

Abstract
A new series of quinoline derivatives of combretastatin A-4 have been designed, synthesised and demonstrated as tubulin polymerisation inhibitors. These novel compounds showed significant antiproliferative activities, among them, 12c exhibited the most potent inhibitory activity against different cancer cell lines (MCF-7, HL-60, HCT-116 and HeLa) with IC50 ranging from 0.010 to 0.042 µM, and with selectivity profile against MCF-10A non-cancer cells. Further mechanistic studies suggest that 12c can inhibit tubulin polymerisation and cell migration, leading to G2/M phase arrest. Besides, 12c induces apoptosis via a mitochondrial-dependant apoptosis pathway and caused reactive oxygen stress generation in MCF-7 cells. These results provide guidance for further rational development of potent tubulin polymerisation inhibitors for the treatment of cancer.
Highlights
A novel series of quinoline derivatives of combretastatin A-4 have been designed and synthesised.
Compound 12c showed significant antiproliferative activities against different cancer cell lines.
Compound 12c effectively inhibited tubulin polymerisation and competed with [3H] colchicine in binding to tubulin.
Compound 12c arrested the cell cycle at G2/M phase, effectively inducing apoptosis and inhibition of cell migration.
Keywords: Combretastatin A-4, quinoline, apoptosis
Graphical Abstract

1. Introduction
Cancer is a disease of an uncontrolled growth and abnormal division of cells, which leads to death. In recent years, targeted antineoplastic agents have become an effective treatment choice for cancer1, with pharmaceutical companies focussing on targeted therapies against different and special cancer types2. Tubulin polymerisation inhibitors represent one of the most well-known and potential examples of such targeted cancer therapies3. Tubulin is a globular protein that performs a substantial function in cell mitosis. Microtubules (MTs), which represent the basic constituents of eukaryotic cell, are cytoskeletons constructed by the association of α- and β-tubulin heterodimers with a head and tail pattern to form hollow cylindrical tubes (nearly 25 nm in diameter)4–7. MTs play a crucial role in many fundamental cellular activities, such as motility, cell formation, cell secretion, signalling, maintenance of cell shape, regulation of intracellular transport and cell division8–10. Due to these multiple functions, microtubule system has become an attractive target for cancer chemotherapy11,12. Disruption of MTs or tubulin dynamics exposes the cell to mitotic arrest of the cell cycle at G2/M phase, and consequently induction of cellular apoptosis13,14. Several microtubule-interfering agents (MIAs) have been identified, e.g. paclitaxel, vincristine, and colchicine that are obtained from the natural products, taxol, vinca and colchicine, respectively. MIAs are known to bind to tubulin at specific binding sites that are classified as taxol, vinca and colchicine sites to either enhance or inhibit tubulin polymerisation.15 For example, microtubule stabilisers, e.g. paclitaxel stimulate microtubule polymerisation16, while microtubule destabilizers, e.g. colchicine, and the vinca alkaloids vinblastine and vincristine inhibit polymerisation of microtubules17.
Significant attention is now focussed on colchicine binding site inhibitors due to their positive impact on ABC-transporter-mediated drug resistance18,19. Combretastatin A-4 (CA-4), (1, Figure 1) has been reported as the most potent antimitotic agent of this family against several tumour cells20. CA-4 was first isolated from the bark of the willow tree Combretum caffrum from South Africa in 198921. CA-4 has a vascular disrupting activity against tumour cell vasculature by preventing blood supply to solid tumour, resulting in apoptosis22–24. Given its structural simplicity, CA-4 has been studied as a lead pharmacophore for deciphering tubulin functions and properties25. Phases II and III clinical studies are currently ongoing with tubulin-targeted drugs24,26. Structure activity relationships (SAR) studies with CA-4 have revealed three important structural features (Figure 1). These include: (i) a 3,4,5-trimethoxy moiety on ring A that is essential for activity; (ii) a cis-configuration of both aromatic rings that is essential for activity (trans-orientation is inactive); (iii) the presence of small substituent on ring B, e.g. methoxy group that is important for activity. The cis-alkene configuration in CA-4 allows the aromatic rings to assume optimal binding orientation for interactions with the colchicine binding site. Unfortunately, the cis configuration of CA-4 has a propensity for undergoing transformation to the inactive trans configuration upon storage and during in vivo metabolism. To overcome this, many structural modifications of CA-4 have been undertaken where the cis double bond is replaced with heterocycles, either monocyclic, such as oxadiazole, isoxazole and imidazole, resulting in compounds, such as 1, 2 and 3 respectively (Figure 1)27–33 or fused heterocyclic, such as pyrazolopyridines34, triazolopyridines35 and triazolothiadiazine derivatives36. These compounds, like CA-4 showed pronounced activity against a panel of cancer cell lines.
Figure 1.

(A) Chemical structure of CA-4 and its analogues; (B) tubulin targeting agents bearing quinoline moiety.
Quinoline derivatives are popular for the treatment of malaria37,38. Moreover, quinoline heterocyclic containing compounds demonstrate potent anticancer activities with different modes of actions, including inhibition of proteasome, tyrosine kinases, and tubulin polymerisation39–41. Previous studies have reported the antiproliferative activity of CA-4, isoCA-4 or chalcone compounds containing quinoline scaffold, either as ring A bioisoster, e.g. 4a23 and 4b42 or ring B bioisoster, e.g. 4c43–45 and 4d46. These compounds demonstrate the potential of the quinoline ring as a template for developing more promising tubulin polymerisation inhibitors and antiproliferative agents.
In this work, we optimised CA-4 into a series of novel hybrid quinoline derivatives as potent tubulin inhibitor, which involves introducing a rigid oxazolone or imidazolone between rings A and B to maintain the cis configuration, as well as targeting the quinolyl moiety (ring B), by varying the electronic substituents effect while maintaining the 3,4,5-trimethoxyphenyl moiety as present in ring A of CA-4 (Figure 2). Following, we synthesised several analogues that constitute two classes of compounds: the oxazolones (Compounds 12a–h) and the imidazolones (Compounds 13a–h). The compounds have been screened for their antiproliferative activities against a variety of cancer cell lines, as well as studied for their mechanism of action. We expect the results to lead to better understanding of the mechanistic mode of the compounds’ activity against tubulin and provide guidance for further development of potent anticancer drugs.
Figure 2.

Structure of CA-4 and our rationalised compounds.
2. Experimental section
2.1. Chemistry
Melting points were determined with a Gallenkamp (London, UK) melting point apparatus and are uncorrected. IR spectra (KBr, cm−1) were recorded on Bruker Vector, 22FT-IR [Fourier Transform Infra-red (FTIR), Germany] spectrometer. Unless otherwise specified, proton (1H) and carbon (13C) NMR spectra were recorded at room temperature in base filtered CDCl3 on a spectrometer operating at 400 & 300 MHz for proton and 100 & 75 MHz for carbon nuclei. The signal due to residual CHCl3 appearing at δ H 7.26 and (CH3)2SO appearing at δ H 2.5 and the central resonance of the CDCl3 “triplet” appearing at δ C 77.0 and for (CD3)2SO “multiplet” appearing at δ C 39.0 were used to reference 1H and 13C NMR spectra, respectively. 1H NMR data are recorded as follows: chemical shift (δ) [multiplicity, coupling constant(s) J (Hz), relative integral] where multiplicity is defined as s = singlet; d = doublet; t = triplet; q = quartette; and m = multiplet or combinations of the above. Elemental analyses were determined using Manual Elemental Analyser Heraeus (Germany) and Automatic Elemental Analyser CHN Model 2400 Perkin Elmer (Waltham, MA, USA) at Microanalytical Centre, Faculty of Science, Cairo University, Egypt. All the elemental analyses results corresponded to calculated values within experimental error. Progress of reactions was monitored by thin-layer chromatography (TLC) using precoated TLC sheets with Ultraviolet (UV) fluorescent silica gel (Merck 60F254), and spots were visualised by iodine vapours or irradiation with UV light (254 nm). All chemicals were purchased from Sigma-Aldrich or Lancaster Synthesis Corporation (UK). Intermediates 6–8a–i were prepared according to reported procedure47,48.
2.1.1. General procedure for preparation of oxazolones (12a–h)
A mixture of N-(3,4,5-trimethoxybenzoyl)glycine (0.30 g, 1.10 mmol) and the appropriate aldehydes 8a–h (1.00 mmol) in acetic anhydride (1 ml) and fused sodium acetate (0.1 g, 1.2 mmol) was heated on an oil bath at 80 °C for 2 h. After cooling down at room temperature the mixture was allowed to stand for 24 h at 0 °C. The precipitate was filtered off and washed three times with ice-cooled ethanol (10 ml), and the product crystallised from ethanol.
2.1.1.1. 4-[(2-Methoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)oxazol-5(4H)-one (12a)
Yellow solid, Yield (81%); m.p. 215–217 °C. IR (KBr): υ = 1776 (C=O), 1621 (C=N), 1599 (C=C) cm−1. 1H NMR (400 MHz, CDCl3) δ: 9.48 (s, 1H, Ar-H), 7.86–7.81 (m, 2H, Ar-H), 7.71–7.67 (m, 2H, Ar-H), 7.45–7.41 (m, 3H, Ar-H), 4.15 (s, 3H, OCH3), 4.00 (s, 9H, 3 OCH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.1, 163.9, 160.0, 153.5, 147.1, 143.1, 142.4, 134.6, 131.5, 128.9, 127.1, 125.2, 124.7, 123.6, 120.1, 118.5, 105.8, 61.1, 56.4, 54.1 ppm. MS (70 eV): m/z (%): 420 (7.79) [M+]; Anal. Calcd for C23H20N2O6: C, 65.71; H, 4.80; N, 6.66. Found: C, 65.64; H, 4.74; N, 6.71
2.1.1.2. 4-[(2-Methoxy-6-methylquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl) oxazol-5(4H)-one (12b)
Pale yellow solid, Yield (79%); m.p. 218–220 °C. 1H NMR (400 MHz, CDCl3) δ: 9.36 (s, 1H, Ar-H), 7.72 (t, J = 4 Hz, 2H, Ar-H), 7.56 (s, 1H, Ar-H), 7.49 (d, J = 8.5 Hz, 1H, Ar-H), 7.41 (s, 2H, Ar-H), 4.11 (s, 3H, OCH3), 3.98 (d, J = 8.0 Hz, 9H, 3OCH3) and 2.51 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.2, 163.8, 159.7, 153.5, 145.7, 143.1, 141.8, 134.4, 134.3, 133.6, 127.8, 126.9, 125.1, 124.1, 120.3, 118.1, 110.0, 105.7, 61.1, 56.4, 53.9, 21.3 ppm. MS (70 eV): m/z (%): 434 (9.79) [M+]; Anal. Calcd for C24H22N2O6: C, 66.35; H, 5.10; N, 6.45. Found: C, 66.29; H, 5.04; N, 6.50.
2.1.1.3. 4-[(2-Methoxy-7-methylquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl) oxazol-5(4H)-one (12c)
Yellow solid, Yield (84%); m.p. 233–235 °C. 1H NMR (400 MHz, CDCl3) δ: 9.50 (s, 1H, Ar-H), 7.76–7.73 (m, 3H, Ar-H),7.45 (s, 2H, Ar-H), 7.30–7.28 (m, 1H, Ar-H), 4.20 (s, 3H, OCH3), 4.00 (d, J = 8.0 Hz, 9H, 3 OCH3), 2.58 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.2, 163.8, 162.6, 160.2, 153.5, 143.1, 142.8, 142.5, 134.3, 128.7, 127.0, 126.3, 123.9, 123.1, 120.2, 117.6, 105.7, 61.1, 56.4, 54.6, 22.1 ppm. MS (70 eV): m/z (%): 434 (6.90) [M+]; Anal. Calcd for C24H22N2O6: C, 66.35; H, 5.10; N, 6.45. Found: C, 66.32; H, 5.05; N, 6.47.
2.1.1.4. 4-[(2-Methoxy-8-methylquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl) oxazol-5(4H)-one (12d)
Yellow solid, Yield (82%); m.p. 236–238 °C. 1H NMR (400 MHz, CDCl3) δ: 9.44 (s, 1H, Ar-H), 7.73 (s, 1H, Ar-H), 7.64 (d, J = 8.0 Hz, 1H, Ar-H), 7.52 (d, J = 6.40 Hz, 1H, Ar-H), 7.39 (s, 2H, Ar-H), 7.32–7.28 (m, 1H, Ar-H), 4.12 (s, 3H, OCH3), 3.98 (s, 9H, 3 OCH3), 2.68 (s, 3H, CH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.2, 163.8, 159.1, 153.7, 146.0, 143.1, 142.7, 135.4, 134.3, 131.6, 126.7, 125.0, 124.3, 124.0, 120.2, 118.0, 105.6, 61.1, 56.3, 53.7, 17.5 ppm. MS (70 eV): m/z (%): 434 (9.50) [M+]; Anal. Calcd for C24H22N2O6: C, 66.35; H, 5.10; N, 6.45. Found: C, 66.30; H, 5.04; N, 6.51.
2.1.1.5. 4-[2,6-Dimethoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)oxazol-5(4H)-one (12e)
Yellow solid, Yield (84%); m.p. 242–244 °C. 1784 (C=O), 1618 (C=N), 1586 (C=C) cm−1. 1H NMR (400 MHz, CDCl3) δ: 9.33 (s, 1H, Ar-H), 7.73 (d, J = 8.0 Hz, 2H, Ar-H), 7.42–7.32 (m, 3H, Ar-H), 7.12 (s, 1H, Ar-H), 4.10 (s, 3H, OCH3), 3.99 (s, 9H, 3 OCH3) 3.93 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.1, 163.8, 158.9, 156.4, 153.5, 143.2, 143.0, 141.1, 134.5, 128.5, 125.7, 124.2, 123.0, 120.2, 118.4, 107.4, 105.9, 61.2, 56.5, 55.6, 53.8 ppm. MS (70 eV): m/z (%): 450 (8.40) [M+]; Anal. Calcd for C24H22N2O7: C, 64.00; H, 4.92; N, 6.22. Found: C, 63.94; H, 4.87; N, 6.28.
2.1.1.6. 4-[(2,7-Dimethoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)oxazol-5(4H)-one (12f)
Yellow solid, Yield (84%); m.p. 221–223 °C. 1H NMR (400 MHz, CDCl3) δ: 9.39 (s, 1H, Ar-H), 7.70–7.66 (m, 2H, Ar-H), 7.38 (s, 2H, Ar-H), 7.16 (s, 1H, Ar-H), 7.04 (d, J = 8.0 Hz, 1H, Ar-H), 4.11 (s, 3H, OCH3), 3.97 (s, 9H, 3OCH3) 3.95 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.4, 163.3, 162.8, 160.7, 153.5, 149.7, 142.8, 142.0, 133.3, 130.2, 124.4, 120.4, 120.1, 117.2, 115.9, 106.4, 105.5, 61.2, 56.2, 55.6, 53.9 ppm. MS (70 eV): m/z (%): 450 (7.30) [M+]; Anal. Calcd for C24H22N2O7: C, 64.00; H, 4.92; N, 6.22. Found: C, 63.96; H, 4.96; N, 6.25.
2.1.1.7. 4-[(7-Isopropoxy-2-methoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxy phenyl)oxazol-5(4H)-one (12g)
Yellow solid, Yield (77%); m.p. 227–229 °C. 1H NMR (400 MHz, CDCl3) δ ppm: 9.41 (s, 1H, Ar-H), 7.74–7.67 (m, 2H, Ar-H), 7.41 (s, 2H, Ar-H),7.16–7.00 (m, 2H, Ar-H), 4.87–4.76 (m, 1H, OCH-), 4.12 (s, 3H, OCH3), 3.98 (s, 9H, 3 OCH3), 1.45 (d, J = 4.0 Hz, 6H, 2 CH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.4, 163.1, 161.2, 160.7, 153.5, 149.5, 142.9, 141.9, 133.2, 130.3, 124.6, 120.4, 119.9, 118.2, 115.7, 107.9, 105.5, 70.3, 61.3, 56.4, 53.9, 22.0 ppm. MS (70 eV): m/z (%): 478 (9.50) [M+]; Anal. Calcd for C26H26N2O7: C, 65.26; H, 5.48; N, 5.85. Found: C, 65.21; H, 5.45; N, 5.91.
2.1.1.8. 4-[(7-(Benzyloxy)-2-methoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxy phenyl)oxazol-5(4H)-one (12h)
Pale yellow solid, Yield (73%); m.p. 247–249 °C. 1H NMR (400 MHz, CDCl3) δ: 9.43 (s, 1H, Ar-H), 7.72 (t, J = 8 Hz, 2H, Ar-H), 7.51–7.35 (m, 6H, Ar-H), 7.28–7.26 (m, 2H, Ar-H), 7.14 (d, 1H, J = 8.0 Hz, 1H, Ar-H), 5.21 (s, 2H, OCH2−), 4.12 (s, 3H, OCH3), 3.98 (s, 9H, 3 OCH3) ppm. 13C NMR (100 MHz, CDCl3) δ: 167.4, 163.3, 161.9, 160.8, 153.5, 149.4, 142.9, 141.9, 136.2, 133.5, 130.2, 128.7, 128.3, 127.7, 124.4, 120.4, 120.3, 117.5, 116.0, 107.5, 105.6, 70.3, 61.3, 56.4, 53.9 ppm. MS (70 eV): m/z (%): 526 (7.30) [M+]; Anal. Calcd for C30H26N2O7: C, 68.43; H, 4.98; N, 5.35. Found: C, 68.39; H, 4.92; N, 5.41.
2.1.2. General procedure for preparation of (13a–h)
The appropriate oxazolones 12a–h (1 mmol) was stirred and heated under reflux in ethanol (10 ml) containing ammonium hydroxide (10 ml), and the reaction monitored by TLC. After completion of the reaction in 24 h, the solvent was concentrated and cooled, and the precipitate was filtered off and crystallised from ethanol.
2.1.2.1. 5-[(2-Methoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)-3,5-dihydro-4H-imidazol-4-one (13a)
Yellow solid, Yield (81%); m.p. 230–232 °C. IR (KBr): υ = 3222 (NH), 1709 (C=O), 1642 (C=N), 1615, 1589 (C=C) cm−1. 1H NMR (400 MHz, DMSO-d6) δ: 12.22 (s, 1H, exch., NH), 9.69 (s, 1H, Ar-H), 8.03 (d, J = 8.0 Hz, 1H, Ar-H), 7.80–7.70 (m, 2H, Ar-H), 7.59 (s, 2H, Ar-H), 7.49 (t, J = 8.0 Hz, 1H, Ar-H), 7.28 (s, 1H, Ar-H), 4.11 (s, 3H, OCH3), 3.94 (s, 6H, 2OCH3), 3.80 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 187.4, 165.0, 164.2, 161.3, 157.9, 153.5, 146.7, 140.9, 132.2, 128.9, 126.7, 125.2, 123.9, 118.4, 108.7, 104.2, 60.3, 56.3, 54.1 ppm. MS (70 eV): m/z (%): 419 (5.40) [M+]; Anal. Calcd for C23H21N3O5: C, 65.86; H, 5.05; N, 10.02. Found: C, 65.81; H, 4.99; N, 10.09.
2.1.2.2. 5-[(2-Methoxy-6-methylquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)-3,5-dihydro-4H-imidazol-4-one (13b)
Yellow solid, Yield (81%); m.p. 223–225 °C. 1HNMR (400 MHz, DMSO-d6) δ: 12.18 (s, 1H, exch., NH), 9.55 (s, 1H, Ar-H), 7.74–7.54 (m, 5H, Ar-H), 7.26–7.24 (d, J = 6.6 Hz, 1H, Ar-H), 4.07 (s, 3H, OCH3), 3.93 (s, 6H, OCH3), 3.80 (s, 3H, OCH3), 2.47 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 172.0, 162.2, 156.0, 153.7, 144.6, 142.6, 142.4, 141.2, 134.4, 133.4, 128.1, 126.8, 125.4, 123.2, 119.3, 116.2, 110.0, 106.2, 60.7, 56.8, 54.2, 21.3 ppm. MS (70 eV): m/z (%): 433 (5.60) [M+]; Anal. Calcd for C24H23N3O5: C, 66.50; H, 5.35; N, 9.69. Found: C, 66.41; H, 5.28; N, 9.75.
2.1.2.3. 5-[(2-Methoxy-7-methylquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)-3,5-dihydro-4H-imidazol-4-one (13c)
Yellow solid, Yield (84%); m.p. 236–238 °C. IR (KBr): υ = 3215 (NH), 1711 (C=O), 1639 (C=N), 1590 (C=C) cm−1. 1H NMR (400 MHz, DMSO-d6) δ: 12.17 (s, 1H, exch. NH), 9.63 (s, 1H, Ar-H), 7.92 (d, J = 8.0 Hz, 1H, Ar-H), 7.58 (s, 3H, Ar-H), 7.32 (d, J = 8 Hz, 1H, Ar-H), 7.27 (s, 1H, Ar-H), 4.09 (s, 3H, OCH3), 3.94 (s, 6H, 2OCH3), 3.80 (s, 3H, OCH3), 2.53 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 162.6, 162.5, 160.3, 153.9, 142.2, 141.5, 129.1, 127.1, 126.3, 123.6, 118.3, 115.8, 105.6, 60.8, 56.5, 54.2, 22.0 ppm. MS (70 eV): m/z (%): 433 (8.40) [M+]; Anal. Calcd for C24H23N3O5: C, 66.50; H, 5.35; N, 9.69. Found: C, 66.44; H, 5.32; N, 9.72.
2.1.2.4. 5-[(2-Methoxy-8-methylquinolin-3-yl)methylene]-2–(3,4,5-trimethoxy phenyl)-3,5-dihydro-4H-imidazol-4-one (13d)
Yellow solid, Yield (82%); m.p. 233–235 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.16 (s, 1H, exch., NH), 9.59 (s, 1H, Ar-H), 7.80 (s, 1H, Ar-H), 7.70 (d, J = 8 Hz, 1H, Ar-H), 7.59–7.56 (m, 3H, Ar-H), 7.29 (s, 1H, Ar-H), 4.10 (s, 3H, OCH3), 3.95 (s, 6H, 2OCH3), 3.80 (s, 3H, OCH3), 2.49 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 161.9, 158.2, 153.3, 142.2, 141.6, 141.5, 134.2, 131.3, 126.7, 124.8, 124.5, 118.6, 115.4, 105.3, 60.2, 56.1, 53.5, 17.1 ppm. MS (70 eV): m/z (%): 433 (7.12) [M+]; Anal. Calcd for C24H23N3O5: C, 66.50; H, 5.35; N, 9.69. Found: C, 66.47; H, 5.30; N, 9.73.
2.1.2.5. 5-[(2,6-Dimethoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)-3,5-dihydro-4H-imidazol-4-one (13e)
Yellow solid, Yield (84%); m.p. 253–255 °C. 1HNMR (400 MHz, DMSO-d6) δ: 12.03 (s, 1H, exchangeable, NH), 9.50 (s, 1H, Ar-H), 7.83–7.77 (m, 1H, Ar-H), 7.53 (s, 2H, Ar-H), 7.48 (s, 1H, Ar-H), 7.20–7.05 (m, 2H, Ar-H), 4.04 (s, 3H, OCH3), 3.90 (s, 9H, 3 OCH3), 3.79 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 164.6, 161.9, 157.1, 156.9, 154.1, 142.8, 141.9, 140.0, 128.4, 125.1, 124.1, 118.9, 110.0, 109.3, 108.0, 105.3, 60.7, 57.0, 56.2, 54.4 ppm. MS (70 eV): m/z (%): 449 (8.35) [M+]; Anal. Calcd for C24H23N3O6: C, 64.16; H, 5.16; N, 9.35. Found: C, 64.11; H, 5.12; N, 9.31.
2.1.2.6. 5-[(2,7-Dimethoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxyphenyl)-3,5-dihydro-4H-imidazol-4-one (13f)
Yellow solid, Yield (86%); m.p. 241–243 °C. 1HNMR (400 MHz, DMSO-d6) δ: 12.10 (s, 1H, exch., NH), 9.57 (s, 1H, Ar-H), 7.86 (d, J = 8.0 Hz, 1H, Ar-H), 7.53 (s, 2H, Ar-H), 7.23 (s, 1H, Ar-H), 7.15–7.08 (m, 2H, Ar-H), 4.07 (s, 3H, OCH3), 3.92 (s, 6H, 2OCH3), 3.79 (s, 3H, OCH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 163.9, 162.6, 161. 5, 158.3, 153.3, 149.0, 140.7, 140.3, 129.9, 118.7, 118.2, 117.4, 106.3, 105.7, 104.1, 60.1, 56.3, 55.7, 54.1 ppm. MS (70 eV): m/z (%): 449 (5.65) [M+]; Anal. Calcd for C24H23N3O6: C, 64.16; H, 5.16; N, 9.35. Found: C, 64.13; H, 5.10; N, 9.29.
2.1.2.7. 5-((7-Isopropoxy-2-methoxyquinolin-3-yl)methylene)-2–(3,4,5-trimethoxy phenyl)-3,5-dihydro-4H-imidazol-4-one (13g)
Yellow solid, Yield (77%); m.p. 238–240 °C. 1HNMR (400 MHz, DMSO-d6) δ: 12.19 (s, 1H, exch.,NH), 9.61 (s, 1H, Ar-H), 7.92 (d, J = 8.0 Hz, 1H, Ar-H), 7.58 (s, 2H, Ar-H), 7.28 (s, 1H, Ar-H), 7.17 (s, 1H, Ar-H), 7.09–7.07 (d, J = 8.0 Hz, 1H, Ar-H), 4.90–4.83 (m, 1H, OCH), 4.09 (s, 3H, OCH3), 3.94 (s, 6H, 2OCH3), 3.79 (s, 3H, OCH3), 1.37 (s, 3H, CH3) 1.36 (s, 3H, CH3) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 164.2, 161.3, 156.7, 154.2, 153.5, 142.0, 140.9, 139.7, 128.1, 124.8, 122.4, 118.6, 109.2, 108.5, 104.3, 69.8, 60.3, 56.3, 53.9, 21.7 ppm. MS (70 eV): m/z (%): 477 (9.25) [M+]; Anal. Calcd for C26H27N3O6: C, 65.40; H, 5.70; N, 8.80. Found: C, 65.35; H, 5.66; N, 8.86.
2.1.2.8. 5-[(7-(Benzyloxy)-2-methoxyquinolin-3-yl)methylene]-2–(3,4,5-trimethoxy phenyl)-3,5-dihydro-4H-imidazol-4-one (13h)
Yellow solid, Yield (73%); m.p. 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.16 (s, 1H, exch., NH), 9.62 (s, 1H, Ar-H), 7.96–7.94 (d, J = 8 Hz, 1H, Ar-H), 7.58–7.28 (m, 10H, ArH), 5.30 (s, 2H, OCH2-), 4.09 (s, 3H, OCH3), 3.94 (s, 6H, 2OCH3), 3.79 (s, 3H, OCH3) ppm. 13C NMR (400 MHz, DMSO-d6) δ: 163.9, 161.6, 158.5, 153.5, 148.8, 140.7, 140.3, 136.6, 130.2, 128.5, 128.0, 127.8, 118.9, 118.5, 117.8, 107.2, 105.9, 104.0, 69.7, 60.4, 56.3, 54.0 ppm. MS (70 eV): m/z (%): 525 (6.75) [M+]; Anal. Calcd for C30H27N3O6: C, 68.56; H, 5.18; N, 8.00. Found: C, 68.51; H, 5.13; N, 8.05.
2.2. Biochemical evaluation of activity
All biochemical assays were performed in triplicate on at least three independent occasions for the determination of mean values.
2.2.1. Cell culture
The four human tumour cell lines MCF-7, HCT-116, HL-60 and HeLa used in this study were obtained from the VACSERA (Giza, Egypt) cell culture unit that were originally acquired from ATCC (Manassas, VA, USA). All the human tumour cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% foetal bovine serum, 2 mM L-glutamine and 100 µg/mL penicillin/streptomycin. Cells were maintained at 37˚C in 5% CO2 in a humidified incubator. All cells were sub-cultured 3 times/week by trypsinisation using TrypLE Express (1X).
2.2.2. Cell viability assay
The quinoline compounds were evaluated for antiproliferative effect using the MTT viability assay of four cancer cell lines (MCF-7, HCT-116, HL-60 and HeLa) and normal breast cells MCF-10A to calculate the relative IC50 values for each compound. Cells were seeded in triplicate in 96-well plates at a density of 10 × 103 cells/ml in a total volume of 200 µl per well. 0.1% of DMSO was used as a vehicle control. Following, the cells were treated with 2 µl test compound (from stock solutions in ethanol) to furnish the concentration range of study, 1 nM to 50 µM, and re-incubated for a further 72 h. The culture medium was then removed, and the cells washed with 100 µL phosphate buffered saline (PBS) and 50 µL MTT added, to reach a final concentration of 1 mg/mL. Cells were incubated for 2 h in darkness at 37 °C. Solubilisation was begun through the addition of 200 ml DMSO, and the cells maintained at room temperature in darkness for 20 min to ensure thorough colour diffusion before reading the absorbance. Plates were incubated for 72 h at 37 °C + 5% CO2. The MTT (5 mg/mL in PBS) was added and incubated for another 4 h, and the optical density was detected with a microplate reader at 570 nm. Results were expressed as percentage viability relative to vehicle control (100%). Dose response curves were plotted and IC50 values (concentration of drug resulting in 50% reduction in cell survival) were obtained using the commercial software package Prism (GraphPad Software, Inc., La Jolla, CA, USA). All the experiments were repeated in at least three independent experiments.
2.2.3. Tubulin polymerisation assay
The assembly of purified bovine tubulin was monitored using a kit, BK006, purchased from Cytoskeleton Inc., (Denver, CO, USA). The assay was carried out in accordance with the manufacturer’s instructions using the standard assay conditions49. Briefly, purified (>99%) bovine brain tubulin (3 mg/mL) in a buffer consisting of 80 mM PIPES (pH 6.9), 0.5 mM EGTA, 2 mM MgCl2, 1 mM GTP and 10% glycerol was incubated at 37 °C in the presence of either vehicle (2% (v/v) ddH2O), CA-4, or the quinoline compounds. Light is scattered proportionally to the concentration of polymerised microtubules in the assay. Therefore, tubulin assembly was monitored turbidimetrically at 340 nm in a Spectramax 340 PC spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). The concentration that inhibits tubulin polymerisation by 50% (IC50) was determined using area under the curve (AUC). The AUC of the untreated controls were considered as 100% polymerisation. The IC50 value for each compound was computed using GraphPad Prism Software.
2.2.4. Colchicine site competitive binding assay
The affinity of compounds 12c to colchicine binding site was determined using Colchicine Site Competitive Assay kit CytoDYNAMIX Screen15 (Cytoskeleton, Inc., Denver, CO, USA) using the standard protocol of the manufacturer to determine Ki. Biotin-labelled tubulin (0.5 µg) in 10 µL of reaction buffer was mixed with [3H]colchicine (0.08 µM, PerkinElmer, Waltham, MA) and the test compounds (positive control colchicine, negative control vinblastine, G-1, fluorescent G-1, or 2-ME) in a 96-well plate (final volume: 100 µL). After incubating for 2 h at 37 °C with gentle shaking, streptavidin-labelled yttrium SPA beads (80 µg in 20 µL reaction buffer, PerkinElmer, Waltham, MA) were added to each well and incubated for 30 min at 4 °C. The plates were then read on a scintillation counter (Packard Instrument, Topcount Microplate Reader) and the percentage of inhibition was calculated50,51.
2.2.5. Cell cycle analysis
MCF-7 cells were seeded at a density of 1 × 105 cells/well in 6-well plates and treated with CA-4 (50 nM) and compound 12c (50 and 250 nM) for 24, 48 and 72 h. The cells were collected by trypsinisation and centrifuged at 800×g for 15 min. Cells were washed twice with ice-cold PBS and fixed in ice-cold 70% ethanol overnight at −20 °C. Fixed cells were centrifuged at 800×g for 15 min and stained with 50 µg/mL of PI, containing 50 µg/mL of DNase-free RNase A, at 37 °C for 30 min. The DNA content of cells (10,000 cells/experimental group) was analysed by flow cytometer at 488 nm using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and all data were recorded and analysed using the CellQuest Software (Becton-Dickinson).
2.2.6. Annexin V/PI apoptotic assay
Apoptotic cell death was detected by flow cytometry using Annexin V and propidium iodide (PI). MCF-7 Cells were seeded in 6 well plated at density of 1 × 105 cells/mL and treated with vehicle (0.1% (v/v) EtOH), positive control (CA-4) or compound 12c (50 and 250 nM) for 24, 48 and 72 h. Cells were then harvested and prepared for flow cytometric analysis. Cells were washed in 1X binding buffer (20X binding buffer: 0.1 M HEPES, pH 7.4; 1.4 M NaCl; 25 mM CaCl2 diluted in dH2O) and incubated in the dark for 30 min on ice in Annexin V-containing binding buffer [1:100]. Cells were then washed once in binding buffer and then re-suspended in PI-containing binding buffer [1:1000]. Samples were analysed immediately using the BD accuri flow cytometer and prism software for analysis the data. Four populations are produced during the assay Annexin V and PI negative (Q4, healthy cells), Annexin V positive and PI negative (Q3, early apoptosis), Annexin V and PI positive (Q2, late apoptosis) and Annexin V negative and PI positive (Q1, necrosis).
2.2.7. Evaluation of expression levels of anti-apoptotic proteins bcl-2, pro-apoptotic proteins bax and caspase 9
The level of the anti-apoptotic marker and apoptotic marker BAX were assessed using Bcl-2 Elisa kit and human Bax ELISA Kit purchased from Zymed laboratories, invitrogen and Cloud-Clone Crop. (Katy, TX, USA), respectively, following the manufacturer’s instructions. Briefly, Treated MCF-7 cell lysate with 250 nM of compound 12c were prepared, and equal amount of cell lysates were loaded and propped with specific antibodies. The samples were measured and analysed at 450 nm in ROBONEK P2000 ELISA reader52. In Vitro Caspase-9 Activation Assay was performed using human active caspase-9 Invitrogen EIA kit according to the manufacturer’s instructions. Compound 12c at concentrations of 50 and 250 nM and CA-4 (50 nM) were prepared in dH2O up to a final volume of 50 µL/well followed by addition of 5 µL of active caspase-9. Following, the cells were mixed and 50 µL of the Master Mix was added to each well and allowed to react at 37 °C for 1 h. The fluorescence intensity of the test samples was recorded and analysed in a fluorescence plate reader at 400 nm excitation and 505 nm emissions. All experiments were conducted in triplicates.
2.2.8. Colony formation assay
MCF-7 cells (600 cells per well) were seeded in 6-well plates and incubated for 24 h before being then treated with different doses of the compound 12c (50 and 250 nM) for 14 days. Following, the cells were washed with PBS twice and subsequently fixed with 4% paraformaldehyde and stained with 0.05% crystal violet for 30 min. Finally, cells were visualised using an inverted microscope.
2.2.9. Wound healing assay
MCF-7 were grown in 6-well plates for 24 h, and scratches were made using pipette tip and washed with PBS to remove non-adherent cell debris. Subsequently, the cells were treated with different concentrations of 12c for 24 h. The migrations across the wound area were photographed under a phase contrast microscopy.
2.2.10. Measurement of mitochondrial depolarisation effect (Δψmt) and ROS levels in cells
Mitochondrial membrane potential (Δψmt) was measured by flow cytometry with DiOC2(3) staining and additional labelling with an annexin V conjugate. After treatment with compound 12c (50 and 250 nM) and CA-4 (50 nM), cells were stained with DiOC2(3) dye for 30 min in the incubator, then harvested and washed with PBS. DiOC2(3)-stained cells were resuspended with 1X annexin binding buffer, followed by addition of annexin V conjugate and incubated at 37 °C for 15 min. The data from the flow cytometry were analysed by Cell Quest software. Production of intracellular reactive oxygen species (ROS) was measured using 2,7–dichlorofluorescin diacetate (H2-DCFDA) dye. MCF-7 cells were seeded and treated either with vehicle (0.1% DMSO) or with compound 12c (50 and 250 nM) or CA-4 (50 nM) for 6, 12 and 24 h. H2O2 was used as a positive control. The amount of ROS generated was estimated after 2 h of selected compound treatment. The cells were collected by centrifugation and washed twice with PBS. Cells were then incubated with DCFDA dye (25 µM) in dark at 37 °C for 1 h. Fluorescence spectra (510 − 600 nm) were monitored using an excitation wavelength of 488 nm53,54.
3. Results and discussion
3.1. Design and chemistry
The natural product, Combretastatin A-4 (CA-4; Figure 1) exhibits significant antiproliferative activities against several tumour cells by binding to the colchicine site of tubulin to inhibit the protein polymerisation20. However, the cis double bond of CA-4 has a propensity to isomerise into the inactive trans configuration, leading to reduction in the molecule’s pharmacologic activity. Several structural modifications of the CA-4 pharmacophore have subsequently been undertaken to overcome this disadvantage, e.g. replacing the cis double bond with a heterocycle, oxadiazole, isoxazole and imidazole, resulting in compounds, such as 1, 2 and 3 respectively (Figure 1)27–32. In this work, we undertook a rational design approach of introducing chalcone system (ring C) in the form of either oxazolone or imidazolone between the two rings A and B, as well as isosterically replace ring B with quinolone. Specifically, the rigidity of the molecules was increased by introducing 1,3-oxazol-5-ones and 1,3-imidazol-4-ones to the cis-olefinic bond of CA-4, which we anticipate would create a desirable conformational and configurational restriction to prevent isomerisation of CA-4 into the inactive trans-isomer, as well as improve on the anticancer activities of these compounds since chalcones are well known for their anticancer properties55,56. The second design step involves varying the electronic substituents effect on the quinolyl moiety (ring B), while maintaining the natural active compound 3,4,5-trimethoxyphenyl moiety, which we anticipate will increase the potency of these compounds.
The syntheses of the proposed quinoline compounds 12a–h (oxazolones) and 13a–h (imidazolones) (Table 1) are shown in Schemes 1 and 2 and involve two core structural components: (i) 2-methoxyquinolyl-3-carbaldehyde nucleus 8a–h, and (ii) 3,4,5-trimethoxyphenyl moiety 11. A concise (three-step) synthesis was used for the synthesis of the first core structure, 2-methoxyquinoline-3-carbaldehyde derivatives 8a–h, as shown in Scheme 1. The synthesis was initiated with acetylation of the starting aniline derivatives 5a–h using acetic anhydride and glacial acetic acid at 0 °C. The produced amides 6a–h were subjected to Vilsmeier–Haack reaction to give the corresponding quinoline-3-aldehyde derivatives 7a–h. Addition of methoxy substituent to 7a–h to give 8a–h was achieved through the use of sodium methoxide at 40 °C in methanol47,48. The synthesis of the second core 3,4,5-trimethoxyphenyl moiety 11 started with acylation of the acid 9 under highly acidic condition using SOCl2 to give acyl benzotriazole 10 (Scheme 2)57,58. Following, treatment of the acyl benzotriazole 10 with glycine in aqueous acetonitrile gave the acyl glycine 11. Condensation of 11 with the appropriate quinoline aldehydes 8a–h in the presence of acetic anhydride and catalytic amount of sodium acetate resulted in the formation of the oxazolones 12a–h. Aminolysis of 12a–h via condensation reaction with ammonia led to the formation of the imidazolones 13a–h. It seems the nucleophilic ammonia attacks the carbonyl group of the oxazolone ring, followed by immediate intramolecular condensation and cyclisation to give the imidazolones 13a–h. In summary, two classes of compounds, 12a–h (oxazolones) and 13a–h (imidazolones) were synthesised and used for further functional and biological studies.
Table 1.
Antiproliferative activity of quinoline analogues against human cancer cell lines (IC50 [µM]). 
| Compound number | X | R | IC50 value (µM)a |
|||
|---|---|---|---|---|---|---|
| HL-60 | MCF-7 | HCT-116 | HeLa | |||
| 12a | O | H | 3.309 ± 0.022 | 1.712 ± 0.040 | 2.012 ± 0.080 | 2.113 ± 0.034 |
| 12b | O | 6-CH3 | 0.197 ± 0.041 | 0.132 ± 0.013 | 0.184 ± 0.007 | 0.117 ± 0.085 |
| 12c | O | 7-CH3 | 0.019 ± 0.059 | 0.010 ± 0.003 | 0.022 ± 0.001 | 0.042 ± 0.001 |
| 12d | O | 8-CH3 | 0.268 ± 0.093 | 0.154 ± 0.071 | 0.191 ± 0.001 | 0.153 ± 0.083 |
| 12e | O | 6-OCH3 | 0.068 ± 0.034 | 0.056 ± 0.027 | 0.031 ± 0.001 | 0.010 ± 0.004 |
| 12f | O | 7-OCH3 | 0.352 ± 0.021 | 0.052 ± 0.0021 | 0.066 ± 0.005 | 0.138 ± 0.026 |
| 12g | O | 7-OCH(CH3)2 | 1.822 ± 0.640 | 1.507 ± 0.220 | 7.880 ± 0.300 | 1.747 ± 0.500 |
| 12h | O | 7-OCH2Ph | 4.660 ± 0.260 | 1.932 ± 0.612 | 1.563 ± 0.720 | 1.054 ± 0.840 |
| 13a | NH | H | 1.561 ± 0.055 | 1.033 ± 0.055 | 8.21 ± 0.0077 | 1.409 ± 0.096 |
| 13b | NH | 6-CH3 | 0.240 ± 0.012 | 0.063 ± 0.0011 | 0.173 ± 0.025 | 0.188 ± 0.013 |
| 13c | NH | 7-CH3 | 0.661 ± 0.026 | 0.223 ± 0.056 | 0.284 ± 0.083 | 0.733 ± 0.091 |
| 13d | NH | 8-CH3 | 0.096 ± 0.006 | 0.137 ± 0.023 | 0.109 ± 0.011 | 0.126 ± 0.044 |
| 13e | NH | 6-OCH3 | 0.272 ± 0.050 | 0.042 ± 0.0025 | 0.085 ± 0.003 | 0.062 ± 0.003 |
| 13f | NH | 7-OCH3 | 0.210 ± 0.098 | 0.092 ± 0.0062 | 0.187 ± 0.009 | 0.101 ± 0.090 |
| 13g | NH | 7-OCH(CH3)2 | 5.114 ± 0.410 | 5.562 ± 0.133 | 1.620 ± 0.950 | 4.439 ± 0.600 |
| 13h | NH | 7-OCH2Ph | 4.931 ± 0.260 | 1.695 ± 0.821 | 2.610 ± 0.430 | 2.070 ± 0.390 |
| CA-4 | – | – | 0.076 ± 0.004 | 0.019 ± 0.004 | 0.026 ± 0.001 | 0.064 ± 0.004 |
aIC50 values are half maximal inhibitory concentrations required to block the growth stimulation of cells. Values represent the mean for three experiments performed in triplicate.
Scheme 1.

Synthetic route for preparation of aldehydes 8a–h. Reagents and conditions: (i) Ac2O, AcOH, 0 °C,1h; (ii) DMF, POCl3, 70–90 °C, 18h; (iii) CH3ONa, MeOH, 40 °C, 3–6h.
Scheme 2.

Synthetic route for preparation of Quinoline targets 12a–h and 13a–h. Reagents and conditions: (i) SOCl2,DCM, r.t; (ii) TEA, MeCN,H2O, r.t; (iii) AcONa, AC2O, 80 °C, 2h; (iv) NH4OH, EtOH, reflux, 18h.
3.2. Biological results and discussion
3.2.1. In vitro antiproliferative activities
All the synthesised compounds (with CA-4 as a positive reference) were evaluated for their antiproliferative activities using MTT assay with four different cancer cell lines – MCF-7 breast adenocarcinoma, HL-60 leukaemia, HCT-116 colorectal carcinoma, and HeLa cervical adenocarcinoma. As shown in Table 1, most of the compounds demonstrated moderate to highly potent antiproliferative activities. In the oxazolone analogues (12a–h), compound 12a without any substituent on the quinoline ring was the least active when compared with quinoline ring substituted compounds. The relative position of the substituent on the quinoline ring also seemed to be critical for antiproliferative activity. Compound 12c with methyl group at the 7-position ring displayed impressive non-selective potency in nanomolar range against HL-60, MCF-7, HCT-116 and HeLa cell lines with IC50 of 0.019, 0.010, 0.022 and 0.042 µM, which compared to 0.076, 0.019. 0.026 and 0.064 µM for CA-4, respectively. In contrast, both the 6-CH3 analog (12b) and 8-CH3 analog (12d) were 3- to 15-fold less active than 12c. The nature of the substituents on the quinoline ring of the oxazolone compounds was also found to significantly influence the biological activity. For example, replacement of the methyl group in 12b and 12c with a stronger electron-releasing methoxy group yielded compounds 12e and 12f, respectively, which resulted in better antiproliferative activities. The methoxy-containing compound 12e was 2.7- to 13-fold more active than the methyl-containing compound 12b with the four cancer cell lines (IC50 of 0.068, 0.056, 0.031 and 0.010 µM in HL-60, MCF-7, HCT-116 and HeLa cancer cell lines, respectively). Compound 12f had a similar effect as 12c against MCF-7 and HCT-116 cells (0.052 and 0.066 µM, respectively), but with reduction in activity against the other two cell lines, HL-60 and HeLa (0.352 and 0.138 µM, respectively). Introducing larger substituents at the quinoline ring as in 12g (7-tert-butyl) and 12h (7-benzyloxy) led to a dramatic decrease in activity compared to their corresponding analog 12f (7-methoxy). Summarily, adding smaller and/or polar groups to the quinoline ring of the oxazolone resulted in significant improvement in the antiproliferative activity.
The imidazolones (Compound 13a–h) also resulted in impressive antiproliferative activity with IC50 values ranging from 0.04–8.21 µM in all four cell lines. In general, the imidazolones showed similar antiproliferative activities as the oxazolones (Table 1), which could be due to similar electronic effects of the oxazolone and imidazolone rings. Like the oxazolone, lack of substituent on the quinolone ring as in compound 13a led to reduction in activity, with IC50 values of more than 1 µM in all four cell lines, similar to the results obtained with the oxazolone derivative 12a. Methyl substitution on the quinoline ring, e.g. 6-CH3 13b, 7-CH3 13c and 8-CH3 13d led to potent activity in submicromolar range in all four cancer cell lines.
The position of the methoxy substituent on the quinoline heterocycle also influenced the antiproliferative activity of the compounds against the cancer cell lines. For example, the antiproliferative activity of 6-methoxy-substituted 13e was better than its analog 7-methoxy-substituted 13f against MCF-7, HCT-116 and HeLa cells with IC50 values of 0.042, 0.085 and 0.062 µM, which compare to 0.092, 0.187 and 0.101 µM for 13f, respectively. However, in HL-60, 13e exhibited less antiproliferative activity with IC50 value of 0.272 µM. In a similar trend as the oxazolone derivatives, bulky substituents on the quinoline ring 13g (7-tert-butyl) and 13h (7-benzyloxy) resulted in drastic decrease in activity in all four cancer cell lines with 14- to 125-fold loss in potency compared to their corresponding 13f (7-methoxy containing) compound.
In summary, both oxazolone and imidazolone compounds displayed potent antiproliferative effects, strengthening our hypothesis that nitrogen-containing heterocycles, such as quinoline, are beneficial surrogates for the ring B of CA-4. The different biological activities of the compounds are likely the result of differences in their mode of interaction with the colchicine binding site. Due to its excellent antiproliferative activity, compound 12c was studied in more details as described below.
3.2.2. In vitro inhibition of tubulin polymerisation and colchicine binding
Trimethoxyphenyl (TMP) containing stilbenoid derived compounds, such as colchicine, resveratrol and CA-4 bind to tubulin at the colchicine binding site, resulting in inhibition of microtubule polymerisation59,60. To confirm whether the quinoline compounds similarly target the tubulin-microtubule system, representative quinoline compounds, including four oxazolone analogues (12a, 12c, 12e and 12g) and two imidazolone analogues (13c and 13e), as well as the reference compound CA-4, were evaluated for their antitubulin polymerisation activities and the results presented in Table 2. The methyl and methoxy substituted oxazolone compounds 12c and 12e, respectively strongly inhibited tubulin assembly with IC50 of 1.21 and 2.26 µM, respectively compared to that of CA-4 (IC50 of 2.17 µM), while the unsubstituted analogue 12a (IC50 of 13.98 µM) and tert-butyl analog 12g (IC50 of 8.23 µM) were 6- and 4-fold less active than CA-4. The imidazolone compound 13e with IC50 of 1.48 µM also showed very potent tubulin polymerisation inhibition compared to CA-4. The methyl analogue 13c was inactive in the tubulin polymerisation assay (IC50 of 20.29 µM), and is 16-fold less active compared to its corresponding oxazolone derivative 12c, which is in agreements with the poor cell growth inhibitory activity of 13c compared to 12c.
Table 2.
Inhibition of Tubulin Polymerisation and Colchicine Binding by quinoline compounds and CA-4.
| Compound number | Tubulin assemblya |
Colchicine bindingb |
|
|---|---|---|---|
| IC50 (µM) | %± SD |
||
| 1 µM drug | 5 µM drug | ||
| 12a | 13.98 | nd | nd |
| 12c | 1.21 | 79 ± 2 | 87 ± 1 |
| 12e | 2.26 | nd | nd |
| 12g | 8.23 | nd | nd |
| 13c | 20.29 | nd | nd |
| 13e | 1.48 | nd | nd |
| CA-4 | 2.17 | 86 ± 0.9 | 97 ± 2 |
aInhibition of tubulin polymerisation. Tubulin was at 10 µM.
bInhibition of [3H] colchicine binding. Tubulin and colchicine were at 1 and 5 µM concentrations, respectively.
Compound 12c was also examined at two different concentrations (1 and 5 µM) for its ability to compete with colchicine for binding to tubulin using a [3H] colchicine binding assay. Compound 12c strongly inhibited colchicine binding to tubulin by 79% and 87% at 1 and 5 µM respectively, which compares with 86% and 97% inhibition by CA-4, respectively. These results suggest that compound 12c is involved in tubulin polymerisation inhibition through the colchicine-binding site.
3.2.3. Cell cycle analysis
Induction of cell cycle arrest at G2/M phase is strongly accompanied with tubulin polymerisation inhibition. It is well established that CA-4 arrests cell cycle at G2M phase61–63. To further gain insight into compound 12c potent antiproliferative activity, cell cycle analysis of MCF-7 cells was performed at two concentrations of 50 nM and 250 nM and at different time points of 0, 24, 48 and 72 h. Figure 3(A) clearly demonstrates that 12c caused a significant arrest in G2/M phase and apoptosis in a dose- and time-dependent manner. After 48 h, the percentage of G2/M phase arrested cells were 28.4% and 38.3% at 50 nM and 250 nM, respectively compared to 9.2% of untreated cells (Figure 3(B)). Moreover, there was an increase in the number of cells in G2/M phase after 72 h (33.0% and 40.8% at 50 nM and 250 nM, respectively) with a concomitant decrease of cells in G0/G1 phase (40.3% and 29.8% at 50 nM and 250 nM, respectively) compared to the control (57.3%). In a comparable finding, CA-4 (50 nM) also significantly arrested G2/M phase at 24, 48 and 72 h (40.3%, 43.8% and 47.7%, respectively). Accordingly, a concomitant decrease of MCF-7 cells was detected in G0 phase (Figure 3(C)). Furthermore, compound 12c induced a gradual increase in apoptosis (16.2%, 23.4% and 32.7%) at 250 nM as the proportion of cells in the sub-G1 phase increased at 24, 48 and 72 h, respectively compared to untreated cells (1.5%) (Figure 3(D)). Similarly, 23.5%, 31.1% and 37.4% increase in apoptosis was observed for CA-4 at 24, 48 and 72 h, respectively. These findings are in agreement with previously reported for a series of related quinoline analogues, which significantly induced apoptosis and G2/M cycle arrest in MCF-7 cells23,42,46,64.
Figure 3.

(A) Effect of compound 12c on the cell cycle and apoptosis in MCF-7 cells. Cells were treated with either vehicle [0.1% ethanol (v/v)], CA-4 (50 nM), 12c (50 nM and 250 nM) for 24 h, 48 h and 72 h. Cells were then fixed, stained with PI, and analysed by flow cytometry. Cell cycle analysis was performed on histograms of gated counts per DNA area (FL2-A). The number of cells with (B) 4 N (G2/M), (C) 2 N(G0G1), and (D) <2 N (sub-G1) DNA content was determined with CellQuest software. Values represent the mean ± SEM for three independent experiments. Statistical analysis was performed using two-way ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001).
3.2.4. Cell apoptosis
We investigated whether cell death induced by compound 12c treatment was related to apoptosis using the Annexin-V/PI double staining flow cytometric assay (Figure 4(A,B)). MCF-7 cells were treated with three different concentrations (0, 50 and 250 nM) of compound 12c at different time points (24, 48 and 72 h). Compound 12c caused a significant accumulation of annexine-V positive cells and induced both early and late apoptosis in a dose- and time-dependent manner compared to the untreated cells. As shown in Figure 4(B), when the cells were treated with 12c (0 and 250 nM) or CA-4 (50 nM) for 48 h, the percentage of Annexin V-staining positive cells significantly increased from 1% in untreated cells to 15%, 21% and 29% respectively. The percentage of early and late apoptotic cells in the presence of 12c increased after 72 h to 17.6% and 29.3% at 1 and 5 µM respectively when compared to the untreated cells (2%). Based on the cell cycle arrest and apoptosis findings (Figure 3(B–D)), it appears that compound 12c could efficiently induce apoptosis cell death in MCF-7 cells in a dose- and time-dependent manner.
Figure 4.

(A) Effect of compound 12c at different time points on apoptosis in MCF-7 cells analysed by flow cytometry after double staining of the cells with Annexin-V-FITC and PI. MCF-7 cells treated with 50 and 250 nM of compound 12c and 50 nM of CA-4 for 24 h, 48 h and 72 h and collected and processed for analysis. (B) Quantitative analysis of apoptosis. Values represent the mean ± SEM for three independent experiments. Statistical analysis was performed using two-way ANOVA (*p < 0.05** and p < 0.01; ***p < 0.001).
3.2.5. Assessment of toxicity to non-tumorigenic human cells
To assess the cytotoxicity and selectivity of 12c towards cancer cells, normal epithelial breast MCF-10A cell viability study was carried out. As shown in Figure 5(A), the IC50 value of 12c was more than 50 µM in MCF-10A cells, which was significantly higher than the IC50 values of 19, 10, 22 and 42 nM in MCF-7, HL-60, HCT-116 and HeLa cancer cell lines, respectively. Remarkably, 12c was found to be less toxic in normal MCF-10A (IC50 >50 µM) when compared to CA-4 (IC50 = 6.1 µM) (Figure 5(B)), suggesting 12c to have better selective toxicity against cancer cells.
Figure 5.
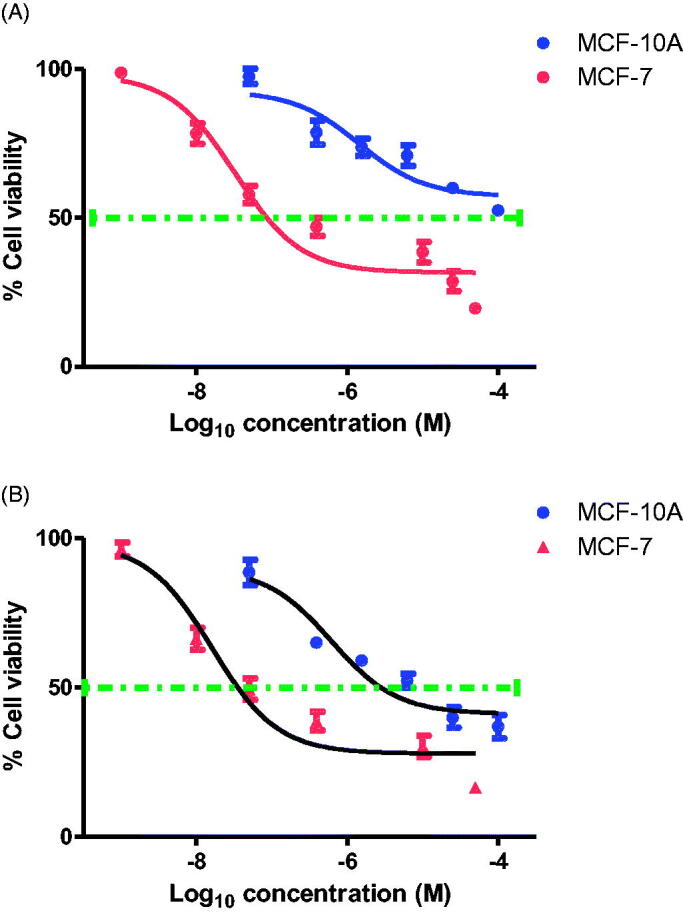
Dose response curve for (A) Compound 12c and (B) CA-4 on the proliferation of breast cancer MCF-7 and normal breast MCF-10A cells. Cells were grown in 96-well plates and treated with serial concentrations of compound 12c or CA-4 for 72 h. Cell viability was expressed as percentage of vehicle control [ethanol 1% (v/v)] treated cells and was measured by MTT assay (average of three independent experiments).
3.2.6. Expression of the apoptotic proteins in MCF-7 cell lines
The previous data clearly demonstrate that 12c is an effective anti-mitotic quinoline compound in MCF-7 cell lines. Herein, the effect of 12c on the expression of apoptosis pathway markers, Bcl-2 anti-apoptotic protein and Bax pro-apoptotic protein was investigated. MCF-7 cells treated with 12c at 250 nM for 48 h decreased the expression level of the anti-apoptotic protein Bcl-2, and correspondingly up-regulated the expression of the pro-apoptotic protein Bax (Figure 6(A,B)).
Figure 6.

Effect of compound 12c on the expression (A) anti-apoptotic protein Bcl2, (B) pro-apoptotic protein BAX and (C) Caspase 9 in MCF-7 cells.
Activation of caspases initiates apoptosis, and in particular caspase-9 is considered an important effector caspase responsible for programmed cell death apoptosis activated by CA-461,65,66. The amount of activated caspase-9 was examined in MCF-7 cells treated with 12c. As observed from Figure 6(C), compound 12c at 50 and 250 nM produced about 8- and 16-fold increases in caspase-9 activation respectively when compared to 14-fold for CA-4 (50 nM). This finding confirms that compound 12c like CA-4 enhanced the rate of apoptosis in MCF-7 cell through caspase-9 activation.
3.2.7. Inhibition of colony formation
Colony formation assay is one of the effective techniques for the determination of long-term cell proliferation upon anticancer drug exposure. The inhibitory potential of 12c on MCF-7 cells colony formation is displayed in Figure 7. Compound 12c suppressed the clonogenic formation potential of MCF-7 cells in a dose dependent manner when compared to CA-4.
Figure 7.

(A) Inhibition of colony formation in MCF-7 cells by 50 and 250 nM of compound 12c and 50 nM of CA-4 for 48 h. (B) Quantitative analysis of colony formation. .
3.2.8. Wound healing assay
Migration and motility of cancer cells are considered as critical factors in tumour progression and metastasis67,68. In order to investigate the effect of compound 12c on the migration of MCF-7, wound healing assay was performed. As illustrated in Figure 8(A,B), the untreated cells migrated to the scraped area while in 12c-treated wells, cell migration was significantly inhibited in a dose-dependent manner. This significant difference in the wound area confirms that 12c suppressed MCF-7 cell migration, an important event in tumour metastasis.
Figure 8.

(A) Inhibition of the migration of MCF-7 cells treated with compound 12c for 48 h in the wound healing assay. (B) Quantitative analysis of wound closure rate and was calculated as mean ± SEM for three independent experiments. Statistical analysis was performed using one-way ANOVA-Bonferroni post-hoc test (***p < 0.001).
3.2.9. Mitochondrial membrane potential
Mitochondria membrane potential plays a crucial role in the propagation of apoptosis. Specifically, loss of mitochondrial membrane potential Δψmt (MMP) is characteristic of early stage of apoptosis69–71. To confirm whether compound 12c could decrease the MMP of MCF-7 cancer cells, MMP was monitored by the fluorescence of the dye DiOC2(3). MCF-7 cells treated with 12c at 50 and 250 nM exhibited significant decrease in MMP in a dose- and time-dependant manner (Figure 9(A)). This depletion in MMP was associated with an increase of annexin-V positive early apoptotic cells. Maximum decrease in MCF-7 MMP was detected after 24 h treatment with 12c in which the percentage of apoptotic cells increased from 1.1% to 24.9% and 31.3% at 50 and 250 nM, respectively (Figure 9(B)). This indicates that compound 12c induces mitochondrial dysfunction in MCF-7, which eventually triggered apoptotic cell death. These results are in agreement with previously reported CA-4 analogues study that were shown to cause apoptosis through the mitochondrial pathway68,72,73.
Figure 9.

Assessment of mitochondrial membrane potential (Δψmt) after treatment of MCF-7 cells with 12c. Cells were treated with indicated concentration of compound 12c for 6, 12 and 24 h and then stained with fluorescent DiOC2(3) for analysis of mitochondrial potential. Cells were then analysed by flow cytometry as described in the experimental section. Data are presented as mean ± SEM of three independent experiments. Statistical analysis was performed using two-way ANOVA (**p < 0.01; ***p < 0.001).
3.2.10. Intracellular reactive oxygen species (ROS) production
The dissipation of mitochondrial potential is strongly associated with mitochondrial production of reactive oxygen species (ROS)71,73. The production of ROS after 12c treatment at 50 and 250 nM, as well as CA-4 (50 nM) with hydrogen peroxide H2O2 was followed with 2,7–dichlorofluorescin diacetate (H2-DCFDA). As shown in Figure 10, after 24 h of 12c treatment, the levels of ROS in MCF-7 cells were 22.7 and 26.6% at 50 and 250 nM, respectively. The level in untreated MCF-7 cells was 1.0%, while it increased only to 18.3% in CA-4-treated cells. This result along with the significant loss of mitochondrial membrane potential above clearly suggests that compound 12c induced apoptosis via the mitochondrial pathway.
Figure 10.
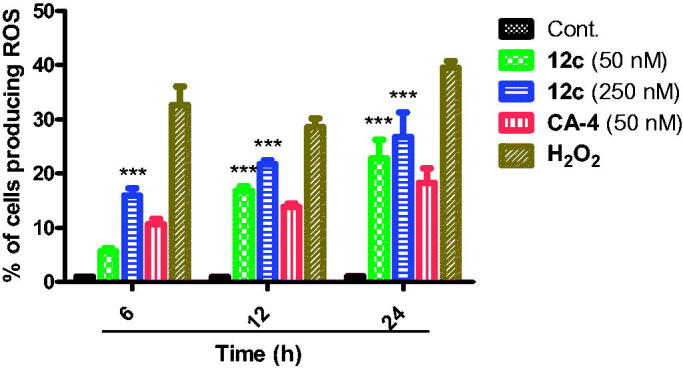
Effect of compound 12c at different time points on ROS production in MCF-7 cells. MCF-7 cells treated without and with 50 and 250 nM of compound 12c and 50 nM of CA-4 and 100 µM H2O2 for 6 h, 12 h and 24 h. Cells were incubated with DCFDA (25 µM) dye for 1 h at 37 °C in dark. Fluorescence intensity per cell at 525 nm was calculated as mean ± SEM for three independent experiments. Statistical analysis was performed using two-way ANOVA (***p < 0.001).
4. Conclusion
In this study, we designed, synthesised and evaluated two classes of novel quinoline compounds combretastatin A-4 derivatives as potential inhibitors of tubulin polymerisation. Several other studies have also reported derivatisation of the CA-4 pharmacophore with varying success27–32. Unlike the previous compounds, we for the first time introduced a chalcone system, including oxazolones and imidazolones to the cis bond of CA-4 to give more rigidity to the required active conformation. The chalcone system is well known for its anticancer activities. Our design also kept the essential natural trimethoxyphenyl pharmacophore (found in CA-4), while varying the electronic substituents effect on the quinolyl moiety (ring B) that were expected to enhance the potency of the compounds. Most of the compounds showed significant and, in some instances, comparable antiproliferative activities against different cancer cell lines as the previously studied combretastatin A-4 compound, CA-4. One of the most promising compound 12c showed potent anti-proliferative activities against HL-60, MCF-7, HCT-116 and HeLA cancer cell lines with IC50 values of 0.019, 0.010, 0.022 and 0.042 µM, respectively, and simultaneously low cytotoxicity towards MCF-10A non-cancer cells. The microtubule polymerisation inhibitory effect of 12c was confirmed with an in vitro tubulin polymerisation and colchicine inhibition assays. Compound 12c effectively block the G2/M phase at the cell cycle and induce MCF-7 cell apoptosis together with significant change of Bax/Bcl expression ratio indicating involvement of mitochondrial apoptosis pathway. Further cellular mechanistic studies confirmed that 12c inhibited MCF-7 cell migration and colony formation. In conclusion, these results highlight our novel quinoline compounds and particularly 12c as promising anti-tubulin agent for the treatment of MCF-7 breast cancer cells. Moreover, the results point to a direction for rational development of potent tubulin polymerisation inhibitors for the treatment of cancer.
Acknowledgements
The authors gratefully acknowledge DSR technical and financial support.
Funding Statement
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant number [RG-18–166-41]. The authors, therefore, gratefully acknowledge DSR technical and financial support.
Disclosure statement
The authors declare no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Decosterd LA, Widmer N, Zaman K, et al. Therapeutic drug monitoring of targeted anticancer therapy. Biomark Med 2015;9:887–93. [DOI] [PubMed] [Google Scholar]
- 3.La Regina G, Coluccia A, Naccarato V, Silvestri R.. Towards modern anticancer agents that interact with tubulin. Eur J Pharm Sci 2019;131:58–68. [DOI] [PubMed] [Google Scholar]
- 4.Stec-Martyna E, Ponassi M, Miele M, et al. Structural comparison of the interaction of tubulin with various ligands affecting microtubule dynamics. Curr Cancer Drug Targets 2012;12:658–66. [DOI] [PubMed] [Google Scholar]
- 5.Devi Tangutur A, Kumar D, Vamsi Krishna K, Kantevari S.. Microtubule targeting agents as cancer chemotherapeutics: An overview of molecular hybrids as stabilizing and destabilizing agents. Curr Top Med Chem 2017;17:2523–37. [DOI] [PubMed] [Google Scholar]
- 6.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer 2010;10:194–204. [DOI] [PubMed] [Google Scholar]
- 7.Coulup SK, Georg GI.. Revisiting microtubule targeting agents: Α-tubulin and the pironetin binding site as unexplored targets for cancer therapeutics. Bioorg Med Chem Lett 2019;29:1865–73. [DOI] [PubMed] [Google Scholar]
- 8.Vindya N, Sharma N, Yadav M, Ethiraj K.. Tubulins – the target for anticancer therapy. Curr Top Med Chem 2015;15:73–82. [DOI] [PubMed] [Google Scholar]
- 9.Dumontet C, Jordan MA.. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 2010;9:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S, Kaur C, Budhiraja A, et al. Chalcone based azacarboline analogues as novel antitubulin agents: design, synthesis, biological evaluation and molecular modelling studies. Eur J Med Chem 2014;85:648–60. [DOI] [PubMed] [Google Scholar]
- 11.Kaur R, Kaur G, Gill RK, Soni R, et al. Recent developments in tubulin polymerization inhibitors: an overview. Eur J Med Chem 2014;87:89–124. [DOI] [PubMed] [Google Scholar]
- 12.Bukhari SNA, Kumar GB, Revankar HM, Qin HL.. Development of combretastatins as potent tubulin polymerization inhibitors. Bioorg Chem 2017;72:130–47. [DOI] [PubMed] [Google Scholar]
- 13.Naumova N, Šachl R.. Regulation of cell death by mitochondrial transport systems of calcium and bcl-2 proteins. Membranes 2020;10:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas E, Gopalakrishnan V, Hegde M, et al. A novel resveratrol based tubulin inhibitor induces mitotic arrest and activates apoptosis in cancer cells. Sci Rep 2016;6:34653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naaz F, Haider MR, Shafi S, Yar MS.. Anti-tubulin agents of natural origin: targeting taxol, vinca, and colchicine binding domains. Eur J Med Chem 2019;171:310–31. [DOI] [PubMed] [Google Scholar]
- 16.Cao YN, Zheng LL, Wang D, et al. Recent advances in microtubule-stabilizing agents. Eur J Med Chem 2018;143:806–28. [DOI] [PubMed] [Google Scholar]
- 17.Cragg GM, Kingston DG, Newman DJ.. Anticancer agents from natural products. Boca Raton (FL): CRC press; 2012. [Google Scholar]
- 18.Gupta SK, Singh P, Ali V, Verma M.. Role of membrane-embedded drug efflux abc transporters in the cancer chemotherapy. Oncol Rev 2020;14:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnst KE, Wang Y, Lei ZN, et al. Colchicine binding site agent dj95 overcomes drug resistance and exhibits antitumor efficacy. Mol Pharmacol 2019;96:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zweifel M, Jayson GC, Reed N, et al. Phase ii trial of combretastatin a4 phosphate, carboplatin, and paclitaxel in patients with platinum-resistant ovarian cancer. Ann Oncol 2011;22:2036–41. [DOI] [PubMed] [Google Scholar]
- 21.Li W, Sun H, Xu S, et al. Tubulin inhibitors targeting the colchicine binding site: a perspective of privileged structures. Future Med Chem 2017;9:1765–94. [DOI] [PubMed] [Google Scholar]
- 22.Kamath PR, Sunil D, Ajees AA.. Synthesis of indole–quinoline–oxadiazoles: their anticancer potential and computational tubulin binding studies. Res Chem Intermed 2016;42:5899–914. [Google Scholar]
- 23.Khelifi I, Naret T, Renko D, et al. Design, synthesis and anticancer properties of isocombretaquinolines as potent tubulin assembly inhibitors. Eur J Med Chem 2017;127:1025–34. [DOI] [PubMed] [Google Scholar]
- 24.Seligmann J, Twelves C.. Tubulin: an example of targeted chemotherapy. Future Med Chem 2013;5:339–52. [DOI] [PubMed] [Google Scholar]
- 25.Piekuś-Słomka N, Mikstacka R, Ronowicz J, Sobiak S.. Hybrid cis-stilbene molecules: novel anticancer agents. Int J Mol Sci 2019;20:1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haider K, Rahaman S, Yar MS, Kamal A.. Tubulin inhibitors as novel anticancer agents: an overview on patents (2013-2018). Expert Opin Ther Pat 2019;29:623–41. [DOI] [PubMed] [Google Scholar]
- 27.Das BC, Tang XY, Rogler P, Evans T.. Design and synthesis of 3,5-disubstituted boron-containing 1,2,4-oxadiazoles as potential combretastatin a-4 (ca-4) analogs. Tetrahedron Lett 2012;53:3947–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biersack B, Effenberger K, Schobert R, Ocker M.. Oxazole-bridged combretastatin a analogues with improved anticancer properties. ChemMedChem 2010;5:420–7. [DOI] [PubMed] [Google Scholar]
- 29.Mahal K, Biersack B, Schruefer S, et al. Combretastatin a-4 derived 5-(1-methyl-4-phenyl-imidazol-5-yl)indoles with superior cytotoxic and anti-vascular effects on chemoresistant cancer cells and tumors. Eur J Med Chem 2016;118:9–20. [DOI] [PubMed] [Google Scholar]
- 30.Biernacki K, Daśko M, Ciupak O, et al. Novel 1,2,4-oxadiazole derivatives in drug discovery. Pharmaceuticals (Basel) 2020;13:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt F, Gosch LC, Dittmer A, et al. Oxazole-bridged combretastatin A-4 derivatives with tethered hydroxamic acids: structure(-)activity relations of new inhibitors of hdac and/or tubulin function. Int J Mol Sci 2019;20:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L, Quan D, Chen J, et al. Design, synthesis, and biological evaluation of 1-substituted-2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur J Med Chem 2019;184:111732. [DOI] [PubMed] [Google Scholar]
- 33.Ibrahim TS, Hawwas MM, Malebari AM, et al. Potent quinoline-containing combretastatin a-4 analogues: design, synthesis, antiproliferative, and anti-tubulin activity. Pharmaceuticals (Basel) 2020;13:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jian XE, Yang F, Jiang CS, et al. Synthesis and biological evaluation of novel pyrazolo[3,4-b]pyridines as cis-restricted combretastatin a-4 analogues. Bioorg Med Chem Lett 2020;30:127025. [DOI] [PubMed] [Google Scholar]
- 35.Yang F, Jian XE, Diao PC, et al. Synthesis, and biological evaluation of 3,6-diaryl-[1,2,4]triazolo[4,3-a]pyridine analogues as new potent tubulin polymerization inhibitors. Eur J Med Chem 2020;204:112625. [DOI] [PubMed] [Google Scholar]
- 36.Ma W, Chen P, Huo X, et al. Development of triazolothiadiazine derivatives as highly potent tubulin polymerization inhibitors: structure-activity relationship, in vitro and in vivo study. Eur J Med Chem 2020;208:112847. [DOI] [PubMed] [Google Scholar]
- 37.Kaur K, Jain M, Reddy RP, Jain R.. Quinolines and structurally related heterocycles as antimalarials. Eur J Med Chem 2010;45:3245–64. [DOI] [PubMed] [Google Scholar]
- 38.Maurya SS, Khan SI, Bahuguna A, et al. Synthesis, antimalarial activity, heme binding and docking studies of n-substituted 4-aminoquinoline-pyrimidine molecular hybrids. Eur J Med Chem 2017;129:175–85. [DOI] [PubMed] [Google Scholar]
- 39.Vaibhav S, Dinesh Kumar M, Rina D.. Synthetic methods of quinoline derivatives as potent anticancer agents. Mini-Rev Med Chem 2017;17:1557–72. [DOI] [PubMed] [Google Scholar]
- 40.Musiol R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin Drug Discov 2017;12:583–97. [DOI] [PubMed] [Google Scholar]
- 41.Solomon VR, Lee H.. Quinoline as a privileged scaffold in cancer drug discovery. Curr Med Chem 2011;18:1488–508. [DOI] [PubMed] [Google Scholar]
- 42.Li W, Xu F, Shuai W, et al. Discovery of novel quinoline-chalcone derivatives as potent antitumor agents with microtubule polymerization inhibitory activity. J Med Chem 2019;62:993–1013. [DOI] [PubMed] [Google Scholar]
- 43.Ma X, Shen L, Zhang J, et al. Novel 4-acrylamido-quinoline derivatives as potent pi3k/mtor dual inhibitors: The design, synthesis, and in vitro and in vivo biological evaluation. Front Chem 2019;7:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu X, Wang Q, Li W.. Recent advances in heterocyclic tubulin inhibitors targeting the colchicine binding site. Anticancer Agents Med Chem 2016;16:1325–38. [DOI] [PubMed] [Google Scholar]
- 45.Nainwal LM, Alam MM, Shaquiquzzaman M, Marella A, et al. Combretastatin-based compounds with therapeutic characteristics: a patent review. Expert Opin Ther Pat 2019;29:703–31. [DOI] [PubMed] [Google Scholar]
- 46.Chaudhary V, Venghateri JB, Dhaked HPS, et al. Novel combretastatin-2-aminoimidazole analogues as potent tubulin assembly inhibitors: exploration of unique pharmacophoric impact of bridging skeleton and aryl moiety. J Med Chem 2016;59:3439–51. [DOI] [PubMed] [Google Scholar]
- 47.Jain KS, Kathiravan MK, Bariwal JB, et al. Novel dual use of formamide-pocl3 mixture for the efficient, one-pot synthesis of condensed 2 h-pyrimidin-4-amine libraries under microwave irradiation. Synth Commun 2013;43:719–27. [Google Scholar]
- 48.Ibrahim TS, Bokhtia RM, AL-Mahmoudy AM, et al. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2,4-dione as HIV-1 fusion inhibitors. Bioorg Chem 2020;99:103782. [DOI] [PubMed] [Google Scholar]
- 49.Kapoor S, Srivastava S, Panda D.. Indibulin dampens microtubule dynamics and produces synergistic antiproliferative effect with vinblastine in mcf-7 cells: implications in cancer chemotherapy. Sci Rep 2018;8:12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai MJ, Ojha R, Lin MH, et al. 1-arylsulfonyl indoline-benzamides as a new antitubulin agents, with inhibition of histone deacetylase. Eur J Med Chem 2019;162:612–30. [DOI] [PubMed] [Google Scholar]
- 51.Du T, Lin S, Ji M, et al. A novel orally active microtubule destabilizing agent s-40 targets the colchicine-binding site and shows potent antitumor activity. Cancer Lett 2020;495:22–32. [DOI] [PubMed] [Google Scholar]
- 52.Mohamed KO, Zaki I, El-Deen IM, Abdelhameid MK.. A new class of diamide scaffold: design, synthesis and biological evaluation as potent antimitotic agents, tubulin polymerization inhibition and apoptosis inducing activity studies. Bioorg Chem 2019;84:399–409. [DOI] [PubMed] [Google Scholar]
- 53.Wu D, Yotnda P.. Production and detection of reactive oxygen species (ros) in cancers. J Vis Exp 2011;21:3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hura N, Sawant AV, Kumari A, et al. Combretastatin-inspired heterocycles as antitubulin anticancer agents. ACS Omega 2018;3:9754–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ayati A, Esmaeili R, Moghimi S, et al. Synthesis and biological evaluation of 4-amino-5-cinnamoylthiazoles as chalcone-like anticancer agents. Eur J Med Chem 2018;145:404–12. [DOI] [PubMed] [Google Scholar]
- 56.Mahapatra DK, Bharti SK, Asati V.. Anti-cancer chalcones: structural and molecular target perspectives. Eur J Med Chem 2015;98:69–114. [DOI] [PubMed] [Google Scholar]
- 57.Zhou Y, Yan W, Cao D, et al. Design, synthesis and biological evaluation of 4-anilinoquinoline derivatives as novel potent tubulin depolymerization agents. Eur J Med Chem 2017;138:1114–25. [DOI] [PubMed] [Google Scholar]
- 58.Agha KA, Abo-Dya NE, Ibrahim TS, et al. N-acylbenzotriazole: Convenient approach for protecting group-free monoacylation of symmetric diamines. Monatsh Chem 2020;151:589–98. [Google Scholar]
- 59.Florian S, Mitchison TJ.. Anti-microtubule drugs. In: Chang P, Ohi R, ed. The mitotic spindle. New York (NY): Humana; 2016:403–421. [DOI] [PubMed] [Google Scholar]
- 60.Ansari M, Shokrzadeh M, Karima S, et al. New thiazole-2 (3h)-thiones containing 4-(3, 4, 5-trimethoxyphenyl) moiety as anticancer agents. Eur J Med Chem 2020;185:111784. [DOI] [PubMed] [Google Scholar]
- 61.Greene LM, Meegan MJ, Zisterer DM.. Combretastatins: More than just vascular targeting agents? J Pharmacol Exp Ther 2015;355:212–27. [DOI] [PubMed] [Google Scholar]
- 62.Agut R, Falomir E, Murga J, et al. Synthesis of combretastatin a-4 and 3′-aminocombretastatin a-4 derivatives with aminoacid containing pendants and study of their interaction with tubulin and as downregulators of the vegf, htert and c-myc gene expression. Molecules 2020;25:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pérez-Pérez MJ, Priego EM, Bueno O, et al. Blocking blood flow to solid tumors by destabilizing tubulin: An approach to targeting tumor growth. J Med Chem 2016;59:8685–711. [DOI] [PubMed] [Google Scholar]
- 64.Shobeiri N, Rashedi M, Mosaffa F, et al. Synthesis and biological evaluation of quinoline analogues of flavones as potential anticancer agents and tubulin polymerization inhibitors. Eur J Med Chem 2016;114:14–23. [DOI] [PubMed] [Google Scholar]
- 65.Mendez G, Policarpi C, Cenciarelli C, et al. Role of bim in apoptosis induced in h460 lung tumor cells by the spindle poison combretastatin-a4. Apoptosis 2011;16:940–9. [DOI] [PubMed] [Google Scholar]
- 66.Tarade D, Ma D, Pignanelli C, et al. Structurally simplified biphenyl combretastatin a4 derivatives retain in vitro anti-cancer activity dependent on mitotic arrest. PLoS One 2017;12:e0171806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Naaz F, Ahmad F, Lone BA, et al. Design and synthesis of newer 1,3,4-oxadiazole and 1,2,4-triazole based topsentin analogues as anti-proliferative agent targeting tubulin. Bioorg Chem 2020;95:103519. [DOI] [PubMed] [Google Scholar]
- 68.Hua S, Chen F, Wang X, Gou S.. Dual-functional conjugates improving cancer immunochemotherapy by inhibiting tubulin polymerization and indoleamine-2,3-dioxygenase. Eur J Med Chem 2020;189:112041. [DOI] [PubMed] [Google Scholar]
- 69.Bortolozzi R, Carta D, Dal Prà M, et al. Evaluating the effects of fluorine on biological properties and metabolic stability of some antitubulin 3-substituted 7-phenyl-pyrroloquinolinones. Eur J Med Chem 2019;178:297–314. [DOI] [PubMed] [Google Scholar]
- 70.Xiong S, Mu T, Wang G, Jiang X.. Mitochondria-mediated apoptosis in mammals. Protein & Cell 2014;5:737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu YR, Jia Z, Liu YJ, Wang XZ.. Novel dibenzoxanthenes compounds inhibit human gastric cancer sgc-7901 cell growth by apoptosis. J Mol Struct 2020;1220:128588. [Google Scholar]
- 72.Romagnoli R, Baraldi PG, Prencipe F, et al. Design and synthesis of potent in vitro and in vivo anticancer agents based on 1-(3',4',5'-Trimethoxyphenyl)-2-Aryl-1H-Imidazole. Sci Rep 2016;6:26602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Romagnoli R, Baraldi PG, Prencipe F, et al. Synthesis and biological evaluation of 2-methyl-4,5-disubstituted oxazoles as a novel class of highly potent antitubulin agents. Sci Rep 2017;7:46356. [DOI] [PMC free article] [PubMed] [Google Scholar]