

Abstract
Selective inhibition of tyrosine kinase 2 (TYK2) may offer therapeutic promise in inflammatory conditions, with its role in downstream pro‐inflammatory cytokine signaling. In this first‐in‐human study, we evaluated the safety, tolerability, and pharmacokinetics (PK) of a novel TYK2 inhibitor, PF‐06826647, in healthy participants. This phase I, randomized, double‐blind, placebo‐controlled, parallel‐group study included two treatment periods (single ascending dose (SAD) and multiple ascending dose (MAD)) in healthy participants and a cohort of healthy Japanese participants receiving 400 mg q.d. or placebo in the MAD period (NCT03210961). Participants were randomly assigned to PF‐06826647 or placebo (3:1). Participants received a single oral study drug dose of 3, 10, 30, 100, 200, 400, or 1,600 mg (SAD period), then 30, 100, 400, or 1,200 mg q.d. or 200 mg b.i.d. for 10 days (MAD period). Safety (adverse events (AEs), vital signs, and clinical laboratory parameters), tolerability, and PK were assessed. Overall, 69 participants were randomized to treatment, including six Japanese participants. No deaths, serious AEs, severe AEs, or AEs leading to dose reduction or temporary/permanent discontinuation were observed. All AEs were mild in severity. No clinically relevant laboratory abnormalities or changes in vital signs were detected. PF‐06826647 was rapidly absorbed with a median time to maximum plasma concentration of 2 hours in a fasted state, with modest accumulation (< 1.5‐fold) after multiple dosing and low urinary recovery. PF‐06826647 was well‐tolerated, with an acceptable safety profile for doses up to 1,200 mg q.d. for 10 days, supporting further testing in patients.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Tyrosine kinase 2 (TYK2) inhibitors offer therapeutic promise for the many patients with inflammatory conditions in which IL‐12/23 signaling is implicated, and who have an inadequate response to existing systemic treatment options.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ PF‐06826647 is an oral TYK2 inhibitor with potency against TYK2‐dependent signaling. We aimed to assess the safety, tolerability, and pharmacokinetics of PF‐06826647 in healthy participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ PF‐06826647 was well‐tolerated, with an acceptable safety profile at a single dose of up to 1,600 mg, or multiple doses up to 1,200 mg daily.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ PF‐06826647 may offer a future oral treatment option for patients with inflammatory and autoimmune conditions in which IL‐12/23 signaling is implicated.
Tyrosine kinase 2 (TYK2), a member of the Janus kinase (JAK) family, is essential for IL‐12/T‐helper cell 1 and IL‐23/T‐helper cell 17 signaling 1 , 2 and IFN type I/II receptor functioning, 2 , 3 and both preclinical and clinical studies have implicated these pathways in the pathogenesis of autoimmune disorders, including psoriasis, inflammatory bowel disease, and systemic lupus erythematosous. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 In large, human, genome‐wide association studies, single nucleotide polymorphisms of the TYK2 gene have been shown to confer protection against psoriasis, ankylosing spondylitis, Crohn’s disease, multiple sclerosis, and ulcerative colitis. 5
Blocking signaling from the pro‐inflammatory cytokines, as well as their downstream pathways, with the TYK2 JH2 domain inhibitor BMS‐986165, 10 the TYK2/JAK1 inhibitor brepocitinib, 11 or with the monoclonal antibodies ustekinumab (anti‐IL‐12 and IL‐23), 12 , 13 , 14 risankizumab (anti‐IL‐23), 14 , 15 secukinumab (anti‐IL‐17A), 16 mirikizumab (anti‐IL‐23), 17 or guselkumab (anti‐IL‐23), 18 , 19 has shown efficacy in the treatment of various autoimmune conditions.
PF‐06826647 is an oral TYK2 inhibitor with potency against TYK2‐dependent signaling (IFNα/IL‐12/IL‐23), but may have dose‐dependent inhibitory activity against other, TYK2‐independent pathways (IFNγ/erythropoeitin). PF‐06826647 is a compound with a low pKa (< 1.7), low solubility across physiological pH (~ 0.3 μg/mL at pH 6.5), and high cellular permeability (~ 17 × 10‐6 cm/s). However, based on preclinical exposure data in rats, using a spray‐dried dispersion formulation, it was expected to be moderately‐to‐well absorbed at the predicted clinically effective dose range in the clinic. In addition, preclinical studies demonstrated limited drug clearance via renal and biliary excretion in rats, and the major human clearance pathway for PF‐06826647 was identified to be cytochrome P450 (CYP)‐mediated (via CYP1A2, CYP2D6, and CYP3A) metabolism. 20 PF‐06826647 has shown minimal inhibition of transporter proteins (i.e., MATE1, MRP1, MRP2, MRP3, sodium/Na+ taurocholate co‐transporting polypeptide, OATP1B1, OATP1B3, and OCT2), with the exception of MATE2 inhibition. 20
In a phase I, first‐in‐human study (NCT03210961), we evaluated the safety, tolerability, and pharmacokinetics (PK) of PF‐06826647 in healthy participants. In this report, we present safety, tolerability, and PK data for escalating single and multiple doses of PF‐06826647. We also present the impact of food on PK parameters, and a comparison of PK parameters in plasma and urine between Western participants and a Japanese cohort during the multiple ascending dose (MAD) period at an expected clinically relevant dose of 400 mg q.d. Doses for the single ascending dose (SAD) and MAD study periods were initially selected based on data from in vitro pharmacologic/toxicologic studies. 20 During the dose escalation, the available safety data (adverse events (AEs), vital signs, electrocardiogram (ECG), clinical laboratory, hematology, and urinalysis) from the ongoing cohort were reviewed and the appropriate dose for the next cohort was selected to provide the projected average exposure (based on available PK data from all doses) being ~ ≤ 3‐fold of the exposure and less or equal to the PK stopping limit.
METHODS
Study design
This was a phase I, within‐cohort, randomized, double‐blind, placebo‐controlled, parallel‐group study with two treatment periods, comprising SAD and MAD periods, in healthy adults (Figure 1 ). The study was conducted between July 14, 2017 and January 25, 2019 at a single clinical research site located in Anaheim, CA, USA. Following a 28‐day screening period, all participants enrolled in the SAD period were admitted to the Clinical Research Unit (CRU) at day –1 until day 4, and subsequently returned to the CRU on day 8 for the final SAD period assessment. Numbers were assigned to each participant sequentially during screening by the investigator, then a randomization schedule provided by Pfizer was used to allocate each participant to corresponding medication containers. In the SAD period, participants were randomized to receive a single oral dose of PF‐06826647 (3, 10, 30, 100, 200, 400, or 1,600 mg) or placebo (3:1) in a fasted state on day 1. The SAD period was followed by a minimum washout period of 7 days, after which participants initiated PF‐06826647 (30, 100, 400, or 1,200 mg q.d.) or placebo q.d. without randomization for 10 days during the MAD period, in a fed state (with standard meal; total daily nutritional composition of 50% carbohydrate, 35% fat, and 15% protein, not to exceed 3,200 kcal). A cohort of Japanese participants was included in the MAD period and randomized to receive PF‐06826647 400 mg q.d. or placebo. Additionally, a separate cohort of participants was randomized to receive PF‐06826647 200 mg or placebo b.i.d. Those in the Japanese and b.i.d. cohorts were dosed in a fed state (with standard meal) and did not participate in the SAD period. As before, participants were admitted to the CRU for the MAD period and discharged on day 14, with an additional end‐of‐study visit on day 28 (± 3 days). Participants and investigators were blinded to study treatment.
Figure 1.
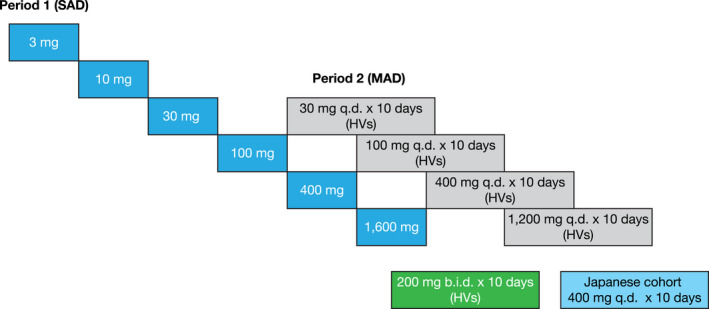
Planned dose‐escalation scheme and dose allocation. HVs, healthy volunteers; MAD, multiple ascending dose; SAD, single ascending dose.
Participants
Eligible participants were women of nonchildbearing potential and men, 18–55 years of age, with a body mass index of 17.5–30.5 kg/m2, weighing > 50 kg, and without active, latent, or inadequately treated Mycobacterium tuberculosis infection (confirmed by a negative IFNγ release assay with the following assays (performed during screening or within 3 months of day 1): QuantiFERON‐TB Gold, QuantiFERON‐TB Gold In‐Tube test (both QIAGEN, Hilden, Germany), T‐SPOT TB (Oxford Immunotec, Abingdon, UK)). Those in the cohort of Japanese participants included in the MAD period were required to have all four biological grandparents born in Japan.
Key exclusion criteria, designed to ensure that only healthy participants were enrolled, included history of: sensitivity to heparin, or heparin‐induced thrombocytopenia; any lymphoproliferative disorder or lymphatic disease; significant infection at screening or ≤ 6 months prior to first dose of study drug, or history of chronic or recurrent infectious disease; symptomatic herpes zoster or herpes simplex within 12 weeks, > 1 episode of local herpes zoster or single episode of disseminated zoster; evidence or history of clinically significant hematological, renal, endocrine, pulmonary, gastrointestinal, cardiovascular, hepatic, psychiatric, neurologic, or allergic disease (including drug allergies, but excluding untreated, asymptomatic, seasonal allergies at time of dosing); any condition possibly affecting drug absorption (e.g., gastrectomy); known infection with human immunodeficiency virus or hepatitis B or C viruses; history of cancer, with the exception of adequately treated basal or squamous cell carcinoma of the skin; other severe acute or chronic medical or psychiatric condition; any clinically significant blood pressure, laboratory, and/or ECG abnormalities at screening; a positive urine drug screen; and significant alcohol or tobacco use.
The study protocol and consent forms were approved by the relevant Institutional Review Board. The study was conducted according to the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Subjects, the International Council for Harmonization Guideline for Good Clinical Practice, and the Declaration of Helsinki. All participants provided written informed consent.
Treatment
Participants were randomized to oral PF‐06826647 (or matching placebo) provided as tablets or as an extemporaneously prepared suspension (3 mg dosing only; Pfizer). During the SAD period, participants received the study drug after an overnight fast of at least 8 hours. During the MAD period, the study drug was administered daily under fed conditions (standard diet within 25 minutes of dose). For those participants receiving b.i.d. dosing, the second dose was administered 12 hours after the first dose. Participants in the PF‐06826647 30, 100, and 400 mg groups received the same blinded treatment assignment for both treatment periods. However, participants randomized to PF‐06826647 1,600 mg during the SAD period received 1,200 mg q.d. in the following MAD period, and participants receiving PF‐06826647 3 or 10 mg during the SAD period did not continue into the MAD period. For the next dose to be initiated in the SAD period, a minimum of 3 days of safety data and 24 hours of PK data were required in at least 6 participants from the previous dose level. MAD dosing commenced at the 30 mg q.d. dose level if adequate single‐dose safety was demonstrated for the next dose level (e.g., 100 mg). Detailed stopping criteria are described in Supplementary Material S1 .
Study end points
Primary end points: Safety
Primary end points included the following safety assessments: number of treatment‐emergent AEs (TEAEs), serious AEs, and AEs leading to treatment discontinuation (monitored for at least 28 days following the last administration of treatment); vital signs; physical examination findings over time; 12‐lead ECG parameters; clinical laboratory parameters; and, during the MAD period, change in 24‐hour creatinine clearance from day –1 to day 10 (serum creatinine and cystatin C were used to assess renal function).
Secondary and exploratory end points
Secondary end points included systemic PK parameters assessed at predose, 0.5, 1, 2, 4, 6, 8, 12, 16, 24, 36, 48, and 72 hours postdose during the SAD period and at 1, 2, 4, 6, 8, 10, 11–14 days postdose during the MAD period. Additional samples for systemic PK parameters were collected on day 10 at predose, 0.5, 1, 2, 4, 6, 8, 12, and 16 hours following PF‐06826647 dosing. Urinary PK parameters were assessed during the MAD period at day − 1 (0–24 hours) and day 10 (0–12 hours and 12–24 hours). The lower limit of quantification for PF‐06826647 in both plasma and urine was 1 ng/mL.
PF‐06826647 area under the concentration–time curve from time zero to 24 hours after single dose (AUC24) and maximum plasma concentration (Cmax) following day 1 dosing in fasted (SAD period) or fed (MAD period) states were assessed to quantify the impact of food on the PK of PF‐06826647.
Statistical analysis
Eight healthy participants (including six participants receiving study drug and two receiving placebo) were planned to be enrolled for each dose in the SAD and MAD periods (total sample size of ~ 48 participants, based on the clinical and pharmacological considerations to provide safety and tolerability information and minimize exposure). This was based on the clinical need to provide safety and PK information, while minimizing exposure to participants at each dose level. An additional nine healthy participants from the b.i.d. cohort were enrolled and evaluated as a separate cohort, but were not included in the SAD or MAD periods. The study also included a subset of 6 Japanese participants receiving 400 mg q.d. or placebo during the MAD period.
Safety, PK data, PF‐06826647 concentration in urine, and renal clearance (CLr) values were summarized descriptively. PK parameters were calculated using noncompartmental analysis software (eNCA, version 2.2.4; Pfizer, Groton, CT, USA). PK parameters were summarized by dose/regimen, with geometric mean and coefficient of variation summary statistics applied for all parameters except for time at which Cmax occurred (Tmax). AUC and Cmax values of PF‐06826647 (normalized to 1 mg) were plotted against dose (logarithmic scale).
RESULTS
Participants
Overall, 69 healthy participants were randomized to treatment (Figure 2 ). One participant receiving PF‐06826647 100 mg discontinued treatment due to urticaria during the SAD period, whereas 3 participants discontinued due to family emergencies during the MAD period (one participant each in the placebo b.i.d., PF‐06826647 30 mg q.d., and PF‐06826647 200 mg b.i.d. groups).
Figure 2.
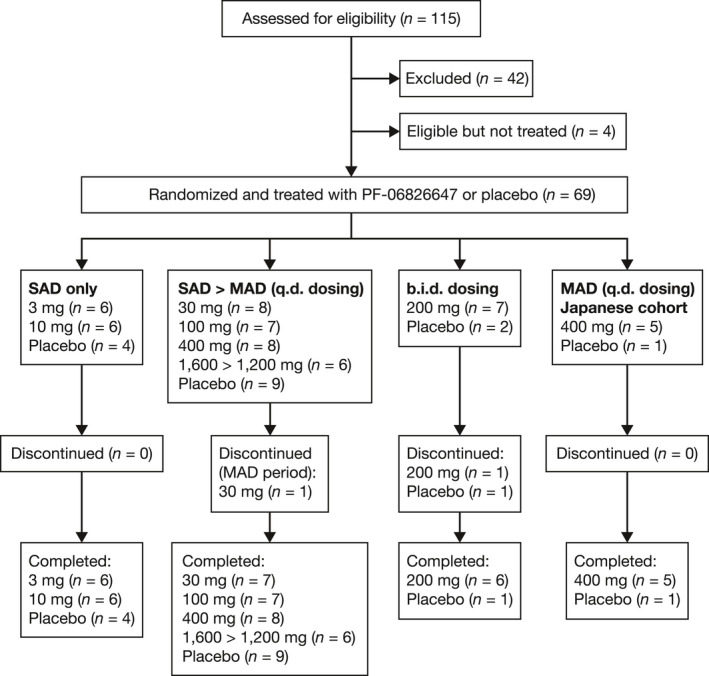
Participant disposition. MAD, multiple ascending dose; n, number of participants; SAD, single ascending dose.
Participant demographics at baseline are summarized in Table 1 . The majority of participants were white, male, and 22–54 years of age (mean age range 31.2–44.3 years). Characteristics were generally similar at baseline, with mean weight ranging from 55.8 to 105.7 kg and mean body mass index ranging from 22.3 to 29.2 kg/m2 between treatment groups.
Table 1.
Participant demographics and baseline characteristics (SAD and MAD periods)
| Treatment period | SAD (q.d. dosing) | SAD > MAD (q.d. dosing) | b.i.d. dosing | MAD (q.d. dosing) Japanese cohort | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 mg (n = 6) | 10 mg (n = 6) | Placebo (n = 4) | 30 mg (n = 8) | 100 mg (n = 7) | 400 mg (n = 8) | 1,600 > 1,200 mg (n = 6) | Placebo (n = 9) | 200 mg (n = 7) | Placebo (n = 2) | 400 mg (n = 5) | Placebo (n = 1) | |
| Age, years | ||||||||||||
| Mean (SD) | 44.3 (11.5) | 40.2 (10.3) | 38.5 (12.2) | 40.5 (11.6) | 36.9 (7.4) | 37.5 (8.2) | 31.2 (5.0) | 42.2 (10.8) | 37.4 (9.3) | 32.5 (5.0) | 39.8 (5.4) | 34.0 (NC) |
| Range | 24–54 | 30–53 | 22–50 | 23–53 | 28–45 | 30–52 | 27–40 | 26–53 | 24–50 | 29–36 | 35–49 | 34–34 |
| 18–44, n (%) | 2 (33.3) | 4 (66.7) | 2 (50.0) | 4 (50.0) | 6 (85.7) | 6 (75.0) | 6 (100.0) | 4 (44.4) | 6 (85.7) | 2 (100.0) | 4 (80.0) | 1 (100.0) |
| 45–64, n (%) | 4 (66.7) | 2 (33.3) | 2 (50.0) | 4 (50.0) | 1 (14.3) | 2 (25.0) | 0 (0) | 5 (55.6) | 1 (14.3) | 0 (0) | 1 (20.0) | 0 (0) |
| ≥ 65, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Sex, n (%) | ||||||||||||
| Male | 6 (100.0) | 5 (83.3) | 4 (100.0) | 8 (100.0) | 7 (100.0) | 7 (87.5) | 6 (100.0) | 8 (88.9) | 7 (100.0) | 2 (100.0) | 5 (100.0) | 1 (100.0) |
| Race, n (%) | ||||||||||||
| White | 3 (50.0) | 1 (16.7) | 3 (75.0) | 6 (75.0) | 3 (42.9) | 6 (75.0) | 5 (83.3) | 6 (66.7) | 4 (57.1) | 1 (50.0) | 0 (0) | 0 (0) |
| Black or African American | 2 (33.3) | 4 (66.7) | 1 (25.0) | 0 (0) | 1 (14.3) | 0 (0) | 0 (0) | 3 (33.3) | 3 (42.9) | 0 (0) | 0 (0) | 0 (0) |
| Asian | 1 (16.7) | 1 (16.7) | 0 (0) | 1 (12.5) | 2 (28.6) | 1 (12.5) | 0 (0) | 0 (0) | 0 (0) | 1 (50.0) | 5 (100.0) | 1 (100.0) |
| Multiracial | 0 (0) | 0 (0) | 0 (0) | 1 (12.5) | 1 (14.3) | 1 (12.5) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Mean height, cm (SD) | 180.60 (7.77) | 169.53 (10.07) | 166.63 (3.84) | 171.21 (5.63) | 174.49 (6.15) | 169.35 (6.41) | 177.85 (4.07) | 173.23 (8.61) | 179.04 (7.47) | 174.15 (6.86) | 172.52 (5.49) | 166.0 |
| Mean weight, kg (SD) | 86.35 (12.82) | 82.10 (12.25) | 74.18 (5.75) | 79.05 (8.41) | 77.61 (11. 07) | 78.40 (10.97) | 82.43 (13.53) | 83.16 (11.47) | 81.66 (6.75) | 88.55 (3.47) | 72.06 (5.95) | 61.50 |
| Mean BMI, kg/m2 (SD) | 26.44 (3.17) | 28.43 (1.29) | 26.77 (2.68) | 26.94 (2.28) | 25.42 (2.81) | 27.25 (2.93) | 25.98 (3.51) | 27.59 (1.85) | 25.5 (2.02) | 29.22 (1.16) | 24.19 (1.33) | 22.32 |
BMI, body mass index; MAD, multiple ascending dose; n, number of participants in dose group; NC, not calculated; SAD, single ascending dose.
Safety
All‐causality and treatment‐related TEAEs are summarized in Table 2 . No deaths, serious AEs, severe AEs, and AEs leading to permanent discontinuation or dose reduction/temporary discontinuation were recorded during either treatment period in any group. During the SAD period, TEAEs occurred postdose in 1 participant receiving PF‐06826647 100 mg (nausea, face edema, edema, headache, pruritis, rash, urticaria mostly on the trunk of the participant (all treatment‐related), periorbital hematoma, and periorbital hemorrhage (not treatment‐related)), 2 participants receiving PF‐06826647 1,600 mg (one participant with peripheral swelling, not treatment‐related, and 1 participant with increased alanine aminotransferase and aspartate aminotransferase, which were attributed to clinically substantial alcohol use), and 1 participant receiving placebo (rash, not treatment‐related) during the SAD period (Table 2 ).
Table 2.
Summary of TEAEs (SAD, MAD, and b.i.d. periods)
| SAD period | ||||||||
|---|---|---|---|---|---|---|---|---|
| PF‐06826647 | Placebo (n = 13) | |||||||
| 3 mg (n = 6) | 10 mg (n = 6) | 30 mg (n = 8) | 100 mg (n = 7) | 400 mg (n = 8) | 1,600 mg (n = 6) | |||
| Participants with AEs, n (%) | 0 | 0 | 0 | 1 (14.3) | 0 | 2 (33.3) | 1 (7.7) | |
| AEs, n | 0 | 0 | 0 | 9 | 0 | 3 | 1 | |
| SAEs, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Severe AEs, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| AEs leading to permanent discontinuation of study drug, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| AEs leading to study drug dose reduction or interruption, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Participants with TEAEs by SOC and PT, n (%) | ||||||||
| Gastrointestinal disorders | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Nausea | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| General disorders and administration site conditions | 0 | 0 | 0 | 1 (14.3) | 0 | 1 (16.7) | 0 | |
| Face edema | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Edema | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Peripheral swelling | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | |
| Injury, poisoning, and procedural complications | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Periorbital hematoma | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Periorbital hemorrhage | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Investigations | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | |
| Alanine aminotransferase increased | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | |
| Aspartate aminotransferase increased | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | |
| Nervous system disorders | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Headache | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Skin and subcutaneous tissue disorders | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 1 (7.7) | |
| Pruritus | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| Rash | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 1 (7.7) | |
| Urticaria | 0 | 0 | 0 | 1 (14.3) | 0 | 0 | 0 | |
| MAD period | b.i.d. cohort | |||||||
|---|---|---|---|---|---|---|---|---|
| PF‐06826647 | Placebo (n = 9) |
PF‐06826647 200 mg b.i.d. (n = 7) |
Placebo b.i.d. (n = 2) | |||||
| 30 mg (n = 6) | 100 mg (n = 6) | 400 mg (n = 6) | 1,200 mg (n = 5) | Japanese cohort 400 mg (n = 5) | ||||
| Participants with AE, n (%) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 1 (20.0) | 1 (20.0) | 2 (22.2) | 3 (42.9) | 0 |
| AEs, n | 2 | 1 | 1 | 1 | 2 | 2 | 3 | 0 |
| SAEs, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Severe AEs, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AEs leading to permanent discontinuation of study drug, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| AEs leading to study drug dose reduction or interruption, n | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Participants with TEAEs by SOC and PT, n (%) | ||||||||
| Gastrointestinal disorders | 2 (33.3) | 0 | 0 | 0 | 0 | 0 | 0 | |
| Constipation | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nausea | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Investigations | 0 | 1 (16.7) | 1 (16.7) | 1 (20.0) | 1 (20.0) | 1 (11.1) | 3 (42.9) | 0 |
| Alanine aminotransferase increased | 0 | 0 | 0 | 0 | 1 (20.0) | 0 | 0 | 0 |
| Blood creatinine increased | 0 | 1 (16.7) | 1 (16.7) | 1 (20.0) | 0 | 1 (11.1) | 3 (42.9) | 0 |
| Nervous system disorders | 0 | 0 | 0 | 0 | 1 (20.0) | 0 | 0 | 0 |
| Headache | 0 | 0 | 0 | 0 | 1 (20.0) | 0 | 0 | 0 |
| Renal and urinary disorders | 0 | 0 | 0 | 0 | 0 | 1 (11.1) | 0 | 0 |
| Dysuria | 0 | 0 | 0 | 0 | 0 | 1 (11.1) | 0 | 0 |
AE, adverse event; MAD, multiple ascending dose; n, number of participants evaluable for adverse events; PT, preferred term; SAD, single ascending dose; SAE, serious adverse event; SOC, system organ class; TEAE, treatment‐emergent adverse event.
During the MAD period, 11 participants experienced 12 TEAEs, 8 of which were treatment‐related: 2 participants receiving placebo (blood creatinine increased (treatment‐related) and dysuria (not treatment‐related), 2 participants receiving PF‐06826647 30 mg q.d. (constipation (treatment‐related) and nausea (not treatment‐related)), 1 participant each receiving PF‐06826647 100 mg, 400 mg, 1,200 mg and placebo q.d. (all blood creatinine increased, all treatment‐related), 3 participants receiving PF‐06826647 200 mg b.i.d. (blood creatinine increased, 2 treatment‐related), and 1 Japanese participant receiving PF‐06826647 400 mg q.d. (alanine aminotransferase increased (treatment‐related) and headache (not treatment‐related)). All TEAEs were mild in intensity (Table 2 ). No events of acne or aphthous ulcers were reported in either treatment period.
Clinical laboratory parameters
Abnormalities in clinical laboratory parameters were recorded in 43 participants; however, none were deemed to be clinically meaningful. Although there appeared to be a trend for an increase in platelet numbers in participants receiving PF‐06826647, compared with placebo, this was not considered to be clinically meaningful by the investigator, and platelet counts remained within the normal range (Figure S1 ). No clinically meaningful changes in lymphocyte (Figure S2 ) or neutrophil (Figure S3 ) counts were detected during the assessment period. A slight dose‐dependent (and exposure‐dependent) decrease in reticulocytes (mean percent change from baseline: –0.06 (90% confidence interval (CI), –0.99 to –0.21)) was detected with dosing at 1,200 mg q.d., but due to the relatively short dosing duration (10 days), the limit of decline was not observed, nor any meaningful effect on hemoglobin (Figure S4 ).
No clinically meaningful changes were observed during the study for serum cystatin C, serum creatinine‐based estimated glomerular filtration rate (Figure S5 ), or serum cystatin C‐based estimated glomerular filtration rate (Figure S6 ). Serum creatinine levels were increased from baseline by up to 0.3 mg/dL in some participants following treatment with PF‐06826647 at isolated timepoints. However, these increases were deemed by the investigator to be due to a variety of reasons, including dehydration, and levels generally returned to baseline at the subsequent visit in each case. Changes in lipid parameters (low‐density lipoproteins, high‐density lipoproteins, and total cholesterol) for the SAD and MAD treatment groups were small, and similar to those observed in the placebo group (Figure S7 ). Finally, no clinically meaningful alteration in the 6‐beta‐hydroxycortisol/cortisol ratio was found in participants receiving PF‐06826647 (Figure S8 ).
Other safety parameters
No clinically significant findings were reported following assessment of vital signs, ECG parameters, or physical examination in participants receiving PF‐06826647, and no relationship was detected between change from baseline in Fredricia‐corrected QT interval and the dose of PF‐06826647.
Pharmacokinetics
The median plasma PF‐06826647 concentration–time profile following single oral doses (fasted state) and multiple doses (fed state) are shown in Figure 3a,b . PK parameters for the SAD period are summarized descriptively in Table 3 and Table S1 . Overall, PF‐06826647 was absorbed rapidly, with a median Tmax of 2 hours in the fasted state (Figure 3a , Table 3 ). The mean terminal half‐lives (t1/2) for the 3 and 10 mg groups were 3.6 and 6.0 hours, respectively. For doses of 30 mg and above, the mean t1/2 ranged from 16.3 to 38.8 hours.
Figure 3.
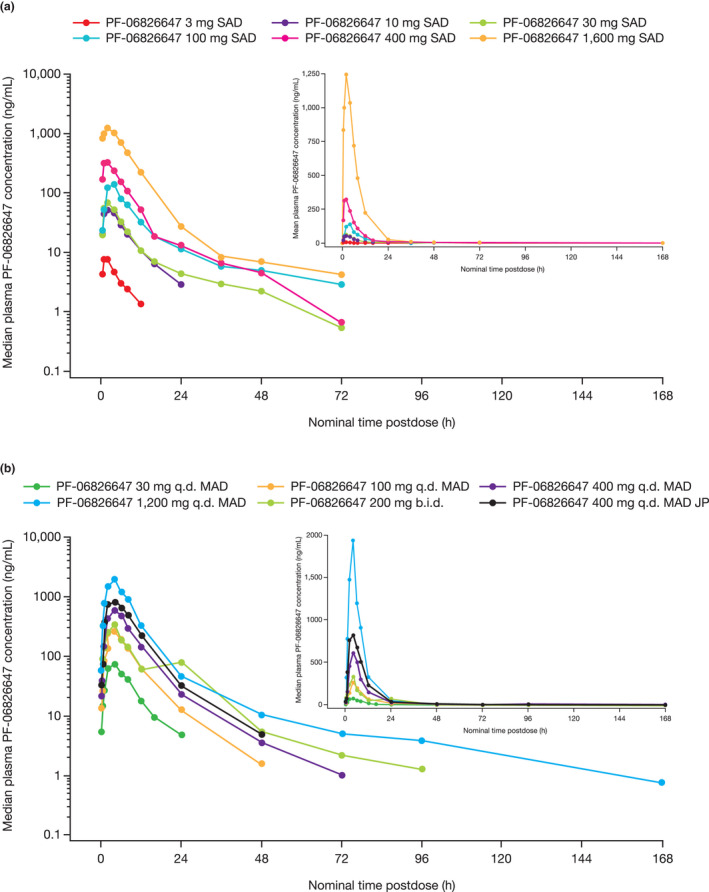
Plasma concentration–time profiles. (a) During the SAD period at day 1. (b) During the MAD period at day 10. PF‐06826647 was administered with standard meal in MAD. JP, Japanese cohort; MAD, multiple ascending dose; SAD, single ascending dose.
Table 3.
Plasma PK parameters (SAD period)
| Parameter | PF‐06826647 | |||||
|---|---|---|---|---|---|---|
| 3 mg (n = 6) | 10 mg (n = 6) | 30 mg (n = 8) | 100 mg (n = 7) | 400 mg (n = 8) | 1,600 mg (n = 6) | |
| Evaluable participants, n2 | 5 | 6 | 6 | 3 | 7 | 4 |
| AUCinf, ng.hour/mL | 47.6 (59) | 369.3 (67) | 848.4 (40) | 1,582.0 (42) | 3,056.0 (65) | 11,790.0 (25) |
| AUCinf (dn), ng.hour/mL/mg | 15.9 (59) | 36.9 (67) | 28.3 (41) | 15.8 (42) | 7.6 (65) | 7.4 (25) |
| Cmax, ng/mL | 7.8 (38) | 47.1 (35) | 77.9 (34) | 166.8 (55) | 404.8 (62) | 1,218.0 (25) |
| Cmax (dn), ng/mL/mg | 2.6 (38) | 4.7 (35) | 2.6 (34) | 1.7 (55) | 1.0 (62) | 0.8 (25) |
| Tmax, hour | 2.0 (1.0–2.0) | 2.0 (1.0–4.0) | 2.0 (1.0–4.0) | 2.0 (2.0–6.0) | 2.0 (0.5–4.0) | 2.0 (0.5–4.0) |
| t1/2, hour | 3.6 (1.3) | 6.0 (1.9) | 19.7 (9.5) | 38.8 (36.0) | 16.3 (9.3) | 22.2 (24.6) |
All values represent geometric mean (coefficient of variation, %) except for Tmax, which is shown as median (range), and t1/2, which is shown as arithmetic mean (± SD).
AUCinf, area under the concentration–time profile from time zero extrapolated to infinite time;Cmax, maximum plasma concentration; dn, dose normalized; n, number of participants in dose group; n2, number of participants contributing to the summary statistics; PK, pharmacokinetics; SAD, single ascending dose; Tmax, time at which Cmax occurred; t1/2, terminal half‐life.
The AUC from time zero extrapolated to infinite time (AUCinf) and Cmax increased in a greater than dose‐proportional manner at PF‐06826647 doses of 3–10 mg, and in a dose‐proportional manner at doses of 400–1,600 mg; however, increases in AUCinf and Cmax were less than dose‐proportional at doses of 10–400 mg (Table 3 ). Variability in exposure based on the geometric coefficient of variation ranged from 25% to 67% for AUCinf and from 25% to 62% for Cmax (Table 3 ).
Median plasma PF‐06826647 concentration–time profiles on day 10 of the MAD period are shown in Figure 3b . PK parameters for the MAD period are summarized descriptively in Table 4 and Table S2 . After dosing with a standard meal, the median Tmax of PF‐06826647 at day 10 was 4 hours with all doses (30–1,200 mg) and the mean t1/2 ranged from 7.4 to 12.7 hours in the PF‐06826647 30–400 mg q.d. dose groups, with no dose‐dependent trend. The mean t1/2 was substantially longer in the PF‐06826647 200 mg b.i.d. and 1,200 mg q.d. dose groups at 26.9 and 34.3 hours, respectively. AUC from time zero to time tau (AUCτ) and Cmax values increased in a dose‐proportional manner for PF‐06826647 400–1,200 mg q.d. dosing, and in a less‐than‐dose‐proportional manner for 30–400 mg q.d. dosing. Variability in exposure ranged from 20% to 70% for AUCτ and from 10% to 67% for Cmax on days 1 and 10, respectively (Table 4 ).
Table 4.
Plasma and urine PK parameters for days 1 and 10 (MAD period)
| Parameter | PF‐06826647 | |||||
|---|---|---|---|---|---|---|
| 30 mg q.d. (n = 6) | 100 mg q.d. (n = 6) | 200 mg b.i.d. (n = 7) | 400 mg q.d. (n = 6) | 1,200 mg q.d. (n = 5) | Japanese cohort 400 mg q.d. (n = 5) | |
| Day 1 | ||||||
| Evaluable participants, n2 | 6 | 6 | 7 | 6 | 5 | 5 |
| AUCτ, ng.hour/mL | 662.8 (25) | 1,803.0 (23) | 1,646.0 (70) | 4,424.0 (67) | 17,090.0 (20) | 6,038.0 (29) |
| AUCτ (dn), ng.hour/mL/mg | 22.1 (25) | 18.0 (23) | 8.2 (70) | 11.1 (67) | 14.2 (20) | 15.1 (29) |
| Cmax, ng/mL | 83.5 (24) | 267.2 (20) | 289.8 (67) | 585.9 (33) | 2,023.0 (10) | 695.5 (19) |
| Cmax (dn), ng/mL/mg | 2.8 (24) | 2.7 (20) | 1.5 (67) | 1.5 (33) | 1.7 (10) | 1.7 (19) |
| Tmax, hour | 3.0 (2.0–6.0) | 4.0 (1.0–4.0) | 4.0 (2.0–6.0) | 3.0 (2.0–6.0) | 2.0 (2.0–4.0) | 4.0 (2.0–4.0) |
| Day 10 | ||||||
| Evaluable participants, n2 | 5 | 6a | 6b | 6c | 5 | 5c |
| AUCτ, ng.hour/mL | 752.9 (32) | 1,952.0 (33) | 2,010.0 (49) | 5,420.0 (57) | 14,890.0 (31) | 7,194.0 (20) |
| AUCτ (dn), ng.hour/mL/mg | 25.1 (32) | 19.5 (33) | 10.0 (49) | 13.6 (57) | 12.4 (31) | 18.0 (19) |
| Cmax, ng/mL | 85.3 (27) | 263.2 (30) | 339.5 (46) | 667.3 (37) | 1,855.0 (21) | 887.0 (11) |
| Cmax (dn), ng/mL/mg | 2.8 (27) | 2.6 (30) | 1.7 (46) | 1.7 (37) | 1.5 (21) | 2.2 (11) |
| Tmax, hour | 4.0 (4.0–6.0) | 4.0 (2.0–4.0) | 4.0 (2.0–4.0) | 4.0 (2.0–6.0) | 4.0 (2.0–4.0) | 4.0 (2.0–6.0) |
| t1/2, hour | 8.8 (5.8) | 12.7 (12.7) | 26.9 (8.1) | 7.5 (2.6) | 34.3 (21.9) | 7.4 (4.8) |
All values represent geometric mean (coefficient of variation, %) except for Tmax, which is shown as median (range), and t1/2, which is shown as arithmetic mean (± SD). Number of participants evaluated for t1/2 = a5, b3, and c4.
AUCτ, area under the concentration–time profile from time zero to time tau (τ), the dosing interval, where tau = 24 hours for q.d. dosing and 12 hours for b.i.d. dosing; Cmax, maximum plasma concentration; dn, dose normalized; MAD, multiple ascending dose; n, number of participants in dose group; n2, number of participants contributing to the summary statistics; PK, pharmacokinetics; Tmax, time at which Cmax occurred; t1/2, terminal half‐life.
Plasma accumulation of PF‐06826647 was < 1.5‐fold, with steady‐state reached by day 2 with both q.d. and b.i.d. dosing. Urinary recovery of PF‐06826647 was low, with < 1.5% of the dose recovered unchanged in urine at day 10 across all doses; CLr ranged from 0.3 to 1.3 L/hour.
During the MAD period, PF‐06826647 PK parameters in plasma and urine were similar between the Japanese cohort receiving 400 mg q.d. and the treatment group that had received 400 mg q.d. during the MAD period, with mean AUCτ and Cmax values on day 10 of 7,194 ng.hour/mL and 887.0 ng/mL, respectively, in the Japanese cohort, compared with 5,420 ng.hour/mL and 667.3 ng/mL in the 400 mg q.d. dose group. Although exposure appeared higher in the Japanese cohort, both the AUCτ and Cmax ranges in Japanese participants were within the ranges observed for the 400 mg q.d. dose group during the MAD period. The t1/2 in Japanese participants was 7.4 hours, compared with 7.5 hours in the 400 mg q.d. MAD treatment group. Likewise, CLr values were similar at 0.4 L/hour for Japanese participants, compared with 0.6 L/hour in the 400 mg q.d. MAD treatment group.
Impact of food on the PK of PF‐06826647
The initial PK assessment (day 1) of PF‐06826647 dosing in the MAD period was done under fed conditions (standard meal), allowing for comparisons with PK assessments in the SAD period done under fasted conditions to demonstrate the effect of food on exposure to a given dose. Administration of PF‐06826647 with food increased the Tmax from a median of 2.0 hours (Table 3 ) to a median of 3.0–4.0 hours at doses of 30–400 mg (Table 4 ). Furthermore, AUC24 and Cmax values were slightly higher under fed vs. fasted conditions. Administration with a meal increased the AUC24 values following PF‐06826647 30 mg, 100 mg, and 400 mg doses by 22%, 56%, and 76% (adjusted geometric mean for fed vs. fasted (90% CI): 122.0 (78.7–189.2), 155.7 (99.1–244.6), and 175.6 (113.3–272.3)), respectively. The corresponding increases in Cmax were 7%, 60%, and 45% (adjusted geometric mean (90% CI) for fed vs. fasted: 107.2 (74.2–154.8), 160.2 (109.7–234.0), and 144.8 (100.2–209.1)), respectively.
DISCUSSION
In this first‐in‐human study of PF‐06826647, we demonstrate that PF‐06826647 single (≤ 1,600 mg), multiple (≤ 1,200 mg q.d.), and twice‐daily dosing (200 mg b.i.d.) was well‐tolerated in healthy participants, with an acceptable safety profile over the 10‐day dosing period.
As expected, based on high permeability, PF‐06826647 was found to be rapidly absorbed. However, the less‐than‐dose‐proportional increase in the exposure between 10 and 400 mg could be due to its poor water solubility. At higher doses, the increase in exposure was approximately linear, which is not yet understood. The t1/2 of PF‐06826647 ranged between 7.5 and 12.7 hours at the lower q.d. dosing regimen. The longer half‐lives observed at 1,200 mg q.d. and 200 mg b.i.d. could be due to better characterization of the terminal phase. However, the modest accumulation suggests a shorter effective half‐life for PF‐06826647.
PF‐06826647 revealed no clinically significant change in the 6‐beta‐hydroxycortisol/cortisol ratio, which serves as a biomarker for CYP3A4, suggesting that the study drug is not likely to be a potent inducer of CYP3A4. Additionally, food increased the absorption of PF‐06826647 in a dose‐dependent manner, with higher doses conferring increased effect (Tables 3 and 4 ), consistent with the characteristically low solubility of PF‐06826647.
All AEs reported in the study were mild in intensity, with no discontinuations or deaths and no evidence of increasing frequency of AEs with increasing dose. Gastrointestinal AEs occurred in only three participants (4%), including two participants with nausea (one in the PF‐06826647 100 mg dose group during the SAD period, and one in the PF‐06826647 30 mg q.d. dose group during the MAD period) and one participant with constipation (receiving PF‐06826647 30 mg q.d. during the MAD period). Several cases of dose‐dependent, mild‐to‐moderate acne, and cases of aphthous ulcer have been reported in patients with psoriasis receiving up to 12 mg total daily dose of the TYK2 inhibitor BMS‐986165 10 ; however, no AEs of acne or aphthous ulcer were reported during this study. This might suggest that such AEs are not specifically correlated with targeting TYK2 signaling, but further study with longer PF‐06826647 exposures would be required to confirm this.
In this study, serum creatinine was used to assess renal function, with the expectation that dose‐dependent elevations in serum creatinine may occur, as they are a known effect of JAK inhibitors. 21 Such elevations are attributable to blockade of renal transporters (e.g., OCT2 and MATE2). 22 , 23 PF‐06826647 is an inhibitor of the MATE2 renal transporter, so in addition to serum creatinine, cystatin C was assessed as a nonbiased biomarker to monitor effects on the renal system. Slight increases in serum creatinine were reported in this study following PF‐06826647 dosing, although creatinine levels returned to baseline at the subsequent dosing visit, and were not dose‐dependent. It may be possible that MATE2K may not be involved in the excretion of creatinine, or another renal transport protein, such as MATE1, could be compensating for the loss of function of MATE2. There is some evidence that inhibition of JAK‐signal transducer and activator of transcription signaling leads to muscle regeneration with increasing age, which could affect serum creatinine levels. 24 , 25 Elevations in serum creatinine are associated with JAK inhibitors in general, 21 and the dosing duration of this study may have been too short to observe longer lasting effects of PF‐06826647 dosing on serum creatinine.
A modest increase in platelet counts was detected following PF‐06826647 dosing, compared with placebo; this was most evident during the SAD period. There was a 28% increase in median platelet count from baseline in patients who received 1,600 mg vs. a 6% decrease in those who received placebo. During the MAD period, the increase in platelet counts was transient, and a return to baseline was observed between days 14 and 28. Inhibition of JAK2 signaling during treatment with baricitinib has also been shown to transiently increase platelet counts, with levels peaking 2 weeks post‐treatment and decreasing toward baseline thereafter. 26 Compared with the general population, patients with autoimmune disease are at an increased risk of thrombosis, 27 , 28 , 29 and an extensive study on the cardiovascular safety of patients with rheumatoid arthritis treated with baricitinib (N = 3,492) found that the risk of venous thromboembolism (VTE) secondary to baricitinib treatment (incidence rate: 0.5 per 100 patient‐years) fell within the expected range of the treatment population (incidence rate: 0.3–0.8 per 100 patient‐years). 30 In this study, platelet counts remained below the upper limit of normal, and increased counts did not appear to be proportional to the extent of PF‐06826647 exposure. Associations between increased platelet counts and risk for VTE in patients receiving JAK inhibitors remains unclear, and any increase in VTE risk may be due to the inflammatory‐mediated pathophysiology of the disease. 30 However, occurrences of thrombosis in patients treated with JAK inhibitors have been reported, and those taking this type of treatment are warned of the risk of these events. 31
This study also included a cohort of Japanese participants, in order to assess the PK and safety of PF‐06826647 in this population at an expected clinically relevant dose level. PK parameters in plasma and urine were generally found to be similar between Japanese and Western populations.
This first‐in‐human study of the oral TYK2 inhibitor PF‐06826647 has several strengths. The use of an ascending dose design, with a minimum of 3 days of safety data prior to initiating the next dose level, was considered an adequate measure to ensure the safety of participants. By virtue of its design, this study was able to also determine food effect at three doses. These results should take into consideration some limitations inherent of a phase I study, including the small sample size per dose, with only a single dose tested in Japanese healthy participants, and the short duration of the multiple‐dosing phase.
The safety, tolerability, and PK data in healthy participants, in conjunction with biomarker data, 32 indicate that PF‐06826647 doses of 100 mg and 400 mg q.d., taken with food, may be appropriate for assessment in patients with psoriasis.
In conclusion, PF‐06826647 is a potent, oral TYK2 inhibitor with an acceptable safety profile and potential for q.d. dosing. These results support the further testing of PF‐06826647 in patients with inflammatory and autoimmune conditions in which IL‐22/23 signaling is implicated.
Funding
This study was sponsored by Pfizer Inc.
Conflict of Interest
R.S.P.S., V.P., E.S.R., M.S., E.K., J.D.G., E.P., M.S.V., A.B., A.F., M.E.D., and C.T. were employees of Pfizer Inc at the time the study was conducted. R.S.P.S., V.P., E.S.R., M.S., E.K., E.P., M.S.V., J.D.G., A.B., A.F., M.E.D., and C.T. hold stock in Pfizer Inc. P.W. is a full‐time employee of Anaheim Clinical Trials.
Author Contributions
R.S.P.S, V.P., E.S.R., M.S., E.K., J.D.G, E.P., M.S.V., A.B., A.F., M.E.D., P.W., and C.T. wrote the manuscript. R.S.P.S., E.S.R., M.S., J.D.G., and M.E.D. designed the research. R.S.P.S., E.S.R., M.S., J.D.G., and C.T. performed the research. R.S.P.S., E.S.R., M.S., J.D.G., M.E.D., and C.T. analyzed the data.
Supporting information
Supplementary Material
Acknowledgments
The authors would like to thank all of the investigators and participants from the contributing sites who made this study possible. The authors would also like to thank Lisa Cohen for managing the study, Kimberley C. Lee for sample analysis, and Hua Wei for noncompartmental analysis. Medical writing support, under the direction of the authors, was provided by Emma Mitchell, PhD, CMC Connect, McCann Health Medical Communications, and Laura George, PhD, on behalf of CMC Connect, and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines. 33
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (i) for indications that have been approved in the US and/or the EU, or (ii) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
References
- 1. Ishizaki, M. et al. Involvement of tyrosine kinase‐2 in both the IL‐12/Th1 and IL‐23/Th17 axes in vivo. J. Immunol. 187, 181–189 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Schwartz, D.M. , Kanno, Y. , Villarino, A. , Ward, M. , Gadina, M. & O'Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16, 843–862 (2017). [DOI] [PubMed] [Google Scholar]
- 3. Prchal‐Murphy, M. et al. TYK2 kinase activity is required for functional type I interferon responses in vivo. PLoS One 7, e39141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shaw, M.H. et al. A natural mutation in the Tyk2 pseudokinase domain underlies altered susceptibility of B10.Q/J mice to infection and autoimmunity. Proc. Natl. Acad. Sci. U S A 100, 11594–11599 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dendrou, C.A. et al. Resolving TYK2 locus genotype‐to‐phenotype differences in autoimmunity. Sci. Transl. Med. 8, 363ra149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Sullivan, L.A. , Liongue, C. , Lewis, R.S. , Stephenson, S.E. & Ward, A.C. Cytokine receptor signaling through the Jak‐Stat‐Socs pathway in disease. Mol. Immunol. 44, 2497–2506 (2007). [DOI] [PubMed] [Google Scholar]
- 7. Mease, P.J. Inhibition of interleukin‐17, interleukin‐23 and the TH17 cell pathway in the treatment of psoriatic arthritis and psoriasis. Curr. Opin. Rheumatol. 27, 127–133 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Torti, D.C. & Feldman, S.R. Interleukin‐12, interleukin‐23, and psoriasis: current prospects. J. Am. Acad. Dermatol. 57, 1059–1068 (2007). [DOI] [PubMed] [Google Scholar]
- 9. Ishizaki, M. et al. Tyk2 is a therapeutic target for psoriasis‐like skin inflammation. Int. Immunol. 26, 257–267 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Papp, K. et al. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N. Engl. J. Med. 379, 1313–1321 (2018). [DOI] [PubMed] [Google Scholar]
- 11. Banfield, C. et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of a TYK2/JAK1 inhibitor (PF‐06700841) in healthy subjects and patients with plaque psoriasis. J. Clin. Pharmacol. 58, 434–447 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Papp, K.A. et al. Risankizumab versus ustekinumab for moderate‐to‐severe plaque psoriasis. N. Engl. J. Med. 376, 1551–1560 (2017). [DOI] [PubMed] [Google Scholar]
- 13. van Vollenhoven, R.F. et al. Efficacy and safety of ustekinumab, an IL‐12 and IL‐23 inhibitor, in patients with active systemic lupus erythematosus: results of a multicentre, double‐blind, phase 2, randomised, controlled study. Lancet 392, 1330–1339 (2018). [DOI] [PubMed] [Google Scholar]
- 14. Bai, F. , Li, G.G. , Liu, Q. , Niu, X. , Li, R. & Ma, H. Short‐term efficacy and safety of IL‐17, IL‐12/23, and IL‐23 inhibitors brodalumab, secukinumab, ixekizumab, ustekinumab, guselkumab, tildrakizumab, and risankizumab for the treatment of moderate to severe plaque psoriasis: a systematic review and network meta‐analysis of randomized controlled trials. J. Immunol. Res. 2019, 2546161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. US Food and Drug Administration . SKYRIZI highlights of prescribing information <https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761105s000lbl.pdf> (2019).
- 16. Langley, R.G. et al. Secukinumab in plaque psoriasis — results of two phase 3 trials. N. Engl. J. Med. 371, 326–338 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Reich, K. et al. Efficacy and safety of mirikizumab (LY3074828) in the treatment of moderate‐to‐severe plaque psoriasis: results from a randomized phase II study. Br. J. Dermatol. 181, 88–95 (2019). [DOI] [PubMed] [Google Scholar]
- 18. Reich, K. et al. Guselkumab versus secukinumab for the treatment of moderate‐to‐severe psoriasis (ECLIPSE): results from a phase 3, randomised controlled trial. Lancet 394, 831–839 (2019). [DOI] [PubMed] [Google Scholar]
- 19. US Food and Drug Administration . TREMFYA highlights of prescribing information. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761061s000lbl.pdf> (2017).
- 20. Gerstenberger, B.S. et al. Discovery of tyrosine kinase 2 (TYK2) inhibitor (PF‐06826647) for the treatment of autoimmune diseases. J. Med. Chem. 10.1021/acs.jmedchem.0c00948. [DOI] [PubMed] [Google Scholar]
- 21. Westhovens, R. Clinical efficacy of new JAK inhibitors under development. Just more of the same? Rheumatology (Oxford) 58(suppl. 1), i27–i33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang, Y. et al. Impact on creatinine renal clearance by the interplay of multiple renal transporters: a case study with INCB039110. Drug Metab. Dispos. 43, 485–489 (2015). [DOI] [PubMed] [Google Scholar]
- 23. Feng, B. & Varma, M.V. Evaluation and quantitative prediction of renal transporter‐mediated drug‐drug interactions. J. Clin. Pharmacol. 56(suppl. 7), S110–S121 (2016). [DOI] [PubMed] [Google Scholar]
- 24. Price, F.D. et al. Inhibition of JAK‐STAT signaling stimulates adult satellite cell function. Nat. Med. 20, 1174–1181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tierney, M.T. et al. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 20, 1182–1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kremer, J. et al. FRI0090 Analysis of neutrophils, lymphocytes, and platelets in pooled phase 2 and phase 3 studies of baricitinib for rheumatoid arthritis. Ann. Rheum. Dis. 76, 512 (2017). [Google Scholar]
- 27. Chung, W.S. et al. Rheumatoid arthritis increases the risk of deep vein thrombosis and pulmonary thromboembolism: a nationwide cohort study. Ann. Rheum. Dis. 73, 1774–1780 (2014). [DOI] [PubMed] [Google Scholar]
- 28. Matta, F. , Singala, R. , Yaekoub, A.Y. , Najjar, R. & Stein, P.D. Risk of venous thromboembolism with rheumatoid arthritis. Thromb. Haemost. 101, 134–138 (2009). [PubMed] [Google Scholar]
- 29. Ungprasert, P. , Srivali, N. , Spanuchart, I. , Thongprayoon, C. & Knight, E.L. Risk of venous thromboembolism in patients with rheumatoid arthritis: a systematic review and meta‐analysis. Clin. Rheumatol. 33, 297–304 (2014). [DOI] [PubMed] [Google Scholar]
- 30. Taylor, P.C. et al. Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol. 71, 1042–1055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pfizer Inc. Highlights of prescribing information: Xeljanz/Xeljanz XR <http://labeling.pfizer.com/ShowLabeling.aspx?id=959> (2020). Accessed June 17, 2020.
- 32. Tehlirian, C. et al. A phase 1, randomised, double‐blind, placebo‐controlled, multiple‐dose study to evaluate the safety, tolerability, efficacy, pharmacokinetics, and pharmacodynamics of the oral TYK2 inhibitor PF‐06826647 in participants with plaque psoriasis. (Manuscript accepted for publication by Lancet Rheumatology). [DOI] [PubMed]
- 33. Battisti, W.P. et al. Good publication practice for communicating company‐sponsored medical research: GPP3. Ann. Intern. Med. 163, 461–464 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information), Pfizer will provide access to individual de‐identified participant data from Pfizer‐sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (i) for indications that have been approved in the US and/or the EU, or (ii) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de‐identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.