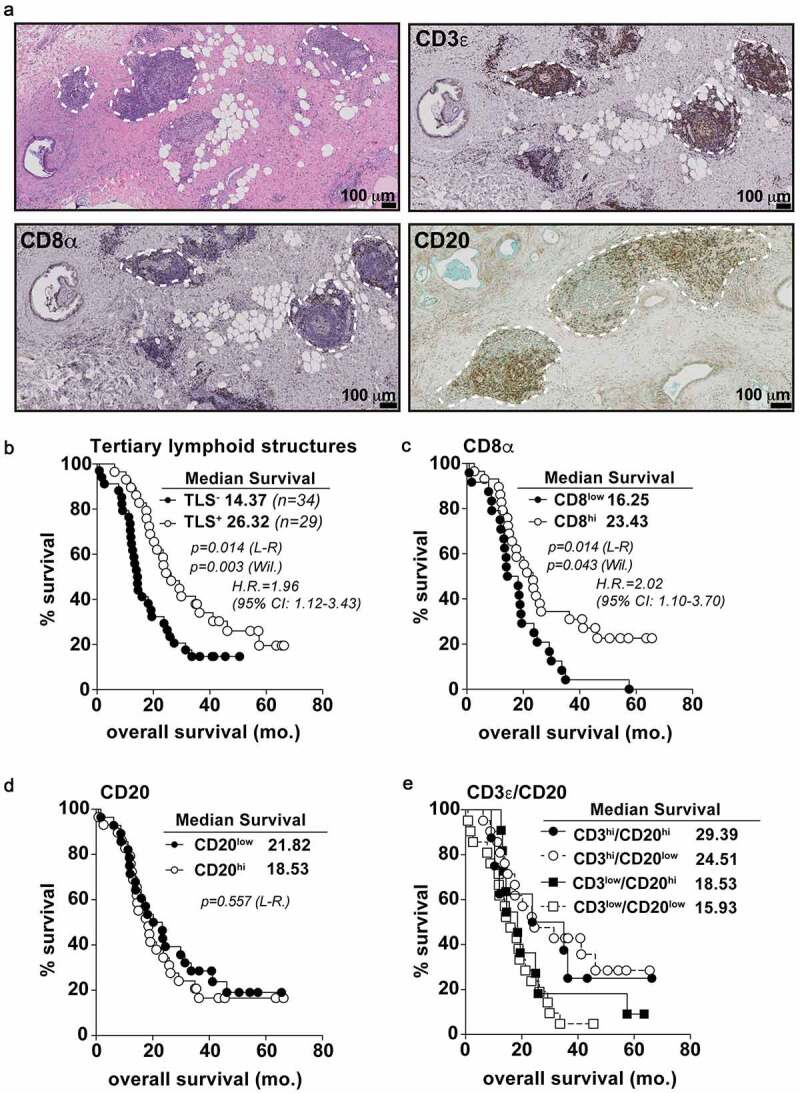
T and B cell aggregates in untreated PDAC tumors predict overall survival. A) Histological micrographs (10x) representative of TLS+ PDAC patients from the PCI cohort. From top left to bottom right: H&E, CD3 IHC, CD8 IHC, and CD20 IHC. TLS regions are outlined in dashed white lines. B) Kaplan-Meier plots displaying overall survival stratified for TLS positivity. Every data point is shown regardless of censor. C-E) CD8 univariate (c), CD20 univariate (d) and CD20/CD3 multivariate (e) analysis for overall survival. Log-rank comparison between CD3hi/CD20hi and CD3hi/CD20low groups non-significant at p = .701. Median cut points were determined for hi vs. low infiltration for all IHC markers. L-R = Log-rank; Wil. = Wilcoxon; H.R. = Hazard ratio; CI: confidence interval. Statistical comparisons were made between all groups. If no p-value is displayed, the comparison was not statistically significant
E-TLS+ PDAC tumors are T cell inflamed with distinctive gene expression patterns
We next evaluated immunophenotypes associated with E-TLS in PDAC tumors by quantifying immune infiltrates by IHC and analyzing transcriptomic signatures from RNA-seq data of whole tumor tissue from FFPE sections as previously described for this patient cohort.43 As expected, tumors with E-TLS contained significantly more CD3+ and CD8+ T cells and CD20+ B cells per/mm2,1 of tumor tissue compared to TLS− tumors (Figure 2a-b). Recent studies in other tumor types have shown that CD103 is a marker of tumor reactive,44 terminally differentiated,45 and/or tissue resident memory46–48 CD8+ T cells that may associate with B cell recruitment in cancer.49 We quantified these cells by immunofluorescent staining for CD103, CD8 and CD3 (Fig. S2A) which demonstrated no difference in the density of CD103+CD8+ T cells in TLS+ tumors (Fig. S2B), but a reduction in the proportion of CD8+ T cells that were CD103+ (Figure 2c). This reduction in CD103-positive CD8+T cells was noted throughout the tumor area as well as directly within lymphocyte conglomerates suggesting no geographical restriction of these cells (Figure 2c). RNA-seq analysis of these tumors showed differentially expressed genes in TLS+ and TLS− sub-types, many of which are associated with T and B cell activity (Figure 2d and S2C). Gene set enrichment analysis revealed significant upregulation of MYC signaling, pathways associated with allograft rejection, and interferon-alpha signaling in TLS+ tumors (Figure 2e). Conversely, TLS− tumors demonstrated gene expression patterns associated with angiogenesis (Figure 2e). Hierarchical clustering based on these gene sets accurately discriminated TLS+ and TLS− tumors (figure 2f). We also performed unsupervised hierarchical clustering using a previously published gene set associated with TLS in melanoma,17 and found these genes failed to group PDAC tumors upon these phenotypic designations, suggesting the process of TLS formation or activity of TLS may differ by tumor type and tissue of origin (Fig. S2D). Interestingly, a recently reported T cell inflamed expression signature43 accurately clustered 14 of 20 TLS+ patients, consistent with the increased T cell infiltrates observed in these tumors (Fig. S2E). These data indicate that TLS formation is associated with greater T and B cell infiltration in PDAC tumors, but unique gene expression patterns accompany early-TLS formation in PDAC.
Figure 2.
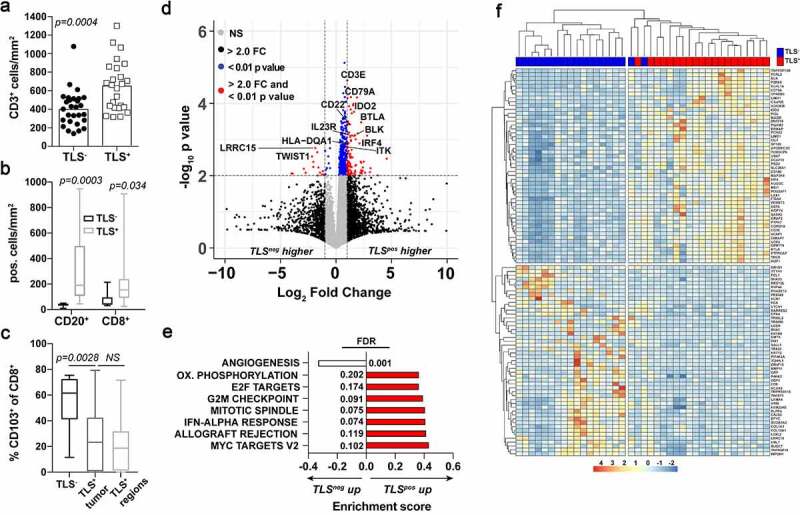
Early stage-TLS+ tumors are T and B cell inflamed with distinctive gene expression signatures. A) CD3+ cell density in TLS+ (n = 30) and TLS− (n = 37) PDAC tumors as determined by IHC. B) CD20+ and CD8+ cell density in surgical tumor sections grouped into TLS+ or TLS− patients as quantified from immunofluorescent imaging. C) Percent positive of CD103+CD8+ cells of total CD8+ cells in TLS+ and TLS− surgical tumor sections. D) Volcano plot comparing differentially expressed genes between TLS+ and TLS− tumors. Cutoffs are indicated by the dashed lines at 2-fold change and p < .01 values. E) Gene set enrichment analysis between TLS+ and TLS− tumors using Hallmark Gene Sets. Shown are the pathways significantly different to a false discovery rate (FDR) of 0.25 or lower. F) Unsupervised hierarchical clustering based on the top 100 most (50 upregulated and 50 downregulated) differentially expressed genes between TLS+ (red) and TLS− (blue) patients
TLS+ tumors are enriched with memory lymphocytes and IgG-switched B cells
To further characterize immune function linked to the presence of TLS, we utilized two previously published RNA deconvolution algorithms, xCell28 and CIBERsort,27 that utilize cell type-specific gene signatures to determine relative enrichments of cell subsets. Both algorithms demonstrated an enrichment in memory B and CD4+ T cells in TLS+ tumors compared to TLS− tumors (Figure 3a-b). In contrast, plasma cells and classical dendritic cells were discordant between deconvolution methods (Fig. S3A-B). To validate these findings using an independent cohort, we utilized the TCGA-pancreatic adenocarcinoma (PAAD, n = 174) dataset. Histological analysis of digital pathology sections allowed for detection of putative TLS, which similarly correlated to a statistically significant benefit in overall survival (Figure 3c). Furthermore, we performed xCell analysis on the TCGA dataset, which demonstrated increased memory B cells, memory CD4+ T cells and classical dendritic cells in TLS+ tumors (Figure 3d and S3C). Together, these data suggest that enhanced adaptive immune memory may be one mechanism by which disease recurrence is limited following surgery in patients whose tumors had TLS.
Figure 3.
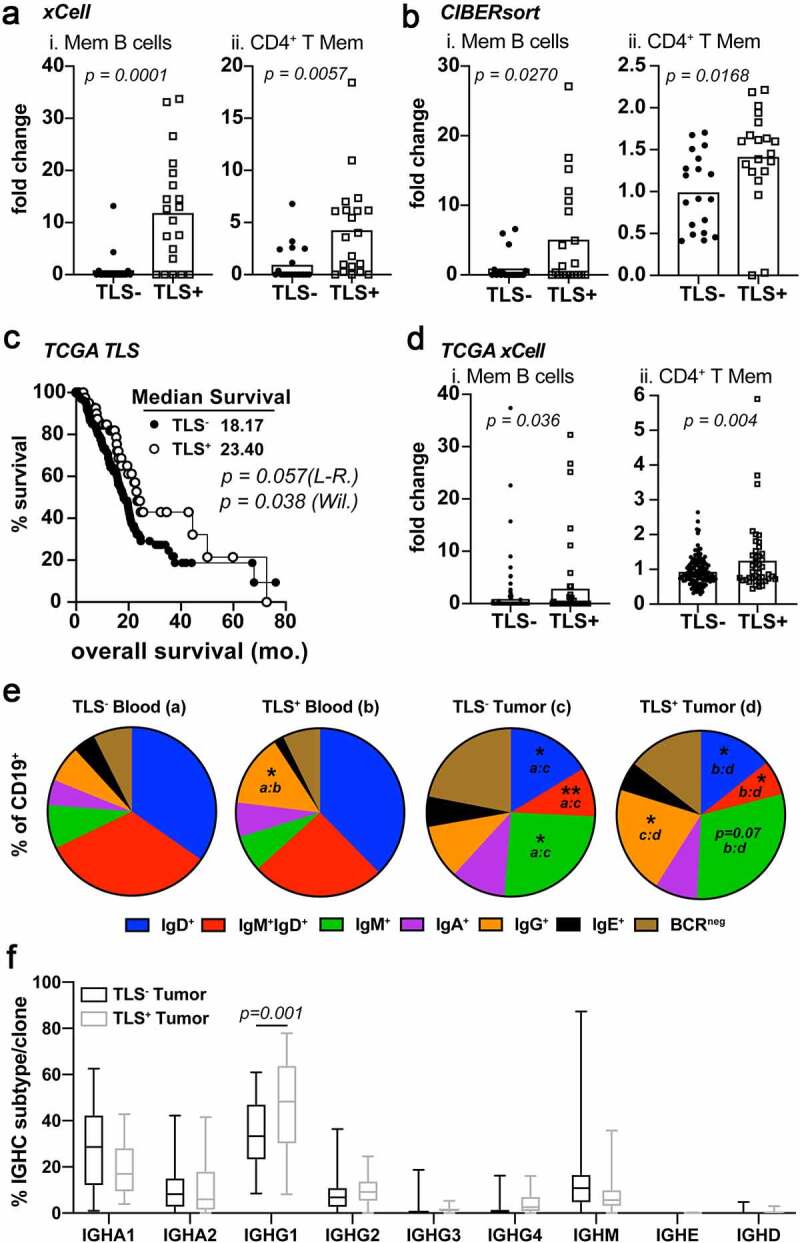
IgG1 class-switched memory B and CD4+ T cells subsets are enriched in TLS+ tumors. A) xCell enrichment scores for Memory B cells (i), CD4+ T memory cells. Deconvolution analysis was generated using TPM normalized RNA-seq data from whole tumor tissue sections. B) CIBERsort enrichment scores for Memory B cells (i), CD4+ T memory cells C) Kaplan-Meier analysis for overall survival and progression free survival stratifying TCGA-PDAC patients (n = 174) based on TLS positivity (TLS+ n = 44). Log-rank and Wilcoxon statistical significance are displayed within the plots. D) xCell enrichment scores for Memory B cells (i) and CD4+ T memory cells (ii) from deconvoluted TCGA RNA-seq data. E) FACS analysis on peripheral blood (groups a and b) and tumors (groups c and d) of PDAC patient samples following surgery. Shown is the mean percentage Ig isotype+ cells of total CD19+ B cells. Statistical significance is indicated within each pie comparing each group as indicated. * p < .05, ** p < .01. F) IGHC analysis from RNA-seq data as determined by the TRUST algorithm. Data is shown as the percent IGHC clone/total clones counted and displayed as box and whisker plots of the mean average and total range of the data. Statistical comparisons were made between all groups. If no p-value is displayed, the comparison was not statistically significant
These alterations in B and CD4+ T cell memory led us to explore more detailed differences in B cell phenotypes. FACS analysis of fresh surgical tissue and matched peripheral blood samples showed a significant increase in tumor-infiltrating IgM+ B cells and a significant decrease in tumor-infiltrating IgM+IgD+ and IgD+ B cells compared to the blood (Figure 3e). Immature B cells express only IgD-isotype B cell receptors and mature, naïve B cells co-express IgM and IgD50 so these data indicate PDAC tumor-resident B cells are enriched for antigen-experienced phenotypes (Figure 3e). Additionally, an increased percentage of B cells with surface IgG expression was observed in tumors and peripheral blood from TLS+ patients (Figure 3e and S3E). Plasmablasts (CD138−CD20−HLADR+) and plasma cells (CD138+CD20−HLADRlo/-) did not change between TLS+ and TLS− tumors and matched peripheral blood samples consistent with the RNA deconvolution subset analysis (Fig. S3A-D). B cell receptor (BCR) sequences determined by TRUST algorithm analysis in our retrospective cohort showed an enrichment in IgG1 subclass heavy chain constant regions in TLS+ tumors (figure 3f). As isotype class switching is an irreversible event driven by antigen engagement of the BCR on naïve B cells, this suggests B cells in TLS+ patients preferentially undergo IgG1 class switching upon antigen recognition. Taken together, these data demonstrate that TLS+ PDAC tumors are enriched with memory CD4+ T cells and memory IgG1 class-switched memory B cells.
Germinal center formation occurs in patients with prolonged survival
Given the increased memory B cell activity in TLS+ PDAC tumors, we sought to further investigate these tumors for evidence of immunologic activity to explain the improved survival observed. In other tumor types, the presence of germinal centers marks, mature TLS (M-TLS) and correlates with long-term survival.12,13 In our PDAC cohort, 12.7% (n = 8) of tumors possessed germinal centers identified both histologically and by IHC evaluation. Germinal centers were defined by clearly marginated borders of large centroblasts in the middle of a tightly compacted TLS with a distinct compartmentalization of CD3+ T cells and CD20+ B cells (Figure 4a-b) and expressing markers of follicular dendritic cells (CD21), and PNAd+ high endothelial venules (HEV) (Figure 4b). B cells contained within the germinal center were highly proliferative (Ki67+) and expressed the gene BCL6, the master transcription factor of germinal center B cells51 (Figure 4b). Patients whose tumors were TLS+ with germinal centers survived significantly longer following surgery versus those with lymphocyte aggregates that lacked germinal centers (56.3 vs. 20.8 months, Log-Rank p = .007; Figure 4c). The 5-year survival rate for patients with GC+ tumors was almost 50%; 4 of which had stage I local disease and 4 had stage II/III regional disease. Tumors with germinal centers did not have significantly increased densities of CD3+, CD8+, or GZMBhi expressing cells (Fig. S4A). Furthermore, no differences were observed in immunosuppressive cells or markers such as PD1/PDL1 expression, CD68+ type 1 or 2 macrophages, or regulatory T cells (Fig. S4B-G). There was also no difference observed in the fibrotic desmoplastic reaction (Fig. S4H-J). However, using the CIBERsort deconvolution algorithm, M-TLS+ tumors were enriched with naïve B cells, activated CD4+ memory T cells and resting NK cells suggesting improved humoral and NK cell immunity in these patients (Figure 4d). M-TLS+ tumors also expressed significant enrichment in pathways related to reactive oxygen species (ROS), MTORC1 signaling and oxidative phosphorylation among others (Fig. S4L). In contrast, we observed a significant downregulation in TGF-beta signaling in GC+ tumors (Fig. S4L-N), a feature strongly associated with immunotherapy-responsive tumors.52–56 These data indicate germinal center reactions within TLS correspond with enhanced features of humoral immunity and diminished TGF-beta signaling associated with improved patient survival in PDAC.
High and low affinity neoantigens are increased in TLS+GC+ patients linked to increased B cell somatic hypermutation
Recent publications have established the link between high tumor mutational burden (TMB) and checkpoint blockade responsiveness in numerous cancer types.8 However, high TMB and T cell inflamed tumors do not correlate in untreated cancer patients.57 In PDAC, TMB quantity does not associate with ICB responsiveness or survival, but rather with the likelihood a mutated antigen induces a T cell response.9 Therefore, we evaluated whether TLS develop in PDAC tumors due to a high TMB and/or expression of de novo neoantigens. Interestingly, tumors with any stage TLS from the TCGA cohort had a significantly lower TMB compared to TLS− tumors (Figure 5a). Survival was similar in patients with high or low TMB, suggesting TMB is not associated with a more favorable immune status in PDAC (Figure 5b), as has been shown for melanoma and soft-tissue sarcoma.17–19,57 We next evaluated the affinity of potential neoantigens for MHC binding to assess whether the quality of neoantigens differed in tumors with TLS. We utilized an in silico neoantigen prediction pipeline where nonsynonymous SNV mutations were filtered at >10% variable allele frequency and combined with all insertions and deletions from each tumor. We then tested the predicted HLA binding affinity using NetMHCpan v4.0 software for the corresponding peptides. This computational pipeline was adopted from one used at our institution to identify tumor antigen-reactive T cells for adoptive T cell therapies.58 TLS+GC+ tumors demonstrated a significant increase in the total number of predicted high affinity and low affinity mutant peptides that bind human leukocyte antigen-A and B (HLA-A, HLA-B) MHC Class I molecules compared to TLS− or TLS+GC− tumors (Figure 5c-D). Interestingly, stratifying patients by HLA-A high- or low-affinity antigens alone did not associate with overall survival (Fig. S5A-B) suggesting quality of neoantigens alone in the absence of TLS did not drive immune-mediated tumor control. Patients expressing a high number of potential high affinity HLA-A antigens demonstrated a significant enrichment in effector memory CD4+ but not CD8+ T cells (Fig. S5C). To link these genetic changes to patterns in the intratumoral B cell repertoire, we performed B cell receptor sequencing using the TRUST algorithm35 on total tumor RNA-seq data. These analyses revealed a numerical decrease in B cell clonality in GC+ tumors but did not meet statistical significance (Figure 5e). In secondary lymphatic organs, germinal center formation is a requirement for B cell somatic hypermutation and affinity maturation during immunological priming and recall responses.59 This process generates new B cell clones from lower affinity progenitors by somatic BCR mutagenesis. By comparing BCR sequences with ≤2 amino acid differences between pair sequences we determined there was a significant increase in the rate of somatic hypermutation in B cells from E-TLS+ and M-TLS+ tumors (figure 5f). However, these mutated clones made up a significantly reduced proportion of the entire B cell repertoire in all TLS+ tumors (Fig. S5D). This is consistent with the concept that most of the mutated clones possess lower antigen affinity following somatic hypermutation prior to affinity maturation and will be eventually negatively selected.50 These data indicate germinal centers in primary tumors may provide a microenvironmental niche whereby increased B cell activity is linked to higher expression of neoantigens, and enrichment of memory CD4+ T cells in untreated pancreatic cancer patients with long-term survival.
Figure 5.
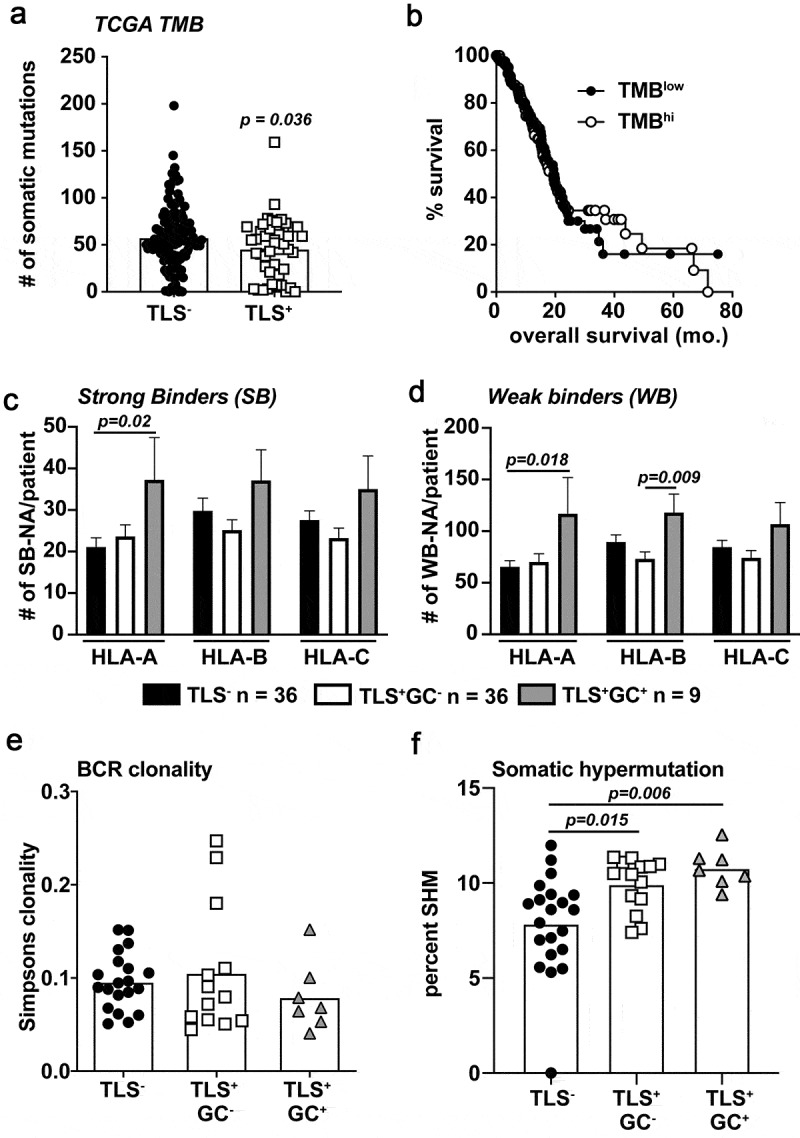
Predicted HLA binding neoantigens are increased in M-TLS+ PDAC patients accompanied by increased rates of B cell somatic hypermutation. A) TCGA tumor mutation burden between TLS− and TLS+ patients from the TCGA-PAAD dataset. B) Kaplan-Meier curve of TMBhi vs. TMBlow determined on a mean cut-point was used to stratify TCGA-PAAD patients for overall survival. C) Predicted strong binding (rank<0.5%) neoantigens/patient partitioned into HLA-A, B, and C specific peptides using a proprietary neoantigen prediction pipeline in conjunction with HLA-peptide affinity predictions determined by NetMHCpan version 4.0 algorithms. D) Predicted weak binding (rank 0.5–2%) neoantigens/patient partitioned into HLA-A, B, and C-specific peptides. E) BCRseq analysis as determined by the TRUST algorithm of tumor RNA-seq data from samples of TLS−, TLS+GC−, and TLS+GC+ patients. Shown is the calculated Simpsons clonality score for each patient. F) The rate of BCR somatic hypermutation is shown calculated by the number of unique mutated B cell clones per total unique B cell clones in each patient tumor. Statistical comparisons were made between all groups. If no p-value is displayed, the comparison was not statistically significant
Discussion
The data reported here provide a deeper understanding of tumor immunity and TLS biology in treatment naïve pancreatic ductal adenocarcinoma. We report that PDAC tumors with lymphocyte aggregates (“early-TLS”) are significantly enriched for CD8+ T cells, memory B cells, IgG1 class switching, and memory CD4+ T cell subsets. These data suggest that improved immune memory is a plausible explanation for the increased survival observed following surgical resection of the primary tumor and the TLS residing in these specimens. However, the mechanism behind how TLS foster immune memory is currently unknown. Evidence is emerging that TLS may support the recruitment or retention of TCF-1 expressing CD8+ T cells with memory precursor phenotypes that are reservoirs for differentiation into effector memory and exhausted cells.17,60,61 Our finding that total CD8+ T cells are increased in TLS+ tumors but the frequency of tissue-resident memory CD103+CD8+ T cells is decreased suggest TLS may support CD8+ T cells with other phenotypes, such as TCF-1+ memory precursor cells. In turn, this pool of progenitor T cells can provide a renewable source to quickly reactivate an anti-tumor immune response following antigen re-exposure or therapeutic intervention as has been shown in other contexts.17,61–63 We submit that the presence of TLS provides a microenvironmental niche in which to promote systemic T cell memory and long-lived humoral immunity. Consequently, these memory lymphocytes could improve the patients’ systemic immune surveillance and thwart local or distant recurrence. Indeed, other groups have shown that the presence of ectopic (a.k.a. tertiary) lymphoid organs in the lungs provides protection against influenza rechallenge, in animals deficient of a spleen or lymph nodes.64 Additionally, murine melanomas over-expressing lymphotoxin-alpha generate TLS subcutaneously and mount effective anti-tumor immunity.65 Another recent study using multiple murine breast cancer models reported that CD4+ T follicular helper cells were required for the production of tumor cell reactive immunoglobulins which mediated checkpoint blockade efficacy but only in cancer cell lines with high mutational burden.66 Our results in human PDAC are consistent with these findings. We conclude that patients who lack TLS have suboptimal immune memory and therefore are more susceptible to cancer relapse following surgery.
The limitations of our study include using mostly archived samples which prevents validation of the predicted biology using ex vivo experiments. Nonetheless, we attempted as much fresh tissue analysis as was afforded us (Figure 3). Ideally, much of these predictions and observations could be tested utilizing an authentic TLS mouse model. Unfortunately, few of these animal models exist and are publicly available. We intend on pursuing these models in subsequent studies. Most of our pathological analysis is presented as univariate analysis which limits confidence in the prognostic value of any single variable. However, the previously aforementioned study by Hiraoka et al. did perform multivariate analysis for all their PDAC patients which validates TLS as an independent prognosticator for PDAC.15 Additionally, a multivariate analysis was previously performed for this cohort of patients and found that CD3+ T cell infiltrate was independently associated with survival.67 Together these data support a role for immune activity in patient survival. Finally, our results are limited in the analysis of why tumors with more MHCI-neoantigens are associated with enhanced humoral immune function. This increase in MHC-I neoantigen expression was also linked with an enrichment in CD4+ effector memory cells (Fig. S5) and a complimentary increase in the rate of B cell somatic hypermutation (Figure 5). As the relationship between CD4+ T and B cell activity is governed by MHCII-restricted antigen presentation,68 this is an interesting finding for future exploration. However, other studies report similar observations that MHCI-antigen responses correspond to superior anti-tumor B cell activity, frequently with IgG-switched BCR.13,19,49,69–71
How and why TLS develop in some patients but not others remains an active field of study. It is not known if TLS-associated transcriptomes are common to all cancer types or if tissue specificity can influence gene expressions patterns and immunological outcomes. Our attempts at reproducing hierarchical clustering using gene expression signatures common to TLS+ patients with other cancer types was unfruitful.17,49,72 However, not all TLS manifest with the same histological architecture and phenotypic features and can vary in density, size and microanatomical localization.73,74 Some have proposed this may represent variable stages of TLS development in the midst of an ongoing maturation process75 and indicate diversity in the immune microenvironment of the tumor. Alternatively, the presence of a TLS may not generate uniform immunological phenotypes but instead be a reservoir for anergic, terminally differentiated, exhausted or antigen-independent T cell activation. It has been well established by other groups that chronic inflammation is a critical factor for TLS neogenesis11 and perhaps may be sufficient for formation in certain contexts.76 However, lymphoid tissue inducer and organizer cells and follicular dendritic cells (FDC) do not naturally exist in peripheral tissues so they must differentiate from preexisting progenitor cells to establish TLS; a process which requires lymphotoxin beta receptor (LTBR) signaling.77 In turn, these newly formed FDC initiate HEV formation and CXCL13-regulated B cell recruitment. Interestingly, type 1 interferons are necessary but not sufficient for inducing lung TLS during influenza infection in mice76 and are potentiated by LTBR agonism.78 As we observed upregulation of type 1 interferon response genes in E-TLS+ patients (Figure 2e), we surmise these pathways may be functional in PDAC as well. It is noteworthy, however, that upregulation of any of these particular pathways may be transient and subsequently downregulated to permit further immunological adaptation such as is seen with type 1 IFN signaling in the E-TLS vs. M-TLS context (Fig. S4L). Finally, results in Fig. S4 exhibit the proportions of various immunosuppressive cell types, such as Tregs, M2 macrophages, and reactive fibroblasts that commonly populate the stroma of pancreatic as well as many other tumors types. While we did not observe any discernable differences in these subsets between TLS+ and TLS− patients, we cannot presently exclude significant alterations in function that could account for the genesis or persistence of TLS. However, the significant downregulation of TGF signaling pathways in GC+ tumors points to genes in this set having a strong association with immunological activity as we and others have previously shown in pancreatic and other solid GI malignancies.55,56,79,80
Despite the positive association of T and B cell clusters with median overall survival in PDAC patients (14 vs. 26 months), the 5-year survival rate remains relatively unchanged for either cohort (~17-19%). However, we identified a smaller subset of patients with germinal center reactions (Figure 4). These follicles expressed GC-specific markers and bore the distinct histological architecture of these microscopic structures indicating these were bonafide M-TLS (Figure 4b). Furthermore, we observed spatially restricted BCL6, CD21, and Ki67 expression in CD20+ zones (Figure 4b) which is a hallmark of germinal center B cells undergoing somatic hypermutation and affinity maturation as has been observed in other cancer types.12,16,81 Overall survival for these patients was 56 months with 37.5% of them surviving past 5 years (Figure 4c); a marked improvement over expected survival rate for these disease stages, despite receiving only standard of care adjuvant-chemotherapy. These patients’ tumors were enriched for subsets of naïve B cells, activated memory CD4+ T cells and NK cells (Figure 4d) but not for CD8+ T or GZMB+ cells (Fig. S4A). While this does not preclude the contributions of cell mediated tumor control, we hypothesize humoral immune function serves a complementary role in preventing disease recurrence in patients with M-TLS+ tumors. Additionally, preferential IgG1 class switching in TLS+ patients can support increased NK cell effector function through antibody dependent cell-mediated cytotoxicity of antibody coated tumor cells,82,83 increased recruitment of cDC1 cells (Fig. S3),84 and correlates with improved survival in melanoma and KRAS-mutated lung cancer patients.36,85 Our data would suggest this caliber of preexisting immunity would portend favorable responses to immunotherapy if PDAC patients with M-TLS+ tumors relapse after surgery.86,87
We also asked if the presence of an immunogenic antigen, in the context of chronic inflammation, was associated with specific aspects of TLS biology and survival. Interestingly, only M-TLS+ patients had a significantly increased number of predicted high and low-affinity neoantigens, specifically for HLA-A and HLA-B (Figure 5). Neither tumor mutational burden nor the increase in neoantigens alone were prognostic for survival (Figure 5b and S5). These data reflect the aforementioned study by Balachandran et al. that demonstrated the total number of possible antigens was not prognostic of disease free survival in PDAC patients but rather the neoantigen “quality” combined with high T cell infiltration was significantly associated with survival.9 These data may also explain why an allogenic whole tumor cell vaccine expressing mesothelin stimulated M-TLS formation and HLA-A restricted peptide responses in 33 of 39 patients in a stage II trial for PDAC patients despite 100% of the patients deemed TLS-negative prior to vaccine intervention.5 Collectively, our data presented here support a model where germinal center formation in M-TLS and high neoantigen expression stimulate cooperation between cell-mediated and humoral immune memory to suppress disease progression leading to longer survival for PDAC patients following surgery. For an immunotherapy unresponsive tumor type with a high mortality rate, these are novel concepts that should have obvious clinical traction.
Supplementary Material
Acknowledgments
We would like to thank members of the Providence HPB surgical team and the Integrated therapies laboratory not listed in the author list that contributed critical data and sample curation toward the completion of this study. We would also like to thank Dr. Walter Urba for critical reading of this manuscript. The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding Statement
A.J.G. was supported in part by the Collins Medical Trust of Oregon. K.H.Y. is funded by the 2016 Sydney Kimmel Foundation Translational Research Scholar Grant. M.R.C. and M.J.G. supported this study through grants: NIH R01 CA182311 (M.J.G.) and NIH R01 CA208644 (M.R.C.);Collins Medical Trust;National Institutes of Health [NIH RO1 CA182311];National Institutes of Health [NIH RO1 CA208644];Adaptive Biotechnologies;Susan G. Komen;
Competing Interests
A.J.G. declares no competing interests. K.H.Y. is the principal investigator of clinical trial NCT02688712 sponsored by Providence Health and Services in collaboration with Eli Lilly and Company. M.J.G. and M.R.C. have research support from Mavupharma, Bristol Myers Squibb, Nanobiotix, and Jounce that is not directly related to the subject of this manuscript. The funders had no part in the subject or content of this manuscript.
Authors’ Contributions
Conceptualization - A.J.G. and K.H.Y.; Methodology - A.J.G., K.H.Y., C.D., B.P., V.R., B.C., M.J.G, T.M., M.S., E.T., J.P.; Board Pathologist – C.B.; Software - C.D., B.P., M.S., V.R., M.J.G., J.P, E.T., A.J.G.; Validation – A.J.G., V.R.; Formal Analysis – A.J.G., M.S., V.R., B.P., C.D., J.P., C.B., K.H.Y.; Investigation – A.J.G, K.H.Y., V.R., B.P., M.S., B.C., J.P., T.M., K.M., M.P., C.D., E.T., M.J.G.; Resources – C.B., P.N., P.H., E.T., B.C., M.R.C., M.J.G., K.H.Y.; Data Curation – M.J.G., K.H.Y., C.B., E.T., A.J.G., V.R.; Writing – A.J.G, K.H.Y.; Visualization – A.J.G., K.H.Y., B.P., M.S.; Supervision – A.J.G., K.H.Y., M.J.G, M.R.C.; Project Administration – A.J.G., K.H.Y., M.J.G., M.R.C.; Funding acquisition – A.J.G., K.H.Y., M.J.G., M.R.C.
Ethics approval and consent to participate
Sample collection from this study was approved by the institutional review board at Providence Portland Medical Center, Portland, OR with the study ID numbers PHS: 06-108 and PHS 10-037. All patients provided informed consent for participation in this study.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding and last author on reasonable request.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Hilmi M, Bartholin L, Neuzillet C.. Immune therapies in pancreatic ductal adenocarcinoma: where are we now? World J. Gastroenterol. 2018;24(20):2137–16. doi: 10.3748/wjg.v24.i20.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie C, Duffy AG, Brar G, Fioravanti S, Mabry-Hrones M, Walker M, Bonilla CM, Wood BJ, Citrin DE, Gil Ramirez EM, et al. Immune checkpoint blockade in combination with stereotactic body radiotherapy in patients with metastatic pancreatic ductal adenocarcinoma. Clin Cancer Res clincanres. 2020;3624:2019. doi: 10.1158/1078-0432.ccr-19-3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Reilly EM, Oh DY, Dhani N, Renouf DJ, Ah Lee M, Sun W, Fisher G, Hezel A, Chang SC, Vlahovic G, et al. Durvalumab with or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: a Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019;5(10):1431–1438. doi: 10.1001/jamaoncol.2019.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, Wallden B, Marincola FM, and Cesano A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J Immunother Cancer. 2018;6(1):1–17. doi: 10.1186/s40425-018-0367-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A, et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunol. Res. 2014;2(7):616–631. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorsson VV, Gibbs DL, Brown SD, wolfe D, Bortone DS, Ou Yang TH, Porta-Padro E, Gao GF, Plaisier CL, Eddy JA, et al. The Immune Landscape of Cancer. Immunity. 2018;48:812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carstens JL, de Sampaio PC, Yang D, Barua S, Wang H, Rao A, Allison JP, LeBleu VS, and Kalluri R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. 2017;8(15095). doi: 10.1038/ncomms15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019;51(2):202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):S12–S16. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Mera M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sautès-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, and Dieu-Nosjean MC. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. 2016;7:1–11. doi: 10.3389/fimmu.2016.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silina K, Soltermann A, Attar FM, Casanova R, Uckeley ZM, Thut H, Wandres M, Isajevs S, Cheng P, Curioni-Fontecedro A, et al. Germinal Centers Determine the Prognostic Relevance of Tertiary Lymphoid Structures and Are Impaired by Corticosteroids in Lung Squamous Cell Carcinoma. Cancer Res. 2018;78(5):1308–1321. doi: 10.1158/0008-5472.CAN-17-1987. [DOI] [PubMed] [Google Scholar]
- 13.Kroeger DR, Milne K, Nelson BH.. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res. 2016;22(12):3005–3015. doi: 10.1158/1078-0432.CCR-15-2762. [DOI] [PubMed] [Google Scholar]
- 14.Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH.. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014;35(11):571–580. doi: 10.1016/j.it.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 15.Hiraoka N, Ino Y, Yamazaki-Itoh R, Kanai Y, Kosuge T, and Shimada K. Intratumoral tertiary lymphoid organ is a favourable prognosticator in patients with pancreatic cancer. Br J Cancer. 2015;112(11):1782–1790. doi: 10.1038/bjc.2015.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, Lepelley A, Becht Et, Katsahian S, Bizouard G, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 2014;189(7):832–844. doi: 10.1164/rccm.201309-1611OC. [DOI] [PubMed] [Google Scholar]
- 17.Cabrita R, Laus M, Sanna A, Donia M, Larsen MS, Mitra S, Johansson I, Phung B, Harbst K , Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 18.Petitprez F, de Reynies A, Keung EZ, Wei-Wu Chen T, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougouin A, et al. B cells are associated to sarcoma survival and immunotherapy response. Nature. 2020;577(7791):556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 19.Helmink B, Reddy SM, Gao J, Zhang S, basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Navai N, Alhalabi O, Siefker-Radtke A, Campbell MT, Tidwell RS, Guo CC, Kamat AM, Matin SF, Araujo JC, et al. Neoadjuvant PD-L1 plus CTLA-4 blockade in patients with cisplatin-ineligible operable high-risk urothelial carcinoma. Nat. Med. 2020;26(12):1845–1851. doi: 10.1038/s41591-020-1086-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting recommendations for tumor marker prognostic studies (REMARK): explanation and elaboration. PLoS Med. 2012;9(5):e1001216. doi: 10.1371/journal.pmed.1001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffel B, Gorjestani S, Liudahl SM, Truitt M, Olson P, et al. Bruton tyrosine kinase–Dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6(3):270–285. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7(1):1–7. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, Lugli A, Zlobec I, Rau TT, Berger MD, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;1–12. doi: 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 25.Metsalu T, ClustVis: VJ. A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43(W1):W566–W570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillete MA, Pzaulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aran D, Hu Z, Butte A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.H. L Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. (2013).
- 30.Cibulskis K, Lawrence MS, Scarter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31(3):213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson DE, Harris CC, Chen K, Koboldt DC, Abbot TE, Dooling D, Ley TJ, Mardis ER, Wilson RK and Ding L. Somaticsniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 33.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK.VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, and Nielsen M. NetMHCpan-4.0: improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol. 2017;199(9):3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu X, Zhang J, Wang J, Fu J, Li T, Zheng X, Wang B, Gu S, Jiang P, Fan J, et al. Landscape of B cell immunity and related immune evaision in human cancer. Nat. Genet. 2019;51(3):560–567. doi: 10.1038/s41588-018-0339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolotin D, Poslavsky S, Davydov ANm, Frenkel FE, Fanchi L, Zolotareva OI, Hemmers S, Ptuintseva EV, Obraztsova AS, Shugay M, et al. Antigen receptor repertoire profiling from RNA-seq data. Nat. Biotechnol. 2017;35(10):908–911. doi: 10.1038/nbt.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raphael BJ, The Cancer Genome Atlast Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BGE, Sumer SO, Aksoy BA, Jacobsen A, Byrne C, Heurer ML, Larsson E, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun R, Limkin EJ, Vakalopoulou M, Dercel L, Champiat S, Han SR, Verlingue Lo, Brandao D, Lancia A, Ammari S, et al. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: an imaging biomarker, retrospective multicohort study. Lancet Oncol. 2018;2045:1–12. [DOI] [PubMed] [Google Scholar]
- 40.Castino GF, Cortese N, Capretti G, Serio S, Di Caro G, Mineri R, Margini E, Grizzi F, Cappello P, Novelli F, et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology. 2016;5(4):1–14. doi: 10.1080/2162402X.2015.1085147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vredevoogd DW, Kuilman T, Ligtenber MA, Boshuizen J, Stecker KE, de Bruijn B, Krijgsman O, Huang X, Kenski JCN, Lacroix R, et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell. 2019;178(3):585–599.e15. doi: 10.1016/j.cell.2019.06.014. [DOI] [PubMed] [Google Scholar]
- 42.Braumüller H, Wieder T, Brenner E, Abmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494(7437):361–365. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 43.Bernard B, Rajamanickam V, Dubay C, Piening B, Alonso E, Jutric Z, Tang E, Newell P, Hansen P, Medler T, et al. Transcriptional and immunohistological assessment of immune infiltration in pancreatic cancer. PLoS One. 2020;15(8):1–19. doi: 10.1371/journal.pone.0238380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermoot JE, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. 2018;9(1). doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amsen D, Van Gisbergen KPJM, Hombrink P, Van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. 2018;19(6):538–546. doi: 10.1038/s41590-018-0114-2. [DOI] [PubMed] [Google Scholar]
- 46.Park SL, Buzzai A, Rautela J, Hor JL, Hockheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature. 2019;565(7739):366–371. doi: 10.1038/s41586-018-0812-9. [DOI] [PubMed] [Google Scholar]
- 47.Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, Cauley LS, Craft J, and Kaech SM. CD4+ T Cell Help Guides Formation of CD103+ Lung-Resident Memory CD8+ T Cells during Influenza Viral Infection. Immunity. 2014;41(4):633–645. doi: 10.1016/j.immuni.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mackay LK, Wynne-Jones E, Freestone D, Pellici DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, et al. T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity. 2015;43(6):1101–1111. doi: 10.1016/j.immuni.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 49.Workel HH, Lubbers JM, Arnold R, Prins TM, van der Vlies P, de Lange K, Bosse T, van Gool IC, Eggink FA, Wouters MCA, et al. A transcriptionally distinct CXCL13+CD103+CD8+ T-cell population is associated with B-cell recruitment and neoantigen load in human cancer. Cancer Immunol Res. 2019;7(5):787-796. doi: 10.1158/2326-6066.CIR-18-0517. [DOI] [PubMed] [Google Scholar]
- 50.Lebien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat. Rev. Immunol. 2015;15(3):137–148. doi: 10.1038/nri3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramento J, Iglesias M, Sevillano M, Ibiza S, Canella A, Hernando-Momblona X, et al. TGF-beta drives immune evasion in genetically reconstituted colon cancer metastasis. Nat Publ Gr. 2018. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 53.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, and Tuveson D. Il1-induced Jak/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9(2):282–301. doi: 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. 2018;9(4692). doi: 10.1038/s41467-018-06654-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gunderson AJ, Yamazaki T, McCarty K, Fox N, Phillips M, Alice A., Blair T, Whiteford M, O'Brien D, Ahmad R, et al. TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 2020;11(1):1–13. doi: 10.1038/s41467-020-15404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanpouille-Box C, Diamond JM, Pilones KH, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, and Dermaria S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015;75(11):2232–2242. doi: 10.1158/0008-5472.CAN-14-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spranger S, Luke JS, Bao R, Zha Y, Hernandez KM, Li Y, Gajewski AP, Andrade J, and Gajewski TF. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. U. S. A. 2016;113(48):E7759–E7768. doi: 10.1073/pnas.1609376113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tran E, Robbins PF, Rosenberg SA. “Final common pathway” of human cancer immunotherapy: targeting random somatic mutations. Nat. Immunol. 2017;18(3):255–262. doi: 10.1038/ni.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mesin L, Ersching J, Victora GD. Germinal Center B Cell Dynamics. Immunity. 2016;45(3):471–482. doi: 10.1016/j.immuni.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity. 2019;51:840–855.e5. doi: 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MH, Cardenas M, Wilikinson S, Lake R, Sowalsky AG, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576(7787):465–470. doi: 10.1038/s41586-019-1836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Im SJ, Hashimoto M, Gerner MY, Lee Ju, Kissick HT, Burger MC, Shan Q, Hale JS,m Lee J, Nasti TH, et al. Defining CD8 + T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Chrristian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen, et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1 − CD8 + Tumor-Infiltrating T Cells. Immunity. 2019;50(181–194.e6):181–194.e6. doi: 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moyron-Quiroz JE, Rangel-Moreno J, Hartson L, Kusser K, Tighe MP, Klonowski KD, Lefrancois L, Cauley LS, Harmsen AG, Lund FE, et al. Persistence and Responsiveness of Immunologic Memory in the Absence of Secondary Lymphoid Organs. Immunity. 2006;25(4):643–654. doi: 10.1016/j.immuni.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 65.Schrama D, thor Straten P, Fischer WH, McLellan AD, Brocker EB, Reisfel RA, and Becker JC. Targeting of lymphotoxin-α to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity. 2001;14(2):111–121. doi: 10.1016/S1074-7613(01)00094-2. [DOI] [PubMed] [Google Scholar]
- 66.Hollern DP, Xu N, Thennavan A, Glodowski C, Garci-Recio S, Mott KR, He X, Garay JP, Carey-Ewend K, and Marron D. B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell. 2019;179(1191–1206.e21):1191–1206.e21. doi: 10.1016/j.cell.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang ES, Newell PH, Wolf RF, Hansen PD, Cottam B, Ballesteros-Merino C, Gough MJ. Association of Immunologic Markers with Survival in Upfront Resectable Pancreatic Cancer. JAMA Surg. 2018;153(11):1055–1057. doi: 10.1001/jamasurg.2018.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bruno TC, Ebner PJ, Moore BL, Squalls OG, Waugh KA, Eruslanov EB, Singhal S, Mitchell JD, Franklin WA, Merrick DT, et al. Antigen-Presenting Intratumoral B Cells Affect CD4+TIL Phenotypes in Non–Small Cell Lung Cancer Patients. Cancer Immunol. Res. 2017;5(canimm.0075.2017):898–907. doi: 10.1158/2326-6066.CIR-17-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hulett TW, Jensen SM, Wilmarth PA, Reddy AP, Ballesteros-Merino C, Afentoulis ME, Dubay C, David LL, and Fox BA. Coordinated responses to individual tumor antigens by IgG antibody and CD8+ T cells following cancer vaccination. J Immunother Cancer. 2018;6(1):1–14. doi: 10.1186/s40425-018-0331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Montfort A, Pearce O, Maniati E, Vincent BG, Bixby L, Bohm S, Dow T, Wilkes EH, Chakravarty P, Thompson R, et al. A strong B-cell response is part of the immune landscape in human high-grade serous ovarian metastases. Clin. Cancer Res. 2017;23(1):250–262. doi: 10.1158/1078-0432.CCR-16-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020;20(5):294–307. doi: 10.1038/s41577-019-0257-x. [DOI] [PubMed] [Google Scholar]
- 72.Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, Schell MKJ , Sondak VK, Weber JS, and Mule JJ. 12-Chemokine Gene Signature Identifies Lymph Node-like Structures in Melanoma: potential for Patient Selection for Immunotherapy? Sci Rep. 2012;2(765). doi: 10.1038/srep00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Colbeck EJ, Ager A, Gallimore A, Jones GW. Tertiary lymphoid structures in cancer: drivers of antitumor immunity, immunosuppression, or Bystander Sentinels in disease? Front Immunol. 2017;8:1–18. doi: 10.3389/fimmu.2017.01830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Astorri E, Bombardieri M, Abba S, Peakman M, Pzzilli P, and Pitzalis C. Evolution of Ectopic Lymphoid Neogenesis and In Situ Autoantibody Production in Autoimmune Nonobese Diabetic Mice: cellular and Molecular Characterization of Tertiary Lymphoid Structures in Pancreatic Islets. J. Immunol. 2010;185(6):3359–3368. doi: 10.4049/jimmunol.1001836. [DOI] [PubMed] [Google Scholar]
- 75.Corsiero E, Delvecchio FR, Bombardieri M, Pitzalis C. B cells in the formation of tertiary lymphoid organs in autoimmunity, transplantation and tumorigenesis. Curr Opin Immunol. 2019;57:46–52. doi: 10.1016/j.coi.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 76.Denton AE, Innocentin S, Carr EJ, Bradford BM, Lafouresse F, Mabbot NA, Morbe U, Ludewig B, Groom JR, Good-Jacobson KL, et al. Type I interferon induces CXCL13 to support ectopic germinal center formation. J. Exp. Med. 2019;216(3):621–637. doi: 10.1084/jem.20181216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, Schwarz P, Armulik A, Browning JL, Tallquist MBush T, et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell. 2012;150(1):194–206. doi: 10.1016/j.cell.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gommerman JL, Browning JL, Ware CF. The Lymphotoxin Network: orchestrating a Type I interferon response to optimize adaptive immunity. Cytokine Growth Factor Rev. 2014;25(2):139–145. doi: 10.1016/j.cytogfr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Young KH, Newell Pi, Cottam B, Friedman D, Savage T, Baird JR, Akporiaye E, Gough MJ, and Crittenden M. TGF Inhibition Prior to Hypofractionated Radiation Enhances Efficacy in Preclinical Models. Cancer Immunol. Res. 2014;2(10):1011–1022. doi: 10.1158/2326-6066.CIR-13-0207. [DOI] [PubMed] [Google Scholar]
- 80.Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Brainbridge TW, Castiglioni A, et al.Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020;10(2):232–253. doi: 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- 81.Cipponi A, Mercier M, Seremet T, Baurain JF, Theate I, van den Oord J, Stas M, Boon T, Coulie PG, and van Baren N. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res. 2012;72(16):3997–4007. doi: 10.1158/0008-5472.CAN-12-1377. [DOI] [PubMed] [Google Scholar]
- 82.Hannani D, Locher C, Yamazaki T, Coin-Minard V, Vetizou M, Aymeric L, Viaud S, Sanchez D, Smyth MJ, Bruhns P, et al. Contribution of humoral immune responses to the antitumor effects mediated by anthracyclines. Cell Death Differ. 2014;21(1):50–58. doi: 10.1038/cdd.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 84.Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, and Reis e Sousa C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172(5):1022–1037. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Isaeva OI, Sharonov GV, Serebrovskaya EO, Turchaninova MA, Zaretsky AR, Shugay M, Chudakov DM. Intratumoral immunoglobulin isotypes predict survival in lung adenocarcinoma subtypes. Journal for ImmunoTherapy of Cancer. 2019;7(1):1–11. doi: 10.1186/s40425-019-0747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16(2):121–126. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding and last author on reasonable request.