

Abstract
Many targeted therapies are administered at or near the maximum tolerated dose (MTD). With the advent of precision medicine, a larger therapeutic window is expected. Therefore, dose optimization will require a new approach to early clinical trial design. We analyzed publicly available data for 21 therapies targeting six kinases, and four poly (ADP‐ribose) polymerase inhibitors, focusing on potency and exposure to gain insight into dose selection. The free average steady‐state concentration (Css) at the approved dose was compared to the in vitro cell potency (half‐maximal inhibitory concentration (IC50)). Average steady‐state area under the plasma concentration‐time curve, the fraction unbound drug in plasma, and the cell potency were taken from the US drug labels, US and European regulatory reviews, and peer‐reviewed journal articles. The Css was remarkably similar to the IC50. The median Css/IC50 value was 1.2, and 76% of the values were within 3‐fold of unity. However, three drugs (encorafenib, erlotinib, and ribociclib) had a Css/IC50 value > 25. Seven other therapies targeting the same 3 kinases had much lower Css/IC50 values ranging from 0.5 to 4. These data suggest that these kinase inhibitors have a large therapeutic window that is not fully exploited; lower doses may be similarly efficacious with improved tolerability. We propose a revised first‐in‐human trial design in which dose cohort expansion is initiated at doses less than the MTD when there is evidence of clinical activity and Css exceeds a potency threshold. This potency‐guided approach is expected to maximize the therapeutic window thereby improving patient outcomes.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The primary objective of most first‐in‐human (FIH) studies is to establish a maximum tolerated dose (MTD). In oncology, the MTD is assumed to be ideal and lower doses are rarely studied.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ How can we best leverage preclinical data to identify doses that exploit the larger therapeutic window expected for next generation targeted therapies?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ At the approved doses of 25 targeted therapies studied, the average free concentration at steady state (Css) was similar to the in vitro cell potency (half‐maximal inhibitory concentration (IC50)). However, 3 of these drugs have Css/IC50 values > 25 suggesting a large therapeutic window. Lower doses of these agent may be equally effective with less toxicity.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ We propose a revised FIH trial design for next generation targeted therapy in which dose cohort expansion is initiated at doses less than the MTD when there is evidence of clinical activity and Css exceeds a threshold informed by in vitro cell potency.
Most often, the primary objective of the first‐in‐human (FIH) trial in oncology is to establish a maximum tolerated dose (MTD). Where targeted therapies are studied in defined patient populations, it is not uncommon to observe meaningful clinical responses during dose escalation. Nonetheless, the MTD is typically assumed to be the ideal therapeutic dose and dose escalation continues with 3–6 patients per dose level until the MTD is reached. An expansion cohort is initiated most often at the MTD to evaluate preliminary efficacy, at which point lower doses are no longer explored. Thus, limited information is collected in these FIH studies that would facilitate a comparison of the efficacy at the MTD with that of lower doses, which may be better tolerated. 1 , 2 , 3
Given the desire to advance the most promising agents to confirmatory trials as rapidly as possible, there has been considerable debate regarding dose selection in oncology. 4 , 5 , 6 It remains a question whether the “MTD approach,” which is well‐established for chemotherapeutics that have a narrow therapeutic window, is equally appropriate for targeted therapies that may have a larger therapeutic window. Analysis of the growing number of approved targeted agents, including preclinical data made public during the regulatory review and approval process, provides unique insights into this question.
A potency‐guided FIH trial leverages quantitative preclinical data regarding the underlying concentration‐response relationship driving therapeutic efficacy. At steady‐state, for cell permeable drugs not subject to active transport processes, the unbound drug concentration in the blood is equal to the unbound concentration in the tumor, where the free drug interacts with its target. Under these conditions, systemic drug concentrations approximating the in vitro potency are expected to elicit the desired pharmacologic response. This hypothesis can be validated using xenograft models in which the inhibition of tumor cell growth is studied in cell culture and in animals under similar conditions. Concordance between in vitro and in vivo potency has been demonstrated for drugs targeting specific genetic abnormalities that drive tumor cell growth. 7 , 8 , 9 , 10
In the present study, the free average steady‐state concentration (Css) of 25 marketed oncology drugs, including 21 kinase inhibitors (5 ABL, 3 ALK, 3 BRAF, 3 CDK4/6, 4 EGFR, and 3 MEK1/2) and 4 poly (ADP‐ribose) polymerase (PARP) inhibitors, has been compared with the in vitro cell line potency (half‐maximal inhibitory concentration (IC50)) of the drug to derive a unitless ratio herein defined as Css/IC50. Many of these therapies have a Css/IC50 value near unity and are administered at their MTD. Drugs that fit these parameters have a relatively narrow therapeutic window where higher doses are not tolerated, and lower doses result in insufficient target engagement. However, for those drugs administered at their MTD that have a Css/IC50 value substantially > 1, a lower dose has the potential to provide similar efficacy with a more favorable safety profile.
FIH studies of mutant‐selective oncogene inhibitors and drugs that leverage synthetic lethal interactions are expected to enroll homogeneous patient populations that are highly sensitive to therapy. We propose that these studies use a revised FIH trial design in which dose cohort expansion is initiated at doses less than the MTD when there is evidence of clinical activity and Css values exceed an IC50 threshold (Figure 1 ). The performance of multiple expansion cohorts can be compared directly before selecting the dose for further evaluation. For medicines, which by design are expected to minimize toxicity to normal tissue and maximize tumor cytotoxicity, this approach should help identify the optimal dose. Where efficacy can be achieved at lower, equally effective doses, we expect less toxicity, better compliance, and, accordingly, increased benefit to patients.
Figure 1.
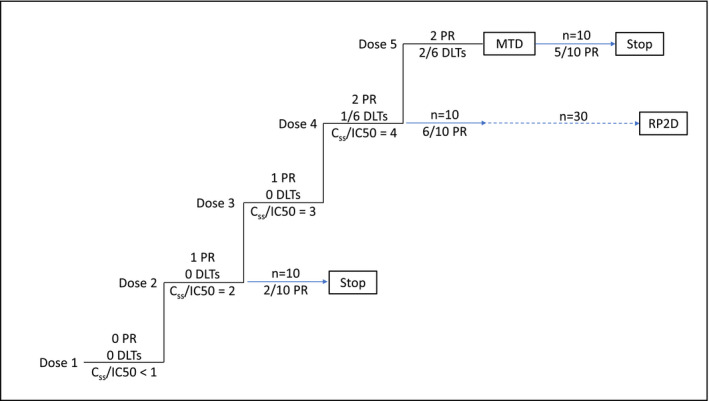
Potency‐guided first‐in‐human trial design, including theoretical outcomes. Dose expansion is initiated at dose level 2 when the steady‐state concentration (Css) value is 2‐fold greater than the half‐maximal inhibitory concentration (IC50) with no DLTs and 1 PR. Dose expansion is also initiated at dose levels 4 and 5 (the maximum tolerated dose (MTD)). Comparison of the first 10 patients in the 3 expansion cohorts suggests dose level 4 is most promising and further enrollment is limited to dose level 4. Dose levels 3 is not selected for expansion as exposure is overlapping, due to pharmacokinetic variability, with adjacent dose levels. Blue arrows represent enrollment into dose expansion cohorts. DLT, dose limiting toxicity; PR, partial response; RP2D, recommended phase 2 dose.
METHODS
Average steady‐state total drug and/or metabolite area under the plasma concentration‐time curve over the dosing interval (AUCtau) for the dose on the drug label, the fraction of unbound drug in plasma (fup), the MTD and in vitro cell potency (IC50) were taken from the US drug labels, US regulatory pharmacology, clinical pharmacology and biopharmaceutics reviews, European regulatory agency assessment reports, and peer‐reviewed journal articles (see Table S1 ). Average unbound Css was calculated by dividing AUCtau by the dosing interval (12 hours for b.i.d., 24 hours for q.d.) then multiplying by fup. When not available otherwise, steady‐state AUCtau was derived from dose and oral clearance. Css was converted to molar units using the molecular weight of the parent drug or metabolite and the unitless ratio Css/IC50 derived. Table 1 summarizes the cell line models and the in vitro assay end points used to estimate drug potency (in most cases, cell proliferation).
Table 1.
Cell line models and the in vitro assay end points used to determine drug potency
| Target | Drug | Cell line | End point |
|---|---|---|---|
| ABL | Bosutinib | K562 | Proliferation |
| Dasatinib | K562 | Apoptosis | |
| Imatinib | K562 | Proliferation | |
| Nilotinib | K562 | Apoptosis | |
| Ponatinib | Ba/F3 | Apoptosis | |
| ALK | Alectinib | KARPAS‐299 | Proliferation |
| Ceritinib | KARPAS‐299 | Proliferation | |
| Crizotinib | KARPAS‐299 | Proliferation | |
| BRAF | Dabrafenib | COLO205 | Proliferation |
| Encorafenib | A375 | Proliferation | |
| Vemurafenib | COLO205 | Proliferation | |
| CDK4/6 | Abemaciclib | EFM‐19 | Proliferation |
| Palbociclib | EFM‐19 | Proliferation | |
| Ribociclib | Not applicable | Not applicable | |
| EGFR | Afatinib | HCC827 | Proliferation |
| Erlotinib | H3255 | Proliferation | |
| Gefitinib | HCC827 | Proliferation | |
| Osimertinib | H1975 | Proliferation | |
| MEK1/2 | Binimetinib | COLO205 | Proliferation |
| Cobimetinib | COLO205 | Proliferation | |
| Trametinib | COLO205 | Proliferation | |
| PARP | Niraparib | CAPAN‐1 | Proliferation |
| Olaparib | ES7 | Viability | |
| Rucaparib | ES7 | Viability | |
| Talazoparib | ES7 | Viability |
In the current analysis, the average free concentration over the dosing interval was used as the exposure metric. It should be noted that fluctuation in the concentration of these drugs over the dosing interval is modest given their half‐lives and regimens (data not shown), therefore, the maximum and minimum concentrations observed are not expected to be substantially different from the average. For these reasons, the conclusions drawn from the present analysis are not expected to differ substantially if maximum concentration (Cmax) or minimum concentration (Cmin) had been used.
PARP inhibitors
Data for the PARP inhibitor analysis was sourced from a single publication, which included a comparison of three of the four inhibitors (olaparib, rucaparib, and talazoparib) in a Ewing’s sarcoma cell line (ES7) sensitive to both PARP enzyme inhibition and DNA trapping. 11 This paper did not include estimates of the IC50s, instead using a few concentrations, which resulted in partial and/or near complete inhibition of colony formation for each agent. Therefore, the estimates of the respective Css/IC50 values used these experimental concentrations. The IC50 for niraparib was taken from a separate publication using a BRCA2‐deficient Capan‐1 pancreatic cell line. 12
CDK4/6 inhibitors
For two of the three CDK4/6 inhibitors, palbociclib and abemaciclib (parent), the cell line EFM‐19 IC50s were available though from different publications. 13 , 14 For ribociclib, data in the EFM‐19 estrogen‐receptor positive breast cancer line was not publicly available. However, a recent paper argues that the potency of ribociclib and palbociclib are very similar across many cell lines. 15 Therefore, the IC50 value of palbociclib in EFM‐19 was also used for ribociclib. Abemaciclib has three active metabolites in addition to parent. Two of the 3 metabolites are equipotent to parent and one is 3 to 20‐fold less potent on cells. 16 The IC50s used in the estimate of Css/IC50 included these assumptions. The relative contribution of each metabolite was estimated considering the relative potency, pharmacokinetic (PK), and protein binding (available for parent and metabolites), and an overall or total effective Css/IC50 was derived by adding the individual values for parent and metabolites (Table 2 ).
Table 2.
Relative contribution of parent and active metabolites to total Css/IC50 a
| Drug | Analyte | AUC0‐12 ng.h/mL | fup |
Css nM |
IC50 nM |
Css/IC50 | Total Css/IC50 | Cell line |
|---|---|---|---|---|---|---|---|---|
| Abemaciclib | Parent | 3,844 | 0.0300 | 18.97 | 19.0 | 1.00 | 4.14 | EFM‐19 |
| M2 | 1,499 | 0.1100 | 28.77 | 19.0 | 1.51 | EFM‐19 | ||
| M18 | 538 | 0.0900 | 8.18 | 190.0 | 0.04 | EFM‐19 | ||
| M20 | 3,152 | 0.0600 | 30.16 | 19.0 | 1.59 | EFM‐19 | ||
| Dabrafenib | Parent | 4,341 | 0.0040 | 2.79 | 6.0 | 0.46 | 1.57 | COLO205 |
| M4 | 47751 | 0.0050 | 35.30 | 320.0 | 0.11 | COLO205 | ||
| M7 | 3907 | 0.0370 | 22.49 | 23.0 | 0.98 | COLO205 | ||
| M8 | 3039 | 0.0010 | 0.50 | 23.0 | 0.02 | COLO205 |
AUC0–12, area under the plasma concentration‐time curve from zero to 12; Css, steady‐state concentration; IC50, half‐maximal inhibitory concentration; fup, fraction of unbound drug in plasma.
M2, M18, and M20 are the N‐desethyl, hydroxy‐N‐desethyl, and hydroxy metabolites, respectively, of abemaciclib. M4, M7, and M8 are the carboxy, hydroxy, and desmethyl metabolites, respectively, of dabrafenib. For abemaciclib, metabolite AUCs were estimated from reported exposures relative to parent in a single dose study. M18 was reported to have activity 3 to 20‐fold less than parent, 15 therefore, a 10‐fold lower potency was used in the table. A 3‐fold lower potency would result in a Css/IC50 value of 0.14 vs. 0.04 for M18 with minimal difference in the total Css/IC50 value.
BRAF, MEK inhibitors
For dabrafenib, PK, protein binding, and cell potency data in the same cell line were available to estimate the relative contribution of parent and active metabolites to the activity and thus to estimate the overall effective Css/IC50 for the drug (Table 2 ). COLO205 cell line potency was used to estimate Css/IC50 for all BRAF and MEK1/2 inhibitors, with the exception of encorafenib, which used A375 cell line data. 17 , 18 , 19 COLO205 is a colorectal adenocarcinoma cell line, whereas A375 is a melanoma cell line. Both have a V600E mutations that constitutively activate the MAP kinase pathway. Vemurafenib and encorafenib have similar potency in COLO205 and A375 cells supporting the comparison across cell lines.
ABL kinase inhibitors
Cell potency of ABL kinase inhibitors was available for 4 of the 5 drugs in this class for the K562 chronic myelogenous leukemia cell line, which expresses the bcr‐abl fusion. 20 , 21 , 22 As ponatinib is the only agent designed to inhibit the T315I gate‐keeper mutation, the engineered Ba/F3 line expressing the mutant enzyme was used to estimate ponatinib Css/IC50. In this cell line, it was demonstrated that the addition of physiological concentrations of human serum albumin did not impact the in vitro cell potency. Therefore, the fup used for ponatinib was 1. Ponatinib protein binding across all species is reported to be in the range 99.92–99.99% (European Medicines Agency (EMA) Assessment Report). If this high binding were clinically relevant (fup ≤ 0.0008), ponatinib would have a Css < 0.1 nM, well below its biochemical potency of 2 nM against ABL T315I. As nilotinib and dasatinib cause apoptosis on the K562 cell line (ponatinib on the Ba/F3 line), for these agents, the IC50 used reflects the apoptosis endpoint. For bosutinib and imatinib, the IC50 assay end point was cell proliferation.
EGFR inhibitors
Three different lung cancer cell lines were used to estimate the Css/IC50 for the four EGFR inhibitors. Gefitinib and afatinib cell line potency were compared head‐to‐head in HCC827 (exon19del) cells in the same laboratory. 23 Because osimertinib targets the T790M resistance mutation, the potency on H1975 (L858R T790M) cells was used for this agent. 24 Erlotinib potency on H3255 cells was used to estimate the Css/IC50 for this drug. 25 This cell line harbors the L858R mutation and similar IC50 values were reported in papers from 2 different laboratories. 26
RESULTS
In the present study, the free average Css of 25 marketed oncology drugs (21 kinase inhibitors and 4 PARP inhibitors) has been compared with the in vitro cell line potency of the drug to derive a unitless ratio, Css/IC50. The kinases included 5 ABL, 3 ALK, 3 BRAF, 3 CDK4/6, 4 EGFR, and 3 MEK1/2 inhibitors. Table 1 summarizes the cell line models used in this analysis by drug target and the in vitro assay end points used to estimate drug potency. The entire dataset was derived from publicly available information including the US drug labels, US regulatory pharmacology, clinical pharmacology and biopharmaceutics reviews, European regulatory agency assessment reports, and peer‐reviewed journal articles (see Table S1 ).
For this dataset, there was a > 2,800‐fold range of steady‐state AUCtau values (208–601,000 ng.h/mL), a > 700‐fold range in the fraction unbound in plasma (fup, 0.0014–1), and a > 2,300‐fold range of in vitro cell line IC50 values (0.5–1,200 nM) demonstrating considerable diversity. The median Css/IC50 value was 1.2 indicating that at the approved doses, the free average steady‐state drug concentrations are very similar to the respective in vitro IC50s (first quartile and third quartile values were 0.97 and 2.48, respectively). The correlation of Css and IC50 is depicted in Figure 2 . Notable outliers include encorafenib, erlotinib, and ribociclib. Individual drug Css/IC50 values are depicted in Figure 3 by drug target. Sevenety‐six percent of the Css/IC50 values were between 0.3 and 3 within – 3‐fold of unity. Assuming coefficients of variation for Css and IC50 of 40 and 100%, respectively, a Css/IC50 value within 3‐fold of unity is plausible considering experimental variability.
Figure 2.
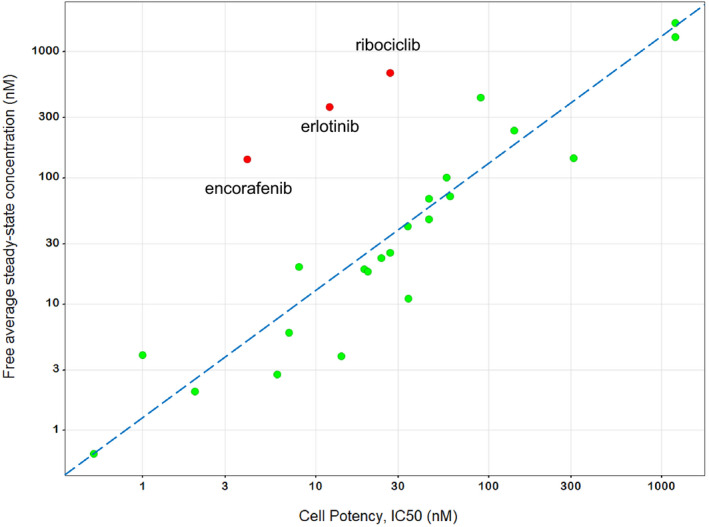
Correlation between free average drug concentration at steady‐state (Css) and in vitro cell potency (half‐maximal inhibitory concentration (IC50)) for 25 targeted therapies. The dashed line is the line of unity. Notable outliers namely, encorafenib, erlotinib and ribociclib, are identified in red. Data are displayed on a log‐log scale to improve readability.
Figure 3.
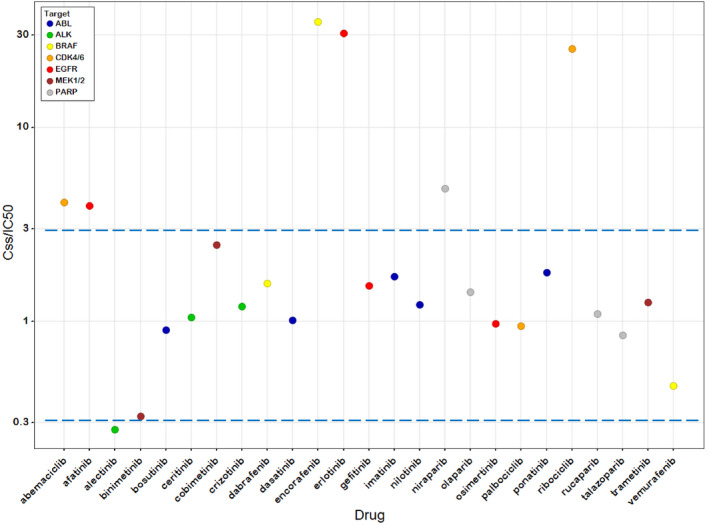
Steady‐state concentration (Css)/half‐maximal inhibitory concentration (IC50) values for 25 targeted therapies. Colors represent drug target. Dashed lines are drawn at ratios of 0.3 and 3 to depict a 3‐fold range of plausible values about unity. Data are displayed on a log‐log scale to improve readability.
Encorafenib
Encorafenib had the highest Css/IC50 value in this dataset with an average free Css > 35 times the in vitro cell potency. The marketed Braf inhibitors vemurafenib and dabrafenib have Css/IC50 values of 0.46 and 1.57, respectively. Assuming dose‐proportionality, doses as low as 13 mg encorafenib (450/35) may be equally effective as the 450 mg dose on the label. In fact, in the FIH study, several impressive responses were observed in patients at the starting dose of 50 mg and the overall response rate in dose escalation was the same as in dose expansion (60%) for Braf inhibitor‐naïve patients. 27 In the Center for Drug Evaluation and Research (CDER) multidisciplinary review of the new drug application (NDA), the minimum effective dose was identified as 50 mg, 9‐times lower than the marketed dose of 450 mg. Administration of encorafenib at the MTD takes no advantage of its apparently large therapeutic window.
Erlotinib
Erlotinib had the second highest Css/IC50 value in this dataset with an average free Css > 30 times the in vitro cell potency. Assuming dose‐proportional PK, a dose of 5 mg is expected to provide a free average Css near the in vitro IC50 (12 nM H3255). The dose on the label is 150 mg. Erlotinib and gefitinib are expected to inhibit wild type EGFR at clinical relevant concentrations, whereas the Css values for afatinib and osimertinib are far below the wild‐type IC50s. 26 As Wild‐type EGFR inhibition is associated with skin rash, 25 the high Css/IC50 value for erlotinib suggests that lower doses might alleviate this dose‐limiting toxicity without compromising L858R or exon19del activity. In fact, doses as low as 25 mg (6‐fold lower than the dose on the label) have demonstrated similar response rates as the 150 mg dose with fewer side‐effects. 28
Ribociclib
Ribociclib had the third highest Css/IC50 value in this dataset with an average free Css > 25 times the in vitro cell potency. The CDK4/6 inhibitors palbociclib and abemaciclib have Css/IC50 values of 0.94 and 4.1, respectively. Considering that over‐proportional increases in ribociclib exposure are observed as the dose is increased, we estimate that doses as low as 60 mg may be equally effective as the 600 mg dose on the label. Given specific concerns for ribociclib cardiac toxicity, the US Food and Drug Administration (FDA) requested that the sponsor conduct a postmarketing study to evaluate lower doses of ribociclib (Ribociclib (Kisqali) Approval Letter), consistent with the conclusions of the present analysis.
Abemaciclib
The Css/IC50 for abemaciclib parent was 1.0. The potency in the EFM‐19 cell line is the same as the free average Css for abemaciclib as it is for palbociclib. However, abemaciclib has two human‐specific metabolites that are equipotent to parent. 16 The Css/IC50 for the N‐desethyl metabolite is 1.5 and the Css/IC50 for the hydroxy metabolite is 1.6. The total effective Css/IC50 is 4.1 (Table 2 ). Therefore, lower doses of abemaciclib (up to 4‐fold) may be expected to be equally efficacious as the recommended dose of 200 mg b.i.d. and may improve drug tolerability. Abemaciclib dose‐limiting toxicity of diarrhea was not observed for ribociclib or palbociclib.
Irreversible inhibitors (afatinib and osimertinib)
Afatinib (Css/IC50 of 3.9) and osimertinib (Css/IC50 of 0.97) are the only two drugs in the set that are irreversible inhibitors. In principle, irreversible inhibitors might be administered intermittently, at doses resulting in average free Css that are lower than the cellular IC50. However, the half‐life of mutant EGFR receptor is reportedly 7–12 hours, 29 so inactivated enzyme may be largely replenished by newly synthesized EGFR between doses. For irreversible inhibitors of targets with longer half‐lives, one might anticipate a different result.
DISCUSSION
Our analysis demonstrates that the desired drug concentration in a patient can be estimated from the in vitro potency for many approved kinase inhibitors used to treat cancer. Similar principles have been applied in immuno‐oncology for checkpoint inhibitors. 30 We propose that this understanding serve as the basis for a revised, potency‐guided approach to the typical escalation‐expansion trial design where the Css/IC50 rather than the MTD is the gating factor for dose cohort expansion. This, in conjunction with an on‐going, real‐time analysis of pharmacodynamics as well as patient efficacy and safety, can help to optimize therapy and decrease clinical development time.
For 13 of the drugs included in this analysis, the approved dose is the MTD; for 7 more the approved dose is < 2 times the MTD (Figure 4 ). The traditional MTD approach has been largely successful in identifying the appropriate dose for these targeted therapies because for many, the therapeutic window is modest. The MTD provides an average free concentration at steady‐state that is similar to the in vitro cellular IC50. Accordingly, higher doses would not be tolerated, and lower doses would result in insufficient target engagement. However, there are several drugs in the dataset that appear to have a substantial therapeutic window and are nonetheless administered at the MTD. Our analysis suggests that doses up to 10‐fold lower than the approved dose may be efficacious. In these cases, patients are potentially exposed to unnecessary toxicity in the absence of substantial evidence of additional therapeutic benefit.
Figure 4.
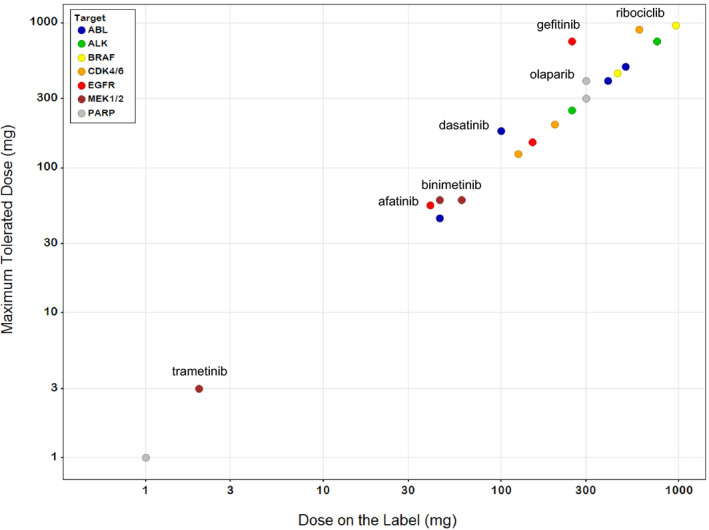
Comparison of the maximum tolerated dose (MTD) to the dose on the label for 20 targeted therapies. All of the drugs are administered at a dose that is < 2 times the MTD; for 13 the recommended dose is the MTD. Drugs for which the MTD is higher than the dose on the label are identified. Drugs represented by the same colored circle inhibit common targets. Five drugs in the dataset are not included because the MTD was not formally identified. Doses are plotted on a log scale to improve readability.
For the next generation of targeted therapies, which by design should have a large therapeutic window, we propose a revised FIH trial design in which dose cohort expansion is initiated prior to reaching the MTD if the Css/IC50 value exceeds a defined threshold (Figure 1 ). Evidence of clinical response in early dose escalation would further strengthen the decision to initiate cohort expansion. This dose would meet the toxicity criteria for continued escalation and therefore would be expected to be well‐tolerated. The study should permit termination of the dose cohort for lack of efficacy or poor tolerability and patients could initiate treatment at a higher or lower dose, respectively.
Unlike the estimation of the rate of dose limiting toxicities, which may require a relatively small number of patients, the expansion cohort should enroll sufficient numbers of patients to be able to distinguish the safety and efficacy of that dose from others that may be studied. As the MTD may not have been reached, escalation should continue in parallel to achieve this objective. Expansion of a second dose level, potentially the MTD, provides an opportunity to compare the performance at this dose with that of the initial expansion cohort. In this case, it is essential to assure that drug exposure at the doses used in these expansions is well‐separated and considers both the steepness of the dose‐response and PK variability. The potency‐guided FIH trial design provides an opportunity for exposure‐response modeling to optimize the dose prior to the initiation of confirmatory trials. Valuable information regarding exposure‐tolerability relationships can be obtained in evaluation of doses up to and including the MTD even if the MTD is not selected as a dose for expansion. An understanding of these relationships will help to put exposure excursions, for example, as a result of drug‐drug interactions, or organ impairment, in perspective.
A critical component of the proposed approach is the selection of the appropriate cell line in which to evaluate in vitro potency. Potency will be cell line‐specific, therefore, Css/IC50 will be dependent on the characteristics of the cell line and how well this cell line represents the typical disease condition. For the most selective compounds, and conditions for which the disease is driven largely by a single, well‐defined genetic alteration, selection of the ideal cell line, and estimation of the efficacious exposure is expected to be most straightforward. BRAF in BRAF mutant melanoma, ABL in Philadelphia chromosome‐positive CML, and ALK in ALK‐positive non‐small cell lung cancer are examples. As is generally the case, potency should be assessed across a panel of disease‐relevant cell lines to understand the underlying factors that drive sensitivity. These analyses will help to establish the most realistic clinical target. When there is a new compound with the same mechanism of action as one already under clinical investigation, the relative potency in cell lines can help to translate effective doses of one agent into effective doses of the other.
The proposed potency‐guided FIH trial design has several important advantages. For medicines, which by design are expected to minimize toxicity to normal tissue and maximize tumor killing, this revised trial design is expected to help identify the optimal dose. This dose is expected to have a better side effect profile resulting in better compliance. A lower dose will be easier to combine with other therapies and there will be fewer dose reductions and interruptions in patients with a complex pharmacopeia. The decision to progress an asset (or not) can be made earlier so that precious resource is applied to the most promising approaches. In sum, we believe that the potency‐guided design for the next generation of precision medicines will maximize the therapeutic window and improve patient outcome.
Funding
No funding was received for this work.
Conflicts of Interest
M.J.G., B.L.W., and C.B.D. are employees and shareholders of Tango Therapeutics. M.J.G. is a shareholder of Biontech and Moderna. C.B.D. is a shareholder of Novartis. B.L.W. is a shareholder of Relay Therapeutics, Cedilla Therapeutics, Fog Pharma and Revolution Medicines, and on the board of directors of Fog Pharma and Revolution Medicines. M.P. is an employee and shareholder of Morphosys AG, and on the board of directors of Tango Therapeutics. All authors declare no competing interests for this work.
Author Contributions
M.J.G. and C.B.D. wrote the manuscript; M.J.G., M.P., B.L.W. and C.B.D. designed the research; C.B.D. performed the research and analyzed the data.
Supporting information
Table S1
References
- 1. Tam, K. Estimating the “first in human” dose – a revisit with particular emphases in oncology drugs. ADMET DMPK 1, 63–75 (2013). [Google Scholar]
- 2. Iasonos, A. & Quigley, J.O. Dose expansion cohorts in phase I trials. Stat. Biopharm. Res. 8, 161–170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou, H. , Yuan, Y. & Nie, L. Accuracy, safety, and reliability of novel phase I trial designs. Clin. Cancer Res. 24, 4357–4364 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Ji, Y. , Jin, J.Y. , Hyman, D.M. , Kim, G. & Suri, A. Challenges and opportunities in dose finding in oncology and immuno‐oncology. Clin. Transl. Sci. 11, 345–351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bullock, J.M. , Rahman, A. & Liu, Q. Lessons learned: dose selection of small molecule‐targeted oncology drugs. Clin. Cancer Res. 22, 2630–2638 (2016). [DOI] [PubMed] [Google Scholar]
- 6. Janne, P.A. et al. Dose finding of small‐molecule oncology drugs: optimization throughout the development life cycle. Clin. Cancer Res. 22, 2613–2617 (2016). [DOI] [PubMed] [Google Scholar]
- 7. Wong, H. et al. Bridging the gap between preclinical and clinical studies using pharmacokinetic‐pharmacodynamic modeling: an analysis of GDC‐0973, a MEK inhibitor. Clin. Cancer Res. 18, 3090–3099 (2012). [DOI] [PubMed] [Google Scholar]
- 8. De Buck, S.S. et al. Population pharmacokinetics and pharmacodynamics of BYL719, a phosphoinositide 3‐kinase antagonist, in adult patients with advanced solid malignancies. Br. J. Clin. Pharmacol. 78, 543–555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fritsch, C. et al. Characterization of the novel and specific PI3K Inhibitor NVP‐BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 13, 1117–1129 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Yamazaki, S. et al. Mechanistic understanding of translational pharmacokinetic‐pharmacodynamic relationships in nonclinical tumor models: a case study of orally available novel inhibitors of anaplastic lymphoma kinase. Drug Metab. Dispos. 43, 54–62 (2015). [DOI] [PubMed] [Google Scholar]
- 11. Gill, S.J. et al. Combinations of PARP inhibitors with temozolomide drive PARP1 trapping and apoptosis in Ewing’s sarcoma. PLoS One 10, 1–17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jones, P. et al. Discovery of 2‐{4‐[(3S)‐piperidin‐3‐yl]phenyl}‐2H‐indazole‐7‐carboxamide (MK‐4827): a novel oral poly(ADP‐ribose)polymerase (PARP) inhibitor efficacious in BRCA‐1 and ‐2 mutant tumors. J. Med. Chem. 52, 7170–7185 (2009). [DOI] [PubMed] [Google Scholar]
- 13. Finn, R.S. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor‐positive human breast cancer cell lines in vitro. Breast Cancer Res. 11, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Torres‐Guzmán, R. et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 8, 69493–69507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim, S. et al. The potent and selective cyclin‐dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 9, 35226–35240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burke, T. et al. Abstract 2830: The major human metabolites of abemaciclib are inhibitors of CDK4 and CDK6. Cancer Res. 76, 2830 (2016). [Google Scholar]
- 17. Choo, E.F. et al. Preclinical disposition of GDC‐0973 and prospective and retrospective analysis of human dose and efficacy predictions. Drug Metab. Dispos. 40, 919–927 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Li, Y. et al. N‐(3‐Ethynyl‐2,4‐difluorophenyl) sulfonamide derivatives as selective raf inhibitors. ACS Med. Chem. Lett. 6, 543–547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamaguchi, T. , Kakefuda, R. , Tajima, N. , Sowa, Y. & Sakai, T. Antitumor activities of JTP‐74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 39, 23–31 (2011). [DOI] [PubMed] [Google Scholar]
- 20. Von Mehren, M. & Widmer, N. Correlations between imatinib pharmacokinetics, pharmacodynamics, adherence, and clinical response in advanced metastatic gastrointestinal stromal tumor (GIST): an emerging role for drug blood level testing? Cancer Treat. Rev. 37, 291–299 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang, X. et al. Discovery of 2‐((3‐Amino‐4‐methylphenyl)amino)‐N‐(2‐methyl‐5‐(3‐(trifluoromethyl)benzamido)phenyl)‐4‐(methylamino)pyrimidine‐5‐carboxamide (CHMFL‐ABL‐053) as a potent, selective, and orally available BCR‐ABL/SRC/p38 kinase inhibitor for chronic myeloid leukemia. J. Med. Chem. 59, 1984–2004 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Golas, J.M. et al. SKI‐606, a 4‐anilino‐3‐quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 63, 375–381 (2003). [PubMed] [Google Scholar]
- 23. Suzawa, K. et al. Antitumor effect of afatinib, as a human epidermal growth factor receptor 2‐targeted therapy, in lung cancers harboring HER2 oncogene alterations. Cancer Sci. 107, 45–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Finlay, M.R.V. et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 57, 8249–8267 (2014). [DOI] [PubMed] [Google Scholar]
- 25. Hirano, T. et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non‐small‐cell lung cancer. Oncotarget 6, 38789–38803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cross, D.A.E. et al. AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046–1061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delord, J.‐P. et al. Phase I dose‐escalation and ‐expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF‐mutant melanoma. Clin. Cancer Res. 23, 5339–5348 (2017). [DOI] [PubMed] [Google Scholar]
- 28. Yeo, W.‐L. et al. Erlotinib at a dose of 25 mg daily for non‐small cell lung cancers with EGFR mutations. J. Thorac. Oncol. 5, 1048–1053 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Greig, M.J. et al. Effects of activating mutations on EGFR cellular protein turnover and amino acid recycling determined using SILAC mass spectrometry. Int. J. Cell Biol. 2015, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Renner, A. , Burotto, M. & Rojas, C. Immune checkpoint inhibitor dosing: can we go lower without compromising clinical efficacy? J. Global Oncol. 5, 1–5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1