

Abstract
The SWItch Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex is a large multi-subunit protein assembly that orchestrates chromatin compaction and accessibility for gene transcription in an ATP-dependent manner. As a key epigenetic regulator, the SWI/SNF complex coordinates gene expression, cell proliferation and differentiation, and its biologic functions, in part, antagonize the polycomb repressive complex 2. The mammalian SWI/SNF complex consists of 15 subunits encoded by 29 genes, some of which are recurrently mutated in human cancers, in the germline or sporadic setting. Most SWI/SNF-deficient tumors share common “rhabdoid” cytomorphology. SMARCB1 (INI1) is the subunit most frequently inactivated in soft tissue neoplasms. Specifically, SMARCB1 deficiency is observed as the genetic hallmark in virtually all malignant rhabdoid tumors, and most cases of epithelioid sarcoma and poorly differentiated chordoma. In addition, subsets of myoepithelial carcinoma (10–40%), extraskeletal myxoid chondrosarcoma (20%), epithelioid schwannoma (40%), and epithelioid malignant peripheral nerve sheath tumor (70%) demonstrate SMARCB1 loss. The gene encoding the SS18 subunit is involved in the SS18-SSX rearrangement, which is pathognomonic of synovial sarcoma and indirectly inactivates SMARCB1. Finally, undifferentiated SMARCA4-deficient thoracic sarcomas are defined by SMARCA4 subunit inactivation, leading to SMARCA4 and SMARCA2 loss. Rarely, inactivation of alternate but biologically equivalent key regulators can substitute for canonical subunit deficiency, such as SMARCA4 inactivation in cases of SMARCB1-retained epithelioid sarcoma. This review briefly highlights SWI/SNF complex biologic functions and its roles in human cancer and provides a detailed update on recent advances in soft tissue neoplasms with canonical SWI/SNF complex deficiency, correlating morphologic, genomic, and immunohistochemical findings.
Keywords: SWI/SNF complex, BAF, Sarcoma, Soft tissue, Mesenchymal, SMARCB1, SMARCA4, SMARCA2
Introduction
As a key epigenetic regulator, the mammalian SWItch Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex coordinates chromatin compaction and accessibility for gene transcription in an ATP-dependent manner. As discussed in detail in other articles in this edition of Seminars in Diagnostic Pathology, a significant subset of human cancers harbor SWI/SNF complex perturbations,1,2 which impact tumor suppressor or oncogenic functions of one or more SWI/SNF complex subunit.3 A variety of mutational evens targeting distinct subunits is observed in specific cancers, indicating tissue-specific protective roles.3
In soft tissue tumors, the SMARCB1 (INI1, BAF47, hSNF5) core subunit encoded by SMARCB1 on 22q11.23 is most frequently targeted by genomic mutations causing loss of tumor suppressor function. These neoplasms, which will be reviewed here in detail, share common “rhabdoid” cytomorphology.4,5 Synovial sarcoma is characterized by the hallmark SS18-SSX gene fusion, which represents another mechanism of indirectly inactivating SMARCB1 function.6,7 Finally, SMARCA4 loss is identified in undifferentiated SMARCA4-deficient thoracic sarcoma, a recently discovered distinct neoplasm, and can occur as alternate event in small subsets of tumors lacking canonical SMARCB1 loss.
This review highlights what is known about the SWI/SNF complex and its roles in soft tissue neoplasms through correlation of characteristic morphologic appearances, genomic mutations, and protein deficiencies in select entities, and demonstrates how these insights enabled discovery of the first targeted treatments for SWI/SNF-deficient neoplasms.
The SWI/SNF chromatin remodeling complex and its roles in cancer
ATP-dependent chromatin modification was initially discovered in yeast,8 and subsequent studies revealed similar mechanisms in Drosophila, vertebrates, and mammals including humans.3 The mammalian SWI/SNF chromatin remodeling complex is a large multi-unit protein assembly reaching a molecular weight of ~2 MDa, and is comprised of 15 highly conserved subunits encoded by 29 genes (Fig. 1).3 The SWI/SNF complex functions as a key epigenetic modifier and coordinates chromatin compaction and decompaction to enable DNA replication and accessibility for selective gene transcription, DNA repair and recombination.3 The catalytic activity of the SWI/SNF complex fosters a state of open chromatin associated with active transcription, and biologically opposes the polycomb repressive complex 2 (PRC2)9 with distinct roles in a variety of cancers, e.g., MPNST.10,11
Fig. 1.
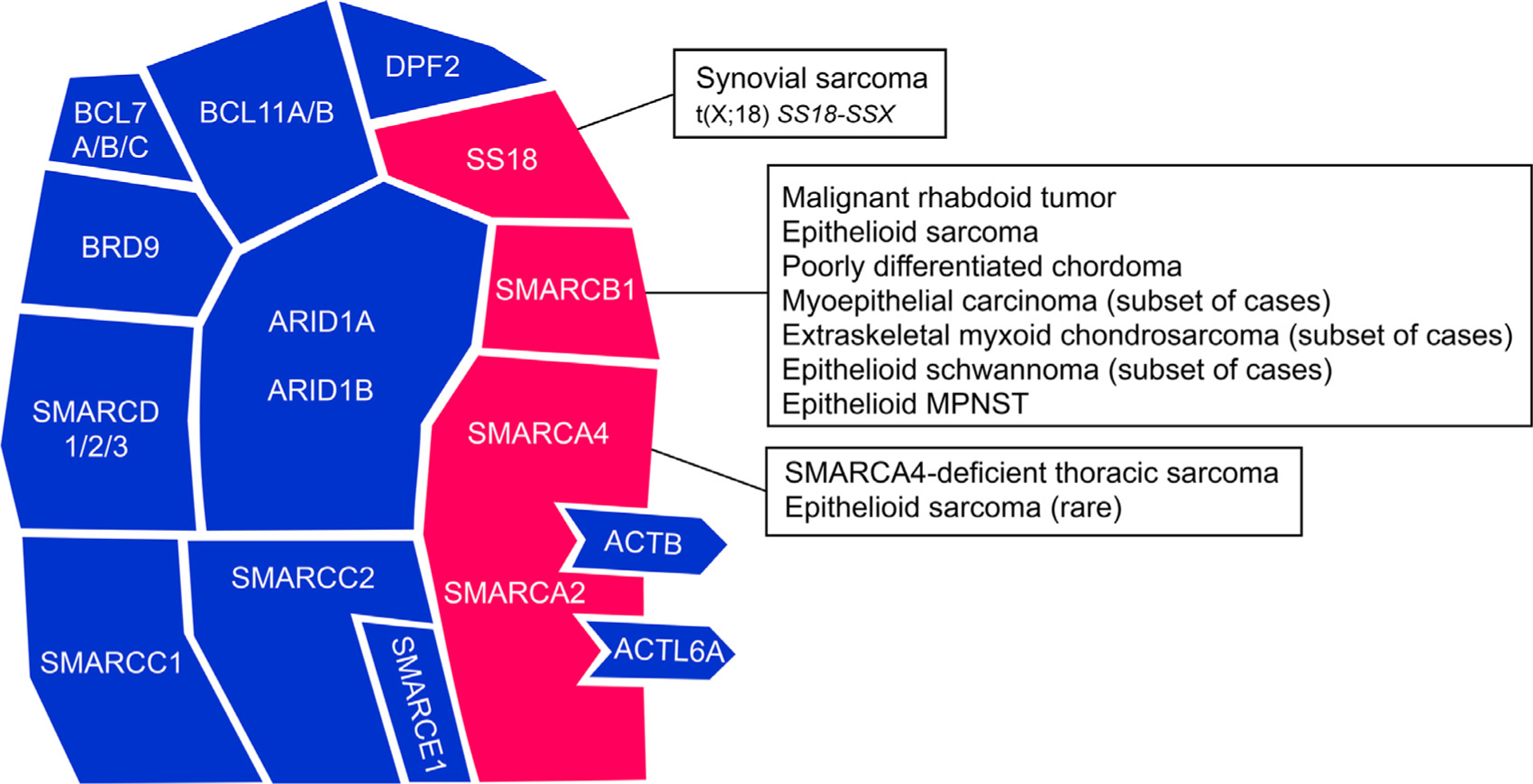
The mammalian SWI/SNF chromatin remodeling complex. This schematic illustrates the assembly of 15 subunits encoded by 29 genes. Key subunits frequently dysregulated in soft tissue neoplasms are highlighted in red; modified from: Kadoch C. and Crabtree G. R., 2015.3
More than 20% of solid tumors and hematologic malignancies in children and adults are characterized by SWI/SNF complex deficiency.1,2 Genomic mutations targeting certain subunits can be either heterozygous or homozygous, occur in the somatic or germ-line context, and include deletions, point mutations, and translocations. SWI/SNF complex dysfunction in cancer can lead to either loss-of-function of tumor suppressor or gain-of-function oncogenic mechanisms.3
SWI/SNF-deficient soft tissue neoplasms
Selected examples of SWI/SNF-deficient soft tissue neoplasms will be discussed here in detail. Most of these tumors are comprised of monomorphic undifferentiated epithelioid cells with prototypical rhabdoid cytomorphology and anaplastic large cell or small round cell features.12 Given its roles in coordinating cellular differentiation and proliferation, inactivation of the SWI/SNF complex often results in an undifferentiated cellular state, which may be responsible for the universal monotonous undifferentiated cytomorphology in most SWI/SNF-deficient neoplasms.4 Because rhabdoid morphology is characteristic of SWI/SNF-deficient neoplasms, its recognition can be helpful in the diagnostic workup of undifferentiated epithelioid (and epithelial) neoplasms, in combination with immunohistochemical testing for SWI/SNF subunit deficiency.
SMARCB1-deficient soft tissue tumors
The SMARCB1 subunit is most frequently targeted by genomic inactivation in soft tissue neoplasms. SMARCB1 deficiency is observed as the genetic hallmark in virtually all malignant rhabdoid tumors and in most cases of epithelioid sarcoma and poorly differentiated chordoma (Table 1). In addition, subsets of myoepithelial carcinoma, extraskeletal myxoid chondrosarcoma, and epithelioid peripheral nerve sheath tumors are SMARCB1-deficient (Table 1). While not discussed here in detail, the hallmark SS18-SSX oncoprotein has been shown to indirectly inactivate SMARCB1 in synovial sarcoma,6 where it leads to gain of SSX to SS18 and competes for assembly with wild-type SS18, forming an altered complex lacking the SMARCB1 tumor suppressor.7 Thereby, synovial sarcoma represents another sarcoma characterized by SWI/SNF abnormalities and loss of SMARCB1 function through impaired subunit assembly.6,7
Table 1.
Overview of mesenchymal neoplasms with SWI/SNF complex deficiency.
| Tumor | Age and sex distribution | Anatomic predilection | SWI/SNF subunit genomic inactivation |
|---|---|---|---|
| Malignant rhabdoid tumor | Infants and children, F = M | Kidney, brain, soft tissue | SMARCB1 |
| Epithelioid sarcoma | Mostly adults, M>F | Distal extremities, trunk | SMARCB1 |
| Poorly differentiated chordoma | Mostly children, F>M | Skull base/clivus, axial spine | SMARCB1 |
| Myoepithelial carcinoma | Children and adults, M>F | Extremities, limb girdle | SMARCB1 |
| Extraskeletal myxoid chondrosarcoma | Mostly adults, M>F | Extremities, limb girdle | SMARCB1 |
| Epithelioid schwannoma | Mostly adults, F = M | Trunk, extremities | SMARCB1 |
| Epithelioid MPNST | Mostly adults, F = M | Trunk, extremities | SMARCB1 |
| SMARCA4-deficient thoracic sarcoma | Mostly adults, M>F | Mediastinum, pleura, lung | SMARCA4 |
F: Female, M: Male, MPNST: Malignant peripheral nerve sheath tumor.
SMARCB1 genomic perturbations do not only occur as somatic events in cancer, but also in the germ-line setting: SMARCB1 mutations have been identified in several hereditary SWI/SNF deficiency syndromes predisposing to development of malignant rhabdoid tumor (OMIM # 609,322), familial schwannomatosis (OMIM # 162,091) or SWI/SNF-related meningiomas (OMIM # 607,174), which will not be further discussed here.5
Malignant rhabdoid tumor
Malignant rhabdoid tumor, a rare tumor occurring almost exclusively in infants and young children, is caused by SMARCB1 biallelic inactivation in virtually all cases.13–15 The discovery of SMARCB1 germline abnormalities in one third of malignant rhabdoid tumor patients shed light on SMARCB1 tumor suppressor functions and provided the earliest evidence for involvement of the SWI/SNF chromatin remodeling complex in cancer.14–16 Inactivating genomic events include point and frameshift mutations, intragenic deletions and duplications, larger deletions including regions both proximal and distal to SMARCB1, 22q deletions or translocations, and monosomy 22.14
Malignant rhabdoid tumor is a highly aggressive neoplasm predominantly arising in kidney, brain, and soft tissue in children <10 years of age.15,17 The tumors are comprised of “rhabdoid cells”, defined by eccentric vesicular nuclei with prominent nucleoli and glassy eosinophilic, inclusion-like cytoplasmic structures that represent para-nuclear aggregates of intermediate filaments on ultrastructural examination (Fig. 2). The tumor cells usually grow in solid sheets or trabeculae separated by fibrous septa, displaying a high mitotic rate and frequent necrosis. Expression of keratins and EMA is usually identified (Table 2), along with SMARCB1 loss in virtually all cases.17,18 CD99, synaptophysin, CD57, neuron-specific enolase (NSE), smooth muscle actin (SMA) and S-100 protein can be positive in subsets of cases; these markers are not diagnostically helpful in this context.
Fig. 2.
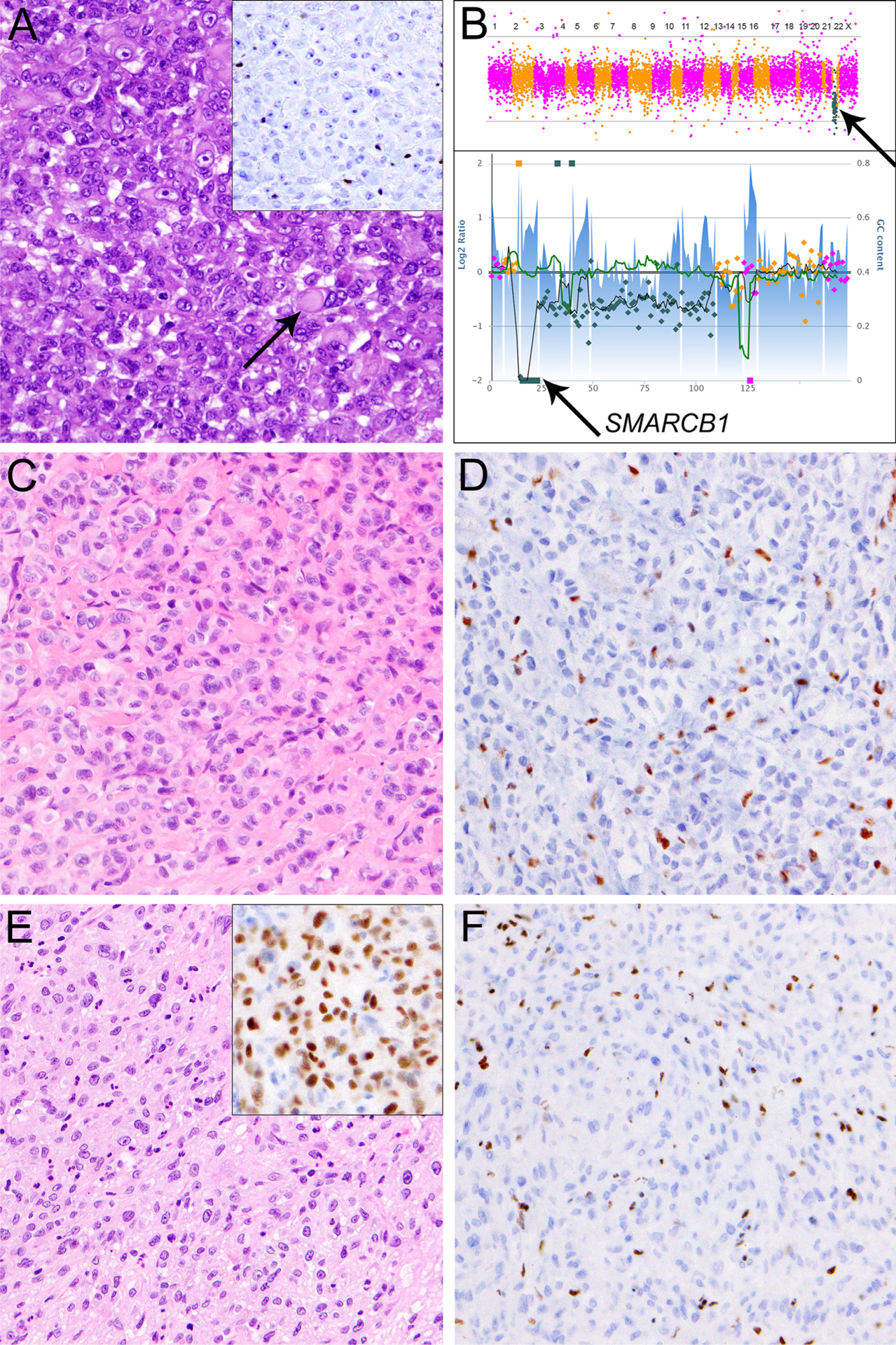
Sarcomas with near-universal SMARCB1 deficiency. A: Malignant rhabdoid tumor arising in the epididymis of a one-year-old boy, comprised of epithelioid cells with large eccentric nuclei with vesicular chromatin and prominent nucleoli, cytoplasmic eosinophilic hyaline inclusion (arrow), and SMARCB1 loss (A, inset). B: Malignant rhabdoid tumors are characterized by SMARCB1 genomic perturbations (top panel, arrow) in an otherwise relatively quiet genomic background. In this case, SMARCB1 was inactivated through homozygous deletion (bottom panel, arrow). C: Epithelioid sarcoma located in the thumb of an adult female, characterized by a proliferation of predominantly epithelioid cells with mild nuclear atypia, vesicular nuclei, small nucleoli and SMARCB1 loss (D). E: A case of poorly differentiated chordoma arising in the clivus of a 3-year-old boy, composed of tumor cells with vesicular nuclei and occasional prominent nucleoli growing in solid sheets. The tumor cells show nuclear expression of brachyury (E, inset) and SMARCB1 loss (F).
Table 2.
Characteristic morphologic appearances and useful diagnostic features and/or immunohistochemical markers in the differential diagnosis of SWI/SNF complex-deficient mesenchymal neoplasms.
| Tumor | SWI/SNF subunit protein deficiency | Positive markers | Other genetic alterations |
|---|---|---|---|
| Malignant rhabdoid tumor | SMARCB1 (100%) | Keratins, EMA, various others | – |
| Epithelioid sarcoma | SMARCB1 (90%) | Keratins, EMA, CD34 (55%) | – |
| Poorly differentiated chordoma | SMARCB1 (100%) | Brachyury (100%), keratins (100%) | – |
| Myoepithelial carcinoma | SMARCB1 (10–40%) | Myoepithelial markers (p63, SMA, GFAP, S100, SOX10), keratins, EMA | t(6;22)(p21;q12) EWSR1-POU5F1 t(19;22)(q13;q12) EWSR1-ZNF444 t(1;22)(q23;q12) EWSR1-PBX1 |
| Extraskeletal myxoid chondrosarcoma | SMARCB1 (17%) | S100 (<50%), EMA (30%), INSM1 (90%) | t(9;22)(q22;q12) EWSR1-NR4A3 t(9;17)(q22;q11) TAF2N-NR4A3 t(9;15)(q22;q21) TCF12-NR4A3 t(3;9)(q11;q22) TFG-NR4A3 t(9;17)(q22;q11) RBP56-NR4A3 |
| Epithelioid schwannoma | SMARCB1 (40%) | S100 (100%), SOX10 (100%), GFAP (40%) | – |
| Epithelioid MPNST | SMARCB1 (70%) | S100 (100%), SOX10 (100%), GFAP (60%) | – |
| SMARCA4-deficient thoracic sarcoma | SMARCA4 (100%), SMARCA2 (100%) | SOX2 (90%), SALL4 (90%), CD34 (50%) | – |
MPNST: Malignant peripheral nerve sheath tumor.
The differential diagnosis includes other tumors with rhabdoid cytomorphology and SMARCB1 deficiency discussed below. One pitfall to avoid is misinterpretation of FISH results in tumors with 22q11–12 deletions involving SMARCB1. As demonstrated in cases of SMARCB1-deficient soft tissue neoplasms, co-deletions of EWSR1 at 22q12.2 may be incorrectly interpreted as equivalent to EWSR1 rearrangement.19 Clinicopathologic correlation and supplementing EWSR1 FISH with other methods may be required in this context.20
Patients with malignant rhabdoid tumor develop distant metastases in 50–80% of cases, with 5-year-survival rates of 15–20%.21,22
Epithelioid sarcoma
Epithelioid sarcoma occurs mostly in the distal extremities of patients with a median age of 30–40 years and slight male predominance. Histologically, this tumor is composed of epithelioid and spindled tumor cells arranged in cellular nodules with occasional central necrosis, resulting in a pseudogranulomatous appearance. The tumor cells show mild nuclear atypia, vesicular nuclei, and small nucleoli with a low mitotic rate and are often surrounded by abundant intercellular collagen (Fig. 2). Perineural and vascular infiltration are common. Epithelioid sarcoma is generally classified into a conventional (“distal”) type with classic cytomorphology and a less frequent, proximal (“large cell”) type displaying larger epithelioid cells with eosinophilic to amphophilic cytoplasm, marked nuclear atypia, vesicular nuclei, and prominent nucleoli; this variant typically affects somewhat older patients and is associated with a more aggressive clinical course.
FISH analysis demonstrates SMARCB1 genomic inactivation through homozygous deletion in 90% of epithelioid sarcomas,23,24 along with SMARCB1 loss in 90% of cases, irrespective of histologic subtype. Most cases show expression of keratins and EMA. CD34 expression in >50% of epithelioid sarcomas is helpful to rule out metastatic carcinoma (Table 2). Rare cases of SMARCB1-retained epithelioid sarcoma may show perturbations of alternate SWI/SNF subunits such as deficiency of SMARCA4, SMARCC2, or SMARCC1.25 However, comprehensive genomic studies in larger case series are required to fully characterize the spectrum of SWI/SNF perturbations in rare epithelioid sarcomas with retained SMARCB1.
More than half of patients with epithelioid sarcoma develop metastases, with 5- and 10-year-survival rates of 70% and 42%, respectively.26 As discussed below, novel therapeutic approaches targeting SWI/SNF complex deficiency across a wide range of neoplasms might provide clinical benefit for some patients with this rare tumor type.
Poorly differentiated chordoma
Chordomas are rare malignant bone tumors of notochordal origin that usually occur in the skull base and clivus, cervical spine, sacrum or coccyx of adult patients with a male predominance. Chordomas can metastasize to soft tissue sites and, very rarely, arise primarily in somatic soft tissue as “chordoma periphericum”.27 Chordomas are classified into conventional, chondroid, dedifferentiated, and poorly differentiated subtypes. The latter variant is extremely rare, occurs predominantly in children (median age, 11 years), and behaves more aggressively than conventional chordoma.28 Poorly differentiated chordoma does not resemble conventional chordoma; this variant shows unusual histologic features that include sheet-like growth with limited stroma and tumor cells with vesicular nuclei, prominent nucleoli, and frequent mitotic figures (Fig. 2).29 As demonstrated recently, virtually all cases of poorly differentiated chordoma show SMARCB1 loss, which is generally retained in conventional chordoma.28 In addition, they express keratins and nuclear brachyury; the latter is highly specific and helps in the distinction from metastatic carcinoma, while S-100 protein is generally not expressed (Table 2).28–30 Homozygous deletions have been identified by genetic analyses as the most important mechanism of SMARCB1 genomic inactivation in poorly differentiated chordoma.31,32
Poorly differentiated chordoma has a mean overall survival of 53% and requires aggressive management with multimodality therapy.28
Myoepithelial carcinoma
Myoepithelial carcinomas of soft tissue develop predominantly in the limbs and limb girdles, and less frequently in the head and neck, trunk, and visceral soft tissues of middle-aged adults with a median age of 40–50 years and slight male predominance. They can present with varied morphologies, including lobulated architecture composed of epithelioid to spindled tumor cells arranged in cords, strands or nests in a hyalinized or chondromyxoid matrix (Fig. 3). The tumor cells may display classic rhabdoid cytomorphology with abundant eosinophilic cytoplasm or a plasmacytoid appearance with prominent hyaline cytoplasmic inclusions. Some myoepithelial carcinomas, particularly when arising in children, contain an undifferentiated round cell component. Expression of one or more myoepithelial markers (e.g., p63, SOX10, SMA, GFAP, and/or S-100 protein) is present to varying extent, as well as keratins and EMA in most cases (Table 2). SMARCB1 loss is identified in up to 40% of cases, especially in children.33 SMARCB1 homozygous deletions have been identified by FISH in a subset of SMARCB1-deficient cases.23
Fig. 3.
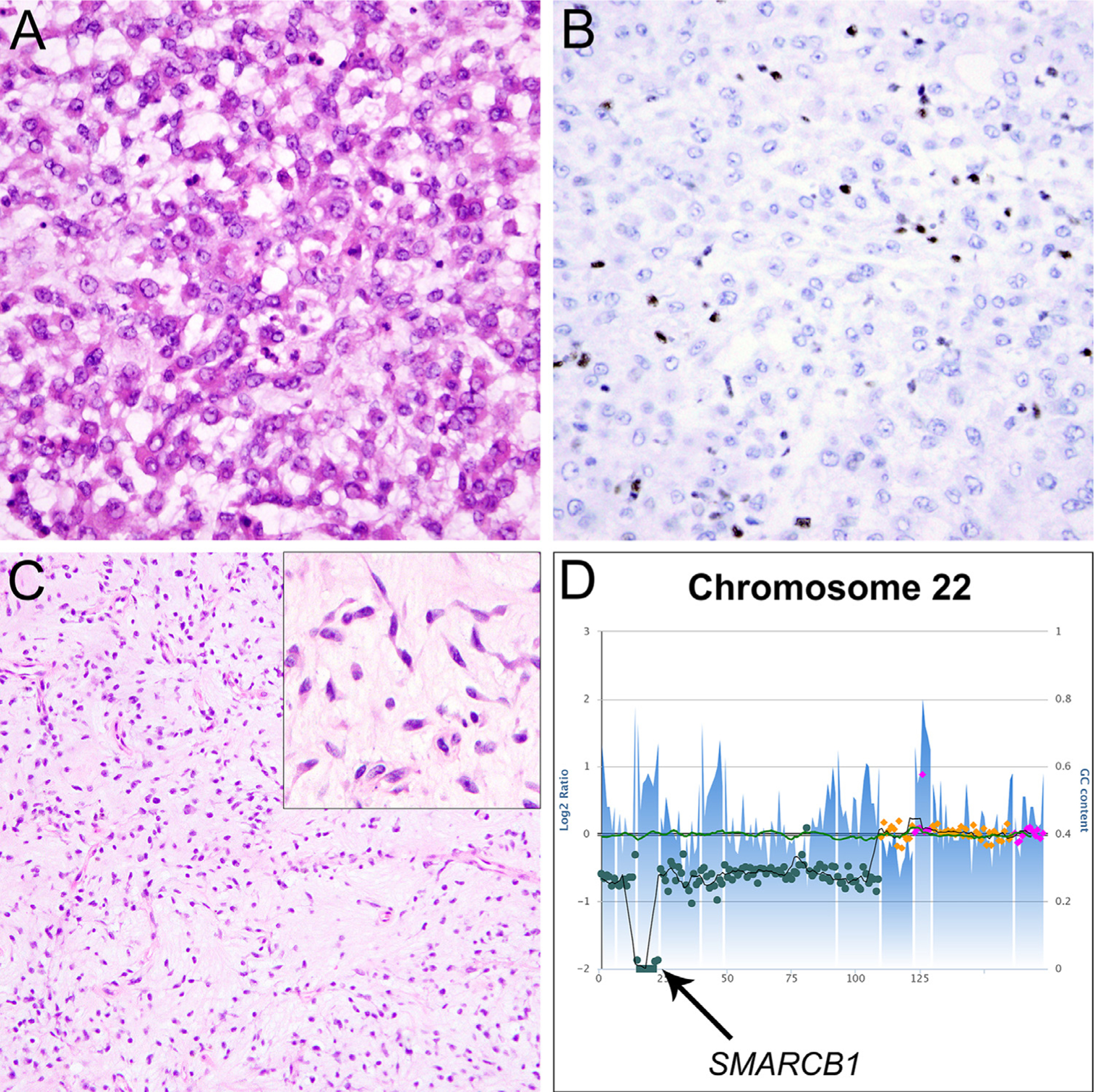
Sarcomas with occasional SMARCB1 deficiency. A: Myoepithelial carcinoma located in the lower leg of an adult female, characterized by epithelioid tumor cells with round nuclei and bright eosinophilic cytoplasm with occasional rhabdoid morphology, arranged in a reticular growth pattern in a background of myxohyaline stroma. B: The tumor cells are negative for SMARCB1. C: This case of extraskeletal myxoid chondrosarcoma arose in the pubic region of an adult female and is comprised of epithelioid to spindled tumor cells with moderate amounts of eosinophilic cytoplasm arranged in cords and strands, embedded in abundant myxoid stroma; in areas, the tumor cells appeared to be interconnected (C, inset). D: Targeted next-generation sequencing (NGS) demonstrated a homozygous deletion of SMARCB1 on chromosome 22 (arrow).
EWSR1 gene fusions are found in about 50% of myoepithelial tumors of soft tissue, including t(1;22)(q23;q12) resulting in EWSR1-PBX1 fusion,34 t(19;22)(q13;q12) resulting in EWSR1-ZNF444 fusion35 or t(6;22)(p21;q12) resulting in EWSR1-POU5F1 fusion,36 which might each be associated with distinct morphologies (Table 2). EWSR1 rearrangement does not appear to be mutually exclusive with SMARCB1 inactivation,37 and given the proximity of both gene loci on 22q, it seems conceivable that both EWSR1 and SMARCB1 would be disrupted simultaneously by a single genomic (translocation) event. However, the association between EWSR1 fusions and SMARCB1 perturbations in myoepithelial carcinomas remains to be characterized.
Local recurrence is observed in approximately 40% of patients with myoepithelial carcinoma, and distant metastases (mostly to lung, lymph nodes and soft tissues) develop in 30% of patients.38 These rates approach 50% in pediatric patients, in whom myoepithelial carcinomas behave more aggressively.
Extraskeletal myxoid chondrosarcoma
Extraskeletal myxoid chondrosarcoma is a rare malignant mesenchymal neoplasm of uncertain differentiation that usually occurs in adults with a median age of 50 years, being more common in males.39 Despite its name, this tumor type has no relationship to cartilage. Most tumors arise in deep soft tissue of the proximal extremities and limb girdles. Histologically, extraskeletal myxoid chondrosarcoma displays a multinodular architecture comprised of hypocellular tumor lobules separated by fibrous septa (Fig. 3). The tumor cells have round to ovoid nuclei with inconspicuous nucleoli and moderate amounts of eosinophilic cytoplasm with a low mitotic rate. They are arranged in clusters and interconnected strands, embedded in a myxoid or chondromyxoid matrix. Extraskeletal myxoid chondrosarcomas express S-100 protein in 20–50% of cases and EMA in about 30% of cases (Table 2).40 In addition, expression of INSM1 has recently been described in 90% of extraskeletal myxoid chondrosarcomas.41 One study demonstrated loss of SMARCB1 expression in 4/24 (17%) cases, of which three cases showed rhabdoid cytomorphology.42 Two of those 4 SMARCB1-deficient extraskeletal myxoid chondrosarcomas revealed SMARCB1 genomic inactivation through homozygous deletion and frameshift mutation, respectively. However larger studies correlating protein expression and genomic findings are required to fully assess the true frequency of SWI/SNF deficiency in this rare sarcoma type.
Extraskeletal myxoid chondrosarcoma is characterized by recurrent translocations involving NR4A3, with t(9;22)(q22;q12) resulting in EWSR1-NR4A3 fusion43 being most common (Table 2), followed by alternate fusions of NR4A3 with TAF2N, TCF12,44 TFG,45 or RBP56.46 It remains to be determined whether EWSR1-NR4A3 fusions may be found predominantly in the subset of cases showing SMARCB1 deficiency, or whether these genetic perturbations occur independently. Extraskeletal myxoid chondrosarcoma develops metastases in ~50% of patients with 5- and 10-year-survival rates of 91% and 84%, respectively.47
A recently reported group of rare primary intracranial neoplasms enters the differential diagnosis of extraskeletal myxoid chondrosarcoma.48 Two cases of primary intracranial myxoid sarcomas with SMARCB1 loss and monosomy 22q have been reported, and in a third case with intact SMARCB1, an EWSR1-CREB1 gene fusion was demonstrated by NGS.48 None of these cases showed NR4A3 rearrangement, suggesting oncogenic pathways distinct from (conventional) extraskeletal myxoid chondrosarcoma. Follow-up studies with large series are required to determine the biologic behavior of these rare neoplasms and aid in their classification.
Benign and malignant epithelioid peripheral nerve sheath tumors
Epithelioid schwannoma and epithelioid malignant peripheral nerve sheath tumor (MPNST) are rare variants of nerve sheath tumors that exhibit distinct morphologic and immunohistochemical features, which clearly distinguish them from their conventional schwannoma and MPNST counterparts. Loss of SMARCB1 expression is demonstrated in 40% of epithelioid schwannomas and 70% of epithelioid MPNST (Table 2).49–51
The epithelioid variant of schwannoma affects mostly adult patients with a median age of 45 years and no gender predilection. The extremities and trunk are the most common sites of involvement; some cases show both conventional and epithelioid morphologies.50 Epithelioid schwannoma shows multilobulated growth of uniform epithelioid cells in sheets and nests or singly dispersed within a frequently myxoid or hyalinized stroma.50 The tumor cells exhibit round vesicular nuclei and abundant pale eosinophilic cytoplasm, without significant pleomorphism or hyperchromasia (Fig. 4).
Fig. 4.
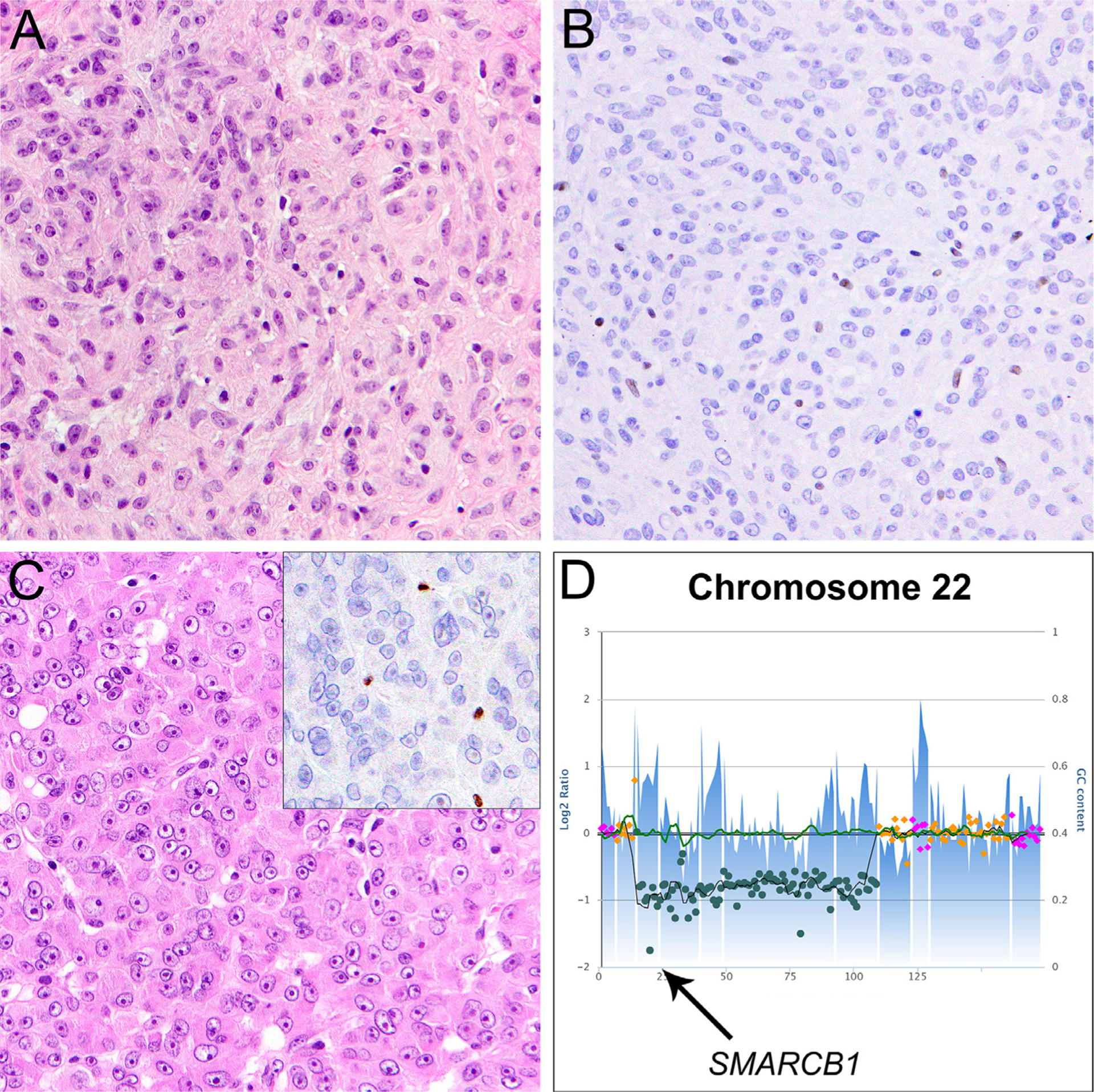
SMARCB1-deficient epithelioid peripheral nerve sheath tumors. A: A case of epithelioid schwannoma located on the arm of a young male. This tumor is comprised of uniform epithelioid or ovoid tumor cells with vesicular nuclei, small nucleoli, and abundant pale eosinophilic cytoplasm without significant mitotic activity, nuclear atypia, or necrosis. B: Expression of SMARCB1 is lost in tumor cells. This tumor harbored homozygous deletion of SMARCB1 exon 1–4 (not shown). C: Epithelioid malignant peripheral nerve sheath tumor (MPNST) arising in the lower leg of a young female. The tumor is composed of monotonous epithelioid tumor cells with prominent nucleoli and abundant cytoplasm with unequivocal nuclear atypia as well as frequent mitoses and necrosis. SMARCB1 expression was lost in tumor cells (C, inset). D: Targeted NGS revealed a localized homozygous deletion on chromosome 22 targeting the SMARCB1 coding region (arrow).
Epithelioid MPNST is a distinctive variant that differs biologically and morphologically from conventional MPNST. While conventional MPNST is associated with neurofibromatosis type 1 in about half of cases, epithelioid MPNST generally does not arise in patients with neurofibromatosis type 1.49 Most patients are adults, with a median age of 44 years and equal sex distribution.49 Epithelioid MPNST usually occur as superficial masses in the extremities and trunk (although some may be deep-seated or in visceral sites).49,52 Occasional origin in a pre-existing schwannoma or neurofibroma has been reported.49, 52, 53 They demonstrate a lobulated growth pattern within a variably myxoid or fibrous stroma and are comprised of a generally uniform but clearly atypical population of polygonal cells with round vesicular nuclei, prominent nucleoli, and abundant amphophilic or pale eosinophilic cytoplasm (Fig. 4).49 Malignant features include the presence of cytologic atypia, significant mitotic activity, and frequent tumor necrosis. Based on these criteria, the distinction of epithelioid schwannoma from epithelioid MPNST is usually straightforward, but occasional epithelioid peripheral nerve sheath tumors cannot be easily classified as benign or malignant; there likely exists a morphologic continuum.50
In addition to frequent SMARCB1 deficiency, both epithelioid schwannoma and epithelioid MPNST are characterized by strong and diffuse positivity for S-100 protein and SOX10 (Table 2) (which distinguishes epithelioid MPNST from conventional MPNST, in which expression of these markers is generally limited in extent or absent).49,50 NGS identified recurrent SMARCB1 genomic inactivation in both entities, through homozygous deletion, nonsense, frameshift or splice site mutations targeting the SMARCB1 gene.54 Epithelioid MPNST lacks canonical aberrations of the PRC2 components SUZ12 and EED, in line with retained H3K27me3 expression in these tumors, and less frequently harbors NF1 mutations than conventional MPNST.54 Perturbations affecting other SWI/SNF subunits have not been identified in the subgroup of SMARCB1-retained epithelioid nerve sheath tumors.54 Additional studies are required to determine whether epithelioid schwannoma can give rise to epithelioid MPNST and which epigenetic/genomic events drive malignant transformation.
Epithelioid schwannomas behave in a benign fashion, and epithelioid MPNST appear to have a low risk for recurrence and metastasis, irrespective of tumor depth, with a much more favorable prognosis than conventional MPNST.49
Given substantial morphologic overlap and characteristic strong and diffuse expression of S-100 protein and SOX10, distinguishing between epithelioid MPNST and metastatic amelanotic melanoma may be challenging. In this context, SMARCB1 deficiency may be useful, as melanomas generally show intact SMARCB1 expression.24 Although heterozygous SMARCB1 missense mutations have been reported in a small subset of melanomas, such mutations do not lead to SMARCB1 loss as they are not completely inactivating.55
SMARCA4-deficient soft tissue tumors
SMARCA4 inactivation was identified by RNA sequencing analyses in a subset of undifferentiated malignancies presenting as compressive mediastinal/pulmonary masses in adults with very aggressive clinical behavior and median survival of 7 months.56 The fact that SMARCA4-deficient thoracic sarcomas shared common morphologic features and gene expression profiles distinct from other SMARCA4-deficient neoplasms led to their recognition as a distinct entity. These tumors occur in adult patients with a median age of 40–50 years (range, 28–90 years) and a marked male predominance; they generally form large compressive masses located in the mediastinum, pleura, or lung.57–59 SMARCA4 genomic inactivation results from nonsense, frameshift, missense, and splice-site mutations.56
SMARCA4-deficient thoracic sarcoma is composed of diffuse sheets of relatively monotonous undifferentiated epithelioid cells with prominent nucleoli57 and frequent rhabdoid features (Fig. 5).58 Concomitant loss of SMARCA4 and SMARCA2 expression with retained SMARCB1 is found in all cases, along with expression of SOX2, CD34, and SALL4 in >80% of cases, and negativity for the tight junction protein claudin-4.57,58 While distinction of undifferentiated SMARCA4-deficient sarcoma from SMARCA4-deficient carcinomas can be challenging, expression of CD34 and lack of claudin-4 help support mesenchymal over epithelial differentiation in this context, although the precise nosologic status of this tumor type remains controversial.
Fig. 5.
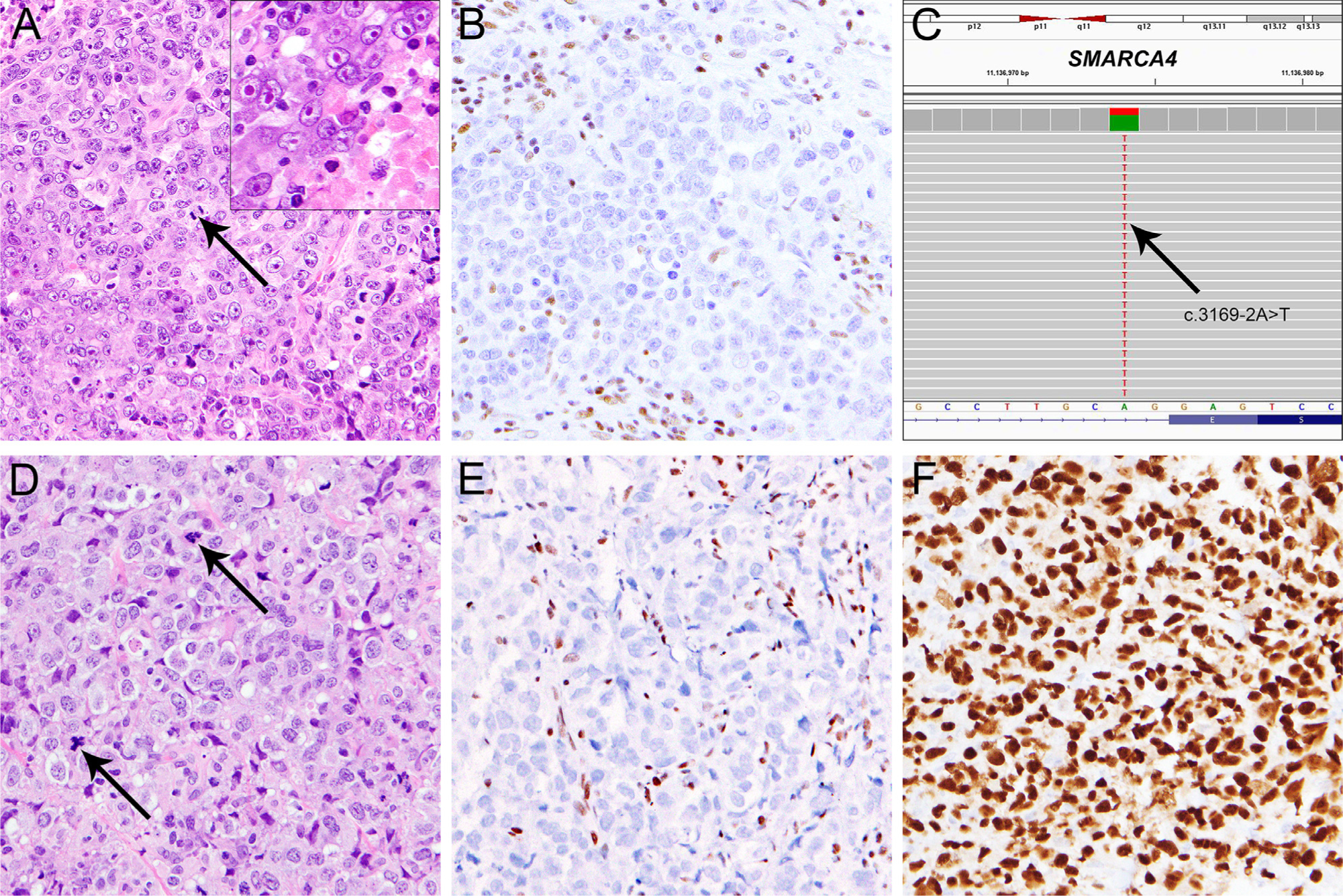
SMARCA4-deficient undifferentiated malignancies. A: SMARCA4-deficient undifferentiated thoracic sarcoma arising in the lung of an adult male, comprised of malignant epithelioid cells growing in sheets with a high degree of mitotic activity (A, arrow), areas of tumor necrosis (A, inset), and occasionally prominent eosinophilic nucleoli (A, inset) with SMARCA4 loss in tumor cells (B). C: Targeted NGS identified a heterozygous splice-site mutation inactivating SMARCA4 (c.3169–2A>T, arrow). D: SMARCA4-deficient undifferentiated sarcoma arising in the base of the tongue of an adult female, comprised of plump tumor cells with epithelioid and pleomorphic cytomorphology, irregularly shaped large nuclei, and moderate amounts of eosinophilic cytoplasm, growing in sheets, with frequent mitoses (D, arrow) and apoptotic bodies. E: Expression of SMARCA4 was lost in tumor cells. The tumor cells expressed strong and diffuse SOX2 (F).
In addition, subsets of undifferentiated sarcomas with similar morphologic features arising in other anatomic locations (Fig. 5) can show SMARCA4/SMARCA2 loss along with SOX2 positivity; their clinical behavior and relationship to SMARCA4-deficient thoracic sarcomas remain to be determined.
Non-mesenchymal neoplasms with SWI/SNF complex inactivation
The SWI/SNF subunit most frequently dysregulated in human cancer is ARID1A, which interacts with DNA to facilitate SWI/SNF binding to target sites.60 ARID1A mutations are demonstrated across a wide range of tumors including ~45% of endometrioid and clear-cell ovarian carcinomas, ~20% of gastric and bladder carcinomas, ~14% of hepatocellular carcinomas, ~10% of colorectal carcinomas and melanomas, and less frequently in lung, pancreatic, and breast carcinomas.2 ARID1A inactivation is also found in pediatric neuroblastoma,61 and dedifferentiated meningioma.62 Genomic inactivation of SMARCA4, ARID1A, ARID1B, ARID2 and PBRM1 is also frequent in cancer and includes mutations of SMARCA4 and SMARCA2 in non-small cell lung carcinomas,63–65 undifferentiated/rhabdoid carcinomas of the gastrointestinal66 and urothelial tract67 and uterus,68 and in rare sinonasal carcinomas,69 which are discussed in detail in other articles in this edition.
Based on significant morphologic and immunohistochemical overlap, the distinction between SWI/SNF-deficient soft tissue tumors and SWI/SNF-deficient undifferentiated carcinomas may be challenging. In this context, staining for the epithelial tight junction protein claudin-4 may be useful, as SWI/SNF-deficient soft tissue neoplasms generally lack claudin-4 expression, whereas some (but not all) SWI/SNF-deficient undifferentiated carcinomas show expression of claudin-4.70
Metastatic SWI/SNF-deficient carcinoma represents a major differential diagnosis of SWI/SNF-deficient soft tissue neoplasms. Detection of a SWI/SNF-deficient malignant neoplasm at unusual sites should therefore prompt additional clinical workup to identify a possible visceral primary tumor. The type of SWI/SNF subunit deficiency may help narrow the differential diagnosis and identify possible or likely primary sites.
In undifferentiated carcinomas with a complex genomic landscape and high mutational burden, SWI/SNF inactivation may have different implications than in soft tissue neoplasms, in which SWI/SNF mutations often represent one of very few genomic perturbations and may have essential functions in tumor development/progression. In many cancers, SWI/SNF perturbations do not necessarily represent oncogenic drivers but are rather passenger mutations.
Therapeutic targeting of SWI-SNF-deficient soft tissue neoplasms
Based on functional antagonism of the SWI/SNF and PRC2 complexes, SMARCB1 deficiency results in loss of inhibition of the EZH2 methyltransferase, and subsequent PRC2-mediated oncogenesis.71,72 Studies in SMARCB1-deficient malignant rhabdoid tumor73–75 and epithelioid sarcoma76 demonstrated early antiproliferative effects of therapeutic EZH2 inhibition with durable responses. A subsequent phase II trial achieved clinical responses in 15% of 67 patients with SMARCB1-deficient epithelioid sarcoma, of which 67% of responses lasted at least 6 months, leading to accelerated FDA approval of the first EZH2-inhibitor tazemetostat in January 2020 for the treatment of patients aged ≥16 years with locally advanced or metastatic epithelioid sarcoma not eligible for complete resection.77–79
Conclusions
SWI/SNF-deficient soft tissue neoplasms share common undifferentiated “rhabdoid” appearances and include malignant rhabdoid tumor as the prototypical example, as well as various other benign and malignant soft tissue tumors in children and adults. The wide range of seemingly unrelated tumor types affected by SWI/SNF mutations, and the spectrum of genomic events targeting distinct subunits, highlight the biologic complexity of the roles of SWI/SNF in cancer. Substantial advances in the past few years have expanded our understanding of SWI/SNF-deficient tumors and resulted in novel therapeutic approaches for a variety of malignancies unified by SWI/SNF-deficiency. Future studies are required to systematically determine the frequencies of SWI/SNF subunit inactivation on the genomic and protein level and to generate insights that help characterize in which biologic context these perturbations foster tumor development and/or progression.
Acknowledgments
The authors would like to thank Dr. Christopher D. M. Fletcher, Department of Pathology, Brigham and Women’s Hospital, for contributing some of the cases illustrated, and the Center for Advanced Molecular Diagnostics, Department of Pathology, Brigham and Women’s Hospital, for providing selected images.
Funding
This work was supported by the National Institutes of Health/National Cancer Institute (I.-M. Schaefer, grant number K08 CA241085-01).
Footnotes
Declaration of Competing Interest
Jason L. Hornick is a consultant to Epizyme.
References
- 1.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE. 2013;8:e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1:e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agaimy A SWI/SNF complex-deficient soft tissue neoplasms: a pattern-based approach to diagnosis and differential diagnosis. Surg Pathol Clin. 2019;12:149–163. [DOI] [PubMed] [Google Scholar]
- 5.Agaimy A, Foulkes WD. Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol. 2018;35:193–198. [DOI] [PubMed] [Google Scholar]
- 6.McBride MJ, Pulice JL, Beird HC, et al. The SS18-SSX fusion oncoprotein hijacks baf complex targeting and function to drive synovial sarcoma. Cancer Cell. 2018;33:1128–1141 e1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stern M, Jensen R, Herskowitz I. Five SWI genes are required for expression of the HO gene in yeast. J Mol Biol. 1984;178:853–868. [DOI] [PubMed] [Google Scholar]
- 9.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57–67. [DOI] [PubMed] [Google Scholar]
- 10.De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. [DOI] [PubMed] [Google Scholar]
- 11.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agaimy A What is new in epithelioid soft tissue tumors. Virchows Arch. 2020;476:81–96. [DOI] [PubMed] [Google Scholar]
- 13.Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. [DOI] [PubMed] [Google Scholar]
- 14.Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011;56:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biegel JA, Fogelgren B, Wainwright LM, Zhou JY, Bevan H, Rorke LB. Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer. 2000;28:31–37. [DOI] [PubMed] [Google Scholar]
- 16.Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. 2014;166C:350–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002;8:3461–3467. [PubMed] [Google Scholar]
- 18.Bourdeaut F, Freneaux P, Thuille B, et al. hSNF5/INI1-deficient tumours and rhabdoid tumours are convergent but not fully overlapping entities. J Pathol. 2007;211:323–330. [DOI] [PubMed] [Google Scholar]
- 19.Huang SC, Zhang L, Sung YS, et al. Secondary EWSR1 gene abnormalities in SMARCB1-deficient tumors with 22q11–12 regional deletions: potential pitfalls in interpreting EWSR1 FISH results. Genes Chromosomes Cancer. 2016;55:767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valencia AM, Collings CK, Dao HT, et al. Recurrent SMARCB1 mutations reveal a nucleosome acidic patch interaction site that potentiates mSWI/SNF complex chromatin remodeling. Cell. 2019;179:1342–1356 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kodet R, Newton WA Jr, Sachs N, et al. Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol. 1991;22:674–684. [DOI] [PubMed] [Google Scholar]
- 22.Fanburg-Smith JC, Hengge M, Hengge UR, Smith JS Jr, Miettinen M. Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immunohistochemical study of 18 cases. Ann Diagn Pathol. 1998;2:351–362. [DOI] [PubMed] [Google Scholar]
- 23.Le Loarer F, Zhang L, Fletcher CD, et al. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer. 2014;53:475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33:542–550. [DOI] [PubMed] [Google Scholar]
- 25.Kohashi K, Yamamoto H, Yamada Y, et al. SWI/SNF chromatin-remodeling complex status in SMARCB1/INI1-preserved Epithelioid Sarcoma. Am J Surg Pathol. 2018;42:312–318. [DOI] [PubMed] [Google Scholar]
- 26.Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218–225. [DOI] [PubMed] [Google Scholar]
- 27.Nielsen GP, Mangham DC, Grimer RJ, Rosenberg AE. Chordoma periphericum: a case report. Am J Surg Pathol. 2001;25:263–267. [DOI] [PubMed] [Google Scholar]
- 28.Shih AR, Cote GM, Chebib I, et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod Pathol. 2018;31:1237–1245. [DOI] [PubMed] [Google Scholar]
- 29.Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol. 2006;30:811–818. [DOI] [PubMed] [Google Scholar]
- 30.Tirabosco R, Mangham DC, Rosenberg AE, et al. Brachyury expression in extra-axial skeletal and soft tissue chordomas: a marker that distinguishes chordoma from mixed tumor/myoepithelioma/parachordoma in soft tissue. Am J Surg Pathol. 2008;32:572–580. [DOI] [PubMed] [Google Scholar]
- 31.Owosho AA, Zhang L, Rosenblum MK, Antonescu CR. High sensitivity of FISH analysis in detecting homozygous SMARCB1 deletions in poorly differentiated chordoma: a clinicopathologic and molecular study of nine cases. Genes Chromosomes Cancer. 2018;57:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih AR, Chebib I, Deshpande V, Dickson BC, Iafrate AJ, Nielsen GP. Molecular characteristics of poorly differentiated chordoma. Genes Chromosomes Cancer. 2019;58:804–808. [DOI] [PubMed] [Google Scholar]
- 33.Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol. 2007;31:1813–1824. [DOI] [PubMed] [Google Scholar]
- 34.Brandal P, Panagopoulos I, Bjerkehagen B, et al. Detection of a t(1;22)(q23;q12) translocation leading to an EWSR1-PBX1 fusion gene in a myoepithelioma. Genes Chromosomes Cancer. 2008;47:558–564. [DOI] [PubMed] [Google Scholar]
- 35.Brandal P, Panagopoulos I, Bjerkehagen B, Heim S. t(19;22)(q13;q12) Translocation leading to the novel fusion gene EWSR1-ZNF444 in soft tissue myoepithelial carcinoma. Genes Chromosomes Cancer. 2009;48:1051–1056. [DOI] [PubMed] [Google Scholar]
- 36.Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010;49:1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thway K, Bown N, Miah A, Turner R, Fisher C. Rhabdoid variant of myoepithelial carcinoma, with EWSR1 Rearrangement: expanding the spectrum of EWSR1-rearranged myoepithelial tumors. Head Neck Pathol. 2015;9:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 2003;27:1183–1196. [DOI] [PubMed] [Google Scholar]
- 39.Meis-Kindblom JM, Bergh P, Gunterberg B, Kindblom LG. Extraskeletal myxoid chondrosarcoma: a reappraisal of its morphologic spectrum and prognostic factors based on 117 cases. Am J Surg Pathol. 1999;23:636–650. [DOI] [PubMed] [Google Scholar]
- 40.Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011;35:e47–e63. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida A, Makise N, Wakai S, Kawai A, Hiraoka N. INSM1 expression and its diagnostic significance in extraskeletal myxoid chondrosarcoma. Mod Pathol. 2018;31:744–752. [DOI] [PubMed] [Google Scholar]
- 42.Kohashi K, Oda Y, Yamamoto H, et al. SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: a special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am J Surg Pathol. 2008;32:1168–1174. [DOI] [PubMed] [Google Scholar]
- 43.Labelle Y, Zucman J, Stenman G, et al. Oncogenic conversion of a novel orphan nuclear receptor by chromosome translocation. Hum Mol Genet. 1995;4:2219–2226. [DOI] [PubMed] [Google Scholar]
- 44.Agaram NP, Zhang L, Sung YS, Singer S, Antonescu CR. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Hum Pathol. 2014;45:1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hisaoka M, Ishida T, Imamura T, Hashimoto H. TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer. 2004;40:325–328. [DOI] [PubMed] [Google Scholar]
- 46.Panagopoulos I, Mencinger M, Dietrich CU, et al. Fusion of the RBP56 and CHN genes in extraskeletal myxoid chondrosarcomas with translocation t(9;17)(q22;q11). Oncogene. 1999;18:7594–7598. [DOI] [PubMed] [Google Scholar]
- 47.Ogura K, Fujiwara T, Beppu Y, et al. Extraskeletal myxoid chondrosarcoma: a review of 23 patients treated at a single referral center with long-term follow-up. Arch Orthop Trauma Surg. 2012;132:1379–1386. [DOI] [PubMed] [Google Scholar]
- 48.Velz J, Agaimy A, Frontzek K, et al. Molecular and clinicopathologic heterogeneity of intracranial tumors mimicking extraskeletal myxoid chondrosarcoma. J Neuropathol Exp Neurol. 2018;77:727–735. [DOI] [PubMed] [Google Scholar]
- 49.Jo VY, Fletcher CD. Epithelioid malignant peripheral nerve sheath tumor: clinicopathologic analysis of 63 cases. Am J Surg Pathol. 2015;39:673–682. [DOI] [PubMed] [Google Scholar]
- 50.Jo VY, Fletcher CDM. SMARCB1/INI1 loss in epithelioid schwannoma: a clinicopathologic and immunohistochemical study of 65 Cases. Am J Surg Pathol. 2017;41:1013–1022. [DOI] [PubMed] [Google Scholar]
- 51.Hart J, Gardner JM, Edgar M, Weiss SW. Epithelioid schwannomas: an analysis of 58 cases including atypical variants. Am J Surg Pathol. 2016;40:704–713. [DOI] [PubMed] [Google Scholar]
- 52.Rekhi B, Kosemehmetoglu K, Tezel GG, Dervisoglu S. Clinicopathologic features and immunohistochemical spectrum of 11 cases of epithelioid malignant peripheral nerve sheath tumors, including INI1/SMARCB1 results and BRAF V600E analysis. APMIS. 2017;125:679–689. [DOI] [PubMed] [Google Scholar]
- 53.Asano N, Yoshida A, Ichikawa H, et al. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology 2017;70:385–393. [DOI] [PubMed] [Google Scholar]
- 54.Schaefer IM, Dong F, Garcia EP, Fletcher CDM, Jo VY. Recurrent SMARCB1 Inactivation in epithelioid malignant peripheral nerve sheath tumors. Am J Surg Pathol. 2019;43:835–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stockman DL, Curry JL, Torres-Cabala CA, et al. Use of clinical next-generation sequencing to identify melanomas harboring SMARCB1 mutations. J Cutan Pathol. 2015;42:308–317. [DOI] [PubMed] [Google Scholar]
- 56.Le Loarer F, Watson S, Pierron G, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet. 2015;47:1200–1205. [DOI] [PubMed] [Google Scholar]
- 57.Yoshida A, Kobayashi E, Kubo T, et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities. Mod Pathol. 2017;30:797–809. [DOI] [PubMed] [Google Scholar]
- 58.Perret R, Chalabreysse L, Watson S, et al. SMARCA4-deficient Thoracic Sarcomas: clinicopathologic study of 30 cases with an emphasis on their nosology and differential diagnoses. Am J Surg Pathol. 2019;43:455–465. [DOI] [PubMed] [Google Scholar]
- 59.Sauter JL, Graham RP, Larsen BT, Jenkins SM, Roden AC, Boland JM. SMARCA4-deficient thoracic sarcoma: a distinctive clinicopathological entity with undifferentiated rhabdoid morphology and aggressive behavior. Mod Pathol. 2017;30:1422–1432. [DOI] [PubMed] [Google Scholar]
- 60.Chandler RL, Brennan J, Schisler JC, Serber D, Patterson C, Magnuson T. ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol Cell Biol. 2013;33:265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet. 2013;45:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abedalthagafi MS, Bi WL, Merrill PH, et al. ARID1A and TERT promoter mutations in dedifferentiated meningioma. Cancer Genet. 2015;208:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xue Y, Meehan B, Fu Z, et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat Commun. 2019;10:557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agaimy A, Fuchs F, Moskalev EA, Sirbu H, Hartmann A, Haller F. SMARCA4-deficient pulmonary adenocarcinoma: clinicopathological, immunohistochemical, and molecular characteristics of a novel aggressive neoplasm with a consistent TTF1(neg)/CK7(pos)/HepPar-1(pos) immunophenotype. Virchows Arch. 2017;471:599–609. [DOI] [PubMed] [Google Scholar]
- 65.Herpel E, Rieker RJ, Dienemann H, et al. SMARCA4 and SMARCA2 deficiency in non-small cell lung cancer: immunohistochemical survey of 316 consecutive specimens. Ann Diagn Pathol. 2017;26:47–51. [DOI] [PubMed] [Google Scholar]
- 66.Agaimy A, Daum O, Markl B, Lichtmannegger I, Michal M, Hartmann A. SWI/SNF complex-deficient Undifferentiated/Rhabdoid carcinomas of the gastrointestinal tract: a series of 13 cases highlighting mutually exclusive loss of SMARCA4 and SMARCA2 and frequent Co-inactivation of SMARCB1 and SMARCA2. Am J Surg Pathol. 2016;40:544–553. [DOI] [PubMed] [Google Scholar]
- 67.Agaimy A, Bertz S, Cheng L, et al. Loss of expression of the SWI/SNF complex is a frequent event in undifferentiated/dedifferentiated urothelial carcinoma of the urinary tract. Virchows Arch. 2016;469:321–330. [DOI] [PubMed] [Google Scholar]
- 68.Strehl JD, Wachter DL, Fiedler J, et al. Pattern of SMARCB1 (INI1) and SMARCA4 (BRG1) in poorly differentiated endometrioid adenocarcinoma of the uterus: analysis of a series with emphasis on a novel SMARCA4-deficient dedifferentiated rhabdoid variant. Ann Diagn Pathol. 2015;19:198–202. [DOI] [PubMed] [Google Scholar]
- 69.Agaimy A, Jain D, Uddin N, Rooper LM, Bishop JA. SMARCA4-deficient sinonasal carcinoma: a series of 10 cases expanding the genetic spectrum of SWI/SNF-driven sinonasal malignancies. Am J Surg Pathol. 2020. [DOI] [PubMed] [Google Scholar]
- 70.Schaefer IM, Agaimy A, Fletcher CD, Hornick JL. Claudin-4 expression distinguishes SWI/SNF complex-deficient undifferentiated carcinomas from sarcomas. Mod Pathol. 2017;30:539–548. [DOI] [PubMed] [Google Scholar]
- 71.Wilson BG, Wang X, Shen X, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kadoch C, Copeland RA, Keilhack H. PRC2 and SWI/SNF chromatin remodeling complexes in health and disease. Biochemistry. 2016;55:1600–1614. [DOI] [PubMed] [Google Scholar]
- 73.Alimova I, Birks DK, Harris PS, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kurmasheva RT, Sammons M, Favours E, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2017:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gounder MM, Stacchiotti S, Schöffski P, et al. Phase 2 multicenter study of the EZH2 inhibitor tazemetostat in adults with INI1 negative epithelioid sarcoma (NCT02601950). J Clin Oncol. 2017;35 11058–11058. [Google Scholar]
- 77.Hoy SM. Tazemetostat: first approval. Drugs. 2020;80:513–521. [DOI] [PubMed] [Google Scholar]
- 78.Italiano A, Soria JC, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19:649–659. [DOI] [PubMed] [Google Scholar]
- 79.First EZH2 Inhibitor Approved-for Rare Sarcoma. Cancer Discov. 2020;10:333–334. [DOI] [PubMed] [Google Scholar]