

Abstract
Atogepant is a potent, selective, oral calcitonin gene–related peptide (CGRP) receptor antagonist in development for migraine prevention. The chemical structure of atogepant is distinct from previous CGRP receptor antagonists, which were associated with elevated serum alanine aminotransferase (ALT) in clinical trials. Here, we report the safety, tolerability, and pharmacokinetics (PKs) of a once‐daily supratherapeutic dose (170 mg) of atogepant for 28 days from a randomized, double‐blind, placebo‐controlled phase I trial in healthy participants. Overall safety, hepatic safety, and plasma PK parameters were evaluated. Thirty‐four participants aged 23–55 years enrolled; 28 (82.4%) completed the study in accordance with the protocol. Multiple doses of 170 mg atogepant for 28 consecutive days were generally well‐tolerated. All adverse events (AEs; reported in 87.0% of the atogepant group; 72.7%, placebo) were mild in severity except one serious AE of subarachnoid hemorrhage due to a bicycle accident and not considered related to treatment. There were two discontinuations due to AEs, both with atogepant, one considered possibly related to treatment. Over 28 days of treatment, no participant receiving atogepant had an ALT elevation above 1.5 × upper limit of normal. Change from baseline in serum ALT levels was not different between atogepant and placebo. Atogepant is rapidly absorbed (median time to maximum plasma concentration, ~ 2 hours) with an apparent terminal half‐life of ~ 11 hours, and no evidence of accumulation after once‐daily dosing. Overall, atogepant at a high oral dose is safe and well‐tolerated in healthy participants with no clinically meaningful elevations in ALT.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Atogepant is a potent, selective, oral calcitonin gene–related peptide (CGRP) receptor antagonist in development for migraine prevention. The impact of atogepant on changes in alanine aminotransferase (ALT) has not yet been evaluated in a dedicated clinical trial.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The potential impact of once‐daily supratherapeutic doses (170 mg) of atogepant for 28 days on ALT levels in healthy adult participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Once‐daily supratherapeutic doses of atogepant for 28 days were found to be safe and well‐tolerated in healthy participants with no clinically meaningful elevations in ALT.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These findings will provide clinicians with the knowledge that atogepant, an oral CGRP receptor antagonist, is unlikely to be associated with drug‐induced liver injury.
Migraine is a highly prevalent and burdensome chronic neurological disease. 1 , 2 It is the second largest cause of disability worldwide, 3 and the leading cause of disability during the most productive ages of 15–49 years. 4 The disabling effects of migraine can also exert negative changes on many aspects of life, including work productivity, quality of life, and finances. 5 , 6 , 7 Effective and safe preventive treatment options are needed to help reduce the burden of migraine.
Inhibition of calcitonin gene−related peptide (CGRP), a potent vasodilatory protein strongly implicated in the pathophysiology of migraine, has emerged as a targeted approach for migraine prevention and treatment. 8 Small‐molecule oral CGRP receptor antagonists (called gepants), such as ubrogepant 9 and rimegepant, 10 have demonstrated efficacy in the acute treatment of migraine attacks and were recently approved by the US Food and Drug Administration, 11 , 12 , 13 , 14 , 15 and rimegepant is being evaluated for prevention in adult patients with migraine (NCT03732638). Monoclonal antibodies that target CGRP or the CGRP receptor are currently available for adults in the United States and Europe. 8 Monoclonal antibodies require parenteral administration (subcutaneous or intravenous injection) and have a long elimination half‐life. 8 Therefore, the development of orally administered CGRP receptor antagonists may provide an alternative for people who prefer an oral route of administration over an injection.
Atogepant is a potent, selective, small‐molecule antagonist of the CGRP receptor that is currently in development for the prevention of migraine, with a half‐life of ~ 11 hours. Atogepant is chemically distinct from prior oral CGRP receptor antagonists, notably telcagepant and MK‐3207, which were discontinued because of drug‐induced liver injury (DILI). 16 The efficacy and safety of atogepant in migraine prevention was demonstrated in a phase IIb/III clinical trial conducted subsequent to this trial in which treatment with atogepant, compared with placebo, significantly decreased monthly migraine days over 12 weeks. 17 Atogepant is in phase III development for migraine prevention (ClinicalTrials.gov NCT03700320, NCT03777059, NCT03855137, and NCT03939312).
This study evaluated the safety, tolerability, and pharmacokinetics (PKs) of multiple oral 170 mg doses of atogepant in healthy adult participants. The 170 mg dose is substantially higher than doses being tested in phase III clinical trials (once daily 10, 30, and 60 mg, and twice daily 30 and 60 mg). The primary objectives were to evaluate the safety and tolerability, and the mean fold change from baseline of alanine aminotransferase (ALT) after 28 days of once‐daily atogepant dosing in healthy participants. The secondary objective was to obtain preliminary plasma PK data following multiple‐dose administration of atogepant. The primary safety end point was mean fold change from baseline in serum ALT.
Methods
Trial design
This was a randomized, double‐blind, placebo‐controlled, single‐site, phase I trial (MSD Protocol MK‐8031 PD004). After screening, participants were randomly assigned in a 2:1 ratio to oral atogepant 170 mg or matching placebo administered once daily for 28 days. For this study, a dose of 170 mg in an oral compressed tablet formulation administered once daily was chosen because it was expected to provide a 3‐fold to 5‐fold margin over the expected highest clinical dose. Follow‐up visits to evaluate safety (including liver function tests) were conducted at 14, 30, and 60 days after the final dose of study medication. This trial was conducted in accordance with the Declaration of Helsinki and the principles of Good Clinical Practice. All participants provided written informed consent prior to initiation of any trial‐specific procedures. The trial was approved by the Thomas Jefferson University Institutional Review Board.
Participants
Eligible participants were adults (18–55 years of age at screening) judged to be in good health, with body mass index between 18 and 32 kg/m2, nonsmokers, and had a fasting glucose value below the upper limit of normal (ULN). Key exclusion criteria included history of clinically relevant medical conditions; estimated creatinine clearance ≤ 80 mL/min based on the Cockcroft‐Gault equation; or major surgery or blood donation or loss within 4 weeks before screening. Participants could not use cytochrome P450 enzyme inhibitors or inducers, P‐glycoprotein inhibitors, or prescription medications (particularly substrates of transporters, such as organic‐anion‐transporting polypeptide or breast cancer resistance protein).
Assessments
Pharmacokinetics
Blood samples for PK assessments were collected before dosing and at 20 and 40 minutes, and at 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, and 36 hours postdose on days 1 and 28 (full PK sampling days), and before dosing on days 7, 14, and 21. The concentration of atogepant was assessed in plasma samples: liquid‐liquid extraction was used to isolate analytes, which were detected and quantified using reversed‐phase chromatographic‐tandem mass spectrometry. The lower limit of quantitation was 1.657 nM with a linear calibration range of 1.657–1657 nM for atogepant.
PK variables included atogepant area under the plasma concentration‐time curve from 0 to 24 hours (AUC0–24hr), maximum plasma concentration (Cmax), plasma concentrations at 2 hours postdose (C2hr) and 24 hours postdose (C24hr), time to maximum plasma concentration (Tmax), the apparent terminal half‐life (t 1/2), and the accumulation ratio of atogepant (day 28/day 1) for AUC0–24hr, Cmax, C2hr, and C24hr.
Overall safety
Safety assessments included reports of treatment‐emergent adverse events (AEs) and serious AEs (SAEs) throughout the trial, and physical examination, vital sign determination, electrocardiogram, and laboratory assessments performed at prespecified times during the trial.
Hepatic safety
Liver function tests, including ALT and aspartate aminotransferase (AST), were conducted on blood samples collected on day –1, day 1 predose, at 24 hours after dosing on days 1, 7, 14, 21, and 28, and at the 3 safety follow‐up visits at ~ 14, 30, and 60 days after the last dose.
Statistical analysis
As an estimate of expected precision, assuming a true total SD for log‐transformed fold‐change from baseline ALT of 0.299 with 20 participants receiving atogepant and 10 receiving placebo, the estimated half‐width of the 95% confidence interval (CI) for the true mean difference (atogepant – placebo) is 0.237 for log‐transformed fold‐change from baseline ALT. For comparison of adverse experience rates, if an adverse experience occurs at a rate of 10%, then the chance of observing such an adverse event among 20 participants receiving atogepant would be 88%. If no AE of a given type is observed in any of the 20 participants receiving atogepant, then with 80% confidence, the true incidence of the adverse experience at that dose is at most 8% (11% with 90% confidence).
AUC0–24hr and Cmax values were natural log‐transformed and analyzed using linear fixed‐effects models with fixed effects for day and sex, day‐by‐sex interaction, and a random effect for participant. Geometric means (GMs) for PK parameters are reported with 95% CIs on day 1 and day 28 and accumulation of atogepant was assessed by construction of a 90% CI for the GM ratio (day 28 value/day 1 value) for PK parameters. Natural log‐transformed fold‐change from baseline values for serum ALT were analyzed in a linear mixed‐effects model with fixed effects for treatment, time and sex, treatment‐by‐sex interaction, treatment‐by‐time interaction, and a random effect for participant within treatment. Least squares mean and 90% CIs for mean treatment differences (atogepant – placebo) in natural log‐transformed change from baseline at each timepoint were calculated from the same model. Treatment differences and CIs were exponentiated to obtain GM treatment fold differences (atogepant/placebo) in fold‐change from baseline at each timepoint. The proportion of participants exceeding 3 × ULN for serum ALT was reported.
RESULTS
Participant disposition and baseline demographics
The study was conducted between March 6, 2013, and July 31, 2013. A total of 34 participants enrolled (atogepant 170 mg, n = 23; placebo, n = 11) and 28 (82.4%) completed the study (Figure 1 ). Two participants (both receiving atogepant) discontinued the trial because of an AE. The mean age was similar between groups (atogepant, 37 years; placebo, 40 years); and most participants were men (atogepant, 19 (83%); placebo, 8 (73%) (Table 1 ).
Figure 1.
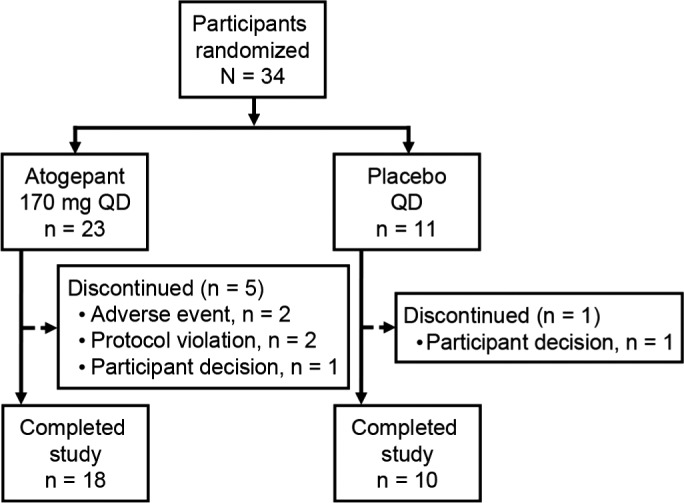
Participant disposition.
Table 1.
Participant characteristics
| Characteristic |
Atogepant 170 mg q.d. (n = 23) |
Placebo q.d. (n = 11) |
|---|---|---|
| Age, mean (SD) [range], years | 37.1 (9.5) [23–55] | 39.8 (9.3) [25–55] |
| Sex, male, n (%) | 19 (82.6) | 8 (72.7) |
| Race, n (%) | ||
| Black/African American | 13 (56.5) | 5 (45.5) |
| White | 9 (39.1) | 6 (54.5) |
| Multiple | 1 (4.3) | 0 |
| Ethnicity, n (%) | ||
| Hispanic or Latino | 0 | 1 (9.1) |
Pharmacokinetics
Oral atogepant was rapidly absorbed (median Tmax of ~ 2 hours) with an apparent t 1/2 of ~ 11 hours (Table 2 ). Consistent with the t 1/2, there was little to no accumulation of atogepant following 28 days of once‐daily dosing, and the ratio of day 28 to day 1 AUC0–24hr was ~ 1. Mean atogepant plasma concentrations over time following once‐daily dosing at 170 mg/d are shown in Figure 2 .
Table 2.
Statistical summary for plasma PK parameters of oral atogepant 170 mg q.d. administered for 28 days in healthy participants
| PK Parameter |
Day 1 GM (95% CI) (n = 23) |
Day 28 GM (95% CI) (n = 18) |
Day 28/Day 1 GMR (90% CI) |
RMSE a |
|---|---|---|---|---|
| AUC0–24, µM•h b | 14.4 (11.5–18.0) | 15.2 (11.7–19.7) | 1.05 (0.88–1.26) | 0.202 |
| Cmax, nM b | 3,170 (2,550–3,930) | 3,090 (2,350–4,070) | 0.98 (0.78, 1.22) | 0.258 |
| C2hr, nM b | 2,270 (1,670–3,100) | 2,830 (1,860–4,310) | 1.24 (0.84–1.85) | 0.468 |
| C24hr, nM b | 43.5 (31.2–60.5) | 76.0 (52.0–111) | 1.75 (1.37–2.24) | 0.273 |
| Tmax, hours c | 2.02 (1.00–6.00) | 1.52 (0.67–4.20) | – | – |
| Apparent t 1/2, hours d | – | 10.6 (4.1) | – | – |
AUC0–24, area under the concentration time curve from 0 to 24 hours; C2hr, plasma concentration at 2 hours postdose; C24hr, plasma concentration at 24 hours postdose; CI, confidence interval; Cmax, maximum plasma concentration; GM, geometric mean; GMR, geometric mean ratio; PK, pharmacokinetic; RMSE, square root of mean squared error; t 1/2, terminal half‐life; Tmax, time to maximum plasma concentration.
RMSE (residual error) was obtained from the linear mixed‐effect model and approximates the within‐subject percentage coefficient of variation on the raw scale when multiplied by 100.
Back‐transformed least squares mean and CI from linear mixed‐effects model, performed on natural log‐transformed values.
Tmax data represent median (minimum, maximum) values.
Apparent t 1/2 data represent mean (SD) based on PK sampling up to 36 hours postdose.
Figure 2.
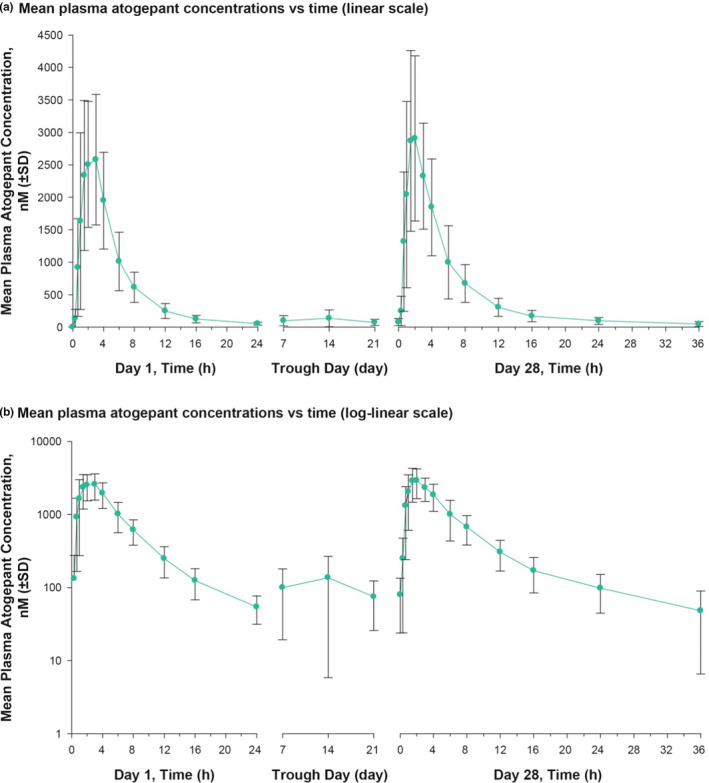
Atogepant concentrations in plasma over time. Arithmetic mean values for atogepant plasma concentration over time following administration of 170 mg q.d. for 28 days in fasted healthy participants are shown on (a) linear scale and (b) log‐linear scale. Error bars represent SDs.
Overall safety
Most participants (atogepant 87%, placebo 73%) reported at least one AE during the trial (Table 3 ). The sole SAE (subarachnoid hemorrhage from a bicycle accident on day 12 after dosing) was not considered to be related to treatment. In addition to the participant with the SAE, 1 other participant discontinued the study because of an event of tachyphrenia (racing thoughts) on day 6 before dosing, which was evaluated by a psychiatrist and considered possibly related to treatment. Tachyphenia was not reported as an AE in the phase IIb/III trial. 17
Table 3.
Participants with AEs
| Participants with event, n (%) |
Atogepant 170 mg q.d. (n = 23) |
Placebo q.d. (n = 11) |
|---|---|---|
| At least 1 AE | 20 (87.0) | 8 (72.7) |
| SAE (subarachnoid hemorrhage due to bicycle accident) | 1 (4.3) | 0 |
| AE leading to trial discontinuation (racing thoughts, subarachnoid hemorrhage due to bicycle accident) | 2 (8.7) | 0 |
| AEs occurring in ≥ 2 participants in a treatment group, n (%) | ||
| Fatigue | 11 (47.8) | 4 (36.4) |
| Headache | 7 (30.4) | 3 (27.3) |
| Dizziness | 5 (21.7) | 1 (9.1) |
| Appetite decreased | 5 (21.7) | 0 |
| Erythema | 3 (13.0) | 1 (9.1) |
| Nausea | 3 (13.0) | 1 (9.1) |
| Vessel puncture site pain | 3 (13.0) | 1 (9.1) |
| Back pain | 3 (13.0) | 0 |
| Pruritus | 3 (13.0) | 0 |
| Hematoma | 2 (8.7) | 2 (18.2) |
| Oropharyngeal pain | 2 (8.7) | 2 (18.2) |
| Scratch | 2 (8.7) | 1 (9.1) |
| Abdominal discomfort | 2 (8.7) | 0 |
| Chills | 2 (8.7) | 0 |
| Constipation | 2 (8.7) | 0 |
| Diarrhea | 2 (8.7) | 0 |
| Feeling hot | 2 (8.7) | 0 |
| Musculoskeletal pain | 2 (8.7) | 0 |
| Neck pain | 2 (8.7) | 0 |
| Weight decreased | 2 (8.7) | 0 |
AE, adverse event; SAE, serious adverse event.
The most commonly reported AEs were fatigue, headache, and dizziness (Table 3 ). Sixteen participants in the atogepant group reported AEs that were considered to be treatment related, most frequently fatigue (n = 9), decreased appetite (n = 5), dizziness (n = 5), and headache (n = 4). Six participants in the placebo group also had treatment‐related AEs, most commonly fatigue (n = 4) and headache (n = 3). All AEs were rated as mild except for the SAE (subarachnoid hemorrhage). No clinically significant abnormalities related to treatment were observed in routine serum chemistry, hematology, urinalysis, electrocardiogram, or vital signs.
Hepatic safety
Mean serum ALT levels were below the ULN for both atogepant and placebo groups throughout the trial (Figure 3 ). The mean serum ALT levels for participants in the atogepant group were lower during the dosing period than at baseline. Point estimates for the model‐based mean fold‐change from baseline in treatment differences were < 1.00 at all times tested during the dosing period (Table 4 ). No participant had an ALT value ≥ 3 × ULN at any timepoint. None of the transaminase elevations were considered to be clinically significant by the investigator.
Figure 3.

Serum ALT concentrations over time. Arithmetic mean values for ALT serum concentration at baseline and following administration of atogepant 170 mg q.d. (black line) or placebo q.d. (gray line) for 28 days in healthy participants. Dotted and dashed lines indicate ALT ULN for men (45 IU/L) and women (30 IU/L), respectively. ALT, alanine aminotransferase; ULN, upper limit of normal.
Table 4.
Fold change from baseline in serum ALT level following administration of oral atogepant 170 mg q.d. a or placebo q.d.
| Timepoint |
Atogepant 170 mg q.d. |
Placebo q.d. |
Mean (90% CI) |
|---|---|---|---|
| Day 1, 24 hours | 0.96 (0.85–1.07) [23] | 0.97 (0.82–1.14) [11] | 0.99 (0.84–1.17) |
| Day 7, predose | 0.89 (0.79–0.99) [22] | 1.05 (0.89–1.23) [11] | 0.85 (0.72–1.00) |
| Day 14, predose | 0.81 (0.72–0.91) [20] | 1.12 (0.95–1.32) [11] | 0.73 (0.61–0.86) |
| Day 21, predose | 0.80 (0.70–0.90) [18] | 1.17 (0.99–1.39) [10] | 0.68 (0.57–0.81) |
| Day 28, predose | 0.78 (0.69–0.88) [18] | 0.99 (0.84–1.18) [9] | 0.79 (0.66–0.94) |
| Day 28, 24 hours | 0.79 (0.70–0.89) [18] | 0.99 (0.84–1.17) [10] | 0.79 (0.67–0.94) |
| Follow‐up visit 1 e | 1.06 (0.94–1.19) [23] | 1.01 (0.85–1.19) [11] | 1.05 (0.89–1.24) |
| Follow‐up visit 2 e | 1.04 (0.93–1.17) [23] | 1.06 (0.90–1.25) [11] | 0.98 (0.83–1.16) |
| Follow‐up visit 3 e | 1.13 (1.01–1.27) [22] | 0.99 (0.84–1.16) [11] | 1.14 (0.97–1.35) |
ALT, alanine aminotransferase; CI, confidence interval; [n], number of participants with data available.
Two participants had elevated serum ALT during follow‐up; both had normal ALT levels during the dosing period. One of these participants had a serum ALT level of 46 IU/mL on day 84 (upper limit of normal (ULN) 45 IU/mL) followed by 28 IU/mL 5 days later, and the other had ALT 57 IU/mL on day 84 that was reported to be in association with physical activity and use of acetaminophen for back pain. One participant had elevated AST (~ 2.3 × ULN) but normal ALT during follow‐up at 10 days after the last atogepant dose, which was attributed to exercise.
Baseline values were calculated as the geometric mean of the day −1 and day 1 predose values.
Data represent back‐transformed least squares mean values; mean values and CIs were based on mixed‐effects model performed on natural log‐transformed values.
Fold treatment difference was calculated as (atogepant/placebo) in fold change from baseline.
Safety follow‐up visits 1, 2, and 3 occurred at 14 days, ~ 30 days, and ~ 60 days, respectively, after last dose.
DISCUSSION
Atogepant 170 mg, a dose 2.8‐fold greater than the highest dose being evaluated in phase III clinical trial participants, administered once daily over a 28‐day period was safe and generally well‐tolerated in healthy adult participants. The mean fold‐treatment differences for change from baseline in serum ALT were less than one throughout the study, suggesting no apparent effect of atogepant on ALT level. Consistent with its known PK profile, atogepant was rapidly absorbed following oral administration, with a median Tmax of ~ 2 hours and mean apparent terminal t 1/2 of ~ 11 hours. Notably, atogepant did not appear to exhibit any features of DILI, namely, increases from baseline in mean ALT or events of markedly elevated ALT as had been seen with the two first‐generation gepants, as described below. Importantly, this trial was conducted to explore the possible occurrence of signals of liver injury prior to conducting the previously reported phase IIb/III trial. 17
Telcagepant was the first gepant to be clinically evaluated, and demonstrated efficacy for the acute treatment of migraine attacks across several clinical trials. 18 , 19 , 20 , 21 , 22 However, a retrospective analysis of data from phase I trials showed that some participants experienced elevated ALT levels and the group mean levels of ALT were elevated following repeated dosing for 14 days or longer (either once daily for 7 days or twice daily for 12 weeks), and these concerns regarding DILI ultimately led to discontinuation of telcagepant development. 21 , 22 MK‐3207, another gepant, was associated with delayed liver test abnormalities in phase I trials (ALT elevations following the discontinuation of MK‐3207 administration), which also led to discontinuation of its clinical development. 23 , 24 , 25 Integrated mechanistic data suggested that DILI associated with telcagepant and MK‐3207 was at least partly attributable to reactive metabolites. 26 In addition, it is believed that the formation of covalent adducts between MK‐3207 and endogenous liver proteins, which does not occur with atogepant, caused the delayed DILI. Atogepant is chemically distinct from telcagepant and MK‐3207, and has characteristics hypothesized to be important for reducing the potential for DILI, such as greater potency and higher target engagement and, therefore, a lower dosing needed for clinical efficacy, and a reduced potential to form reactive metabolites. 16
In a phase IIb/III trial, all tested doses of atogepant (10 mg q.d., 30 mg q.d., 60 mg q.d., 30 mg b.i.d., and 60 mg b.i.d.) were superior to placebo in reducing mean monthly migraine days across 12 weeks of treatment. 17 Nausea was the only treatment‐related treatment‐emergent AE occurring in at least 5% of participants in multiple dose atogepant groups. During the trial, no treatment‐related SAEs were reported with atogepant. After daily dosing for 12 weeks, the overall percentage of participants who had ALT/AST elevations at least 3 × ULN ranged from 0.6–2.2% with atogepant compared with 1.1% for placebo. There were no cases of Hy’s Law of concurrent ALT/AST elevations at least 3 × ULN or bilirubin elevation at least 2 × ULN. The phase IIb/III trial results, combined with results from the present study and other ongoing and completed 27 atogepant phase I studies, all of which have not observed incidences of DILI, support the notion that the toxicity observed with telcagepant and MK‐3207 was due to the chemical structures of those particular molecules and is not inherent to all gepants. 16 Nonetheless, the potential for DILI has been instrumental in the design and implementation of the clinical development program for atogepant, and the safety profile of atogepant continues to be closely monitored across the phase III clinical trials for migraine prevention (ClinicalTrials.gov NCT03700320, NCT03777059, NCT03855137, and NCT03939312).
The present trial has several limitations. Comparisons in ALT levels between groups were not adjusted for multiplicity for the numerous timepoints tested. The trial had a relatively small sample size, although this is a limitation of phase I studies in general and only healthy participants were included. Additionally, most of the participants were men and, because migraine is more prevalent in women, these data may not be generalizable to the overall population of people with migraine. Participants received atogepant for only 28 days, which is shorter than the proposed long‐term, once‐daily dosing regimen for migraine prevention. Data from larger randomized clinical trials are needed to more fully evaluate the overall safety and risk of DILI associated with atogepant in the target therapeutic population, people with migraine.
Strengths of the trial include the use of a supratherapeutic dose of atogepant: a 170 mg dose is ~ 3‐fold higher than the highest once‐daily dose administered in the dose‐ranging phase IIb/III study, 17 which was safe and well‐tolerated when used once daily for 28 days. These results are supported by the lack of serious DILI found in the phase IIb/III clinical trial, further adding to the evidence supporting a lack of atogepant‐associated DILI.
In conclusion, atogepant was rapidly absorbed (median Tmax of 2 hours), had a t 1/2 of ~ 11 hours, and exhibited little to no accumulation following 28 days of once‐daily dosing. Multiple doses of atogepant 170 mg administered once daily had no clinically meaningful effect on ALT levels in healthy adults.
Funding
This study was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Conflicts of Interest
K.C.M., J.X., and L.M. were employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, with stock in Merck & Co., Inc., Kenilworth, NJ, USA, at the time of this study. W.K.K. served as a consultant to Merck in 2019 for a topic unrelated to atogepant. P.B., F.C.‐G., W.L., D.P., M.F.D., and C.Z.M. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, with stock in Merck & Co., Inc., Kenilworth, NJ, USA. R.B. is an employee of AbbVie, and may hold AbbVie stock.
Author Contributions
All authors wrote the manuscript. K.C.M., P.B., F.C.‐G., J.X., L.M., and M.F.D. designed the research. K.C.M. and W.K.K. performed the research. K.C.M., F.C.‐G., W.L., J.X., D.P., L.M., M.F.D., C.Z.M., and R.B. analyzed the data.
Acknowledgments
This study was sponsored by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Medical writing and editorial assistance was provided to the authors by Peloton Advantage, an OPEN Health company, Parsippany, NJ, and was funded by AbbVie. The authors thank the study participants, as well as the site/clinical research unit personnel, including clinical and data management staff and the biostatisticians. The authors acknowledge Wendy Ankrom, PhD, for her contributions to this publication.
Data Availability Statement
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.
References
- 1. Pietrobon, D. & Moskowitz, M.A. Pathophysiology of migraine. Annu. Rev. Physiol. 75, 365–391 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Burch, R.C. , Buse, D.C. & Lipton, R.B. Migraine: epidemiology, burden, and comorbidity. Neurol. Clin. 37, 631–649 (2019). [DOI] [PubMed] [Google Scholar]
- 3. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steiner, T.J. , Stovner, L.J. , Vos, T. , Jensen, R. & Katsarava, Z. Migraine is first cause of disability in under 50s: will health politicians now take notice? J. Headache Pain 19, 17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lipton, R.B. , Bigal, M.E. , Diamond, M. , Freitag, F. , Reed, M.L. & Stewart, W.F. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology 68, 343–349 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Lipton, R.B. , Manack Adams, A. , Buse, D.C. , Fanning, K.M. & Reed, M.L. A Comparison of the Chronic Migraine Epidemiology and Outcomes (CaMEO) Study and American Migraine Prevalence and Prevention (AMPP) Study: demographics and headache‐related disability. Headache J. Head Face Pain 56, 1280–1289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buse, D.C. et al. Life with migraine: Effects on relationships, career, and finances from the Chronic Migraine Epidemiology and Outcomes (CaMEO) study. Headache 59, 1286–1299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Charles, A. & Pozo‐Rosich, P. Targeting calcitonin gene‐related peptide: a new era in migraine therapy. Lancet 394, 1765–1774 (2019). [DOI] [PubMed] [Google Scholar]
- 9. Ubrelvy [package insert]. (Allergan USA, Inc., Madison, NJ, 2020). [Google Scholar]
- 10. Nurtec ODT [package insert]. (Biohaven Pharmaceuticals, Inc., New Haven, CT, 2020). [Google Scholar]
- 11. Voss, T. et al. A phase IIb randomized, double‐blind, placebo‐controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia 36, 887–898 (2016). [DOI] [PubMed] [Google Scholar]
- 12. Marcus, R. , Goadsby, P.J. , Dodick, D. , Stock, D. , Manos, G. & Fischer, T.Z. BMS‐927711 for the acute treatment of migraine: a double‐blind, randomized, placebo controlled, dose‐ranging trial. Cephalalgia 34, 114–125 (2014). [DOI] [PubMed] [Google Scholar]
- 13. Lipton, R.B. et al. Rimegepant, an oral calcitonin gene‐related peptide receptor antagonist, for migraine. N. Engl. J. Med. 381, 142–149 (2019). [DOI] [PubMed] [Google Scholar]
- 14. Dodick, D.W. et al. Ubrogepant for the treatment of migraine. N. Engl. J. Med. 381, 2230–2241 (2019). [DOI] [PubMed] [Google Scholar]
- 15. Lipton, R.B. et al. Effect of ubrogepant versus placebo on pain and the most bothersome associated symptom in the acute treatment of migraine: the ACHIEVE II randomized clinical trial. JAMA 322, 1887–1898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hargreaves, R. & Olesen, J. Calcitonin gene‐related peptide modulators ‐ the history and renaissance of a new migraine drug class. Headache 59, 951–970 (2019). [DOI] [PubMed] [Google Scholar]
- 17. Goadsby, P.J. et al. Safety, tolerability, and efficacy of orally administered atogepant for the prevention of episodic migraine in adults: a double‐blind, randomised phase 2b/3 trial. Lancet Neurol. 19, 727–737 (2020). [DOI] [PubMed] [Google Scholar]
- 18. Ho, T.W. et al. Randomized controlled trial of an oral CGRP receptor antagonist, MK‐0974, in acute treatment of migraine. Neurology 70, 1304–1312 (2008). [DOI] [PubMed] [Google Scholar]
- 19. Ho, T.W. et al. Efficacy and tolerability of MK‐0974 (telcagepant), a new oral antagonist of calcitonin gene‐related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo‐controlled, parallel‐treatment trial. Lancet 372, 2115–2123 (2008). [DOI] [PubMed] [Google Scholar]
- 20. Connor, K.M. et al. Randomized, controlled trial of telcagepant for the acute treatment of migraine. Neurology 73, 970–977 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ho, T.W. et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology 83, 958–966 (2014). [DOI] [PubMed] [Google Scholar]
- 22. Ho, T.W. et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for prevention of headache in women with perimenstrual migraine. Cephalalgia 36, 148–161 (2016). [DOI] [PubMed] [Google Scholar]
- 23. Hewitt, D.J. et al. Randomized controlled trial of the CGRP receptor antagonist MK‐3207 in the acute treatment of migraine. Cephalalgia 31, 712–722 (2011). [DOI] [PubMed] [Google Scholar]
- 24. Merck updates status of clinical development programs for investigational CGRP receptor antagonist treatments for acute migraine; MK‐3207 clinical development discontinued [press release]. BusinessWire, 2009. <https://www.businesswire.com/news/home/20090910005709/en/Merck‐Updates‐Status‐Clinical‐Development‐Programs‐Investigational>. Accessed December 10, 2018.
- 25. Edvinsson, L. The CGRP pathway in migraine as a viable target for therapies. Headache 58 (suppl. 1), 33–47 (2018). [DOI] [PubMed] [Google Scholar]
- 26. Smith, B. et al. Mechanistic investigations support liver safety of ubrogepant. Toxicol. Sci. 177, 84–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ankrom, W. et al. Atogepant has no clinically relevant effects on the pharmacokinetics of an ethinyl estradiol/levonorgestrel oral contraceptive in healthy female participants. J. Clin. Pharmacol. 60, 1157–1165 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.