

Abstract
Chemotherapy‐induced peripheral neuropathy (CIPN) is a common and dose‐limiting toxicity to widely used chemotherapeutics. Although the exact molecular mechanism of chemotherapy‐induced peripheral neuropathy remains elusive, there is consensus that it is caused by damage to the peripheral nervous system leading to sensory symptoms. Recently developed methodologies have provided evidence of expression of drug transporters in the peripheral nervous system. In this literature review, we explore the role for drug transporters in CIPN. First, we assessed the transport of chemotherapeutics that cause CIPN (taxanes, platins, vincristine, bortezomib, epothilones, and thalidomide). Second, we cross‐referenced the transporters implicated in genetic or functional studies with CIPN with their expression in the peripheral nervous system. Several drug transporters are involved in the transport of chemotherapeutics that cause peripheral neuropathy and particularly efflux transporters, such as ABCB1 and ABCC1, are expressed in the peripheral nervous system. Previous literature has linked genetic variants in efflux transporters to higher risk of peripheral neuropathy with the taxanes paclitaxel and docetaxel and the vinca alkaloid vincristine. We propose that this might be due to accumulation of the chemotherapeutics in the peripheral nervous system due to reduced neuronal efflux capacity. Thus, concomitant administration of efflux transporter inhibitors may lead to higher risk of adverse events of drugs that cause CIPN. This might prove valuable in drug development where screening new drugs for neurotoxicity might also require drug transporter consideration. There are ongoing efforts targeting drug transporters in the peripheral nervous system to reduce intraneuronal concentrations of chemotherapeutics that cause CIPN, which might ultimately protect against this dose‐limiting adverse event.
Chemotherapy‐induced peripheral neuropathy (CIPN) is a dose‐limiting toxicity that is correlated with a number of widely used chemotherapeutic drugs. CIPN presents predominantly as a sensory polyneuropathy with paresthesia sometimes accompanied by pain, and patients often describe the symptoms as “walking on needles.” These symptoms follow a symmetric, distal, stocking and glove distribution. CIPN can be very serious, but even mild to moderate symptoms commonly persist for up to 2 years after treatment cessation, significantly impairing quality of life. 1 , 2 With more patients surviving cancers due to improved pharmacological treatment, this severe and long‐lasting adverse reaction is particularly concerning and of public health interest. The risk and incidence of CIPN depends on chemotherapeutic agent, cumulative dose, chemotherapy regimen, duration of exposure, and a number of both genetic and nongenetic factors. Clinical strategies for reducing these debilitating symptoms include dose reduction/delay, alternative chemotherapy when feasible, increasing duration of infusion, and experimental approaches, such as cryotherapy. 3 A number of pharmacological agents have been tested to treat or prevent CIPN, but results have been disappointing, leaving this clinical challenge unresolved.
Numerous effective chemotherapeutic drugs can cause CIPN. The incidence of CIPN varies between chemotherapeutics and depends on the specific drug and indication for treatment. Recent evidence indicates that cancer itself may exacerbate CIPN. 4 In this review, we have focused on the most widely used chemotherapeutics that produce substantial risk of developing CIPN. The taxanes paclitaxel and docetaxel both cause CIPN, with paclitaxel being slightly more neurotoxic than docetaxel. 4 Taxane‐induced peripheral neuropathy will normally improve 4–6 months after treatment cessation, but some patients may experience varying degrees of peripheral neuropathy up to 2 years later. 1 Platinum‐based chemotherapeutics (cisplatin, oxaliplatin, and carboplatin) also cause CIPN. Symptoms of cisplatin‐induced and oxaliplatin‐induced peripheral neuropathy may continue to worsen after cessation of treatment and some patients may experience initial symptoms after concluding treatment. The widely used vinca alkaloid vincristine causes peripheral neuropathy in nearly all patients, and, in contrast to the abovementioned chemotherapeutics, may cause motor neuropathy and autonomic neuropathy with symptoms such as constipation and abdominal pain. The proteasome inhibitor bortezomib, the microtubule‐stabilizing epothilone ixabepilone and the angiogenesis inhibitor thalidomide also cause CIPN. 5
The pathogenesis of CIPN differs between chemotherapeutics. Primary axonopathy of peripheral neurons have been described along with demyelination of large myelinated sensory fibers during taxane treatment. 6 Mechanisms, such as mitochondrial dysfunction and production of oxidative stress and immune‐mediated toxicity, have also been linked to CIPN. 7 Exposure of the peripheral neurons to chemotherapeutic agents is also considered a critical determinant of the risk of CIPN, and variation in both drug metabolizing enzymes and transporters have been investigated as potential mechanisms underlying this dose‐limiting toxicity.
Over the last decade, a number of clinical pharmacogenetic studies have associated polymorphisms in transporter genes with risk of CIPN (Table 1 ), although these associations are not consistent and are rarely validated. This effect was assumed to be caused by altered pharmacokinetics, but polymorphisms in transporter proteins only cause minor pharmacokinetic differences. 8 Consistent with the findings in humans, Mdr1a/Mdr1b knockout mice only exhibit 30% higher area under the paclitaxel concentration‐time curve after intravenous administration. 9 The peripheral nervous system is not protected by the blood‐brain barrier, 10 which protects the central nervous system from xenobiotics. This may explain why chemotherapeutics cause peripheral‐ and not central nerve damage. Although it is well‐known that drug transporters play a key role in regulating tumor toxicity of chemotherapeutics, their role in regulating tissue‐specific toxicity has only recently been elucidated. A number of studies have highlighted the role of drug transporters in chemotherapy‐induced ototoxicity, nephrotoxicity, and cardiotoxicity. 11 , 12 , 13 Due to the high number of studies linking CIPN to transporter polymorphisms, we reviewed the literature to find plausible mechanisms underlying these associations.
Table 1.
Clinical pharmacogenetic studies correlating risk of chemotherapy‐induced peripheral neuropathy to genetic variants in genes encoding drug transporters
| Class | Drug | Transporter (gene) | dbSNP | Outcome | Odds ratio [95% confidence interval] | Ref. |
|---|---|---|---|---|---|---|
| Taxanes | Paclitaxel | P‐gp (ABCB1) | rs3213619 | NCI CTCAE version 2. Grade ≥ 2 neurotoxicity. | 0.5 [0.3–0.8] | 71 |
| rs1045642 | NCI CTCAE version 4. Grade ≥ 2 neurotoxicity. | 2.8 [1.2–6.5] | 72 | |||
| rs1128503 | NCI CTCAE version 4. Grade ≥ 2 neurotoxicity. | 2.4 [1.1–5.4] | 73 | |||
| MRP2 (ABCC2) | rs17222723 | NCI CTCAE version 2 Grade ≥ 2 neurotoxicity. | 0.6 [0.4–0.9] | 71 | ||
| Docetaxel | P‐gp (ABCB1) | rs2032582 | NCI CTCAE version 2. Time to onset of peripheral neuropathy. | 1.9 months for reference genotypes vs. 0.7 months for carriers of variant | 27 | |
| Platinum‐based | Oxaliplatin | BCRP (ABCG2) | rs3114018 | NCI CTCAE version 2. Grade ≥ 2 neurotoxicity when combined with CCNH rs2230641. | 2.5 [1.2–5.1] | 74 |
| Vinca alkaloids | Vincristine | MRP1 (ABCC1) | rs3887412 | NCI CTCAE version 3. Grade 2‐3 peripheral neuropathy vs. no peripheral neuropathy after two‐three cycles of vincristine. | 3.4 [1.5–7.7] | 75 |
| rs2644983 | 4.2 [1.7–10.5] | |||||
| rs11864374 | Obtained from medical records. Grade 1‐4 peripheral neuropathy vs. no peripheral neuropathy. | 0.4 [0.2–0.8] | 76 | |||
| rs3743527 | 0.3 [0.1–0.8] | |||||
| rs1967120 | 0.4 [0.2–0.8] | |||||
| rs17501331 | 2.5 [1.1–5.7] | |||||
| rs12923345 | 2.4 [1.1–5.3] | |||||
| rs11642957 | 0.4 [0.2–1.0] | |||||
| rs3784867 | NCI CTCAE version 4. Grade ≥ 2 peripheral neuropathy. | 4.9 [2.0–12.1] | 77 | |||
| MRP2 (ABCC2) | rs3740066 | Obtained from medical records. Grade 1‐4 peripheral neuropathy vs. no peripheral neuropathy. | 0.2 [0.1–0.5] | 76 | ||
| rs12826 | 0.2 [0.1–0.5] | |||||
| P‐gp (ABCB1) | rs4728709 | NCI CTCAE version 3. Grade 1‐2 peripheral neuropathy vs. no peripheral neuropathy. | 0.3 [0.1–0.9] | 78 |
dbSNP, single nucleotide polymorphism database identifier; NCI CTCAE, National Cancer Institute Common Terminology Criteria for Adverse Events.
TRANSPORTER‐MEDIATED DISTRIBUTION OF CHEMOTHERAPEUTICS THAT CAUSE PERIPHERAL NEUROPATHY
Below, we have assessed the role for membrane transporters in the uptake and efflux of chemotherapeutic agents that lead to substantial risk of CIPN. Direct evidence for transport (i.e., genetic overexpression or knockdown of transporters) is summarized in Table 2 . Although thalidomide treatment is associated with CIPN, 14 there is no evidence for its transport by SLC or ABC transporters and this chemotherapeutic was not considered further.
Table 2.
Summary of direct in vitro evidence (i.e., genetic overexpression or knockdown) of drug transport of chemotherapeutics that cause peripheral neuropathy
| Transporter | Gene | Paclitaxel | Docetaxel | Oxaliplatin | Cisplatin | Carboplatin | Vincristine | Bortezomib | Ixabepilone |
|---|---|---|---|---|---|---|---|---|---|
| MRP1 | ABCC1 | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ | * | ‐ |
| MRP2 | ABCC2 | ✓ | ✓ | ‐ | ‐ | ‐ | ✓ | * | ‐ |
| MRP7 | ABCC10 | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| BCRP | ABCG2 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | * | * |
| P‐gp | ABCB1 | ✓ | ✓ | ‐ | ‐ | ‐ | ✓ | ✓ | ✓ |
| OCT2 | SLC22A2 | ‐ | ‐ | ✓ | ✓ | * | ‐ | ‐ | ‐ |
| OCTN1 | SLC22A4 | ‐ | ‐ | ✓/* | ‐ | ‐ | ‐ | ‐ | ‐ |
| OCTN2 | SLC22A5 | ‐ | ‐ | ✓/* | ‐ | ‐ | ‐ | ‐ | ‐ |
| OAT2 | SLC22A7 | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| MATE1 | SLC47A1 | ‐ | ‐ | ✓ | ‐ | ‐ | ‐ | ‐ | ‐ |
| OATP1B1 | SLCO1B1 | ✓ | ✓ | ‐ | ‐ | ‐ | ✓ | ✓ | ‐ |
| OATP1B3 | SLCO1B3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ‐ | ‐ |
| CTR1 | SLC31A1 | ‐ | ‐ | ✓ | ✓ | ✓ | ‐ | ‐ | ‐ |
| References | ‐ | 17, 19, 20, 21, 22, 23, 26 | 22, 28, 29 | 35, 39, 40, 41, 43, 45 | 34, 35, 43, 45 | 34, 35, 43, 45 | 17, 46, 47, 49, 50, 51, 52 | 53, 56 | 57 |
✓ Direct evidence of transport (i.e., genetic overexpression or knockdown).
‐ Literature not available.
*Evidence against transport.
Taxanes
Both the uptake and efflux of taxanes have been characterized in vitro and their contribution to pharmacokinetics and peripheral neuropathy studied in rodents. Paclitaxel is a well‐known substrate for P‐gp (MDR1, ABCB1) 9 , 15 , 16 , 17 and P‐gp inhibition enhances oral absorption of the drug. 18 Multidrug resistance associated protein 2 (MRP2; ABCC2) 9 , 19 and MRP7 (ABCC10) 20 , 21 have been shown to efflux paclitaxel in vitro but their contribution to paclitaxel distribution outside the tumor cell is not well‐characterized. Paclitaxel uptake by organic anion transporting polypeptide 1B3 (OATP1B3; SLCO1B3) and OATP1B1 (SLCO1B1) was highly dependent on the taxane solubilizer. 22 , 23 Additionally, paclitaxel is an OATP1B1 and OATP1B3 inhibitor at clinically relevant concentrations. 24 , 25 Human OAT2 transports paclitaxel but OAT2‐mediated paclitaxel transport is saturated at nM concentrations, which might limit its role at pharmacological concentrations. 26 Another widely used taxane, docetaxel, is also a substrate for P‐gp. 27 , 28 Similar to paclitaxel, docetaxel is also a substrate for MRP2. 29 Docetaxel is transported by OATP1B1 and OATP1B3, 22 , 29 , 30 , 31 and in vivo evidence suggests that these uptake transporters play a more central role for the pharmacokinetics of docetaxel compared with paclitaxel. In Slco1b2(‐/‐) mice, the clearance of docetaxel is 66–83% lower 30 , 32 compared with only 30% lower clearance for paclitaxel. 33 Interestingly, despite minor changes in paclitaxel pharmacokinetics, Slco1b2(‐/‐) mice are protected against paclitaxel‐induced neurotoxicity. 33 Whether this translates to humans is unknown.
Platins
The DNA‐binding platinum‐based chemotherapeutics cisplatin, carboplatin, and oxaliplatin were shown to be substrates of copper carriers, such as copper uptake protein 1 (CTR1), 34 , 35 although a role for CTR1‐mediated platinum cellular uptake appears limited. 36 In mouse models, CTR1 contributes to the basolateral uptake of cisplatin in the kidneys and regulates cisplatin‐induced nephrotoxicity. 37 In rat dorsal root ganglion (DRG), oxaliplatin appears to damage primarily CTR1‐expressing neurons, 38 consistent with its importance in regulating oxaliplatin distribution to these cells. Although there are conflicting reports for transport of oxaliplatin by the organic anion transporters novel 1 and 2 (OCTN1/2), a careful analysis in a tightly regulated expression system suggests that OCTN1 does not transport this platinum agent to any significant extent. 39 , 40 This lack of transport of oxaliplatin by OCTN1 limits the interpretation of findings from oxaliplatin‐induced peripheral neuropathy studies under conditions of OCTN1 inhibition in rats and highlights the sensitivity of both in vitro transport assays and in vivo neuropathy studies to experimental systems and species differences. 41 , 42 Additionally, oxaliplatin and cisplatin are substrates for OCT2, 43 which was shown to regulate the neurotoxicity of oxaliplatin in mice. 44 Multidrug and toxin extrusion 1 (MATE1; SLC47A1) may play a role in the efflux of oxaliplatin. 41 Knockdown of MATE1 using siRNA leads to increased oxaliplatin‐mediated neuropathy in rats. 41 Transport of carboplatin is not well‐characterized; carboplatin is not transported by OCT2, 43 although its uptake might be mediated by CTR1. 35 All three platinum‐based chemotherapeutics are substrates for OATP1B3 and cisplatin is a substrate for OATP1B1. 45
Vincristine
Among the antimitotic vinca alkaloids, vincristine is the most widely used chemotherapeutic. Vincristine is a substrate for P‐gp 46 and high expression of P‐gp results in increased resistance to vincristine in cancer cells. 47 Concomitant treatment of patients with vincristine and the known P‐gp/CYP3A4 inhibitor cyclosporin A leads to exacerbated CNS toxicity, suggesting potent inhibition of P‐gp at the blood‐brain barrier. 48 In addition to P‐gp, vincristine is a substrate for MRP1 17 and knockout of this gene in mice reduces the maximum tolerated dose of vincristine, particularly when combined with genetic deletion of P‐gp. 49 MRP2 can also transport vincristine in cells but the significance of this transport in vivo is not known. 50 Similarly, vincristine has been shown to be transported by OATP1B1 and OATP1B3 in vitro, 51 , 52 although their contribution to vincristine disposition or neurotoxicity are not characterized.
Bortezomib
There is limited information on the transport of the proteasome inhibitor bortezomib. Bortezomib cytotoxicity is influenced by the level of P‐gp, but not BCRP or MRP1 53 expression in cancer cell lines but direct evidence of transport by P‐gp is lacking. Polymorphisms in ABCB1 and ABCC1 were linked to treatment response, 54 although the significance of these associations require validation. Interestingly, bortezomib appears to inhibit proteasome‐mediated internalization of CTR1 caused by platinum‐based chemotherapy in tumor cells, enhancing cisplatin uptake and cell killing both in vitro and in vivo. 55 Transporter‐mediated uptake of bortezomib by OATP1B1 was measurable but contributed insignificantly to intracellular levels of the drug. 56
Epothilones
In vitro studies showed that the epothilone ixabepilone is a P‐gp substrate, but not a BCRP substrate. 57 Thus, P‐gp inhibitors reverse MDR1‐mediated drug resistance to ixabepilone in MDCK cells. Additionally, P‐gp inhibition leads to reduced efflux of ixabepilone in LLC‐MDR1 cells. 57
EXPRESSION OF DRUG TRANSPORTERS IN HUMAN DORSAL ROOT GANGLION
The expression of drug transporters in the peripheral nervous system was reported in several recent studies. Two studies utilized RNA sequencing of human DRG (hDRG), 58 , 59 whereas another study reported a proteomic analysis of hDRG lysate. 60 To assess transporter expression, we cross‐referenced these datasets for expression of drug transporters that transport chemotherapeutics with significant CIPN, as outlined in the previous section. The expression of key transporters in hDRG that are relevant for chemotherapeutics that cause CIPN are summarized in Table 3 . Expression of drug transporters was confirmed if FPKM was > 1 for RNA sequencing datasets. At the protein level, expression of drug transporters was defined as detectable protein expression in at least two samples from the proteomic dataset (table S1 from Schwaid et al. 60 ).
Table 3.
Data from RNA sequencing and proteomic studies show expression of drug transporters involved in transport of chemotherapeutics that cause peripheral neuropathy
| Transporter (gene/transporter) | RNAseq hDRG (> 1 FPKM cutoff) | Proteomics hDRG | ||
|---|---|---|---|---|
| Flegel et al. (2015) | Ray et al. (2018) | Schwaid et al. (2018) | Stage et al. (2020) | |
| ABCC1/MRP1 | + | + | + | + |
| ABCC2/MRP2 | − | − | n.d. | − |
| ABCC10/MRP7 | + | + | n.d. | n.d. |
| ABCG2/BCRP | + | + | n.d. | n.d. |
| ABCB1/P‐gp | + | + | + | + |
| SLC22A2/OCT2 | − | − | n.d. | n.d. |
| SLC22A4/OCTN1 | + | + | n.d. | n.d. |
| SLC22A5/OCTN2 | + | + | n.d. | n.d. |
| SLC22A7/OAT2 | − | − | n.d. | n.d. |
| SLC47A1/MATE1 | + | + | n.d. | n.d. |
| SLCO1B1/OATP1B1 | − | − | n.d. | n.d. |
| SLCO1B3/OATP1B3 | − | − | n.d. | n.d. |
| SLC31A1/CTR1 | + | + | n.d. | n.d. |
+ Expression confirmed.
− No expression
hDRG, human dorsal root ganglion; n.d., not determined.
The efflux transporters P‐gp (ABCB1), BCRP (ABCG2), MRP1 (ABCC1), and MATE1 (SLC47A1) are expressed in hDRG, whereas MRP2 (ABCC2) is not. High expression of efflux transporters in the peripheral nervous system makes sense from an evolutionary standpoint. The peripheral nervous system is not protected by the blood‐brain‐barrier and thus expression of efflux transporters is crucial to confer protection against neurotoxic agents. The uptake transporters OCTN1/OCTN2 (SLC22A4/SLC22A5) and CTR1 (SLC31A1) are expressed in hDRG. These transporters mediate cellular uptake of endogenous substances, such as OCTN1/OCTN2‐mediated uptake of ergothioneine or CTR1‐mediated copper transport. The transcriptomic and proteomic data did not support expression of other SLC uptake transporters, such as OATP1B1 and OATP1B3, OAT2, or OCT2. This is in contrast to a previous report showing expression of OCT2 mRNA in human DRG. 44 We were unable to find an exhaustive source for protein expression data in hDRG as the published datasets did not contain comprehensive evaluation of drug transporters (Table 3 ). Finally, these data highlight expression of drug transporters in the hDRG but do not provide evidence for localization or function. Further evaluation of these properties will be critical for the field.
DISCUSSION AND FUTURE PERSPECTIVES
This review highlights a number of important features of chemotherapy transport in sensory neurons. Although there is very little direct human evidence of the importance of chemotherapy transport in the peripheral nervous system, the number of clinical associations between polymorphisms in transporter genes with CIPN and expression of key transporters in the dorsal root ganglion supports their role in the disposition of chemotherapeutics to the peripheral nervous system (Figure 1 ).
Figure 1.
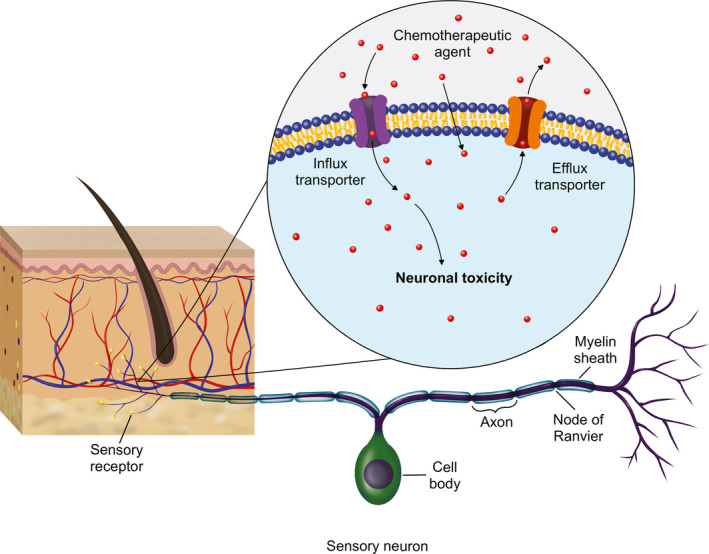
Drug transporters regulate influx and efflux of chemotherapeutics in the peripheral nervous system likely modulating toxicity and peripheral neuropathy.
P‐gp overexpression is a well‐established cause of tumor resistance to chemotherapeutics and intestinal P‐gp limits the oral absorption of drugs, such as paclitaxel. 61 Inhibition of this efflux transporter has been investigated as an approach to counter both intestinal and tumor efflux of chemotherapeutics. 62 , 63 , 64 Although organ or tumor level inhibition of P‐gp could improve the bioavailability and tumor distribution of chemotherapeutics, inhibition of efflux transporters may cause increased accumulation of chemotherapeutics in the peripheral nervous system. Several pharmacogenetic studies have correlated polymorphisms in efflux transporters with risk of CIPN (Table 1 ). Additionally, a recent clinical study among patients with breast cancer showed that miR‐451a regulates P‐gp expression and may be a risk factor for paclitaxel‐induced peripheral neuropathy. 65 We recently showed that inhibition of P‐gp leads to increased neuronal accumulation of paclitaxel in cultured neurons and in patients with breast cancer and ovarian cancer. 66 Interestingly, several widely prescribed drugs that inhibit P‐gp, including the prominent cholesterol‐lowering drug atorvastatin, led to substantially increased risk of paclitaxel‐induced peripheral neuropathy when used in combination with paclitaxel. Collectively, this supports the proposal that inhibition of efflux transport might cause increased accumulation and toxicity of paclitaxel in sensory neurons. This is further supported by a study showing that concomitant administration of paclitaxel with the well‐established P‐gp inhibitor valspodar causes a minor shift in paclitaxel plasma pharmacokinetics. 63 Despite apparently unaltered plasma concentrations, this combination leads to higher risk of paresthesia and pain, 62 supporting a protective role of P‐gp in the peripheral nervous system. These interesting clinical findings warrant further evaluation. Additional investigation is also needed to understand why P‐gp activity in the peripheral nervous system is more sensitive to changes in transport expression and function than P‐gp at the blood‐brain‐barrier. The relative expression levels of P‐gp at peripheral and central nerve barriers and the contribution of other uptake and efflux transporters at these sites should also be evaluated.
CTR1 might play a key role for disposition of platinum‐based chemotherapy. This transporter is highly expressed in many tissues, including peripheral neurons and tumor tissue and expression of CTR1 in tumor cells correlates with response to platinum‐based chemotherapy. 67 Although the attempt to increase CTR1 activity to increase platinum uptake in tumor tissue could improve chemotherapy response, the wide tissue distribution of CTR1 might also lead to increased risk of off‐target toxicity due to increased permeability of platins. An additional concern to targeting CTR1 includes interfering with cell copper balance, which may result in unwanted toxicity. Clinical attempts to target CTR1 to increase tumor uptake of platinum‐based chemotherapy should only be done with significant consideration to adverse effects and toxicity.
Both paclitaxel and ixabepilone were shown to activate the nuclear pregnane X receptor, 68 which is a known mechanism for induction of ABCB1 expression. Activation of this receptor by St. John’s wort, rifampicin, or dicloxacillin causes upregulation of a number of important pharmacogenes, including CYP3A4 and ABCB1. 69 Increased expression of ABCB1 and corresponding increases in P‐gp function could influence chemotherapeutic distribution to peripheral neurons. Induction of drug‐metabolizing enzymes or drug transporters through pregnane X receptor typically requires prolonged exposure to the inducer and as both paclitaxel and ixabepilone treatment are given weekly or every third week, the clinical impact of potential induction of ABCB1 will require further investigation.
Published transcriptomic data do not support expression of OATP1B transporters in hDRG. This is in contrast to rodent studies that have shown both gene and protein expression of OATP1B2. 33 , 70 Additionally, RNAseq data does not support OCT2 expression, although Sprowl et al. showed that OCT2 is expressed in hDRG using quantitative polymerase chain reaction. 44 The expression of drug transporters in the peripheral nervous system should be supported by quantitative protein determination by liquid chromatography‐tandem accurate mass spectrometry in hDRG before being dismissed. Species differences in transporter expression in the peripheral nervous system require consideration in the design and interpretation of drug transporter studies.
Very little is known about the role of membrane transporters in drug disposition to the human peripheral nervous system. Recent transcriptomic approaches and extensive rodent studies have shed important light on expression and activity of drug transporters in these tissues, although their function and impact in human models is unknown. There are two ongoing clinical studies (clinicaltrials.gov identifiers NCT04205903 and NCT04164069) aiming to inhibit uptake transporters of oxaliplatin and paclitaxel to reduce paclitaxel‐induced and oxaliplatin‐induced peripheral neuropathy in patients with cancer. These studies and continued laboratory efforts may lead to an increased understanding of the disposition of neurotoxic drugs that could also inform drug development. Cellular influx and efflux mechanisms are critical for drug distribution and should be considered when attempting to target drugs to the peripheral nervous system. For example, P‐gp and MRP1 are highly expressed in sensory neurons and drugs that are substrates of these efflux transporters may not reach sufficient therapeutic concentrations in the peripheral nervous system. Expression and activity of transporters in supporting cells, such as Schwann cells or satellite cells, is currently unknown and should be assessed in the future.
As discussed previously, it seems tempting to try to upregulate efflux transporters to promote efflux of chemotherapy agents from sensory neurons. As this would also increase efflux of chemotherapeutics from tumor cells, this is not a compelling therapeutic strategy. However, using antibody‐directed delivery of transport modulators to peripheral sensory neurons might prove a useful tool to modulate chemotherapy transport in the peripheral nervous system. This approach might be useful to inhibit uptake transporters or induce efflux transporters in the human DRG, ensuring low sensory neuron concentrations of chemotherapeutics, which are expected to limit CIPN. Future development is needed to test this approach in preclinical models and in humans.
It is important to note that CIPN is a complex phenotype. In this review, we have attempted to highlight the role of membrane transporters in the peripheral nervous system and their putative influence on CIPN. The evidence linking drug transporters to CIPN is primarily based on in vitro or animal studies and thus more work in human models is warranted. Although drug transporters may present a piece of the puzzle to help explain variability in CIPN, there are many other factors that should be taken into account. Predicting and understanding CIPN will require collaboration across research disciplines to build models to provide a deeper understanding of this dose‐limiting toxicity.
In conclusion, drug transporters, particularly efflux transporters, are highly expressed in the human peripheral nervous system. Although there is limited direct evidence supporting a functional role of these transporters in sensory neurons in humans, an increasing body of evidence supports their involvement in regulating disposition of chemotherapeutics to the peripheral nervous system and thus drug transporters may play a role in CIPN.
Funding
This work was supported by grants from the Independent Research Fund Denmark (5053‐00042B), Danish Cancer Society (R231‐A13918), and the National Institutes of Health (R01CA192156).
Conflicts of Interest
A provisional patent application related to chemotherapy‐induced peripheral neuropathy has been filed by University of Southern Denmark with Tore Bjerregaard Stage listed as inventor. All other authors declared no competing interest for this work.
Disclaimer
As Deputy Editor‐in‐Chief of Clinical and Translational Science, Deanna L. Kroetz was not involved in the review or decision process for this paper.
References
- 1. Hershman, D.L. et al. Association between patient reported outcomes and quantitative sensory tests for measuring long‐term neurotoxicity in breast cancer survivors treated with adjuvant paclitaxel chemotherapy. Breast Cancer Res. Treat. 125, 767–774 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Bennett, B.K. , Park, S.B. , Lin, C.S.‐Y. , Friedlander, M.L. , Kiernan, M.C. & Goldstein, D. Impact of oxaliplatin‐induced neuropathy: a patient perspective. Support Care Cancer 20, 2959–2967 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Hanai, A. et al. Effects of cryotherapy on objective and subjective symptoms of paclitaxel‐induced neuropathy: prospective self‐controlled trial. J. Natl. Cancer Inst. 110, 141–148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chon, H.J. et al. Docetaxel versus paclitaxel combined with 5‐FU and leucovorin in advanced gastric cancer: combined analysis of two phase II trials. Cancer Res. Treat. 41, 196–204 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Argyriou, A.A. , Bruna, J. , Marmiroli, P. & Cavaletti, G. Chemotherapy‐induced peripheral neurotoxicity (CIPN): an update. Crit. Rev. Oncol. Hematol. 82, 51–77 (2012). [DOI] [PubMed] [Google Scholar]
- 6. Cashman, C.R. & Höke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 2, 33–50 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Flatters, S.J.L. , Dougherty, P.M. & Colvin, L.A. Clinical and preclinical perspectives on chemotherapy‐induced peripheral neuropathy (CIPN): a narrative review. Br. J. Anaesth. 119, 737–749 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Gréen, H. et al. Pharmacogenetic studies of paclitaxel in the treatment of ovarian cancer. Basic Clin. Pharmacol. Toxicol. 104, 130–137 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Lagas, J.S. et al. Multidrug resistance protein 2 is an important determinant of paclitaxel pharmacokinetics. Clin. Cancer Res. 12(20 Pt 1), 6125–6132 (2006). [DOI] [PubMed] [Google Scholar]
- 10. Allen, D.T. & Kiernan, J.A. Permeation of proteins from the blood into peripheral nerves and ganglia. Neuroscience 59, 755–764 (1994) <http://www.sciencedirect.com/science/article/pii/0306452294901929>. [DOI] [PubMed] [Google Scholar]
- 11. Filipski, K.K. , Mathijssen, R.H. , Mikkelsen, T.S. , Schinkel, A.H. & Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin‐induced nephrotoxicity. Clin. Pharmacol. Ther. 86, 396–402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. More, S.S. , Akil, O. , Ianculescu, A.G. , Geier, E.G. , Lustig, L.R. & Giacomini, K.M. Role of the copper transporter, CTR1, in platinum‐induced ototoxicity. J. Neurosci. 30, 9500–9509 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang, K.M. , Hu, S. & Sparreboom, A. Drug transporters and anthracycline‐induced cardiotoxicity. Pharmacogenomics 19, 883–888 (2018). [DOI] [PubMed] [Google Scholar]
- 14. Kocer, B. , Sucak, G. , Kuruoglu, R. , Aki, Z. , Haznedar, R. & Erdogmus, N.I. Clinical and electrophysiological evaluation of patients with thalidomide‐induced neuropathy. Acta Neurol. Belg. 109, 120–126 (2009). [PubMed] [Google Scholar]
- 15. Fellner, S. et al. Transport of paclitaxel (Taxol) across the blood‐brain barrier in vitro and in vivo. J. Clin. Invest. 110, 1309–1318 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brooks, T.A. et al. Taxane‐based reversal agents modulate drug resistance mediated by P‐glycoprotein, multidrug resistance protein, and breast cancer resistance protein. Mol. Cancer Ther. 2, 1195–1205 (2003). [PubMed] [Google Scholar]
- 17. Allen, J.D. , Brinkhuis, R.F. , van Deemter, L. , Wijnholds, J. & Schinkel, A.H. Extensive contribution of the multidrug transporters P‐glycoprotein and Mrp1 to basal drug resistance. Cancer Res. 60, 5761–5766 (2000). [PubMed] [Google Scholar]
- 18. Woo, J.S. , Lee, C.H. , Shim, C.K. & Hwang, S.‐J. Enhanced oral bioavailability of paclitaxel by coadministration of the P‐glycoprotein inhibitor KR30031. Pharm. Res. 20, 24–30 (2003). [DOI] [PubMed] [Google Scholar]
- 19. Huisman, M.T. , Chhatta, A.A. , van Tellingen, O. , Beijnen, J.H. & Schinkel, A.H. MRP2 (ABCC2) transports taxanes and confers paclitaxel resistance and both processes are stimulated by probenecid. Int. J. Cancer 116, 824–829 (2005). [DOI] [PubMed] [Google Scholar]
- 20. Hopper‐Borge, E.A. et al. Contribution of Abcc10 (Mrp7) to in vivo paclitaxel resistance as assessed in Abcc10−/− mice. Cancer Res. 71, 3649–3657 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hopper‐Borge, E. , Chen, Z.‐S. , Shchaveleva, I. , Belinsky, M.G. & Kruh, G.D. Analysis of the drug resistance profile of multidrug resistance protein 7 (ABCC10): resistance to docetaxel. Cancer Res. 64, 4927–4930 (2004). [DOI] [PubMed] [Google Scholar]
- 22. Smith, N.F. , Acharya, M.R. , Desai, N. , Figg, W.D. & Sparreboom, A. Identification of OATP1B3 as a high‐affinity hepatocellular transporter of paclitaxel. Cancer Biol. Ther. 4, 815–818 (2005). [DOI] [PubMed] [Google Scholar]
- 23. Nieuweboer, A.J.M. et al. Influence of drug formulation on OATP1B‐mediated transport of paclitaxel. Cancer Res. 74, 3137–3145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mori, D. et al. Alteration in the plasma concentrations of endogenous OATP1B‐biomarkers in non‐small cell lung cancer patients treated with paclitaxel. Drug Metab. Dispos. 48, 387–394 (2020). [DOI] [PubMed] [Google Scholar]
- 25. Stage, T.B. , Bergmann, T.K. & Kroetz, D.L. Clinical pharmacokinetics of paclitaxel monotherapy: an updated literature review. Clin. Pharmacokinet. 57, 7–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kobayashi, Y. , Ohshiro, N. , Sakai, R. , Ohbayashi, M. , Kohyama, N. & Yamamoto, T. Transport mechanism and substrate specificity of human organic anion transporter 2 (hOat2 [SLC22A7]). J. Pharm. Pharmacol. 57, 573–578 (2005). [DOI] [PubMed] [Google Scholar]
- 27. Sissung, T.M. et al. ABCB1 genetic variation influences the toxicity and clinical outcome of patients with androgen‐independent prostate cancer treated with docetaxel. Clin. Cancer Res. 14, 4543–4549 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu, Y. et al. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol. Cancer Ther. 12, 1829–1836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baker, S.D. et al. Pharmacogenetic pathway analysis of docetaxel elimination. Clin. Pharmacol. Ther. 85, 155–163 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee, H.H. , Leake, B.F. , Teft, W. , Tirona, R.G. , Kim, R.B. & Ho, R.H. Contribution of hepatic organic anion‐transporting polypeptides to docetaxel uptake and clearance. Mol. Cancer Ther. 14, 994–1003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Graan, A.‐J.M. et al. Influence of polymorphic OATP1B‐type carriers on the disposition of docetaxel. Clin. Cancer Res. 18, 4433–4440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iusuf, D. et al. Human OATP1B1, OATP1B3 and OATP1A2 can mediate the in vivo uptake and clearance of docetaxel. Int. J. Cancer 136, 225–233 (2015). [DOI] [PubMed] [Google Scholar]
- 33. Leblanc, A.F. et al. OATP1B2 deficiency protects against paclitaxel‐induced neurotoxicity. J. Clin. Invest. 128, 816–825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishida, S. , Lee, J. , Thiele, D.J. & Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 99, 14298–14302 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holzer, A.K. , Manorek, G.H. & Howell, S.B. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 70, 1390–1394 (2006). [DOI] [PubMed] [Google Scholar]
- 36. Ivy, K.D. & Kaplan, J.H. A re‐evaluation of the role of hCTR1, the human high‐affinity copper transporter, in platinum‐drug entry into human cells. Mol. Pharmacol. 83, 1237–1246 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pabla, N. , Murphy, R.F. , Liu, K. & Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Renal Physiol. 296, F505–F511 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ip, V. , Liu, J.J. , Mercer, J.F.B. & McKeage, M.J. Differential expression of ATP7A, ATP7B and CTR1 in adult rat dorsal root ganglion tissue. Mol. Pain 13, 53 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jong, N.N. , Nakanishi, T. , Liu, J.J. , Tamai, I. & McKeage, M.J. Oxaliplatin transport mediated by organic cation/carnitine transporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 338, 537–547 (2011). [DOI] [PubMed] [Google Scholar]
- 40. Tschirka, J. , Kreisor, M. , Betz, J. & Gründemann, D. Substrate selectivity check of the ergothioneine transporter. Drug Metab. Dispos. Biol. Fate Chem. 46, 779–785 (2018). [DOI] [PubMed] [Google Scholar]
- 41. Fujita, S. , Hirota, T. , Sakiyama, R. , Baba, M. & Ieiri, I. Identification of drug transporters contributing to oxaliplatin‐induced peripheral neuropathy. J. Neurochem. 148, 373–385 (2018). [DOI] [PubMed] [Google Scholar]
- 42. Nishida, K. , et al. Ergothioneine ameliorates oxaliplatin‐induced peripheral neuropathy in rats. Life Sci. 15, 516–524 (2018). [DOI] [PubMed] [Google Scholar]
- 43. Burger, H. et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2). Br. J. Pharmacol. 159, 898–908 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sprowl, J.A. et al. Oxaliplatin‐induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. USA 110, 11199–11204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lancaster, C.S. , Sprowl, J.A. , Walker, A.L. , Hu, S. , Gibson, A.A. & Sparreboom, A. Modulation of OATP1B‐type transporter function alters cellular uptake and disposition of platinum chemotherapeutics. Mol. Cancer Ther. 12, 1537–1544 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Horio, M. et al. Transepithelial transport of drugs by the multidrug transporter in cultured Madin‐Darby canine kidney cell epithelia. J. Biol. Chem. 264, 14880–14884 (1989). [PubMed] [Google Scholar]
- 47. Takara, K. et al. Cytotoxic effects of 27 anticancer drugs in HeLa and MDR1‐overexpressing derivative cell lines. Biol. Pharm. Bull. 25, 771–778 (2002). [DOI] [PubMed] [Google Scholar]
- 48. Bertrand, Y. , Capdeville, R. , Balduck, N. & Philippe, N. Cyclosporin A used to reverse drug resistance increases vincristine neurotoxicity. Am. J. Hematol. 40, 158–159 (1992). [DOI] [PubMed] [Google Scholar]
- 49. Johnson, D.R. , Finch, R.A. , Lin, Z.P. , Zeiss, C.J. & Sartorelli, A.C. The pharmacological phenotype of combined multidrug‐resistance mdr1a/1b‐ and mrp1‐deficient mice. Cancer Res. 61, 1469–1476 (2001). [PubMed] [Google Scholar]
- 50. Chen, Z.S. et al. Effect of multidrug resistance‐reversing agents on transporting activity of human canalicular multispecific organic anion transporter. Mol. Pharmacol. 56, 1219–1228 (1999). [DOI] [PubMed] [Google Scholar]
- 51. Marada, V.V. , Flörl, S. , Kühne, A. , Burckhardt, G. & Hagos, Y. Interaction of human organic anion transporter polypeptides 1B1 and 1B3 with antineoplastic compounds. Eur. J. Med. Chem. 92, 723–731 (2015). [DOI] [PubMed] [Google Scholar]
- 52. Nicolaï, J. et al. Role of the OATP transporter family and a Benzbromarone‐SensitiveEfflux transporter in the hepatocellular disposition of vincristine. Pharm. Res. 34, 2336–2348 (2017). [DOI] [PubMed] [Google Scholar]
- 53. O’Connor, R. et al. The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 71, 1357–1368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Buda, G. et al. Polymorphisms in the multiple drug resistance protein 1 and in P‐glycoprotein 1 are associated with time to event outcomes in patients with advanced multiple myeloma treated with bortezomib and pegylated liposomal doxorubicin. Ann. Hematol. 89, 1133–1140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jandial, D.D. , Farshchi‐Heydari, S. , Larson, C.A. , Elliott, G.I. , Wrasidlo, W.J. & Howell, S.B. Enhanced delivery of cisplatin to intraperitoneal ovarian carcinomas mediated by the effects of bortezomib on the human copper transporter 1. Clin. Cancer Res. 15, 553–560 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Clemens, J. et al. Cellular uptake kinetics of bortezomib in relation to efficacy in myeloma cells and the influence of drug transporters. Cancer Chemother. Pharmacol. 75, 281–291 (2015). [DOI] [PubMed] [Google Scholar]
- 57. Shen, H. , Lee, F.Y. & Gan, J. Ixabepilone, a novel microtubule‐targeting agent for breast cancer, is a substrate for P‐glycoprotein (P‐gp/MDR1/ABCB1) but not breast cancer resistance protein (BCRP/ABCG2). J. Pharmacol. Exp. Ther. 337, 423–432 (2011). [DOI] [PubMed] [Google Scholar]
- 58. Flegel, C. et al. RNA‐seq analysis of human trigeminal and dorsal root ganglia with a focus on chemoreceptors. PLoS One 10, e0128951 (2015) <https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0128951>. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ray, P. et al. Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA‐seq‐based resource for pain and sensory neuroscience research. Pain 159, 1325–1345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schwaid, A.G. , Krasowka‐Zoladek, A. , Chi, A. & Cornella‐Taracido, I. Comparison of the rat and human dorsal root ganglion proteome. Sci. Rep. 8, 13469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sparreboom, A. et al. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P‐glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA 94, 2031–2035 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lhommé, C. et al. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J. Clin. Oncol. 26, 2674–2682 (2008). [DOI] [PubMed] [Google Scholar]
- 63. Fracasso, P.M. et al. Phase I study of paclitaxel in combination with a multidrug resistance modulator, PSC 833 (Valspodar), in refractory malignancies. J. Clin. Oncol. 18, 1124–1134 (2000). [DOI] [PubMed] [Google Scholar]
- 64. Fracasso, P.M. et al. Phase II study of paclitaxel and valspodar (PSC 833) in refractory ovarian carcinoma: a gynecologic oncology group study. J. Clin. Oncol. 19, 2975–2982 (2001). [DOI] [PubMed] [Google Scholar]
- 65. Noda‐Narita, S. et al. Peripheral neuropathy from paclitaxel: risk prediction by serum microRNAs. BMJ Support Palliat Care. https://doi.org/ 10.1136/bmjspcare-2019-001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stage, T.B. et al. P‐glycoprotein inhibition exacerbates paclitaxel neurotoxicity in neurons and cancer patients. Clin. Pharmacol. Ther. https://doi.org/ 10.1002/cpt.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kilari, D. et al. Copper transporter‐CTR1 expression and pathological outcomes in platinum‐treated muscle‐invasive bladder cancer patients. Anticancer Res. 36, 495–501 (2016). [PubMed] [Google Scholar]
- 68. Mani, S. et al. Activation of the steroid and xenobiotic receptor (human pregnane X receptor) by nontaxane microtubule‐stabilizing agents. Clin. Cancer Res. 11, 6359–6369 (2005). [DOI] [PubMed] [Google Scholar]
- 69. Geick, A. , Eichelbaum, M. & Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 276, 14581–14587 (2001). [DOI] [PubMed] [Google Scholar]
- 70. Feurstein, D. , Kleinteich, J. , Heussner, A.H. , Stemmer, K. & Dietrich, D.R. Investigation of microcystin congener‐dependent uptake into primary murine neurons. Environ. Health Perspect. 118, 1370–1375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Abraham, J.E. et al. Replication of genetic polymorphisms reported to be associated with taxane‐related sensory neuropathy in patients with early breast cancer treated with Paclitaxel. Clin. Cancer Res. 20, 2466–2475 (2014). [DOI] [PubMed] [Google Scholar]
- 72. Kus, T. et al. Polymorphism of CYP3A4 and ABCB1 genes increase the risk of neuropathy in breast cancer patients treated with paclitaxel and docetaxel. OncoTargets Ther. 9, 5073–5080 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tanabe, Y. et al. Paclitaxel‐induced sensory peripheral neuropathy is associated with an ABCB1 single nucleotide polymorphism and older age in Japanese. Cancer Chemother. Pharmacol. 79, 1179–1186 (2017). [DOI] [PubMed] [Google Scholar]
- 74. Custodio, A. et al. Pharmacogenetic predictors of severe peripheral neuropathy in colon cancer patients treated with oxaliplatin‐based adjuvant chemotherapy: a GEMCAD group study. Ann. Oncol. 25, 398–403 (2014). [DOI] [PubMed] [Google Scholar]
- 75. Broyl, A. et al. Mechanisms of peripheral neuropathy associated with bortezomib and vincristine in patients with newly diagnosed multiple myeloma: a prospective analysis of data from the HOVON‐65/GMMG‐HD4 trial. Lancet Oncol. 11, 1057–1065 (2010). [DOI] [PubMed] [Google Scholar]
- 76. Lopez‐Lopez, E. et al. Vincristine pharmacokinetics pathway and neurotoxicity during early phases of treatment in pediatric acute lymphoblastic leukemia. Pharmacogenomics 17, 731–741 (2016). [DOI] [PubMed] [Google Scholar]
- 77. Wright, G.E.B. et al. Pharmacogenomics of vincristine‐induced peripheral neuropathy implicates pharmacokinetic and inherited neuropathy genes. Clin. Pharmacol. Ther. 105, 402–410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ceppi, F. et al. Polymorphisms of the vincristine pathway and response to treatment in children with childhood acute lymphoblastic leukemia. Pharmacogenomics 15, 1105–1116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]