

Abstract
The inhibitor of anaplastic lymphoma kinase (ALK) crizotinib significantly increases survival in patients with ALK‐positive non‐small cell lung cancer (NSCLC). When evaluating crizotinib pharmacokinetics (PKs) in patients taking the standard flat oral dose of 250 mg b.i.d., interindividual PK variability is substantial and patient survival is lower in the quartile with the lowest steady‐state trough plasma concentrations (Cmin,ss), suggesting that concentrations should be monitored and doses individualized. We investigated whether the CYP3A inhibitor cobicistat increases Cmin,ss of the CYP3A substrate crizotinib in patients with low exposure. Patients with ALK‐positive NSCLC of our outpatient clinic treated with crizotinib were enrolled in a phase I trial (EudraCT 2016‐002187‐14, DRKS00012360) if crizotinib Cmin,ss was below 310 ng/mL and treated with cobicistat for 14 days. Crizotinib plasma concentration profiles were established before and after a 14‐day co‐administration of cobicistat to construct the area under the plasma concentration‐time curve in the dosing interval from zero to 12 hours (AUC0–12). Patients were also monitored for adverse events by physical examination, laboratory tests, and 12‐lead echocardiogram. Enrolment was prematurely stopped because of the approval of alectinib, a next‐generation ALK‐inhibitor with superior efficacy. In the only patient enrolled, cobicistat increased Cmin,ss from 158 ng/mL (before cobicistat) to 308 ng/mL (day 8) and 417 ng/mL (day 14 on cobicistat), concurrently the AUC0–12 increased by 78% from 2,210 ng/mL*h to 3,925 ng/mL*h. Neither safety signals nor serious adverse events occurred. Pharmacoenhancement with cobicistat as an alternative for dose individualisation for patients with NSCLC with low crizotinib exposure appears to be safe and is cost‐effective and feasible.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Crizotinib is an oral inhibitor of anaplastic lymphoma kinase, prolonging progression‐free survival and overall survival in patients with non‐small cell lung cancer. There is considerable variability in exposure and the lowest exposure quartile is associated with worse outcome.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Does inhibition of CYP3A, the enzyme metabolizing crizotinib, significantly increase crizotinib exposure and is it safe?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Experience from a single case suggests that cobicistat can substantially and cost‐effectively boost crizotinib exposure in patients with low plasma concentrations.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These data expand the concept of pharmacoenhancement into the indication of oncology. Inhibition of CYP3A‐mediated drug metabolism can increase plasma crizotinib concentration and hence possibly improve outcome.
Non‐small cell lung cancer (NSCLC) is the predominant type of lung cancer and a leading cause of death worldwide. Anaplastic lymphoma kinase (ALK) is a druggable driver mutation of NSCLC. 1 Targeting ALK with the small molecule inhibitor crizotinib demonstrated significant clinical benefit improving progression‐free survival, overall response rate (ORR), lung cancer symptoms, and global quality of life. 2 , 3 Oral crizotinib exhibits substantial variability in steady‐state plasma trough concentrations (Cmin,ss), associated with a concentration‐dependent variability in ORR 4 ; the ORR was only 24–47% in the quartile with the lowest Cmin,ss (< 310 ng/mL) as opposed to 60–75% in the quartile with the highest Cmin,ss. 4 The benefit in ORR translates into longer progression‐free survival of patients with higher exposure. The model suggested a higher hazard in the lowest quartile vs. the combined upper three quartiles with a hazard ratio of 3.2 (90% confidence interval: 1.62–6.36). 4 The high rate of isolated intracranial disease progression under crizotinib has been linked to reduced local central nervous concentrations due to poor penetration. 5 , 6 Based on preclinical and clinical considerations, a lower effective plasma concentration limit of 235 ng/mL for Cmin,ss has been proposed. 7 Interestingly, no exposure‐response relationship was observed for adverse events related to respiratory or liver function, albeit a dose reduction strategy is recommended in the drug label. 8
In patients experiencing disease progression, an ALK mutation explaining crizotinib resistance is found in only 30%. 9 Subtherapeutic exposure is a possible explanation for some of the remaining cases, suggesting that fixed standard doses might not meet the needs of these patients and exposure should be monitored early in therapy. Crizotinib is the primary active circulating moiety. 8 , 10 Its lactam metabolite (PF‐06260182), which consists of two diastereoisomers, is formed by CYP3A‐dependent oxidation but does not contribute significanty to pharmacological activity despite ALK‐inhibiting activity (4–8%). 8 , 10 Crizotinib exposure critically depends on the highly variable CYP3A activity: 8 , 10 , 11 Co‐administration of CYP3A‐inducing rifampin decreases the area under the concentration‐time curve (AUC) by 72% and, conversely, the strong CYP3A inhibitor ketoconazole increases crizotinib AUC by 220%. 8 , 12
Therefore, we screened patients with ALK‐positive metastasized NSCLC for subtherapeutic drug levels. Patients within the lowest quartile of exposure (i.e., Cmin,ss < 310 ng/mL) were eligible to participate in a pharmacokinetic (PK) drug trial and were to receive the CYP3A inhibitor cobicistat to determine whether pharmacoenhancement will boost crizotinib exposure in the range of patients previously reported to have a better outcome.
METHODS
Study design
We measured Cmin,ss in patients with metastasized ALK‐positive NSCLC who had received crizotinib for at least 2 weeks as standard of care. Patients with crizotinib Cmin,ss < 310 ng/mL were eligible to participate in the trial. We obtained a steady‐state crizotinib PK profile before starting oral cobicistat 150 mg q.d. (day 1) and after treatment with cobicistat for 14 days (day 15). Additionally, crizotinib Cmin,ss was measured after 7 days of treatment (day 8; Figure 1 ).
Figure 1.
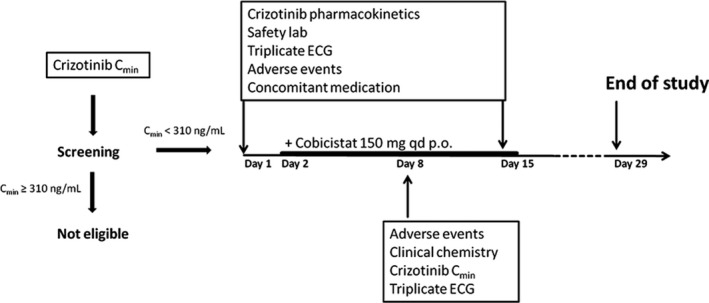
Crizotinib trough concentrations (Cmin,ss) were measured at the start of the trial. Patients with Cmin,ss < 310 ng/mL were eligible for cobicistat treatment. On day 1 and day 15 we collected pharmacokinetic (PK) samples over the 12‐hour dosing interval, safety laboratory, and triplicate echocardiogram (ECG). Oral treatment with 150 mg cobicistat q.d. was started on day 2. A safety visit was scheduled on day 8 and day 29.
Regulatory and ethical requirements
Before enrollment of the first participant, the trial protocol was approved by the national competent authority (Bundesinstitut für Arzneimittel und Medizinprodukte, Bonn, Germany) and the responsible Ethics Committee of the Medical Faculty of Heidelberg University. The trial was equally registered in the European clinical trial database (EudraCT 2016‐002187‐14) and the German clinical trial register (DRKS00012360). The trial was conducted at the Department of Clinical Pharmacology and Pharmacoepidemiology, which is certified according to DIN EN ISO 9001. We followed the principles of the Declaration of Helsinki (version 2013; Fortaleza, Brazil), the guidelines of the International Conference on Harmonisation, the principles of good clinical practice, the corresponding European regulation, and all national legal requirements and regulations.
Study population
Patients of either sex, ≥ 18 years with histologically confirmed diagnosis of ALK‐positive NSCLC were eligible if they were prescribed a standard of care treatment with oral crizotinib (Xalkori; Pfizer), 250 mg b.i.d. for at least 14 days by their treating oncologist. Only patients with an Eastern Cooperative Oncology Group (ECOG) performance score of 0–2, a life expectancy > 12 weeks, and adequate renal, hematologic, and liver functions were considered. Patients with brain or leptomeningeal metastases were allowed, if asymptomatic. Patients had to be able to understand and comply with the trial interventions and voluntarily sign the informed consent form after full explanation of the trial before enrollment.
Main exclusion criertia were therapy‐associated grade 3 or higher toxicity that has not shown improvement and was considered to interfere with current study medication, patients with corrected QT Fridericia’s formula (QTcF) > 470 ms, patients with symptomatic bradycardia or heart rate < 50 bpm without symptoms, aspartate aminotransferase and/or alanine aminotransferase > 2.5‐fold the upper limit of normal (> 5‐fold upper limit of normal in the presence of hepatic metastases), estimated glomerular filtration rate according to the Cockcroft and Gault formula of ≤ 30 mL/min, contraindication against crizotinib, as stated in the German Xalkori drug label, especially the intake of sensitive CYP3A substrates or CYP3A inhibitors/inducers, co‐administration of QT‐prolonging drugs, and known intolerance of crizotinib or any additives. 8
Investigational medicinal products
The patients took the commercially available 250 mg crizotinib (Xalkori; Pfizer) tablets provided by their local pharmacy and the corresponding batch number was registered. Commercially available cobicistat (Tybost; Gilead) 150 mg tablets were provided by the Pharmacy of Heidelberg University Hospital.
Blood sampling and blood handling
Blood was drawn after catheterisation of an antecubital vein predose, and 0.5, 1, 2, 2.5, 3, 4, 6, 8, 10, and 12 hours after oral intake of crizotinib. Samples were centrifuged (3,600 rpm, 10 minutes, 4°C) within 10 minutes after sampling, and the supernatant was stored at −20°C until quantification.
Quantification of crizotinib
Crizotinib plasma concentrations were analyzed using high‐performance liquid chromatography (HPLC) coupled to tandem mass spectrometry (Thermo Surveyor LC plus coupled to a Thermo TSQ 7000 triple quadrupole mass spectrometer, Dreieich, Germany). Human plasma samples (100 µL) were spiked with the internal standard 13C2,2H5‐crizotinib. Borate buffer (0.2 M/pH 9.0) was added. After vortexing, tert‐butyl methyl ether was added and the samples were placed on a rotating shaker. Subsequently the samples were centrifuged (3,000 g). The supernatants were transferred into glass tubes and dried under nitrogen. The residues were reconstituted in HPLC mobile phase solution and analyzed by HPLC coupled to tandem mass spectrometry. Extracts were chromatographed on a reversed‐phase column (Synergi Max‐RP; Phenomenex, Aschaffenburg, Germany) using an eluent gradient composed of acetonitrile and aqueous ammonium acetate. After positive electrospray ionization, mass‐to‐charge transitions of m/z 450 > 259 (crizotinib) and m/z 457 > 266 (13C2,2H5‐crizotinib) were monitored for quantification in the range of 1 to 5,000 ng/mL. The assay fulfilled all criteria of the US Food and Drug Administration (FDA) 13 and the European Medicines Agency (EMA) 14 guidelines for bioanalytical method validation. The lower limit of quantification of crizotinib was 1 ng/mL. Samples were analyzed with within‐batch and batch‐to‐batch accuracy (precision) varying between 95.2 and 104.6% (1.1 to 5.7%).
Electrocardiography
Triplicate 12‐lead echocardiograms were recorded predose at day 1 (before cobicistat), day 8, and day 15 (during cobicistat). We used QTcF.
Pharmacokinetics and statistics
The crizotinib AUC0–12 was constructed using Kinetica 5.0 (Thermo Fischer Scientific, Waltham, MA) through noncompartmental analysis. Descriptive statistics of the crizotinib baseline measurements during screening were calculated using Prism 6.07.
RESULTS
Population
From a total of 7 screened patients, 5 had Cmin,ss < 310 ng/mL (Figure 2a ); 2 of them declined to participate, 2 could not be included because of pre‐existing conditions leaving one 66‐year‐old male patient who participated in the trial. Diagnosis of metastasized (bone and retroperitoneal lymph nodes) ALK‐positive adenocarcinoma occurred 13 months prior to inclusion and first‐line treatment with crizotinib was started 10 months after undergoing palliative‐analgetic radiotherapy. No further patient was enrolled and the trial was prematurely stopped because of changes in the standard of care after reports of superior responses to the next‐generation ALK‐inhibitor alectinib. 15
Figure 2.
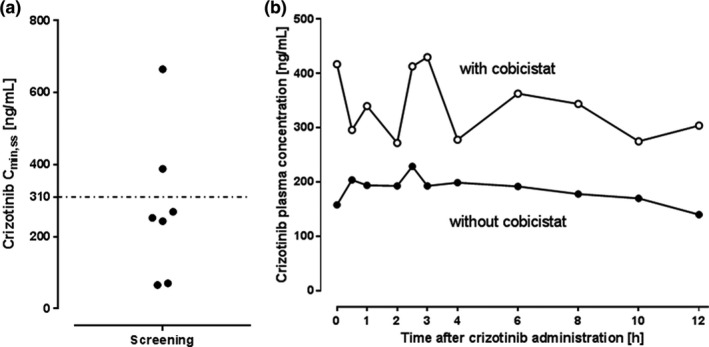
Crizotinib plasma concentrations before and during intervention. (a) Plasma crizotinib trough concentrations (Cmin,ss) of seven patients with anaplastic lymphoma kinase (ALK)‐positive non‐small cell lung cancer (NSCLC) treated with crizotinib. The dashed line at 310 ng/mL denotes the limit of plasma levels considered too low. (b) Plasma concentration‐time profile of crizotinib with (solid circles) and without (open circles) cobicistat in a 66‐year‐old male patients with ALK‐positive NSCLC at steady‐state.
Crizotinib plasma concentrations
Mean crizotinib Cmin,ss at screening was 279 ± 77 ng/mL (Figure 2 ). In the enrolled patient, crizotinib Cmin,ss increased from 158 ng/mL (day 1) to 308 ng/mL (day 8) and 417 ng/mL (day 15) after adding cobicistat to the treatment regimen. The concentration‐time profile is shown in Figure 2b ; the crizotinib AUC0–12 at baseline was 2,210 ng/mL*h and 3,925 ng/mL*h after 14 days of cobicistat treatment (AUC0–12 ratio: 1.78).
Safety
No safety signals occurred, no significant laboratory changes except a grade I increase in transaminases were observed, heart rate was 56 bpm on day 1 and 54 bpm on day 15. QTcF was 394 ms ± 1.7 before cobicistat treatment, 410 ms ± 6.9 on day 8, and 413 ms ± 8.2 on day 15 of cobicistat treatment.
DISCUSSION
In our unselected NSCLC population, underexposed patients were remarkably frequent (71%), suggesting that therapeutic drug monitoring soon after treatment initiation might help identifying suboptimum exposure and reaching effective concentrations more quickly. No inducers of crizotinib metabolism were identified in their comedication, so that reasons may be incomplete adherence to the drug regimen, differences between our unselected NSCLC population and the population from the reported trials, or coincidence. Our data also demonstrate that pharmacoenhancement with cobicistat is feasible and cost‐effective, and may be safe. Instead of increasing dosage to achieve target levels, as proposed for other tyrosine kinase inhibitors (e.g., sorafenib, sunitinib, pazopanib, or axitinib), 7 we chose to inhibit CYP3A‐dependent metabolism. This strategy increased Cmin,ss by 164% and AUC by 78%. This order of magnitude is congruent with the 220% AUC increase observed in a single dose healthy volunteer drug interaction trial with ketoconazole. 12 The monthly costs of using cobicistat are ~ 1% of the monthly costs of a 75% crizotinib dose increase needed to achieve a comparable increase in AUC (~ 45 EUR vs. 5,400 EUR).
We did not observe clinically significant adverse events or exaggerated crizotinib toxicity in the single patient tested. Because of the favorable safety profile, cobicistat appears more suitable for pharmacoenhancement than clarithromycin or azole antimycotics. 16
Concurrently, the potential risks of chronic CYP3A inhibition in patients with cancer must be considered because this population is often exposed to supportive medication, such as opioids or non‐opioid analgesics, antiemetics, and anticoagulants that can be boosted as well. Although, in general, CYP3A‐based drug interactions caused by a CYP3A inhibitor are manageable, as known from the population of patients with HIV, it certainly adds complexity to patient management.
Limitations
This clinical trial was stopped early for ethical reasons after approval of the next‐generation ALK inhibitor alectinib. We did not amend the trial to incorporate alectinib, because the exposure‐response analysis found no statistically significant relationship between exposure and overall survival following alectinib treatment. 17 In addition, whereas CYP3A contributes 40–50% to alectinib clearance, the resulting metabolite M4 is equipotently active and has a similar protein binding. Moreover, the administration of the CYP3A inhibitor posaconazole leads to no relevant PK interaction because the combined AUC of alectinib and the M4 metabolite only increases by 36%. 18 Therefore, all available evidence currently suggests that pharmacoenhancement of alectinib is not promising.
Hence, we can only report one case, which clearly confirms the concept but cannot provide insight into consistency and extent of this interaction or its safety. This concept could be applied to other tyrosine kinase inhibitors in cases where the victim drug’s clearance significantly depends on CYP3A activity, major active metabolites are not critically reduced by inhibition of this pathway, and where a close relationship between plasma exposure and response is established (e.g., in the cases of imatinib, sunitinib, or axitinib). While conducting our trial, a similar approach was reported in a patient with renal cancer under the treatment with axitinib, in whom excessive cobicistat doses helped achieving therapeutic axitinib exposure. 19 In addition, whereas we demonstrated feasibility and safety of pharmacoenhancement, the 14‐day period was too short to answer the question of whether increasing drug exposure will improve antitumor reponse to the drug.
In conclusion, administration of cobicistat may be a promising and nonexpensive strategy to increase subtherapeutic exposure of crizotinib suggesting that this approach might also work with other tyrosine kinase inhihibitors because many of them are substantially cleared by CYP3A. 20
Funding
This project received funding from the Stiftung für Krebs‐ und Scharlachforschung (Grant No. AZ3542.2 to N.H.). The project was partly funded by the Dietmar‐Hopp‐Stiftung (PROMISE: PRedictability of Outcome based on iMmunological sIgnatureS in lung cancEr – an explorative investigation).
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
N.H., F.B., P.C., M.T., A.B., G.M., J.B., and W.E.H. wrote the manuscript. N.H., F.B., M.T., and W.E.H. designed the research. N.H., F.B., P.C., M.T., A.B., G.M., and W.E.H. performed the research. N.H., F.B., P.C., M.T., A.B., G.M., J.B., and W.E.H. analyzed the data. J.B. contributed new reagents/analytical tools.
Acknowledgments
The authors would like to thank Marlies Schnetz, Peter Rose, Kathrin Foerster, and Andrea Deschlmayr for valuable assistance during the trial and Claudia Marquart for proof reading. Open access funding enabled and organized by ProjektDEAL.
Data Availability Statement
Data are available for sharing upon reasonable request.
References
- 1. Kwak, E.L. et al. Anaplastic lymphoma kinase inhibition in non‐small‐cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shaw, A.T. et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N. Engl. J. Med. 368, 2385–2394 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Frampton, J.E. Crizotinib: a review of its use in the treatment of anaplastic lymphoma kinase‐positive, advanced non‐small cell lung cancer. Drugs 73, 2031–2051 (2013). [DOI] [PubMed] [Google Scholar]
- 4. U.S. Food and Drug Administration . Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000ClinPharmR.pdf (2011). Accessed July 9, 2020.
- 5. Chun, S.G. et al. Isolated central nervous system progression on crizotinib: an achilles heel of non‐small cell lung cancer with EML4‐ALK translocation? Cancer Biol. Ther. 13, 1376–1386 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Camidge, D.R. Taking aim at ALK across the blood‐brain barrier. J. Thorac. Oncol. 8, 389–390 (2013). [DOI] [PubMed] [Google Scholar]
- 7. Verheijen, R.B. et al. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin. Pharmacol. Ther. 102, 765–776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. European Medicines Agency . European public assessment report (EPAR) for Xalkori https://www.ema.europa.eu/en/medicines/human/EPAR/xalkori#product‐information‐section (2020). Accessed July 9, 2020.
- 9. Shaw, A.T. & Engelman, J.A. ALK in lung cancer: past, present, and future. J. Clin. Oncol. 31, 1105–1111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnson, T.R. et al. Metabolism, excretion and pharmacokinetics of [14C]crizotinib following oral administration to healthy subjects. Xenobiotica 45, 45–59 (2015). [DOI] [PubMed] [Google Scholar]
- 11. Dresser, G.K. , Spence, J.D. & Bailey, D.G. Pharmacokinetic‐pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin. Pharmacokinet. 38, 41–57 (2000). [DOI] [PubMed] [Google Scholar]
- 12. Xu, H. et al. The effects of ketoconazole and rifampin on the single‐dose pharmacokinetics of crizotinib in healthy subjects. Eur. J. Clin. Pharmacol. 71, 1441–1449 (2015). [DOI] [PubMed] [Google Scholar]
- 13. U.S. Food and Drug Administration . Bioanalytical method validation https://www.fda.gov/downloads/drugs/guidances/ucm070107.pdf (2018). Accessed June 30, 2020.
- 14. European Medicines Agency . Guideline on bioanalytical method validation http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (2012). Accessed June 30, 2020. [DOI] [PubMed]
- 15. Peters, S. et al. Alectinib versus crizotinib in untreated ALK‐positive non‐small‐cell lung cancer. N. Engl. J. Med. 377, 829–838 (2017). [DOI] [PubMed] [Google Scholar]
- 16. Greenblatt, D.J. & Harmatz, J.S. Ritonavir is the best alternative to ketoconazole as an index inhibitor of cytochrome P450–3A in drug‐drug interaction studies. Br. J. Clin. Pharmacol. 80, 342–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morcos, P.N. et al. Exposure‐response analysis of alectinib in crizotinib‐resistant ALK‐positive non‐small cell lung cancer. Cancer Chemother. Pharmacol. 82, 129–138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morcos, P.N. et al. Clinical drug‐drug interactions through cytochrome P450 3A (CYP3A) for the selective ALK inhibitor alectinib. Clin. Pharmacol. Drug Dev. 6, 280–291 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Lubbermann, F.J.E. et al. Boosting axitinib exposure with a CYP3A4 inhibitor, making axitinib treatment personal. Acta Oncol. 56, 1238–1240 (2017). [DOI] [PubMed] [Google Scholar]
- 20. Mikus, G. & Foerster, K.I. Role of CYP3A4 inkinase inhibitor metabolism and assessment of CYP3A4 activity. Transl. Cancer Res. 6 (suppl. 10), S1592–S1599 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available for sharing upon reasonable request.