

ABSTRACT
Testicular germ cell tumours (TGCTs) are heterogeneous neoplasms mostly affecting young-adult men. Despite high survival rates, some patients with disseminated disease acquire cisplatin resistance, entailing the need for less toxic therapies. Epigenetic alterations constitute an important feature of TGCTs, which are also implicated in resistance mechanism(s). These alterations might be used as potential targets to design epigenetic drugs. To date, several compounds have been explored and evaluated regarding therapeutic efficacy, making use of pre-clinical studies with in vitro and in vivo models, and some have already been explored in clinical trials. This review summarizes the several epigenetic mechanisms at play in these neoplasms, the current challenges in the field of TGCTs and critically reviews available data on ‘epidrugs’ in those tumours.
KEYWORDS: Testicular germ cells tumours, epigenetics, methylation, histone modifications, microRNA, targeted therapies
Introduction
The testis is composed by different types of cells, and each cell type may give rise a neoplasm, justifying the wide variety of tumours within this organ. However, around 95% of testicular cancers are derived from germ cells, which fail to complete their normal differentiation, and these are called testicular germ cell tumours (TGCTs) [1,2].
Overall, TGCTs are not prevalent cancers. According to worldwide statistics for 2018 (Globocan) [3], they represent the 21st most incident neoplasm in men worldwide, with 71,105 new cases diagnosed in 2018. Nonetheless, from an Epidemiological perspective, they constitute a real concern, since they are the most incident solid tumour arising specifically in young-adult men (aged until 34 years), a figure only surpassed by the liquid tumours (lymphomas/leukaemias) [3]. Indeed, the ‘genvironmental model’ of understanding this disease fits both with the changing epidemiology of this cancer type and the intricate association with disorders of sex development [1].
Over the years, there has been better and more complete understanding of the biology of these tumours. A developmental perspective of these cancers has led to an integrated classification, universal to all types of germ cell tumours (GCTs) and genders (both testicular, ovarian and extragonadal), based on the biology of the cell of origin; this is a mostly due to its epigenetic status [4]. Among the seven subclasses of GCTs, type II GCTs of the testis are the most frequent and, at the same time, the most clinically challenging, due to malignant behaviour, being the focus of this Review. TGCTs are derived from the precursor lesion germ cell neoplasia in situ (GCNIS), which emerges from primordial germ cells/gonocytes that get arrested in differentiation, and are categorized into two major groups: the more homogeneous seminomas (SEs) and the heterogeneous group of non-seminomas (NSs), which includes embryonal carcinoma, yolk-sac tumour, choriocarcinoma and teratoma, as well as tumours composed of mixtures of two or more components, the so-called mixed tumours [1,5]. Clinical behaviour (and hence therapeutic strategy) is distinct and morphology is strikingly different, despite the low mutational rate and paucity of genomic aberrations (similar to other pediatric/young-onset tumours), the most frequent being the (almost) universal presence of isochromosome 12p [6]. The contribution of epigenetics is remarkable [7], and dictates phenotypic and clinical variety [5].
Clinical challenges remain in TGCT field, and research efforts should be directed to them. Overall, about 70% of patients are diagnosed with localized disease (approximately 80% of SE and 60% of NS) [1,8,9], and the cure rate is around 95%, making TGCTs a model of curable cancer, even in the case of disseminated disease. Whereas for a substantial proportion of patients (around 75%) orchiectomy alone is curative, for some patients, adjuvant chemotherapy should be given to avoid disease relapse [10]. The major challenge nowadays is to uncover biomarkers (for instance, epigenetic) with high accuracy for determining the subset of patients that will never relapse and may be spared cisplatin-based treatments and its associated long-term side effects [11]. On the other side of the spectrum, patients presenting with (or developing) metastases receive cisplatin-based chemotherapy, the mainstay drug for treatment of GCTs and responsible for the major drop in mortality of these patients since its incorporation around the 1970s [12]. Despite its remarkable efficacy, due to the extreme sensitivity of TGCTs to DNA damage, a subset of patients develop resistance to cisplatin. The major relevance of this event relates to the absence of validated therapeutic options for these patients, which are the ones experiencing morbidity and mortality from disease at present [1,13]. Several studies attempted to elucidate the mechanisms of resistance to cisplatin [14–17], but a specific mechanism is still lacking. Again, there is evidence that epigenetic deregulation may also provide insight on this matter [18–20].
Epigenetics focuses on reversible changes in gene expression, which occur without altering DNA sequences, and is an expanding research field, namely (but not only) in cancer. Three major epigenetic mechanisms are known: DNA methylation, histone post-translational modifications and chromatin remodelling, and regulation by non-coding RNAs [21]. Importantly, and because these alterations are reversible, they can be therapeutically targeted with ‘epidrugs’, some of them already approved for cancer treatment or under investigation in clinical trials [22]. Given the parallelism between development/TGCTs/epigenetics, it is expected that targeting TGCTs’ epigenetic landscape may help reverse the cisplatin-resistant phenotype, which would be of major clinical impact [18].
The main goal of this Review is to briefly summarize the main epigenetic mechanisms implicated in TGCTs, and to highlight and discuss the current status of epigenetic-based therapies for these patients, namely its achievements and also areas where improvement is still lacking. Finally, we intend to provide rationale for researchers to pursue, and move forward within the field of epigenetic targeting in TGCTs, creating new opportunities.
Brief summary of DNA methylation in TGCTs
DNA methylation is the most well studied epigenetic mechanism, and methylation-based biomarkers are expanding as promising non-invasive means of diagnosis and follow-up of cancer patients. As mentioned, TGCTs tumorigenesis is tightly linked to epigenetic phenomena; specifically, the methylation profiles of SE and NS types (and even within NS subtypes) are remarkably distinct (see below) [2,6]. Altogether, these data seem to indicate that DNA methylation could be explored to uncover both TGCT-specific biomarkers and alternative treatment options (‘epidrugs’).
The covalent addition of a methyl group to the 5ʹ carbon cytosine of DNA (occurring mainly at CpG sites) and is catalysed by a family of enzymes, the DNA methyltransferases (DNMTs). There are three main DNMTs: DNMT1, DNMT3A and DNMT3B; the former is responsible for the maintenance of parental cell DNA methylation in a replication-dependent manner, whereas the remainder are associated with de novo methylation, which occurs during embryogenesis/germ cell development and ensures re-establishment of parental imprinting marks [23,24]. The DNA methylation process is dynamic and may be reversed through DNA demethylation. The so-called ‘active demethylation’ process consists of removal of the methyl group from DNA [25], which involves the action of ten-eleven translocation enzymes (TETs; including TET1, TET2 and TET3), which catalyse oxidation of 5-methylcytosine to 5-hydroximetylcytosine [23,25]. Active demethylation occurs, for instance, in the paternal genome in the zygote, while the maternal genome undergoes ‘passive demethylation’, due to failure of DNMTs in establishing methylation during replication, creating a CpG methylation dilution effect [26].
Concerning TGCTs, SEs, in general, display global hypomethylation and erasure of imprinting marks, resembling their originating cell – primordial germ cells. Hypomethylation and the initial phenomenon of polyploidization lead to instability and tumour progression. Studies have shown erasure of methylated peaks in SE samples after correcting for methylation provided by infiltrating lymphocytes [2,6]. Conversely, there is locus-specific hypermethylation in NSs [2,27], including several gene promoters (e.g., MGMT, CALCA, HIN1 (SCGB3A1), RASSF1A, HOXA9, CRIPTO, MCAM, MLH1, S100A2, SSBP2, APC, VGF and PGP9.5) [7,19,28,29]. Data from genome-wide studies also showed distinct methylation patterns among NS subtypes, for instance with non-canonical methylation (CpH methylation) occurring in embryonal carcinoma (and correlating with DNMT3A/B expression), a pattern followed by embryonic stem cells, whereas more differentiated components lose such non-CpG methylation and display patterns approximating somatic cancers [6], indicating shifts in methylation that follow tumour differentiation. Importantly, MLH1, RASSF1A and HIC1 hypermethylation was associated with cisplatin resistance [19,30], and CALCA, MGMT, HOXA-9 and SCGB3A1 hypermethylation was associated with poor prognosis in TGCT patients [7,31]. Demethylation of specific DNA segments is also typical of TGCTs biology, namely hypomethylation of LINE1 sequences and XIST, the latter related to the phenomenon of X-chromosome inactivation that is patent in TGCTs, which show extra copies of X-chromosome [2,32]. A summary of DNA methylation patterns of TGCTs is illustrated in Figure 1.
Figure 1.

Methylation pattern of (a) normal somatic cells, (b) seminomas and (c) non-seminomas. In normal somatic cells, gene promoters are unmethylated in general, while repetitive sequences, like LINE1 and ALU are densely methylated. The XIST promoter (a long non-coding RNA involved in X-chromosome inactivation) is methylated in normal somatic male cells (due to XY chromosome constitution) but demethylated overall in TGCTs, both seminomas and non-seminomas (due to the initial step of polyploidization and universal gain of X chromosome). Seminomas show an overall unmethylated profile. Non-seminomas present locus-specific methylation of gene promoters and methylated ALU sequence
SEs: Seminomas; XIST: X–inactive specific transcript; LINE1: Long interspersed nuclear element 1.
Overall, deregulation and specific expression patterns of DNMTs and/or TETs may be expected in TGCTs, considering the shifting methylation profile across subtypes. Several studies (both in vitro, in vivo and using patient samples) have showed this, with NSs displaying higher DNMTs expression levels and lower/variable TETs expression levels [25,33–35]. This should be taken into account when treating patients with specific inhibitors (see below).
Overview of ‘epidrugs’ over time: DNMT inhibitors in TGCTs
In the last decades, some DNMT inhibitors (DNMTis) have shown therapeutic efficacy, effectively contributing to tumour cell death [23]. Overall, DNMTis block DNMTs action, leading to global hypomethylation and, consequently, to re-expression of genes (especially tumour suppressor genes), reversing or attenuating the malignant phenotype. According to the mode of action, DNMTis can be divided into two main groups: nucleoside and non-nucleoside analogues; the former incorporate directly into DNA, during S phase of cell cycle, disrupting replication; the latter bind to the catalytic site of DNMTs, preventing their activity [23].
5-azacytidine and 5-aza-2ʹdeoxytidine are two nucleoside analogues approved by Food and Drug Administration (FDA) and European Medicines Agency (EMA) for treatment of patients with haematological cancers [23,25]. Although not yet approved for treatment of solid cancers, many studies have demonstrated the cytotoxic effect of those drugs in a pre-clinical setting [25,36]. It is acknowledged that sensitivity to 5-azacytidine depends on high DNMT3B expression [37]. Thus, it has been hypothesized that these agents might be useful for treatment of TGCT patients, mostly for one of the most aggressive NS – embryonal carcinoma – which overexpresses that enzyme [25].
Indeed, some studies have explored DNMTis in TGCTs (Table 1). The first study dates already from 1994, when Juttermann et al [38] exposed embryonic stem cells to 5-aza-2ʹ-deoxycytidine, observing demethylation and re-expression of tumour suppressor genes, culminating in apoptosis, direct or indirectly mediated by this drug. Subsequently, Lind et al [28] demonstrated that TGCT cell lines cultured with 5-aza-2ʹ-deoxycytidine reversed the aberrant epigenetic gene silencing pattern, specifically the SCGB3A1 gene promoter [7].
Table 1.
Studies with epidrugs in testicular germ cell tumours
| Drug | Category | Phase of study | Cell lines/Patients | Dosage | Assays/Endpoints | Main Results | Notes | Ref. |
|---|---|---|---|---|---|---|---|---|
| 5-aza-2ʹdeoxycytadine | DNMT inhibitor | Pre-clinical | TERA1, TERA2 and NCCIT | 10 µM | MSP and bisulphite sequencing (methylation) | Can reverse aberrant DNA methylation, mainly in SCGB3A promoter | [29] | |
| 5-aza-2ʹdeoxycytidine | DNMT inhibitor | Pre-clinical | NT2/D1, 833 K and their cisplatin-resistant clones, TERA-1, 577 M and 2102EP | 10 nM | Cell-TiterGlo (viability) | Pre-treatment with low doses of this compound restores cisplatin cytotoxic response by inducing p53 and/or re-expressing genes (MGMT, RASSF1A, and HOXA-9) | The effect of 5-aza-2´deoxytidine depends on DNMT3B expression, but cisplatin sensitivity not | [40] |
| 5-aza-2ʹdeoxycytadine | DNMT inhibitor | Pre-clinical | 2102EP, TERA1, NTERA2 and H7 | 10 nM | Flow cytometry (apoptosis) qRT-PCR (transcript levels) Western Blot (protein levels) |
There is an induction of apoptosis and differentiation | First study that shows that DNMT3B can act as oncogene | [42] |
| 5-azacytidine | DNMT inhibitor | Pre-clinical | TCam-2 | 10 µM | qRT-PCR (transcript levels) MTT (viability) MSP and bisulphite sequencing (methylation) |
Restores sensitivity to cisplatin, causing changes in methylation pattern and downregulation of pluripotency genes (NANOG and OCT3/4) | [28] | |
| 5-aza | DNMT inhibitor | Pre-clinical | NT2/D1 and its cisplatin-resistant clone | 10 nM | Cleavage of PARP1 and Western Blot (apoptosis) MSP and bisulphite sequencing (methylation) |
Low doses of 5-aza lead to DNA damage and apoptosis by activation of p53 targets, global hypomethylation and downregulation of pluripotency genes | First study that shows that induction of p53 is not associated with increased p53 mRNA, but with p53 stability | [43] |
| Guadecitadine (SGI-110) |
DNMT inhibitor | Pre-clinical | NT2/D1 and its cisplatin-resistant clone | 5 nM | Cell-TiterGlo (viability) | Exposure to low concentration results in a decrease of tumour cells growth, induces p53 target genes, re-expresses RASSF1A and SOX15 and, consequently, restores cisplatin sensitivity | Using in vivo (mouse) models, combination of SGI-110 with cisplatin causes complete tumour regression | [44] |
| 5-aza | DNMT inhibitor | Pre-clinical | 2102EP and NCCIT and their cisplatin-resistant clones | 10–20 nM | Trypan Blue (viability) Cleavage of Caspase 3 and PARP1 Western Blot Flow cytometry (apoptosis) |
Anti-tumour activity as a single agent at low concentration, but more effectively when combined with cisplatin | Demonstrated that the effect of 5-aza is independent of TP53 mutational status | [17] |
| 5-aza alone or in combination with TSA | DNMT inhibitor and HDAC inhibitor | Pre-clinical | JEG-3 and primary choriocarcinoma stem-like cells | 75 µM (5-aza) 100 nM (TSA) |
qRT-PCR (transcript levels) Western Blot (protein levels) |
5-aza as a single agent leads to decreased DNMT1 and DNMT3B. In combination with TSA it also reduces the expression of pluripotency genes (NANOG, OCT3/4, SOX2, and ABCG2) | This study introduces a natural compound (curcumol) for treatment of choriocarcinoma cells with satisfactory results | [36] |
| 5-azacytidine | DNMT inhibitor | Clinical trial, phase II | 17 patients with advanced germ cell tumours | 150 mg/m2/day | Response to treatment Disease-free survival |
All patients progressed; 16/17 died | [45] | |
| 5-azacytidine | DNMT inhibitor | Clinical trial, phase II | 4 patients with testicular cancer | 150–225 mg/m2 | Response to treatment | Two of four patients presented partial responses | [46] | |
| Hydralazine in combination with valproate | DNMT inhibitor and HDAC inhibitor | Clinical trial, phase II | One patient with non-seminoma | 83–182 mg (hydralazine) 700 mg (valproate) |
Response to treatment Disease-free survival Overall survival |
Patient with stable disease | [47] | |
| TSA | HDAC inhibitor | Pre-clinical | P19 | 10–100 ng/mL | TUNEL and flow cytometry (apoptosis) | TSA inhibits cell progression but alone does not induce differentiation (only in combination with retinoic acid) | [61] | |
| CBB | KDM inhibitor | Pre-clinical | F9, NCCIT, NTERA2, HELA, 293, NIH3TS | 5,27–11,16 µM | Spectrometry | CBB blocks demethylation activity of LSD1 on mono- and- di-methylated H3K4, inducing differentiation | To mimic in vivo models, authors treated mouse F9 embryonic stem cells and obtained similar results in in vitro analysis | [62] |
| CBB3001 | KDM inhibitor | Pre-clinical | F9 | 21,25 µM | Spectrometry | CBB3001 inhibits specifically LSD1, reducing cell growth and downregulates SOX2 and OCT3/4 | [63] | |
| TSA and vorinostat as a single agent | HDAC inhibitor | Pre-clinical | GH | Various concentrations | qRT-PCR (transcript levels) Cleavage of PARP-1 and caspase 3 and Western Blot (apoptosis) |
Both drugs restore GTAp63 and induce apoptosis. Combination with cisplatin causes complete cell death | First study that reports the induction of GTAp63 as relevant for treatment | [66] |
| Romidepsin | HDAC inhibitor | Pre-clinical | TCam-2, 2102EP-R, NCCIT-R, NT2/D1-R, JAR and JEG-3 | 1–10 nM | XTT (viability) Cleavage of PARP-1 and Western Blot and flow cytometry (apoptosis) qRT-PCR (transcript levels) |
Romidepsin is highly toxic at low concentration, inducing stress, apoptosis and cell cycle arrest | Used in vivo models (mouse). Romidepsin kills SurePath cells by inducing apoptosis | [67] |
| JQ1 | Bromodomain inhibitor | Pre-clinical | NCCIT, NT2/D1, 2102EP and their cisplatin-resistant clones, TCam-2, FS1 and MPAF | 100–500 nM | XTT (viability) Western Blot (protein levels) Flow cytometry (apoptosis) qRT-PCR (transcript levels) |
JQ1 not only increases G1 arrest and apoptosis, but also differentiation, and inhibits angiogenesis | First study that used bromodomain inhibitor in TGCTs | [58] |
| Animacroxam | HDAC inhibitor and cellular cytoskeletal dynamics inhibitor | Pre-clinical | 2102EP and resistant clone, NCCIT | 0,1–3,2 µM | ELISA iCELLigence AC-DEVD-AMC Flow cytometry |
Animacroxam has an anti-proliferative effect and inhibits cell migration | Authors used CAM as in vivo model, observing reduction of tumour growth | [69] |
DNMT: DNA methyltransferase; MSP: Methylation-specific PCR; qRT-PCR: Real-Time Quantitative Polymerase Chain Reaction; TSA: trichostatin A; HDAC: Histone deacetylase; KDM: Histone demethylases; ELISA: Enzyme-Linked Immunosorbent Assay; CAM: Chick Chorioallantoic Membrane
Since platinum-based therapies are extremely effective in treating these patients (and not likely to be replaced), studies rapidly focused on the ability of epidrugs to restore sensitivity to cisplatin, which is a major clinical issue in current times. Pre-treatment with DNMTis was indeed reported to rescue sensitivity to chemotherapy with cisplatin in TGCT cell lines. Beyrouthy and colleagues [37] demonstrated that 5-aza-2´deoxycitidine not only affected cisplatin-resistant embryonal carcinoma cells, but also cells pre-treated with cisplatin. They also demonstrated that this effect might be caused by induction of p53 target genes, like GDF15 and BTG2, or by re-expression of other genes (MGMT, RASSF1A and HOXA-9) through demethylation of respective promoters. Moreover, low doses of 5-aza-2´deoxycitidine increased activated ATM and phosphorylated H2AX levels, inducing DNA damage. This study also reports that the effect of this compound depends on DNMT3B expression, as previously mentioned. Indeed, Wongtrakoongate and co-workers [39] demonstrated that DNMT3B behaves as an oncogene, and that 5-aza-2´deoxycitidine treatment of TGCT cell lines causes a reduction of DNMT3B, which associates with increased apoptosis.
In 2010, Wermann et al [27], treated SE-like cell line (TCam-2) with 5-azacytidine and showed that the increased sensitivity to cisplatin was not only caused by changes in methylation pattern, but also through downregulation of pluripotency genes (NANOG, OCT3/4). Biswal and co-workers [40] reported similar results with 5-aza treatment in NS cell lines (NT2/D1 and NT2/D1-R1), and demonstrated that p53 induction was not associated with increased p53 transcript levels, but rather with p53 stability.
Second generation demethylating agents were also explored; in 2016, Albany et al [41] demonstrated that low concentrations of guadecitadine (SGI-110) had similar effects to 5-aza in TGCT cell lines. Combination therapies started to be documented as the most promising. Using in vivo mouse models, authors observed that 0.5 mg/kg of guadecitabine as single agent resulted only in incomplete tumour inhibition, whereas combination with cisplatin resulted in complete tumour regression at 48 hours, without significant toxicity. Furthermore, they observed an early and extensive activation of p53 pathway and induction of immune tumour cell recognition components, like HLA class I and cancer testis antigens. This deserves further investigation, especially due to the potential for combination with immunotherapies (see below).
Very recently, in 2018, the findings of Oing et al [18] reinforced the idea that the anti-tumour effect is more prominent when DNMTis are used in combination with cisplatin. Authors demonstrated that the effect of this ‘epidrug’ is independent of TP53-mutational status in TGCT cell lines. Combination of several ‘epidrugs’ was also investigated to explore potential synergistic mechanisms. Peng et al [35] combined DNMTi 5-azacytidine with histone deacetylase inhibitor (HDACi) trichostatin A (TSA), as well as the natural compound curcumol (an active component of Curcuma zedoria, used in traditional Chinese Medicine for treatment of gynaecological tumours). Choriocarcinoma cell lines (JEC-3 and primary choriocarcinoma stem-like cells), an aggressive tumour type, were treated with 5-azacytidine alone, TSA alone, 5-azacytidine in combination with TSA and curcumol. With 5-azacytidine treatment as single agent there was a decreased DNMT1 and DNMT3B expression, whereas the combination with TSA resulted in a decrease of pluripotency genes, such as NANOG, OCT4, SOX2 and ABCG2. Curcumol’s effects were studied in cell lines and in vivo models, with cell lines losing the stem-like phenotype and treated mice surviving longer than the control group. The regulatory mechanism of this compound, however, is not well understood at present. This study shows that natural compounds may have potential as anticancer therapy, eventually through modification of the epigenetic background of tumour cells. This supports research on this kind of compounds, and should also trigger investigation on repurposing drugs [42]. A summary of the mechanisms of action of DNMTis in TGCTs is illustrated in Figure 2.
Figure 2.
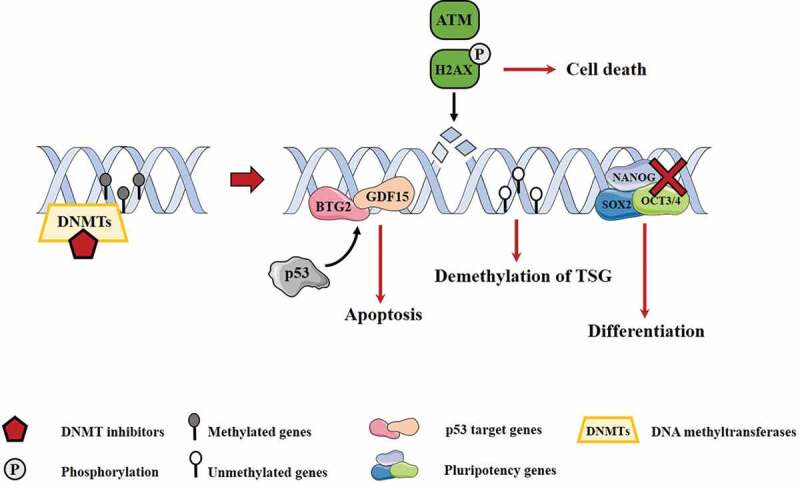
Main molecular mechanisms associated with DNMT inhibitors. DNMT inhibitors can induce p53 targets, leading to apoptosis. On the other hand, they lead to increase of ATM and pH2AX, associated with DNA damage, conducting to cell death. Besides this, there is re-expression of tumour suppressor genes by demethylation process. Oppositely, pluripotency genes are downregulated, leading to differentiation of cells
DNMTs: DNA methyltransferases TSG: tumour suppressor genes.
According to these results in pre-clinical studies, DNMTis have also been tested in patients. However, clinical data so far has not been satisfactory, and these drugs have not moved forward to the clinic. In 1993, Roth and colleagues [43] enrolled 17 patients with advanced GCTs undergoing four weeks of cisplatin therapy. Subsequently, patients were treated with 5-azacytidine, 150 mg/m2/day, for three weeks (with one patient refusing therapy beyond the first course). All patients progressed after the therapy and 16/17 died, 15 from progressive disease and one from sepsis. The only surviving patient was reported as disease-free at 38.5 months. Furthermore, patients experienced toxicity: nine patients with granulocytopenia and three with anaemia and thrombocytopenia. Quagliana and colleagues [44] treated 214 patients with solid tumours, including four with testicular cancer cases (with no further specifications), with the same drug. First, they received 225 mg/m2, but owing to associated toxicities, it was decreased to 175 mg/m2 and then, 150 mg/m2. Remarkably two of those four patients disclosed partial response. However, drug toxicity limited prolonged use. Overall, these studies constitute a mismatch to pre-clinical findings, in which demethylating drugs were quite effective. The clinical setting, namely giving the drug in monotherapy to patients with advanced GCTs, may not have been the most appropriate setting. Combination strategies with cisplatin or other drugs should be better explored (discussed below).
Combinations of several ‘epidrugs’ were also tested. A study combined hydralazine, a drug used for treating hypertension and repurposed for its action as weak non-nucleoside DNMTi (demonstrated by us and others [45]), and magnesium valproate, a histone deacetylase inhibitor (HDACi) [46]. In that study, only one patient with testicular cancer was included and although no disease regression was achieved, no progression was observed, either. Nevertheless, in general, there was increased sensitivity to chemotherapy, with stable clinical response, with a 5.6 month progression-free survival and overall survival of 5.7 months. We consider that further studies are needed and preferentially should test other ‘epidrugs’, such as zebularine and procainamide, already evaluated in other urological cancers [23]. Moreover, some studies suggested TET inhibitors’ therapeutic effect for treatment of TGCTs [47].
Brief summary of histone post-translational modifications in TGCTs
Histones are proteins that provide structural support to chromatin. Importantly, histones possess a flexible N-terminal tail, which can undergo post-translational modifications, such as methylation, acetylation (the most well studied) and others. These mechanisms alter chromatin pattern with implications on the accessibility of transcription factors to DNA and, consequently, altering gene expression without changing the genetic code [25]. All these modifications are mediated by histone-modifying enzymes. Acetylation is controlled by the balanced activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs), which add/remove acetyl groups on lysine residues of histones [25,48]. In general, acetylation of histones is associated with chromatin unfolding and gene transcription [48], whereas HDACs are seen as transcription co-repressors [25]. Histone methylation (transfer of methyl groups to histone lysine or arginine residues) is mediated by histone methyltransferases (KMTs), and its removal is catalysed by histone demethylases (KDMs). These can associate with transcriptional activation or repression, depending on the modified amino acid and its position (Figure 3). In addition, a heterogeneous family of chromatin remodelling complexes (ChRCs) can alter the nucleosome structure, again affecting DNA accessibility and gene expression; therapeutic targeting of these complexes is less well developed compared to histone (de)acetylation, and could be alternative therapies to explore, as discussed below [25,49].
Figure 3.
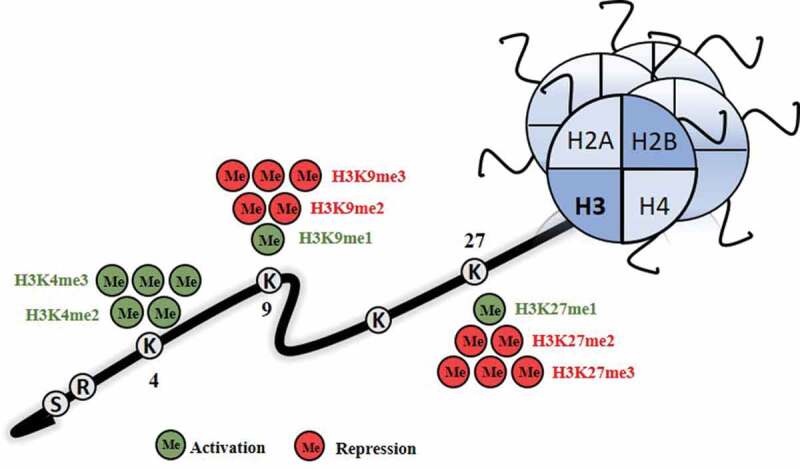
Repression and activation marks associated with methylation of histones. H3K4me2/3, H3K9me1 and H3K27me1 are activation marks, while H3K9me2/3 and H3K27me2/3 are repression marks
ME: methylation; H3: histone 3.
In TGCTs, differential expression of HATs and HDACs between SEs and NSs has been reported. An in silico analysis of The Cancer Genome Atlas (TCGA) dataset disclosed that SEs present higher HATs expression levels, whereas NSs display HDACs overexpression [25]. Moreover, HDACs isoforms’ immunoexpression profile significantly differs among TGCTs histological subtypes. Whereas HDAC2 and 3 were shown to be highly expressed in all types of TGCTs, HDAC1 displayed low levels. Choriocarcinomas, an aggressive tumour subtype, presented high expression of all HDACs isoforms [50].
Studies on histone methylation are more conflicting. Hinz and colleagues [51] did not observe significant differences in activating or repressive modifications between SEs and NSs. However [25], in an in silico analysis we showed that SEs depict higher expression levels of enzymes primordially implicated in establishment of activations marks, like KDM4D, KDM3A, KMT2B/C/D and SETD1A, whereas overexpression of enzymes that catalyse addition of repressive marks, like EHMT2 and EZH2, was found in NSs. Almstrup and co-workers [52] reported that SEs disclose high levels of selective repressive modifications. Lambrot et al [53], and other authors [54,55], observed that SMYD3, an H3K4 methyltransferase, was upregulated in NS-like NT2 and NCCIT cell lines.
Altered ChRCs dynamics have seldom been addressed in TGCTs, with Jostes et al [56] recently reporting that BRD2, BRD3 and BRD4 are overexpressed in TGCTs.
Overview of ‘epidrugs’ over time: histone-modifying enzymes and bromodomain inhibitors in TGCTs
Histone-modifying enzyme inhibitors, such as HDACis, KMT inhibitors (KMTis), KDM inhibitors (KDMis) and bromodomain inhibitors, have been synthetized and explored in several tumour types. Until now, FDA has approved four HDACis for treatment of haematological malignancies: vorinostat (SAHA), romidepsin (FK288), panobinostat (LBH589) and belinostat (PXD101) [57,58], although many more compounds are under testing in clinical trials and for several malignancies. An advantage of these inhibitors (for instance, HDACis) is its specificity, with some agents being pan-inhibitors (i.e., inhibiting all classes of HDACs, from I to IV), such as vorinostat, and others being more specific (such as entinostat, a class I HDACi) [58]. Moreover, some drugs are amenable to be repurposed for targeting HDACs and treat cancer, such as valproic acid (a class I and IIa HDACi), which is routinely used for treating epilepsy [57]. For the time being, no histone-modifying enzyme inhibitors have been approved for treatment of TGCT patients, yet, but they have been a subject of scientific interest, and some studies are already exploring these agents (Table 1).
The first study using histone-modifying enzyme inhibitors for cancer treatment dates from 1997 when Minucci et al [59] exposed P19 cell lines to TSA in combination with retinoic acid (which induces differentiation). Authors found TSA to synergistically promote differentiation when combined with retinoic acid, activating the transcription of retinoic acid responsive promoters [35]. Interestingly, Wang and collaborators [60] synthesized a KDM-inhibitor (CBB), which specifically blocked LSD1 activity, but not other demethylases, such as LSD2 and JARID1A. Again, the compound could induce the expression of genes responsible for differentiation in TGCT cell lines. Indeed, high LSD1 levels associated with overexpression of OCT3/4 and SOX2, two well-known pluripotency genes. In the same line, Hoang and co-workers [61] synthesized another drug targeting LSD1 (CBB3001) and verified that F9 cell lines developed growth arrest and downregulation of SOX2 and OCT3/4.
Subsequently, in 2011, GTAp63 was shown to be downregulated during the development of TGCTs [62]. GTAp63 is a transcriptionally active isoform of p63, localized in the endogenous retrovirus type 9 (ERV9)-LTR which is able to induce apoptosis upon genotoxic stress [63]. In TGCTs, this process can be restored by HDACs’ inhibition. TGCT cell lines firstly treated with TSA and SAHA endured cell death when exposed to cisplatin, mediated by induction of GTAp63 transcription (demonstrating a synergistic effect between HDACis and cisplatin) [64].
More recently, pre-clinical studies have focused on more newly approved HDAC inhibitors, namely romidepsin. Nettersheim and co-workers [65] used this drug, already shown to induce apoptosis in TCam-2 cells [66], for treating cisplatin-resistant TGCT cells lines with distinct p53 mutational status, and demonstrated a reduction of cell viability at low nanomolar concentrations, supported by tumour growth inhibition in an in vivo mouse model. The proposed mechanism of action comprises a chromatin remodeller, ARID1A, whose inhibition consequently triggers apoptosis and induces cell cycle arrest. Mechanistically, high DUSP1 levels in response to romidepsin blocked the MAPK/ERK cascade, leading to decreased pRBS5 levels and, subsequently, to cell cycle arrest and apoptosis. Importantly, authors also tested the effects in non-GCT cell lines (fibroblasts and Sertoli cell lines), in which apoptosis was not triggered, suggesting that toxicity in other tissues is likely to be low. Since class I HDACs seem to be the particularly relevant in TGCT, future studies using class I-specific HDACi would be of interest. Moreover, combination of the latter with immunotherapies could be particularly effective, following the immunomodulatory supportive data on a phase I/II clinical trial on renal cell carcinoma [67]. In this line, follow-up on studies, including one on lung cancer which used newly synthetized inhibitors that overcame drug resistance, could be relevant for TGCT treatment, particularly given the inhibitory interaction with the pluripotency factor SOX2, fitting well with the TGCT model [68].
Recently, the same group mentioned above [56] reported promising effects of the bromodomain inhibitor JQ1. JQ1 is a small molecule that inhibits the binding pocket of bromodomains, mainly BRD4, which interferes with the histone code. TGCT cells treated with JQ1 endured increased apoptosis and growth arrest, along with differentiation. Importantly, in mice, JQ1 treatment associated with reduction of tumour size and blood vessel density, suggesting an anti-angiogenic effect; we hypothesize that combination with anti-angiogenic agents, such as VEGF inhibitor sunitinib, should be explored, considering the previously reported effects of this drug in TGCTs, including cisplatin-resistant tumours [69]. Moreover, this might also allow for a dose reduction, avoiding the reported apoptosis observed in a Sertoli cell line (but not in fibroblasts), which suggests toxicity for testis microenvironment.
Drugs that have dual effects on cells could also be of use. Steinemann et al [70] tested a single drug (animacroxam) that targets two distinct molecules: HDACs and cellular cytoskeletal dynamics. As an HDACi, animacroxam induced apoptosis and G0-G1 phase arrest in TGCT cell lines, along with induction of cytoskeletal fibres’ stress. The latter reduced cell migration by 96%, suggesting that it might reduce the propensity for metastization. Drugs like animacroxam or combinations between different agents/treatments with similar properties have shown promising results and should be further explored.
Non-coding RNAs in TGCTs: are these possible therapeutic targets?
In the past few years, non-coding RNAs (ncRNAs), mainly microRNAs (miRNAs), have gained special attention, as they have been acknowledged as key gene expression regulators [71]. Specifically, miRNAs are very attractive, since they can be easily detected in biofluids in a cost-effective manner, allowing for patient diagnosis and monitoring. They modulate gene expression post-transcriptionally by targeting specific messenger RNA (mRNA) molecules, functioning as oncomiRNAs or as tumour supressor miRNAs [72].
In TGCTs, the remarkable clinical impact of a set of embryonic miRNAs, especially miR-371a-3p, as a disease biomarker must be acknowledged. Whereas in several malignancies issues like tissue/cell specificity have been of concern in liquid biopsy studies, TGCTs (and GCTs in general) are fortunate since a cluster of miRNAs (miR-371-3 and miR-367-3p, in particular) are involved in regulation of embryonic development. Hence high levels are detectable across TGCT subtypes (except mature teratoma), but low or completely absent in healthy males or carriers of other conditions, as shown in several clinical settings and in large retrospective and prospective multicentric studies [73,74] (for a review see [75]). Based on its performance, miR-371a-3p is deemed to be available for clinical use within the near future.
However, if miR-371a-3p (and miRNAs, in general), are currently revolutionizing the field owing to their biomarker capabilities, authors also foresee the potential of these molecules to be used as therapeutic vehicles [76]. Because miRNAs can target many mRNA segments simultaneously, they might affect the transcript levels of several players and target several pathways; because the mechanisms of cisplatin resistance are most likely multifactorial, miRNA modulation may more efficiently target the cisplatin resistant phenotype. This field of research is growing, with novel ways of conceiving synthetic miRNAs and, especially, of delivering them selectively to tumour cells under active development (like via nanoparticles) [77]. Given the set of embryonic miRNAs regulating TGCTs biology, there is a rationale for pursuing miRNAs as an additional form of epigenetic-based therapy. Indeed, miRNA-based therapies are real, considering the phase II study with delivery of an anti-miR-122 (miravirsen) to hepatitis C patients, demonstrating prolonged reduction of viral RNA levels [78]. On the other hand, instead of using anti-miRs, replenishing the downregulated miRNAs might also be envisioned and has been already attempted, with delivery of miR-16 to patients with lung cancer and pleural malignant mesothelioma [79]. Murray et al report an example of this: a mimic of let-7 (which is downregulated in TGCTs) delivered to TGCT cells was able to activate anti-tumour mRNA targets and reduce tumour growth [80]. Indeed, miRNA-based therapies are already being pursued by several groups (and taken over by specific companies), using both the miRNA inhibition or miRNA restoration strategies, with published works in both pre-clinical and clinical settings, including solid cancers (renal cell carcinoma, melanoma, hepatocellular carcinoma), as summarized in [81].
Overall, miR-125b, miR-302a, b, c and d, miR-371, miR-372, miR-373 and miR-375 have been implicated in TGCT tumorigenesis. MiR-125b is considered a tumour suppressor miRNA that regulates several mechanisms, like proliferation, apoptosis and, importantly, pluripotency. In TGCTs, low miR-125b levels associated with tumour growth. Importantly, miR-125b also led to pro-tumorigenic macrophage recruitment to the microenvironment [82] and to inhibition of tumour-derived chemokines, contributing to decreased tumour growth [82]. This way, strategies for replenishing miR-125b within these tumours could be have therapeutic effect and potentiate the action of immunotherapies (an expanding field in itself) by recruiting immune cells to the microenvironment and making it more ‘inflamed’.
MiR-375 has been recently considered a promising marker for teratoma (the histology left undetected by miR-371a-3p, and for which clinical attitude may be different) [6], but this has been contradicted in liquid biopsies [73]. Remarkably, miR-375 and miR-302a/b/c/d may disrupt the TP53 pathway, leading to development of TGCTs and may, thus, constitute valuable therapeutic targets [83]. Of notice, whereas miRNAs of the 371–3 cluster disrupt the p53 pathway (by targeting LATS2) [84], miR-885-5p (a p53 activator) was shown to be highly expressed in mature teratomas, which are, by definition, resistant to cisplatin. A miRNA switch (371a-3p to 885–5p) was proposed that might be involved in the process of differentiation, possibly amenable for therapeutic targeting (as seen for the above-mentioned ‘epidrugs’, many of them influencing differentiation) [73]. Also, this might be explored together with other agents aimed at targeting the p53 pathway, namely Nutlin-3, a Mdm2 inhibitor (with Mdm2, in its turn, responsible for targeting p53 for degradation) [16]. Moreover, miR-302 is overexpressed in TGCTs, acting as an oncogene, inducing SPRY4 expression and, consequently, activating MAPK/ERK pathway, which is relevant in TGCTs. Of note, it has been demonstrated that its inhibition results in decreased proliferation of TGCT cell lines [85]. Finally, Chen and collaborators [86] also demonstrated that miRNAs could also influence the methylation status of relevant genes (mentioned above in the DNMTi section); mir-199a-3p overexpression restored APC and MGMT expression in NT2 cells, affecting the methylation status of their promoters, disclosing the crosstalk between epigenetic mechanisms. Overall, therapies with miRNAs still face multiple challenges, related to long-term effects, toxicities and need for better delivery options; however, we do believe that the dependence of these tumours on a more specific cluster of miRNAs constitutes an ideal tumour model for therapeutic targeting using these strategies.
Future perspectives: in which direction are TGCTs epigenetic-based treatments moving?
Over the last years, epigenetic therapies have gained special attention for cancer treatment and have been increasingly studied, with the ultimate goal of enhancing specificity and selectivity, while decreasing the side effects (Figure 4). Moreover, it is acknowledged that ‘epidrugs’ exhibit less toxicity than conventional chemotherapy [23]. To date, only a limited number of trials have included TGCT patients, which should deserve more attention, especially those acquiring cisplatin resistance for which no curative treatments are available. Although in pre-clinical studies, these drugs used in monotherapy appear to have an anti-cancer effect, no significant efficacy was demonstrated in the clinical trials. This could be due to several reasons, including: epigenetic therapies do not have an immediate effect, since these drugs cause reprogramming of cancer cells, initiating a long-term anti-neoplastic action; the appropriate dosage may not have been achieved; and the clinical context and population characteristics may not have been the most adequate (e.g., refractory and advanced stage disease). To our view, this should not discourage work in the field; on the contrary, it should prompt researchers for tuning their experimental settings, think on combination therapies and find the optimal treatment doses and schemes.
Figure 4.

Overview of current challenges in testicular germ cell tumours and the putative role of epidrugs
Combination therapies (not only with epigenetic agents, but also with these drugs and other therapies) may be the key issue, by triggering a synergistic effect. Indeed, the combination of different ‘epidrugs’ was considered very promising [35,46]. Steele et al demonstrated that the combination of belinostat with decitabine enhanced the effect of rescuing sensitivity to cisplatin in ovarian cancer cell lines [87], suggesting that the same may be possible for TGCTs. Indeed, belinostat was well tolerated in a phase II trial enrolling women with platinum-resistant ovarian cancer [88]. Dual inhibitors could also be envisioned, such as HDAC1 and LSD1 dual inhibitor Corin, which was shown to reduce tumour growth in melanoma cell lines and in mouse models [89]. On the other hand, the combination between ‘epidrugs’ and cisplatin, in addition to increase the therapeutic sensitivity to this chemotherapeutic agent, allows for the reduction of cisplatin dose, avoiding intense treatments and reducing the associated toxicities [90]. This should be the way to go, in a field where cisplatin will continue to be the mainstay drug for treating TGCT patients, but also at the cost of building on short-term and long-term side effects, including metabolic syndrome, cardiovascular disease, hearing loss, renal toxicity, secondary malignancies, etc [11].
The immune landscape of tumours is also epigenetically regulated [91]. Combinations of ‘epidrugs’ with immunotherapies, which have witnessed remarkable progress in several tumours, might be advantageous, as mentioned above. Indeed, the inflammatory infiltrate present in TGCTs is very rich and distinct among subtypes [92,93]. Notwithstanding that recent clinical trials with immunotherapies have not shown impressive results [93,94], combinations with epigenetic-based treatments may be beneficial, as illustrated by the results of combining HDACis with immunotherapies in urological malignancies [95,96]. In other words, epigenetic ‘priming’ of tumours, facilitating acquisition of a more inflamed tumour microenvironment, may be the required pre-treatment for taking maximal advantage of immune checkpoint inhibitors (and this is currently being explored in multiple clinical trials, summarized in [97]).
Natural compounds are also a source of potential anti-cancer agents. They may influence several biological processes, including epigenetic mechanisms, through which they might exert the anti-neoplastic properties. Importantly, they are usually associated with low toxicity, as many are included in the diet [23]. In TGCTs, the encouraging results of curcumol upon tumour cells [35], pave the way for more intense research concerning the efficacy for treatment of TGCT patients. Recently, CRISPR-Cas9 technology for epigenetic silencing of aberrantly demethylated epigenetic mediators was indicated as promising therapy [98], but more studies are required to confirm its efficacy, safety and reliability.
Considering the promising results in pre-clinical studies, and the still reduced number of clinical trials and of study participants with TGCT, a more robust approach is needed. Hence, studies with larger patient cohorts and with different previous treatments and comorbidities, are needed to elucidate and validate these results. Importantly, these studies should include different TGCTs types, as well as detailed clinical and pathological characterization and appropriate endpoints. Indeed, it should be recalled that TGCTs are quite heterogeneous neoplasms, each subtype bearing distinct epigenetic alterations that should be considered when designing and selecting ‘epidrugs’.
With the increasing understanding of epigenetic regulation mechanisms, we believe that there are still many opportunities for targeted treatment with epigenetic-based strategies. For instance, the expanding field of proteolysis targeting chimeras (PROTACs) could be useful for epigenetics research [99], by inducing degradation of specific proteins (like the androgen receptor, through use of a non-steroidal androgen receptor ligand connected to the Mdm2 ligand Nutlin, which leads to ubiquitination of the receptor and consequent degradation [100]). Remarkably, Mdm2 (which is frequently amplified in TGCTs with cisplatin resistance) is one of the E3 ubiquitin ligases used in this technology. Indeed, ubiquitination is nothing less than a post-translational modification, still seldom explored in TGCTs; our in silico analysis of TCGA database disclosed differential expression of ubiquitin ligases and deubiquitinating enzymes according to TGCT subtype, and some have shown impact on patient survival [25], meaning that these could also be explored in the future. Moreover, Oing et al demonstrated that a distinct histone mark, monoubiquitination of Lys120 (H2Bub1), was associated with cisplatin resistance, and targeting it with a specific inhibitor like LDC000067 (a CDK9 inhibitor) was shown to increase sensitivity to DNA damage by cisplatin and radiation, meaning that ubiquitination should be another post-translational modification to be explored in TGCTs [101]. Importantly, most PROTACs aimed at targeting epigenetic players also target chromatin remodelling complexes, such as BRD4, the same complex targeted by JQ1, already shown to be effective in vitro and in vivo in TGCTs (see above). It remains to be seen if the higher specificity of this therapy may overcome the toxicity demonstrated by JQ1 [56]. More PROTACs targeting non-BRD4 epigenetic proteins are being explored, such as those involved in other chromatin remodellers (SWI/SNF and SMARC2/4 complexes) and other enzymes such as HDACs (like for SIRT2 [102] and HDAC6 [103]) and PCAF/GCN5 [104].
In another setting, ncRNAs might also be envisioned as therapeutic opportunities, including in chemotherapy resistance, considering their involvement in several pathways frequently triggered upon the resistant phenotype is reached [105]. This was already mentioned for miRNAs, with miR-34a being the first of these therapies introduced in the clinic, as this ‘all-around’ miRNA targets many tumour-prone pathways, including cyclin-dependent kinases, SIRT1 and SOX2 and is efficiently delivered in liposomal nanoparticles. Currently, long non-coding RNAs (lncRNAs) are also in clinical trials for cancer treatment (summarized in [105]). These have been less explored in TGCTs, and deserve further studies, also exploring novel ways of delivery (lipid nanoparticles, but also carriers and oncolytic adenoviruses). Moreover, one of most expanding fields in recent years has been the niche of RNA modifications, namely methylation of adenosine 6 (m6A) [106]. There is a current competitive race to effectively drug and target these modifications, which are fundamental for cancer development across all tumour types [71]. We and others have explored the role of these modifications and respective enzymes in TGCTs [107,108], showing that they are related to differentiation. Currently, synthesis of novel small inhibitors of the m6A writer METTL3 was already achieved and effectively used for treating acute myeloid leukaemia cells [109]; in a fast-progressing field, it is expected that novel inhibitors are uncovered, and given the high expression of these players in TGCTs, they might be optimally tested in this tumour model (Figure 5).
Figure 5.

Overview of future directions in epigenetic-based therapies for testicular germ cell tumour patients
The spectrum of available tools for modulating and studying epigenetic landscape is developing at high pace and leading to discovery of versatile ways to probe chromatin, including chemical biology tools such as fluorescent ligands, chemical dimerizers, phase separation disruptors, among others. All in all, there are still a lot of opportunities within the ‘chemical biology toolkit’ for taking advantage of epigenetic features and looking at them as therapeutic opportunities in TGCTs [110].
Acknowledgements
AC and JL collected the information and drafted the manuscript. VMG, RH and CJ supervised the work and reviewed the manuscript. AC drafted the figures.
Funding Statement
The authors would like to acknowledge the support of the Programa Operacional Competitividade e Internacionalização (POCI), in the component FEDER, and by national funds (OE) through FCT/MCTES, in the scope of the project EpiMarkGermCell (PTDC/MECONC/29043/2017). JL is recipient of a fellowship from FCT - Fundação para a Ciência e Tecnologia—(SFRH/BD/132751/2017). VM-G contract is funded by POCI-01-0145-FEDER-29043.
Disclosure of interest
The authors declare that they have no conflicts of interest.
References
- [1].Lobo J, Gillis AJM, Jeronimo C, et al. Human germ cell tumors are developmental cancers: impact of epigenetics on pathobiology and clinic. Int J Mol Sci. 2019;20(2):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rijlaarsdam MA, Tax DM, Gillis AJ, et al. Genome wide DNA methylation profiles provide clues to the origin and pathogenesis of germ cell tumors. PLoS One. 2015;10:e0122146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- [4].Oosterhuis JW, Looijenga LHJ.. Human germ cell tumours from a developmental perspective. Nat Rev Cancer. 2019;19:522–537. [DOI] [PubMed] [Google Scholar]
- [5].Costa AL, Lobo J, Jeronimo C, et al. The epigenetics of testicular germ cell tumors: looking for novel disease biomarkers. Epigenomics. 2017;9:155–169. [DOI] [PubMed] [Google Scholar]
- [6].Shen H, Shih J, Hollern DP, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018;23:3392–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Costa LA, Barbosa CM, Lobo J, et al. DNA methylation profiling as a tool for testicular germ cell tumors subtyping. Epigenomics. 2018;10(12):1511–1523. [DOI] [PubMed] [Google Scholar]
- [8].Moch HUT, Reuter V, Eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. 4th ed. Lyon: IARC; 2016. [Google Scholar]
- [9].Wagner T, Toft BG, Engvad B, et al. Prognostic factors for relapse in patients with clinical stage i testicular cancer: protocol for a danish nationwide cohort study. BMJ Open. 2019;9:e033713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lobo J, Stoop H, Gillis AJM, et al. Interobserver agreement in vascular invasion scoring and the added value of immunohistochemistry for vascular markers to predict disease relapse in stage i testicular nonseminomas. Am J Surg Pathol. 2019;43:1711–1719. [DOI] [PubMed] [Google Scholar]
- [11].Chovanec M, Abu Zaid M, Hanna N, et al. Long-term toxicity of cisplatin in germ-cell tumor survivors. Ann Oncol. 2017;28:2670–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moul JW, Dodge RK, Robertson JE, et al. The impact of the “cisplatin era” of treatment on survival in testicular cancer. World J Urol. 1991;9:45–50. [Google Scholar]
- [13].Schmidtova S, Kalavska K, Kucerova L. Molecular mechanisms of cisplatin chemoresistance and its circumventing in testicular germ cell tumors. Curr Oncol Rep. 2018;20:88. [DOI] [PubMed] [Google Scholar]
- [14].Cierna Z, Miskovska V, Roska J, et al. Increased levels of xpa might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer. 2020;20:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Honecker F, Wermann H, Mayer F, et al. Microsatellite instability, mismatch repair deficiency, and braf mutation in treatment-resistant germ cell tumors. J Clin Oncol. 2009;27:2129–2136. [DOI] [PubMed] [Google Scholar]
- [16].Bauer S, Muhlenberg T, Leahy M, et al. Therapeutic potential of mdm2 inhibition in malignant germ cell tumours. Eur Urol. 2010;57:679–687. [DOI] [PubMed] [Google Scholar]
- [17].Jacobsen C, Honecker F. Cisplatin resistance in germ cell tumours: models and mechanisms. Andrology. 2015;3:111–121. [DOI] [PubMed] [Google Scholar]
- [18].Oing C, Verem I, Mansour WY, et al. 5-azacitidine exerts prolonged pro-apoptotic effects and overcomes cisplatin-resistance in non-seminomatous germ cell tumor cells. Int J Mol Sci. 2018;20(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brait M, Maldonado L, Begum S, et al. DNA methylation profiles delineate epigenetic heterogeneity in seminoma and non-seminoma. Br J Cancer. 2012;106:414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Singh R, Fazal Z, Freemantle SJ, et al. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019;2:580–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kelly AD, Issa JJ. The promise of epigenetic therapy: reprogramming the cancer epigenome. Curr Opin Genet Dev. 2017;42:68–77. [DOI] [PubMed] [Google Scholar]
- [22].Azad N, Zahnow CA, Rudin CM, et al. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol. 2013;10:256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Marques-Magalhaes A, Graca I, Henrique R, et al. Targeting DNA methyltranferases in urological tumors. Front Pharmacol. 2018;9:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sharif J, Muto M, Takebayashi S, et al. The sra protein np95 mediates epigenetic inheritance by recruiting dnmt1 to methylated DNA. Nature. 2007;450:908–912. [DOI] [PubMed] [Google Scholar]
- [25].Lobo J, Henrique R, Jeronimo C. The role of DNA/histone modifying enzymes and chromatin remodeling complexes in testicular germ cell tumors. Cancers (Basel). 2018;11(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guo F, Li X, Liang D, et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell. 2014;15:447–459. [DOI] [PubMed] [Google Scholar]
- [27].Wermann H, Stoop H, Gillis AJ, et al. Global DNA methylation in fetal human germ cells and germ cell tumours: association with differentiation and cisplatin resistance. J Pathol. 2010;221:433–442. [DOI] [PubMed] [Google Scholar]
- [28].Lind GE, Skotheim RI, Fraga MF, et al. Novel epigenetically deregulated genes in testicular cancer include homeobox genes and scgb3a1 (hin-1). J Pathol. 2006;210:441–449. [DOI] [PubMed] [Google Scholar]
- [29].Spiller CM, Gillis AJ, Burnet G, et al. Cripto: expression, epigenetic regulation and potential diagnostic use in testicular germ cell tumors. Mol Oncol. 2016;10:526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Koul S, McKiernan JM, Narayan G, et al. Role of promoter hypermethylation in cisplatin treatment response of male germ cell tumors. Mol Cancer. 2004;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Martinelli C, Lengert AVH, Carcano FM, et al. Mgmt and calca promoter methylation are associated with poor prognosis in testicular germ cell tumor patients. Oncotarget. 2017;8:50608–50617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lobo J, Nunes SP, Gillis AJM, et al. Xist-promoter demethylation as tissue biomarker for testicular germ cell tumors and spermatogenesis quality. Cancers (Basel). 2019;11(9):1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Benesova M, Trejbalova K, Kucerova D, et al. Overexpression of tet dioxygenases in seminomas associates with low levels of DNA methylation and hydroxymethylation. Mol Carcinog. 2017;56:1837–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nettersheim D, Heukamp LC, Fronhoffs F, et al. Analysis of tet expression/activity and 5mc oxidation during normal and malignant germ cell development. PLoS One. 2013;8:e82881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Peng Z, Zhou W, Zhang C, et al. Curcumol controls choriocarcinoma stem-like cells self-renewal via repression of DNA methyltransferase (dnmt)- and histone deacetylase (hdac)-mediated epigenetic regulation. Med Sci Monit. 2018;24:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang X, Lay F, Han H, et al. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Beyrouthy MJ, Garner KM, Hever MP, et al. High DNA methyltransferase 3b expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 2009;69:9360–9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Juttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2ʹ-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A. 1994;91:11797–11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wongtrakoongate P. Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries. World J Stem Cells. 2015;7:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Biswal BK, Beyrouthy MJ, Hever-Jardine MP, et al. Acute hypersensitivity of pluripotent testicular cancer-derived embryonal carcinoma to low-dose 5-aza deoxycytidine is associated with global DNA damage-associated p53 activation, anti-pluripotency and DNA demethylation. Plos One. 2012;7:e53003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Albany C, Hever-Jardine MP, von Herrmann KM, et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget. 2017;8:2949–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moreira-Silva F, Camilo V, Gaspar V, et al. Repurposing old drugs into new epigenetic inhibitors: promising candidates for cancer treatment? Pharmaceutics. 2020;12(5):410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Roth BJ, Elson P, Sledge GW, et al. 5-azacytidine (nsc 102816) in refractory germ cell tumors. Invest New Drugs. 1993;11:201–202. [DOI] [PubMed] [Google Scholar]
- [44].Quagliana JM, O’Bryan RM, Baker L, et al. Phase ii study of 5-azacytidine in solid tumors. Cancer Treat Rep. 1977;61:51–54. [PubMed] [Google Scholar]
- [45].Graca I, Sousa EJ, Costa-Pinheiro P, et al. Anti-neoplastic properties of hydralazine in prostate cancer. Oncotarget. 2014;5:5950–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Candelaria M, Gallardo-Rincon D, Arce C, et al. A phase ii study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol. 2007;18:1529–1538. [DOI] [PubMed] [Google Scholar]
- [47].Chua GNL, Wassarman KL, Sun H, et al. Cytosine-based tet enzyme inhibitors. ACS Med Chem Lett. 2019;10:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Iizuka M, Smith MM. Functional consequences of histone modifications. Curr Opin Genet Dev. 2003;13:154–160. [DOI] [PubMed] [Google Scholar]
- [49].Langst G, Manelyte L. Chromatin remodelers: from function to dysfunction. Genes (Basel). 2015;6:299–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fritzsche FR, Hasler A, Bode PK, et al. Expression of histone deacetylases 1, 2 and 3 in histological subtypes of testicular germ cell tumours. Histol Histopathol. 2011;26:1555–1561. [DOI] [PubMed] [Google Scholar]
- [51].Hinz S, Magheli A, Weikert S, et al. Deregulation of ezh2 expression in human spermatogenic disorders and testicular germ cell tumors. World J Urol. 2010;28:631–635. [DOI] [PubMed] [Google Scholar]
- [52].Almstrup K, Nielsen JE, Mlynarska O, et al. Carcinoma in situ testis displays permissive chromatin modifications similar to immature foetal germ cells. Br J Cancer. 2010;103:1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lambrot R, Kimmins S. Histone methylation is a critical regulator of the abnormal expression of pou5f1 and rassf1a in testis cancer cell lines. Int JAndrology. 2011;34:110–123. [DOI] [PubMed] [Google Scholar]
- [54].Buljubasic R, Buljubasic M, Bojanac AK, et al. Epigenetics and testicular germ cell tumors. Gene. 2018;661:22–33. [DOI] [PubMed] [Google Scholar]
- [55].Litchfield K, Levy M, Orlando G, et al. Identification of 19 new risk loci and potential regulatory mechanisms influencing susceptibility to testicular germ cell tumor. Nat Genet. 2017;49:1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jostes S, Nettersheim D, Fellermeyer M, et al. The bromodomain inhibitor jq1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J Cell Mol Med. 2017;21:1300–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yoon S, Eom GH. Hdac and hdac inhibitor: from cancer to cardiovascular diseases. Chonnam Med J. 2016;52:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Eckschlager T, Plch J, Stiborova M, et al. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7):1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Minucci S, Horn V, Bhattacharyya N, et al. A histone deacetylase inhibitor potentiates retinoid receptor action in embryonal carcinoma cells. Proc Natl Acad Sci U S A. 1997;94:11295–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang J, Lu F, Ren Q, et al. Novel histone demethylase lsd1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011;71:7238–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hoang N, Zhang X, Zhang C, et al. New histone demethylase lsd1 inhibitor selectively targets teratocarcinoma and embryonic carcinoma cells. Bioorg Med Chem. 2018;26:1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Beyer U, Moll-Rocek J, Moll UM, et al. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc Natl Acad Sci U S A. 2011;108:3624–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu M, Eiden MV. Role of human endogenous retroviral long terminal repeats (ltrs) in maintaining the integrity of the human germ line. Viruses. 2011;3:901–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Beyer U, Kronung SK, Leha A, et al. Comprehensive identification of genes driven by erv9-ltrs reveals tnfrsf10b as a re-activatable mediator of testicular cancer cell death. Cell Death Differ. 2016;23:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nettersheim D, Jostes S, Fabry M, et al. A signaling cascade including arid1a, gadd45b and dusp1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget. 2016;7:74931–74946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nettersheim D, Gillis A, Biermann K, et al. The seminoma cell line tcam-2 is sensitive to hdac inhibitor depsipeptide but tolerates various other chemotherapeutic drugs and loss of nanog expression. Genes Chromosomes Cancer. 2011;50:1033–1042. [DOI] [PubMed] [Google Scholar]
- [67].Pili R, Quinn DI, Hammers HJ, et al. Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: A multicenter, single-arm, phase i/ii trial (nci-ctep#7870). Clin Cancer Res. 2017;23:7199–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bora-Singhal N, Mohankumar D, Saha B, et al. Novel hdac11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing sox2. Sci Rep. 2020;10:4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Castillo-Avila W, Piulats JM, Garcia Del Muro X. Sunitinib inhibits tumor growth and synergizes with cisplatin in orthotopic models of cisplatin-sensitive and cisplatin-resistant human testicular germ cell tumors. Clin Cancer Res. 2009;15:3384–3395. [DOI] [PubMed] [Google Scholar]
- [70].Steinemann G, Dittmer A, Kuzyniak W, et al. Animacroxam, a novel dual-mode compound targeting histone deacetylases and cytoskeletal integrity of testicular germ cell cancer cells. Mol Cancer Ther. 2017;16:2364–2374. [DOI] [PubMed] [Google Scholar]
- [71].Lobo J, Barros-Silva D, Henrique R, et al. The emerging role of epitranscriptomics in cancer: focus on urological tumors. Genes (Basel). 2011;26(11):552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kasinski AL, Slack FJ. Epigenetics and genetics. Micrornas en route to the clinic: progress in validating and targeting micrornas for cancer therapy. Nat Rev Cancer. 2011;11:849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lobo J, Gillis AJM, van den Berg A, et al. Identification and validation model for informative liquid biopsy-based microrna biomarkers: insights from germ cell tumor in vitro, in vivo and patient-derived data. Cells. 2019;8(12):1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nappi L, Thi M, Lum A, et al. Developing a highly specific biomarker for germ cell malignancies: plasma mir371 expression across the germ cell malignancy spectrum. J Clin Oncol. 2019;37:3090–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Almstrup K, Lobo J, Morup N, et al. Application of mirnas in the diagnosis and monitoring of testicular germ cell tumours. Nat Rev Urol. 2020;4:201–213. DOI: 10.1038/s41585-020-0296-x [DOI] [PubMed] [Google Scholar]
- [76].Murray MJ, Coleman N. Microrna dysregulation in malignant germ cell tumors: more than a biomarker? J Clin Oncol. 2019;37:1432–1435. [DOI] [PubMed] [Google Scholar]
- [77].Rupaimoole R, Slack FJ. Microrna therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–222. [DOI] [PubMed] [Google Scholar]
- [78].Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR . Treatment of hcv infection by targeting microrna. N Engl J Med. 2013;368:1685–1694. [DOI] [PubMed] [Google Scholar]
- [79].van Zandwijk N, Pavlakis N, Kao SC, et al. Safety and activity of microrna-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017;18:1386–1396. [DOI] [PubMed] [Google Scholar]
- [80].Murray MJ, Saini HK, Siegler CA, et al. Cclg. Lin28 expression in malignant germ cell tumors downregulates let-7 and increases oncogene levels. Cancer Res. 2013;73:4872–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Shah MY, Ferrajoli A, Sood AK, et al. Microrna therapeutics in cancer - an emerging concept. EBioMedicine. 2016;12:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Batool A, Wang YQ, Hao XX, et al. A mir-125b/csf1-cx3cl1/tumor-associated macrophage recruitment axis controls testicular germ cell tumor growth. Cell Death Dis. 2018;9:962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Boublikova L, Buchler T, Stary J, et al. Molecular biology of testicular germ cell tumors: unique features awaiting clinical application. Crit Rev Oncol Hematol. 2014;89:366–385. [DOI] [PubMed] [Google Scholar]
- [84].Voorhoeve PM, le Sage C, Schrier M, et al. A genetic screen implicates mirna-372 and mirna-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. [DOI] [PubMed] [Google Scholar]
- [85].Das MK, Evensen HSF, Furu K, et al. Mirna-302s may act as oncogenes in human testicular germ cell tumours. Sci Rep. 2019;9:9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Chen BF, Gu S, Suen YK, et al. Microrna-199a-3p, dnmt3a, and aberrant DNA methylation in testicular cancer. Epigenetics. 2014;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Steele N, Finn P, Brown R, et al. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br J Cancer. 2009;100:758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Mackay HJ, Hirte H, Colgan T, et al. Phase ii trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (lmp) ovarian tumours. Eur J Cancer. 2010;46:1573–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kalin JH, Wu M, Gomez AV, et al. Targeting the corest complex with dual histone deacetylase and demethylase inhibitors. Nat Commun. 2018;9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Oing C, Skowron MA, Bokemeyer C, et al. Epigenetic treatment combinations to effectively target cisplatin-resistant germ cell tumors: past, present, and future considerations. Andrology. 2019;7:487–497. [DOI] [PubMed] [Google Scholar]
- [91].Lobo J, Jeronimo C, Henrique R. Targeting the immune system and epigenetic landscape of urological tumors. Int J Mol Sci. 2020;21(3):829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lobo J, Rodrigues A, Guimaraes R, et al. Detailed characterization of immune cell infiltrate and expression of immune checkpoint molecules pd-l1/ctla-4 and mmr proteins in testicular germ cell tumors disclose novel disease biomarkers. Cancers (Basel). 2019;11(10):1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Mego M, Svetlovska D, Chovanec M, et al. Phase ii study of avelumab in multiple relapsed/refractory germ cell cancer. Invest New Drugs. 2019;37:748–754. [DOI] [PubMed] [Google Scholar]
- [94].Necchi A, Giannatempo P, Raggi D, et al. An open-label randomized phase 2 study of durvalumab alone or in combination with tremelimumab in patients with advanced germ cell tumors (apache): results from the first planned interim analysis. Eur Urol. 2019;75:201–203. [DOI] [PubMed] [Google Scholar]
- [95].Orillion A, Hashimoto A, Damayanti N, et al. Entinostat neutralizes myeloid-derived suppressor cells and enhances the antitumor effect of pd-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res. 2017;23:5187–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Shen L, Ciesielski M, Ramakrishnan S, et al. Class i histone deacetylase inhibitor entinostat suppresses regulatory t cells and enhances immunotherapies in renal and prostate cancer models. PLoS One. 2012;7:e30815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Sun W, Lv S, Li H, et al. Enhancing the anticancer efficacy of immunotherapy through combination with histone modification inhibitors. Genes (Basel). 2018;9(12):633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Vojta A, Dobrinic P, Tadic V, et al. Repurposing the crispr-cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Vogelmann A, Robaa D, Sippl W, et al. Proteolysis targeting chimeras (protacs) for epigenetics research. Curr Opin Chem Biol. 2020;57:8–16. [DOI] [PubMed] [Google Scholar]
- [100].Schneekloth AR, Pucheault M, Tae HS, et al. Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett. 2008;18(22):5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Oing C, Dyshlovoy S, Honecker F, et al. Ep-1618: monoubiquitinylated histone h2b as a potential target in treatment resistant germ cell tumors. Radiother Oncol. 2018;127:S871. [Google Scholar]
- [102].Schiedel M, Herp D, Hammelmann S, et al. Chemically induced degradation of sirtuin 2 (sirt2) by a proteolysis targeting chimera (protac) based on sirtuin rearranging ligands (sirreals). J Med Chem. 2018;61(2):482–491. [DOI] [PubMed] [Google Scholar]
- [103].Wu H, Yang K, Zhang Z, et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J Med Chem. 2019;62:7042–7057. [DOI] [PubMed] [Google Scholar]
- [104].Bassi ZI, Fillmore MC, Miah AH, et al. Modulating pcaf/gcn5 immune cell function through a protac approach. ACS Chem Biol. 2018;13:2862–2867. [DOI] [PubMed] [Google Scholar]
- [105].Wang WT, Han C, Sun YM, et al. Noncoding rnas in cancer therapy resistance and targeted drug development. J Hematol Oncol. 2019;12:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Esteve-Puig R, Bueno-Costa A, Esteller M. Writers, readers and erasers of rna modifications in cancer. Cancer Lett. 2020;474:127–137. [DOI] [PubMed] [Google Scholar]
- [107].Nettersheim D, Berger D, Jostes S, et al. N6-methyladenosine detected in rna of testicular germ cell tumors is controlled by mettl3, alkbh5, ythdc1/f1/f2, and hnrnpc as writers, erasers, and readers. Andrology. 2019;7:498–506. [DOI] [PubMed] [Google Scholar]
- [108].Lobo J, Costa AL, Cantante M, et al. M(6)a rna modification and its writer/reader virma/ythdf3 in testicular germ cell tumors: A role in seminoma phenotype maintenance. J Transl Med. 2019;17:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Tzelepis K, De Braekeleer E, Yankova E, et al. Pharmacological inhibition of the rna m6a writer mettl3 as a novel therapeutic strategy for acute myeloid leukemia. Blood. 2019;134:403. [Google Scholar]
- [110].Cermakova K, Hodges HC. Next-generation drugs and probes for chromatin biology: from targeted protein degradation to phase separation. Molecules. 2018;23(8):1958. [DOI] [PMC free article] [PubMed] [Google Scholar]