

Abstract
Small cell lung cancer (SCLC) is a leading cause of cancer death worldwide, with few treatment options. Rovalpituzumab tesirine (Rova‐T) is an antibody‐drug conjugate that targets delta‐like 3 on SCLC cells to deliver a cytotoxic payload directly to tumor cells. In this study, the cardiac safety profile of Rova‐T was assessed by evaluating changes in QT interval, electrocardiogram (ECG) waveform, heart rate, and proarrhythmic adverse events (AEs) after treatment with Rova‐T in patients with previously treated extensive‐stage SCLC. Patients underwent ECG monitoring for 2 weeks after each of 2 i.v. infusions of 0.3 mg/kg Rova‐T over 30 minutes, administered 6 weeks apart. Forty‐six patients received at least one dose of Rova‐T. At the geometric mean Rova‐T maximum serum concentration of 7,940 ng/mL, ECG monitoring showed no significant changes in the Fridericia‐corrected QT (QTcF) interval; the upper limit of the 2‐sided 90% confidence interval did not exceed 10 msec for any time point. There were no clinically significant changes in QRS or PR intervals, ECG waveforms, or heart rate after Rova‐T administration. All patients experienced a treatment‐emergent AE (TEAE); 78% had a grade ≥ 3 TEAE, 59% had a serious TEAE, and 41% had a cardiac‐related TEAE. The TEAEs that might signal proarrhythmia tendencies were uncommon. Confirmed partial responses were observed in 24% of patients. Based on the evaluation of ECG data collected in this study from patients treated with Rova‐T at 0.3 mg/kg i.v. administered every 6 weeks, a QTcF effect of clinical concern can be excluded.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ There currently are no clinical data regarding the electrocardiographic (ECG) effects of rovalpituzumab tesirine (Rova‐T).
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study was conducted to address questions regarding cardiac safety of this agent during its clinical drug development cycle to meet the US Food and Drug Administration (FDA) requirements.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study confirmed that targeting delta‐like 3 using Rova‐T at the 0.3 mg/kg dose that was utilized in the phase II/III studies did not result in any clinically significant changes in the ECG throughout several time points.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This knowledge will facilitate future development of tesirine‐containing antibody‐drug conjugates by ameliorating concerns of potential ECG effect, even if Rova‐T itself did not meet efficacy end points for drug approval.
Lung cancer is one of the most common and deadly cancers, with 228,000 new diagnoses per year and 143,000 deaths per year in the United States. 1 Small cell lung cancer (SCLC) accounts for 10% to 15% of lung cancers 1 and is a leading cause of cancer death worldwide. 2 , 3 The prognosis of patients with SCLC is poor, with a 5‐year survival rate of < 5%. 4 , 5 SCLC is categorized into limited‐stage and extensive‐stage (ES) disease based on the extent of the disease, with ES disease accounting for 65% of cases. 6 Treatment options are limited for ES disease, with platinum doublet chemotherapy along with anti‐PD‐L1 checkpoint blockade (atezolizumab or durvalumab) as the preferred first‐line treatment. There are few effective therapies approved for second‐line treatment of ES SCLC 7 ; median overall survival in patients treated with topotecan is only 26 weeks. 8 Recent studies of single‐agent cytotoxic agents and immunotherapy have yielded only modest improvements. 7
The Notch‐family ligand delta‐like 3 (DLL3) is highly expressed on SCLC cells but not expressed in most normal tissue, making it a tractable drug target for SCLC. 9 Rovalpituzumab tesirine (Rova‐T) is a first‐in‐class antibody‐drug conjugate (ADC) that targets DLL3 to deliver a cytotoxic agent directly to SCLC cells. Rova‐T is composed of a monoclonal DLL3 antibody linked to a DNA‐intercalating payload (pyrrolobenzodiazepine (PBD)) via a protease‐cleavable linker. The safety and efficacy of Rova‐T were initially evaluated in 82 patients in the first‐in‐human phase I study SCRX16‐001 (74 patients with SCLC and 8 patients with large‐cell neuroendocrine carcinoma). Treatment‐related cardiac adverse events (AEs) were uncommon. 10 The change in Fredericia‐corrected QT interval (QTcF) remained below a 10‐msec increase relative to baseline at 30 minutes after the end of infusion, when maximum Rova‐T serum concentrations were observed (unpublished data). Effects on QTcF at later time points have not been evaluated. In the phase 2 TRINITY study of patients with DLL3‐expressing relapsed/refractory SCLC, 12% of patients had a confirmed objective response to Rova‐T, and a manageable safety profile was observed. 11 Further development of Rova‐T has since been halted because two phase III studies showed a lack of clinical benefit of Rova‐T in the frontline maintenance and second‐line settings. 12 , 13
Determining cardiovascular safety is a key part of drug characterization. A delay in cardiac repolarization results in an electrophysiological environment that may lead to development of potentially fatal cardiac arrythmias. Typically, a thorough QT/corrected QT (QTc) study is conducted after the initial clinical study to determine whether an investigational agent meets a threshold level of effect on cardiac repolarization as detected by QT/QTc prolongation. 14 The cytotoxic component of Rova‐T precluded a thorough conventional QT/QTc study, which usually includes a crossover with the therapeutic dose, a supratherapeutic dose, a placebo, and a positive control in healthy volunteers. Additionally, because patients with SCLC require active treatment, the use of a placebo and/or a positive control would not be ethical. 15 Therefore, an alternative, intensive QT/QTc study design was determined to be appropriate, with the primary objective of determining the effect of Rova‐T on QTcF in patients with SCLC during the first two cycles of Rova‐T administration.
METHODS
Patients
Previously treated patients aged ≥ 18 years with ES SCLC and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2 were included in this study. Patients could not have any clinically significant cardiac abnormalities at enrollment (including baseline QRS duration of > 120 msec, and QTcF > 470 msec for women and > 450 msec for men), congenital long QT syndrome, or family history of long QT syndrome. Patients could not have had prior exposure to PBD‐containing drugs, including Rova‐T.
Study design and objectives
This was a multicenter, open‐label study that enrolled patients across 13 sites in the United States and Canada. Patients were enrolled in this study from October 2016 to September 2018. The study is registered at clinicaltrials.gov (NCT02874664) and was conducted according to the Declaration of Helsinki and all applicable laws, rules, and regulations within the relevant jurisdictions of the investigators. All patients provided written informed consent. The primary end point was the effect of treatment with Rova‐T on QTcF. Secondary end points included the effect of Rova‐T on RR and PR intervals, QRS duration, waveform composition, and the relationship between Rova‐T and proarrhythmic AEs, safety, and efficacy.
Treatment and assessments
Patients were treated with 0.3 mg/kg Rova‐T i.v. over 30 minutes on day 1 of every 6‐week cycle, omitting dosing every third cycle; dose reductions of Rova‐T were allowed. Patients were eligible to continue treatment until unacceptable toxicity, disease progression, investigator decision, withdrawal of consent, or study termination. Premedication with dexamethasone (8 mg) was administered orally twice daily on the day before, the day of, and the day after treatment with Rova‐T. Echocardiograms were performed at baseline and before treatment on day 1 of cycle 2 and onward to assess for any pericardial effusions and changes in cardiac function as assessed by left ventricular ejection fraction.
Patients underwent intensive electrocardiogram (ECG) monitoring during cycles 1 and 2 using an ambulatory 12‐lead Holter monitor. Predose monitoring was conducted during screening and on day 1 before infusion. Postdose monitoring was conducted for 4 hours after the end of infusion on day 1 of cycles 1 and 2. On days 2, 3, 4, 8, and 15 of cycles 1 and 2, monitoring was conducted for at least 1 hour. Triplicate 10‐second ECG tracings were extracted from the continuous Holter recordings at screening, with 2 time points at least 1 hour apart; on day 1 of cycles 1 and 2, with 3 time points at predose, 30 minutes postdose, and 4 hours postdose; and on days 2, 3, 4, 8, and 15 of cycles 1 and 2, with 1 time point at approximately the same time of day as the day 1 predose quantitative ECG collection period.
Disease assessments were performed with computed tomography or magnetic resonance imaging every 6 weeks for 24 weeks and every 12 weeks thereafter until disease progression. Tumor response was assessed by investigators per Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. AEs were summarized using Medical Dictionary for Regulatory Activities (MedDRA) version 13.1 preferred terms and graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Treatment‐emergent AEs (TEAEs) were defined as any AE that started or worsened on or after the first dose date and before subsequent anticancer therapy. TEAEs that might signal a dysrhythmia event included syncope, pre‐syncope, palpitations, dizziness, lightheadedness, tachycardia, unexplained seizure, and unexplained fall. Patients were followed for survival until study termination.
Drug concentration measurements and methods
Blood samples were collected before each dose, 30 minutes after dosing, 4 hours after dosing, and on days 2, 3, 4, 8, 15, and 29 of cycles 1 and 2 and on day 42 of cycle 1. Plasma concentrations of Rova‐T were measured as concentrations of ADC (including the Rova‐T antibody bound to at least 1 out of 2 molecules of toxin) using a validated electrochemiluminescent method. The lower limit of quantification for the assay was 39.1 ng/mL.
Statistical analysis
ECG analyses were conducted in all patients who received at least 1 dose of Rova‐T and had both baseline ECG measurements and at least one post‐baseline ECG measurement. Baseline ECG values were the mean value from all the data extracted from the screening ECGs and the day 1 of cycle 1 predose time point. Baseline values were used to calculate the mean change in QTcF (ΔQTcF) for each time point in cycles 1 and 2 using averages of triplicate measurements. Additional analyses included categorical summaries of QTcF data and other ECG parameters, such as heart rate, RR interval, PR interval, and QRS duration, as well as morphological analyses of ECG waveforms. Efficacy was assessed in all patients who received at least one dose of Rova‐T. Duration of response, progression‐free survival, and overall survival were evaluated using the Kaplan–Meier method. Safety analyses were performed in all patients who received at least one dose of Rova‐T.
Data sharing
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan and execution of a Data Sharing Agreement. Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.
RESULTS
Patient demographics, baseline characteristics, and disposition
Forty‐seven patients were enrolled, and 46 received treatment with at least 1 dose of Rova‐T. One patient had elevated liver function tests before dosing on day 1 of cycle 1 and was withdrawn from the study before treatment. Twenty‐eight patients (61%) were women, the median age was 64 years (range 45–83 years), and 44 of 46 patients (96%) had ECOG performance status of 0 or 1 (Table 1 ). Most patients (n = 44; 96%) were previous or current smokers. All patients had received at least 1 prior line of therapy, with 37 patients (80%) having received either 1 or 2 prior therapies. All patients had received at least 1 platinum‐containing therapy, with 24 (52%) having received prior immunotherapy.
Table 1.
Patient demographics and baseline characteristics
| Characteristic | All treated patients (N = 46) |
|---|---|
| Median age (range), year | 64 (45–83) |
| Male sex, n (%) | 18 (39) |
| ECOG PS, n (%) | |
| 0 | 12 (26) |
| 1 | 32 (70) |
| 2 | 2 (4) |
| Smoking status, n (%) | |
| Never | 2 (4) |
| Previous | 36 (78) |
| Current | 8 (17) |
| Prior lines of therapy, n (%) | |
| 1 | 14 (30) |
| 2 | 23 (50) |
| 3 | 7 (15) |
| 4 | 2 (4) |
| Prior platinum‐containing therapy, n (%) | 46 (100) |
| Prior topotecan, n (%) | 7 (15) |
| Resistant/refractory to first‐line therapy, n (%) | 12 (26) |
| DLL3 status at baseline, n/n (%) a | |
| High (≥ 75%) | 21/37 (57) |
| Positive (≥ 25%) | 28/37 (76) |
| Negative (0–24%) | 9/37 (24) |
DLL3, delta‐like 3; ECOG PS, Eastern Cooperative Oncology Group performance status.
Thirty‐seven patients were evaluated for DLL3 status.
The median treatment duration was 43 days (range 1–414 days). Patients were treated with a median of 2 cycles of therapy (range 1–7 cycles), with 12 patients (26%) having received only 1 cycle of Rova‐T. Eight patients required dose reductions because of AEs, and three patients required dose interruptions (2 because of AEs and 1 because of a dose miscalculation). Primary reasons for treatment discontinuation included disease progression in 19 patients (41%), AEs in 16 patients (35%), investigator decision in 8 patients (17%), withdrawn consent in 2 patients (4%), and other reasons in 1 patient (2%).
ECG analyses
One patient did not have ECG measurements at baseline and was excluded from the ECG analysis group. Based on a linear mixed‐effects model, the mean ΔQTcF at the geometric mean of the Rova‐T maximum serum concentration (Cmax) of 7,940 ng/mL was estimated to be −1.1 msec, with an upper limit of the 2‐sided 90% confidence interval (CI) of 2.2 msec (Figure 1 ). The mean Rova‐T concentration over time during the ECG monitoring period through cycles 1 and 2 is shown in Table 2 and Figure S1 . The mean ΔQTcF from baseline over this period were mostly decreases, except for small increases in ΔQTcF on day 1 of cycle 1, 30 minutes postdose, and on day 1 of cycle 2 predose and 30 minutes and 4 hours postdose (Table 3 ). In cycle 1, the largest mean ΔQTcF increase was 2.9 msec (90% CI 0.4–5.5) on day 1, 30 minutes postdose, and in cycle 2 was 2.4 msec (90% CI −1.7 to 6.4) on day 1, 4 hours postdose. The 1‐sided 95% CI did not exceed 10 msec for any time point. No patients showed QTcF values that exceeded 480 msec at any time.
Figure 1.
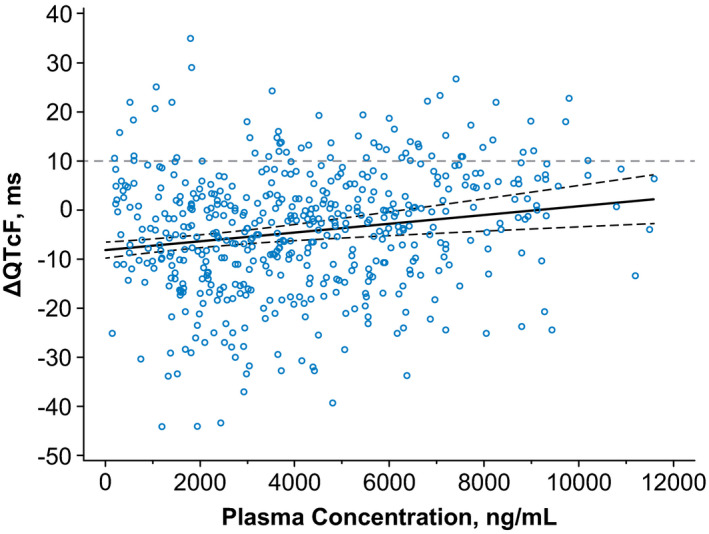
Change in QTcF vs. rovalpituzumab tesirine (Rova‐T) plasma concentration. The relationship between the change in QTcF and Rova‐T plasma concentrations was evaluated using a linear‐effects model with a fixed effect for intercept and random concentration (slope) per patient. The dotted lines represent the 90% confidence interval (CI). The regression line had an intercept of −8.1430, with a slope of 0.00089 (90% CI 0.00139–0.00139). ΔQTcF, change in Fridericia‐corrected QT interval.
Table 2.
Summary of Rova‐T plasma concentrations over time
| Visit | 0.3 mg/kg Rova‐T | Dose reduced 0.2 mg/kg Rova‐T | ||||||
|---|---|---|---|---|---|---|---|---|
| n | CV, % | Mean (SD) | Median (range) | n | CV, % | Mean (SD) | Median (range) | |
| Cycle 1 | ||||||||
| Day 1, 30‐minute postdose | 41 | 25.0 | 7.6 (1.9) | 7.7 (2.6–11.6) | – | – | – | – |
| Day 1, 4‐hour postdose | 43 | 18.7 | 7.1 (1.3) | 7.0 (4.7–10.9) | – | – | – | – |
| Day 2 | 44 | 17.8 | 5.5 (1.0) | 5.7 (3.6–7.7) | – | – | – | – |
| Day 3 | 44 | 20.4 | 4.3 (0.9) | 4.4 (2.7–5.9) | – | – | – | – |
| Day 4 | 43 | 21.1 | 3.6 (0.8) | 3.6 (2.2–5.1) | – | – | – | – |
| Day 8 | 45 | 29.9 | 2.2 (0.7) | 2.2 (1.0–4.5) | – | – | – | – |
| Day 15 | 42 | 28.1 | 1.5 (0.4) | 1.4 (0.7–2.3) | – | – | – | – |
| Cycle 2 | ||||||||
| Day 1 predose | 26 | 37.7 | 0.5 (0.2) | 0.5 (0.2–0.9) | 8 | 44.3 | 0.3 (0.1) | 0.3 (0.1–0.6) |
| Day 1, 30‐minute postdose | 24 | 21.4 | 7.9 (1.7) | 7.5 (4.7–10.8) | 8 | 20.8 | 5.2 (1.1) | 5.6 (3.4–6.2) |
| Day 1, 4‐hour postdose | 24 | 19.2 | 7.5 (1.4) | 7.2 (5.4–9.7) | 7 | 27.7 | 5.2 (1.4) | 5.6 (3.1–6.7) |
| Day 2 | 26 | 20.4 | 5.8 (1.2) | 5.7 (4.2–8.1) | 8 | 17.1 | 3.8 (0.7) | 3.8 (2.5–4.8) |
| Day 3 | 26 | 21.0 | 4.7 (1.0) | 4.8 (3.3–6.6) | 8 | 21.4 | 3.0 (0.6) | 3.1 (2.1–3.9) |
| Day 4 | 26 | 22.3 | 3.9 (0.9) | 4.0 (2.9–5.9) | 8 | 21.4 | 2.5 (0.5) | 2.5 (1.6–3.4) |
| Day 8 | 26 | 23.7 | 2.6 (0.6) | 2.7 (1.5–4.0) | 8 | 17.2 | 1.8 (0.3) | 1.8 (1.2–2.1) |
| Day 15 | 22 | 27.6 | 1.8 (0.5) | 1.7 (1.1–3.3) | 6 | 23.0 | 1.2 (0.3) | 1.3 (0.7–1.5) |
CV, coefficient of variation; Rova‐T, rovalpituzumab tesirine.
Table 3.
QTcF and ΔQTcF over time
| Visit | QTcF, msec | ΔQTcF, msec | ||||
|---|---|---|---|---|---|---|
| N | Mean (SD) |
2‐Sided 90% CI |
N | Mean (SD) |
2‐Sided 90% CI |
|
| Baseline | 45 | 411.8 (16.38) | 407.7–415.9 | — | — | — |
| Cycle 1 | ||||||
| Day 1, 30‐minute postdose | 41 | 415.3 (18.29) | 410.5–420.1 | 41 | 2.9 (9.57) | 0.4 to 5.5 |
| Day 1, 4‐hour postdose | 39 | 410.5 (18.09) | 405.6–415.3 | 39 | −0.6 (10.66) | −3.5 to 2.2 |
| Day 2 | 44 | 404.8 (16.90) | 400.5–409.1 | 43 | −6.3 (9.84) | −8.8 to −3.8 |
| Day 3 | 41 | 407.0 (15.16) | 403.0–411.0 | 41 | −5.6 (10.45) | −8.3 to −2.8 |
| Day 4 | 41 | 406.1 (16.01) | 401.9–410.3 | 41 | −7.7 (11.53) | −10.7 to −4.6 |
| Day 8 | 43 | 402.4 (17.38) | 398.0–406.9 | 42 | −8.3 (10.52) | −11.1 to −5.6 |
| Day 15 | 40 | 407.0 (13.79) | 403.4–410.7 | 40 | −7.0 (14.37) | −10.8 to −3.2 |
| Cycle 2 | ||||||
| Day 1 predose | 32 | 416.5 (15.23) | 412.9–422.7 | 31 | 1.2 (10.18) | −1.9 to 4.3 |
| Day 1, 30‐minute postdose | 32 | 417.8 (16.35) | 412.9–422.7 | 31 | 2.1 (12.47) | −1.7 to 5.9 |
| Day 1, 4‐hour postdose | 27 | 416.7 (15.78) | 411.5–421.8 | 26 | 2.4 (12.08) | −1.7 to 6.4 |
| Day 2 | 31 | 411.3 (15.11) | 406.7–415.9 | 31 | −3.9 (13.00) | −7.9 to 0.1 |
| Day 3 | 32 | 410.2 (14.94) | 405.7–414.7 | 31 | −5.1 (12.27) | −8.8 to −1.3 |
| Day 4 | 30 | 407.7 (15.10) | 403.0–412.4 | 29 | −8.3 (14.50) | −12.8 to −3.7 |
| Day 8 | 31 | 403.7 (15.30) | 399.0–408.4 | 30 | −11.9 (14.01) | −16.2 to −7.5 |
| Day 15 | 26 | 406.7 (21.57) | 399.4–413.9 | 25 | −5.9 (15.40) | −11.1 to −0.6 |
CI, confidence interval; QTcF, Fridericia‐corrected QT interval; ΔQTcF, change in Fridericia‐corrected QT interval.
There were no clinically significant changes in QRS or PR intervals. In cycle 1, the mean change in QRS interval from baseline (ΔQRS) was within ±2 msec until day 4, and the largest mean ΔQRS was −2.6 msec on day 8. In cycle 2, ΔQRS changes were negative at most time points and varied between −3.9 and 0.8 msec. The mean change in PR interval from baseline was within ±2 msec on day 1, and subsequent PR intervals generally decreased. The largest decrease in mean change in PR interval from baseline was −9.8 msec in cycle 1 and −11.5 msec in cycle 2.
Mean changes in heart rate from baseline over time were not clinically significant. During cycle 1, the largest increase in heart rate was 3.7 beats per minute, and the largest decrease was −4.4 beats per minute. During cycle 2, the largest increase was 9.8 beats per minute, and the largest decrease was −2.1 beats per minute.
ECG waveforms were classified by a cardiologist as normal, abnormal and clinically insignificant, or abnormal and potentially clinically significant (APCS) without comparison to baseline or previous ECGs. Twelve patients had ECGs that were considered APCS. No ECGs categorized as APCS were considered to be treatment emergent except for one patient who had high lateral Q waves that may have been indicative of myocardial infarction during cycle 2. After the ECG collection period, this patient experienced a worsening of pre‐existing pericardial effusion that led to withdrawal of study treatment and a new AE of atrial flutter reported after the ECG collection period; no clinical AE of myocardial infarction was reported. Two patients had clinically significant ST and T wave changes. One patient had nonspecific ST and T wave abnormalities at baseline, and on day 15 of cycle 1, that patient had new ST segment depression and anterior T wave inversion that were considered APCS and possibly evidence of ischemia by the over‐reading cardiologist. Review by a second cardiologist of these ECGs confirmed new ST and T wave changes of unclear clinical significance. The second patient had ST depression and T wave inversion on day 8 of cycle 1 and on days 3, 4, and 8 of cycle 2. There were no baseline ECGs available for this patient. Given that the inferior and anterolateral T wave inversions were present on multiple visits weeks apart, these findings were considered unlikely to represent myocardial ischemia. No patients had a U wave noted on any ECG.
There were no cases of torsade de pointes, ventricular fibrillation/flutter, atrial fibrillation/flutter, or supraventricular tachycardia detected in ECG findings. TEAEs that might signal proarrhythmia, including syncope, presyncope, palpitations, dizziness, lightheadedness, tachycardia, unexplained seizure, and unexplained fall, were assessed within the first 2 cycles during which ECG measurements were taken. Twelve patients experienced a TEAE that might signal proarrhythmia potential, including one patient with tachycardia, six with falls, four with dizziness, one with seizure, and one with syncope. Based on a lack of ECG findings of clinical relevance at the time points these TEAEs occurred, it was determined that there was no relationship between this subset of clinically reported TEAEs of interest and abnormal ECG findings occurring from days 1 through 15 of cycles 1 and 2.
Clinical safety
All patients treated with Rova‐T had at least one TEAE, with the most common being fatigue (57%), pleural effusion (46%), and photosensitivity reaction (41%). Thirty‐six patients (78%) had a grade ≥ 3 TEAE, with the most common being thrombocytopenia (24%) and anemia and malignant neoplasm progression (both 20%; Table 4 ). Serious TEAEs were reported by 59% of patients, with the most common being malignant neoplasm progression (20%), pleural effusion (11%), and pneumonia (11%). Overall, cardiac TEAEs were reported in 19 patients (41%), with 5 (11%) reporting grade ≥ 3 cardiac events. The most common cardiac TEAE was pericardial effusion, reported by 16 patients (35%), of which 13 cases (28%) were considered treatment‐related. Three patients (7%) had grade ≥ 3 events of pericardial effusion, of which 2 cases (4%) were considered treatment‐related. Other grade ≥ 3 treatment‐emergent cardiac events included 2 patients with grade ≥ 3 atrial fibrillation (1 treatment‐related), and 1 patient with grade ≥ 3 supraventricular tachycardia that was unrelated to Rova‐T.
Table 4.
TEAEs in ≥ 15% of patients at any grade or ≥ 5% of patients at grade ≥ 3
| Preferred term, n (%) | Any grade | Grade ≥ 3 | ||
|---|---|---|---|---|
| All | Related | All | Related | |
| Any TEAE | 46 (100) | 40 (87) | 36 (78) | 26 (57) |
| Fatigue | 26 (57) | 14 (30) | 2 (4) | 1 (2) |
| Pleural effusion | 21 (46) | 15 (33) | 6 (13) | 3 (7) |
| Photosensitivity reaction | 19 (41) | 17 (37) | 4 (9) | 4 (9) |
| Dyspnea | 19 (41) | 11 (24) | 2 (4) | 1 (2) |
| Nausea | 18 (39) | 11 (24) | 1 (2) | 1 (2) |
| Thrombocytopenia | 16 (35) | 13 (28) | 11 (24) | 10 (22) |
| Pericardial effusion | 16 (35) | 13 (28) | 3 (7) | 2 (4) |
| Peripheral edema | 15 (33) | 10 (22) | 0 (0) | 0 (0) |
| Anemia | 15 (33) | 9 (20) | 9 (20) | 4 (9) |
| Decreased appetite | 15 (33) | 6 (13) | 1 (2) | 1 (2) |
| Constipation | 14 (30) | 2 (4) | 0 (0) | 0 (0) |
| Vomiting | 10 (22) | 6 (13) | 0 (0) | 0 (0) |
| Hypokalemia | 9 (20) | 0 (0) | 2 (4) | 0 (0) |
| Urinary tract infection | 9 (20) | 0 (0) | 2 (4) | 0 (0) |
| Malignant neoplasm progression | 9 (20) | 0 (0) | 9 (20) | 0 (0) |
| Weight decreased | 9 (20) | 1 (2) | 0 (0) | 0 (0) |
| Asthenia | 8 (17) | 4 (9) | 0 (0) | 0 (0) |
| Diarrhea | 8 (17) | 5 (11) | 0 (0) | 0 (0) |
| Hypoalbuminemia | 7 (15) | 3 (7) | 0 (0) | 0 (0) |
| Pyrexia | 7 (15) | 2 (4) | 0 (0) | 0 (0) |
| Pneumonia | 7 (15) | 2 (4) | 3 (7) | 1 (2) |
| Aspartate aminotransferase increased | 5 (11) | 3 (7) | 3 (7) | 1 (2) |
| Lipase increased | 4 (9) | 1 (2) | 3 (7) | 1 (2) |
| Blood alkaline phosphatase increased | 4 (9) | 2 (4) | 3 (7) | 2 (4) |
| Fluid overload | 3 (7) | 3 (7) | 3 (7) | 3 (7) |
| Lymphopenia | 3 (7) | 0 (0) | 3 (7) | 0 (0) |
TEAE, treatment‐emergent adverse event.
TEAEs led to discontinuation in 13 patients (28%), dose reduction in 8 patients (17%), and dose interruption in 2 patients (4%). A total of 38 patients (81%) died, with most deaths related to underlying disease. Deaths from TEAEs were reported in 11 patients (24%). Nine patients died due to malignant neoplasm progression and one patient died due to hepatic failure; none of these deaths were considered related to Rova‐T. One death from gastrointestinal hemorrhage was reported that was considered related to Rova‐T. This patient had concomitant therapy with warfarin for prior deep vein thrombosis and pulmonary embolism. On day 15, the patient experienced a serious AE of grade 3 gastrointestinal hemorrhage, as well as events of grade 4 thrombocytopenia and grade 3 anemia. The patient received a transfusion with packed red blood cells, and warfarin was discontinued. Further treatment was declined, and the patient entered hospice care and died on day 19 from the event of gastrointestinal hemorrhage. Overall, the TEAEs observed in this study are consistent with the known Rova‐T safety profile, and no new safety signals were observed.
Efficacy
All 46 patients who received at least 1 dose of Rova‐T were included in the efficacy analyses. Partial responses were reported in 14 patients (30%), and 11 patients (24%) had confirmed responses (Table 5 ). Most patients (11 of 14) responded at the first post‐baseline efficacy assessment at ~ 6 weeks. The median duration of response in 11 patients with confirmed responses was 3.3 months (95% CI 1.9–4.3 months). The median progression‐free survival was 3.5 months (95% CI 2.6–4.1 months), and the median overall survival was 5.4 months (95% CI 4.1–7.1 months).
Table 5.
Efficacy
| Outcome | All patients (N = 46) |
|---|---|
| Overall response rate, n (%) (95% CI) | 14 (30) (17.7–45.8) |
| CR, n (%) | 0 (0) |
| PR, n (%) | 14 (30) |
| Objective response rate, n (%) (95% CI) a | 11 (24) (12.6–38.8) |
| CR, n (%) | 0 (0) |
| PR, n (%) | 11 (24) |
| SD, n (%) | 20 (43) |
| PD, n (%) | 10 (22) |
| Missing, n (%) b | 2 (4) |
| Median DOR (95% CI), months c | 3.3 (1.9–4.3) |
| Median PFS (95% CI), months | 3.5 (2.6–4.1) |
| Median OS (95% CI), months | 5.4 (4.1–7.1) |
CI, confidence interval; CR, complete response; DOR, duration of response; OS, overall survival; PD, progressive disease; PFS, progression‐free survival; PR, partial response; SD, stable disease.
CR or PR requires confirmation of at least 4 weeks from the initial determination of response per Response Evaluation Criteria in Solid Tumors, version 1.1.
Because of death before disease assessment.
DOR presented for confirmed responses.
DISCUSSION
The tesirine payload attached to the anti‐DLL3 antibody rovalpituzumab is derived from a dimer of the PBD family of DNA minor‐grove binding agents, based on the anthramycin group of antitumor antibiotics. Early clinical experience with the naturally occurring PBD monomer anthramycin was limited due to dose‐limiting cardiotoxicity, and whereas human data are not available, ECG changes induced by anthramycin have been demonstrated in chronically treated rats. 16 , 17 Prior functional characterization in structural experiments demonstrated that cardiotoxicity of PBDs may arise from the presence of a hydroxy (‐OH) group at the C9 position mediated through free radical production, which can damage cardiac tissue 18 ; however, tesirine is not hydroxylated at the C9 position. Due to minimal to no DLL3 expression in normal tissue, cardiotoxicity directly related to the drug target of Rova‐T was not anticipated. 9
Consistent with the above expectation of the electrophysiological safety of Rova‐T, the results of this study did not show significant effects of Rova‐T on QTcF in patients with SCLC. The mean ΔQTcF at the mean Rova‐T Cmax was estimated to be −1.1 msec, with an upper bound of the 2‐sided 90% CI limit of 2.2 msec. These results are in line with the by‐time‐point analyses showing that QTcF increases from baseline were small in cycles 1 and 2, with the largest increase across both cycles being 2.9 msec (90% CI 0.4–5.5). Heart rate increases were larger in cycle 2 than in cycle 1 (9.8 beats per minute on day 15 of cycle 2 and 3.7 beats per minute on day 1 of cycle 1). There were no clinically relevant effects of Rova‐T on cardiac conduction based on review of PR and QRS intervals. Morphological analyses of ECG waveforms did not show any clinically relevant abnormalities with Rova‐T exposure except in one patient with a new high lateral Q relationship observed during cycle 2, which may have indicated myocardial infarction, although myocardial infarction as an AE was not officially reported for this patient and was attributed to worsening pericardial effusion by the treating physician.
Despite the lack of previous preclinical or clinical evidence of QT prolongation, this study was conducted to provide an appropriate, rigorous assessment of the potential for Rova‐T to prolong cardiac ventricular repolarization. ECG recordings from ambulatory Holter monitoring allowed for evaluation of potential effects of Rova‐T on the QTc interval, including clinically relevant time points, such as the Rova‐T ADC Cmax. For cytotoxic therapeutics, such as Rova‐T, approaches incorporating intensive ECG Holter monitoring, such as those used in this study, provide a feasible alternative to a conventional thorough QT/QTc study.
The results of this study of Rova‐T administered at 0.3 mg/kg in 6‐week cycles are consistent with the findings from the first‐in‐human phase I study of Rova‐T that reported no clinically meaningful increases in QT interval after exposure to Rova‐T at doses up to 0.8 mg/kg administered in 3‐week or 6‐week cycles (unpublished data). The TEAEs reported in this study were similar to those observed previously with Rova‐T. 10 No relationship between AEs that might signal proarrhythmia occurring in the first 2 weeks of cycles 1 and 2 and ECG results could be determined. In conclusion, based on evaluation of ECG data, a clinical effect of Rova‐T given at 0.3 mg/kg i.v. every 6 weeks on QTc interval can be excluded.
Funding
Rovalpituzumab tesirine (Rova‐T) was developed by AbbVie. AbbVie provided financial support for the study.
Conflict of Interest
J.G. received research funding from Stemcentrx and AbbVie. J.P. had a consultancy/advisory role for AbbVie, AstraZeneca, Genentech, and Takeda and received research funding to institution from Bristol Myers Squibb. A.W. had a consultancy/advisory role for Boehringer Ingelheim, AstraZeneca, Premier, Inc., Lilly Oncology, Karyopharm, and Epizyme, served on a data safety monitoring board for Odonate Therapeutics, BeyondSpring Pharmaceuticals, and HUYA Bioscience International, and received research funding from Boehringer Ingelheim. A.D. had a consultancy/advisory role for Takeda, AbbVie, Seagen, AstraZeneca, and Bristol Myers Squibb and received research funding from Loxo, Bayer, Incuron, Takeda, Regeneron, Tesaro, Amgen, Seagen, Symphogen, AbbVie, and Ipsen. A.S. had a consultancy/advisory role with Sandoz and Bayer, and received honorarium from Bristol Myers Squibb. T.O. had a consultancy/advisory role for AbbVie. W.E. had a consultancy/advisory role for Chimerix. S.L. received research funding to institution from AbbVie. D.D.C., S.L., M.K., and D.H. are employed by AbbVie and may hold stock or other options with AbbVie. M.K. is employed by Regeneron and may hold stock or other options with Regeneron and was a former employee of AbbVie. G.D. had a consultancy/advisory role for and honorarium from AbbVie. All other authors declared no competing interests for this work.
Author Contributions
J.W.G., M.B., J.D.P., A.W., A.D., A.S., T.K.O., W.E., S.A.L., D.D., S.L., M.K., M.P.K., D.H., and G.K.D. wrote the manuscript. S.L. and D.D. designed the research. J.W.G., M.B., J.D.P., A.W., A.D., A.S., T.K.O., W.E., S.A.L., and G.K.D performed the research. J.W.G., M.B., J.D.P., A.W., A.D., A.S., T.K.O., W.E., S.A.L., D.D., S.L., M.K., M.P.K., D.H., and G.K.D. analyzed the data.
Supporting information
Fig S1
Acknowledgments
The authors thank the patients and their families, study coordinators, and support staff. AbbVie provided financial support for this study and participated in the design, study conduct, and analysis and interpretation of data, as well as the writing, review, and approval of this manuscript. Medical writing assistance was provided by Allison Cherry, PhD, of Bio Connections, LLC, and funded by AbbVie. AbbVie provided financial support for this study and participated in the design, study conduct, and analysis and interpretation of the data, as well as the writing, review, and approval of the article. All authors had full access to the study data and were involved in the data gathering, analysis, review, interpretation, and article preparation and approval. No honoraria or payments were made for authorship.
References
- 1. American Cancer Society . Cancer facts & figures 2019 (American Cancer Society, Atlanta, GA: ). <https://www.cancer.org/research/cancer‐facts‐statistics/all‐cancer‐facts‐figures/cancer‐facts‐figures‐2019.html> (2019). Accessed May 19, 2020. [Google Scholar]
- 2. Kalemkerian, G.P. et al. Small cell lung cancer. J. Natl. Compr. Canc. Netw. 11, 78–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Torre, L.A. , Siegel, R.L. & Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 893, 1–19 (2016). [DOI] [PubMed] [Google Scholar]
- 4. Demedts, I.K. , Vermaelen, K.Y. & van Meerbeeck, J.P. Treatment of extensive‐stage small cell lung carcinoma: current status and future prospects. Eur. Respir. J. 35, 202–215 (2010). [DOI] [PubMed] [Google Scholar]
- 5. Stinchcombe, T.E. & Gore, E.M. Limited‐stage small cell lung cancer: current chemoradiotherapy treatment paradigms. Oncologist 15, 187–195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernhardt, E.B. & Jalal, S.I. Small cell lung cancer. Cancer Treat. Res. 170, 301–322 (2016). [DOI] [PubMed] [Google Scholar]
- 7. Yang, S. , Zhang, Z. & Wang, Q. Emerging therapies for small cell lung cancer. J. Hematol. Oncol. 12, 47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Brien, M.E. et al. Phase III trial comparing supportive care alone with supportive care with oral topotecan in patients with relapsed small‐cell lung cancer. J. Clin. Oncol. 24, 5441–5447 (2006). [DOI] [PubMed] [Google Scholar]
- 9. Saunders, L.R. et al. A DLL3‐targeted antibody‐drug conjugate eradicates high‐grade pulmonary neuroendocrine tumor‐initiating cells in vivo. Sci. Transl. Med. 7, 302ra136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rudin, C.M. et al. Rovalpituzumab tesirine, a DLL3‐targeted antibody‐drug conjugate, in recurrent small‐cell lung cancer: a first‐in‐human, first‐in‐class, open‐label, phase 1 study. Lancet Oncol. 18, 42–51 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgensztern, D. et al. Efficacy and safety of rovalpituzumab tesirine in third‐line and beyond patients with DLL3‐expressing, relapsed/refractory small‐cell lung cancer: results from the phase II TRINITY study. Clin. Cancer Res. 25, 6958–6966 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. AbbVie discontinues rovalpituzumab tesirine (Rova‐T) research and development program. North Chicago, IL: AbbVie Inc.; August 29, 2019 <https://news.abbvie.com/news/press‐releases/abbvie‐discontinues‐rovalpituzumab‐tesirine‐rova‐t‐research‐and‐development‐program.htm>. Accessed October 23, 2019. [Google Scholar]
- 13. Phase 3 trial of Rova‐T as second‐line therapy for advanced small‐cell lung cancer (TAHOE Study) Halted. North Chicago, IL: AbbVie Inc.; December 5, 2018 <https://news.abbvie.com/news/phase‐3‐trial‐rova‐t‐as‐second‐line‐therapy‐for‐advanced‐small‐cell‐lung‐cancer‐tahoe‐study‐halted.htm>. Accessed October 23, 2019. [Google Scholar]
- 14. E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐anti‐arrhythmic drugs. US Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), and Center for Biologicals Evaluation and Research (CBER) <https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/e14‐clinical‐evaluation‐qtqtc‐interval‐prolongation‐and‐proarrhythmic‐potential‐non‐antiarrhythmic‐0> (2005). Accessed April 6, 2020. [Google Scholar]
- 15. Clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐anti‐arrhythmic drugs questions and answers (R3) guidance for industry. US Department of Health and Human Services Food and Drug Administration. Center for Drug Evaluation and Research (CDER) and Center for Biologicals Evaluation and Research (CBER) <https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/e14‐clinical‐evaluation‐qtqtc‐interval‐prolongation‐and‐proarrhythmic‐potential‐non‐antiarrhythmic‐1> (2017). Accessed April 6, 2020. [Google Scholar]
- 16. Cargill, C. , Bachmann, E. & Zbinden, G. Effects of daunomycin and anthramycin on electrocardiogram and mitochondrial metabolism of the rat heart. J. Natl. Cancer Inst. 53, 481–486 (1974). [DOI] [PubMed] [Google Scholar]
- 17. Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 20, 733–744 (2011). [DOI] [PubMed] [Google Scholar]
- 18. Mantaj, J. , Jackson, P.J. , Rahman, K.M. & Thurston, D.E. From anthramycin to pyrrolobenzodiazepine (PBD)‐containing antibody‐drug conjugates (ADCs). Angew Chem. Int. Ed. Engl. 56, 462–488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1