

Abstract
In previous work, participants with a G970R mutation in cystic fibrosis transmembrane conductance regulator (CFTR) (c.2908G>C) had numerically lower sweat chloride responses during ivacaftor treatment than participants with other CFTR gating mutations. The objective of this substudy was to characterize the molecular defect of the G970R mutation in vitro and assess the benefit of ivacaftor in participants with this mutation. This substudy assessed sweat chloride, spirometry findings, and nasal potential difference on and off ivacaftor treatment in three participants with a G970R/F508del genotype. Intestinal organoids derived from rectal biopsy specimens were used to assess ivacaftor response ex vivo and conduct messenger RNA splice and protein analyses. No consistent or meaningful trends were observed between on‐treatment and off‐treatment clinical assessments. Organoids did not respond to ivacaftor in forskolin‐induced swelling assays; no mature CFTR protein was detected in Western blots. Organoid RNA analysis demonstrated that 3 novel splice variants were created by G970R‐CFTR: exon 17 truncation, exons 13–15 and 17 skipping, and intron 17 retention. Functional and molecular analyses indicated that the c.2908G>C mutation caused a cryptic splicing defect. Organoids lacked an ex vivo response with ivacaftor and supported identification of the mechanism underlying the CFTR defect caused by c.2908G>C. Analysis of CFTR mutations indicated that cryptic splicing was a rare cause of mutation misclassification in engineered cell lines. This substudy used organoids as an alternative in vitro model for mutations, such as cryptic splice mutations that cannot be fully assessed using cDNA expressed in recombinant cell systems.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Participants with the G970R mutation in the CFTR gene have lower sweat chloride responses to ivacaftor treatment than do participants with G551D or other non–G551D‐CFTR gating mutations.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This substudy characterized the molecular defect of the G970R‐CFTR mutation in vitro and assessed the benefit of ivacaftor in participants with this mutation.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The functional and molecular analyses revealed that the G970R‐CFTR mutation is not responsive to ivacaftor treatment and identified the mechanism of the CFTR defect as a cryptic splicing defect.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ For mutations that cannot be fully assessed using recombinant cell systems, such as cryptic splicing defects, organoid in vitro assessments and RNA analyses can be used as alternatives to characterize mutations.
Cystic fibrosis (CF) is an autosomal recessive hereditary disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene that result in decreased quantity and/or function of the CFTR protein. 1 , 2 The CFTR protein is an epithelial chloride channel aiding in the regulation of salt and fluid absorption and secretion that is located in the epithelia of multiple organs, including the lungs, pancreas, intestinal tract, liver, and vas deferens. 3 , 4 , 5 , 6 CF is a multisystemic disease with clinical manifestations, including lung function decline, chronic airway infections, pancreatic insufficiency, and malnutrition. 7
Ivacaftor (Kalydeco(R); Vertex Pharmaceuticals, Boston, MA) is a CFTR modulator that increases chloride transport by potentiating the channel open probability (P0 (or gating)) of the CFTR protein at the epithelial cell surface. 8 Currently, ivacaftor is indicated in the United States for people aged ≥ 4 months with CF with ≥ 1 CFTR mutation that is responsive to ivacaftor based on clinical and/or in vitro assay data 9 ; the approved genotypes and ages vary in other regions. 10 , 11 , 12 In clinical studies of people with CF with eligible genotypes, ivacaftor treatment has been shown to achieve improvements in CFTR function, lung function, risk of pulmonary exacerbations, pancreatic function, and nutritional status. 13 , 14 , 15 , 16 , 17
Among > 340 known CF‐causing CFTR mutations identified, 10 have historically been classified as class III mutations that result in severe defects in CFTR channel gating: G551D, G178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P, and G1349D. 2 , 18 , 19 , 20 , 21 In Fischer rat thyroid (FRT) cells engineered to individually express complementary DNAs (cDNAs) that express CFTR proteins coding each of these mutations, chloride transport as determined by electrophysiological studies was < 5% of normal CFTR, despite the presence of mature CFTR protein at levels similar to those of normal CFTR. Ivacaftor increased the channel P0 of these 10 CFTR forms and caused an increase in CFTR‐mediated chloride transport to levels equivalent to > 10% above baseline. These in vitro data supported investigation of the potential clinical benefit with ivacaftor in people with CF with severe gating mutations. 8 In a clinical study of ivacaftor in participants with CF who had a non‐G551D severe gating mutation, participants with a G970R‐CFTR mutation had a numerically lower treatment response than participants with the other gating mutations. 14
Given this result, the molecular defect of the G970R mutation was further evaluated as part of the completed open‐label extension study KONTINUE. 15 Organoids, a cell culture technology platform, 22 enabled generation of an in vitro model from each participant that expresses the CFTR protein based on the genomic DNA rather than on a cDNA construct. These organoids were used for in vitro functional CFTR experiments to evaluate the response with ivacaftor and for further mechanistic studies to help clarify the range of functional defects caused by the G970R mutation.
METHODS
Clinical substudy design
The G970R‐CFTR mutation substudy was part of the KONTINUE (VX12‐770‐112; NCT01707290) open‐label study to evaluate the long‐term safety and efficacy of ivacaftor in participants with CF who completed the studies KONDUCT (VX11‐770‐110 (NCT01614457); participants with an R117H mutation), 15 KONNECTION (VX12‐770‐111 (NCT01614470); participants with a non‐G551D severe gating mutation), 14 and VX12‐770‐113 (NCT01685801; participants with phenotypic or molecular evidence of residual CFTR function). 23 Participants with a G970R mutation who had participated in KONNECTION and were enrolled in the ivacaftor treatment arm of KONTINUE had the option to participate in the G970R substudy. Participants received treatment with ≥ 12 weeks of ivacaftor 150 mg every 12 hours in KONTINUE before beginning the substudy.
The substudy included 1 on‐ivacaftor treatment visit (visit A) and 1 off‐ivacaftor treatment visit (visit B), separated by a ≥ 14‐day ivacaftor washout before and after which spirometry, sweat chloride levels, and nasal potential difference (NPD) were assessed. Rectal biopsies were performed at one visit (either visit A or B) for creation of intestinal organoid cell cultures. Ivacaftor treatment resumed after substudy visit B. The substudy was conducted in compliance with institutional review board regulations, International Conference on Harmonisation Good Clinical Practice guidelines, and the Declaration of Helsinki. All participants provided written informed consent or assent, as appropriate. Substudy visits were conducted at a single site (Universitair Ziekenhuis Gasthuisberg, Leuven, Belgium).
Outcome measures
The sweat chloride test was administered within 1 hour after the start of the NPD assessments. Sweat was collected using a Macroduct device (Wescor, Logan, UT), and samples were evaluated in a blinded fashion by ICON (Farmingdale, NY) using coulometric titration quantification.
Spirometry was performed according to the American Thoracic Society guidelines 24 before bronchodilator use and before ivacaftor dosing at visit A. Visit B spirometry was performed before ivacaftor treatment was restarted. NPD measurements were performed according to the Cystic Fibrosis Foundation Therapeutics TDN SOP 528.01 “Standardization of Measurement of Nasal Membrane Transepithelial Potential Difference (NPD).” NPD measurements were conducted 3 hours after ivacaftor dosing on visit A and before ivacaftor treatment was restarted at visit B. Tracings were evaluated in a blinded fashion at the University of Alabama at Birmingham.
Intestinal organoids
Rectal biopsy specimens were collected at either visit A or B and shipped overnight at 4°C to Hubrecht Organoid Technology (Utrecht, The Netherlands). Primary intestinal organoid cell cultures were generated as previously described. 22 As control samples to assess ivacaftor response, organoids from the Hubrecht Organoid Technology biorepository that were derived from participants with CF with the following CFTR mutations were used: positive controls were two organoid lines with the S1251N/F508del genotype (identified as S1251N/F508del (A) and S1251N/F508del (B)) and one organoid line with the G551D/F508del genotype; negative controls were three organoid lines with a stop codon/F508del genotype (identified as W1282X/F508del, Y1092X/F508del, and R1162X/F508del). Additionally, an organoid line with the genotype F508del/F508del and a CFTR wild‐type (WT) line were included as controls for Western blot and RNA analysis. All experiments using human tissues for organoid generation described herein were approved by the ethical committee at University Medical Center Utrecht (TcBio 14‐008). Informed consent for tissue collection and generation, storage, and use of the organoids was obtained from the participants.
Forskolin‐induced swelling assay
Forskolin‐induced swelling (FIS) assays were performed on the organoids as previously described. 25 A forskolin titration was performed using forskolin 0, 0.008, 0.02, 0.05, 0.12, 0.32, 0.8, 2, and 5 µmol/L with and without ivacaftor 3 µmol/L. An ivacaftor titration was performed using ivacaftor 0, 0.01, 0.03, 0.1, 0.3, 1, 3, and 10 µmol/L with forskolin 0.32 µmol/L.
Western blot
For CFTR protein detection, Western blots were performed as previously described. 25
RNA analysis
Bulk RNA sequencing data were generated by Genewiz, USA, from a single biological replicate of each organoid sample (G970R/F508del and F508del/F508del). Standard bulk extraction from organoid cell pellets was followed with a reverse transcriptase step primed off of the poly‐A with oligo‐dT to select for messenger RNA (mRNA). Sequencing data were aligned using STAR 26 and analyzed using StringTie 27 and IRFinder 28 to identify novel splice isoforms and intron retention. Abundance estimates for CFTR splice variants were made using reads overlapping splice junctions (Figure S1 ).
In addition, RNA was isolated with an RNeasy Mini Kit (Qiagen, Venlo, The Netherlands) at Hubrecht Organoid Technology and shipped to Vertex (San Diego, CA) for processing. RNA samples were incubated for 30 minutes at 37°C in the presence of RNase‐Free DNase (Qiagen) to avoid contamination with genomic DNA and repurified using an RNeasy Mini Kit prior to conversion to cDNA using an Omniscript RT Kit (Qiagen). Samples from healthy control (WT), F508del/F508del genotype, and W1282X/F508del genotype were included as controls. Primer sequences were as follows: forward primer 5′‐TCACCAGCACCAGTTCGTAT‐3′, 62 base pairs downstream of the 5′ splice junction of exon 17 (within exon 17), and reverse primer 5′‐AAGCCAGCACTGCCATTAGA‐3′, 69 base pairs downstream of the 5′ intron 17 splice site (within intron 17; Figure 1 ). Polymerase chain reaction (PCR) was performed using a ProFlex PCR System (Applied Biosystems, Foster City, CA). Amplification conditions were as follows: initial denaturation at 95°C for 5 minutes, denaturation at 95°C for 30 seconds, annealing at 56°C for 15 seconds, extension at 72°C for 15 seconds, and final extension at 72°C for 5 minutes. To visualize the resulting PCR product, one‐tenth of each PCR product was electrophoresed on a 3% agarose gel with ethidium bromide. Images were captured with a GelDoc‐It Imaging System (Analytik Jena, Upland, CA).
Figure 1.
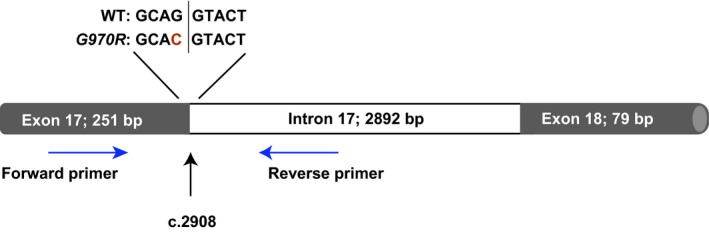
Schematic indicating primer binding locations. The primer set indicated by the blue arrows amplified a 278‐bp product, and a product would only be generated from messenger RNA that retained the canonical exon 17 donor site. The position of the variant encoding the G970R mutation (c.2908) is indicated in red text. bp, base pair; WT, wild type.
CFTR sequencing
Organoid samples were provided to Cergentis (Utrecht, The Netherlands) for targeted locus amplification and sequencing of the CFTR gene. DNA was analyzed according to methods previously published. 29 Biallelic sequencing confirmed the c.1521_1523delCTT (F508del) and c.2908G>C (G970R) mutations. No additional exonic mutations, which might indicate the presence of a complex allele, were found.
Statistical analysis
Given the small number of participants in this substudy, no statistical analysis of the clinical data was performed.
RESULTS
Clinical assessment of participants with CF with a G970R‐CFTR mutation
Three participants with the G970R mutation (all with the G970R/F508del genotype) enrolled in this G970R substudy of KONTINUE (NCT01707290). This G970R substudy was conducted after the week 12 visit for all participants. The participants, all of whom were men, were 10, 25, and 39 years of age at baseline in the prior study. Sweat chloride levels, percent predicted forced expiratory volume in 1 second (ppFEV1), and NPD results for individual participants are summarized in Table 1 .
Table 1.
Substudy sweat chloride, ppFEV1, and NPD results
| Participant | Time point | Sweat chloride | ppFEV1 | NPD: ΔTCC (zero chloride + isoproterenol) | |||
|---|---|---|---|---|---|---|---|
| Value, mmol/L | Absolute change from visit A to B, mmol/L | Value, % predicted | Absolute change from visit A to B, percentage points | Value, mV | Absolute change from visit A to B, mV | ||
| 001 |
On treatment Off treatment |
109.0 112.0 |
3 |
69.4 67.1 |
‒2.3 |
‒3.0 ‒3.4 |
‒0.4 |
| 002 |
On treatment Off treatment |
122.0 111.0 |
‒11 |
53.7 52.4 |
‒1.3 |
‒1.1 1.1 |
2.2 |
| 003 |
On treatment Off treatment |
121.0 113.5 |
‒7.5 |
58.8 59.3 |
0.5 |
‒3.9 ‒7.6 |
‒3.7 |
ΔTCC, change in total chloride conductance; NPD, nasal potential difference; ppFEV1, percent predicted forced expiratory volume in 1 second.
From visit A (on ivacaftor treatment) to visit B (off ivacaftor treatment), two participants had a decrease in sweat chloride levels (‒7.5 and ‒11 mmol/L), and one participant had an increase in sweat chloride (+ 3 mmol/L). Two participants had a decrease in ppFEV1 (‒1.2 and ‒2.3 percentage points), and one participant had an increase in ppFEV1 (0.5 percentage points; Table 1 ). The change in total chloride conductance as assessed by NPD varied between ‒3.7 and + 2.2 mV (Table 1 ). No consistent or meaningful trends were observed between the on‐treatment and off‐treatment values across assessments.
Characterization and functional assessment of intestinal organoids
Intestinal organoid cultures were generated from the three participants enrolled in the substudy. 22 In vitro organoid studies were conducted to evaluate the in vitro phenotype of cells taken from these three participants. In the FIS assay, activation of CFTR by forskolin led to accumulation of fluid in the organoid lumen—a fully CFTR‐dependent process that can be quantified. 25 In the positive control organoids (heterozygous for F508del and a gating mutation known to respond to ivacaftor (S1251N/F508del and G551D/F508del)), concentration‐dependent FIS was enhanced in the presence of ivacaftor 3 µmol/L (Figure S2 ). No substantial swelling was observed in the organoids derived from participants with G970R‐CFTR mutations or from the negative controls (Figure 2 and Figure S3 ). Thus, swelling observed in organoids from three participants carrying the G970R mutation was comparable to that observed in organoids from people with mutations for which no CFTR protein was produced.
Figure 2.
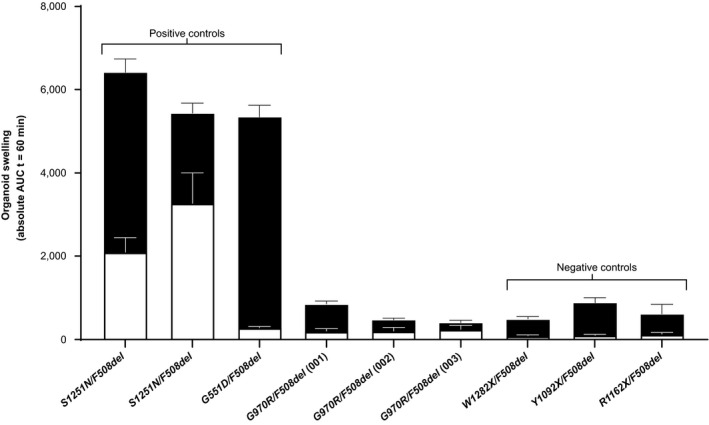
Swelling of rectal organoids at 5 µmol/L forskolin (mean AUC ± SD at t = 60 minutes) with (solid bars) and without (hollow bars) 3 µmol/L ivacaftor. AUC, area under the curve; t, time.
Western blots were performed to determine whether mature CFTR protein was present in the G970R/F508del organoids (Figure 3a ). The positive controls (healthy control and G551D/F508del genotype) showed abundant mature protein (C band) and less abundant immature protein (B band). The G970R/F508del organoids had a small amount of immature protein (B band) but no apparent mature protein (C band), similar to the F508del/F508del organoid.
Figure 3.
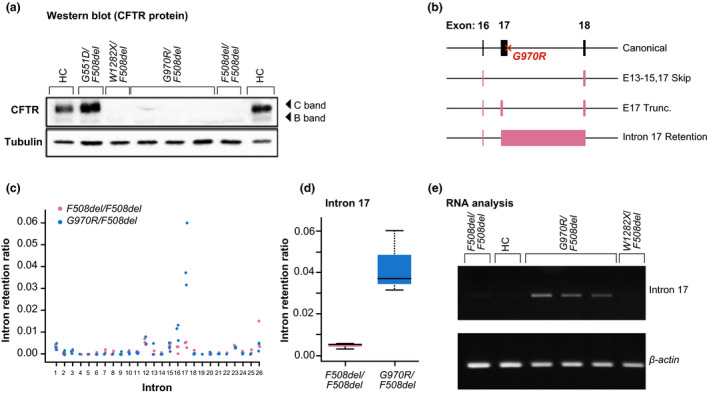
Protein and RNA expression of G970R‐CFTR. (a) Western blot showing expression of cystic fibrosis transmembrane conductance regulator (CFTR) protein (C band, mature protein; B band, immature protein) in participant‐derived intestinal organoids with wild‐type (WT) CFTR (HC) and G551D/F508del‐CFTR, W1282X/F508del‐CFTR, G970R/F508del‐CFTR, and F508del/F508del‐CFTR mutations—with tubulin as a loading control. (b) Gene diagram representing CFTR splice variants detected in G970R/F508del organoids: exons 13–15 and 17 skipping (E13–15,17 Skip), exon 17 truncation (E17 Trunc.), and intron 17 retention. Intron retention ratio calculated with IRFinder for all canonical introns in CFTR (c) and for intron 17 (d). (e) Reverse‐transcriptase polymerase chain reaction (PCR) of messenger RNA from organoids with F508del/F508del‐CFTR mutations, WT CFTR (healthy controls (HCs)), G970R/F508del‐CFTR mutations, and W1282X/F508del‐CFTR mutations—with β‐actin as a loading control; shows retention of the 3′ splice junction of exon 17 (labeled intron 17).
RNA sequencing studies showed that 3 novel splice variants are created by G970R‐CFTR: exon 17 truncation, as reported by Amato et al. 30 ; exons 13–15 and 17 skipping; and intron 17 retention (Figure 3b–d and Figure S1 a). Exon 17 truncation and exon 17 skipping appear to be the predominant splice variants, together representing 44% of total CFTR transcripts in G970R/F508del organoids (Figure S1 c). The presence of the retained canonical 3′ exon‐intron boundary of exon 17 was, however, further confirmed through the analysis of mRNA derived from these G970R/F508del organoids (Figure 3e ). Normal CFTR splicing at this junction was observed in organoids with F508del/F508del‐CFTR, WT CFTR, and W1282X/F508del‐CFTR. Together, these data showed that, in organoids, the G970R genotype was associated with a splicing defect that resulted in little to no mature CFTR at the cell surface and limited response to ivacaftor.
DISCUSSION
More than 340 CF‐causing CFTR mutations have been identified and have been classified into 6 categories or classes based on a reduction in either the amount or function of CFTR protein. 31 , 32 , 33 The G970R protein is encoded by a single‐nucleotide polymorphism of the last nucleotide in exon 17 (c.2908G>C)—first identified in a screen of Belgian participants with CF. 34 , 35 It is a rare CF‐causing mutation, with only 11 people with the mutation identified worldwide at the time of this writing. 32
The G970R‐CFTR mutation has traditionally been considered a mutation that results in a class III gating defect. This classification was attributed to the protein‐coding mutation based on conceptual translation and on in vitro studies in recombinant cell systems using a CFTR cDNA containing the G970R mutation. 19 , 20 , 36 Seibert et al. 19 and Caputo et al. 36 showed that G970R‐CFTR cDNA stably expressed in Chinese hamster ovary and COS‐7 cells, respectively, produced fully mature, glycosylated CFTR protein that reached the cell surface but was incapable of forskolin‐induced iodide transport, and the protein had a lower mean P0. In a study that analyzed G970R‐CFTR by using cDNA‐expressing FRT cells, Yu et al. 8 confirmed the production of mature CFTR protein and the reduced P0 and demonstrated responsiveness to ivacaftor that was comparable to the responsiveness of other CFTR mutations that result in a gating defect. In this study, a > 10‐fold increase in CFTR‐mediated chloride transport was seen in these cells in response to the addition of ivacaftor. 8
The in vitro demonstration of a severe gating defect was consistent with the severe clinical phenotype of participants with CF with a G970R‐CFTR mutation who exhibit high sweat chloride levels (average of 101 mmol/L) and pancreatic insufficiency (91% of individuals). 32 Clinical benefit in 9 of the 10 CFTR mutations that result in a gating defect was established in multiple phase II and phase III ivacaftor clinical studies. 13 , 14 , 16 , 37 However, participants with the G970R‐CFTR mutation had a lower sweat chloride response during treatment with ivacaftor than did participants with CF with other gating mutations. In this substudy in three participants with CF with the G970R/F508del genotype, no consistent or meaningful trends were observed between the on‐ivacaftor and off‐ivacaftor treatment values for NPD, sweat chloride, or ppFEV1, thus calling into question the molecular phenotype of the G970R mutation.
Cuppens et al. 34 previously noted that the location of the G970R mutation may affect mRNA splicing; however, the recombinant cell systems initially used to evaluate the mutation were based on expression of a mutant CFTR cDNA and presumed normal splicing in the native context. 8 , 19 , 36 Primary cell systems, such as human bronchial epithelial cells, provide a physiologically relevant cell system that captures the full biological impact of CFTR mutations on mRNA splicing, protein processing and trafficking, and chloride channel function; however, these cells are often not available for rare mutations, including G970R. The relatively recent development of conditions for the long‐term propagation of human intestinal epithelial cell cultures, or organoids, derived from rectal biopsy specimens provides an alternative source of primary cells for the study of rare CFTR mutations and allows for assessment of mRNA splicing from the native genomic context. 22 Organoids can be readily generated from any person with CF. 38 Because organoids can be expanded long term, they can be used for mechanistic studies in vitro.
In an FIS assay, intestinal organoid cultures from three participants with a G970R/F508del genotype showed only minimal response to stimulation in the presence of ivacaftor, similar to negative control organoids derived from participants with F508del on one allele and a mutation resulting in the production of no protein on the second allele. Western blot analysis demonstrated the lack of mature CFTR protein production. The G970R mutation is caused by a G>C mutation in the last position of CFTR exon 17. 35 , 39 On the basis of results of studies of experimentally confirmed splice junctions, the last nucleotide of an exon is usually G and is rarely C (≈ 0.7% of exons), 39 suggesting that the c.2908G>C mutation could affect splicing efficiency.
RNA sequencing indicated that the G970R‐CFTR mRNA creates 3 novel splice variants: exon 17 truncation, as reported by Amato et al. 30 ; exons 13–15 and 17 skipping; and intron 17 retention. The presence of 3 different splice variants was unexpected and emphasizes the importance of unbiased approaches when characterizing splice variants. These data are also compatible with the previously reported gating defect seen with a fully translated G970R‐CFTR protein expressed from cDNA in engineered cell lines and indicate that any correctly spliced mRNA transcribed from the c.2908G>C‐encoding CFTR allele would have the gating defect caused by the G970R amino acid change. With the methods used here, we can only estimate, not definitively quantify, the amount of correctly spliced c.2908G>C mRNA.
A limitation of in vitro model systems using recombinant cell lines and cDNA constructs is that they will not capture cryptic splicing events. Our work shows that primary organoids, like human bronchial epithelial cells, are a physiologically relevant cell system in which to study the effects of CFTR mutations on mRNA splicing, protein processing and trafficking, and chloride channel function. Primary cell systems and traditional recombinant cell systems, such as FRT cells, play an important and complementary role in CF research.
The US Food and Drug Administration expanded the approved use of ivacaftor and tezacaftor/ivacaftor (Symdeko(R); Vertex) for an additional 28 and 21 mutations, respectively, based on the data from in vitro studies performed in the FRT cell model. This novel approach to drug approval was based on a robust and validated laboratory assay model and comprehensive understanding of the molecular defects caused by CFTR mutations. In addition, verification of data integrity by the regulatory agency—as well as the previously established correlation between in vitro cell system responses in the FRT model at Vertex with clinical benefit in large, randomized trials and well‐established safety profiles—contributed to this approach. 40
The results of this substudy highlight the challenge of using recombinant cell models expressing cDNA constructs to identify CFTR mutations that may cause aberrant splicing of CFTR. Of the > 2,000 CFTR mutations identified, there are rare examples of exonic cryptic splice variants that can result in no or reduced amounts of CFTR protein in native cells. 41 , 42 The molecular consequence of these CFTR variants may be misclassified when expressed in recombinant cell models using cDNA. 42 Of the 1,423 unannotated CFTR mutations evaluated in silico, 33 exonic missense and 6 exonic synonymous mutations were identified as potential splice variants. 42 Only 6 mutations identified are present in the CFTR2.org database (R560K, R560T, G970R, G970D, I1234V, and R1239R). 32 For the G970R mutation, their analysis suggested that the G>C change in the last position of the canonical 5′ splice donor site of exon 17 weakens the likelihood that this position will be recognized as a splice donor site. 42 Here, we experimentally validated their prediction and recommend that the G970R mutation be reclassified primarily as a splice mutation. Similarly, Lee et al. 42 predict that the c.2909G>A change (G970D) on the first nucleotide of exon 18 will weaken the canonical acceptor. However, experimental data by Amato et al. 30 demonstrated correct splicing between exon 17 and exon 18 for this mutation using primary, patient‐derived tissue. Thus, whereas computational tools can help identify potential cryptic splice mutations, experimental validation using patient tissues is critical for confirmation of splicing defects.
Functional and molecular analyses using participant‐derived organoids indicated that the c.2908G>C mutation causes a splicing defect and should no longer be categorized as a CFTR gating mutation. Although there is a robust correlation between the FRT system and data from clinical trials using CFTR modulators, G970R appears to be an exception based on its previously undocumented role as a splice mutation. This substudy used organoids as a complementary in vitro model to characterize cryptic splice mutations that cannot be assessed fully in recombinant cell systems expressing cDNA constructs.
Funding
This study was supported by Vertex Pharmaceuticals Incorporated. Vertex was involved in the study design, RNA analysis, and analysis and interpretation of the data, and contributed to the decision to submit this paper for publication.
Conflict of Interest
M.C.F., A.B., M.H., M.M.Z., P.N., and F.v.G. are employees of Vertex Pharmaceuticals Incorporated and may own stock or options in the company. J.C.S. is a former employee of Vertex and may own stock or options in the company. M.S., S.F.B., and R.G.J.V. are employees of Hubrecht Organoid Technology and report grants from Vertex during the conduct of the study and grants from companies involved in cystic fibrosis research and drug development outside the submitted work, for which Hubrecht Organoid Technology performs contract research organizational work in the area of cystic fibrosis drug development. A.M. reports personal fees from Vertex outside the submitted work. K.D.B. reports support from Boehringer, Protalix, Raptor, Teva, NovaBiotics, Eloxx, Vertex, and Galapagos. All authors received nonfinancial support (assistance with manuscript preparation) from ArticulateScience LLC.
Author Contributions
M.C.F. wrote the manuscript. M.C.F., A.B., J.C.S., M.S., S.F.B., R.G.J.V., A.M., M.H., M.M.Z., P.N., F.v.G., and K.D.B. designed the research. S.F.B. and R.G.J.V. performed the research. M.C.F., A.B., J.C.S., M.S., S.F.B., R.G.J.V., A.M., M.H., M.M.Z., P.N., F.v.G., and K.D.B. analyzed the data.
Supporting information
Fig S1
Fig S2
Fig S3
Table S1
Supplementary Material
Acknowledgments
The authors acknowledge the participants involved in this study. Tejendra Patel, PharmD, formerly of Vertex Pharmaceuticals Incorporated, provided editorial coordination and assistance. Additional editorial support was provided by Karen Kaluza Smith, PhD, of ArticulateScience LLC, which received funding from Vertex Pharmaceuticals Incorporated.
Data Availability Statement
Vertex is committed to advancing medical science and improving the health of people with cystic fibrosis. This includes the responsible sharing of clinical trial data with qualified researchers. Proposals for the use of these data will be reviewed by a scientific board. Approvals are at the discretion of Vertex and will be dependent on the nature of the request, the merit of the research proposed, and the intended use of the data. Please contact CTDS@vrtx.com if you would like to submit a proposal or need more information.
References
- 1. Riordan, J.R. et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 (1989). [DOI] [PubMed] [Google Scholar]
- 2. Welsh, M.J. & Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73, 1251–1254 (1993). [DOI] [PubMed] [Google Scholar]
- 3. Anderson, M.P. et al. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 253, 202–205 (1991). [DOI] [PubMed] [Google Scholar]
- 4. Anderson, M.P. , Rich, D.P. , Gregory, R.J. , Smith, A.E. & Welsh, M.J. Generation of cAMP‐activated chloride currents by expression of CFTR. Science 251, 679–682 (1991). [DOI] [PubMed] [Google Scholar]
- 5. Rowe, S.M. , Miller, S. & Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 352, 1992–2001 (2005). [DOI] [PubMed] [Google Scholar]
- 6. Denning, G.M. , Ostedgaard, L.S. , Cheng, S.H. , Smith, A.E. & Welsh, M.J. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J. Clin. Invest. 89, 339–349 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Sullivan, B.P. & Freedman, S.D. Cystic fibrosis. Lancet 373, 1891–1904 (2009). [DOI] [PubMed] [Google Scholar]
- 8. Yu, H. et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 11, 237–245 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Kalydeco® USPI . Vertex Pharmaceuticals Incorporated (2020).
- 10. Kalydeco® SmPC . Vertex Pharmaceuticals (Ireland) Limited (2019).
- 11. Kalydeco® IS prescribing information . Vertex Pharmaceuticals (U.K.) Limited (2019).
- 12. Kalydeco® CA product monograph . Vertex Pharmaceuticals (Canada) Incorporated (2019).
- 13. Ramsey, B.W. et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365, 1663–1672 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Boeck, K. et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non‐G551D gating mutation. J. Cyst. Fibros. 13, 674–680 (2014). [DOI] [PubMed] [Google Scholar]
- 15. Moss, R.B. et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His‐CFTR mutation: a double‐blind, randomised controlled trial. Lancet Resp. Med. 3, 524–533 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davies, J.C. et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 187, 1219–1225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rosenfeld, M. et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to less than 24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single‐arm study. Lancet Resp. Med. 6, 545–553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anderson, M.P. & Welsh, M.J. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide‐binding domains. Science 257, 1701–1704 (1992). [DOI] [PubMed] [Google Scholar]
- 19. Seibert, F.S. et al. Cytoplasmic loop three of cystic fibrosis transmembrane conductance regulator contributes to regulation of chloride channel activity. J. Biol. Chem. 271, 27493–27499 (1996). [DOI] [PubMed] [Google Scholar]
- 20. Seibert, F.S. et al. Disease‐associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J. Biol. Chem. 271, 15139–15145 (1996). [DOI] [PubMed] [Google Scholar]
- 21. Sosnay, P.R. et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 45, 1160–1167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato, T. et al. Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 141, 1762–1772 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Nick, J.A. et al. Ivacaftor in cystic fibrosis with residual function: lung function results from an N‐of‐1 study. J. Cyst. Fibros. 19, 91–98 (2020). [DOI] [PubMed] [Google Scholar]
- 24. Miller, M.R. et al. Standardisation of spirometry. Eur. Respir. J. 26, 319–338 (2005). [DOI] [PubMed] [Google Scholar]
- 25. Dekkers, J.F. et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 19, 939–945 (2013). [DOI] [PubMed] [Google Scholar]
- 26. Dobin, A. et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol. 33, 290–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Middleton, R. et al. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol. 18, 51 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Vree, P.J.P. et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat. Biotechnol. 32, 1019–1025 (2014). [DOI] [PubMed] [Google Scholar]
- 30. Amato, F. et al. Two CFTR mutations within codon 970 differently impact on the chloride channel functionality. Hum. Mutat. 40, 742–748 (2019). [DOI] [PubMed] [Google Scholar]
- 31. Castellani, C. et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 7, 179–196 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. The Clinical and Functional TRanslation of CFTR (CFTR2). <https://www.cftr2.org> (2011). Accessed July 27, 2020.
- 33. Veit, G. et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 27, 424–433 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cuppens, H. , Marynen, P. , De Boeck, C. & Cassiman, J.‐J. Detection of 98.5% of the mutations in 200 Belgian cystic fibrosis alleles by reverse dot‐blot and sequencing of the complete coding region and exon/intron junctions of the CFTR gene. Genomics 28, 693–697 (1993). [DOI] [PubMed] [Google Scholar]
- 35. Cystic Fibrosis Mutation Database: detailed view of exon 17. <http://genet.sickkids.on.ca/PicturePage.html?domain_id=17> (2011). Accessed July 27, 2020.
- 36. Caputo, A. et al. Mutation‐specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J. Pharmacol. Exp. Ther. 330, 783–791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Accurso, F.J. et al. Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation. N. Engl. J. Med. 363, 1991–2003 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dekkers, J.F. et al. Characterizing responses to CFTR‐modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 8, 344ra384 (2016). [DOI] [PubMed] [Google Scholar]
- 39. Yeo, G. , Hoon, S. , Venkatesh, B. & Burge, C.B. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl. Acad. Sci. USA 101, 15700–15705 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Durmowicz, A.G. , Lim, R. , Rogers, H. , Rosebraugh, C.J. , & Chowdhury, B.A . The U.S. Food and Drug Administration's experience with ivacaftor in cystic fibrosis. Establishing efficacy using in vitro data in lieu of a clinical trial. Ann. Am. Thorac. Soc. 15, 1–2 (2018). [DOI] [PubMed] [Google Scholar]
- 41. Cystic Fibrosis Mutation Database: CFMDB statistics <http://www.genet.sickkids.on.ca/StatisticsPage.html> (2011). Accessed July 27, 2020.
- 42. Lee, M. et al. Systematic computational identification of variants that activate exonic and intronic cryptic splice sites. Am. J. Hum. Genet. 100, 751–765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1
Supplementary Material
Data Availability Statement
Vertex is committed to advancing medical science and improving the health of people with cystic fibrosis. This includes the responsible sharing of clinical trial data with qualified researchers. Proposals for the use of these data will be reviewed by a scientific board. Approvals are at the discretion of Vertex and will be dependent on the nature of the request, the merit of the research proposed, and the intended use of the data. Please contact CTDS@vrtx.com if you would like to submit a proposal or need more information.