

Abstract
Zanubrutinib is a potent, second‐generation Bruton’s tyrosine kinase inhibitor that is currently being investigated in patients with B‐cell malignancies and recently received accelerated approval in the United States for treatment of relapsed/refractory mantle cell lymphoma. The objective of this analysis was to develop a population pharmacokinetic (PK) model to characterize the PKs of zanubrutinib and identify the potential impact of intrinsic and extrinsic covariates on zanubrutinib PK. Data across nine clinical studies of patients with B‐cell malignancies and data of healthy volunteers (HVs) were included in this analysis, at total daily doses ranging from 20 to 320 mg. In total, 4,925 zanubrutinib plasma samples from 632 subjects were analyzed using nonlinear mixed‐effects modeling. Zanubrutinib PKs were adequately described by a two‐compartment model with sequential zero‐order then first‐order absorption, and first‐order elimination. A time‐dependent residual error model was implemented in order to better capture the observed maximum concentration variability in subjects. Baseline alanine aminotransferase and health status (HVs or patients with B‐cell malignancies) were identified as statistically significant covariates on the PKs of zanubrutinib. These factors are unlikely to be clinically meaningful based on a sensitivity analysis. No statistically significant differences in the PKs of zanubrutinib were observed based on age, sex, race (Asian, white, and other), body weight, mild or moderate renal impairment (creatinine clearance ≥ 30 mL/minute as estimated by Cockcroft‐Gault), baseline aspartate aminotransferase, bilirubin, tumor type, or use of acid‐reducing agents (including proton pump inhibitors). These results support that no dose adjustment is considered necessary based on the aforementioned factors.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Zanubrutinib has demonstrated promising activity for the treatment of multiple B‐cell malignancies and has recently received accelerated approval in the United States for treatment of relapsed/refractory mantle cell lymphoma.
WHAT QUESTION DID THIS STUDY ADDRESS?
What are the potential intrinsic and extrinsic covariates that can impact the pharmacokinetics (PKs) of zanubrutinib?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This is the first report on the population PK analysis of zanubrutinib. No dose adjustments for zanubrutinib are warranted due to age, body weight, creatinine clearance, sex, or the use of acid‐reducing agents.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This report described a sequential zero‐order followed by a first‐order absorption kinetics and time‐dependent residual error model in order to better capture the observed maximum concentration (Cmax) variability in subjects. This approach may be applicable to the analysis of other compounds.
B‐cell receptor (BCR) signaling is essential for normal B‐cell development, differentiation, function, and survival. 1 , 2 Aberrant activation of protein kinases within the BCR signaling pathway consequently results in the development of various B‐cell malignancies, including mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL), follicular lymphoma, Waldenström’s macroglobulinemia (WM), and diffuse large B‐cell lymphoma. 3 Bruton’s tyrosine kinase (BTK), a member of the Tec family of protein kinases, is a key component within the BCR signaling cascade, and suppression of BCR signaling—notably through inhibition of BTK—has emerged as a promising strategy for the targeting and management of B‐cell malignancies. 4
Zanubrutinib (BRUKINSA™; also known as BGB‐3111) is a potent, specific, and irreversible second‐generation BTK inhibitor that was granted an accelerated approval by the US Food and Drug Administration (FDA) in November 2019 for the indication of MCL in adult patients who have received at least one prior therapy. Zanubrutinib was also approved in June 2020 by the China National Medical Products Administration for CLL or small lymphocytic lymphoma and MCL in adult patients who have received at least one prior therapy. 5 , 6 Similar to ibrutinib, zanubrutinib forms an irreversible covalent bond at Cys481 within the ATP binding pocket of BTK. However, zanubrutinib was designed to have favorable pharmacokinetic (PK) and pharmacodynamic properties to maximize BTK occupancy and minimize off‐target inhibition. Zanubrutinib has been found to be more selective than ibrutinib for the inhibition of BTK, as evidenced by less off‐target inhibition of epidermal growth factor receptor, Janus tyrosine kinase 3, human epidermal growth factor receptor 2, Tec protein tyrosine kinase, and inducible tyrosine kinase. 7 After adjusting for plasma protein binding, zanubrutinib exposure at a total daily dose of 320 mg is ~ 8‐fold higher than that observed with ibrutinib 560 mg daily. 8 Zanubrutinib is currently being investigated in ongoing phase I, II, and III studies in patients with B‐cell malignancies.
Zanubrutinib was first evaluated in a global phase I/II, multicenter, open‐label, dose‐escalation/cohort‐expansion study (NCT02343120) in patients with B‐cell malignancies at doses ranging from 40 mg to 320 mg. The median steady‐state BTK occupancy in peripheral blood mononuclear cells was maintained at 100% for over 24 hours at a total daily dose of 320 mg in patients with B‐cell malignancies, regardless of dosing schedule (either 320 mg q.d. or 160 mg b.i.d.). 8 , 9 , 10 In addition, the median steady‐state BTK occupancy in lymph node biopsies was 94% and 100% after the 320 mg q.d. and 160 mg b.i.d. doses, respectively. These results indicate efficient inactivation of BTK in target tissues by zanubrutinib throughout the recommended dosing interval and are consistent with high rates of objective response in patients with B‐cell malignancies, including WM, MCL, and CLL in clinical studies of zanubrutinib. 11 , 12 , 13 The recommended dose of zanubrutinib is a total daily dose of 320 mg (either as 160 mg b.i.d. or 320 mg q.d. with or without food).
Previous PK data demonstrate that zanubrutinib is rapidly absorbed and eliminated after oral administration in patients with B‐cell malignancies. 13 Zanubrutinib is primarily metabolized by cytochrome P450 3A (CYP3A4) enzymes and no major active metabolites are found in circulation. The mean terminal elimination half‐life (t 1/2) of zanubrutinib is ~ 2–4 hours and median time to peak zanubrutinib plasma concentration (Tmax) is 2 hours. After multiple‐dose administration of zanubrutinib at doses ranging from 40 mg to 320 mg q.d. and 160 mg b.i.d., limited systemic accumulation is observed, which is consistent with the observed t 1/2. A dose‐proportional increase in maximum concentration (Cmax) and area under the plasma concentration‐time curve (AUC) from time 0 extrapolated to infinity (AUC0–∞) is observed at doses from 40 mg to 320 mg. 13
The objective of the current analysis was to develop a population PK model to characterize and identify the potential impact of intrinsic and extrinsic covariates on the PK profile of zanubrutinib. We report findings of the population PK analysis, including the individual and mean model‐derived parameters and associated interindividual variabilities (IIVs), as well as the covariates impacting zanubrutinib exposure and variability. The results of this population PK analysis were used to assess whether dose adjustment was considered necessary based on baseline covariates in patients with B‐cell malignancies.
METHODS
Clinical Data
Zanubrutinib PK data across nine clinical studies of patients with B‐cell malignancies and healthy volunteers (HVs) were included in the analysis. A summary of the studies and patient population is provided in Table 1 . All studies were conducted in accordance with the Declaration of Helsinki and Guidelines for Good Clinical Practice; approval from institutional review boards or independent ethics committees were obtained for each study. 14 , 15 Written informed consent was obtained from all study participants.
Table 1.
Summary of studies included in the population PK analysis
| Study no. | Dose regimen | N | Study description | PK sampling design |
|---|---|---|---|---|
| BGB‐3111‐AU‐003 (NCT02343120) |
40 mg, 80 mg, 160 mg, and 320 mg q.d. 160 mg b.i.d. |
337 | A phase I, open‐label, multiple‐dose, dose escalation and expansion study to investigate the safety and pharmacokinetics of the BTK inhibitor BGB‐3111 in patients with B‐cell lymphoid malignancies |
Part 1: W1D1: Predose, 0.5, 2, 3, 4, 8, 24 hours W2D1: Predose, 0.5, 2, 3, 4, 7, 8 hours W5D1 and W9D1: Predose Part 2: W1D1 and W2D1: Predose, 2 hours |
| BGB‐3111‐1002 (NCT03189524) |
320 mg q.d. 160 mg b.i.d. |
44 | A phase I clinical study to investigate the safety, tolerability, and PKs/pharmacodynamics of the BTK inhibitor BGB‐3111 in Chinese patients with B‐cell lymphoma |
Part 1: W1D1: Predose, 0.5, 1, 2, 3, 4, 8, 12, 24 hours (W1D2 predose) W2D1: Predose, 0.5, 1, 2, 3, 4, 8 hours W5D1 and W9D1: Predose Part 2: W1D1: Predose, 0.5, 1, 2, 3, 4, 6, 8, 12, 24 hours (W1D2 predose) W2D1: Predose, 0.5, 1, 2, 3, 4, 6, 8, 12 hours W5D1 and W9D1: Predose |
| BGB‐3111‐205 (NCT03206918) | 160 mg b.i.d. | 13 | A single‐arm, open‐label, multicenter phase II study to evaluate safety and efficacy of BGB‐3111, a BTK inhibitor in relapsed or refractory CLL/SLL |
C1D1: Predose, 2, 4–6 hours C2D1: Predose, 2, 4–6 hours |
| BGB‐3111‐206 (NCT03206970) | 160 mg b.i.d. | 20 | A single‐arm, open‐label, multicenter phase II study to evaluate the efficacy and safety of BGB‐3111, a BTK inhibitor, in patients with relapsed or refractory MCL |
C1D1: Predose, 2, 4–6 hours C2D1: Predose, 2, 4–6 hours |
| BGB‐3111‐103 (NCT04163523) | 320 mg q.d. | 18 | A single‐center, phase I, open‐label, randomized, crossover study to evaluate the effect of food on the PKs of a single dose of 320 mg BGB‐3111 given orally to healthy adult subjects | Predose, 0.5, 1, 2, 3, 4, 6, 8, 12, 24 hours (under each of high‐fat, low‐fat, and fasted conditions) |
| BGB‐3111‐104 (NCT03301181) |
20 mg q.d. 320 mg q.d. |
38 | A phase I, open‐label, parallel‐group, fixed‐sequence study to investigate the effect of the CYP3A inducer rifampin and the CYP3A inhibitor itraconazole on the PKs of BGB‐3111 in healthy subjects |
Part A: D1 and D10: Predose, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48 hours Part B: D1 and D6: Predose, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48 hours |
| BGB‐3111‐105 (NCT04163783) | 320 mg q.d. | 6 | A phase I study to investigate the absorption, metabolism, and excretion of [14C] BGB‐3111 following a single oral administration in healthy male subjects | Predose, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144 hours |
| BGB‐3111‐106 (NCT03432884) |
160 mg q.d. 480 mg q.d. |
28 | A two‐part study consisting of a randomized, placebo‐controlled, single dose safety and tolerability study (part A) evaluating a supratherapeutic dose of zanubrutinib followed by a randomized, placebo and positive‐controlled, crossover study (part B) to evaluate the effect of zanubrutinib on cardiac repolarization in HVs | Predose, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 24, 36, 48 hours |
| BGB‐3111‐302 (NCT03053440) | 160 mg b.i.d. | 128 | A study comparing BGB‐3111 and ibrutinib in subjects with WM |
C1D1: Predose 2, 3–6 hours C2D1: Predose 2, 3–6 hours |
BTK, Bruton’s tyrosine kinase; C, cycle; CLL, chronic lymphocytic leukemia; D, day; HV, healthy volunteers; MCL, mantle cell lymphoma; PK, pharmacokinetics; SLL, small lymphocytic lymphoma; W, week; WM, Waldenström’s macroglobulinemia.
Zanubrutinib was administered at doses ranging from 20 to 320 mg q.d. or b.i.d., with most subjects receiving a dose of 160 mg b.i.d. Measurements of zanubrutinib concentrations in plasma were performed using a validated high‐performance liquid chromatography tandem mass spectrometry assay with a lower limit of quantitation of 1 ng/mL. Subjects were considered to be evaluable for PK analysis if they had at least one dose of zanubrutinib and a corresponding PK sample collection after drug administration. A total of 4,925 zanubrutinib plasma samples from 632 subjects were used in the analysis. Methodologies on how the data were handled, with respect to outliers and missing data, are provided in the Supplementary Material .
Population PK Modeling
The population PK analysis was performed using a nonlinear mixed‐effects modeling with the first‐order conditional estimation with interaction method. 16 Model parameter estimation and model evaluation were implemented with NONMEM 7 17 (version 7.4.0; ICON Development Solutions, Ellicott City, MD) with Intel Fortran Compiler (version 10.1.021), Perl‐speaks‐NONMEM (version 3.2.12; Uppsala University, Sweden) 18 , 19 and R (version 3.5.3).
Base model and random‐effects model development
Based on a graphical assessment of zanubrutinib plasma concentration‐time profiles, an initial structural model was selected and tested with various modifications. Goodness‐of‐fit of the models was evaluated and symmetry of the individual parameters around the estimated median parameter was assessed graphically. Model diagnostics provided direction for further model modifications and/or refinements.
Assuming a log‐normal distribution, the IIV in PK parameters was described by an exponential model:
| (1) |
where θi is the parameter for the i th subject, θT is natural logarithm of the typical value of the parameter in the population, and ηi (individual random effects) is a random interindividual effect with mean 0 and variance ω 2 . The ω values are the diagonal elements of the IIV‐covariance matrix (Ω), which was initially modeled as diagonal (DIAG option in the NM‐TRAN $OMEGA record), thus assuming no covariance between the random effects. A non‐diagonal Ω matrix was finally implemented using the BLOCK option to estimate correlation between random effects in both the base PK model and the final population PK model. If required, interoccasion variability was modeled.
Residual error was described using the combined additive and proportional error model:
| (2) |
where C(t)ij is the jth observed plasma concentration of individual i; Ĉ(t)ij is the jth model predicted value (plasma concentration) for individual i; and ɛpij and ɛaij are normally distributed residual random errors with mean of 0 and variances of and , respectively. Additive ( = 0) or proportional ( = 0) residual error models or combination of both error models were tested during model building.
Covariate model development
After base model development, all covariates likely to impact zanubrutinib exposure were explored for a possible correlation with key zanubrutinib post hoc PK parameters. Covariates were selected based upon industry standards, clinical judgment, mechanistic plausibility, and prior knowledge with zanubrutinib. The impact of baseline age, body weight, sex, race (white, Asian, and other), creatinine clearance (CrCL), bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase, tumor type (MCL, CLL/small lymphocytic lymphoma, WM, and other B‐cell malignancies), health status (HVs or patients with B‐cell malignancies), and use of acid‐reducing agents (ARAs) on the PKs of zanubrutinib was investigated. ARAs used by patients in clinical studies included H2 receptor antagonists (H2RA; e.g., cimetidine, famotidine, and ranitidine) and proton pump inhibitors (PPIs; e.g., esomeprazole, omeprazole, and pantoprazole). In the analysis, subjects that took either a PPI or H2RA were categorized together, while subjects that did not use either PPIs or H2RAs were categorized as “other.”
Correlations between the PK parameters and the covariates were explored graphically, followed by linear regression (continuous covariates) and analysis of variance (ANOVA) testing (categorical covariates) using R software.
Continuous covariates were modeled using a power model, as described in Eq. 3:
| (3) |
Categorical covariates were modeled using the general equation:
| (4) |
where θ is a model parameter, Cov is a continuous covariate, X is a categorical variable, i is an index for each subject, pop is an index describing the usual value of this covariate in the population, and kcov is a coefficient describing the strength of the covariate effect.
These analyses were conducted on individual random effects for apparent clearance (CL/F) and apparent central volume (Vc/F). Only covariates that showed a significant (P < 0.01) effect on the random effect, and that could be meaningfully explained from both a clinical and scientific perspective, were examined further using NONMEM.
Selection of the final covariate model (final population PK model) was determined for its significance based on the likelihood ratio test at the P < 0.01 level for forward inclusion and P < 0.001 for backward deletion.
Model evaluation
The final population PK model was developed after utilizing multiple internal model validation assessments, including goodness‐of‐fit diagnostics, prediction‐corrected visual predictive check (pcVPC) plots, 20 a numerical predictive check, 21 bootstrap analysis, 22 , 23 and evaluation of shrinkage.
In a pcVPC, variability arising from binning across independent variables is removed by normalizing the observed and simulated dependent variable based on the typical population prediction for the median independent variable in the bin. The pcVPC plots show the time course of the predicted mean and spread of concentrations (2.5th to 97.5th percentile) vs. the observed data. A total of 1,000 trial replicates were simulated using the observed covariates and dose regimens for each subject, the final model parameter estimates, and simulated subject‐specific random effects and residual errors.
Sensitivity analyses
Sensitivity analyses were performed to examine the influence of statistically significant covariates on the expected steady‐state exposure of zanubrutinib, including steady‐state area under the concentration vs. time curve (AUCss), steady‐state maximum concentration (Cmax,ss), and steady‐state trough concentration (Cmin,ss). The simulated exposure of patients with extreme covariate values (5th and 95th percentiles) was compared with a typical patient with median covariate values for each of the statistically significant covariates in the final model.
Zanubrutinib, at 160 mg, administered orally b.i.d., was selected as the recommended dose for phase II and phase III studies and, therefore, individual zanubrutinib exposures (AUCss, Cmax,ss, and Cmin,ss) were simulated for multiple doses at 160 mg b.i.d.
RESULTS
Clinical data
A summary of the demographics, consisting of categorical and continuous covariates, is provided in Tables S1a and S1b . Due to a less than dose‐proportional increase in exposure at the 480‐mg dose, data from 9 HVs in study BGB‐3111‐106 (NCT03432884) were excluded from the analysis. During model development, data points were classified as outliers using conditional population weighted residuals. Data points with conditional population weighted residuals > 5 were considered potential outliers and were excluded from the PK analysis. Furthermore, subjects (n = 2) with extreme PK parameters were also excluded from the PK analysis. Observations below the lower limit of quantitation, which made up 6.05% (317/5,242) of the data points, were omitted in the analysis. The final analysis dataset included 4,925 zanubrutinib plasma concentrations from 632 subjects. Of these, a total of 90 HVs were included in the analysis.
Base model development and covariate assessment
The PKs of zanubrutinib in the dose range tested were best described by a two‐compartment model with sequential zero‐order then first‐order absorption, as well as first‐order elimination from the central compartment and redistribution from the peripheral compartment, as illustrated in Figure 1 . The model development process is summarized in Table S2 of the Supplementary Material . The population PK model was parameterized in terms of CL/F, Vc/F, apparent clearance from the central to the peripheral compartment (Q/F), apparent volume of the peripheral compartment (Vp/F), absorption rate constant, and the duration (D1) of zero‐order absorption into the depot compartment. In order to better explain the variability observed between occasions (single dose vs. after repeated dosing), interoccasion variability was sequentially evaluated on the PK parameters and was included on select parameters (CL/F, Vc/F, and D1) based on a statistically significant change in objective function value (P < 0.05). Occasion, defined as different scheduled visits and associated dose/sampling times, was represented as < 7 (for single‐dose data) and ≥ 7 days (after repeated dosing). Among different residual error models evaluated, both an additive error model and the combined additive and proportional error model were included in the final model (an additive error model utilized < 5 hours postdose, and combined additive and proportional error model afterward). This was done by establishing a cutoff point to divide the data into two groups (time after previous dose < 5 and time after previous dose ≥ 5 hours). The improvement of fit, especially around description of Cmax, was observed based on goodness‐of‐fit criteria compared with other error models evaluated.
Figure 1.
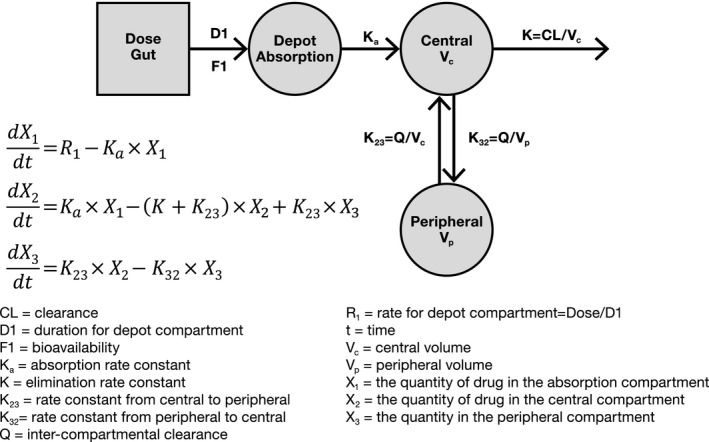
Population pharmacokinetic (PK) model diagram for zanubrutinib.
Results from the covariate screening analysis are presented in Figure S1 . Based on an examination of PK parameter‐covariate relationships, ALT, health status, and tumor type were statistically significant at P < 0.01 and were thus carried forward to the nonlinear mixed‐effects covariate modeling search in NONMEM. Screening of other covariates showed that age (19–90 years), sex, race (Asian, white, and other), body weight (36–144 kg), mild or moderate renal impairment (CrCL ≥ 30 mL/minute as estimated by Cockcroft‐Gault), baseline aspartate aminotransferase, bilirubin, tumor type, or use of ARAs (including PPIs and H2RAs) were not statistically significant covariates on zanubrutinib CL/F and Vc/F. In particular, post hoc PK estimates were similar among patients receiving PPI or H2RA treatment and patients who did not receive ARAs (P > 0.074, ANOVA, Figure S2 ).
Final population PK model
The key model parameters (CL/F and Vc/F) are defined in Eqs. 5 and 6. In the final population PK model, baseline ALT and health status were found to be statistically significant covariates on zanubrutinib’s CL/F (Figure S3 ).
The parameter‐covariate relationships were described as follows:
| (5) |
| (6) |
The estimated CL/F was 170 L/hour, Vc/F was 112 L, Q/F was 26.5 L/hour, Vp/F was 345 L, absorption rate constant was 0.526 hour‐1, and D1 was 1.13 hours for a typical patient with ALT of 18 U/L. The geometric mean elimination t 1/2 was 3.44 hours with a coefficient of variance of 40.0%. The IIV estimated for zanubrutinib CL/F and Vc/F was 36.7% and 37.1%, respectively.
Bootstrapping of 1,000 datasets resulted in median parameter estimates and 95% confidence intervals, which was similar to estimates from the original dataset (Table 2 ), indicating that the final population PK model provided good precision for parameter estimation. General goodness‐of‐fit plots showed good agreement between predicted and observed concentrations of zanubrutinib (Figure 2a ), with no apparent bias in the residual plots over time and across predicted concentrations (Figure 2b ). Distributions of IIV and their correlations are shown in Figures S4 and S5 in the Supplementary Material . The pcVPC plots (Figure 3 ) and numerical predictive check (data not shown) showed that the final population PK model could adequately reproduce the central tendency and variability of the zanubrutinib plasma concentrations across all studies.
Table 2.
Summary of final population PK parameters and bootstrap results
| Parameter | Parameter description |
Final model estimate (95% CI) |
Bootstrap estimate median (2.5–97.5 percentiles) |
|---|---|---|---|
| exp(θ 1) | CL/F (L/hour, patient) | 170 (169, 170) | 169 (162, 178) |
| exp(θ 10) | CL/F (L/hour, HV) | 118 (107, 130) | 119 (110, 127) |
| θ 11 | Influence of ALT on CL/F | −0.189 (−0.197, −0.181) | −0.190 (−0.262, −0.115) |
| exp(θ 2) | Vc/F (L) | 112 (111, 113) | 115 (88.4, 151) |
| exp(θ 3) | Q/F (L/hour) | 26.5 (26.3, 26.7) | 25.6 (19.9, 32.3) |
| exp(θ 4) | Vp/F (L) | 345 (343, 347) | 363 (274, 513) |
| exp(θ 5) | Ka (1/hour) | 0.526 (0.525, 0.526) | 0.524 (0.481, 0.591) |
| exp(θ 6) | D1 (hour) | 1.128 (1.125, 1.131) | 1.114 (0.974, 1.265) |
| ω Cl,,Vc | Covariance (CL/F, Vc/F) | 0.129 (0.0972, 0.160) | 0.13 (0.0757, 0.183) |
| IIV | CL/F | 36.7 (32.9, 40.2) | 36.6 (32.6, 40.2) |
| Vc/F | 37.1 (NA, 53.1) | 39.2 (25.9, 57.8) | |
| Q/F | 102 (97.8, 106) | 103 (83.9, 118) | |
| Vp/F | 86.4 (71.3, 99.3) | 80.4 (58.9, 94.6) | |
| D1 | 62.3 (57.1, 67.1) | 62.2 (51.9, 72.4) | |
| IOV | CL/F | 28.6 (27.9, 29.3) | 28.7 (24.4, 33.0) |
| Vc/F | 67.5 (66.8, 68.3) | 65.9 (49.4, 76.8) | |
| D1 | 62.3 (57.1, 67.1) | 62.2 (51.9, 72.4) | |
| θ 9 | Additive residual error (ng/mL, TFDS < 5 hour) | 8.69 (8.68, 8.69) | 8.69 (8.14, 9.10) |
| θ 7 | Additive residual error (ng/mL, TFDS ≥ 5 hour) | 0.633 (0.632, 0.634) | 0.656 (0.380, 1.45) |
| θ 8 | Proportional residual error (%) | 44.9 (44.8, 45.0) | 45.2 (42.4, 48.4) |
ALT, alanine aminotransferase; CI, confidence interval, CL/F, apparent clearance of the drug from plasma after oral administration; D1, the duration of zero‐order absorption into the depot compartment; HV, healthy volunteers; IIV, interindividual variability; IOV, interoccasion variability; Ka, absorption rate constant; NA, not available; PK, pharmacokinetic; Q/F, apparent clearance from the central to the peripheral compartment; TFDS, time after previous dose; Vc/F, apparent central compartment volume; Vp/F, apparent peripheral distribution volume.
Figure 2.
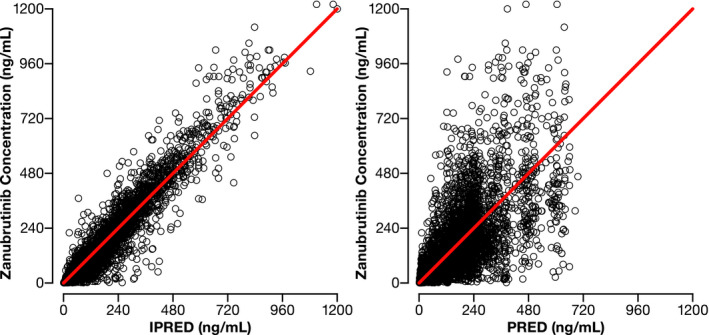
Diagnostic plots for the final population pharmacokinetic (PK) model. (a) Observed vs. individual predicted (IPRED) concentrations (left) and observed vs. population predicted (PRED) concentrations (right) for the final population PK model. Points are individual data and red lines represent the line of unity. (b) Conditional weighted residuals (CWRES) vs. time (left) and population PRED (right). Points are individual data. Red solid lines represent the unit line at zero. Dotted lines represent CWRES of 5.
Figure 3.
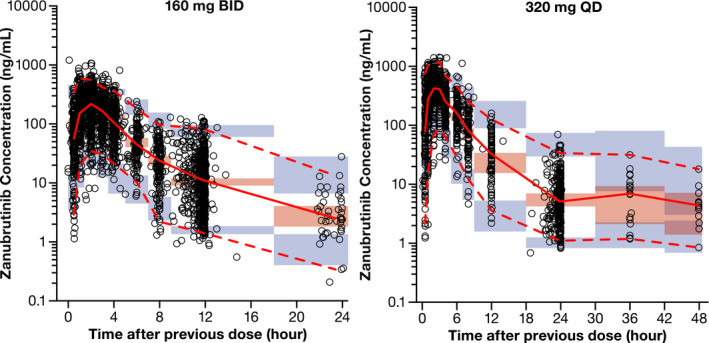
The pcVPC of zanubrutinib plasma concentration–time profiles across all studies. Points are observed concentrations, solid red line represents the median observed value, and dashed red lines represent 2.5th percentile and 97.5th percentile of the observed values. Pink shaded area represents the spread of the median predicted values (2.5th to 97.5th percentile), and purple shaded areas represent the spread (2.5th and 97.5th percentiles) of the 2.5th and 97.5th predicted percentile concentrations. pcVPC, prediction‐corrected visual predictive check.
The η‐shrinkage for CL/F, Vc/F, and Q/F was 24.2%, 27.8%, and 25.9%, respectively (< 30%), which suggests that the model provides reliable empirical Bayes estimates for these parameters. The Vp/F had an η‐shrinkage of 51.3%, which may result in individual estimates approaching the population estimate. 24 The IIV on Vp/F should therefore be interpreted with caution as the current data may be insufficient to adequately characterize the IIV on Vp/F.
Sensitivity analysis
The isolated influence of statistically significant covariates on the expected steady‐state exposure of zanubrutinib was evaluated in a sensitivity analysis (Figure 4 ). Predicted Cmax,ss and AUCss after repeat‐dose administration of 160 mg b.i.d. were 254 ng/mL and 943 ng·hour/mL in a typical patient; this corresponds to a total daily AUC of 1,886 ng·hour/mL. Sensitivity analyses (Figure 4 ) suggested that the magnitude of effect of ALT on zanubrutinib AUCss (< 15%) and Cmax,ss (< 10%) was low and moderate on zanubrutinib Cmin,ss (< 30%), respectively. Of note, Cmin,ss was associated with greater variability than AUCss and Cmax,ss. Given the observed IIV of key zanubrutinib PK parameters (CL/F, 36.7%; Vc/F, 37.1%), baseline ALT alone is not expected to have clinically meaningful effects on zanubrutinib exposure in patients with B‐cell malignancies. The impact of health status (HVs vs. patients) on zanubrutinib CL/F and Vc/F resulted in 43.7% higher AUCss and 26.8% higher Cmax,ss in HVs compared with patients with B‐cell malignancies.
Figure 4.
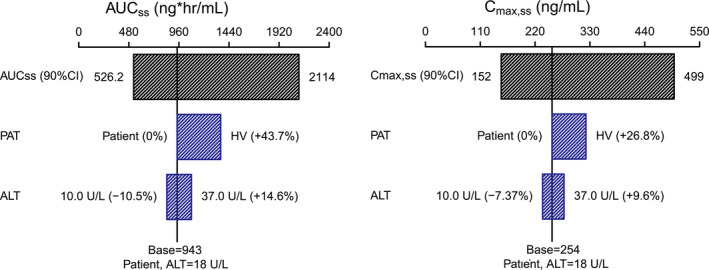
Sensitivity analysis plot comparing the effect of covariates on zanubrutinib steady‐state exposure (AUCss, Cmax,ss). Base, as represented by the black vertical line and values, refers to the predicted exposure (AUCss and Cmax,ss) of zanubrutinib in a typical male patient after repeated 20 doses of 160 mg twice a day. The black‐shaded bar with value at each end shows the 5th to 95th percentile exposure range across the entire population. Each blue‐shaded bar represents the influence of covariates on the exposure. The label at the left end of the bar represents the covariate being evaluated. The upper and lower values for each covariate capture 80% of the plausible range in the population. The length of each bar describes the potential impact of the covariates on zanubrutinib exposure, with the percentage value in the parentheses at each end representing the percent change of exposure from the base. The most influential covariates are at the top of the plot for each exposure parameter. ALT, alanine aminotransferase; AUCss, steady‐state area under the plasma concentration‐time curve; CI, confidence interval; Cmax,ss, steady‐state maximum observed plasma concentration; HV, healthy volunteer(s); PAT, patient.
DISCUSSION
This population PK analysis of data from nine studies adequately characterized the PK profile of zanubrutinib and assessed the impact of potential covariates. In order to better capture observed Cmax variability in subjects, a sequential zero‐order followed by first‐order absorption kinetics and implementation of time‐dependent residual error model was incorporated into the two‐compartment model. Zanubrutinib is primarily eliminated hepatically via CYP3A4, and the impact of drug‐drug interactions (e.g., drug‐drug interactions with CYP3A4 inhibitors/inducers) and hepatic impairment on the PKs of zanubrutinib have previously been characterized. 25 The current analysis indicated that other patient baseline characteristics, such as age, body weight, race, sex, CrCL, tumor type, and the use of ARAs, including PPIs, did not show a statistically significant impact on the PKs of zanubrutinib.
The current analysis indicated a lack of significant impact for the use of PPIs and other ARAs on zanubrutinib PKs. This attribute could aid in the management of clinically relevant drug interactions, as the prevalence of patients with cancer receiving ARAs (some are over‐the‐counter) could range from 20% to 55%. 26 In the work of Parrott et al., it was shown that gastric ARAs, such as PPIs, can increase the stomach pH to ~ 4.5 compared with the typical values of 1 to 2 measured in untreated individuals. 27 Given that zanubrutinib exhibits pH‐dependent solubility and the use of gastric ARAs were allowed in zanubrutinib clinical studies, the potential effects of ARAs on the PKs of zanubrutinib were assessed in this population PK analysis. Results of the analysis showed that co‐administration of zanubrutinib with PPIs and other ARAs did not appear to significantly impact the PKs of the drug. The current work further corroborates the results from a physiologically‐based PK model of zanubrutinib and a clinical drug‐drug interaction study, indicating that zanubrutinib exposures are not anticipated to be affected by ARAs. 28
A population PK model for zanubrutinib was developed using a two‐compartment model to describe first‐order elimination from the central compartment and redistribution from the peripheral compartment. The absorption phase was described with sequential zero‐order then first‐order absorption kinetics. The sequential linked zero‐order then first‐order model has been previously described 29 and can be interpreted mechanistically with drug concentrations reaching a solubility threshold in the gut at the onset of absorption. Subsequentially, drug absorption leads to concentrations declining below the solubility limit initially reached, converting absorption from a zero‐order to a first‐order input. 29 Although zanubrutinib concentrations were largely accounted for by this absorption model, an underprediction of Cmax was present in several cases. This may have been due to the variability of zanubrutinib concentrations observed during the absorption phase.
After establishing a mechanistically plausible structural model, the characterization of zanubrutinib absorption was evaluated by implementation of various residual error models. Residual variability (within or IIV) is the remaining, unexplained variability that may arise from multiple potential sources of error such as assay variability, dosing or sampling errors, and model misspecification. Data used from two populations, such as HVs and patients, may also introduce residual variability. 30 This may, in part, be due to better control of dosing and sampling times as well as the homogeneity of HVs, resulting in lower residual variability in comparison to patients. Additionally, potential sources of variability, such as in dosing and sampling history, may be time‐dependent. For example, time‐dependent residual error may result in residual variability being greater in the absorption phase of a drug in comparison to the elimination phase. 30 In cases where time‐dependent errors may be a factor toward higher residual variability, such as with zanubrutinib data particularly during the absorption phase, a general residual error model can incorporate time‐varying variables or a threshold model where one error model accounts for residual variability up to time t followed by another model. 30 Karlsson et al. proposed such alternative residual error models to evaluate errors from structural model misspecification or time‐dependent error due to inaccurate sample timing. 31 In the current analysis, a threshold for a time‐dependent residual error model (an additive error model utilized < 5 hours postdose, and combined additive and proportional error model afterward) was implemented and significantly improved the model description of Cmax. A cutoff of 5 hours was selected based on data exploration and goodness‐of‐fit plots. PK data showed that the absorption phase (within ~ 5 hours after the dose) had a higher residual error than the elimination phase. Improvement of model fit was observed based on goodness‐of‐fit plots after adding the time‐varying residual error. Without this implementation, there appeared to be underestimation of Cmax using combined additive and proportional error model throughout. By incorporating such a time‐varying residual error model, the variability of Cmax in subjects was more accurately characterized.
In evaluating sources of IIV through the covariate analysis, baseline ALT and health status (patients with B‐cell malignancies vs. HVs) were identified as statistically significant covariates impacting the PKs of zanubrutinib. Additionally, a sensitivity analysis indicated that health status was the most influential covariate on the PKs of zanubrutinib as its impact on zanubrutinib CL/F resulted in a 43.7% higher AUCss and 26.8% higher Cmax,ss in HVs compared with patients with B‐cell malignancies. This finding is consistent with a cross‐study comparison of intensive PK data in HVs and patient studies by noncompartmental analysis. However, it is currently unknown what contributed to lower zanubrutinib exposure in patients relative to exposure in HVs. This difference was not likely due to the effect on drug clearance, because higher exposure would be expected due to a reduction in CYP3A4 abundance in the liver and gut noted in the cancer population. 32 , 33 The median age of patients included in this analysis was 66 years, whereas the median age of subjects in HV studies was 43 years. It is plausible that the absorption of zanubrutinib could be impacted by age‐related physiological changes in elderly patients with cancer, such as reduced gastrointestinal motility and splanchnic blood flow. 34 , 35 Alternatively, it could, in part, be due to the high PK variability in elderly patients with comorbidities who often receive multiple concomitant medications.
The impact of baseline ALT on zanubrutinib exposure, although statistically significant on the drug’s apparent clearance, was relatively small compared with the overall variability of the population; this aligns with observed clinical data in a dedicated hepatic impairment study in which comparable exposure levels between subjects with mild or moderate hepatic impairment and those with normal hepatic function were observed, whereas dose modifications are recommended in cases of severe hepatic impairment. 25 Race was not found to be a statistically significant covariate on zanubrutinib PKs. The model predicted steady‐state zanubrutinib exposures were similar among Asians, whites, and subjects of other races. These results align with findings from a clinical study evaluating intensive PK data between subjects of both Asian and non‐Asian descent, which showed no marked PK difference between the two ethnic groups. 36
In summary, zanubrutinib PK was adequately described by a two‐compartment model with sequential zero‐order then first‐order absorption. The overall impact of covariates that have an influence on the variability of zanubrutinib exposures were identified and assessed. Baseline covariates, including age, body weight, race, CrCL, sex, tumor type, and use of ARAs did not show a statistically significant impact on the PK of zanubrutinib, and therefore no dose adjustments would be warranted based on these covariates. These results are reflected in the current recommendations in the BRUKINSA™ United States Prescribing Information. 5
Funding
This study was funded by BeiGene, Ltd.
Conflicts of Interest
Y.C.O., B.T., Z.T., and S.S. are employees of and own stock in BeiGene, Inc. K.W., L.L., and C.Y.G. are employees of Shanghai Qiangshi Information Technology Co., Ltd. A.J. was an employee and owned stock in BeiGene, Inc. at the time the work was performed.
Author Contributions
Y.O., S.S., B.T., Z.T., and A.J. wrote the manuscript; Y.O. and S.S. designed the research; Y.O., L.L., B.T., K.W., A.J, Z.T., Y.G and S.S. performed the research, K.W., L.L., and Y.G. analyzed the data.
Supporting information
Supplementary Material
Acknowledgments
The authors wish to thank all the subjects, study coordinators, and support staff who contributed to the clinical data collection used in this analysis. Editorial assistance was provided by Open Health Medical Communications (Chicago, IL), and was funded by BeiGene, Inc.
References
- 1. Dal Porto, J.M. et al. B cell antigen receptor signaling 101. Mol. Immunol. 41, 599–613 (2004). [DOI] [PubMed] [Google Scholar]
- 2. Niiro, H. & Clark, E.A. Regulation of B‐cell fate by antigen‐receptor signals. Nat. Rev. Immunol. 2, 945–956 (2002). [DOI] [PubMed] [Google Scholar]
- 3. Rickert, R.C. New insights into pre‐BCR and BCR signalling with relevance to B cell malignancies. Nat. Rev. Immunol. 13, 578–591 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Niemann, C.U. & Wiestner, A. B‐cell receptor signaling as a driver of lymphoma development and evolution. Semin. Cancer Biol. 23, 410–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. BRUKINSA™ (zanubrutinib) [package insert]. BeiGene USA, Inc., San Mateo CA (2019) <https://www.brukinsa.com/prescribing‐information.pdf>. Accessed May 19, 2020.
- 6. BeiGene . Press Releases. BeiGene announces the approval of BRUKINSA™ (zanubrutinib) in China for patients with relapsed/refractory chronic lymphocytic leukemia or small lymphocytic lymphoma and relapsed/refractory mantle cell lymphoma <http://ir.beigene.com/news‐releases/news‐release‐details/beigene‐announces‐approval‐brukinsatm‐zanubrutinib‐china>. Accessed June 26, 2020.
- 7. Guo, Y. et al. Discovery of zanubrutinib (BGB‐3111), a novel, potent, and selective covalent inhibitor of Bruton's tyrosine kinase. J. Med. Chem. 62, 7923–7940 (2019). [DOI] [PubMed] [Google Scholar]
- 8. Tam, C.S. et al. The BTK inhibitor, BGB‐3111, is safe, tolerable, and highly active in patients with relapsed/ refractory B‐cell malignancies: initial report of a phase 1 first‐in‐human trial. Blood 126, 832 (2015). [Google Scholar]
- 9. Tam, C.S. et al. Twice daily dosing with the highly specific BTK inhibitor, BGB‐3111, achieves complete and continuous BTK occupancy in lymph nodes, and is associated with durable responses in patients (pts) with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). Blood 128, 642 (2016). [Google Scholar]
- 10. Tam, C.S. et al. Safety and activity of the highly specific BTK inhibitor BGB‐3111 in patients with indolent and aggressive non‐Hodgkin’s lymphoma. Blood 130, 152 (2017). [Google Scholar]
- 11. Dimopoulos, M.A. et al. Major responses in MYD88 wildtype (MYD88WT) Waldenström macroglobulinemia (WM) patients treated with Bruton tyrosine kinase inhibitor zanubrutinib (BGB‐3111). Annual Congress of the European Hematology Association (2019) <https://library.ehaweb.org/eha/2019/24th/266287/meletios.a.dimopoulos.major.responses.in.myd88.wildtype.%28myd88wt%29.waldenstrm.html>. Accessed May 19, 2020.
- 12. Song, Y. Zanubrutinib in patients with relapsed/refractory mantle cell lymphoma . 15th International Conference on Malignant Lymphoma (2019) <https://www.lymphcon.ch/icml/website/doc/15‐ICM>. Accessed August 21, 2020.
- 13. Tam, C.S. et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B‐cell malignancies and safety and efficacy evaluation in CLL. Blood 134, 851–859 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. World Medical Association . World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. Bull. World. Health. Organ. 79, 373–374 (2001). [PMC free article] [PubMed] [Google Scholar]
- 15. The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) <https://www.ich.org/>. Accessed May 20, 2020. [DOI] [PMC free article] [PubMed]
- 16. Lindstrom, M.L. & Bates, D.M. Nonlinear mixed effects models for repeated measures data. Biometrics 46, 673–687 (1990). [PubMed] [Google Scholar]
- 17. Beal, S.L. , Sheiner, L.B. , Boeckmann, A.J. & Bauer, R.J. NONMEM User's Guide. Part I‐VII (1989‐2018) <https://nonmem.iconplc.com/nonmem743/guides>. Accessed August 26, 2020.
- 18. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Methods Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 19. Lindbom, L. , Ribbing, J. & Jonsson, E.N. Perl‐speaks‐NONMEM (PsN)–a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 75, 85–94 (2004). [DOI] [PubMed] [Google Scholar]
- 20. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harling, K. & Ueckert, S.A.H. VPC and NPC user guide. 2019‐01‐27 PsN 5.0.0 <https://github.com/UUPharmacometrics/PsN/releases/download/5.0.0/vpc_npc_userguide.pdf>. Accessed May 19, 2020.
- 22. Ette, E.I. Stability and performance of a population pharmacokinetic model. J. Clin. Pharmacol. 37, 486–495 (1997). [DOI] [PubMed] [Google Scholar]
- 23. Bootstrap User Guide. 2011. [updated 2011] <http://psn.sourceforge.net/pdfdocs/bootstrap_userguide.pdf>. Accessed May 19, 2020.
- 24. Savic, R.M. & Karlsson, M.O. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 11, 558–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ou, Y.C. et al. A phase 1, open‐label, single‐dose study of the pharmacokinetics of zanubrutinib in subjects with varying degrees of hepatic impairment. Leuk. Lymphoma 61, 1355–1363 (2020). [DOI] [PubMed] [Google Scholar]
- 26. Budha, N.R. et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH‐dependent solubility the Achilles heel of targeted therapy? Clin. Pharmacol. Ther. 92, 203–213 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Parrott, N.J. , Yu, L.J. , Takano, R. , Nakamura, M. & Morcos, P.N. Physiologically based absorption modeling to explore the impact of food and gastric pH changes on the pharmacokinetics of alectinib. AAPS J. 18, 1464–1474 (2016). [DOI] [PubMed] [Google Scholar]
- 28. NDA/BLA Multi‐disciplinary Review and Evaluation NDA 213217 BRUKINSA™ (zanubrutinib) <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213217Orig1s000MultidisciplineR.pdf>. Accessed October 6, 2020.
- 29. Holford, N.H. , Ambros, R.J. & Stoeckel, K. Models for describing absorption rate and estimating extent of bioavailability: application to cefetamet pivoxil. J. Pharmacokinet. Biopharm. 20, 421–442 (1992). [DOI] [PubMed] [Google Scholar]
- 30. Bonate, P.L. Nonlinear mixed effects models: theory. In Pharmacokinetic‐Pharmacodynamic Modeling and Simulation 2nd edn., 238–245 (Springer, New York, 2011). [Google Scholar]
- 31. Karlsson, M.O. , Beal, S.L. & Sheiner, L.B. Three new residual error models for population PK/PD analyses. J. Pharmacokinet. Biopharm. 23, 651–672 (1995). [DOI] [PubMed] [Google Scholar]
- 32. Coutant, D.E. et al. Understanding disease‐drug interactions in cancer patients: implications for dosing within the therapeutic window. Clin. Pharmacol. Ther. 98, 76–86 (2015). [DOI] [PubMed] [Google Scholar]
- 33. Schwenger, E. et al. Harnessing meta‐analysis to refine an oncology patient population for physiology‐based pharmacokinetic modeling of drugs. Clin. Pharmacol. Ther. 103, 271–280 (2018). [DOI] [PubMed] [Google Scholar]
- 34. Cusack, B.J. Pharmacokinetics in older persons. Am. J. Geriatr. Pharmacother. 2, 274–302 (2004). [DOI] [PubMed] [Google Scholar]
- 35. Klotz, U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab. Rev. 41, 67–76 (2009). [DOI] [PubMed] [Google Scholar]
- 36. Mu, S. et al. Effect of rifampin and itraconazole on the pharmacokinetics of zanubrutinib (a Bruton's tyrosine kinase inhibitor) in Asian and non‐Asian healthy subjects. Cancer Chemother. Pharmacol. 85, 391–399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material