

ABSTRACT
Accumulating evidence has demonstrated that N6-methyladenosine (m6A) plays important roles in various cancers, making it essential to profile m6A modifications at a transcriptome-wide scale in colorectal cancer (CRC). In the present study, we performed high-throughput sequencing to determine the m6A methylome in CRC. We obtained six pairs of CRC samples and tumour-adjacent normal tissues from Peking University People’s Hospital. We used MeRIP-seq to determine that compared to the tumour-adjacent normal tissues, the CRC samples had 1343 dysregulated m6A peaks, and 625 m6A peaks were significantly upregulated and 718 m6A peaks were significantly downregulated. Genes with altered m6A peaks play critical roles in regulating glucose metabolism, RNA metabolism, and cancer stem cells. Furthermore, we identified 297 hypermethylated m6A peaks and 328 hypomethylated m6A peaks in mRNAs through conjoint analyses of MeRIP-seq and RNA-seq data. After analysing these genes with differentially methylated m6A peaks and synchronously differential expression, we identified four genes (WDR72, SPTBN2, MORC2, and PARM1) that were associated with prognosis of colorectal cancer patients by searching The Cancer Genome Atlas (TCGA). Our study suggests that m6A modifications play important roles in tumour progression and survival of CRC patients. The results also indicate that modulating m6A modifications may represent an alternative strategy to predict the survival of cancer patients and interfere with tumour progression in the future.
KEYWORDS: M6A, MeRIP sequencing, colorectal cancer
Introduction
N6-methyladenosine (m6A), as an epigenetic regulatory mechanism, is the most abundant modification of mRNA in eukaryotic cells [1]. Numerous studies show that m6A modification is regulated reversibly by ‘writers’, ‘erasers’, and ‘readers’. The methyltransferase complex, called ‘writers’, consists of METTL3 (methyltransferase-like 3), METTL14 (methyltransferase-like 14), METTL16 (methyltransferase-like 16) and its additional adaptor molecules (such as WTAP, KIAA1429, VIRMA, ZC3H13, HAKAI, and RBM15/15B) [2]. Demethylases are categorized as ‘erasers’, including FTO (fat mass and obesity-associated protein) and ALKBH5 (alkB homolog 5) [3,4]. ‘Readers’ are m6A-binding proteins, such as YTHDC1/2 (YTH domain containing proteins 1/2), YTHDF1/2/3 (YTH-family proteins 1/2/3), and IGF2BP1/2/3 (insulin-like growth factor 2 mRNA binding proteins 1/2/3) [5].
As the most prevalent modification of mRNA, m6A modification affects various fundamentally biological processes of mRNA, such as decay, stability, translation, transport, and splicing of mRNAs [1]. Recently, accumulating evidence has demonstrated that m6A plays important roles in different cancers [6–9]. It is possible to profile the transcriptome-wide scale of m6A modifications with methylated RNA immunoprecipitation sequencing (MeRIP-seq), which has rapidly developed in recent years [10]. However, the transcriptome-wide m6A methylome in multiple cancers has not been fully characterized.
Colorectal cancer (CRC) is the third leading cause of cancer-related death worldwide [11]. Recent studies have demonstrated the vital roles of m6A modification of mRNA in CRC. For instance, Li et al. revealed that METTL3 facilitated CRC progression by preventing SOX2 mRNA degradation in an m6A-dependent manner in CRC cells [12]. Uddin et al. indicated that METTL3 methylated p53 R273H (a point-mutated codon 273 (G > A)) pre-mRNA, which increased the expression of p53 R273H mutant protein and further promoted multidrug resistance of colon cancer cells [13]. Many studies elaborate the important roles of m6A modification in CRC. However, the transcriptome-wide m6A methylome of CRC has not been determined to date.
In this study, we performed high-throughput sequencing to determine the transcriptome-wide m6A methylome of CRC. Using MeRIP-seq, we found differentially methylated peaks within mRNAs by comparing CRC samples and tumour-adjacent normal tissues. Using RNA-seq, we identified differentially expressed mRNAs. We carried out further analysis on combined MeRIP-seq and RNA-seq data. Finally, we analysed the relationship between gene expression regulated by m6A modification and clinical features in TCGA database.
Materials and methods
Patients and samples
Six pairs of CRC samples and tumour-adjacent normal tissues were obtained from Peking University People’s Hospital, and all tissues were histopathologically confirmed by pathologists. After surgery, the CRC samples and tumour-adjacent normal tissues were immediately collected and separated into centrifuge tubes. All tissues were stored at −80°C before use. This study was approved by the Ethics Committee of Peking University People’s Hospital.
MeRIP sequencing and RNA sequencing
Total RNA from each sample was isolated using TRIzol reagent (Invitrogen) and then quantified using a NanoDrop ND-1000 instrument. mRNA was isolated first from total RNA samples using the Arraystar Seq-StarTM poly(A) mRNA Isolation Kit and then fragmented into 100-nucleotide-long fragments. The size of fragmented mRNAs was confirmed using agarose electrophoresis. m6A-methylated mRNA fragments were enriched by immunoprecipitation with an anti-m6A antibody (Synaptic Systems, 202,003). A portion of the original fragmented mRNAs was kept as input. RNA-seq libraries of m6A antibody-enriched mRNAs and input mRNAs were prepared using the KAPA Stranded mRNA-seq Kit (Illumina) as follows: 1) first strand cDNA synthesis; 2) second strand cDNA synthesis; 3) A-tailing; 4) adapter ligation; 5) PCR amplification; and 6) library purification. Quality control of the completed libraries was performed with an Agilent 2100 Bioanalyzer. Libraries were mixed in equal amounts according to the quantification results by qPCR and used for sequencing on the instrument. The DNA fragments in well-mixed libraries were denatured with 0.1 M NaOH to generate single-stranded DNA molecules and loaded onto the reagent cartridge at the appropriate loading concentration. Clustered libraries were then sequenced on the Illumina NovaSeq 6000 following the NovaSeq 6000 S4 Reagent Kit (300 cycles) protocol.
Data analysis
Illumina HiSeq raw sequencing read data that passed the Illumina chastity filter were used for the following analysis. The IP and trimmed input reads (with 5’- and 3’-adaptor bases removed) were aligned to genome reference sequences using HISAT2 software (v2.1.0) [14]. For MeRIP-seq, aligned reads were used for peak calling of the MeRIP regions, and significant MeRIP-enriched regions (peaks) were identified for each sample at a p-value threshold of 0.05. The MeRIP-enriched regions (peaks) were annotated by the overlapping gene using the newest UCSC RefSeq database. Statistical analysis was performed on the alignment to retain the valid sequences for the subsequently identified significant methylated regions. Differentially expressed genes were identified using the RNA-seq data (the corresponding MeRIP-seq input library data) by the R package Ballgown [15]. GO and KEGG pathway analyses were performed based on the differentially methylated protein coding genes and differentially expressed genes [16].
Public databases and analysis
RNA sequencing data were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). A total of 568 CRC patients were selected for analysing differentially expressed mRNAs. Then, 489 CRC patients were selected for further study because these patients had complete clinical information, including primary tumour, lymph node metastasis, distant metastasis, and clinical stage and were followed up for at least one month. The basic clinical characteristics of 489 CRC patients are shown in Supplementary Table 1. Kaplan-Meier analysis was used to construct survival curves, and log-rank tests were utilized to estimate the significance of the differences. A univariate Cox regression model was adopted to calculate the hazard ratios (HRs) of the genes.
Results
Overview of the m6A methylation map in CRC
We performed MeRIP-seq analysis of tumour tissues and tumour-adjacent normal tissues from six CRC patients. A total of 1733 m6A peaks were detected in these two groups by R package exomePeak (v2.13.2). As shown in Figure 1(a), compared with normal tissues, tumour tissues had 625 significantly upregulated m6A peaks, which corresponded to transcripts of 265 genes, and 718 significantly downregulated m6A peaks, which represented transcripts of 311 genes (| log2(fold change) | > 0.585 and p < 0.05). The top 20 altered m6A peaks are listed in Table 1. Then, we investigated the distribution of m6A peaks in normal and tumour tissues. Figure 1(b-c) shows that m6A peaks both in tumour and normal tissues were primarily enriched in the coding sequence near the stop codon. However, m6A peaks in tumour tissues showed a distinct pattern from peaks in normal tissues with a relative increase in the number of m6A peaks in the coding sequence (CDS) (48.16% vs 45.05%) and 5’ untranslated region (5’UTR) (4.85% vs 2.54%) and a relative decrease in the 3’ untranslated region (3’UTR) (22.79% vs 27.97%). By analysing the distribution of m6A peaks per mRNA or gene, we found that the majority of mRNAs or genes had one m6A peak (mRNAs with upregulated peaks: 504/553; mRNAs with downregulated peaks: 600/653; genes with upregulated peaks: 140/265; genes with downregulated peaks: 157/311; Figure 1(d,e)). All altered m6A peaks were mapped to human chromosomes. Dysregulated m6A peaks were found in all chromosomes, except chrY, and were particularly found in chr1, chr2, chr10, and chr12 (figure 1(f)). In addition, the sequence logo showed the top five m6A motifs enriched from altered m6A peaks (Figure 1(g)).
Figure 1.

Characteristics of m6A methylation in CRC
(A) Volcano plots showing the significantly differential m6A peaks. (B) Accumulation of the region of average m6A peaks along all transcripts in normal and tumour tissues. (C) Pie charts showing the distribution of m6A peaks in normal and tumour tissues. (D) The distribution of altered m6A peaks per mRNA.(E) The distribution of altered m6A peaks per gene. (F) The distributions of altered m6A peaks in human chromosomes. (G) The top five m6A motifs enriched from the altered m6A peaks.
Table 1.
Top 20 altered m6A peaks in CRC
| Chromosome | Peak start | Peak end | Gene name | Regulation | Fold change | Peak region | P value |
|---|---|---|---|---|---|---|---|
| chr1 | 168,665,559 | 168,665,708 | DPT | up | 125.366 | 3’UTR | 5.012E-24 |
| chr10 | 71,176,027 | 71,176,177 | TACR2 | up | 87.4266 | 5’UTR, CDS | 2.512E-14 |
| chr1 | 42,621,345 | 42,621,495 | GUCA2B | up | 53.4456 | 3’UTR | 2.512E-55 |
| chr1 | 116,243,696 | 116,243,846 | CASQ2 | up | 51.6251 | 3’UTR | 2.63E-09 |
| chr4 | 174,450,931 | 174,451,081 | HAND2 | up | 44.3235 | 5’UTR | 5.495E-08 |
| chr5 | 40,945,422 | 40,947,776 | C7 | up | 37.7918 | CDS | 2.57E-08 |
| chr19 | 4,513,330 | 4,513,540 | PLIN4 | up | 36.5044 | CDS | 5.012E-08 |
| chr7 | 128,475,484 | 128,475,605 | FLNC | up | 36.5044 | CDS | 7.943E-46 |
| chr1 | 160,112,843 | 160,112,934 | ATP1A2 | up | 30.2738 | 3’UTR | 1.445E-05 |
| chr12 | 41,967,276 | 41,967,457 | PDZRN4 | up | 29.8571 | CDS | 1.778E-06 |
| chr12 | 50,359,286 | 50,359,464 | AQP5 | down | 0.0028398 | 3’UTR | 3.162E-18 |
| chr12 | 50,358,990 | 50,359,168 | AQP5 | down | 0.0036955 | 3’UTR | 1.585E-15 |
| chr10 | 54,076,879 | 54,077,000 | DKK1 | down | 0.0074943 | 3’UTR | 5.129E-05 |
| chr12 | 76,419,286 | 76,419,586 | PHLDA1 | down | 0.0306069 | 3’UTR | 1E-134 |
| chr12 | 76,419,286 | 76,419,587 | PHLDA1 | down | 0.0306069 | 3’UTR | 1E-134 |
| chr1 | 169,691,870 | 169,692,050 | SELE | down | 0.032352 | 3’UTR | 1.778E-06 |
| chr1 | 169,691,870 | 169,692,050 | SELE | down | 0.032352 | 3’UTR | 1.778E-06 |
| chr20 | 23,666,276 | 23,666,421 | CST4 | down | 0.0325771 | 3’UTR | 4.571E-07 |
| chr1 | 169,691,870 | 169,692,050 | SELE | down | 0.0337259 | 3’UTR | 2.754E-06 |
| chr1 | 169,691,870 | 169,692,050 | SELE | down | 0.0337259 | 3’UTR | 2.754E-06 |
3’UTR: 3’ untranslated region; 5’UTR: 5’ untranslated region; CDS: coding sequence
Differentially methylated mRNAs are involved in important signalling pathways
To investigate the biological significance of m6A modification in human CRC, we performed GO and KEGG pathway analyses of differentially methylated mRNAs. GO analysis classified genes into three functional groups: biological process (BP), and cellular component (CC), molecular function (MF). Figure 2(a) shows the top 10 significantly enriched BPs, CCs, and MFs of genes with upregulated m6A peaks, while GO analysis of genes with downregulated m6A peaks were shown in Figure 2(b). For KEGG pathway analysis, we found that genes with upregulated m6A peaks in CRC were significantly associated with the glucagon signalling pathway, calcium signalling pathway and MAPK signalling pathway (Figure 2(c)). Genes with downregulated m6A peaks were significantly associated with the AGE-RAGE signalling pathway in diabetic complications, spliceosome and RNA transport (Figure 2(d)).
Figure 2.
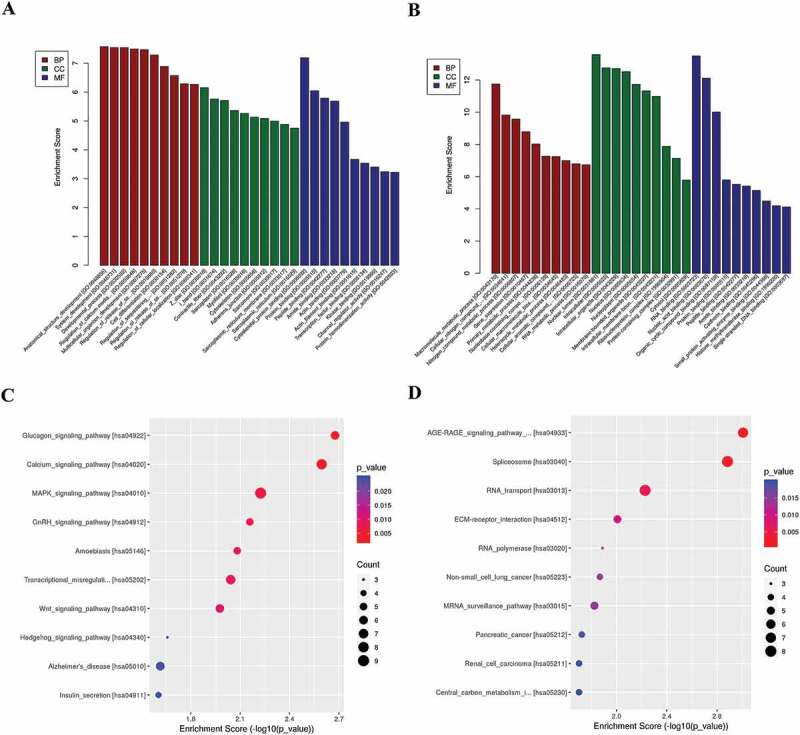
GO and KEGG pathway analyses of differentially methylated mRNA
(A) The top 10 GO terms of genes with upregulated m6A peaks. (B) The top 10 GO terms of genes with downregulated m6A peaks. (C) The top 10 KEGG pathways of genes with upregulated m6A peaks. (D) The top 10 KEGG pathways of genes with downregulated m6A peaks.
Conjoint analysis of MeRIP-seq and RNA-seq data
Using RNA-seq (MeRIP-seq input library), we detected the transcriptome profiles of tumour tissues versus tumour-adjacent normal tissues from six CRC patients. Differentially expressed genes were detected by R package Ballgown. Compared with normal tissues, tumour tissues had 2207 significantly upregulated genes and 1615 significantly downregulated genes (| log2(fold change) | > 0.585 and p < 0.05; Figure 3(a,b)). The top 20 differentially expressed genes are listed in Table 2. The top 10 GO and KEGG pathways of differentially expressed genes are displayed in Supplementary Figure 1. Then, all differentially methylated m6A peaks with differential mRNA levels (625) were divided into four groups by conjoint analysis of the MeRIP-seq and RNA-seq data. We identified 297 hypermethylated m6A peaks in mRNAs that were significantly upregulated (3; hyper-up) or downregulated (294; hyper-down), while 328 hypomethylated m6A peaks in mRNAs that were significantly upregulated (322; hypo-up) or downregulated (6; hypo-down) (Figure 3(c)). We performed GO and KEGG pathway analyses to investigate the biological significance of those genes (294) with differentially methylated m6A peaks and synchronously differential expression. GO analysis indicated that these genes were mainly enriched in the canonical Wnt signalling pathway (GO term: BP), focal adhesion (GO term: CC), and protein binding (GO term: MF) (Figure 3(d)). KEGG pathway analysis revealed that these genes were primarily enriched in ECM-receptor interaction, the PI3K-Akt signalling pathway, and pathways in cancer (Figure 3(e)).
Figure 3.

Conjoint analysis of MeRIP-seq and RNA-seq data
(A) Volcano plots showing the differentially expressed genes in CRC tissues compared with those in adjacent normal tissues. (B) Heatmap plots showing the differentially expressed genes in CRC tissues vs those in adjacent normal tissues. (C) Four-quadrant plots showing the distribution of genes with significant changes in both the m6A modification and mRNA levels. (D) The top 10 GO terms of genes with significant changes in both the m6A modification and mRNA levels. (E) The top 10 KEGG pathways of genes with significant changes in both the m6A modification and mRNA levels.
Table 2.
Top 20 differentially expressed genes in CRC
| Gene Name | Locus | Regulation | Fold Change | p value |
|---|---|---|---|---|
| DPEP1 | chr16:89,679,716–89,704,864 | up | 25.46142173 | 0.000105456 |
| MMP1 | chr11:102,660,651–102,668,891 | up | 16.81681753 | 3.14839E-05 |
| MMP3 | chr11:102,706,532–102,714,534 | up | 16.61511963 | 0.000184952 |
| CA9 | chr9:35,673,853–35,681,156 | up | 12.43408505 | 7.70013E-05 |
| CXCL8 | chr4:74,606,223–74,609,433 | up | 11.77081368 | 4.55228E-05 |
| CEMIP | chr15:81,071,684–81,244,117 | up | 10.84886762 | 4.74106E-07 |
| FOXQ1 | chr6:1,312,675–1,314,983 | up | 9.33757516 | 3.92868E-06 |
| ETV4 | chr17:41,605,212–41,656,988 | up | 9.33315578 | 1.67595E-05 |
| CST1 | chr20:23,728,190–23,731,905 | up | 9.002222678 | 6.92064E-05 |
| ASCL2 | chr11:2,289,725–2,292,182 | up | 8.931964119 | 6.55196E-05 |
| GUCA2B | chr1:42,619,092–42,621,495 | down | 0.012583824 | 2.48721E-09 |
| CLCA4 | chr1:87,012,761–87,046,437 | down | 0.014892602 | 3.52331E-06 |
| GUCA2A | chr1:42,628,362–42,630,389 | down | 0.017519091 | 5.95593E-06 |
| CD177 | chr19:43,857,811–43,867,324 | down | 0.017997201 | 2.27529E-07 |
| AQP8 | chr16:25,227,052–25,240,261 | down | 0.018263723 | 0.000182041 |
| CLCA1 | chr1:86,934,051–86,965,972 | down | 0.026155601 | 0.000742252 |
| CA1 | chr8:86,239,837–86,291,243 | down | 0.028354301 | 1.4233E-06 |
| CA7 | chr16:66,878,282–66,888,056 | down | 0.028662033 | 2.60639E-10 |
| BEST4 | chr1:45,249,257–45,253,377 | down | 0.02915239 | 2.00868E-10 |
| ZG16 | chr16:29,789,561–29,794,294 | down | 0.031832317 | 3.08934E-05 |
Gene expression regulated by m6A modification correlates with clinical parameters of CRC patients
To assess the clinical significance of gene expression regulated by m6A modification, TCGA database was explored. We validated the expression of 294 m6A-regulated genes by R package limma in TCGA database. And we identified 261 differently expressed genes, which commonly appeared in our study and TCGA database (Figure 4(a)). Next, we performed univariate Cox regression analysis of the expression of the 261 differentially expressed genes to identify prognostic genes. The results showed that the expression of 10 candidate genes was significantly related to the prognosis (p < 0.05) of CRC patients via univariate Cox regression analysis, which included 3 protective genes and 7 risk genes (Figure 4(b)). Furthermore, we combined univariate Cox regression analysis (p < 0.05) and the log-rank test (p < 0.05) to detect four genes (WDR72, SPTBN2, MORC2, and PARM1) correlated with the prognosis of patients with CRC. Overall survival curves and disease-specific survival curves of these four genes are shown in Figure 4(c-f) and Supplementary Figure 2a-2d. This result showed that high expression of WDR72, SPTBN2 and MORC2 was associated with worse prognosis of CRC patients, and high expression of PARM1 was associated with better prognosis of CRC patients. Finally, we analysed the relationship between these four genes and clinical features. Three genes expression (WDR72, SPTBN2, MORC2) were found to be significantly high in patients with distant metastasis (Figure 4(g-i)), lymph node metastasis (Figure 4(j)) and late clinical stage (Figure 4(k)), while PARM1 was low expressed in patients with lymph node metastasis (Figure 4(l)) and late clinical stage (Figure 4(m)).
Figure 4.

The relationship between gene expression regulated by m6A modification and clinical parameters in TCGA database
(A) The intersection of genes in our study and TCGA database. (B) 10 prognostic genes identified via univariate Cox regression analysis. (C) (D) (E) (F) Overall survival curves of WDR72, SPTBN2, MORC2 and PARM1, respectively. (G) The relationship between WDR72 expression and M.(H) The relationship between SPTBN2 expression and M.(I) (J) (K) The relationships between MORC2 expression and M, N, and clinical stage. (L) (M) The relationships between PARM1 expression and N and clinical stage. (N: lymph node metastasis; M: distant metastasis; OS: overall survival)
Discussion
As the most abundant modification of mRNA, m6A modification has been reported to be associated with various cancers. Many studies found that m6A modifications play critical roles in CRC [12,13], but the transcriptome-wide m6A methylome of CRC has not been characterized. In this study, we performed high-throughput sequencing to reveal the m6A landscape in CRC. By using MeRIP-seq, we identified 1343 aberrant m6A peaks in tumour tissues, of which 625 m6A peaks were significantly upregulated and 718 m6A peaks were significantly downregulated. Moreover, we performed GO and KEGG pathway analyses to reveal the potential functions of differentially methylated transcripts. Additionally, by combining MeRIP-seq and RNA-seq data, we discovered genes with differentially methylated m6A peaks and synchronously differential expression and genes enriched in cancer-related pathways. Finally, four genes were further proven to be associated with the prognosis of CRC patients in TCGA database.
In the present study, we detected many differentially methylated mRNAs related to many important biological pathways. KEGG pathway analysis illustrated that glucose metabolism was regulated by genes with upregulated m6A-modified sites in CRC, such as the glucagon signalling pathway and insulin secretion. Many studies have indicated the relationship between glucose metabolism and human CRC. For instance, researchers found that in the earliest stages of CRC, MPC (mitochondrial pyruvate carrier) was downregulated, which disturbed glucose metabolism and contributed to cancer initiation [17]. Li et al. demonstrated that KDM4B (histone lysine demethylase 4B) promoted CRC progression by enhancing glucose metabolism [18]. Our results suggested that m6A modification might suppress or promote tumours by regulating glucose metabolism.
Moreover, genes with upregulated m6A-modified sites were related to MAPK signalling pathway. MAPK signalling pathway played important roles in CRC. SNU-C1, a CRC cell line with MEK1 F53L mutation, showed strong sensitivity to MEK1 inhibitors [19]. p38α MAPK contributed to proliferation of CRC cell lines, playing a potential pro-tumorigenic role [20]. RAF signalling is downstream of RAS in the MAPK signalling pathway. BRAF activating mutations were associated with a poor prognosis of CRC patients [21]. One molecular therapeutic approach for tumour was to target MAPK signalling pathway with specific inhibitors [22]. According to our results, we believe that modulating m6A modifications of MAPK signalling pathway may be a possible therapy for CRC in the future.
Additionally, genes with upregulated m6A-modified sites were enriched in signalling pathways regulating cancer stem cells in CRC, such as the Wnt signalling pathway and Hedgehog signalling pathway (shown in the top 10 KEGG pathways). Increasing evidence has demonstrated that m6A modifications are key regulators of cancer stem cells in different kinds of cancers. In glioblastoma stem-like cells, m6A modifications regulate the growth, self-renewal, and tumorigenesis of glioblastoma stem-like cells [23,24]. MeRIP-seq analysis found that high expression of ALKBH5 enhanced FOXM1 expression via m6A modifications, which promoted glioblastoma stem-like cell tumorigenesis [23]. In acute myeloid leukaemia, YTHDF2 overexpression inhibited apoptosis of leukaemic stem cells in an m6A-dependent manner, which contributed to the overall integrity of leukaemic stem cell function [25]. In CRC, researchers demonstrated that m6A-modified SOX2, a cancer stem cell marker, was upregulated, thereby promoting cell self-renewal and stem cell frequency [12].
In contrast to genes with upregulated m6A-modified sites, genes with downregulated m6A-modified sites were mainly enriched in RNA metabolism, such as spliceosome, RNA transport, and RNA polymerase. m6A modifications have been reported to regulate RNA transport and alternative splicing [26–30]. The m6A ‘reader’ YTHDF1 has been suggested to regulate alternative splicing events and expedite mRNA export from the nucleus by recognizing m6A sites [26,29,30]. HNRNPA2B1 also regulated alternative splicing events by binding to m6A sites [28]. Based on the results of our study, we hypothesize that methylation of mRNAs associated with RNA metabolism could influence their expression, leading to subsequent changes in global gene expression, which may be a novel mechanism of the regulation of gene expression.
Genes with downregulated m6A-modified sites were also involved in ECM-receptor interaction. Consistent with our results, plenty of studies implied that ECM-receptor interaction regulated the progression of CRC. For example, integrin subunit β like 1 (ITGBL1) facilitated tumour cell migration and invasion, and CRC patients with high expression of ITGBL1 had shortened survival [31]. Hyaluronic acid (HA) promoted proliferation and blocked apoptosis by binding to TLR4 and CD44 in CRC [32]. In aggressive CRC, collagen-I induced tumour-specific mesenchymal gene expression, facilitating tumour cell invasion [33].
By combining analysis of MeRIP-seq and RNA-seq data, we discovered 294 genes with differentially methylated m6A peaks and synchronously differential expression in CRC. The expression of these genes may be regulated by m6A modification of mRNAs. Of these 294 genes, 4 genes (WDR72, SPTBN2, MORC2, and PARM1) were associated with the prognosis of CRC patients in TCGA. We demonstrated that WDR72, SPTBN2, and MORC2 were protective factors and that PARM1 was a risk factor for CRC. It has been reported that some of these genes regulate tumour progression in various types of cancer. For example, MORC2 was reported as an oncogene promoting migration and invasion by inhibiting NDRG1 in CRC [34] and promoting cancer stemness and tumorigenesis by regulating the Hippo signalling pathway in hepatocellular carcinoma [35]. PARM1 was involved in lymph node metastasis and tumour size in CRC [36]. These genes regulated by m6A modifications may play critical roles in the development of CRC. However, detailed molecular mechanisms are still unknown and further exploration deserves careful consideration in the future.
Previous studies indicated that inhibition of m6A demethylases ALKBH5 and FTO may be a potential strategy to target tumour cells [6,7]. With the development of m6A sequencing, it is possible to develop alternative therapeutic strategy by directly modulating m6A-modified mRNAs or m6A-regulated genes. Using MeRIP-seq, we profiled the transcriptome-wide scale of m6A modifications of mRNAs in CRC. Our study suggested that m6A modifications in mRNAs regulated gene expression in CRC and affected tumour progression and survival of CRC patients. Based on the results of the present study, directly modulating m6A modifications would be alternative therapeutic strategy for CRC in the future.
Conclusions
In the present study, we performed high-throughput sequencing to reveal the m6A transcriptome-wide map of human CRC. We performed conjoint analysis of MeRIP-seq and RNA-seq data and discovered many genes with differentially methylated m6A peaks and synchronously differential expression, indicating a potential link between m6A methylation and gene expression. In addition, several genes were further suggested to be associated with the prognosis of CRC patients.
Supplementary Material
Acknowledgments
We thank the patients involved in the study and our colleagues for their helpful suggestions.
Funding Statement
This work was supported by the National Key Research and Development Program of China (2017YFC1308800); and the National Natural Science Foundation of China (Grant No. 81672375; 81972240; 81871962).
Disclosure statement
The authors declare no conflicts of interest for this article.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Yang Y, Hsu PJ, Chen Y-S, et al. Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28(6):616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shi H, Wei J, He C, et al. Where, when, and How: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74(4):640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zheng G, Dahl J, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bi Z, Liu Y, Zhao Y, et al. A dynamic reversible RNA N(6) -methyladenosine modification: current status and perspectives. J Cell Physiol. 2019;234(6):p. 7948–7956. [DOI] [PubMed] [Google Scholar]
- [6].Chen XY, Zhang J, Zhu JS.. The role of m(6)A RNA methylation in human cancer. Mol Cancer. 2019;18(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lan Q, Liu P-Y, Haase J, et al. The critical role of RNA m(6)A methylation in cancer. Cancer Res. 2019;79(7):1285–1292. [DOI] [PubMed] [Google Scholar]
- [8].Lobo J, Barros-Silva D, Henrique R, et al. The emerging role of epitranscriptomics in cancer: focus on urological tumors. Genes (Basel). 2018;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hu BB, Wang X-Y, Gu X-Y, et al. N(6)-methyladenosine (m(6)A) RNA modification in gastrointestinal tract cancers: roles, mechanisms, and applications. Mol Cancer. 2019;18(1):178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. [DOI] [PubMed] [Google Scholar]
- [11].Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- [12].Li T, Hu P-S, Zuo Z, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Uddin MB, Roy KR, Hosain SB, et al. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharmacol. 2019;160:134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frazee AC, Pertea G, Jaffe AE, et al. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat Biotechnol. 2015;33(3):243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Huang Da, Sherman BT, Lempicki RA.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- [17].Bensard CL, Wisidagama DR, Olson KA, et al. Regulation of tumor initiation by the mitochondrial pyruvate carrier. Cell Metab. 2020;31(2):284–300.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li H, Lan J, Wang G, et al. KDM4B facilitates colorectal cancer growth and glucose metabolism by stimulating TRAF6-mediated AKT activation. J Exp Clin Cancer Res. 2020;39(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gannon HS, Kaplan N, Tsherniak A, et al. Identification of an “exceptional responder” cell line to MEK1 inhibition: clinical implications for MEK-targeted therapy. Mol Cancer Res. 2016;14(2):207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chiacchiera F, Simone C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy. 2009;5(7):1030–1033. [DOI] [PubMed] [Google Scholar]
- [21].Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009;361(25):2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Burotto M, Chiou VL, Lee J-M, et al. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120(22):3446–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang S, Zhao B-S, Zhou A, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cui Q, Shi H, Ye P, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Paris J, Morgan M, Campos J, et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019;25(1):137–148.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roundtree IA, Luo G-Z, Zhang Z, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017;6. DOI: 10.7554/eLife.31311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Adhikari S, Xiao W, Zhao Y-L, et al. m6A: signaling for mRNA splicing. RNA Biol. 2016;13(9):756–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Alarcon CR, Goodarzi H, Lee H, et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ye F, Chen ER, Nilsen TW. Kaposi’s sarcoma-associated herpesvirus utilizes and manipulates RNA N(6)-adenosine methylation to promote lytic replication. J Virol. 2017;91(16). DOI: 10.1128/JVI.00466-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–519. [DOI] [PubMed] [Google Scholar]
- [31].Qiu X, Feng J-R, Qiu J, et al. ITGBL1 promotes migration, invasion and predicts a poor prognosis in colorectal cancer. Biomed Pharmacother. 2018;104:172–180. [DOI] [PubMed] [Google Scholar]
- [32].Makkar S, Riehl TE, Chen B, et al. Hyaluronic acid binding to TLR4 promotes proliferation and blocks apoptosis in colon cancer. Mol Cancer Ther. 2019;18(12):p. 2446–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Vellinga TT, den Uil S, Rinkes IH, et al. Collagen-rich stroma in aggressive colon tumors induces mesenchymal gene expression and tumor cell invasion. Oncogene. 2016;35(40):5263–5271. [DOI] [PubMed] [Google Scholar]
- [34].Liu J, Shao Y, He Y, et al. MORC2 promotes development of an aggressive colorectal cancer phenotype through inhibition of NDRG1. Cancer Sci. 2019;110(1):135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang T, Qin Z-Y, Wen L-Z, et al. Epigenetic restriction of Hippo signaling by MORC2 underlies stemness of hepatocellular carcinoma cells. Cell Death Differ. 2018;25(12):2086–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu YR, Hu Y, Zeng Y, et al. Neurexophilin and PC-esterase domain family member 4 (NXPE4) and prostate androgen-regulated mucin-like protein 1 (PARM1) as prognostic biomarkers for colorectal cancer. J Cell Biochem. 2019;120(10):18041–18052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.