

Abstract
The principle of vinylogy states that the electronic effects of a functional group in a molecule are possibly transmitted to a distal position through interposed conjugated multiple bonds. As an emblematic case, the nucleophilic character of a π-extended enolate-type chain system may be relayed from the legitimate α-site to the vinylogous γ, ε, ..., ω remote carbon sites along the chain, provided that suitable HOMO-raising strategies are adopted to transform the unsaturated pronucleophilic precursors into the reactive polyenolate species. On the other hand, when “unnatural” carbonyl ipso-sites are activated as nucleophiles (umpolung), vinylogation extends the nucleophilic character to “unnatural” β, δ, ... remote sites. Merging the principle of vinylogy with activation modalities and concepts such as iminium ion/enamine organocatalysis, NHC-organocatalysis, cooperative organo/metal catalysis, bifunctional organocatalysis, dicyanoalkylidene activation, and organocascade reactions represents an impressive step forward for all vinylogous transformations. This review article celebrates this evolutionary progress, by collecting, comparing, and critically describing the achievements made over the nine year period 2010–2018, in the generation of vinylogous enolate-type donor substrates and their use in chemical synthesis.
1. Introduction
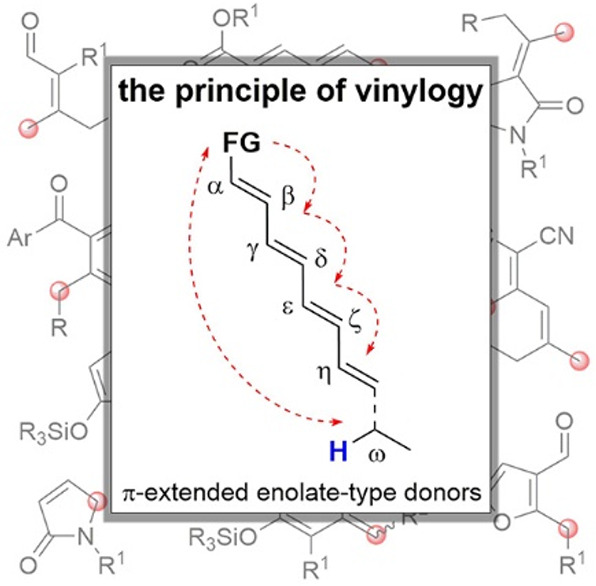
The design and development of selective C–H activation reactions at carbon sites remotely positioned from a leading functional group has evolved into an exciting research topic of contemporary synthetic chemistry.1,2 The principle of vinylogy, originally formulated by Fuson in 1935,3 states that the electronic effects of a functional group in a molecule can be transmitted, via interposed conjugated multiple bonds, to a distal position in the molecule (Scheme 1).
Scheme 1. Depiction of the Principle of Vinylogy Applied to π-Extended Carbonyl Compounds I/II.
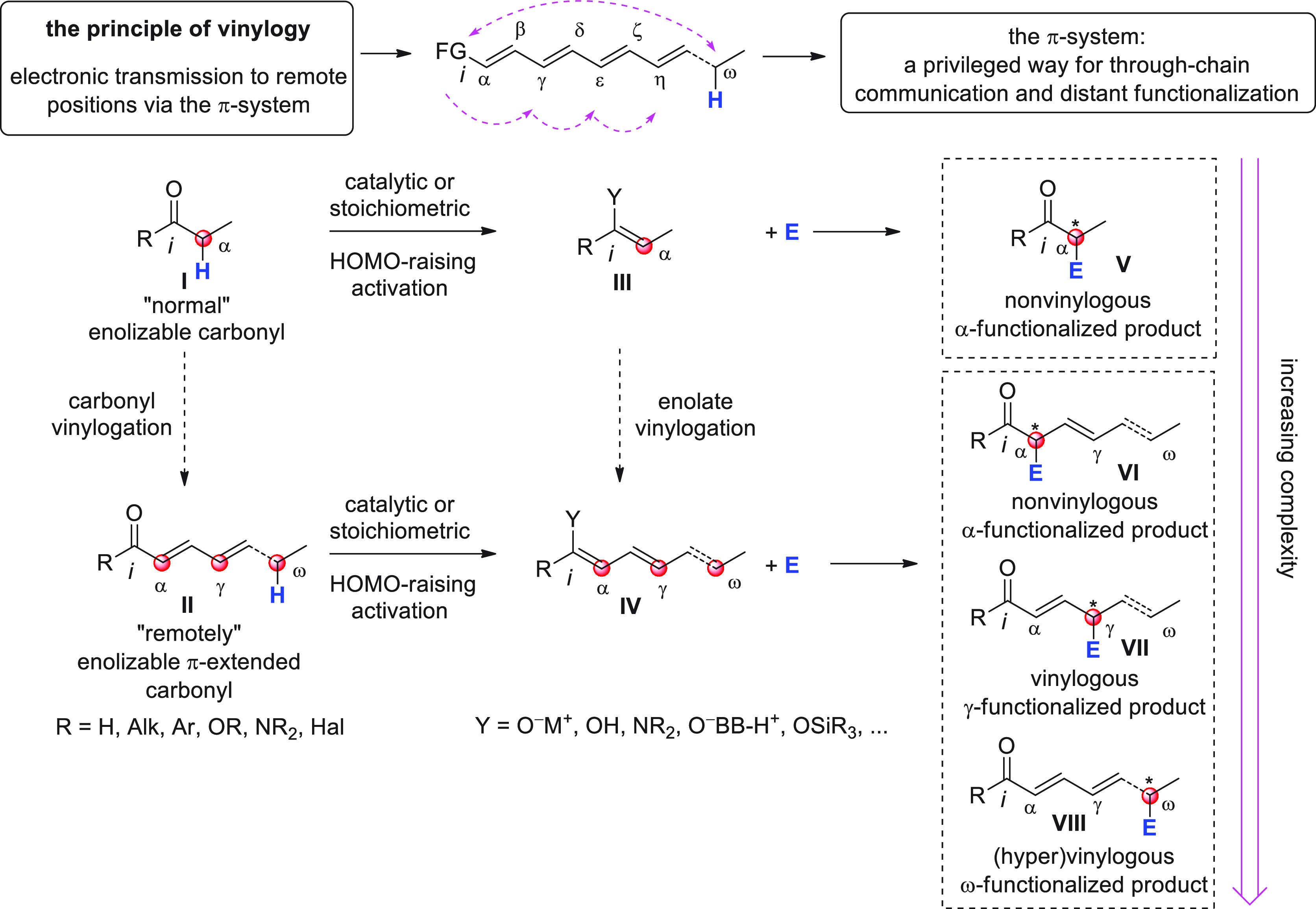
[FG = functional group; E = electrophile; catalytic or stoichiometric HOMO-raising activation refer to covalent or noncovalent activation strategies inducing the formation of metal enolates (Y = O–M+), enols (Y = OH), enamines (Y = NR2), enolates with protonated Brønsted base counterions (Y = O–BB–H+), or silyl enol ethers (Y = OSiR3). Red circles indicate pronucleophilic (compounds I/II) and nucleophilic (compounds III/IV) carbon sites.
Enolizable, π-extended carbonyl systems of general formula II (Scheme 1) (generally including aldehydes, ketones, and carboxyl-level functionalities such as esters/lactones, amides/lactams, acyl halides) can be considered to be emblematic examples of this principle, whereby the electronic properties of the carbonyl functional group are relayed along the carbon chain to remote carbon positions through the conjugated π-system, which effectively represents a privileged means of communication between distant sites. For example, the conventional electrophilic character of the C=O group (ipso position, i) is “usurped” by the conjugated β, δ, etc. carbon sites and thus a typical 1,2-nucleophilic addition to carbonyl compounds becomes a 1,4-, 1,6-, etc. conjugate addition reaction (not shown). On the other hand, the prototypical pronucleophilic character at the α-position of “normal” enolizable carbonyl compounds I is propagated long-range to the vinylogous (and hypervinylogous) γ, ε, ..., ω carbon sites, via in situ-formed or preformed polyenolate-type intermediates IV—the vinylogous versions of enolates III—using suitable catalytic or stoichiometric HOMO-raising activation procedures (here and throughout this review, hypervinylogous sites refer to those carbon atoms along the π-chain which are separated from the leading functional group by more than one unsaturated linkage). These inherently polydentate donor systems may be engaged in useful enolate-based chemistry with suitable electrophilic partners (e.g., C=O, C=N, activated C=C bonds and other electrophiles) ultimately providing, at least in principle, a plethora of diverse synthetic pathways and products of increasing structural complexity vis-à-vis their simple nonvinylogous counterparts.
While these concepts are generally a well established part of a chemist’s repertoire, far from obvious is how to simultaneously maintain chemo-, regio-, and stereocontrol of the multisite reactivity present in these vinylogous substrates. For example, which of the several possible competing regioisomeric products VI–VIII (α vs γ, ... vs ω site selectivity) emerges as being the preferred is a multifactorial issue depending on (1) the intrinsic electronic bias of the nucleophilic carbon sites (HOMO coefficient, in turn dependent on the type of metal/counterion such as Li+, SiR3, NR4+, ...), (2) the electrophilic susceptibility of the coupling partner (LUMO coefficient), (3) the presence of strategically placed biasing/bulky substituents along the chain (steric effects), (4) the thermodynamic stability of the products (when the reaction is thermodynamically controlled), and (5) the type and mechanism of the employed catalyst (if any).
Other parameters may add to the complexity of this matter: the use of ketones or branched molecular substrates with a conspicuous number of enolizable positions, the coupling of a vinylogous C–C/C–X bond-forming event to cascade processes of cyclization, and stereochemical issues, concerning the E/Z geometry of the emerging olefins within the products, as well as the simple and facial stereocontrol of the newly forged stereocenters.
Given this state of affairs, the advantage of vinylogous transformations over “normal” reactions, namely, increased product complexity with simultaneous formation of multiple functional groups and stereogenic elements, can be brought effectively to fruition, provided that two fundamental conditions are met. First, suitable activation strategies and/or catalytic modalities are selected to chemoselectively activate either or both partner substrates, while inducing maximal regio- and stereocontrol, and second, the vinylogous substrates must remain coplanar in their reactive conformations, in order to preserve the electronic transmission through the conjugated π-system.
One of the greater successes in the development of vinylogous enolate-based chemistry over the past decades has been the use of preformed silyl enol ethers (or silyl ketene acetal polyenolates) from the corresponding π-extended carbonyl precursors, whose innate electronic predisposition to react at remotely positioned carbon sites has been certified and widely exploited in synthesis.4 In the new millennium, the remarkable development of novel covalent and noncovalent, HOMO raising and LUMO lowering activation strategies using chiral organo- and metal-based catalysis has shaped the concept of and the way of conducting both old and new chemical reactions. Merging the principle of vinylogy with activation modalities and concepts such as iminium ion/enamine organocatalysis, NHC-organocatalysis, cooperative organo/metal catalysis, bifunctional organocatalysis, dicyanoalkylidene activation, and organocascade reactions truly represents an impressive step forward for vinylogous transformations.
A critical survey of the contributions published in the literature over the past recent years and our own experience in this dynamic field of research made us realize that (1) the palette of pronucleophilic species used as direct or indirect sources of vinylogous donor species has been greatly enriched and diversified since the pre-2010 era; (2) these electron-rich species trigger a spectrum of reactions including the “traditional” aldol/Mannich/Michael addition reactions, but also “new” connections such as [4 + 2], [3 + 2], [n + m] annulations, nitro-Henry additions, Rauhut–Curier reactions, amination and alkylation reactions, and others yet; (3) the activation of such pronucleophiles often includes direct catalytic modalities (e.g., HOMO-raising organocatalytic enamine, NHC activation), while indirect activation of these matrices (e.g., via silyl enol ether preformation) is losing ground although still used in target-oriented synthesis; (4) when strategies are involved that activate the “unnatural” carbonyl ipso-site as a nucleophile (umpolung), vinylogation transmits the nucleophilic characteristic to “unnatural” β, δ, ... remote sites.
Our intention here is to celebrate these evolutionary improvements of the past few years, by collecting, comparing, and critically describing the achievements made, over the nine year period 2010–2018, in the generation of vinylogous enolate-type donor substrates and their use in chemical synthesis.
2. About this Review
In 2000, a review article was published in this journal, dealing with vinylogous aldol addition reactions and chronicling their development and application in organic synthesis from the onset to the end of 1999.5 About ten years later, a sequel to this article was published, covering the topic of vinylogous aldol domain and related vinylogous Mannich and Michael reactions emerging from research carried out in the first decade of the new millennium (January 2000-April 2010).6
Given the interest shown in this topic by the chemical community7 and the continual, ever-growing number of papers published in this field, we have compiled a comprehensive and critical review article about the exploitation of vinylogous enolate-type donor substrates in chemical synthesis over the most recent period January 2010-December 2018.
In order to emphasize the structure and vinylogous reactivity of the pronucleophilic species focus of this article, this review article is subdivided into main sections, according to the functional group responsible for vinylogous reactivity, namely, vinylogous aldehydes, ketones, esters/lactones, amides/lactams, nitriles, and others (generally unsaturated and saturated acyl halides and carboxylic acids, nitro (hetero)aromatic compounds, vinylphenols). In this way, the reader can readily visualize the main classes of provinylogous substrates (which will also be presented collectively by appropriate figures at the beginning of each main section) and observe how they act in different ways to generate the active nucleophilic species and fare in subsequent homologation reactions.
Each main section is organized into subsections, according to the nature of the electrophilic substrate involved in the vinylogous coupling, namely, additions to C=O (aldol-type additions and related cascade cyclization reactions), C=N (Mannich-type additions, 1,3-dipolar cycloadditions and related cascade cyclization reactions), electron-poor C=C bonds (Michael-type additions and related cascade cyclization reactions), and miscellaneous electrophiles (alkyl halides, amination reagents, and others). A further distinction between direct procedures (in situ activation of pronucleophiles by suitable organo- and/or metal-catalysts) and indirect procedures (use of preformed and isolated silyl-derived nucleophiles) has been made. For each subchapter, the contributions are grouped according to the acyclic vs cyclic nature of vinylogous donors, where the term “cyclic donor” denotes those substrates where the conjugated π-system, responsible for the vinylogous transmission, is partially or totally included in one, or more, carbo- or heterocyclic ring. Far from being a fictitious subdivision, this classification reflects the profound differences between the two classes, attributed mainly to different steric and electronic properties, especially when aromatic compounds are involved. In the absence of pertinent studies, the relevant subsection is not treated.
As already stated, given that the principle of vinylogy, per se, includes practically all existing functional groups which are “vinylogated”, the field has been restricted here, by choosing vinylogous pronucleophiles containing carbonyl, as well as carboxyl-level and miscellaneous, functional groups which may act, upon suitable activation, as electron-rich donor components in polar, enolate-type reactions to forge new remote C–C and C–X (X = N, O, S, halogen, H) connections. Special attention is paid to enantioselective reactions yielding chiral nonracemic products.
For these reasons, the review will not cover the following topics: (1) vinylogous electrophiles (e.g., Michael acceptors and higher homologues, which are the subject of focused reviews);8−10 (2) condensation reactions where the newly created stereocenters are promptly lost;11 (3) examples where the vinylogous multidentate donors react at nonvinylogous positions (α-attack);12,13 (4) reactions involving reactive π-extended radical species; and (5) simple Friedel–Crafts reactions where the π-extended conjugate system remains within an aromatic ring and no leading “carbonyl-type” functionalities are present.14
On a technical note, conceptually similar articles are reviewed sequentially or under tabular format, and are often preceded by general, explanatory schemes. Throughout the work, reactive nucleophilic or pronuleophilic vinylogous sites in the structural formulas are denoted by red circles, electrophilic sites by blue circles, and the newly formed linkages are colored in red. For the sake of the reader, dashed lines connecting the electronically complementary reactive sites within substrates are used when complex annulation reactions are described. Particular emphasis is placed on the mechanistic investigations made by the authors, especially when vinylogous enolate-type donors are employed, as diene components, in cycloaddition reactions; most of the alleged cycloadditions under review either were not mechanistically studied thoroughly or they revealed their actual stepwise, vinylogous nature.
During the 2010–2018 period considered here, a considerable number of authoritative reviews, accounts, and highlights appeared in the literature that focused on particular aspects of vinylogy.7 Many of them accounted for specific HOMO-raising catalytic activation modalities of pronucleophiles (amino-organocatalysis,15−18 NHC-organocatalysis,19,20 noncovalent organocatalysis);21−23 some dealt with selected classes of vinylogous functional groups (aldehydes,24,25 nitriles,26,27 alkylidene carbo- or heterocycles28,29), while others were focused on either specific reaction types (Michael additions,30,31 Mukaiyama-type C–C bond forming reactions,32 organocascade reactions33) or selected target categories (polyketides,34 γ-butenolides35,36).
An updated, comprehensive and critical review article embracing all these allied subjects under one common underlying principle—the principle of vinylogy—was missing, and our intention here is to fill that void.
3. Vinylogous Aldehydes
The aldehyde function occupies a cardinal position among the most popular polar functional groups that can be “vinylogated”. The possibility of readily obtaining remotely enolizable enals and higher-order homologues via synthesis makes this class of compounds a qualified source of carbon pronucleophiles for use in fruitful additions to electronically complementary partners, especially carbonyl acceptors or electron-poor alkenes. Due to the invention and powerful exploitation of catalytic activation modalities, particularly suited for the aldehyde functional group, the majority of the examples in this section command direct procedures, where conjugated aldehydes are activated in situ to unveil their remote carbon nucleophilicity at either “natural” γ, ε, ... sites, or “unnatural” β sites, according to umpolung polarity reversal (vide infra). On the other hand, indirect procedures, using preformed stable enolates (e.g., silicon extended polyenolates), are limited to a rather restricted number of examples, reflecting the general trend in organic synthesis toward the use of direct, catalytic, and stereoselective methods. Pronucleophilic aldehyde substrates, reported in this chapter under the section “direct procedures”, are depicted in Figure 1, subdivided into acyclic and cyclic representatives, and with their reactive pronucleophilic remote sites indicated. The molecular structure of one aldehyde-derived dienolate nucleophile, used in indirect procedures, is also portrayed in this same figure.
Figure 1.
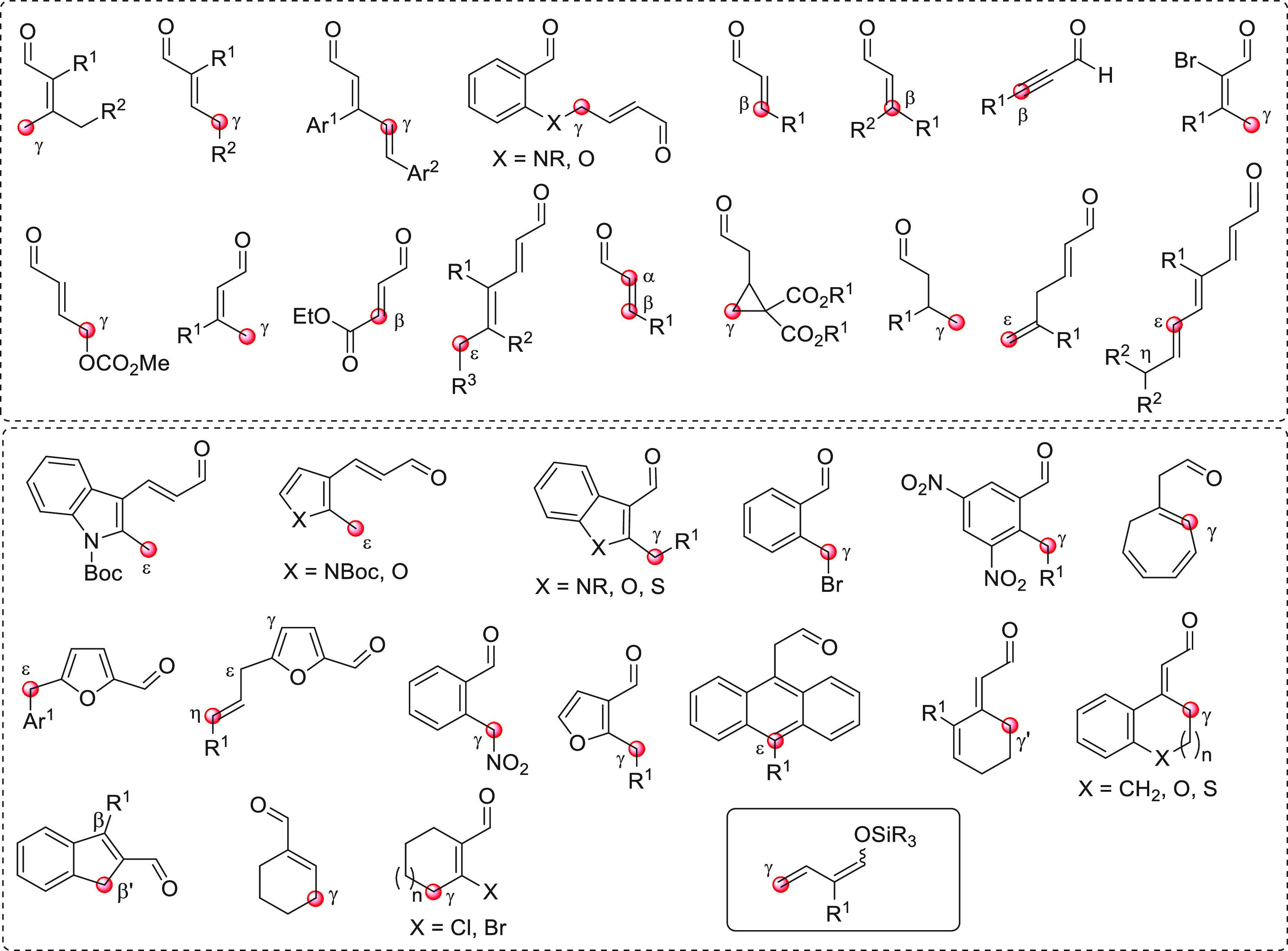
Collection of acyclic (above) and cyclic (below) pronucleophilic aldehydes at work in this section using the direct procedures. In the plain box the only type of nucleophilic aldehyde-derived silyl dienol ether used in indirect procedures. Red circles denote the reactive (pro)nucleophilic carbon site.
3.1. Additions to C=O Bonds
3.1.1. Direct Procedures
3.1.1.1. Acyclic Pronucleophiles
After the launch of chiral secondary amines as useful organocatalysts to be systematically exploited in either the α-pronucleophilic activation of enolizable aldehydes (according to the HOMO-raising principle) or β-electrophilic activation of α,β-unsaturated aldehydes (LUMO-lowering principle) via enamine and iminium ion intermediates, respectively,37 it was promptly recognized that such principles could be efficiently translated and exploited in the realm of vinylogy. In particular, the covalent activation of a remotely enolizable conjugated aldehyde IX (Scheme 2), through condensation of the carbonyl group with a chiral secondary amine, can give rise to an extended polyenamine species of type XI (dienamine, trienamine, tetraenamine, ...) by isomerization of the polyunsaturated iminium ion X, thus unveiling multiple and often competing nucleophilic carbon centers to be successfully engaged in, for example, aldol-type addition reactions (Scheme 2, E = carbonyl). In the event, the ω-coupling product XIII is formed after the original carbonyl restoration and catalyst recycling; alternatively and more frequently, variously forged cyclized carbonyl products (e.g., compounds XIII′) are obtained, when additional electronically complementary functional groups in both reacting partners trigger intramolecular cascade reactions.
Scheme 2.

The Woggon and Bräse groups were among the first to recognize that merging the concepts of asymmetric amine-based covalent organocatalysis with vinylogy and cascade reactions could have enormous potential in the synthesis of natural products. By following the chemical strategies they had already adopted prior to 2010,38,39 these two groups independently exploited terpenoid-related γ-enolizable α,β-unsaturated aldehydes 1a or 1b as useful γ-donor precursors (Scheme 3). When farnesal 1a (R1 = geranyl)40 or prenal 1b(41) (R1 = H) was reacted with salicylaldehyde 2 in the presence of diarylprolinol silyl ethers A1 or A2 as the key amine organocatalysts (the same reaction conditions were used in both cases, i.e. 30 mol % catalyst and benzoic acid additive, in toluene), the tricyclic chiral lactols 3a or 3b were respectively obtained in useful yields and moderate to good enantioselectivities. Both authors hypothesized, but did not experimentally prove, that these products were the result of an organocatalytic domino sequence33,42−44 encompassing an initial vinylogous γ-aldol addition reaction (γ-VAR) between the activated dienamine 1′a/1′b (formed upon condensation of the catalyst and enal 1 with subsequent iminium ion/enamine isomerization) with aldehyde 2, followed by intramolecular oxa-Michael closure of the phenolic OH on the nascent unsaturated iminium ion and final spontaneous acetalization. Thus, the pronucleophilic aldehyde initiators 1a/1b provided the γ/β carbon sites in a stepwise [4 + 2] annulation process. Lactol 3a was subsequently used for the enantioselective entry to anti-HIV active daurichromenic acid and confluentin,40 while 3b and related analogues served to obtain naturally occurring diversonol and other tetrahydroxanthone and chromone lactone families.45
Scheme 3.

To be precise, in both studies, terpenoid enals 1a and 1b possess two nonequivalent, remotely enolizable positions, namely, the γ and γ′ sites, which, in principle, could give rise to diverse competing regioisomeric products. Although this issue did not emerge in these studies, as only products 3a and 3b were reported, it was rebooted some years later by the Liu group,46 who studied the same reaction extensively and investigated how diverse substituents to the aromatic ring of salicylaldehyde precursors of type 2 could influence the γ/γ′ regioselectivity of the process, thus opening a doorway to novel non-natural chroman derivatives embedded with quaternary stereocenters (not shown).
The first study of a direct, enantioselective VAR, involving acyclic enolizable enals yielding isolable and stable aldol products, uninvolved in cascade sequences, was reported by the Melchiorre group in 2012.47 It was found that different α,γ-dialkyl substituted enals 4 reacted directly with isatins 5 upon in situ HOMO-raising activation by the bulky chiral secondary amine A3 catalyst through formation of dienamine 4′ (Scheme 4). Access to the enantioenriched vinylogous aldol-oxindole products 7 was secured (exclusive γ-attack was observed) as variable mixtures of isolable diastereoisomers at C3′ in high yields and moderate to good enantioselectivities.
Scheme 4.

Of note, it was observed that the chemical behavior of enals 4 strictly depended upon the nature of the α-R2 substituent: when R2 was aryl instead of alkyl or benzyl, coupling of 4 with 5 under the same reaction conditions gave bicyclic acetal products 8 with reverted stereochemistry at Cγ as a mixture of separable C3′ diastereoisomers. This behavior was probably the result of a pericyclic [4 + 2] cycloaddition (hetero-Diels–Alder, HDA) involving s-cis dienamine conformer 4″, as demonstrated by NMR-based experiments and control reactions (Scheme 4). In this last instance, given the concerted nature of the HDA coupling, the transformation cannot be strictly regarded as a vinylogous procedure, since no electronic transmittal from a given functional group is relayed along a conjugated system to a remote site; however, it could be classified as a vinylogous process if an asynchronous cycloaddition mechanism was invoked; this was not proven nor excluded by the authors.
Nonenolizable polyenals of type 11 may well serve as γ-donor components in aldol-type additions, provided that suitable activation modalities are implemented with timely precision (Scheme 5). This did not remain unnoticed by the Melchiorre group when they exploited β,δ-diaryl-substituted dienals 11 in a three-component organocatalyzed cascade coupling reaction with α-enolizable aldehydes 9 and isatin-derived activated alkenes 10.48 Using prolinol silyl ether A4 in 20 mol % loading secured the prompt formation of highly valuable spirooxindole cyclohexane derivatives 12 bearing six contiguous stereocenters with excellent enantiocontrol (99% ee in all cases). Given the multisite, electronically complementary reactivity of the three reaction partners, different reaction pathways could be, in principle, operative. Indeed, a well orchestrated activation sequence was in motion, encompassing an initial enamine-triggered Michael addition of aldehyde 9 to acceptor 10, a second intermolecular δ-selective 1,6-conjugate addition of the emerging enolate to iminium ion-activated dienal 11 to forge spirocyclic dienamine 11′, and a final intramolecular VAR providing the final products (which were isolated as alcohol derivatives 12 after aldehyde reduction with NaBH4). Key to the success of the reaction in terms of both regio- and chemoselectivity were the judicious positioning of biasing aryl β-substituents within 11, in preventing parasitic 1,4-conjugate additions; and one-pot, sequential additions of the starting materials (9 + 10 + A4 at the beginning, followed by addition of 11) to preclude the formation of unwanted aldol condensation products.
Scheme 5.

Ad hoc prepared γ-enolizable dialdehydes 13 served as the starting materials for the enantioselective entry to various diheteroaryl alkanals 15, according to a one-pot sequential reaction involving indoles 14 under silyl prolinol organocatalysis (Scheme 6).49 To account for the observed chemical behavior, the authors hypothesized that an initial intramolecular γ-VAR occurs through the intermediacy of dienamine 13′, formed after condensation of the Hayashi–Jørgensen catalyst ent-A4 with the enal-aldehyde function, followed by subsequent iminium–enamine isomerization. In the event, the aldol condensation product 13″ is obtained, with no new stereocenters formed. An intermolecular Friedel–Crafts reaction then occurs between indole 14 and the formed iminium ion 13″, under classical “steric shielding control” by the appended catalyst, to afford the targeted heterocycles 15 in good yields and notable enantiomeric excesses. Antiproliferative assays of the new products on cancer cell lines revealed interesting cytotoxic activity, in some instances, comparable or even superior to that of cisplatin.
Scheme 6.

The HOMO-raising activation of enals, via catalytic dienamine formation, could also be effectively applied to an unsual yet very interesting [5 + 2] formal cycloaddition reaction involving α,β-unsaturated aldehydes and oxidopyrylium ylides generated in situ from 1-acetoxyisochroman-4-ones (not shown).50 These dienamine intermediates showed exclusive γ,β-reactivity and provided direct asymmetric entry to oxabicyclooctane-containing products by using a bifunctional secondary amine-squaramide organocatalyst.
Besides the HOMO-raising activation modality of enals involving amine organocatalysts, another successful activation mode is represented by covalent N-heterocyclic carbene (NHC) organocatalysis. The classic NHC-catalyzed a1 → d1 umpolung reactions of aldehydes, where benzoin products are formed by the C-ipso (i) pronucleophilic site of the Breslow intermediate attacking a carbonyl acceptor (Scheme 7), have found their vinylogous counterparts in “homoenolate” chemistry introduced in 2004 by the pioneering and independent works of the Bode and Glorius groups.51,52 In this instance, the direct covalent activation of the starting enal, via NHC catalyst, generates the key homoenolate species–a vinylogous Breslow intermediate–whose pronucleophilic β-carbon site (d3) attacks the carbonyl acceptor providing γ-lactone products after tautomerization to a catalyst-bound carboxylate and ring closure by the formed alkoxide with concomitant catalyst recycling (Scheme 7). A formal [3 + 2] annulation is thus attained, where the β/ipso carbon sites of the starting enal close onto the C=O bond of the carbonyl acceptor. Complete β vs ipso regioselectivity was attained within the reactive homoenolate by means of the judicious choice of the NHC catalyst, whose electronic and, above all, steric properties can allow for complete depletion of the C-ipso reactivity in favor of the β-position, as well as the premature proton quenching of the catalyst-bound intermediate.
Scheme 7.

Following these pillar studies, intense research in this field was perfomed by diverse groups during 2004–2009, where assorted electrophiles (i.e., C=O, C=N, and activated C=C bonds, vide infra) were also used, and by the new decade (from 2010 onward), the time was right for extending the horizon of this process toward highly diastereo- and enantioselective variants for the asymmetric construction of a wide range of hetero- and carbocyclic structures. In particular, chiral 1,3,4-triazol-2-ylidene catalysts were used for this scope, often combined with additives.
Among the first reports of efficient and enantioselective [3 + 2] annulations involving NHC-homoenolates was that by Ye et al., who employed enals and isatins as starting substrates (Scheme 8).53 It was found that L-pyroglutamic acid-derived triazolium salt B1 (5 mol % loading), bearing a free hydroxyl group appendage, was the NHC precursor best to succeed in the transformation. Hence, a series of aromatic enals of type 16 coupled with isatins 17 to give the corresponding spirocyclic oxindolo-γ-butyrolactones 18 with very good yields and stereoselectivities, except for the single case of β-n-propyl-substituted enal 16 (R2 = n-Pr), which afforded product 18 in a rather poor 38% yield. One limitation of the procedure was that only isatin carbonyls proved to be suitable substrates, since other aldehydes, such as benzaldehyde or propionaldehyde, failed to react under the reported conditions. Given the strong influence of the free hydroxyl group on the activity of the catalyst, the authors proposed a plausible transition state where H-bonding operated between the catalyst and isatin and was probably responsible for enhancing the carbonyl reactivity and directing the homoenolate addition along the indicated trajectory (see 16′).
Scheme 8.

Shortly after this study, three other works were independently published by the Scheidt,54 Coquerel,55 and Glorius56 groups, who assayed the prototypic reaction between enals and isatins exploiting the concept of dual organocatalysis: the first group applied an NHC/Lewis acid activation, the second exploited bifunctional NHC/thiourea organocatalysts, and the third group employed NHC/Brønsted acid catalysis.
In the first contribution,54 the ultimate goal was to demonstrate whether the use of a mild Lewis acid additive together with an NHC catalyst would be able to enhance the reaction performance in terms of both yield and stereoselectivity, as had been previously reported for related additions to activated C=N and C=C systems (vide infra). After extensive screening of diverse Lewis acid/chiral azolium salt couples, it was found that treatment of variously substituted isatins 19 smoothly reacted with β-aryl enals 20 in the presence of triazolium precatalyst B2 (5 mol %), DBU, and, importantly, lithium chloride (2 equiv), to afford the corresponding spirocyclic oxindoles 21 in good yields and enantioselectivities, as separable mixtures of diastereoisomers at C3′ (poor to very good dr were observed, Scheme 9, eq 1).
Scheme 9.

Curiously, the Lewis acid additive (LiCl) had a detrimental effect on the reaction involving less reactive β-alkyl substituted enals 20 (R3 = Me, n-Pr), and new optimization conditions were needed. In this instance, bicyclic triazolium salt B3 was chosen as the best precatalyst, giving products ent-21 in good yields and opposite facial selectivity (Scheme 9, eq 2). To account for the observed diastereoselectivity in β-aryl substituted enals, a highly coordinated model (see 20′) was postulated, where the lithium cation coordinates both the enal oxygen atom of the homoenolate and the 1,2-dicarbonyl of isatin; for alkyl derivatives, on the other hand, no definitive explanation was given. Interestingly, one spirocyclic product 21 (R1 = R3 = Me, R2 = H) was transformed to the anticancer agent maremycin B via a five-step procedure, thus emphasizing the synthetic utility of the disclosed [3 + 2] annulation.
An alternative approach for the same reaction type was devised by Coquerel and co-workers, who synthesized a small library of bifunctional organocatalysts where a chiral 1,3-imidazolin-2-ylidene NHC core is covalently connected to a hydrogen-bond donor group.55 The H-bond donor would be able to control the approach of the incoming electrophile and concomitantly stabilize the E-configuration of the vinylogous Breslow intermediate, thus anticipating a good overall stereocontrol. Following this line of thought, an extensive initial study of diverse bifunctional NHC catalysts bearing either hydroxyl, guanidine, thiourea, or urea moieties was carried out on several model reactions, to demonstrate the viability of this concept especially in view of the challenging possibility of avoiding self-quenching of the active carbene by intramolecular acid–base reactions. While the reactions involving enals with nitrosobenzene or chalcone acceptors met with only partial success, the γ-lactonization reaction of enal 20a with isatin 19a (Scheme 10, eq 1) proceeded efficiently by using the NHC-urea catalyst from B4: spiroxindole 21a was thus obtained in a high yield and diastereomeric ratio, though with modest enantiomeric excess, probably via the hypothesized highly coordinated transition state 20a′.
Scheme 10.

A NHC/Brønsted acid dual catalytic system was exploited to perform similar [3 + 2] annulations, involving isatins 19 and scantly used β,β-disubstituted enals 22 (Scheme 10, eq 2).56 The delicate issue to be faced here was to activate properly the pronucleophilic, sterically congested β-position of 22 through the expected homoenolate-type reactivity, while suppressing a possible competing γ-site nucleophilic activation toward [4 + 2] annulation (vide infra). The presence of a Brønsted acid cocatalyst (t-BuCO2H) flanking the catalysis of NHC from B5 was beneficial for the purpose, providing efficient access to a collection of racemic spirocyclic oxindoles 23 bearing two congested and contiguous quaternary centers with moderate to excellent diastereomeric ratios, probably via a postulated tight transition state where two stabilizing hydrogen bonds arise between the carboxylic acid function of the cocatalyst and the two reacting partners, namely, the isatin carbonyl and the NHC-bound homoenolate hydroxyl (not shown). A couple of examples involving the use of less active, fully aliphatic enal substrates 22 were also reported, but a change in the triazolium precatalyst was needed.
An enantioselective version of the reaction was also performed starting from isatin and β-methylcinnamaldehyde 22 (R3 = Ph, R4 = Me) and using bicyclic azolium B3 and the o-fluorobenzoic acid agent (1 equiv) (not shown); the reaction gave the corresponding enantioenriched products of type 23 in promising though not excellent results (83% yield, 5:1 dr, 84% ee).
If, instead of being coupled to α-ketolactams such as isatins, an NHC-β-activated enal of the types previously mentioned (e.g., compounds 16 and 20) is reacted with α-ketolactones such as benzofuran-2,3-diones, a similar [3 + 2] annulation will occur, and entry to interesting spiro-bis-lactones is secured. This strategy was devised and exploited independently by the Cheng57 and Nair58 research groups in 2013 and 2014.
In the first report, β-aryl- or β-alkyl enals 25 were treated with achiral imidazolium salt B6 (20 mol %) in t-BuOK and were reacted with diverse benzofuran-2,3-diones 24 (Scheme 11, eq 1) giving rise to the expected [3 + 2] annulation products (±)-26 as single diastereoisomers, accompanied by variable quantities of the regioisomeric spirocycles (±)-27, which were the result of a rather unexpected attack of the NHC-bound homoenolate on the lactone carbonyl (Scheme 11, eq 1).57 Using different NHC precursors (B6 vs B7), solvents, and reaction temperatures, optimized conditions were found to regiodivergently access predominantly either (±)-26 or (±)-27 (Scheme 11, eq 1 vs eq 2), though in the latter case the regioisomeric selectivity remained poor. Access to (±)-26 was also attained using an alternative thiazolium salt precatalyst (not shown), in which case the regioselectivity in favor of 26 was excellent (26/27 > 20:1) but isolated yields were moderate (40–62% range).
Scheme 11.

In the second work (Scheme 11, eq 3), the reaction was performed using NHC precatalyst B5′; in this case, however, despite similar reaction conditions, only spiro-bis-lactones of type (±)-26 were reported, without any mention of possible byproducts of type (±)-27.58 Isolated yields of 26 were good, though no diastereomeric ratios were documented. In both examples, no attempts to translate the transformation into a chiral nonracemic format were made.
The asymmetric NHC-catalyzed formal [3 + 2] annulation of α,β-unsaturated aldehydes 29 with acyclic α-ketophosphonates 28 was documented by Scheidt et al. en route to the synthesis of unprecedented enantioenriched γ-butyrolactones 30 bearing a phosphonate moiety (Scheme 12).59 Initial DFT-based computational investigation of a model reaction guided the design of tailored C1-symmetric NHC precatalyst with the intention of selecting the optimal catalyst structure, capable of maximizing the stabilization of the transition state leading to the major enantiomer product, while maximizing the destabilization of the minor enantiomer. Azolium precatalyst B8 resulted in the best candidate, which was used in the realization of the product panel 30. The scope of the reaction was fairly broad, including aryl- and heteroaryl-substituents on both the donor and acceptor components. Many diverse products 30 were isolated in good yields and enantiomeric excesses, though cis/trans diastereoselection was poor or moderate. As a limitation, alkyl-substituted substrates did not provide good levels of efficiency. Based on computational modeling, a highly organized transition state was proposed, featuring H-bonding between the ketone oxygen within 28 and the enol function of the extended Breslow intermediate 29′.
Scheme 12.

The asymmetric NHC-catalyzed homoenolate-mediated reaction of α,β-unsaturated enals 32 was cleverly exploited in the reaction with racemic β-halo α-keto esters of type (±)-31 to produce the corresponding chiral nonracemic γ-butyrolactones 33 via dynamic kinetic resolution (Scheme 13).60
Scheme 13.

Catalyst optimization was performed in order to channel the reaction path toward the intended [3 + 2] annulation and prevent possible competitive side reactions such as cross-benzoin coupling. Apart from a few exceptions, deploying the catalyst from B9 generally furnished lactones 33 in good yields and stereoselectivities.
In strict analogy with the disclosed homoenolate-based [3 + 2] annulations involving enal substrates, replacement of the enal with a linear conjugated ynal of type 34 (Scheme 14) and subsequent NHC activation may lead to the corresponding homoenolate 34′—an allenolate equivalent—which may conveniently be coupled in a vinylogous fashion (β-position) to an electrophile such as a ketone, giving rise to valuable γ,γ-disubstituted butenolide structures. Putting this concept into practice may encounter difficulties, due to the weak nucleophilicity of the allenolate and the competitive, facile redox transfer through protonation of the homoenolate/allenolate to forge an α,β-unsaturated acyl azolium of type 34‴ with reverted (electrophilic) ipso/β polarity.61 This issue was cleverly handled independently by three research groups in the 2013–2014 biennium, by employing cooperative NHC-Lewis acid or NHC-Brønsted acid catalysis in order to enhance the reactivity of both substrates and hopefully coordinate them in organized transition states.62−64
Scheme 14.

The first study, performed in a nonasymmetric setting, reported the reaction of ynals 34 with unsaturated α-keto esters 35 under NHC catalysis from B10 (20 mol % loading) together with the mediation of LiCl (1 equiv) as an indispensable Lewis acid ingredient (Scheme 15, eq 1).62 The butenolide products (±)-36 were obtained in generally good yields with both aromatic and aliphatic ynals, while the keto ester component was restricted to nonenolizable aryl (or phenylvinyl)-substituted ketoesters 35.
Scheme 15.

The utility of this [3 + 2] annulation in an intramolecular, diastereoselective setting was soon after demonstrated by Snyder and co-workers, who successfully forged the fused polycyclic core, shared by securinega alkaloids, in one step starting from ad hoc-prepared enynal ketone precursors 37 and 39 (Scheme 15, eq 2).63 The challenge here was even harder than in the previous example, since an enolizable ketone moiety is present in the starting substrates (37 and 39), and competitive intramolecular addition reactions or even hypervinylogous versions involving the δ-site could, in principle, occur. Nonetheless, conditions were found to perform the transformation successfully, namely, slow addition (over 8 h) of 37 or 39 to a preformed suspension of precatalyst B11 and Ti(Oi-Pr)4 (2 equiv) in toluene, which ensured preparation of the corresponding tricyclic butenolides 38 or 40 in 47% and 31% yields, respectively. It was demonstrated that the efficiency of this annulation strongly depended upon the conformational bias of the enynal starter, since carrying out the same reaction on a simplified model (e.g., des-allyl compound 37) produced the butenolide product in 91% yield (not shown). Compound 38 was easily elaborated in two steps to the 3-deshydroxy-secu’amamine A target, an analogue of natural secu’amamine A.
The enantioselective version of this transformation was developed by the Scheidt group, who coupled aromatic or heteroaromatic alkynyl aldehydes 34 with aromatic α-ketoesters 41, to give enantioenriched butenolides 42 through chiral NHC/chiral phosphate cooperative catalysis (Scheme 16).64
Scheme 16.

Key to the success of this endeavor was the new mode of cooperative catalysis centered on the combination of three components: (i) the Hoveyda C1-symmetric biaryl saturated imidazolium salt B12 as the chiral NHC-precatalyst, (ii) the lithium tert-butoxide base which activates the NHC precatalyst while furnishing the coordinating lithium ion, and (iii) a chiral Brønsted acid L1 (20 mol %) serving as a cocatalyst. Though the specific roles of each component of the catalyst triad were not fully delineated, it was proposed that the lithium ion acts as a Lewis acid capable of coordinating both the BINOL-derived phosphate and the α-ketoester (as shown in 34″), thus ensuring the observed yields and enantioselectivities of the butenolide products.
Besides the β-sp2-CH nucleophilic activation of enals (and ynals) via in situ generated homolenolates (Scheme 7), the inherently multifaceted chemistry, triggered by NHC catalysis, may well allow for the nucleophilic activation of γ-sp3-CH bonds starting from, for example, γ-enolizable enals or α-haloenals, via oxidatively generated vinylogous enolates (Scheme 17). Upon interception of such extended enolates with an electrophile—e.g. an activated carbonyl—and subsequent ring closure, δ-lactone products are accessed according to a formal [4 + 2] annulation, while the NHC is recovered for the next catalytic cycle.
Scheme 17.

Significant regioselectivity issues are posed here, which are seen to depend on the switching from the homoenolate pathway (unveiling Cβ- or even Cα donor sites)65 to the vinylogous enolate pathway, which unmask the competitive Cγ/Cα nucleophilic sites. In addition, pointing on the γ-functionalization may pose questions about effective stereocontrol, given the distance between the NHC chiral inducer and the remote γ-site.
Capitalizing on outstanding precedents on the γ-functionalization of enals via dienamine catalysis (vide supra) as well as NHC-catalyzed γ-activation of α,β-unsaturated ketenes, Chi et al. documented, for the first time, in 2012 the enantioselective γ-addition of remotely enolizable enals 43 to activated ketones 44 via oxidatively generated NHC-bound vinylogous enolate intermediates in cooperation with Lewis acid cocatalysis (Scheme 18, eq 1).66 To skip the homoenolate pathway, biased enal substrates were used, bearing bulky and nonenolizable β-aryl (or heteroaryl, arylvinyl) substituents. The combined use of NHC from B13 (20 mol %) and the two Lewis acids Sc(OTf)3 and Mg(OTf)2, together with potassium carbonate as the base and quinone 45 as the external oxidant, provided the optimal conditions to consign the δ-lactone products 46 in good yields and modest-to-good enantioselectivities. Mechanistic studies were not performed, but given the indispensable role of the Lewis acids in drastically improving enantiofacial discrimination, the authors proposed that a multisite coordination as in 43′ might occur, where the scandium ion is capable of bringing the ketone acceptor in close proximity to the chiral NHC dienolate.
Scheme 18.

A similar formal [4 + 2] annulation was carried out by Yao and co-workers,67 who utilized α-bromo-α,β-unsaturated aldehydes 47 as γ-enolizable starter molecules.68 In this case, debromination from the extended Breslow intermediate is operative (see general Scheme 17), providing the key dienolate intermediate without the need of an external oxidant reagent. Thus, treating enals 47 with isatins 48 in the presence of catalytic NHC precursor B14, catalytic quantities of lanthanium triflate as a cocatalyst, and the carbonate base, afforded spirocyclic oxindole-dihydropyranones 49 with good efficiency in terms of both yields and enantioselectivities (Scheme 18, eq 2). As in the previous work by Chi, a tightly coordinated transition state was proposed, where the lanthanium ion coordinates the carbonyl acceptor thus enhancing the chiral induction exerted by the NHC catalyst.
A similar reaction scheme was performed some years later by Yang, Zhong, and colleagues using β-phenylcrotonaldehyde donors and N-deprotected isatin acceptors (not shown).69 In this case, a cooperative catalysis between NHC and Brønsted acid (pivalic acid) resulted effective in producing the corresponding spiroindoline pyrans similar to compounds 49 in generally good yields and moderate to high enantiomeric excesses. A further study on this subject was developed soon after, focusing on the chiral NHC-catalyzed [4 + 2] annulation between β-aryl or β-methyl crotonaldehydes and isatins (not shown).70 In the absence of any additional cocatalyst and under oxidative conditions, the reactions proceeded successfully giving, again, the corresponding spiroxindole δ-lactones of type 49 in good yields and enantioselectivities.71
As already stated, the NHC-catalyzed formal [4 + 2] annulation involving the γ/ipso carbon sites of γ-enolizable enals proved viable when the vinylogous enolate pathway prevails over the homoenolate pathway, and this was attained by placing biasing β-substituents in the starting enals (e.g., Scheme 18). As a clever alternative, Ye and collaborators were able to perform this chemistry by exploiting β-unsubstituted-γ-preoxidized enals, which are able to generate the expected unsubstituted dienolates in situ, en route to the formation of the δ-lactone targets (Scheme 19).72
Scheme 19.

After careful experimentation, optimal conditions were found [NHC precatalyst B13, Mg(Ot-Bu)2] to channel the coupling reaction of enal carbonate 50 with isatins 51 toward the desired spirocyclic oxindolo dihydropyranones 52, while completely suppressing the competitive homoenolate-mediated [3 + 2] annulation. The efficacy of the reaction was quite good for many diversely substituted isatins 51, with the sole exception of N-Boc and N-Cbz protected isatins, which proved completely unreactive under these reaction conditions.
The cooperative oxidative catalysis by a chiral NHC and a Lewis acid was also exploited for the dynamic kinetic resolution of racemic α-ketoesters to forge enantioenriched δ-lactone products (Scheme 20, eq 1).73
Scheme 20.

The optimized reaction conditions (precatalyst B2, scandium triflate cocatalyst, cesium acetate as the base, and quinone 45 as the external oxidant) were applied to a considerable number of substrates, where both the β-substituted crotonaldehydes 53 and the racemic ketoester components (±)-54 could tolerate several substituent variables. The corresponding δ-lactones 55 were obtained in good isolated yields, very high diastereomeric excesses, and good levels of enantioselectivity on almost all occasions. Just a few cases met with limited success, namely, the reactions using fluoroderivatives 54 (X = F) or methyl-crotonaldehyde 53 (R1 = Me).
The NHC-catalyzed [4 + 2] annulation strategy was also recently applied for the enantioselective entry to 2-pyranylphosphonates 57 by coupling γ-enolizable enals 53 to α-ketophosphonates 56 (Scheme 20, eq 2) (for the analogous NHC-catalyzed [3 + 2] annulation involving α-ketophosphonates, see Scheme 12).74 In this case, the use of NHC from B13 and quinone oxidant 45 was sufficient for triggering the reactions with high efficiency and enantioface discrimination without the need for additional cocatalysts, probably due to the unique stereoelectronic properties of the ketophosphonate substrates. With the exception of alkyl derivatives (R1 = cyclohexyl or methyl within enals 53, or alkyl phosphonates of type 56, not depicted in the scheme), all other substrates 53 and 56 were successfully coupled giving very good results. The chiral δ-lactone products 57 were assayed in vitro for their antibacterial and antiviral activities and some of them showed promising results for potential use in plant protection.
3.1.1.2. Cyclic Pronucleophiles
The set of cyclic unsaturated aldehydes for use in direct vinylogous aldol additions (VARs) is much smaller than that of their acyclic counterparts, and the reported studies are more recent. This may be due to the particular difficulties of catalytically activating cyclic substrates in situ, especially when they possess aromatic properties.
The only example dealing with nonaromatic substrates is the work by Gong and co-workers, who documented how cyclic β-haloenals of type 58 could undergo TBAF-triggered direct VAR with aromatic aldehydes 59, giving racemic δ-hydroxy-β-haloenals (±)-60 (Scheme 21).75
Scheme 21.

Among the tested bases (DBU, DABCO, Et3N, DMAP, and others), only fluoride ion (as in TBAF) or hydroxide (TMAH) proved competent reagents, and indeed almost stoichiometric quantities of TBAF at 18 °C were needed to afford the products in appreciable yields and syn-diastereoselectivities. Raising the reaction temperature to 28–32 °C led to decreased isolated yields of the aldol products in favor of dehydrated aldol condensation byproducts. The syn diastereopreference of the products was hypothesized as deriving from the transition state 58′ with an endo-antiperiplanar disposition of the two reacting partners. The minor anti-configured diastereoisomers were seen to derive from an antiperiplanar disposition in the transition state where unfavorable steric interactions between the X and R1 substituents occurred (not shown). No mention, however, was made by the authors for any possible, favorable synclinal transition states alternative to 58′ (with R1 pointing away from the cyclohexene ring) giving the observed syn-products 60. It is worth noticing that complete γ-site selectivity was attained in all cases, as opposed to the α-selectivity observed in another study by the same researchers,76 where identical substrates were coupled to Michael acceptors under TBAF agency, indicating that the γ/α regioselectivity of the nucleophilic addition for these cyclic dienolates is strongly dependent on the electrophilic counterpart involved in the process.
The benzylic C(sp3) sites of ortho-methyl substituted aromatic carbaldehydes of type XIV (n = 0, Scheme 22) or extended polyenals XIV (n ≥ 1) may be envisaged as remotely enolizable sites of the vinylogous aldehyde system so much so that useful functionalization chemistry with suitable acceptor components may be anticipated. However, deprotonation of such benzylic positions, especially when carbocyclic aromatic rings are involved, is far from trivial, since it generates highly reactive and unstable orthoquinodimethane species (oQDM), particular polyenolate donors, where the aromatic character of the original ring is temporarily lost. To enhance the acidity of protons at these benzylic positions, clever solutions were devised, either by strategically placing electron-withdrawing groups within the aromatic ring [strategy (i), Scheme 22], by activating the carbonyl function via covalent iminium/enamine organocatalysis [strategy (ii)], or by NHC-organocatalysis [strategy (iii)]. In either instance, the corresponding HOMO-raised polyenolate-oQDM dearomatized species (e.g., XVIII, XIX, XX) are formed, which can engage in remote benzylic functionalization with suitable electrophiles, often leading to cycloannulation products. The driving force of these processes is the thermodynamic stability of the products, which recover the aromatic properties, while further being stabilized within new 5- or 6-membered rings. In all cases, be they concerted or stepwise annulations, the regioselectivity of the processes is granted by the transmission of the electronic effects of the carbaldehyde through the conjugated π-system, exquisitely according to the vinylogy principle. The examples that follow in this subsection will deal with the exploitation of these concepts (strategies i and iii) for additions to C=O bonds, while additions to C=N and C=C bonds (strategies i, ii, and iii) will be treated in the competent subsections.
Scheme 22.

Capitalizing on precedent works on NHC-based activation of aldehydes and oQDM generation via trienamine activation (vide infra), Chi and co-workers successfully realized for the first time the functionalization of benzylic C(sp3)–H bonds of heteroaryl aldehydes through NHC organocatalysis.77 They started from 2-methyl-3-carboxaldehydes of indole, benzofuran, and benzothiophene of type 61 (Scheme 23) and coupled them with activated ketones such as trifluoromethyl ketones 62 (or isatins, not shown here) in the presence of NHC precatalyst ent-B13 (20 mol %) under oxidative conditions. The corresponding δ-lactone products 63 were obtained, as emerged by the formal [4 + 2] annulation involving an NHC-bound acylazolium of type XVII and oQDM intermediate of type IXX (see Scheme 22).
Scheme 23.

The experimental conditions were slightly adapted depending upon the nature of ketones 62 (R2 = aryl vs alkyl), affording the heterocyclic products 63 in good yields (apart from a couple of recalcitrant acceptors) and enantiomeric excesses. It is worth noticing that this procedure could not be extended to simple aromatic aldehydes (e.g., 2-methylbenzaldehyde) which were oxidized under the oxidative conditions of the reaction, nor could it be adapted to prostereogenic indole substrates where the methyl group at C2 within 61 was replaced by benzyl or CH2CO2Et groups.
Almost the same concept was applied by these authors using indole substrates with “inverted” substituents, namely, starting from 3-methyl 2-formylindoles (not shown).78 The reaction of these substrates with ketones such as trifluoromethyl ketones and α-ketoesters under NHC-catalysis delivered the corresponding racemic hydropyranoindoles in useful yields. Unfortunately, any attempts to translate this transformation into a nonracemic setting using chiral NHC did not yield appreciable results.
As already mentioned, in situ formation of oQDM species, derived from benzene precursors, is much more difficult than that of corresponding heteroaromatic precursors, given the higher degree of aromaticity to be temporally broken.
In 2016, Glorius et al. succeeded in the realization of this goal, by using NHC catalysis and ortho-bromomethylbenzaldehyde 64 as the starting materials (Scheme 24).79 In this instance, the presence of a leaving group (the bromine atom) facilitated the in situ generation of the NHC-bound dienolate oQDM 64″ through elimination of HBr from the Breslow intermediate 64′, thus circumventing the need for oxidation to acylazolium species and γ-deprotonation.
Scheme 24.

Coupling with activated ketones such as aryl glyoxylates (eq 1), isatins (eq 2), or trifluoromethyl ketones (eq 3) under uniformed reaction conditions (NHC from B15 or B16 and cesium carbonate) invariably produced the [4 + 2] annulation products 66, 68, or 70, respectively, in low to moderate yields in a racemic format. An attempt to use chiral NHC was performed on trifluoromethyl acetophenone (not shown), leading to a 48% ee of the δ-lactone product.
Almost during the same period, Rovis and Chen developed a chiral version of this reaction using chiral NHC/Brønsted acid cooperative catalysis.80 Thus, starting with bromomethyl benzaldehydes 64 and perfluoroalkyl aryl ketones 71 as the substrates, the combined use of chiral NHC from B14 and BINOL-derived chiral phosphoric acid L2 gave the desired products 72 with good results (Scheme 25). As for the ketone scope, electron-withdrawing substituents on the R2 aryl group generally decreased enantioselectivities, while alkyl derivatives failed to give any products.
Scheme 25.

Given the substantial role of the Brønsted acid cocatalyst in improving the enantioselectivity in a matched sense (a combination of ent-B14/L2 was less effective), a mechanism was proposed, where the Breslow intermediate undergoes transformation into an oQDM-dienol 64‴ with the formation of an ion pair between chiral triazolium NHC and chiral phosphate counterion, which dictates the enantioface discrimination of the incoming carbonyl. In the absence of mechanistic studies, the actual role of the acid as either a Brønsted acid, chiral counterion, or phase transfer promoter for the NHC catalyst remained undefined.
According to the concepts reported above (Scheme 22, strategy i), the introduction of electron-withdrawing groups such as nitro groups at ortho- and/or para-positions of 2-methylbenzaldehyde should enhance the acidity of the methyl protons, thus facilitating remote deprotonation and generation of highly reactive oQDM species. In 2017, Li et al. developed a domino asymmetric benzylation/aldol hemiacetalization reaction involving 2-methyl-3,5-dinitrobenzaldehyde (73) and β-aryl- (or heteroaryl)-substituted α,β-unsaturated trifluoromethyl ketones 74 (Scheme 26).81 The reaction was catalyzed by the tertiary amine/thiourea bifunctional organocatalyst C1 (10 mol %) and provided the corresponding 3,4-dihydroisocoumarin products 75 after oxidation (PCC). It was found that both electron-withdrawing and electron-donating substituents on Ar1 were well tolerated, but yields and enantioselectivities decreased when bulky (e.g., naphthyl) or ortho-positioned groups were used. Based on the experimental results, the authors proposed a transition state model of type 73′ where the benzylic pronucleophilic site of 73 is deprotonated by the tertiary amine catalyst forming a chiral ion pair, while the ketone carbonyl of 74 is activated and positioned by hydrogen bond interaction with the thiourea moiety of the catalyst. Unfortunately, control experiments using benzylic substrates without either nitro groups or carbaldehyde groups were not performed, which would have furnished further information about the actual role of these groups in the reaction.82
Scheme 26.

3.1.2. Indirect Procedures
The vinylogous addition of silicon-stabilized dienolates to C=O bonds, namely, the vinylogous Mukaiyama aldol reaction (VMAR), was introduced by Mukaiyama in 1975 using crotonaldehyde-derived silyl enol ether and cinnamaldehyde dimethyl acetal with TiCl4 as the Lewis acid promoter.83 Since then, research in this field has flourished, centered upon the application of stereoselective VMAR especially in the synthesis of polyketide-related natural products. Indeed, before the advent of direct HOMO-raising catalytic modalities triggering regioselective remote functionalization (see direct procedures), the VMAR represented the most common and orthodox way to carry out the aldol addition reaction in a vinylogous fashion.4 During the period covered by this review article, most of the silyl-based vinylogous reactions are associated with ester- or amide-derived nucleophiles, while a few examples deal with the use of aldehyde- or ketone-derived acyclic silyl enol ethers.
3.1.2.1. Acyclic Nucleophiles
The first enantioselective VMAR of aldehyde-derived dienolates was documented by Kalesse and Gieseler in 2011.84 Based on precedent results on the use of amino acid-derived oxazaborolidinones (OXB) in VMAR with ester-derived ketene acetals,85 they carried out the VMAR between preformed silyl dienol ethers 77 and aldehydes 76 utilizing the chiral OXB promoters of type M1 (Scheme 27, eq 1).
Scheme 27.

Various aldehyde substrates could be used successfully, including both aliphatic (i.e., cyclohexyl, iodoalkenyl, silyloxyalkyl, ...) and aromatic aldehydes, giving the corresponding δ-hydroxy-α-methyl enals 78 in satisfactory yields and variable levels of enantioselectivities. In any case, the chiral OXB Lewis acid had to be added in stoichiometric (or more) quantities (1.0–1.4 equiv range). The VMAR products 78 could be used as the precursors in various transformations. For example, the conjugate addition of hydrides followed by internal stereoselective α-protonation could give rise to hemiacetals 79.86
The sequence of VMAR followed by hydride reduction and α-protonation was exploited by the same authors twice during the elegant total synthesis of the naturally occurring antiobiotic (−)-angiolam A (Scheme 27, eq 2).87 In this instance, the two key VMARs involving first 76a + 77 and then 80 + 77 proceeded with two different OXB promoters (M2 and M1) and afforded the corresponding aldol products 78a and 81 in good yields and high stereoselectivities. The addition of trimethylborate as a competitor for binding the product to the chiral Lewis acid led to improved turnover numbers and allowed a catalytic loading of the OXB.
In the previous examples, the chiral metal-based catalyst (or promoter) acts as a Lewis acid capable of lowering the LUMO of the acceptor component, while not intervening with the silyl enolate substrate. Over the past decade, organocatalyzed asymmetric VMARs have been introduced and widely exploited, where a metal-free organocatalyst is able to activate either the acceptor or donor component, or both, as in the case of bifunctional organocatalysts. Despite the numerous reports in this field dealing with ester-derived silyl ketene acetals, we had to wait until 2018 to witness the first (and sole) enantioselective vinylogous aldol addition reaction involving aldehyde-derived silyl dienol ether donors and isatin electrophiles under bifunctional organocatalytic conditions (Scheme 28).88
Scheme 28.

Alemán and co-workers found that variously substituted isatins 82 could efficiently couple to silyl dienol ethers 83 using cinchona-thiourea catalyst C2 (10 mol %) and a controlled quantity of water (3 equiv) affording the respective products 84 with exclusive γ-site selectivity and good-to-very good levels of enantioselectivity. The role of water was crucial: in the absence of water, almost no conversion occurred, whereas an excess of water (6 equiv) led to hydrolysis of 83 and overall decrease of reaction efficiency. The authors affirmed that water played an important role in triggering the aldol reaction and in the catalyst interaction, but they did not enter into the specific study of the reaction mechanism. They did, however, refer to a previous work they had conducted dealing with the nonvinylogous, α-regioselective coupling of silyl dienol ethers 83 with nitroolefins under bifunctional organocatalysis in the presence of water as an indispensable ingredient (not shown).89 In that case, experimental studies and DFT calculations corroborated the conclusion that hydrolysis of the silyl dienolate occurs in the rate-determining step and is followed by the C–C formation, due to the appropriate orientation of both reagents in the transition state by the catalyst. The reasons for the striking difference in α vs γ regioselectivity of the two reactions compared remain to be clarified.
3.2. Additions to C=N Bonds
In this chapter, additions of remotely activated aldehyde-derived carbon nucleophiles to aldimines, ketimines, hydrazides, hydrazones, nitrones, and azomethine imines have been grouped together. All the examples refer to direct procedures involving acyclic pronucleophiles, and the HOMO-raising activation modalities parallel those encountered in the additions to C=O bonds, namely, activation of the remote β- or γ-sites via NHC organocatalysis and activation of remote γ- or ε-sites via enamine organocatalysis, often in cooperation with complementary LUMO-lowering Lewis/Brønsted acid catalysis.
3.2.1. Direct Procedures
3.2.1.1. Acyclic Pronucleophiles
As previously disclosed (Scheme 7), the covalent combination of α,β-unsaturated aldehydes with N-heterocyclic carbene (NHC) catalysts generates homoenolate equivalents in which the electron density of the heterocyclic ring is relayed to the vinylogous β-carbon via the diene portion of the molecule. When coupled to C=N bonds, these substrates may give rise to precious γ-lactam products after intramolecular closure according to a formal [3 + 2] annulation. Despite the numerous reports on this chemistry since its introduction in 2005,90 the enantioselective versions of these transformations had to wait until 2010 for their debut.91
Scheidt and collaborators recognized that the addition of homoenolate equivalents generated by enals 86 to hydrazones 85 would furnish the desired [3 + 2] annulation products with stereochemical induction, if the simultaneous activation of both reaction partners, by distinct catalysts, was at hand (Scheme 29).92 Indeed, treating α,β-unsaturated aldehydes 86 bearing aryl, furyl, or alkyl moieties with aryloyl hydrazones 85 in the presence of azolium salt B2 (5 mol %), a strong base (triazabicyclodecene, TBD), and catalytic magnesium di-tert-butoxide (5 mol %) effectively produced cis-disposed γ-lactam products 87 in good isolated yields and high enantioselectivities. When varying the imine portion of the substrate, glyoxylate-derived hydrazones and even an oxazolidinone-containing substrate reacted efficiently; whereas hydrazones from aromatic aldehydes (benzaldehyde) were still not electrophilic enough to undergo this annulation even with Mg(II) activation.
Scheme 29.

The presence of a hard, oxophilic Lewis acid such as the Mg(II) salt proved strategically indispensable for the overall good of the reaction, since it was able to enhance the electrophilicity of the hydrazone substrate, while not disturbing the catalytic activity of the NHC. After some preliminary kinetic studies, the authors proposed a mechanism according to which the vinylogous Breslow intermediate 86′ attacks the Lewis acid-coordinated hydrazone; subsequent intramolecular acylation of the magnesium-bound nitrogen closes the ring with regeneration of both the NHC and Mg(II) catalysts (via 86″).
An efficient and enantioselective [3 + 2] annulation between α,β-unsatutated aldehydes and unactivated imines was developed by Rovis and co-workers, using cooperative NHC and Brønsted acid catalysis.93 The focal idea was that the conjugate acid of the base, used to generate the carbene species, could be useful in activating basic functionalities such as that of an unactivated imine. The judicious tuning of the electronic and steric nature of the catalysts (i.e., weak basicity of the carbene induced by electron-withdrawing groups, steric hindrance of the carbene, weak basicity of the carbene-forming base) led the researchers to find the right combination of catalysts for an optimal reaction. Thus, β-ethoxycarbonyl-substituted enal 89 was treated with preformed α,β-unsaturated imines 88 in the presence of catalytic, chiral triazolium salt B17 and sodium o-chlorobenzoate 91 (20 mol % each), leading to γ-lactams 90 having an unprecedented trans-disposition in the predominant diastereoisomers (Scheme 30).
Scheme 30.

The best solvents were acrylonitrile or CH2Cl2, depending on the electronic nature of the Ar1 aryl group of aldimines 88. It was found that under the reaction conditions, other β-substituted enals of type 89 (e.g., β-keto- or β-aryl-substituted) could prove to be competent substrates (not shown), whereas changing the imine component (e.g., N-Bn, N-Ts unsaturated imines or N-phenyl aldimine from p-bromobenzaldehyde) gave modest results, if any. Based on control experiments, the authors proposed a mechanism in which the vinylogous Breslow intermediate 89′ attacks the acid-activated imine via hydrogen bonding; subsequent proton transfer would produce an acyl carboxylate eventually affording the annulated targets.
Compared to aldimines, ketimines are generally less reactive, and high stereoinduction is more difficult to achieve. The group of Chi developed an efficient protocol for the addition of NHC-activated enals 93 to isatin-derived ketimines 92, to afford spirocyclic oxindole-γ-lactams 94 according to the previously disclosed formal [3 + 2] annulation path (Scheme 31).94 The aminoindanol-derived triazolium salt B13 was chosen as the best NHC precatalyst, together with cesium carbonate as the base. A broad range of N-protected or even N-deprotected isatins 92 can successfully participate in the coupling reaction to diverse β-aryl or β-alkyl enals 93, giving products 94 in moderate to good yields and very good enantioselectivities.
Scheme 31.

Almost during the same period, a similar study was reported, dealing with the NHC-catalyzed homoenolate additions of enals with N-aryl oxindole-derived ketimines (not shown). The γ-lactam products of type 94 were obtained in good yields in a racemic format, though some preliminary attempts using chiral NHC were performed, obtaining modest enantioinduction.95,96
The concept of enabling both nucleophilic and electrophilic activation by combining metal catalysis and organocatalysis was exploited by Wu and co-workers in an inspiring work dating back to 2010.97 The researchers discovered that 2-alkynylbenzylidene hydrazides of type 95 could be easily converted to highly electrophilic isoquinolinium ions of type 95′ via 6-endo-cyclization in the presence of a suitable metal catalyst (AgOTf); in situ interception of these electrophiles by NHC-activated homoenolates from enals 96 could lead to 2-amino-1,2-dihydroisoquinolines (±)-97 after the intervention of methanol which liberates the catalyst, as depicted in Scheme 32. After identification of the best reaction conditions (AgOTf/B7, 5 mol % each, cesium carbonate as the base), the scope of this one-pot three-component reaction was analyzed, by exploring differently substituted substrates. Aromatic groups attached to the C≡C bond within 95 gave good results, while aliphatic groups (n-Bu or cyclopropyl) proved detrimental to the reaction.
Scheme 32.

Another one-pot, three-component reaction was proposed by Siddiqui and collaborators during the synthesis of 1,3-diazepanes 101 (Scheme 33).98 NHC-activated homoenolates from α,β-unsaturated aldehydes 100 were coupled to aryl imines, which in turn were generated in situ by condensation of aryl aldehydes 98 and urea (or thiourea) derivatives 99. A formal [4 + 3] annulation took place, likely through intramolecular closure (via 100′), giving racemic (±)-101 in good isolated yields.
Scheme 33.

The NHC-homoenolate component unveils a remote β-carbon nucleophilic site which may well act as a “dipolarophile” in a stepwise 1,3-dipolar cycloaddition with suitable dipole components. For example, if a nitrone is used in combination with a β-carbon NHC-activated enal, a formal [3 + 3] annulation may occur. Along this line, sugar-derived cyclic nitrones 102 were found to react cleanly with enals 103 under NHC catalysis, giving polyhydroxylated pyrrolidine and piperidine (and even azepane) derivatives of type 105 (Scheme 34).99 In the event, NHC-activated homoenolate from 103 coupled to nitrones 102 according to a formal [3 + 3] annulation, affording intermediates 104 (otherwise stable and storable), which were quenched in situ with NaOMe/MeOH in a one-pot operation, to furnish the desired products 105. The large collection of products 105 was then easily transformed to the corresponding pyrrolizidine and indolizidine alkaloids (not shown), which were eventually assayed against various glycosidase enzymes to give important clues to the structure–activity relationship of this new compound class.
Scheme 34.

γ-Enolizable enals 107 served as the four-carbon component in the [3 + 4] 1,3-dipolar cycloaddition with azomethine imines 106 (Scheme 35).100 Activation of enals 107 at the remote γ-carbon via oxidative NHC-activation (for the general concept of γ-carbon activation via NHC, see Scheme 17) afforded vinylogous enolates 107′ as the reactive dipolarophile components, which reacted with racemic azomethine substrates (±)-106 to forge the first C–C bond, as depicted in structure 107″. Intramolecular closure then yielded the expected dinitrogen-fused seven-membered heterocycles 108 with high optical purities and moderate-to-good yields. The approach also provided effective kinetic resolution of the starting racemic azomethine imines.
Scheme 35.

The enantioselective NHC-catalyzed [4 + 2] annulation reaction of γ-enolizable enals and aldimine or ketimine acceptors was developed by diverse authors to obtain highly enantioenriched δ-lactam products. As a first example, Chi and co-workers considered the reaction between enal 110 and cyclic sulfonyl imine 109 in the presence of the chiral NHC precatalyst B18 and quinone oxidant 45 as the model reaction for studying the general reaction mechanism of these types of annulation (Scheme 36, eq 1).101 It was noted that the reaction proceeded effectively, producing the δ-lactam product 111 in a good yield and enantioselectivity, without having to add a base, since the counteranion of the azolium salt behaved as a weak base.
Scheme 36.

Experimental kinetic studies monitored in situ by 1H NMR, as well as deuterium labeling and kinetic isotope effect studies, revealed that the rate-determining step of this oxidative catalysis is the formation of the vinylogous Breslow intermediate between the enal substrate 110 and the catalyst from B18, whereas the subsequent steps, namely, oxidation of the Breslow intermediate, γ-carbon deprotonation of the unsaturated azolium ester intermediate, and the vinylogous addition of the γ-carbon to the imine, are facile steps (the depiction of these steps for analogous addition to C=O is given in Scheme 17). The reaction scope of this transformation was then extended to a series of diverse enal/sulfonyl imine pairs (not shown), producing a collection of sulfonyl amides of type 111 which were duly evaluated in vitro for their antibacterial activity.102
The cooperative catalysis using NHC from ent-B18 and Brønsted acid 114 (2-bromobenzoic acid) was used for the effective dual activation of γ-enolizable enal 113 and sulfonyl imines 112 for the asymmetric synthesis of δ-lactams 115 (Scheme 36, eq 2).103 Instead of using oxidative conditions, enals 113 with an appropriate leaving group (p-nitrophenoxy) were utilized to grant the formation of the NHC-bound dienolate intermediates. All the Mannich reactions proceeded uneventfully, giving a diversified array of δ-lactam products in effective yields and good-to-excellent enantiomeric excesses.
The oxidative NHC-catalyzed [4 + 2] annulation reaction involving β-methyl enals 117 and cyclic trifluoromethyl ketimines 116 was developed by Enders and collaborators in 2017 en route to the enantioselective synthesis of dihydroquinazolinone products 118 (Scheme 36, eq 3).104 The optimization of the reaction conditions demonstrated that nitro-substituted NHC precatalyst B19 (20 mol %) together with the cesium acetate base and quinone oxidant 45 were the best choice to furnish the expected products in high yields and stereoselectivities. Of note, besides the β-aryl-substituted enals 117, aliphatic citral was also tolerated, delivering the target lactam in moderate yield and enantioinduction.
Besides NHC organocatalysis, aminocatalysis offers a powerful activation modality of remotely enolizable polyenals, and dienamine- or polyenamine-mediated additions of these donor substrates to C=N bond acceptors may trigger Mannich-initiated cascade reactions or cycloadditions eventually evolving to annulated products incorporating the γ/β, γ/β/ipso-, ε/β-, ... carbon portions of the starting enal (for a depiction of the general concept, see Scheme 2).
The first aminocatalytic, asymmetric, γ-selective, and Mannich-initiated cascade reaction was reported by Jørgensen and Albrecht in 2014.105 γ-Enolizable α,β-unsaturated aldehydes 121, anilines 120, and salicylaldehydes 119 were treated together according to a one-pot, three-component procedure in the presence of the Jørgensen–Hayashi organocatalyst A2 (20 mol %), directly affording bridged benzoxazocines 122 incorporating the γ/β/ipso carbon skeleton of the starting enal (Scheme 37).
Scheme 37.

Tuning the electronic and steric properties by careful choice of the R1/Ar1 substituents of the salicylaldehydes 119 and anilines 120 was important to increase the rate of their initial condensation to forge an imine in situ, while not being detrimental to the subsequent coupling reaction with 121. Thus, electron-withdrawing R1 groups of 119 and electron-rich anilines 120, bearing variously substituted phenyl-, naphthyl, and anthracyl groups (including p-ethynylphenyl), were revealed to be good substrates for affording the desired products effectively in generally good yields and with high diastereo- and enantioselectivities. A plausible reaction mechanism was proposed according to which an initial condensation between 119 and 120 occurs, giving the corresponding imine in situ. The s-cis-dienamine 121′ derived from condensation of enal 121 with the catalyst A2 and iminium-to-enamine isomerization then couples to the imine component, giving the catalyst-linked [4 + 2] cycloalkene intermediate 121″ and hence the highly reactive iminium ion 121‴ after elimination of the amine catalyst. Finally, an intramolecular oxa-Michael addition within 121‴ consigns the benzoxazocines targets 122. Neither a truly pericyclic cycloaddition nor an asynchronous—or even stepwise—mechanism was proven by further investigations, though the authors affirmed that employment of electron-rich N-aryl anilines might favor a [4 + 2] cycloaddition pathway. Whatever is the case, the vinylogous transmission of the nucleophilic character of the dienamine functionality to the remote γ-site through the π-system is operative, thus accounting for the observed regiocontrol.
Dienamine-mediated enantioselective [3 + 2] cycloaddition reactions were independently reported in the same year by Du, Wang, et al.106 and Alemán, Fraile, et al.,107 based on the 1,3-dipolar addition of γ-enolizable α,β-unsaturated aldehydes to C,N-cyclic azomethine imines (Scheme 38).
Scheme 38.

In the first work,106 γ-aryl-substituted enals 124 reacted with N-aroyl azomethine imines 123 using chiral prolinol silyl ether A2 (20 mol %) and 2,4-dinitrobenzoic acid (125) as an additive (Scheme 38, eq 1). After reduction with NaBH4, the corresponding cycloadducts 126 were recovered in high isolated yields as the sole products and with excellent enantiocontrol. The N–N bond of 126 could be easily cleaved with SmI2 to give precious tetrahydroisoquinoline products (not shown). The authors affirmed that products 126 were the result of a 1,3-dipolar cycloaddition (whether stepwise or concerted was not specified) involving dienamine-activated γ-carbon of 124 attacking the C=N bond and closure of the terminal nitrogen atom of the azomethine ylide to the enal β-carbon (γ/β carbon sites of the enal substrate involved, Scheme 38, eq 3). Interestingly, aliphatic α,β-unsaturated aldehydes 124 gave completely different results when treated in the same reaction conditions; LUMO-lowered iminium ion activation of 124 occurred in this case which, added to the 1,3-dipole 123 of reverted polarity, gave β/α-functionalized [3 + 2] cycloadducts of type 127.
The same ambivalent behavior of dienamine/iminium ion activable enals and azomethine imine dipoles was experienced by Alemán and Fraile,107 who also annotated some discrepancies between the results of the above cited paper and their own results. Considerable efforts were devoted to finding experimental conditions capable of chemoselectively channelling the reaction paths in either direction, namely, via the dienamine path (inverse electron demand character of the 1,3 dipolar cycloaddition) toward products of type 126 or via the reverted iminium ion-driven path (normal electron demand 1,3-dipolar cycloaddition) toward products of type 127 (Scheme 38, eqs 2 and 3). Thus, the optimized protocol addressing products 126 consisted in treating 123′, the hydrated hemiaminal form of 123, with γ-aryl-substituted 124 in the presence of amine catalyst A4 in CH2Cl2 at 0 °C; after reduction, targets 126 were obtained with complete exo-diastereoselectivity in variable yields and good enantiomeric excesses (Scheme 38, eq 2). A key role in determining the chemoselectivity of the reaction was played by the electronic/steric properties of the amine catalyst, the azomethine imine nature (dipole form vs hydrate form), and the presence or absence of additives (TBAB favoring the iminium ion path). Sustained by in-depth NMR-based and DFT calculations and the body of experimental results, the authors proposed that products 126 were formed as the result of a concerted 1,3-dipolar cycloaddition between in situ dehydrated azomethine imine from 123′ and dienamine-activated 124 thus accounting for the observed diastereo- and enantiocontrol (Scheme 38, bottom). Finally, the researchers found that the products they possessed, resulting from the iminium ion path of type 127, had the opposite configuration of those reported by Du and Wang.
The HOMO activation strategy of ε-enolizable conjugated dienals of type 129 via trienamine organocatalysis was exploited by Chen and collaborators to perform regio- and stereoselective [4 + 2] cycloaddition reactions involving 2-aryl-3H-indol-3-ones 128 (Scheme 39).108
Scheme 39.

Using prolinol silyl ether A4 (20 mol %) and salicylic acid (130) as a Brønsted acid cocatalyst, the addition reactions of different 2,4-hexadienals (and one 2,4-heptadienal) 129 to indolone derivatives 128 went to completion affording, after reduction, highly functionalized tricyclic products 131 efficiently and with good stereoselectivities. The good performance of the reaction was also due to the ad hoc-placed R2-R4 substituents of the dienal starters 129; almost all successful examples concerned γ-phenyl-substituted hexadienals (R2 = Ph), while the use of unsubstituted 2,4-hexadienal or 2,4-heptadienal gave aza-Baylis–Hillman-type products (not shown).109 The authors referred to the overall reaction as a normal-electron-demand aza-Diels–Alder reaction, and they did not enter into details about either the concerted or Mannich-initiated stepwise nature of the mechanism.
3.3. Conjugate Additions to Electron-Poor C=C Bonds
Paralleling the functionalization chemistry of remote carbon sites of unsaturated aldehydes in addition reactions to C=O and C=N bonds, even in the case of additions to electron-poor alkenes, the majority of examples are associated with direct procedures (no indirect examples were indeed found in the literature over the period covered) and mainly involve the organocatalytic activation of pronucleophilic substrates via either (poly)enamine or NHC catalysis. Most of the documented studies give rise to cycloaddition products emerging from Michael type-initiated additions to the electron poor olefins, followed by intramolecular closure as testified by diverse computational and/or experimental evidence.
3.3.1. Direct Procedures
3.3.1.1. Acyclic Pronucleophiles
α,β-Unsaturated aldehydes may explicit their β-pronucleophilicity via NHC activation to form the corresponding vinylogous Breslow intermediates (see previous sections) which may react with electron-poor alkenes via conjugate addition (vinylogous β-donor on vinylogous β-acceptor). The fate of the resulting coupling—i.e. product type and stereochemical result—depends mainly upon the functional groups of the acceptor component and the catalyst/cocatalyst/additive used.
Following the fundamental work by Nair110 and Bode,111 Scheidt and collaborators discovered how β-aryl enals 133 could productively react with a series of chalchones 132, giving cis-configured 1,3,4-trisubstituted cyclopentenes 134 (Scheme 40, eq 1).92 Key to the success of the transformation was the integrated use of NHC catalysis (chiral aminoindanol-derived triazolium salt B2 was the best precatalyst) and Lewis acid catalysis (Ti(OiPr)4 bearing the donating metal alkoxide ligands, proved to be the best choice) exploiting the concept of simultaneous activation of the two reacting partners via cooperative catalysis. Using the NHC/LA cocktail, together with catalytic i-PrOH as an addititve and DBU as the base, ensured preparation of the targeted cyclopentenes 134 in high isolated yields, excellent cis-diastereoselectivity, and optimal enantioselectivity. Carrying out the reaction without the Lewis acid led to the formation of the trans-isomers as the major products, pointing to the key role exerted by the Lewis acid during the stereodetermining step. A catalytic pathway was proposed, where the titanium-coordinated homoenolate 133′ adds via conjugate addition to the titanium-coordinated chalcone acceptor, generating the cis-disposed species. Protonation/tautomerization of the chalcone moiety then predisposes the second C–C bond-forming event, namely, the intramolecular aldol addition involving the α-carbon of the starting enal and the chalcone carbonyl. The cyclopentane ring 133‴ is thus forged, which quickly converts to the cyclopentene target after intramolecular acylation, with NHC release and decarboxylation (the ipso carbon is lost as CO2). A global [3 + 2] annulation occurs, involving the β/α carbon donor sites of the starting enal.112,113
Scheme 40.

Merging the Lewis acid activation strategy with NHC Lewis base catalysis was soon after exploited by the same research group in the enantioselective dimerization of enals of type 133 to give cyclopentene esters 135 (Scheme 40, eq 2).114 In this case, the challenge was to channel the reaction path toward a [3 + 2] annulation involving 1,4-conjugate addition of the β-donor homoenolate (β to β), while bypassing the competing and often prevailing 1,2-addition path (β to ipso). It was demonstrated that the presence of the Ti(OiPr)4 cocatalyst was essential in driving the reaction toward the desired cyclopentenes. The reaction pathway proposed is very similar to the previous one (via 133′ and 133″); in this case, however, the isopropoxide ligand promotes the final acylation with liberation of the NHC catalyst, while the excess of TBD base promotes water elimination to provide the alkene moiety.
Besides chalchones and enals, another class of electron-poor alkenes could be conveniently added to NHC-bound homoenolates under cooperative Lewis acid/NHC catalysis, namely, β,γ-unsaturated α-ketoesters.115 As depicted in Scheme 41, several keto esters 136 were analyzed, bearing aryl, heteroaryl, cyclopropyl, and alkynyl γ-substituents, all giving the corresponding cyclopentane bis-esters 137 with good results (only alkyl and alkenyl R1 groups did not work, data not shown). Again, a Michael-type addition of vinylogous homoenolate from 133 to keto esters 136 occurs, followed by intramolecular aldol addition of the Cα site within 133 to the ketone acceptor (see a similar mechanism in Scheme 40, via intermediates 133′–133‴). In this case, however, intermolecular acylation and transesterification by isopropanol/isoproxide ligand occurs, liberating the NHC catalyst. No intramolecular acylation nor water elimination to cyclopentene products was observed.
Scheme 41.

A highly regio- and stereoselective addition of NHC-activated enals with benzodienone acceptors was reported by Chi and co-workers, highlighting a Michael–Michael-type cascade annulation, and furnishing multifunctionalized polycyclic products (not shown).116
Similarly, β-NHC-activated 2-aroylvinylcinnamaldehydes were reacted with 2-aroylvinylchalchones, providing access to complex indane products (as racemates), via triple Michael-type addition and lactonization cascade.117 Some years later, analogous 2-aroylvinylcinnamaldehydes were added to α,β-unsaturated sulfonyl imines under asymmetric NHC catalysis (homoenolate chemistry) giving rise to controlled regio- and stereodivergent pathways depending upon the employed catalysts (not shown).118
The reaction of cis-configured enals of type cis-138 activated by NHC catalyst with α,β-unsaturated imines was thoroughly investigated by Chi et al. in 2013 (Scheme 42).119 It was known from previous studies by Bode,120 that trans-enals react with these electron-deficient alkenes under NHC catalysis giving bicyclic β-lactams of type 141 (Scheme 42, left side), presumably as the result of a first Michael-type addition of the β-site of the corresponding trans-homoenolate (trans-138′) to the conjugated sulfonyl imine and subsequent intramolecular Mannich-type closure and N-acylation.120 Accordingly, the β/α/ipso carbon atoms of the starting enal are inserted within the lactam bicycle.
Scheme 42.

Quite unexpectedly, in treating cis-138 under optimized conditions (precatalyst B21, DIPEA base and triphenol additive 142) aimed at preserving its cis-stereointegrity, a completely diverse reaction path was unveiled, and coupling with unsaturated sulfonyl imines 139 led to alkylidene cyclopentenone products 140 in high yields, complete diastereoselectivity, and with negligible, if any, presence of the bicyclic lactam counterparts 141. In this case, it was proposed and partially proved by control experiments that cis-homoenolate cis-138′ was formed, which underwent a first Michael-type addition followed by intramolecular Claisen reaction (Scheme 42, right side). This work represents a nice example of how two diastereomeric intermediates (dashed boxes in the scheme) trigger divergent reaction pathways toward different target chemotypes.121
In 2013, Scheidt and McCusker developed a new NHC-catalyzed formal [4 + 2] annulation reaction between enals and α,β-unsaturated imidazolidinone acceptors. In this case, however, a nonvinylogous path was triggered, where the α-position of the unsaturated enal acted as a donor site instead of the vinylogous homoenolate-linked β-position (not shown).122
Among electron-poor alkenes, nitroalkenes were also usefully used in Michael-type addition reactions with α,β-unsaturated aldehydes via NHC-activated homoenolates, to furnish linear δ-nitro esters (Scheme 43).
Scheme 43.

Following a pioneering report by Nair who initially applied this chemistry in a racemic format,123 Liu and collaborators succeeded in performing this transformation in an asymmetric context.124 Thus, as shown in Scheme 43 (eq 1), a large series of nitroalkenes 144, including nitrodienes, nitroenynes, and nitrostyrenes, efficiently reacted with enals 143 (mainly aromatic, but also acrolein, crotonaldehyde, and furyl-substituted enals were reported), using NHC precatalyst B18 and furnishing, after acylation by external methanol, the corresponding nitromethyl esters 145 in generally good yields, anti-diastereoselectivity, and moderate to high enantiocontrol. Interestingly, the reactions proved completely regioselective (β-carbon donor on β-carbon acceptor), and no 1,6-conjugate additions (β-donor on δ-acceptor) or Stetter coupling (ipso donor on β-acceptor) was witnessed. As a limitation, aliphatic nitroalkenes did not prove to be suitable substrates in this transformation.
Complementary to this work were the studies by Rovis et al., who found conditions to perform the same type of transformation to deliver syn-configured nitro esters, while extending the scope to aliphatic nitroalkenes (Scheme 43, eq 2).125 Key to the success of the reaction was the rational design of the NHC-precatalyst to be used, whose stereoelectronic properties were carefully tuned in order to bestow a positive impact on the regio- and stereocontrol of the overall reaction. Thus, using bis-n-butyl O-TMS triazolium precatalyst B22 in combination with sodium acetate and ethanol, aryl-substituted enals 143 were added to a large number of nitroalkenes 144 giving syn-disposed nitroethyl esters 146 (Scheme 43, eq 2). Enantio- and diastereoselectivities were typically greater for alkyl- and cycloalkyl nitroalkenes, though bulky derivatives such as t-Bu did not work (not shown). Interestingly, alkyl-substituted enals 143 were not productive and gave mainly the undesired Stetter products (not shown). In both studies,124,125 the nitro ester products were easily manipulated into valuable nitrogen-containing cyclic products.
α,β-Unsaturated esters were also competent electron-poor substrates to be added to β-NHC-activated enals. For example, Nair and collaborators developed a NHC-catalyzed process featuring the intramolecular Michael-initiated cascade reaction involving 2-O-alkenoate cinnamaldehydes and culminating in the formation of racemic coumarin derivatives (not shown).126
In 2013, Scheidt et al. devised a dual activation strategy where two reactive, transient species were concomitantly generated: a NHC-bound vinylogous β-donor of type 147′ and o-quinone methide (oQM) acceptor 148′ (Scheme 44, eq 1), derived from the respective precursors, enals 147 and silyl phenols 148.127 The optimal reagent combination (CsF/18-crown-6 for oQM generation and n-Bu4NOAc as a mild base for NHC generation from precatalyst B2) provided formal [4 + 3] benzoxopinone products 149 in moderate/good yields and with acceptable to excellent enantioselectivities. The scope of the reaction was explored: various cinnamaldehydes 147 were well tolerated, while highly reactive acrolein gave a totally diverse [4 + 2] product (not shown). As for oQM precursors, both bromides and chlorides could be used as good leaving groups, and in general R2 electron-donating groups gave the best results. Also, prostereogenic silyl phenols precursors were exploited (R1 ≠ H), extending the [4 + 3] process to vicinally substituted products. In these cases, however, low diastereoselectivities were observed. Based on DFT calculations (and other data), it was proposed that the vinylogous Breslow intermediate 147′ would intercept the transient oQM 148′ through a Michael-type C–C-bond-forming addition in an open transition state. The formed species 147″ would then undergo tautomerization and intramolecular O-acylation by the phenoxide anion, thereby producing the lactone product with concomitant release of the NHC catalyst. Overall, the driving force to induce the formation of a 7-membered ring was the rearomatization of the oQM species. The β-vinylogous regioselectivity strictly depended upon the stability of the extended Breslow intermediate 147′; in the absence of β-aryl substitution (e.g., acrolein, crotonaldehyde), protonation of the β-site becomes competitive and nonvinylogous [4 + 2] annulation occurs via NHC-enolate C-α conjugate addition/O-acylation (not shown).
Scheme 44.

Similar chemistry was developed by Ye and collaborators over the same period.128 In this instance, the fairly stable and electron-rich PMP- or styryl-substituted oQM species 151 were directly used as electrophilic substrates without the necessity of generating them in situ (Scheme 44, eq 2). These species were reacted with enals 150, which were activated in situ by the NHC catalyst derived from carbinol B23 and potassium acetate base. The corresponding [4 + 3] annulated products 152 were obtained, as emerging from the vinylogous conjugate addition of the β-carbon donor 150 to the oQM 151, followed by intramolecular acylation (see mechanism in the same scheme, eq 1). The authors affirmed that the bulkiness of the NHC catalyst, together with its hydrogen-bonding properties, covered a role in governing the vinylogous regioselectivity and the enantiocontrol as well. Interestingly, not only β-aryl and β-heteroaryl enals but also β-alkyl enals of various chain length (R1 = ethyl, n-propyl to n-heptyl) could be efficiently used to give, in these last cases, remarkable diastereoselectivities and enantioselectivities.
A catalyst-controlled chemodivergent reaction between α,β-unsaturated aldehydes 154 and cyclic enones 153 was developed by Zhao et al., leading to the formation of either [4 + 3] lactone products 155 (Scheme 45, eq 1) or spirocycles 156 (eq 2).129 In the first instance, cooperative catalysis was adopted, using NHC precatalyst B13/DBU and Ti(Oi-Pr)4 as a Lewis acid. The substrate scope of this catalytic system was broad and included enals 154 bearing alkyl, ether-containing alkyl, and phenyl groups, while the Michael acceptor component 153 included both oxygen and nitrogen heterocycles.
Scheme 45.

In almost all cases, the corresponding coumaranone or indole-fused derivatives 155 (X = O, NH) were obtained with excellent cis diastereoselectivity and good chemoselectivity (155:156 = 5:1 to 15:1). When the precatalyst B21/DABCO combination was used instead, the reaction between 153 (X = O) and 154 gave [2 + 3] annulation to furnish mainly spirocycles 156 (156:155 = 4:1 to 7:1). A stepwise mechanism was proposed, according to which both targets are generated by a common intermediate precursor 153′ (the product of the conjugate addition of NHC-bound homoenolate to the Michael acceptor), which closes either to the [4 + 3] product 155 by O-acylation or to the [2 + 3] product 156 by C-acylation. The authors speculated that the backbone of the azolium catalyst played a dramatic effect on this chemodivergent path and postponed a more rigorous mechanistic discussion to future work.
Similar chemistry was cleverly developed by Glorius and co-workers, where Michael acceptors of type 153 were coupled to enals under NHC catalysis, leading to the exclusive formation of [2 + 3] products with high optical purity (not shown).130
In line with the chemistry disclosed in the previous sections, the direct HOMO-raising remote activation of α,β-unsaturated aldehydes by NHC catalysis may involve not only the β-site (vinylogous with respect to the ipso carbon) but also the γ-site (vinylogous with respect to the α-carbon), opening new synthesis perspectives toward precious carbocyclic and heterocyclic targets.
As a clever example of this concept, Chi et al. devised a very simple and efficient NHC-catalyzed formal [3 + 3] cycloaddition reaction to address multifunctionalized benzenes in one step by starting from α,β-unsaturated enals and unsaturated ketones.131 As illustrated in Scheme 46, a very easy procedure was implemented, according to which γ-enolizable β-aryl (or even β-heteroaryl) enals 157 reacted with a wide collection of enones 158 using achiral NHC precatalyst B6 under oxidative conditions, directly giving benzenes 159 in useful isolated yields. The variability of the R1-R3 substituents within enones 158 granted access to a wide collection of aromatic targets ranging from alkyl, aryl, and trifluoroalkyl ketones, to esters and nitro derivatives.
Scheme 46.

Though no in depth studies on the reaction mechanism were executed, based on precedents of NHC-γ-activation (see Scheme 17), a plausible mechanistic path of this formal [3 + 3] cycloaddition was proposed. This would encompass a Michael addition step involving vinyl enolates 157′, in turn obtained from oxidation/γ-deprotonation of the extended Breslow intermediate, and enones 158, to furnish intermediate 157″. Subsequent γ-deprotonation of 157″ and intramolecular α-aldol addition would lead to 157‴ which collapses to aromatic products 159 by O-acylation with NHC release, decarboxylation, and final oxidative aromatization. This straightforward and versatile procedure greatly outperformed the previously reported traditional syntheses of these targets, which justifies the legitimate collocation of this work in the context of the vinylogous realm, notwithstanding the fact that achiral products are obtained.
γ-Enolizable enals could also be suitable substrates for formal [4 + 2] cycloaddition reactions with suitable Michael acceptors. Along this line, Yao et al. used γ-methyl α-bromo-α,β-unsaturated aldehydes and 3-alkylideneoxindoles to perform the diastereoselective synthesis of spirocarbocyclic oxindoles under NHC catalysis (not shown).132
One year later, Xu, Liu, et al. documented a similar transformation in an enantioselective context (Scheme 47, eq 1).133 In this case, β-methyl enals 161 were reacted with oxindoles 160 using NHC catalyst from ent-B18 and LiCl as a useful Lewis acid additive. With the exception of heteroaryl and cyclohexyl derivatives 161, which gave products 162 in low yields and enantiomeric excesses, the other enal/enone substrates performed quite well, affording the corresponding all-carbon spirocyclic oxindoles 162 with good results. A plausible mechanism of this reaction entails a stepwise process: an initial γ-regioselective Michael addition of NHC-bound dienolate from 161 to 160, followed by intramolecular closure of the emerging enolate to the C-ipso carbon (C-acylation) with the liberation of the NHC catalyst.
Scheme 47.

3-Methyl-4-nitro-5-vinyl isoxazoles 163 also served as interesting Michael acceptors in formal [4 + 2] cycloadditions with enals 164 under NHC catalysis (Scheme 47, eq 2).134 A varied collection of cyclohexenone products 165 was assorted in generally good yields, excellent diastereoselectivities, and moderate to good enantioselectivities. The proposed mechanism is similar to the previously disclosed examples; in this specific case, the NHC-activated γ-dienolate from 164 is engaged in a 1,6-conjugate addition to the electron-poor nitrodiene 163 (vinylogous γ-donor on bis-vinylogous δ-acceptor), and the emerging γ-nitronate attacks, again in a vinylogous sense, the NHC-bound C-ipso site to consign the cycloadducts 165. The same procedure was also extended to other electron-deficient alkenes, namely, 2,4-diene from antipyrine and 2,4-dienes from malononitrile, benzoylacetonitrile, and Meldrum’s acid, affording the respective products (e.g., compound 166).
The HOMO-raised dienamine species derived from an amine catalyst and γ-enolizable α,β-unsaturated aldehydes may act as good 2π dienophile species in various [4 + 2] cyclization reactions with electron-deficient diene partners in what could be (at least formally) categorized as inverse-electron-demand Diels–Alder (IEDDA) or hetero Diels–Alder (IED-HAD) reactions (Scheme 48).
Scheme 48.

Since 2010, various strategies were proposed by different research groups, to regioselectively steer such cyclizations either toward nonvinylogous processes involving α,ipso carbon sites of the initial enal,135 or toward vinylogous processes, involving the remote γ,β carbon atoms.
A first attempt in this latter direction was made by Chen and co-workers, who chose allylidene malononitriles or cyanoacetates 168 as 4π-component, and crotonaldehyde 167 as 2π-component in their aminocatalytic all-carbon IEDDA-type reactions (Scheme 49, eq 1).136 The combination of catalytic secondary amine A4 and benzoic acid additive ensured formation of the desired cyclohexene products 169 in moderate-to-high yields and generally good stereoselectivities. In the absence of direct evidence about the actual reaction mechanism yet considering the very good enantiocontrol at the remote γ,β positions, the authors speculated that this reaction proceeded via a concerted [4 + 2] cycloaddition pathway. An endo-selectivity emerging from the attack of the Si face of the s-cis-dienamine intermediate from 167 to the diene 168 under steric-shielding catalyst control seemed to be responsible for the observed stereoinduction within the products. Nevertheless, as stated by the authors themselves, more investigations to elucidate the actual mechanism remained to be carried out.
Scheme 49.

In order to expand the scope of such IEDDA reactions beyond the simple crotonaldehyde substrate, the same group developed an asymmetric all-carbon IEDDA-type cycloaddition involving γ,β-functionalization of γ-enolizable β,β-disubstituted enals and chromone-fused dienes via dienamine organocatalysis. Multifunctional caged or fused heterocyclic products were obtained in high optical purity and with high efficiency as the result of a sequence encompassing an IEDDA reaction, followed by deprotonation-isomerization and vinylogous aldol closure (not shown).137
A remotely γ,β-regioselective and stereoselective IED oxa-DA reaction was reported by the Jørgensen group, by applying H-bond directing aminocatalysis.138 As shown in Scheme 49 (eq 2), various unsaturated oxoesters 170 were selected as electron-deficient components, anticipating their possible activation in H-bonding aminocatalysis. These substrates were reacted with γ-enolizable enals 171 in the presence of catalytic secondary amine-squaramide dual catalyst A5 (16 mol %) and N,N-diethylacetamide (DEA), affording the desired [4 + 2] cycloadducts 172. The generality of the reaction with respect to both reacting substrates was assayed, demonstrating that dihydropyran derivatives 172 could be assembled in various assortments with generally good efficiency and moderate-to-good stereocontrol. The rationalization accounting for the stereochemical outcome was provided. Although no calculations were performed, yet in accordance with earlier calculations in related [2 + 2] cycloadditions, a stepwise mechanism was invoked (i.e., a vinylogous Michael-initiated addition followed by O-closure) involving s-trans dienamine 171′ and H-bonding activated keto esters 170, with possible beneficial π-stacking interactions between the aromatic moieties of the reacting partners.139
By employing the same bifunctional squaramide-amine catalyst, an asymmetric IED oxa-DA was developed by the same authors, who employed γ-enolizable α,β-unsaturated aldehydes and α,β-unsaturated acyl phosphonates as starting substrates. Useful dihydropyran rings were obtained in good yields and optical purity (not shown).140
Inspired by these IED-HDA reactions using H-bond-directing dienamine-mediated strategies, Pericàs and collaborators developed [4 + 2] cycloadditions between γ-enolizable enals 174 and alkylidene pyrazolones 173 (Scheme 50, eq 1).141
Scheme 50.

The corresponding chiral tetrahydropyranopyrazoles 175 were produced in good yields, with high enantioselectivity and satisfying levels of diastereoselectivity. Based on previous DFT calculations, an E,s-trans,E-dienamine 174′ was postulated to be the active conformer, which approaches the heterodiene component along an exo-trajectory with an H-bonding between the OH of the diarylprolinol and the pyrazolone carbonyl.
A novel approach to optically active benzothiophene-dihydropyran ring-fused compounds 178 was devised by Albrecht et al., based on the γ,β-regioselective IED oxa-DA reaction between 2-alkylidenebenzothiophenones 176 and γ-enolizable enals 177 (Scheme 50, eq 2).142 The reaction proceeded under aminocatalytic conditions using protected prolinol A3 (20 mol %) with the participation of a key dienamine intermediate derived by condensation/isomerization between the starting enals and the amine catalyst. The driving force of the process was judged to be the aromatization of the thiophene moiety within the targets, which were isolated as dibromoalkene derivatives after olefination of the crude cycloadducts (CBr4, PPh3).
Besides [4 + 2] cyclizations, HOMO-raised dienamine species derived from γ-enolizable enals and amine catalysts may be conveniently engaged in [2 + 2] cycloaddition reactions involving electron-poor alkene partners such as nitroalkenes (Scheme 51). While the nonvinylogous α-functionalization leads to linear Michael-type products,143 the vinylogous γ-functionalization of such dienamines may lead to stable cyclobutane products (formal [2 + 2] cycloaddition), provided that suitable activating catalysts and product-stabilizing tricks are invented. In 2012, two research groups independently and almost simultaneously addressed this issue with success.
Scheme 51.

The Jørgensen group purposely designed a bifunctional organocatalyst which would be able to both efficiently activate the two reacting partners and provide an appropriate distance between them in the transition state in order to steer the reaction toward the intended, remote vinylogous functionalization (Scheme 52, eq 1).144 Thus, treating enals 179 and nitroalkenes 180 with H-bond directing squaramide-amine catalyst A5 (20 mol %) in the presence of N,N-diethylacetamide (DEA) as catalyst-solubilizing agent and controlled quantities of water (2.8 equiv) in CH2Cl2 cleanly provided cyclobutanes 181 in good yields and exceptional levels of diastereo- and enantioselectivities. A variety of nitrostyrenes (R3 = Ar) and even heteroaryl or alkyl-substituted nitroalkenes were used, which were coupled with diverse enals 179 with equal success. Computational studies supported a stepwise mechanism for this [2 + 2] cycloaddition: at first, a vinylogous Michael-type addition occurs between the γ-carbon of the dienamine-activated enal 179′ and the β-carbon of the squaramide-activated nitroolefin. The nitronate intermediate, thus formed, then intramolecularly attacks the vinylogous iminium ion to furnish the cyclobutane product.
Scheme 52.

To perform similar [2 + 2] cyclizations, Vicario and collaborators started from enolizable enals 182 and hydroxymethyl nitrostyrenes 183, and used chiral secondary amine A4/achiral thiourea H1 as an efficient catalytic couple (Scheme 52, eq 2).145 Dual activation of both reagents occurred (covalent activation of 182 via dienamine and hydrogen bonding activation of the nitro group by the thiourea) triggering a Michael/Michael/hemiacetalization reaction cascade to the final bicyclic products 184. Even in this case, complete diastereoselectivity was witnessed and the products were isolated in high yields (except for the case of alkyl derivatives, R1 = Et) and high optical purity as 1:1 mixture of anomers. Of note, the reaction of 182 with nitrostyrene under the reaction conditions failed, pointing to the conclusion that the hydroxymethyl appendage within 183 was indispensable to steer the reaction toward hemiacetal derivatives 184, thereby providing a thermodynamic driving force for the reaction to proceed to completion.
On the basis of similar H-bond-directing dienamine activations, the asymmetric synthesis of spirocyclobutyl oxindoles was carried out by Wang et al. via formal [2 + 2] cycloaddition between enolizable enal donors and alkylidene oxindole acceptors (not shown).146
In 2015, Jørgensen and co-workers discovered a conceptually novel organocatalytic enamine-activation mode of cyclopropanes and exploited this concept in stereoselective cycloaddition reactions (Scheme 53).147
Scheme 53.

Based on preliminary robust computational studies, they first demonstrated that treating racemic diester-substituted cyclopropylacetaldehyde (±)-185a with prolinol silyl ether catalyst A4 and benzoic acid promoted smooth ring opening to α,β-unsaturated aldehyde 186 via HOMO-activated enamine 185a′ (eq 1). Hence, they proceeded to investigate whether it was possible to intercept the organocatalytically activated cyclopropane intermediates of type 185a′ in reactions with electron-deficient alkenes. Indeed, it turned out to be the case. Cyclopropanes (±)-177 were reacted with olefinic oxindoles (or benzofurans) 187 in the presence of amine catalyst A4 (10 mol %) and benzoic acid affording (after one-pot Wittig-type olefination) spirocyclobutane oxindoles (or benzofuranones) 188 with good results in terms of scope generality, efficiency, and stereoselectivity. Two mechanistic pathways were proposed, namely, a [3 + 2] cycloaddition followed by ring-contracting rearrangement (unlikely) or a dienamine-mediated formal [2 + 2] cycloaddition (preferred). According to this last proposal, catalyst-induced ring opening of the cyclopropane within 185 produces iminium ion 185′ and hence dienamine 185″, which undergoes γ,β-regioselective cyclization with the electron-poor substrate 187. In practice, the amine-activated cyclopropylacetaldehyde substrates acted as useful dienamine surrogates to be used, as in these cases, in vinylogous processes.
HOMO-raised dienamine species obtained by condensation/isomerization of enolizable enals with amine organocatalysts could be cleverly used as 4π components in [4 + 2] cycloaddition reactions, either stepwise or concerted, with electron poor 2π alkenes148,149 to give precious six-membered rings. Inherent challenges in this strategy include possible double participation of the amine catalyst in activating both substrates (e.g., dienamine and iminium ion), difficulty in efficient catalyst release and recycling, and efficient catalyst-to-substrate stereoinduction. Several reports dealing with this subject were chronicled in the 2010–2018 period, which are grouped and briefly commented on in the following schemes.
In a first example, Vicario et al. exploited the potential of dienamine catalysis (using amine catalyst A4 and 4-nitrobenzoic acid cocatalyst 192) to promote the coupling reaction between enolizable enals 189 and racemic acyloxy-substituted dihydropyranones (±)-190 (Scheme 54, eq 1).150 Enantioenriched isochromanes 191 were obtained regioselectively as single diastereoisomers in moderate-to-good yields as the result of [4 + 2]/elimination cascade reaction in a dynamic kinetic resolution process. No mention was made of the concerted vs stepwise nature of the cyclization.
Scheme 54.

In another study by the Jørgensen group, dienamine-activated enals 193 added to 1,4-benzo- or 1,4-naphthoquinones 194, affording dihydronaphtho- and dihydroanthraquinones 195 (Scheme 54, eq 2).151 Excellent levels of enantioinduction and complete regioselectivity (γ-carbon donor attacking the more hindered quinone carbon acceptor) were uniformely observed. Computational studies supported the notion that a stepwise mechanism was involved; an initial vinylogous Michael-type addition of the dienamine along an endo pathway, to forge the zwitterionic enolate/iminium ion intermediate of type 193′, which is internally stabilized by favorable electrostatic interactions, after which intramolecular aldol closure to 193″ and catalyst elimination consign the targeted compounds.
Several years later, a closely related [4 + 2] cycloaddition reaction was performed by Gong and co-workers, by adopting a completely different C–H activation strategy.152 The basic idea was just as simple as it was powerful: treatment of unbiased saturated aldehydes 197 (Scheme 55, eq 1) with a chiral amine catalyst would easily produce enamine 197′, which could be converted to unsaturated iminium ion 197″ by Pd-catalyzed Saegusa-type oxidation. Clean iminium ion-enamine isomerization of γ-enolizable 197″ would then produce the active dienamine species 197‴, which is able to participate in a plethora of asymmetric vinylogous (or nonvinylogous) coupling reactions with suitable electrophiles. A mixed metal/organo cooperative catalysis would thus enable the enantioselective functionalization of inactive C(sp3)–H bonds at the γ-position of saturated aldehydes by directly transforming them in situ into the corresponding HOMO-raised dienamine species. The feasibility of this concept was proven by treating different saturated β-substituted aldehydes 197 with methylquinones (or methylnaphthoquinones) 198 in the presence of catalytic Pd(OAc)2, chiral amine ent-A4, and acid additive 196 in DMSO under oxygen atmosphere (to ensure reoxidation of Pd(0) for the next catalytic cycle) at 40 °C (Scheme 55, eq 2). Further addition of a Lewis acid such as benzene complex of copper(II) triflate was found to be beneficial for the overall efficiency of the reaction, especially when less reactive methylnaphthoquinones substrates were involved. The expected [4 + 2] cycloadducts 199 were obtained with good optical purity in variable yields, mainly depending on the electronic properties of the R1-R3 substituents within the substrates.
Scheme 55.

Yang et al. documented an intramolecular version of dienamine-based eliminative [4 + 2] cycloadditions, by starting from bis-enal substrates to forge highly enantioenriched dihydrobenzofurans (not shown).153
In another study, coumarin-3-carboxylic acids 200 and enolizable enals 201 were involved in a catalytic decarboxylative and eliminative [4 + 2] cycloaddition reaction to forge cyclohexadiene lactones 202 (Scheme 56, eq 1).154 In this case, in situ decarboxylation enabled release of the amine catalyst, allowing the transformation to proceed in a high yield and with high enantio- and diastereoselectivity. Subsequent one-pot hydride reduction of 202 and acid-catalyzed intramolecular cyclization provided access to chiral bridged tricyclic benzopyrans (not shown).
Scheme 56.

A similar aminocatalytic, decarboxylative, and eliminative [4 + 2] cycloaddition reaction was devised by Albrecht and Bojanowski for the synthesis of enantioenriched dihydroxanthones (not shown).155 In this instance, β,β-disubstituted γ-enolizable enals and chromone-3-carboxylic acids were used as substrates, while methyl-protected (S)-diphenylprolinol was employed as the amine organocatalyst.
The asymmetric synthesis of a collection of tricyclic derivatives 204 was developed by Alemán and collaborators via an aminocatalyzed desymmetrization reaction involving ad hoc prepared cyclohexadienone/enals 203 (Scheme 56, eq 2).156 DFT-based calculations and control experiments showed that this transformation proceeds via an asynchronous eliminative [4 + 2] cycloaddition (endo transition state) and not a stepwise Michael/aldol/elimination cascade reaction.
An asymmetric dienamine-mediated eliminative [4 + 2] cycloaddition between sulfone-enones (i.e., sulfonyl Nazarov reagents) and enolizable enals was developed by Díez et al. leading to highly functionalized cyclohexa-1,3-dienes (not shown).157
The multifaceted reactivity of dienamine species from α,β-unsaturated aldehydes toward electron-poor alkenes (e.g., γ,β-[2 + 2]-, γ,ipso-[4 + 2]-cyclizations, vide supra) could be extended further to include γ,α-[3 + 3] formal cycloadditions with suitable 1,3-bis-electrophilic partners (Scheme 57).158 When Chen and co-workers treated enolizable enals 205 with 2-nitroallylic acetates 206 using catalyst A8, cyclohexenal products 207 were obtained, as the result of a reaction cascade presumably comprising a vinylogous Michael addition of dienamine 205′ to the nitroalkenes, followed by a second intramolecular α-regioselective Michael addition involving 205″. After catalyst screening, bifunctional secondary amine-thiourea catalyst A8 was identified as the best catalyst in terms of reaction efficiency and enantioselectivity. However, due to low cis/trans diastereoselection in all assayed experimental circumstances, the product diastereomeric mixtures were subjected to base-promoted epimerization to afford the trans-isomers 207 exclusively.
Scheme 57.

While the remote activation of enals using dienamine-mediated covalent catalysis was well established and widely exploited in synthesis, the noncovalent asymmetric and catalytic strategies based on the use of chiral Brønsted bases lagged behind, probably due to the inherent low reactivity in dienolate formation with mild bases. This task was cleverly faced by Xu et al., who succeeded in the HOMO-raising noncovalent activation of α-aryl-α,β-unsaturated aldehydes 208 by using bifunctional Brønsted base catalyst C2 (Scheme 58).159
Scheme 58.

Such a catalyst would induce base-promoted enolization of the enal, while providing hydrogen-bonding activation of the reacting partners. The corresponding [4 + 2] cyclization products were obtained in high yields and with good-to-excellent enantioselectivities. During optimization studies, it was found that using catalyst C3,160all-cis-disposed products 210 could be accessed predominantly, accompanied by variable amounts of the corresponding C5-epimers. When an alternative squaramide-tertiary amine catalyst was used, instead, the C5-epimers of 210 were chiefly recovered, thus providing a general diastereodivergent route to the cyclohexene targets. Based on control experiments and previous reports, the authors proposed a working mode to rationalize the observed stereoinduction. Accordingly, after enal deprotonation, the ammonium ion portion of the catalyst would stabilize the nitro function of the acceptor as in 208′, while the thiourea moiety would be able to stabilize the developing dienolate oxygen.
The condensation/isomerization of optically active secondary amines with enolizable fully or partially conjugated enals generates reactive trienamine species, whose intrinsic vinylogous nucleophilicity may be propagated through the π-system to the α, γ, until the remote ε-positions (Scheme 59).
Scheme 59.

In the presence of electron-deficient alkenes, these species can readily participate in ε,β-regioselective [4 + 2] cycloaddition processes as activated diene components, providing direct access to highly complex chiral cyclohexene frameworks. The viability of trienamine activation was for the first time established in a collaborative project between the Chen and Jørgensen groups in 2011,161 using either olefinic oxindoles or alkylidene cyanoacetates as suitable dienophile acceptors (Table 1, eq 1). On that occasion, the authors were able to prove with NMR studies the existence of the in situ generated all-trans trienamine intermediate as the result of the reaction between the starting polyenal substrate and the prolinol silyl ether catalyst (eq 1). The complete ε,β-regioselectivity of the [4 + 2] cycloaddition (overriding the possibly competing γ,ipso-[4 + 2] closure) could be rationalized by computational studies considering both the favorable rotation barrier for the formation of the reactive s-trans,s-trans,s-cis-trienamine conformation required for the cycloaddition and the energy of the frontier molecular orbitals set up for the interaction with the olefin. High endo-selectivity was explained by invoking secondary orbital interactions between the interacting partners, and face-selectivity was nicely secured by the proven ability of the chiral amine catalyst to govern the enantioface discrimination by steric factors. The authors hypothesized a plausible pericyclic Diels–Alder mechanism based on the absence of any observable Michael-addition type intermediates and high stereochemical induction indicative of a highly concerted closure.
Table 1. Dienals in Action with Electron-Poor Alkenes in ε,β-Regioselective Remote Cycloaddition Reactions.
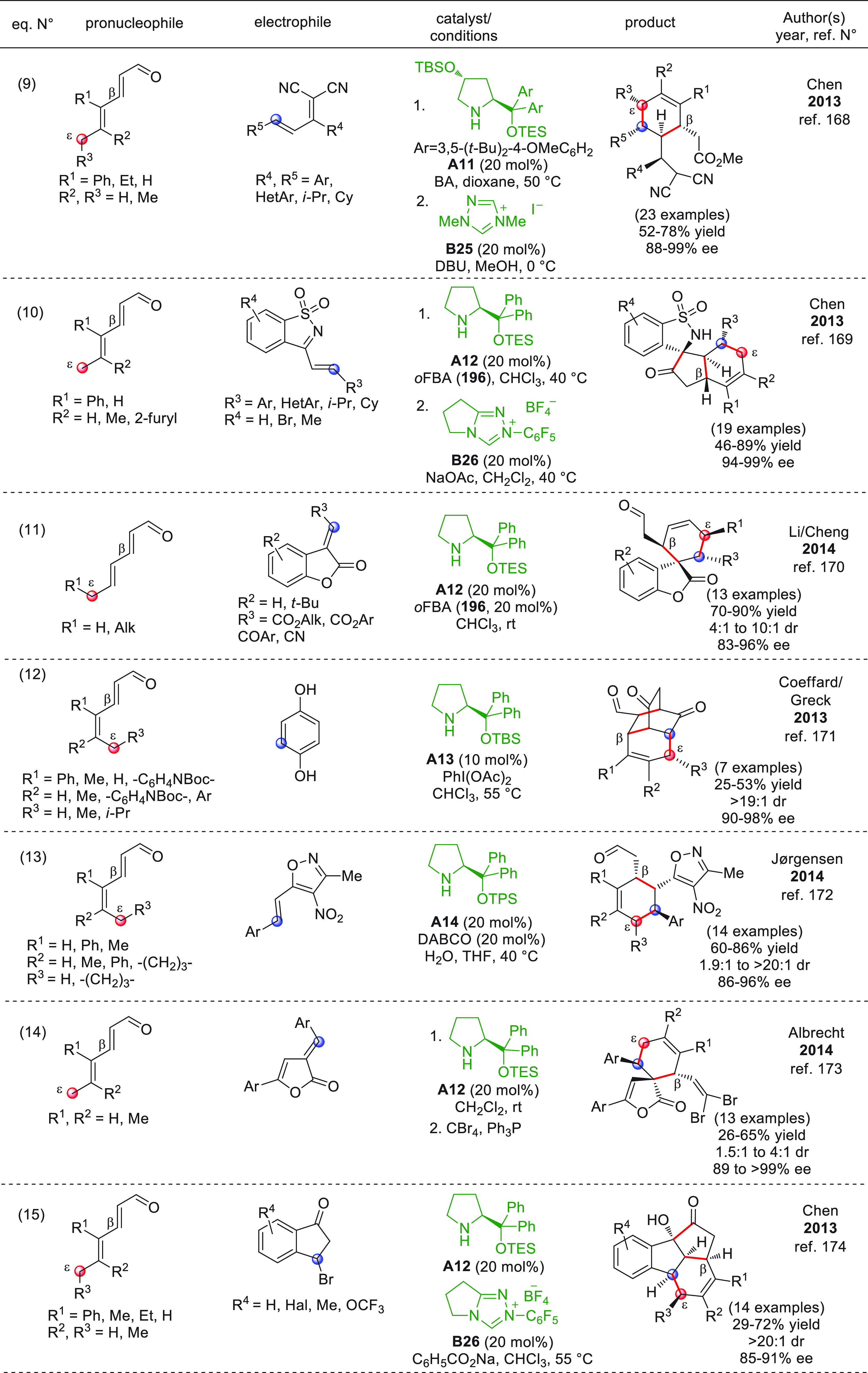
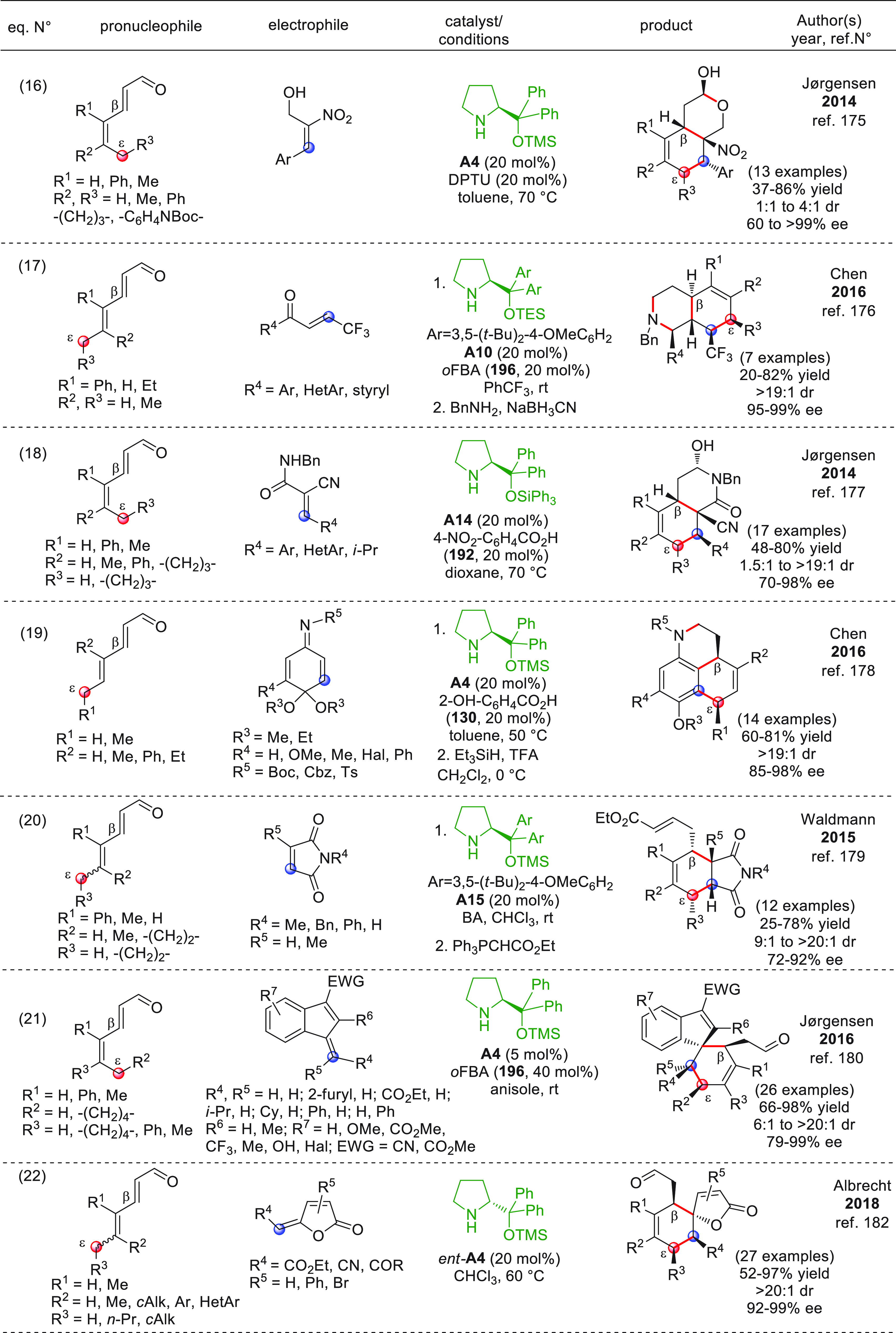
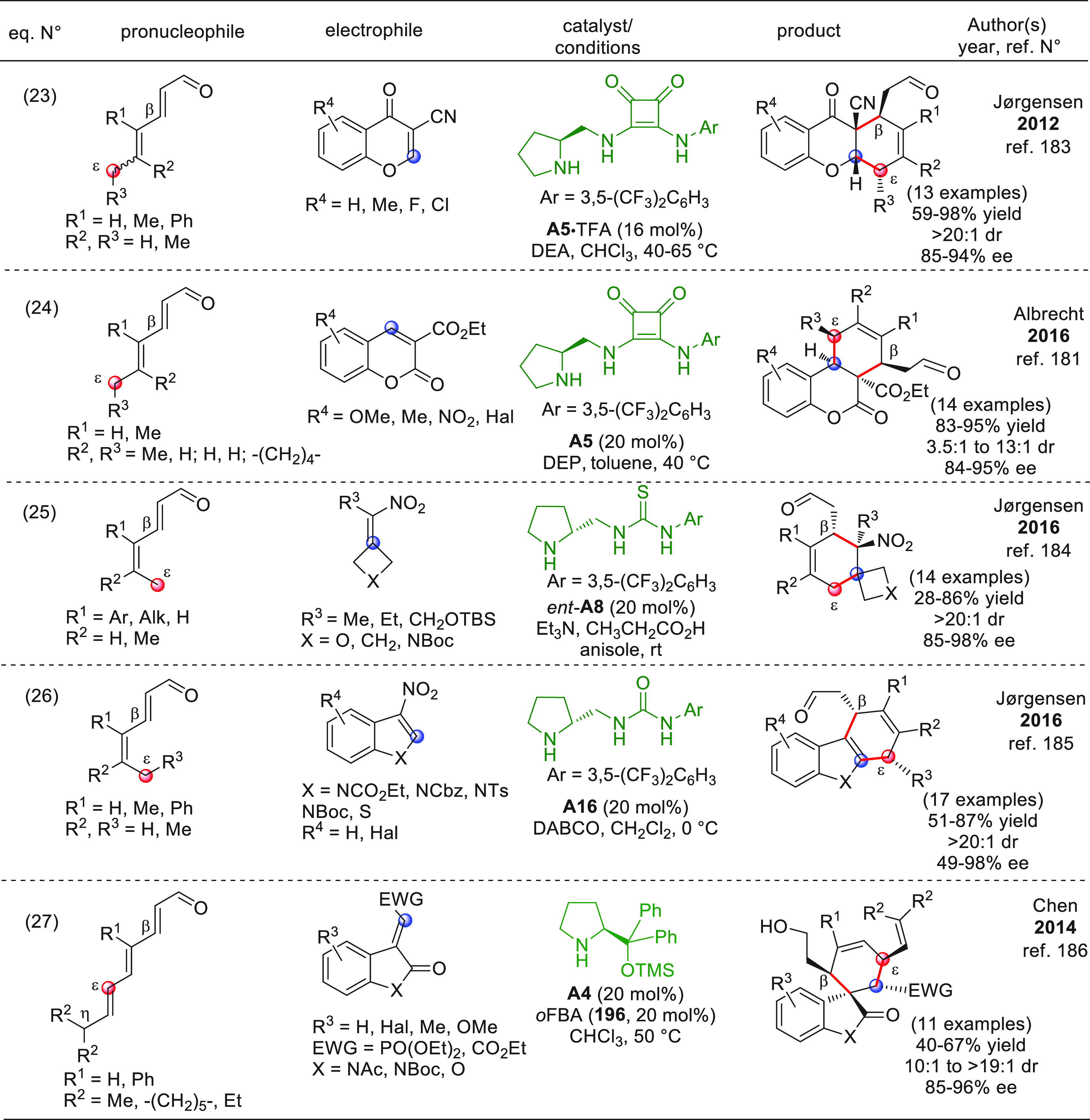
Henceforth, an impressive, flourishing number of reports appeared by these and other research groups, which focused on the ε,β-regioselective [4 + 2] cycloaddition between polyenals and a wide array of diverse dienophiles ranging from nitroalkenes, alkylidene heterocycles, to azadienes, enones, etc. For an immediate glance at this varied reaction panorama, a table is given (Table 1), collectively depicting these contributions.162−186
The richness of the cyclohexene products obtained appears evident, with hundreds of structurally and stereochemically diverse new C(sp3)-rich chemotypes becoming available with extraordinary simplicity and immediacy. In some cases (e.g., eq 4), ad hoc-placed electron-donating substituents at the γ,δ carbon sites of the starting enals ensured further HOMO activation while granting full ε,β-regioselectivity; in other instances (eq 5), nonconjugated polyunsaturated aldehydes were used as substrates to increase their reactivity during the condensation with the amine catalyst. The molecular diversity of the [4 + 2] products was increased even further by their transformation into secondary targets in either sequential or one-pot domino sequences, as in the case of amine/NHC cascade catalytic processes (eqs 9, 10, 15, 17, 19, and 20). In most examples, the immediate derivatization/transformation of the cycloadducts proved indispensable for product stabilization and/or isolation. Interesting examples within Table 1 also include in situ-activated electrophilic species, as in the case of methylene cyclic carbonyls from methiodide salts (eq 7), dearomatized quinones from phenols (eq 12), and indenones from 3-bromoindanones (eq 15). In one case (eq 20), the ε,β-[4 + 2] methodology was functional for accessing collections of products to be assayed in biology-oriented studies. In the majority of reports, the amine catalyst exerted its stereochemical induction via steric shielding control (eqs 1–22), while in some cases (eqs 23–26), a H-bond-directing approach was exploited using bifunctional amine catalysts.
From a mechanistic point of view, apart from the pioneering studies by Chen and Jørgensen,161 no experimental/computational studies were performed by the authors in order to substantiate the concerted vs stepwise nature of these [4 + 2] cyclizations; in one case (eq 14), a stepwise Michael/Michael reaction mechanism was openly postulated, while in another example (eq 26), calculations pointed to an asynchronous or stepwise mechanism. Computational work by Houk et al. indicated that, for a cyclic extended trienamine system (see later, cyclic systems), a stepwise mechanism might be operative.187
Lastly, a nice example of HOMO-activation strategy extended to highly conjugated enolizable 2,4,6-trienal substrates was reported, with the formation of multidentate tetraenamine intermediates under amine catalysis (Table 1, eq 27). In this case, the reaction with alkylidene oxindole acceptors gave the trienamine-type ε,β-locked [4 + 2] cycloaddition products regioselectively, as a testimony of the fact that the middle-positioned Cβ-Cε diene system of the reactive tetraenamine was involved in the coupling event. The alternative remote closure, involving the η,δ-diene, was not observed probably due to steric hindrance of ad hoc-placed substituents at the η-position.
3.3.1.2. Cyclic Pronucleophiles
As disclosed in the previous sections (Scheme 22 and related text), asymmetric functionalization of benzylic C–H bonds of (hetero)aryl aldehydes or extended polyenal variants is one major focal point of the chemistry currently powering the field of vinylogy, and different remote activation strategies have been devised, especially in direct, organocatalytic approaches.
Considering the special focus of this subsection, namely, the conjugate addition to electron-poor alkenes, we opted to group the research contributions into three categories, depending of the structural features of the pronucleophilic aldehydes.
In a first scenario, the aldehyde carbonyl group is distanced from the heteroaromatic ring by one carbon–carbon double bond, which is positioned ortho to the enolizable benzyl site, as outlined by examples in Table 2 (eqs 1–5).188,189
Table 2. Functionalization of Benzylic C–H Bonds of π-Extended Heteroaryl Aldehydes: ε,β-Regioselective Remote Cycloaddition Reactions with Electron-Poor Alkenes.
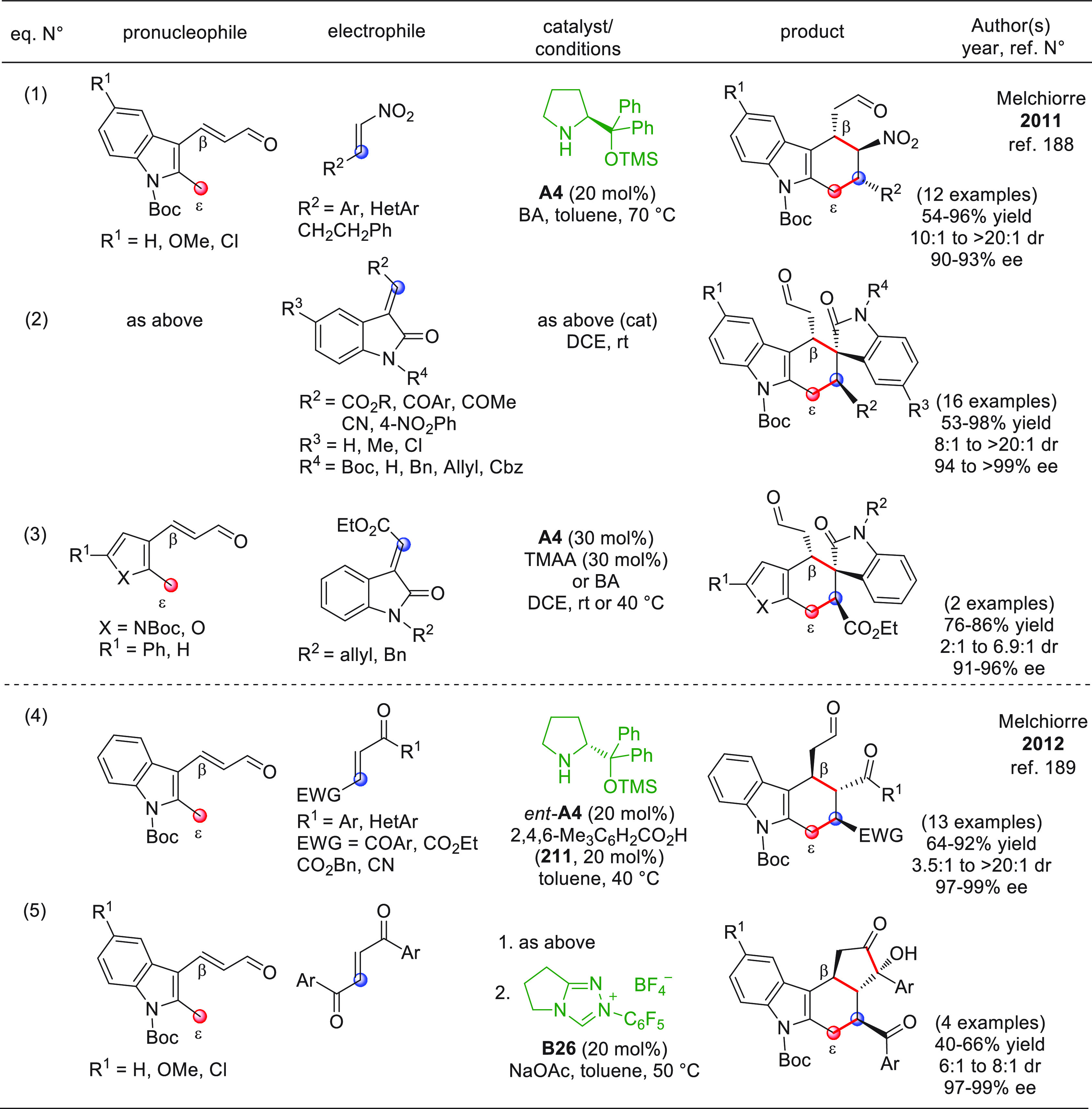
The group of Melchiorre first devised a clever strategy according to which amine-catalyzed activation of the starting pronucleophile leads to the formation of a HOMO-raised trienamine intermediate (see species XX in Scheme 22), a temporarily dearomatized oQDM species that can serve as a highly reactive s-cis-locked terminal diene in useful [4 + 2] cycloaddition reactions with suitable dienophiles. In the event, ε,β-closure of the starting polyenal occurs, producing highly functionalized ring-fused cyclohexene products bearing an exocyclic acetylaldehyde moiety, with the possibility of complete catalyst recovery and recycling. 2-Methyl-substituted indole, furan, and pyrrole-based enals were cleanly reacted with diverse dienophiles, including nitroalkenes, alkylidene oxindoles, and enones, affording the corresponding sp3-rich polycycles in generally good yields and stereoselectivities. One-pot, multicatalytic approaches could also be exploited, as exemplified by the one-pot Diels–Alder/benzoin cascade reaction reported in eq 5 (Table 2). In all instances, the regioselectivity of the [4 + 2] cyclizations was strictly dictated by the favorable frontier orbital interaction between the reacting diene/dienophiles partners, with full vinylogous transmission of the pronucleophilic character along the trienamine π-system to the remotely positioned benzylic Cε-site. The [4 + 2] cyclization was considered to be a true pericyclic Diels–Alder reaction, though no further supporting evidence was given at the time.
A second class of pronucleophilic species is represented in Table 3, where the aldehyde function is directly attached to the (hetero)aromatic ring and the enolizable benzylic site is positioned away (a sort of “para”-substitution) from the formyl function. The covalent activation by a chiral secondary amine catalyst would thus produce trienamine (structure XXI, related to eqs 1–2, Table 3) or tetraenamine (XXII, eq 3, Table 3) intermediates, which are temporarily dearomatized and chiralized species.
Table 3. Regioselective Remote Functionalization of Benzylic or More Remote C–H Bonds of “p-Substituted” Heteroaryl Aldehydes with Electron-Poor Alkenes.
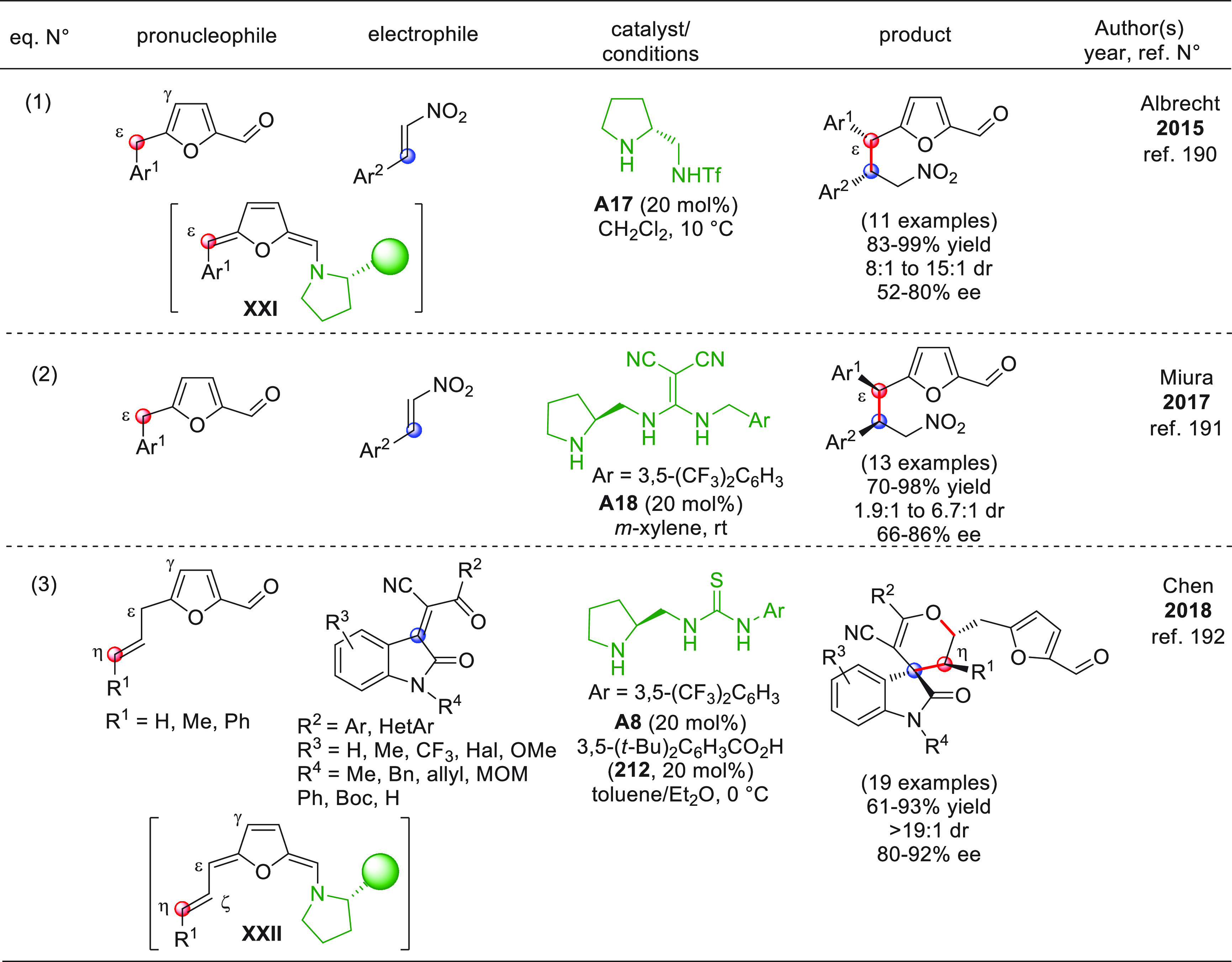
In the first two examples, independently authored by Albrecht et al. and Miura et al. in different years (Table 3, eqs 1–2),190,191 a ε-regioselective bisvinylogous Michael addition occurs on nitroalkene acceptors, since the conformationally locked trienamine intermediate XXI prevented any pericyclic cycloadditions involving the β,ε-sites of the terminal diene.
In a third, recent example, instead (Table 3, eq 3), authored by Chen et al.,192 the remotely positioned ζ,η-C=C double bond of the tetraenamine intermediate XXII is free to act as HOMO-raised 2π-component in IED-HDA [4 + 2] cycloadditions with alkylidene oxindole oxadienes acting as 4π-partners, to furnish the corresponding spirocyclic oxindole products incorporating dihydropyran-furfural moieties. In the three works, the remote asymmetric induction, exterted by the dual catalysts, could consistently count on additional activation of the electrophile via advantageous H-bonding interactions.
A third structural scenario is given by ortho-alkyl-substituted (hetero)aryl aldehydes, as represented in Table 4.
Table 4. Regioselective γ- or γ,ipso-Remote Functionalization of Benzylic C–H Bonds of ortho-Alkyl-Substituted (Hetero)aryl Aldehydes with Electron-Poor Alkenes.
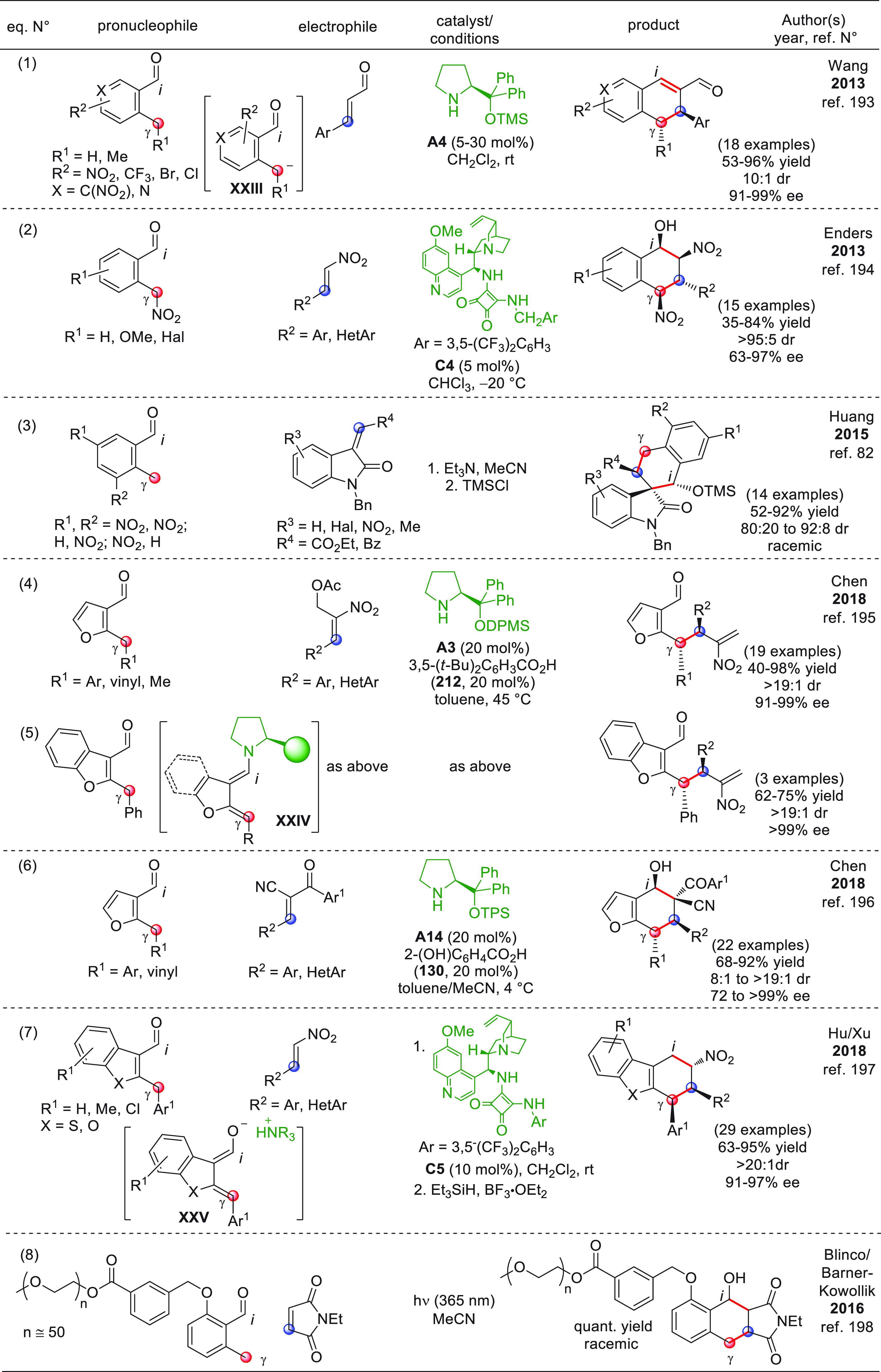
In the independent works by Wang,193 Enders,194 and Huang82 (Table 4, eqs 1–3), ad hoc placed electron-withdrawing groups (e.g., nitro) within the benzene ring provided enough C–H acidity at the benzylic position for this to be easily deprotonated by mild Brønsted bases. The resulting carbanionic active intermediates of type XXIII (resonating as oQDM/nitronate species, not shown) undergo [4 + 2] cycloadditive reactions with activated alkenes, providing the corresponding six-membered carbocycles. An eliminative stepwise vinylogous Michael/aldol cascade reaction was supposed to intervene in the first case (eq 1), with the covalent activation of the electrophile component via iminium ion formation. In the second instance (eq 2), dual catalysis by the tertiary amine/squaramide catalyst ensured the concomitant activation of the coupling substrates via stepwise nitroalkene-Michael/Henry cascade. In the third work (eq 3), an achiral tertiary amine afforded the appropriate deprotonation–cyclization reaction to access the silyl-stabilized racemic cycloaldol products.
In a recent work, Chen et al. developed a rare example of covalent amine-activation of enolizable o-alkyl furfural (or benzofuran analogues) (Table 4, eqs 4–5).195 The supposed catalyst-tethered dearomatized oQDM intermediate XXIV was engaged in Michael-type addition to nitroallylic acetates to furnish, after acetic acid elimination, the benzylic alkylation products.
This research group exploited the same catalyst-bound oQDM activation strategy in a subsequent work (eq 6),196 using α-cyanochalchones as electrophilic partners. The corresponding [4 + 2] cyclohexanol products were conveniently obtained, as the result of a postulated stepwise vinylogous Michael/intramolecular aldol cascade. The stepwise nature of this addition made hydrolysis of the iminium ion intermediate possible after the first step and thus allowed for catalyst recycling.
An interesting and unprecedented strategy was devised by Hu, Xu, et al. (Table 4, eq 7),197 who exploited the dual tertiary amine/squaramide catalyst C5 for the noncovalent activation of heteroaryl aldehydes. The in situ-generated oQDM-dienolate species of type XXV were intercepted by nitroalkene acceptors to afford the cyclic products via stepwise vinylogous Michael/nitroaldol cascade followed by reductive OH elimination.
Finally, a similar γ,i-selective [4 + 2] cycloaddition was exploited by Blinco, Barner-Kowollik, and colleagues to demonstrate how photochemically induced γ-enolization of funtionalized o-methylbenzaldehydes could undergo fast and chemoselective cycloaddition to maleimide, even in the presence of an amine competitor (eq 8)198 (for the light-induced formation of oQDM dienolates from carbonyl compounds, see the next section on ketone substrates). In the event, irradiation of the starting benzaldehyde at λmax = 365 nm led to the quantitative conversion of the starting material to the Diels–Alder product after 7 min, whereas when the irradiation was switched off, benzaldehyde reacted exclusively, under otherwise identical experimental conditions, with the amine coreagent, giving the corresponding imine (not shown) in 23% yield. The orthogonality of the reaction path was then cleverly exploited for the selective synthesis of block copolymers.
In 2012 and 2013, the group led by Jørgensen disclosed a new activation concept for anthracene derivatives by using covalent aminocatalysis.199,200 The basic idea was as clever as it was simple: treating α-enolizable 9-acetylaldehyde-substituted anthracenes 213 (Scheme 60) with suitable secondary amine catalyst would provide the corresponding enamine (e.g., 213′) with direct extended conjugation to the π-system of the central anthracene ring. The aromaticity of this ring was calculated (by nucleus-independent chemical shift, NICS-method) to be less than that of the same ring in anthracene or the parent aldehyde-anthracene 213; on the other hand, the HOMO of this special “trienamine” system was higher in energy compared to 213. These calculations predicted that “trienamines” of type 213′ could be favorably engaged in ε,β-selective [4 + 2] Diels–Alder cycloaddition reactions with suitable electron-poor alkenes and emphasized the role of the catalyst in accelerating aromaticity-breaking toward the targets.
Scheme 60.

In the first contribution (Scheme 60, eq 1),199 anthracene aldehydes 213 were reacted with aryl- or alkyl-substituted nitroalkenes 214, using bifunctional secondary amine/squaramide catalyst A5, affording, after carbonyl reduction, cycloadducts 215 with efficiency and high enenatioselectivities. Here, the excellent enantiofacial discrimination was dictated by the strict H-bonding control exerted by the bifunctional catalyst A5, as depicted in 213′. In a subsequent report (Scheme 60, eq 2),200 the same anthracene aldehydes 213 were treated with maleimides 216 (and even maleic anhydride or substituted cyclopentenes, not shown) under the guidance of C2-symmetric amine catalyst A19. In this instance, the good stereoselectivities observed in the formation of cycloadducts 217 could be explained by both experimental and computational studies. It was observed that analogous reactions, using nonsymmetric aminocatalysts, produced lower selectivities, pointing to the notion that symmetry-breaking could be cleverly effected by C2-symmetric catalysts. DFT studies explained that the discrimination between the two possible enantiofaces of the dienophile was not the result of a steric shielding effect, but was instead due to the extended (or prevented) π-conjugation along the trienamine chain in the preferred vs disfavored transition state structures. In any case, calculations suggested that the reaction pathway was concerted and highly exothermic.
Among cyclic pronucleophilic polyenals, the class of cycloalkenylidene acetaldehydes (e.g., structure XXVI, Scheme 61) has enjoyed notable success, especially thanks to the studies performed by the Jørgensen group.
Scheme 61.

The covalent HOMO-raising activation of these substrates via amine organocatalysis may lead to the corresponding extended or cross-conjugated trienamines (or both) which can couple, at least in principle, to suitable electron-poor olefins during diverse and competitive reaction itineraries, including “simple” vinylogous (at γ′) or hypervinylogous (at ε) Michael-type additions, as well as ε,β-[4 + 2], γ′,δ-[4 + 2], γ′,i-[4 + 2], and γ,β-[2 + 2] cycloadditions (Scheme 61) or even γ′,β-[4 + 2] HAD cycloadditions (not shown). Carrying out regio- and stereocontrolled reactions in such a multifaceted scenario can thus result in a challenging venture.
In 2012, Jørgensen and co-workers faced this task by reacting 2,4-dienals 218 with 3-olefinic oxindoles 219 under prolinol silyl ether catalysis (catalyst A4, 20 mol %) (Scheme 62, eq 1).201 The spiro-polycyclic products 220 were exclusively obtained (isolated as unsaturated esters after Horner–Wadsworth–Emmons olefination), as the result of a γ′,δ-regioselective [4 + 2] cycloaddition to the dienophiles 219 via cross-conjugated trienamine (see also Scheme 61). The generality of the reaction was proven, and wide structural variations in both the reacting partners were well tolerated, producing cycloadducts 220 in good yields and excellent stereoselectivities. The same cross-conjugated trienamine concept was successfully applied to aryl-substituted olefinic azlactone dienophiles, affording [4 + 2] products, again with exclusive γ′,δ-regioselectivity (not shown). When, instead, 1,1-bis(phenylsulfonyl)ethylenes 221 were employed as electrophiles (eq 2), γ′-substituted products 222 were isolated, as the result of γ′-regioselective vinylogous Michael addition reaction.
Scheme 62.

To clarify the reason why cycloadditions and Michael additions occurred via the cross-conjugated trienamine, a combination of both computational and NMR studies were performed. Calculations were carried out on a simplified model system related to the two possible trienamines (extended vs cross-conjugated) and showed that deprotonation at the most remote ε-position is favored, since it provides a fully conjugated, thermodynamically stable π-system. Despite furnishing the linear trienamine in higher concentrations, the authors reasoned that this reaction system would progress toward the cross trienamine-triggered γ′,δ-pathway since it would be able to give the thermodynamically more stable products 220 (thermodynamically controlled reaction) via a highly asynchronous, concerted Diels–Alder mechanism.
Having suspects about the actual concerted nature and the thermodynamic control of this cycloaddition, Houk and co-workers reanalyzed and rationalized the behavior of this reaction by employing in-depth DFT calculations.187 Interestingly, they concluded that a stepwise mechanism for this formal [4 + 2] cycloaddition is probably operative, entailing a first, thermodynamically and kinetically favored vinylogous Michael addition of the γ′-site donor to the olefin acceptor to produce a zwitterionic intermediate of type 218′ (Scheme 62), after which, such an intermediate would close, under thermodynamic control, to give the experimentally observed γ′,δ-cycloadducts, overriding the alternative, yet possible, competitive closures (e.g., γ′,i-[4 + 2], γ,β-[2 + 2] cycloadditions, Scheme 61).
Carbocyclic and heterocyclic aromatic cycloalkylidene aldehydes 223 were the key substrates utilized by the Jørgensen group during a synthesis campaign involving amine-catalyzed coupling reactions with various electron-deficient olefin types (Scheme 63).202 In a first group of reactions (Scheme 63, eq 1), enals 223 were treated with phosphonate olefins 224 using catalyst A4 and benzoic acid as an indispensable acid additive. After the one-pot addition of cesium carbonate as the base, products 225 were isolated in good yields and stereoselectivities. These products were the result of a one-pot, two-step cascade reaction entailing a first γ-regioselective vinylogous Michael addition of 223 to 224 to furnish 223′, followed by intramolecular base-promoted HWE olefination. Furthermore, the reaction employing cyclohexene-derived enal behaved impeccably (not shown) while, as a limitation, β-substituted vinyl phosphonates did not perform well under these experimental conditions. Alternative nitroalkene acceptors were also used (eq 2) in reactions with carbocyclic donors 223 (X = CH2) by using bifunctional organocatalyst ent-A5 and DIPEA as an additive. Again, the cyclohexadiene products 225 were obtained, derived by (at least formal) eliminative γ,i-[4 + 2] cycloaddition to the nitroalkenes, where the DIPEA base was thought to play a beneficial role during the E1CB-elimination of the amine catalyst. Of note, while in the first group of reactions (eq 1), a steric shielding effect by the catalyst was invoked to account for the observed stereoselectivity, hydrogen-bonding assistance by the dual-action catalyst ent-A5 was hypothesized for the latter group of reactions (eq 2).
Scheme 63.

Cyclic dienal substrates similar to 223 were also cleverly employed in γ,i-regioselective [4 + 2] cycloaddition reactions with either cyclopentadiones or quinone-based dienophiles, to access important 14β-steroids and D-homosteroids, respectively (not shown).203 In all cases, secondary amine catalyst A4 was employed, which ensured excellent stereocontrol via the usual dienamine intermediate.
The eliminative γ,i-regioselective [4 + 2]-cycloaddition of racemic 2-cyclohexenylidene acetaldehydes with benzoquinones under asymmetric organocatalysis was also exploited by the same research group to achieve dynamic resolution of the starting aldehydes (not shown).204
Cycloheptatrienyl acetaldehyde 226 was the carbonyl substrate chosen by Jørgensen and collaborators to demonstrate the viability of the first asymmetric organocatalyzed [4 + 2] cycloaddition via tetraenamine intermediate (Scheme 64).205 Treatment of deconjugated aldehyde 226 with amine catalyst A2 led to the formation of fully conjugated tetraenamine 226′, as demonstrated by NMR analysis. This HOMO-activated species shows multidentate nucleophilicity at the α, γ, ε, and η sites; nevertheless, when it was reacted with alkylidene oxindoles (or benzofuran analogues) of type 227, a highly regio- and stereoselective formal γ,i-[4 + 2] cycloaddition occurred, giving the new class of spiroxindole products 228 incorporating fused 7/6-membered rings and four contiguous stereocenters. Though the annulation was limited to trienyl aldehyde 226, the generality of the coupling reaction was wide with respect to the acceptor component 227, since different R1, R2, X substituents could confer good to optimal reactivity, with production of the corresponding compounds 228. NMR studies together with calculations and further experiments leading to the isolation of useful intermediates all sustained a stepwise mechanism entailing a first γ-vinylogous Michael addition between the reactive s-cis tetraenamine conformer 226″ and the acceptor 227 to provide the zwitterionic intermediate 226‴. Subsequently, hydrolytic release of the catalyst, isomerization, and cyclization consigned the targeted compounds. Notably, intramolecular closure involving directly 226‴ could indeed occurr, which would entrap the catalyst and prevent catalytic recycle. However, this unproductive cyclization step was calculated to be reversible and therefore uninfluential to the overall efficiency of the process.
Scheme 64.

Trienamine organocatalysis was the central theme of an interesting work by Anderson et al., who reported how regioisomeric azacycle-tethered exocyclic dienals, deriving from palladium-catalyzed cycloisomerization of enynamides, could take part in ε,β-regioselective [4 + 2] cycloaddition processes with electron-poor alkenes, to give hexahydroindole products (not shown).206
To conclude this section, we will comment about a couple of recent, brilliant works on higher-order cycloadditions. Higher-order cycloadditions are cycloaddition reactions involving more than 6π electrons. Though they have been conceptualized and recognized for their high synthetic potential since the times of Woodward and Hoffmann more than 50 years ago,207 their full exploitation in asymmetric synthesis has been rising satisfactorily only over the past recent years. One of the major concerns about these reactions is the difficulty of judiciously channeling the pericyclic reaction toward a selected pathway (periselectivity) among the many competing itineraries; asymmetric aminocatalysis was envisioned to be an optimal tool to promote both peri- and enantioselectivity in this intriguing class of cycloadditions. The research groups of Jørgensen,208 Chen,209 and Hayashi210 contributed with excellent works to this field, and we report here only those studies that are conceptually connected to the focus of this review (for examples involving ketone pronucleophiles, see the next section).
In a recent communication, Jørgensen et al. documented the first aminocatalytic [8 + 2] cycloaddition between indene-2-carbaldehydes 229 and nitroalkenes 230 (Scheme 65, eq 1).211C2-Symmetric 2,5-diphenylpyrrolidine A19 (20 mol %) was identified as the best amine catalyst to promote the intended β′,β-selective [8 + 2] cycloaddition via the catalytic formation of amino isobenzofulvene intermediate 229′ acting as the 8π component. Investigation of the scope of the nitroolefins demonstrated that both aromatic and heteroaromatic substrates 230 could productively afford cycloadducts 232 in good yields and selectivities, with the exception of ortho-substituted aromatic moieties giving poor results. Furthermore, an alkyl-substituted derivative (R2 = n-Bu) gave poor results in terms of conversion but maintained high stereocontrol. Among the other olefins assayed, β-substituted unsaturated nitriles gave successful results (not shown), while electron-rich olefins did not work. Quantum mechanical calculations suggested a kinetically controlled, stepwise mechanism involving initial reversible formation of the zwitterionic species 229″ derived by the attack of the β′ site of semiaromatic polyenamine 229′ onto the nitroolefin. Subsequently, irreversible closure of the nitronate onto the Cβ-site provided an enamine intermediate and hence the observed product after catalyst hydrolysis (and NaBH4 carbonyl reduction). Of note, during calculations, an alternative [10 + 4] cycloaddition pathway was identified as a possible route (e.g., deriving by attack of the nitronate oxygen on the iminium ion within 229″), even if no [10 + 4] adducts were observed with the employed olefins under these experimental conditions.
Scheme 65.

Intrigued by this unexplored, yet possible alternative, the same group soon after developed a strategy where such rare [10 + 4] couplings could be successfully realized in an aminocatalytic asymmetric context.212
As outlined in Scheme 65 (eq 2), various 3-aryl-substituted indenes 233 were used as 10π components (3-unsubstituted congeners of type 229 gave, instead, complex product mixtures), which were reacted with electron-poor cyclopentenone (or furanone) dienes 234 as 4π partners, giving rise to tetracyclic products 236 with generally good yields, excellent diastereocontrol, and appreciable enantioselectivities. Amine catalyst ent-A13 was found to be the best aminocatalyst (20 mol %), together with the indispensable presence of p-methoxybenzoic acid additive. Experimental and computational evidence suggested that the observed stereoselectivity could derive from the kinetically controlled formation of an amino isobenzofulvene intermediate similar to 229′, which could undergo stepwise eliminative closure to give the overall β′,ipso-selective [10 + 4] cycloaddition.
3.4. Other Reactions
The exploitation of the vinylogy concept goes well beyond the “conventional” addition reaction domain covered in the previous chapters, but it usefully includes examples of other reaction types such as alkylation (including cylopropanation, allylation), amination, nitrosation, thia-Diels–Alder cycloaddition, aziridination and protonation reactions. Indeed, we must remember that the first example of remote (γ) functionalization of enals via dienamine catalysis was an amination reaction.213 Most of the studies appearing in the 2010–2018 period involve the direct remote activation of vinylogous acyclic pronucleophiles, but a few examples with cyclic substrates have also been documented. To the best of our knowledge, no examples of indirect procedures were found.
3.4.1. Direct Procedures
3.4.1.1. Acyclic Pronucleophiles
A first group of contributions deal with the direct asymmetric γ-alkylation reaction of linear α,β-unsaturated aldehydes via dienamine, NHC, and/or metal catalysis, as outlined in Table 5.
Table 5. Remote Functionalization of Enals in Alkylation Reactions.
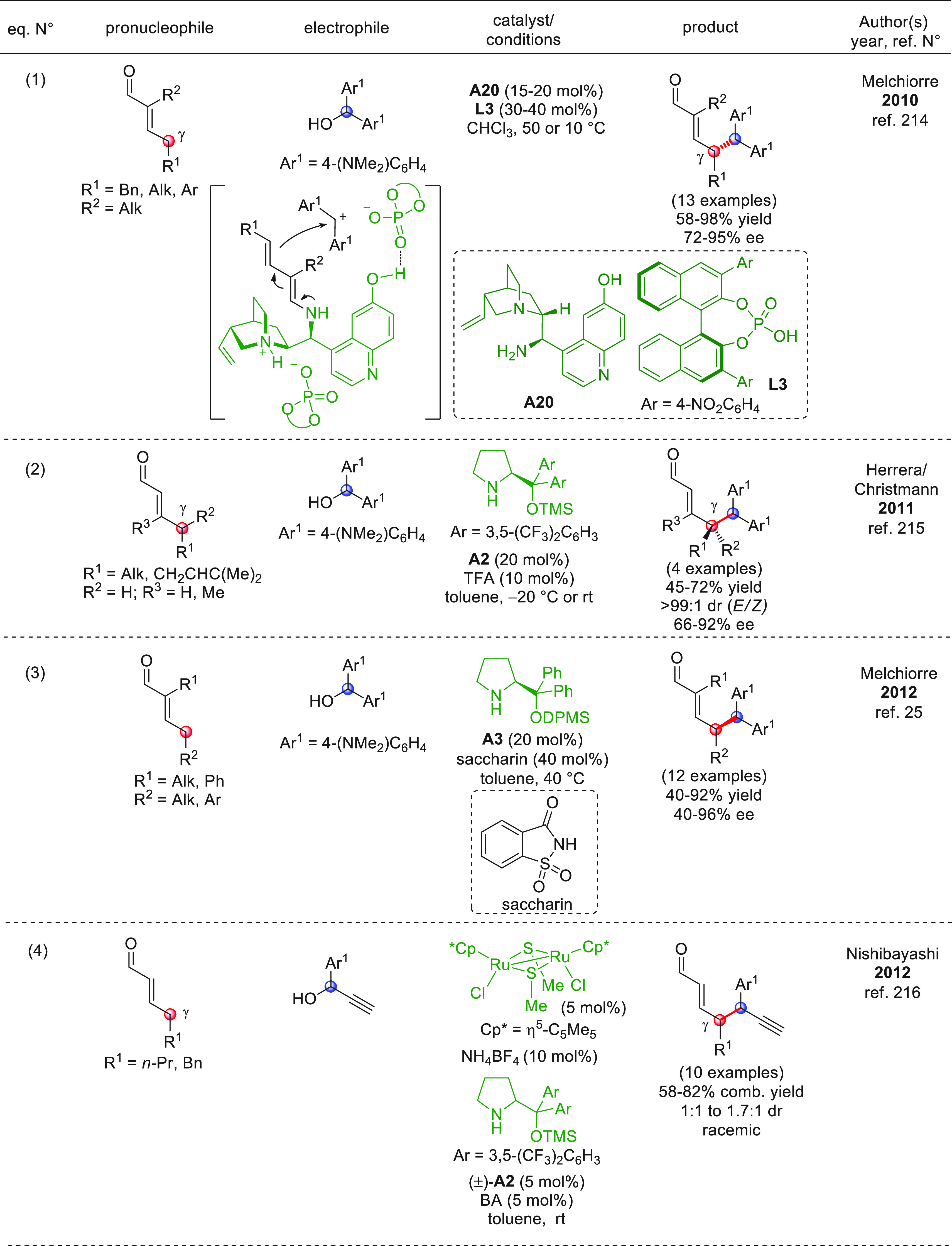
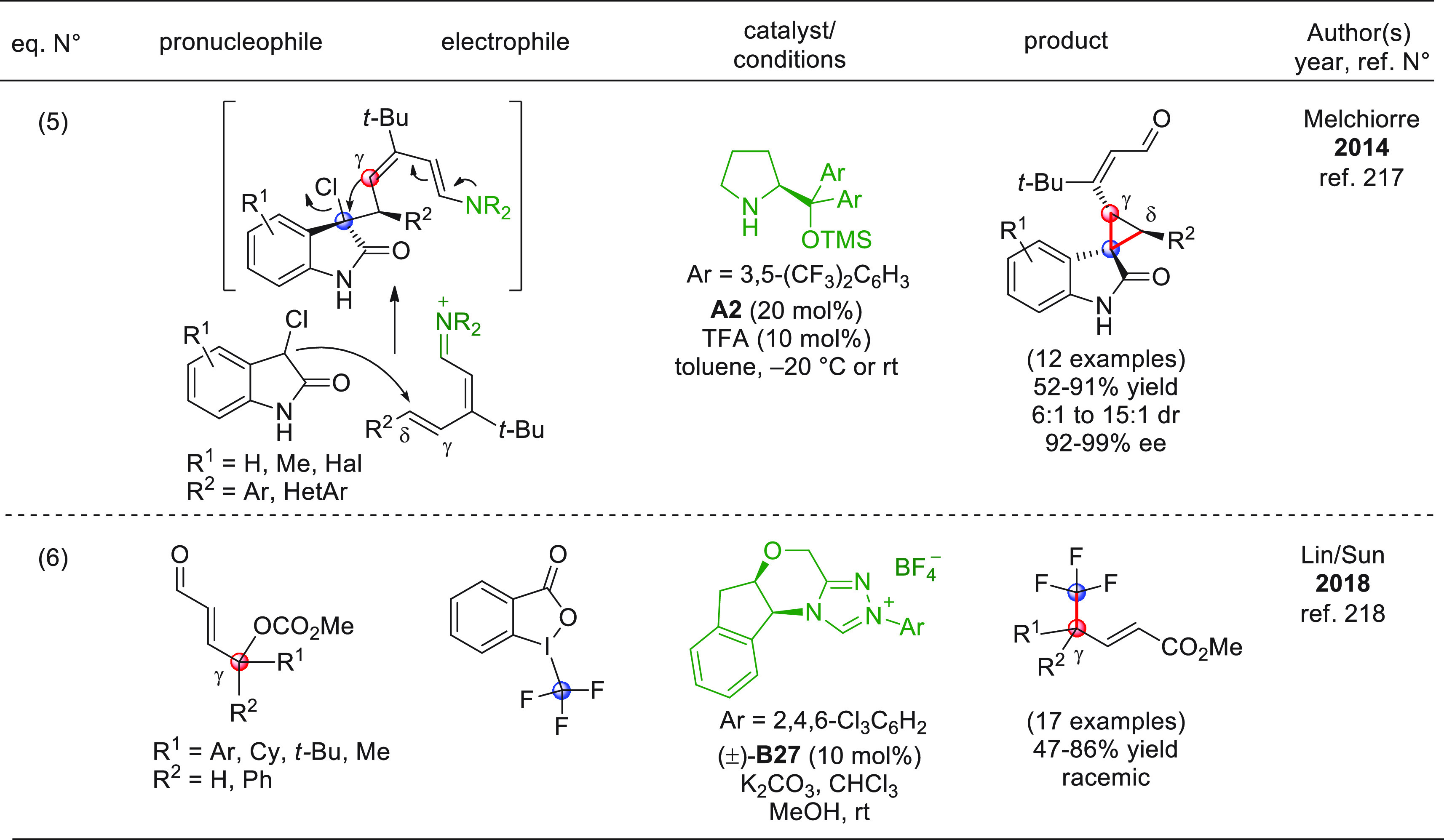
Melchiorre et al. developed an asymmetric γ-alkylation process of α-substituted enals, by using bis(4-dimethylaminophenyl)methanol as the electrophilic source and exploiting the cooperative catalysis exerted by primary amine catalyst A20 with phosphoric acid L3 (Table 5, eq 1).214 Interception of the in situ generated benzhydryl carbocation under acidic conditions with dienamine intermediate from the pronucleophile gave the γ-alkylation products with generally good results (SN1 pathway). A tightly organized transition state was proposed (depicted in eq 1) where the chiral phosphate acts synergistically with the dienamine chiral inducer as a counterion for both the carbocationic electrophile and the protonated quinuclidine of the amine catalyst.
Soon after, Herrera, Christmann, et al.215 studied the same alkylation reaction starting from α-unsubstituted enals bearing diverse substitution patterns: linear unbranched and β-substituted enals favored the vinylogous alkylation path (Table 5, eq 2); γ,γ-disubstituted enals (R1, R2 ≠ H) privileged the nonvinylogous α-attack (not shown), and α-substituted enals did not work at all under these experimental conditions. Notably, the E vs Z diastereomeric ratio within the products was under thermodynamic control (exclusive E isomers), and the stereodefining step of the process was the final protonation of the dienamine-linked products before catalyst release by hydrolysis.
Similar γ-alkylation reactions of α-branched linear enals were carried out,25 using secondary amine catalyst A3 and achiral saccharin as a Brønsted acid additive (Table 5, eq 3). Complete γ-regioselectivity and high enantioselectivities were attained in the corresponding products.
γ-Regioselective propargylation of enolizable α,β-unsaturated aldehydes was realized by Nishibayashi et al. using the cooperative catalyst system consisting of a thiolate-bridged diruthenium complex and racemic secondary amine (±)-A2 (Table 5, eq 4).216 In the event, the corresponding propargyl allylated products were regioselectively obtained in moderate-to-high yields as mixtures of racemic diastereoisomers. It was proposed that the ruthenium-based catalyst activated the propargyl carbinol via the formation of an allenylidene complex (not shown), while the secondary amine would simultaneously activate the starting enal via dienamine formation. Though carried out in a racemic context, this is a quite rare and interesting example of the exploitation of cooperative transition metal- and organocatalysis in the vinylogous domain.
The asymmetric synthesis of cyclopropane spiroxindole was carried out by Melchiorre et al., featuring a highly regio- and stereoselective vinylogous organocatalytic cascade (Table 5, eq 5).217 This time, the dienal substrates initially act as iminium ion-activated vinylogous acceptors, which are attacked at their remotest δ-position by chlorooxindole, to furnish dienamine intermediates (between square brackets, eq 5). Intramolecular vinylogous SN2 of these dienamines then affords the targeted cyclopropanes with good efficiency and stereocontrol. Key to the success of the reaction was the positioning of the biasing bulky t-Bu group at the β-site of the starting dienals, allowing exclusive δ-regioselective 1,6-addition in the first step of the cascade.
The first example of NHC-catalyzed vinylogous trifluoromethylation reaction of α,β-unsaturated aldehydes was recently documented by Lin, Sun, and co-workers, as a general route for the remote installation of C(sp3)-CF3 bonds (Table 5, eq 6).218 Enals bearing a γ-leaving group were elected as NHC-activatable substrates (for the general γ-activation concept of enals under NHC catalysis, see Schemes 17 and 19 and related text), while the benziiodoxole Togni reagent was chosen as the electrophilic trifluoromethyl source. Using racemic indane-based triazolium salt precatalyst (±)-B27, potassium carbonate, and methanol, the efficient formation of racemic trifluoromethylated methyl ester products was obtained, with exclusive γ-regioselectivity. Control experiments supported the notion that the NHC-bound vinylogous enolate intermediate served as the actual nucleophilic species. The strict vinylogous regioselectivity could be explained by DFT calculations, indicating that the γ-pathway is both kinetically and thermodynamically favored compared to the α-pathway. In fact, beneficial electrostatic interactions were thought to be present in the transition state leading to the γ-products, involving the hypervalent iodine moiety of the trifluoromethylating reagent and the indane NHC motif; in the α-pathway, these favorable interactions would be disrupted.
In the previous sections, we discussed the prolific chemistry emerging from the (often formal) [4 + 2] oxa- and aza-Diels–Alder reactions between HOMO-raised dienamine- or trienamine-derived dienes and carbon-centered C=O or C=N dienophiles. Much rarer are examples of catalytic asymmetric thia-Diels–Alder cycloadditions, especially in the vinylogous realm. In 2014, Jørgensen et al. faced this challenge, using remotely enolizable dienals as the diene source via trienamine activation and various thioesters as the dienophile components (Table 6, eq 1).219 The corresponding ε,β-locked dihydrothiopyran cycloadducts were obtained with wide tolerance of substituents in both reaction partners. Of note, the regioselectivity of the reaction emphasized that the remote ε-site of the trienamine intermediate from the dienal attacked the sulfur atom and not the carbon atom of the thiocarbonyl function. To rationalize the reaction outcome, DFT calculations were performed, which pointed to a stepwise mechanism involving zwitterionic intermediates; electronic factors, particularly those exerted by the ester group adjacent to the thiocarbonyl acceptor, were considered responsible for the observed regioselectivity, while endo/exo diastereoselectivity is most likely to be kinetically controlled.
Table 6. Remote Functionalization of (Poly)enals in Thia-Diels–Alder Cycloaddition, Amination, Nitrosation, Aziridination, and Protonation Reactions.
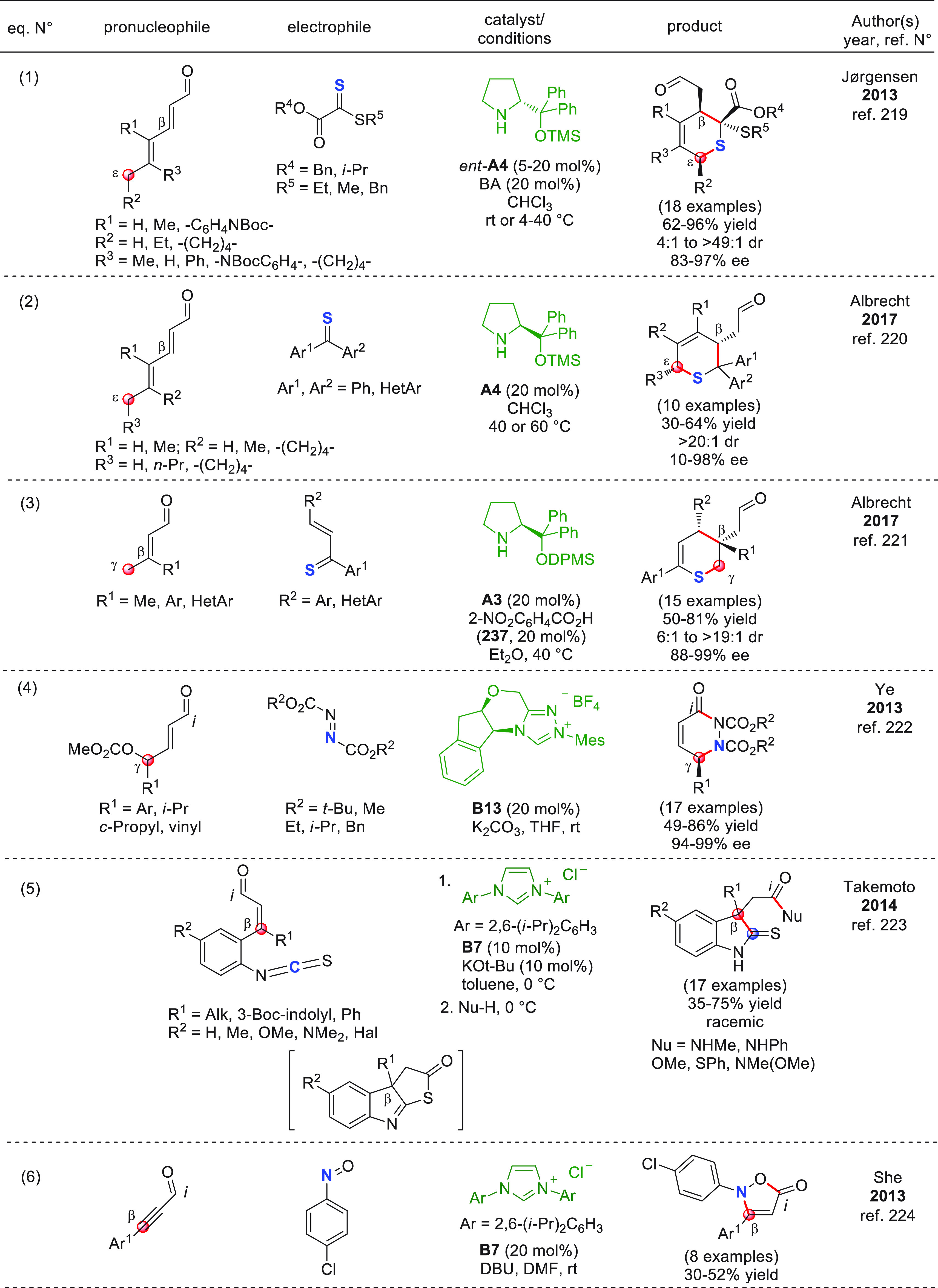
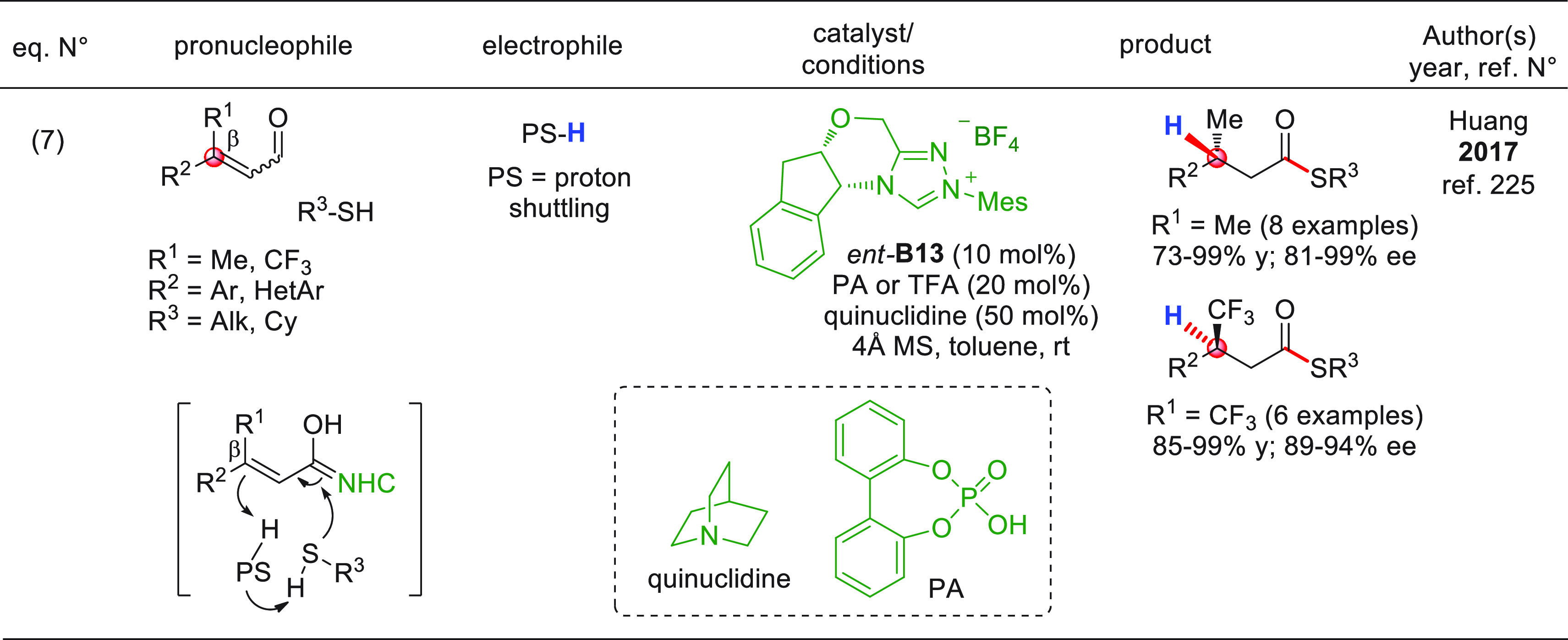
Remotely enolizable dienals served also as 4π-components in ε,β-regioselective thia-Diels–Alder reactions with thioketone heterodienophiles (Table 6, eq 2).220 The aminocatalytic trienamine-mediated cyclization provided 5,6-dihydro-2H-thiopyranes, probably via either concerted asynchronous [4 + 2] cycloaddition or nonorthodox diradical process, depending upon the substituent attached to the C=S bond (aryl vs heteroaryl).
An ortho-regioselective IED-HDA reaction was explored by Albrecht et al. featuring regio- and stereoselective coupling between γ-enolizable enals, acting as electron-rich dienophiles, and thiochalcones, acting as heterodienes (Table 6, eq 3).221 The aminocatalytic [4 + 2] cycloaddition resulted in the formation of enantioenriched 3,4-dihydro-2H-thiopyrans.
The NHC-catalyzed γ,ipso-regioselective [4 + 2] annulation between α,β-unsaturated aldehydes, bearing a γ-leaving group, and azodicarboxylate acceptors was reported by Ye et al., affording highly enantioenriched dihydropyridazinone products (Table 6, eq 4).222 A plausible mechanistic rationale was proposed, where the NHC-bound dienolate (derived from HOMO activation of the enal) attacks the azodicarboxylate, affording an acyl azolium adduct, which finally releases the catalyst and consigns the targeted compounds.
A tandem, NHC-catalyzed reaction was developed by Takemoto and co-workers, entailing the synthesis of racemic indoline thiones starting from β,β-disubstituted α,β-unsaturated aldehydes carrying an isothiocyanate moiety (Table 6, eq 5).223 Initially, covalent activation of the enal by the NHC catalyst gives a vinylogous Breslow intermediate, whose β-vinylogous carbon site intramolecularly attacks the isothiocyanate to furnish a thienoindolone structure (between brackets). Subsequent nucleophilic opening of this intermediate provides the targeted products.
NHC-catalyzed formal [3 + 2] annulation involving alkynyl aldehydes and nitrosobenzenes was reported by She and collaborators in 2013 (Table 6, eq 6).224 Reaction conditions were found to regioselectively steer the reaction path either along a vinylogous β,ipso-annulation to give isoxazol-5(2H)ones (eq 6) or along nonvinylogous ipso,β-annulation to give regioisomeric isoxazol-3(2H)ones (not shown) (for general β,ipso NHC-catalyzed annulations, see also Scheme 14).
The β-nucleophilic activation of α,β-unsaturated aldehydes via NHC-catalyzed homoenolate formation was also cleverly exploited by Huang and collaborators in a quite rare example of enantioselective vinylogous protonation of enals in the absence of any directing groups (Table 6, eq 7).225−227 The careful choice of a Brønsted base (a chiral bridgehead tertiary amine such as quinuclidine) together with a strong Brønsted acid cocatalyst (phosphoric acid PA or TFA) provided a smart proton-shuttle system (the quinuclidinium ion, PS-H in the table, eq 7) capable of ensuring β-protonation of the NHC-bound homoenolate in a highly enantioselective manner. Since no match/mismatch was observed when using chiral quinine or quinidine with chiral NHC, it was proposed that the chiral influence came mainly from the NHC moiety, while the effect of the quinuclidine is mostly steric. Thus, using various thiol nucleophiles, the corresponding saturated thioesters with a β-stereocenter were prepared, in very good yields and enantioselectivities.
3.4.1.2. Cyclic Pronucleophiles
During a broad, brilliant study focusing mainly on the asymmetric dienamine-catalyzed vinylogous Michael addition of cyclic enones to nitroalkenes (see infra, section on ketone pronucleophiles), a brief exploration was also dedicated to the asymmetric vinylogous amination of a cyclic enal (Scheme 66, eq 1).228
Scheme 66.

Thus, using primary amine catalyst A21, the reaction between 238 and tert-butylazodicarboxylate 239 proceeded smoothly, giving aminated product 240 with exclusive γ-regioselectivity and excellent enantioselectivity.
An iminium ion-dienamine catalytic cascade was fruitfully exploited by Jørgensen et al. in a rare example of asymmetric remote aziridination reaction (Scheme 66, eq 2).229 Cyclic 2,4-dienals having diverse aliphatic substituents in the γ-position (R1 had to be strictly different from H) were treated with 242 using secondary amine ent-A2 catalyst, furnishing the corresponding aziridines 243 with exclusive δ,γ-regioselectivity (>95:5 δ,γ: β,α), optimal E:Z geometric selectivity (>95:5 E:Z), and moderate-to-good enantioselectivities. The reaction mechanism features a δ-selective hypervinylogous 1,6-Michael addition of the nitrogen nucleophile 242 onto the iminium ion from 241, followed by SN2 closure of the formed γ-dienamine into the nitrogen atom favored by the OTs leaving group.
The Jørgensen group also developed asymmetric γ-allylation reactions of cycloalkylidene acetaldehydes by merging vinylogous aminocatalysis with transition metal catalysis (Scheme 67).230,231 The direct asymmetric allylation of enals of type 223 could pose, in principle, a series of challenges including regioselectivity issues of both the donor (α vs γ) and acceptor (branched, b vs linear, l) components, as well as stereoselectivity concerns, i.e. E vs Z olefin geometry, syn vs anti diastereoselectivity, and enantioselectivity within the products. The wise orchestration of both aminocatalytic activation of the pronucleophile 223 and electrophilic activation of the π-allyl system within either 244 or 246 allowed for the regio- and stereoselective entry to either branched allyl products 245 (eq 1) or linear allyl products 247 (eq 2).
Scheme 67.

Treatment of variously substituted enals 223 with allyl benzyl alcohols 244 using the combination of triphenylsilyl-protected prolinol A14 (providing dienamine activation of 223) and iridium-based catalyst having phosphoramidite ligand L4 produced the branched γ-allyl products 245 in good yields and with excellent levels of regioselectivity (>20:1 γ/α; >20:1 b/l) as well as stereoselectivity (>20:1 E/Z) (eq 1). By switching, instead, to palladium-based catalyst with achiral ligand L5 and using TMS-protected l-prolinol A4, enals 223 coupled to allylic acetates 246 to furnish linear vinylogous γ-allyl products 247 (eq 2), again with admirable levels of site- and stereoselectivity (>20:1 γ/α; >20:1 l/b; >20:1 E/Z). Of note, control experiments revealed that the branched vs linear regioselectivity exclusively depended upon the metal catalyst of choice and not on the nature of the allylic alcohol in use. By employing ent-A14 instead of A14, syn-configured products were formed (not shown), thus demonstrating the possibility to selectively synthesize all six isomers of vinylogous allylated products.
Conceptually similar synergistic combination of metal- and organocatalysis was exploited by Gong and collaborators in the hypervinylogous, ε-regioselective asymmetric allylation of furfural derivatives (Scheme 68).232 The focal idea was that the remote ε-site of the furfural substrates 248 would be activated via trienamine catalysis, and the palladium-ligand complex would concomitantly activate the allylic alcohols 249. Thus, bulky secondary amine ent-A14 together with palladium(II) complex using TADDOL-based phosphoramidite ligand L6 and a phosphoric acid additive turned out to be the best “cocktail” for promoting the ε-allylation reaction between 248 and 249 and for allowing the preparation of products 250 in high yields and very good stereocontrol. Nonbenzylic ε-alkyl substituted furfurals were also examined (not shown), but they gave inferior results in terms of reaction efficiency. The use of ent-L4 (under otherwise identical conditions) led to the enantiomeric products with lower stereocontrol, and this suggested that stereochemical induction was mainly dependent on the configuration of the chiral ligand of palladium, while the chiral amine catalyst played a role of assistance in stereochemical control.
Scheme 68.

4. Vinylogous Ketones
In comparison to the high number of documents involving catalytic γ-selective (or β-, ε-, etc.) activation of enals, reviewed in the previous section, the corresponding reactions embracing enones as γ-selective pronucleophiles are definitely encountered to a lesser extent. Controlling the γ-regioselectivity of reactions with vinyl ketones as pronucleophiles is not straightforward, because of the low electron density at the γ-position233 of the corresponding dienolates, which tends to favor nonvinylogous α-selective reactions234−236 (α vs γ regioselectivity). Moreover, ketones bearing an α′ enolizable site possess an additional pronucleophilic position,237,238 and this makes the regiocontrol of the reaction (γ vs α′) particularly challenging.
In these years, different strategies have been adopted to overcome these issues, for example by using cyclic enones, using deconjugated ketones, or even placing a bulky group at the nonvinylogous α position, with the aim to apply to vinylogous ketones the catalytic enantioselective strategies developed for vinylogous aldehydes (Figure 2).
Figure 2.

Collection of pronucleophilic ketones (above) at work in this chapter using the direct procedures. Below, the nucleophilic ketone-derived silyl dienol ethers used in indirect procedures. Red circles denote the reactive (pro)nucleophilic carbon sites.
A limited number of examples dealing with vinylogous ketones as donors in addition to C=O and C=N bonds were reported in the last 8 years; in fact, most of the studies concern the addition to activated C=C bonds, often followed by cyclization reactions. The interest was focused on developing stereoselective procedures, and organocatalysis was the main activation strategy of pronucleophilic vinylogous ketones, often assuring high levels of diastereo- and enantioselectivity. However, even if outstanding results were reached in recent past years, the use of these vinylogous procedures in the synthesis of target molecules is still limited, and none of the reported examples utilizes enantioselective organocatalytic activation modalities.
4.1. Additions to C=O Bonds
4.1.1. Direct Procedures
4.1.1.1. Acyclic Pronucleophiles
In 2013 Jiang and co-workers, inspired by previous work of Shibasaki,239−241 where acyclic allyl cyanides were used as donors in direct asymmetric vinylogous aldol reactions, developed the first example of application of allyl ketones in catalytic asymmetric reactions.242 The enantioselective direct vinylogous aldol reaction between allyl ketones 251 and isatins 252 (Scheme 69) was catalyzed by the l-valine-derived bifunctional tertiary amine/thiourea catalyst (C6), that is supposed to first deprotonate the allyl ketone at the α position and then bring the resulting enolate and the isatin electrophile together to form the hydrogen-bonded complex for the C–C bond formation. The reaction is highly enantio- and E-selective, and the best reaction outcome was achieved with unprotected isatins. Good stereocontrol was maintained even when the reaction was performed on a gram scale. The products are valuable R-configured 3-hydroxy-2-oxindole derivatives of type 253. Computational studies, based on the density-functional theory (DFT), strongly supported that the observed stereochemistry, and preference for γ- over α-alkylation, is the result of favorable secondary π–π* and H-bonding interactions in the transition state.
Scheme 69.

Subsequent to this report and because of their easy accessibility, allyl ketones were also employed as vinylogous pronucleophiles in addition reactions to C=C (vide infra), C=N (vide infra), and C=O. In 2016, the Jiang group reported the first catalytic, asymmetric vinylogous aldol reaction (VAR) of allyl aryl ketones 251 to activated acyclic compounds 254 (Scheme 70, eq 1).243 The screening of four bifunctional tertiary amine/thiourea catalysts revealed that C7 (10 mol %) was the most efficient agent and that the addition of basic Na3PO4 (2 equiv) improved both yields and enantioselectivity. These conditions were applicable to diverse activated acyclic ketones with electron-withdrawing groups, including trifluoromethyl ketones, α-ketoesters, and α-keto phosphonates, thus furnishing compounds 255 in good yields and enantioselectivities. The authors showed how, starting from compounds 255, it was possible to prepare, by suitable transformations, chiral EWG-(α,δ)-tertiary hydroxy-based carboxylic acids, key structural motifs in important bioactive molecules.
Scheme 70.

Two more works, where allyl ketones were used as donor components in direct VARs, were published in 2018. In the first one, Wang and co-workers reported the addition of 251 to silyl glyoxylates 256 (Scheme 70, eq 2),244 opening a new route for the enantioselective preparation of α-hydroxysilanes 257. The reaction was organocatalyzed by the bifunctional catalyst C8, which may activate the carbonyl of 256 via strong hydrogen bonding. The steric hindrance of the silicon moiety in the acylsilane favors the attack of the vinylogous donor at the less bulky γ-position (γ-selectivity). Moreover, the reaction conditions avoided possible [1,2]-Brook rearrangement (the silyl migration from carbon to oxygen) and ensured the formation of the desired products 257 with moderate to good yields and good enantioselectivity levels. A mechanism based on DFT calculations was proposed. Interestingly, the authors verified that using the conjugated ketone 1-phenylbut-2-en-1-one as a donor, only a trace amount of the product was formed, emphasizing the importance of deconjugated allyl ketones as vinylogous precursors.
In the second study, Mukherjee and Ray reported the first example of the use of pyrazole-4,5-diones 258 as acceptors in vinylogous reactions (Scheme 70, eq 3).245 The VAR of 251 was catalyzed by the quinine-derived bifunctional tertiary amine/amide catalyst C9, a single H-donor catalyst. The reaction proceeded exclusively in γ- and E-selective manner to give products 259 in good yields, but with moderate to discrete enantioselectivities. The lowest enantioselections were registered with allyl ketones 251 bearing ortho-substituted aryl substituents, probably because of the steric hindrance of the substituent. Two examples of alkyl allyl ketones as donors were also reported, furnishing the products in good yields albeit with a moderate enantiomeric excess.
4.1.1.2. Cyclic Pronucleophiles
In 2013, the Melchiorre group reported the direct VAR of 3-methyl 2-cyclohexen-1-one (260) with α-keto esters 261 to furnish the aldol adducts 262 (Scheme 71).246 The reaction was catalyzed by the bifunctional primary amine-thiourea A22, which could simultaneously activate both the enone, by forming a nucleophilic dienamine, and the electrophilic acceptor by an H-bond-directing activation. This dual activation strategy, with the presence of benzoic acid, secured the access to aldol products 262 with good stereocontrol and perfect γ-site selectivity.
Scheme 71.

If a wide variability of R1 and R2 of the α-keto ester was well tolerated, the limitation of the system was the enone structure; in fact, with 3-methyl 2-cyclopenten-1-one, a complete loss of reactivity was registered.
The same year, Melchiorre et al. reported the first example of a vinylogous organocascade catalysis, where a 1,6- addition/aldol sequence between cyclic dienones 263 and oxindoles 264 carried to the formation of spirocyclopentane oxindoles 266 bearing four contiguous stereocenters.247 The bifunctional primary amine–thiourea catalyst A22, in the presence of 2,6-bis(trifluoromethyl)benzoic acid 265 as cocatalyst, operates through a dual activation strategy; in fact, it activates the dienone as iminium ion that reacts at first as vinylogous electrophile to give 263′ (Scheme 72). Then, dienamine activation in 257′ allows the intramolecular VAR that eventually carries to the spirocyclic product.
Scheme 72.

The last example of a direct cyclic VAR, again from the Melchiorre group, is the recent application of photoactivation of 2-alkyl benzophenone substrates 267 to give highly reactive hydroxy-o-quinodimethanes (E)-267′′′, that react with 2-substituted-2-fluorocyclopentane-1,3-diketones 268.248 The photoenol generation was obtained by irradiation at λ = 365 nm. The mechanism of formation of the reactive photoenol (E)-267′′′ was already in depth studied,249 and it is reported in Scheme 73. Irradiation of 2-alkyl benzophenone 267 triggers the formation of a singlet excited state S1-267′ that, upon intersystem crossing, decays to a triplet state T1-267′. The 1,5-hydrogen transfer generates the diradical intermediate (Z)-267′′, which then undergoes rotation to give the reactive enol (E)-267′′′. The aldol acceptor was an achiral 1,3-diketone of type 268, and good stereocontrol was achieved with the chiral amido–thiourea catalyst C10, which is able to activate one of the enantiotopic carbonyl groups. Overall, the process is a rare example of a light-driven organocatalytic aldol desymmetrization in which two stereocenters are simultaneously generated, one of them being a fluorine-containing quaternary stereocenter.
Scheme 73.

In order to assemble the tetracyclic core of rhodexin A, Jung and Guzaev developed a formal inverse electron demand Diels–Alder reaction (IEDDA) between hindered dienone 270 and silyl enol ether 271, catalyzed by the strong Lewis acid aluminum triflimide complex, Me2AlNTf2 (Scheme 74).250 The authors sustained that the overall [4 + 2] process consists of a stepwise cascade with an initial Mukaiyama Michael reaction, followed by a vinylogous Mukaiyama aldol reaction, which would proceed through the intermediacy of in situ-formed silyl dienol ether 270′.
Scheme 74.

4.1.2. Indirect Procedures
4.1.2.1. Acyclic Nucleophiles
Indirect procedures, in which the vinylogous enolates of ketones are preformed as stable silyl derivatives, are almost absent in the literature of this period, probably for reasons of atom economy and the desire to develop simple and expedient synthetic procedures. A sole example was reported by the Alemán group in 2018, dealing with an enantioselective organocatalytic vinylogous Mukaiyama aldol reaction (VMAR) of silyloxy dienes and isatins (Scheme 75).88 The reaction was catalyzed by the bifunctional organocatalyst C2 and gave 3-hydroxy-2-oxindole derivatives 253. Most of the silyloxy dienes used in the paper derived from aldehydes, and the work has already been reviewed in section 3.1.2.1 (Scheme 28). However, two examples of the panel presented by Alemán are ketone-derived silyloxy dienes (273, Scheme 75). Interestingly, the reaction is the indirect version of the direct one presented in Scheme 69, in which Huang and collaborators used allyl ketones as pronucleophiles. The direct methodology afforded compounds 253 definitely in higher yields and enantioselectivity (86% versus 73% yield, 92% versus 60% ee for R = Me; 93% versus 79% yield, 98% versus 70% ee for R = Bn). Moreover, in the indirect procedure, the stereocontrol with N-unprotected isatins was modest and the methodology was developed with N-substituted isatins, while in the direct procedure the best stereocontrol was registered with N-unprotected isatins.
Scheme 75.

4.1.2.2. Cyclic Nucleophiles
The sole example of indirect vinylogous reaction between a silyloxy diene derived from a “special” cyclic ketone (namely, silyl phenol 275) and a carbonyl group was reported by Onyango and Jacobi,251 where the intramolecular vinylogous Mukaiyama aldol-type reaction was the key step in the synthesis of the core structure of viridin, a furanosteroid able to inhibit phosphatidylinositol-3-kinase (Scheme 76).
Scheme 76.

The reaction was catalyzed by TiCl4 and furnished the syn-adduct 276 preferentially, with dearomatization of the reactive silyl phenol ring. The authors found out that the substituents at C17 have a crucial impact on the cyclization. In fact, only 275, where C17 is a CH2, underwent cyclization, whereas the analogous precursors with X = O(CH2)2O, H and OH, or H and OAc, did not afford any cyclized products.
4.2. Additions to C=N Bonds
4.2.1. Direct Procedures
4.2.1.1. Cyclic Pronucleophiles
In 2015, Chi and collaborators described an organocatalytic activation of C–C bonds through the addition of an N-heterocyclic carbene (NHC) catalyst to cyclobutenones of type 277.252 The key step is the C–C single bond cleavage, and the generation of an NHC-bound intermediate of type 277′, a vinyl enolate, that reacts in a chemo- and stereoselective manner with an imine in a formal [4 + 2] cyclization, to form lactam products with two contiguous stereogenic centers. The reaction was reported with both sulfonyl imines (278, Scheme 77, eq 1) and isatin imines (280, Scheme 77, eq 2), while other imines, such as N-tosyl imine derived from benzaldehyde or aryl trifluoroacetone, did not lead to any product formation. In this strategy, all atoms of the substrates end up in the products, fulfilling the atom economy principle, and the overall reaction is redox-neutral. The reaction between 277 and 278 provided lactams 279 after 72 h in moderate to good yields, with excellent diastereoselectivities and good to excellent enantioselectivities. The stereoselectivity of the reaction was ensured by the use of aminoindanol-derived triazolium salts B28 or B29. The authors disclosed how the electronic properties of both the imine substrates and NHC catalysts significantly affected the enantioselectivity of the reaction. Lactams 281 derived from isatin imines were generally obtained with lower diastereoselectivities but with high ee, apart from the case when an electron-donating substituent (R5 = OMe) was present on the isatin aromatic ring.
Scheme 77.

In 2016, the Melchiorre group developed a photochemical organocatalytic strategy for the direct enantioselective Mannich-type reaction of 2-alkylbenzophenones 282 and cyclic imines 283, affording enantioenriched chiral sulfamates 284 (Scheme 78).253 Light irradiation (λ = 365 nm) generates the transient hydroxy-o-quinodimethane (see Scheme 73 for the discussion of the mechanism), that can be trapped by an imine acceptor. The reaction did not work with linear imines. A wide screening of diverse sets of organocatalysts revealed that the best stereoselection was achieved using dimeric cinchona alkaloid derivative C11 in an apolar solvent such as cyclohexane. The authors demonstrated that the catalyst controls the stereochemical outcome of the reaction by solely interacting with the imine substrate 283 and not with the donor. However, the stereocontrol was moderate, revealing the overall difficulty of developing an efficient asymmetric organocatalytic procedure, due to the high reactivity and fleeting nature of the photoenol species.
Scheme 78.

4.2.2. Indirect Procedures
4.2.2.1. Acyclic Nucleophiles
In 2015, Yang et al. reported for the first time the use of 1,3-bis-trimethylsilyl enol ether 285 as a vinylogous nucleophile in a Mannich-type reaction.254 They developed a stereoselective three-component vinylogous Mannich-type reaction between 285 and in situ-formed aldimines to access chiral 2,3-dihydropyridinones of type 286 (Scheme 79). The reaction was catalyzed by Sn(OTf)2, and the stereoselectivity was ensured by the use of the chiral α-methyl benzylamine substrate; after manipulation of the product, the auxiliary group could be removed by TFA treatment or hydrogenolysis. The authors exampled the utility of this methodology by preparing, from cyclic adducts 286, some bioactive natural alkaloids which incorporate cis-2,6-dialkylpiperidine as the core structure. Beyond the synthesis of simple piperidine compounds, the method also provided a rapid route for chiral quinolizidine construction.
Scheme 79.

4.3. Conjugate Additions to Electron-Poor C=C Bonds
4.3.1. Direct Procedures
4.3.1.1. Acyclic Pronucleophiles
In 2012, the Chen group first applied the trienamine catalysis strategy, already developed for 2,4-dienals (see section 3.3.1.1),161 to 2,4-dienones in order to generate a chiral electron-rich triene system in situ, which could perform as a diene component in Diels–Alder (DA) reactions with electron-deficient dienophiles.255 The possibility of applying this procedure was limited to δ,δ-disubstituted 2,4-dienones, and the presence of an aryl (or 2-styryl) group at the α position was required to suppress the formation of an alternative, unreactive trienamine intermediate. Apart from these limitations, the authors developed a straightforward asymmetric DA cycloaddition reaction, catalyzed by the primary amine 9-amino-9-deoxyepiquinine A23 and trifluoroacetic acid, in which the triene system 287′, generated from 2,4-dienones 287, reacted with dienophiles 288 with exclusive ε,β-regioselectivity (Scheme 80). Cycloadducts 289 were isolated as single endo diastereomers and with excellent enantioselectivity.
Scheme 80.

The authors did not investigate whether the mechanism was concerted or asynchronous, but they proposed a transition state to justify the observed stereoselection, in which a π–π interaction between the quinoline moiety of the catalyst and the arene ring (R1) of 2,4-dienone would block the Re face of the resulting trienamine intermediate. Then, an endo-selective cycloaddition would occur from the Si face of the triene system (Scheme 80). Finally, the generality of this methodology was demonstrated by applying it to other electron-deficient dienophiles, in particular to 3-allylideneoxindole, benzylidenecyanoacetate, and nitrostyrene acceptors to furnish multifunctional cyclohexene derivatives 290, 291, and 292, respectively, with very good enantioselectivities.
After this seminal work, where a chiral primary amine was used for HOMO-raising activation of 2,4-dienones, in 2014 the Chen group also applied this strategy to activate the remote ε-position of deconjugated linear 3,5-dienones of type 294,256 by capitalizing on a recent work by themselves (see Scheme 93), in which a δ,ε-positioned C=C bond of interrupted cyclic 2,5-dienones acted as an inducing group for the formation of linear trienamines.257 Thus, a series of 3,5-dienones 294 were reacted with 3-alkylidene 2-oxindoles 293 via trienamine catalysis, producing spirocyclic oxindoles 295 as [4 + 2] cycloadducts in very good yields, remarkable diastereoselectivity (dr >19:1), and excellent enantioselectivity (Scheme 81). The reactions were conducted in toluene in the presence of catalytic amounts of 9-amino-9-deoxyepiquinine A23 and salicylic acid (130).
Scheme 93.

Scheme 81.

In the same year, Chen et al. applied the HOMO-raising activation strategy exerted by chiral primary amines to activate the γ-position of deconjugated ketones.258 Dienamine catalysis, in fact, could not be applied to activate the γ-position of linear conjugated ketones since, as shown in Scheme 82, cross-conjugated dienamines were preferably generated when the α′-CH group was present.238,259−261
Scheme 82.

The vinylogous Michael addition of allyl alkyl ketones 296 to maleimide 297 was efficiently catalyzed by the commercially available (R,R)-1,2-diphenylethanediamine A24, furnishing γ-products 298 exclusively, in very good yields and with excellent enantioselectivity (Scheme 83). Linear or branched alkyl-substituted allyl ketones exhibited similar high reactivity. The procedure was simple and reliable also on a gram scale.
Scheme 83.

In 2016, the Chen group exploited the HOMO-raising activation by dienamine catalysis to activate the γ-position of allyl ketones in an inverse-electron-demand oxa-Diels–Alder (IED-oxa-DA) cycloaddition reaction with α-cyano-α,β-unsaturated ketones.262 Remote β,γ-regioselective IED-oxa-DA reactions with α,β-unsaturated aldehydes and β,γ-unsaturated-α-ketoesters through dienamine catalysis had already been developed by the Jørgensen group,138,140 but Chen was able to extend the strategy to linear enones. The dienophiles of the asymmetric IED-oxa-DA developed by Chen and co-workers were represented by electron-rich dienamines derived from condensation/isomerization of allyl ketones 299 with the cinchona-derived primary amine A25, while α-cyano-α,β-unsaturated ketones 300 were used as the diene counterparts (Scheme 84). Densely substituted dihydropyran products 301 were obtained in good yields, with excellent enantioselectivities and fair to outstanding diastereoselectivities. Oxadienes 300 with diverse β-aryl, heteroaryl, and 2-styryl groups were well tolerated, and various α′-substitutions were compatible with the reaction. The lowest dr was registered with R4 = CF3. Allyl ketones bearing diverse α′-alkyl groups showed similar reactivity, while the α′-phenyl group lowered the ketone reactivity, even if the corresponding product was obtained with excellent stereocontrol. Other oxadiene partners (not shown in Scheme 84), such as 3-benzoyl-2H-chromen-2-one and an α-nitro-α,β-unsaturated ketone, were assembled with allyl ketones 299, enriching the palette of tetrahydropyran derivatives that could be prepared with this protocol.
Scheme 84.

Another strategy for direct vinylogous Michael addition reactions (VMcR) of linear allyl ketones as donors to α,β-unsaturated aldehydes was proposed by Xu and co-workers in 2014.263 They reported the combined use of two catalysts, namely, the diphenylprolinol trimethylsilyl ether ent-A4 and the bis(sulfonamide) H2, to trigger the VMcR between donors 302 and enals 303 (Scheme 85), by exploiting what they called “multifunctional supramolecular iminium ion catalysis” (SIC).264 Prolinol derivative ent-A4 would activate the aldehyde by forming a conjugated iminium ion species, whose Si face is hindered from the attack of the nucleophile; the acidic −NHs of the cocatalyst H2 would activate the vinylogous donor, stabilizing the dienolate by anion-binding interactions and favoring the γ-attack by shielding the α position. In the SIC strategy, the separation of the iminium–enolate ion pair by the cocatalyst H2 increases the turnover number and allows a lower catalyst loading. The chiral 1,7-dioxo product compounds 304 were generated with good yields and excellent regio- and enantioselectivities. The reaction worked well only with α,β-unsaturated aldehyde 303 bearing aromatic substituents (both electron rich and electron poor groups). As for allyl ketones 302, aromatic substituents bearing both electron-withdrawing and electron-donating groups were well tolerated, while aliphatic substituents furnished the products in lower yields and γ/α ratios.
Scheme 85.

In 2016, Brenner-Moyer and co-workers published the first example of an organocatalyzed direct vinylogous Michael addition of linear conjugated ketones 305 to enals 306, activated by the prolinol derivative ent-A4 via iminium ion formation (Scheme 86).265 To achieve good reaction outcome in terms of both yield and enantioselectivity, the addition of Et3N (1 equiv) and carboxylic acid 307 (20 mol %) was necessary. With these conditions, after 3 days of reaction, the γ-alkylated products 308 were obtained. Sterically congested R2 groups such as a branched alkyl group hampered the reaction, while electron-withdrawing or -releasing substituents on cinnamaldehydes had minimal impact on product yield or enantioselectivity. No reaction was observed with an aliphatic enal. The authors applied these conditions to other acceptors to clarify which factors influenced the α- vs γ-alkylation, and interestingly, they could conclude that steric hindrance of the Michael acceptors, and not electronic influence, played a major role in regioselectively directing the alkylation. In fact, enals with larger R3 groups favored γ-alkylation, while enals with smaller R3 groups favored α-alkylation of these linear vinylogous Michael donors.
Scheme 86.

An alternative catalytic strategy for the site-selective and stereocontrolled γ-functionalization of linear enones of type 309 was reported by the Wang group in 2013 (Scheme 87).266 The complex L7-Mg, where the magnesium ion is coordinated by a salen-type chiral ligand, could direct γ-deprotonation of linear α,β-unsaturated ketones, thanks to the presence of basic groups within the ligand and contemporary hindrance of the α-position of the donor by suitable substituents. The β,β-disubstituted α,β-unsaturated ketones 309 gave a Michael-type addition to nitroalkenes 310, followed by cyclization of the nitronate intermediate to the ipso carbonyl, leading to a variety of optically active cyclohexene frameworks 311. Aliphatic nitroalkenes did not undergo this transformation, while different aryl groups at either the β- or α′-positions of the vinyl ketones were compatible.
Scheme 87.

The following year, Wang et al. designed a combinational magnesium catalyst for the stereocontrolled cross reaction of enones.267 The stereocontrol of the reaction between 312 and 313 (Scheme 88) was particularly challenging, since the reaction partners have a very similar substitution pattern, but the use of the phosphoric acid L8, together with quinidine C12 and MgBu2 in p-xylene was able to ensure the formation of γ,ipso-[4 + 2] cyclization products 314 in modest to good yields and high diastereo- and enantioselectivities. Nucleophilic enones 312 at first coordinate to the metal center of the preformed combinational catalyst (Scheme 88), that determines the attack direction to electrophilic enones 313. Several control experiments were performed by the authors, concluding that the absolute configuration of the products is mainly induced by the phosphoric acid, while the cinchona alkaloid in the combinational catalysis plays the role of reaction promoter.
Scheme 88.

Very recently, Hong and co-workers exploited the vinylogous reactivity of deconjugated ketones 315 toward nitroolefins 316 to prepare fully substituted cyclobutanes of type 317 (Scheme 89) via a γ,β-regioselective [2 + 2] annulation.268 The reaction was catalyzed by cinchona squaramide catalyst C5, that favors the enolization of enone 315 through the squaramide hydrogen-bonding activation and deprotonation at the α-position by the quinuclidine moiety and subsequent activation of the nitroolefin by hydrogen-bonding. Cyclobutanes 317 were obtained in good yields and excellent diastero- and enantioselectivities. Lower yields were registered with nitroalkenes with aliphatic substituents on the β position. The authors underlined how the α vs γ regioselectivity was governed not only by the organocatalyst but also by the substituents on both the nitroolefins and the vinylogous donors. Small amounts of Michael-type products from the donor α-site were however formed, even under the optimized conditions.
Scheme 89.

The first use of α-benzoyloxy ketones of type 318 as dienolate precursors was reported by Fang and co-workers in 2017 in an enantioselective all-carbon [4 + 2] annulation (Scheme 90).269 The in situ formed dienolate 318′ is able to react with α,β-unsaturated acyl azolium species 319′, in turn in situ generated from α-bromoenals 319(68,270,271) under NHC catalysis, giving the vinylogous Michael adduct 318′′. Isomerization and intramolecular aldol reaction then lead to the formation of intermediate 318′′′, that after lactonization provides a β-lactone with the release of the chiral carbene B30. Methanolysis of β-lactone species furnishes the cyclohexene products 320. This strategy, thanks to an efficient isomerization process, not observed in the previous reports of intermolecular annulations,272 allowed the synthesis of cyclohexenes 320, inaccessible from conventional Diels–Alder reactions, in modest to good yields, as single diastereoisomers and with good enantioselectivities. Lower yields were registered when aliphatic ketones (R2 = Alk), alkyl esters (R1 = Alk), or carbonate (R1 = OAlk) were used. Moreover, when R4 was an aliphatic group, the annulation reaction did not occur under the optimal conditions.
Scheme 90.

4.3.1.2. Cyclic Pronucleophiles
In 2010, Melchiorre and co-workers were the first who developed a procedure for a direct intermolecular vinylogous Michael addition of unmodified β-substituted cyclohexenone derivatives to nitroalkenes via dienamine catalysis.228 Cyclohexenones activated as dienamines have multiple potential nucleophilic sites, the α′-position in the kinetic cross-conjugated dienamine XXVII, the α- and γ′-positions in the endo extended dienamine of type XXVIII, and the α- and γ-positions in the exo extended dienamine of type XXIX (Scheme 91). Theoretical calculations accounted for a thermodynamically driven site-selective formation of an exo cyclic dienamine favored over the other two. The bifunctional epi-quinine-derived catalyst A26 (10 mol %) and 2-fluorobenzoic acid (196, 20 mol %) as cocatalyst in toluene were the best combination to catalyze the reaction between 3-methylcyclohexenone (321, R1 = H) and nitroalkenes 322, to give products 323 in high yields, complete γ-regioselectivity, and excellent enantiomeric excesses. Different substituents at the aromatic moiety of β-nitrostyrene derivatives were well-tolerated, regardless of their electronic properties, while aliphatic nitroalkenes, as well as a different cyclic scaffold of the nucleophilic component (i.e., 3-methyl-2-cyclopenten-1-one), resulted in a complete loss of reactivity. A modified catalyst salt combination was used to catalyze the reaction of prostereogenic 321 (R1 ≠ H) to give products 324 with two contiguous stereogenic centers, in good yield and enantioselectivity, with variable levels of diastereoselectivity in favor of anti-adducts.
Scheme 91.

Some years later, the same activation mode was exploited by Bencivenni and collaborators to catalyze the vinylogous Michael addition of 3-alkyl cyclohexenones 325 toward N-(2-t-butylphenyl)maleimides 326 (Scheme 92).273 The cinchona alkaloid-derived amine catalyst A23, forming a dienamine intermediate, was able not only to transfer its stereochemical information to the prochiral γ-position of the donor but also to the prochiral axis of the acceptor, directing the attack from the side not shielded by the tert-butyl group. The authors registered an efficient control in the desymmetrization of maleimides 326, isolating adducts 328 with low diastereomeric ratios but high enantiomeric excesses.
Scheme 92.

The reaction of cyclic enones of type 325 with alkylidene, allylidene, and alkynylidene malononitriles catalyzed by a quinidine-derived catalyst was developed by Chen and co-workers.261 With alkylidenemalononitriles, γ-regioselective VMcR occurred, providing the corresponding products with very low enantioselection. Instead, using allylidene or alkynylidene malononitriles, α′,β-regioselective [4 + 2] bicyclo[2.2.2]octane adducts generated by the cross-conjugated dienamine of type XXVII (Scheme 91) were exclusively produced. Interestingly, the authors proved that the reaction, an apparently concerted Diels–Alder cycloaddition, actually proceeded by a stepwise Michael–Michael cascade.
In 2013, the Chen group introduced the use of interrupted 3-allylcyclohexen-2-ones 329 as bisvinylogous donors by trienamine catalysis (Scheme 93).257 In fact, the use of 2,4-dienones was hampered, because of their tendency to enolize at the α′-position to give cross-conjugated trienamines. Using the deconjugated dienones 329, instead, polyconjugated linear trienamines were formed, with the possibility of the transmission of the HOMO raising activation at the remote ε position, through the conjugated π system. Thanks to the activation exerted by the epiquinidine catalyst A27, and using sialic acid 130 as cocatalyst, compounds 329 reacted with electron deficient 1-azadienes 330 (in particular 3-vinyl-1,2-benzoisothiazole-1,1-dioxides (n = 0), or analogs with a 1,2,3-benzoxathiazine-2,2-dioxide motif (n = 1)) in an asymmetric inverse-electron-demand aza-Diels–Alder reaction. Cycloadducts 331 were obtained in high yields, with exclusive ε,δ-regioselectivity, and excellent stereocontrol. The reaction was efficient also on an acyclic N-tosyl-1-azadiene.
The following year, the exploitation of this trienamine catalysis-based strategy permitted to further disclose the bisvinylogous reactivity of 2,5-dienones 332 in direct enantioselective 1,4- or 1,6-additions.274 The reaction of 332 with nitroalkenes 333 provided linear Michael adducts 334 (Scheme 94, eq 1), with remarkable remote ε-regioselectivity, while cyclized ε,β-cycloaddition products were not observed. The enantioselectivity given by the use of the epiquinine catalyst was good, even if not excellent. Nitroalkenes bearing diversely substituted aryl or heteroaryl groups were well tolerated, while alkyl-substituted nitroalkenes gave the worst results in terms of both yield and enantioselection. The reaction could not be applied to ε-substituted 2,5-dienones, that were almost inert as donors.
Scheme 94.

Bifunctional primary amine-thiourea catalyst A28 was necessary to favor the 1,6-addition of 332a to α,α-dicyanodienes 335 forming adducts 336 (Scheme 94, eq 2).275 In fact, when cinchona-derived catalysts were tested, δ,ε-Diels–Alder cycloadducts were the major products. With the optimized conditions, linear products 336 were obtained in modest yields, but with complete ε-regioselectivity and high enantioselection. The reaction was not applicable to both α,α-dicyanodienes without β-substitution (R5 = H in 335), and ε-substituted cyclic 2,5-dienones, which proved to be almost inert.
In 2014, the Chen group carried out the investigation of HOMO-activation of the remote C=C bond via trienamine catalysis and applied this strategy to other nucleophiles and reaction types. For example, this methodology was used to activate 2-vinyl heteroarenes, such as β-(2-benzofuryl)methyl enones 337 in the coupling reaction with maleimides 338 (Scheme 95, eq 1).276 The conditions were similar to those previously reported, i.e. 9-amino-9-deoxyepiquinine A23 (20 mol %) as chiral catalyst and sialic acid 130 (40 mol %) as acidic cocatalyst, in toluene. Treating the reaction crude in acidic conditions, the authors isolated cycloadducts 339 in good yields and diastereo- and enantioselectivities. However, under these conditions, 2,5-dienone substrates of type 337 bearing α′-enolizable groups did not react at all with dienophile 338a; thus the authors prepared deconjugated 3,5-dienone-type substrates of type 340. Benzofuryl derivatives were not stable in the reaction conditions, while 2-benzothiophenyl enones 340 reacted with maleimide 338a, directly furnishing, without acidic treatments, products 341 possessing four contiguous stereocenters (Scheme 95, eq 2). This asymmetric dearomatizative Diels–Alder protocol was also applied to 3-benzofuryl and 3-benzothiophenyl derivatives and, even in these cases, non-α′-enolizable 2,5-dienones 342 produced the corresponding ε,β-locked cycloadducts 344 (Scheme 95, eq 3), while when R2 in the α′-position was an enolizable alkyl group (R2 = Me, Et) the reaction had to be performed starting from deconjugated 3,5-dienones of type 343. The generality of this method was proven by reacting 343 (R1 = H, R2 = Me) with other dienophiles, such as 3-alkylidene oxindoles and benzylidenecyanoacetate, obtaining the corresponding ε,β-cycloadducts in fair to good yields and excellent enantioselectivities.
Scheme 95.

Some years later, the same asymmetric dearomatizative [4 + 2] reaction via trienamine catalysis was carried out on 2-(3-vinylbenzofuran-2-yl)ethan-1-ones 345, activated by epiquinine A23 to form the trienamine reactive species 345′, and using 3-olefinic 7-azaoxindoles 346 as the dienophile partners (Scheme 96). The reaction products consisted of fused spirocycles of type 347, bearing two vicinal tetrasubstituted stereogenic centers. They were obtained in fair yields and with good to excellent stereocontrol. Several protecting groups on the oxindole nitrogen were tolerated, and lower yield and enantioselectivity were registered with NH-free 346.
Scheme 96.

When applying the trienamine-activation strategy to 2,4-dienones 348 in coupling reactions to benzoyl-bearing activated alkenes 349, Chen and collaborators did not isolate the expected α′,β-locked [4 + 2] cycloadducts (vide supra),261 but they witnessed the regioselective formation of γ′,δ-locked [4 + 2] products 351, due to the formation of unprecedented cross-conjugated trienamine intermediates of type 348′ (Scheme 97).277 The combination of cinchona-derived catalyst A27 and 2-mercaptobenzoic acid 350 secured the formation of 351 in good yields, with excellent diastereo- and enantioselectivities. Different aryl and heteroaryl groups at the β-position of acceptors 349 were compatible with this procedure, but the reaction did not furnish the desired products when R1 (in the donor) and R3 (in the acceptor) were alkyl substituents. This methodology was applied to other acceptors, such as alkenes 352 derived from Meldrum’s acid and isoxazolones 354, obtaining good results in terms of yields, but with lower enantiomeric excesses. The analysis of the absolute configuration of the stereocenters in 355 suggested that the [4 + 2] reaction does not proceed along a concerted Diels–Alder pathway, but rather via a stepwise vinylogous Michael–Michael mechanism.
Scheme 97.

The use of α′-alkylidene-2-cyclopentenones 356 as vinylogous donors, via trienamine catalysis, was cleverly disclosed by the Chen group in 2018.209 Activation of 356 with 2′-tert-butyl-9-amino-9-deoxyepicinchonidine (A32) in toluene carried to the in situ formation of HOMO-raised cross-conjugated trienamine 356′ that, depending on the acceptor nature, gave rise to either an asymmetric γ,β′-regioselective [6 + 2] cycloaddition or a γ,β-regioselective [2 + 2] cycloaddition (Scheme 98). With the highly electrophilic 3-olefinic (7-aza)oxindoles 357, under the catalysis of A30 or A32 and salicylic acid (130) in toluene at 50 °C for 72 h, the reaction furnished [6 + 2]-cycloadducts 358 with five contiguous stereogenic centers, in good yields and with high levels of diastereo- and enantiocontrol. When the same conditions were applied to the reaction of 356 with maleimides 359, fused cyclobutanes of type 360, derived by a [2 + 2] cycloaddition, were obtained with good yields and excellent stereoselectivities. DFT computational calculations were performed, showing that the [6 + 2] cycloaddition likely proceeds in a stepwise vinylogous Michael–Michael reaction, while the [2 + 2] cycloaddition might involve a process via an γ,ipso-[4 + 2]-cycloaddition, followed by a concerted ring-opening and ring-closure process, to form the products 360.
Scheme 98.

An extension of this strategy was reported the following year again by Chen and collaborators.278 They developed an asymmetric four-component [5 + 1+1 + 1] formal cycloaddition reaction between 3-substituted 2-cyclopentenones 361, aryl aldehydes 362, and 3-methylisoxazolones 363, catalyzed by the cinchona-derived primary amine A30 and salicylic acid (130) in toluene (Scheme 99). Products 364 were obtained in modest yields, but with a high level of enantioselectivity. Moderate enantiomeric excesses were registered when 3-phenylisoxazolone or thiophene-2-carboxaldehyde were used (78% and 79% ee, respectively); otherwise, ee higher than 91% was measured. The authors investigated the reaction mechanism and proposed a cascade process, in which the 3-methylisoxazolone acts also as a cocatalyst in the formation of the key dienone 356. Next, a domino vinylogous Michael–Michael addition between 356′ and 363′ takes place carrying to products 364. Other types of activated methylene nucleophiles different from isoxazolones 363 were used, expanding this strategy to the construction of chiral frameworks with increased structural diversity.
Scheme 99.

In 2017, Jørgensen and co-workers published the first example of organocatalytic [6 + 4] and [8 + 2] cycloadditions, together with a [4 + 2] pathway.208 Cyclic enones 365, activated as dienamines by cinchona-based primary amine catalysts, reacted with tropone 366a or cyanoheptafulvenes 366b and 366c (Scheme 100). These cycloadditions were periselective and exhibited both diastereo- and enantioselectivity, affording complex bicyclic structures 367–369 with up to four contiguous stereocenters. To rationalize the observed peri- and stereoselectivity, several control reactions were carried out and potential energies of all products were calculated relative to their starting compounds. The α′,β-locked [6 + 4] cycloadduct 367, formed from cyclopentenone 365a (n = 1), is favored when the cross-conjugated dienamine 365a′ reacts with tropone 366a (X = O). In this case, the dienamine intermediate 365a′ reacts at the α′-position along a nonvinylogous pathway. Conversely, [8 + 2] cycloadditions to γ,β-locked products 368 are favored when linear dienamines 365b′ (n > 1) react with cyanoheptafulvenes 366 (X = C(CN)2, C(CN)CO2Et). The pathway to γ,β-locked [4 + 2] cycloadducts 369 was explained as a rapid rearrangement of unobserved [8 + 2] enamine intermediates, by an intramolecular vinylogous Michael type reaction (Scheme 100, bottom). Interestingly, the authors concluded that all these higher-order cycloadditions might proceed through stepwise mechanisms.
Scheme 100.

In 2017, Xu and co-workers applied the multifunctional supramolecular iminium catalysis (SIC), already optimized for linear donors (see Scheme 85),263 to perform the vinylogous Michael reaction of deconjugated 3-cyclohexenones 370 with α,β-unsaturated aldehydes 371 to give chiral 1,7-dioxo adducts 372 (Scheme 101).279 The combination of the Jørgensen–Hayashi catalyst ent-A4 for the acceptor activation as iminium ion, with cocatalyst H2 for dienolate stabilization, in a low polar solvent such as toluene, provided the almost exclusive γ-attack (γ/α 18:1), a diastereomeric ratio >20:1, and excellent enantioselection. Compounds 372 were the products of the first step of two sequential reactions. After the optimization process, the author did not separate 372 anymore, but after the evaporation of the solvent, they exploited the NHC-precatalyst B31 in chloroform to catalyze an intramolecular Stetter reaction, obtaining products 373 with the Hajos–Wiechert ketone skeleton.
Scheme 101.

The asymmetric organocatalytic domino vinylogous Michael/aldol reaction between 3-(trifluoroacetyl)-2-methylindoles 374 and enals of type 375 was reported by the Enders group in 2018.280 The dienolate species 374′, derived from 374 by γ-methyl deprotonation, regioselectively attacks the β-position of iminium ion-activated acceptors 375′ (Scheme 102). The VMcR is followed by an intramolecular aldol reaction, to afford trifluoromethylated tetrahydrocarbazoles of type 376, bearing three vicinal stereocenters. Products 376 were produced in high yields, as single diastereomers, and with high enantiomeric excesses. Protecting groups different from Boc on the indole nitrogen (such as Bz, Ac, Me), as well as the use of unprotected indoles, were not compatible with this strategy. The reaction did not succeed also when a methyl group was present in place of a trifluoromethyl group.
Scheme 102.

The pronucleophilic character of 2-alkylidene-1H-indene-1,3-(2H)-diones 377 in asymmetric vinylogous Michael reactions was discovered by Lin and colleagues in 2016.281 The high γ-proton acidity of 377, due to the strong electron-withdrawing effect of the 1,3-indandione moiety, allows for the facile γ-deprotonation and formation of vinylogous nucleophiles able to attack nitroolefins 378. One of the two carbonyl groups in the indandiones 377 intercepts the emerging Michael products, giving an intramolecular Henry reaction. This tandem vinylogous Michael addition/Henry reaction cascade was promoted by the bifunctional squaramide-cinchonidine catalyst C5 in xylenes and furnished tetrahydrofluoren-9-ones 379 in very good yields and enantiomeric excesses (Scheme 103, eq 1). In each case, despite four contiguous stereocenters being formed, only one diastereoisomer is obtained. The reaction did not work with aliphatic nitroolefins, with the exception of R3 = c-hexyl, which anyway had scarce efficiency. It is noteworthy that when arylethylidene indanones were used (377a, R2 = H), the predominant formation of the vinylogous Michael addition products 380 was observed (Scheme 103, eq 2). Since the authors were confident that the overall cyclization consisted of a stepwise vinylogous Michael/Henry cascade, they explained the formation of linear Michael products 380, by assuming that Henry cyclization did indeed take place, followed by a retro-Henry opening to the thermodynamically more stable products 380.
Scheme 103.

Albrecht and colleagues were the first who disclosed the vinylogous pronucleophilic character of 2-benzyl substituted-1,4-naphthoquinones of type 381, which were used in an organocatalytic cascade reaction with nonenolizable enals 382, to give carboannulated naphthalen-1(4H)-one derivatives 384 (Scheme 104).282 Under the optimized conditions, the dienolate species 381′ promoted a vinylogous Michael attack to enals activated as iminium ions 382′ by the diphenylprolinol trimethylsilyl ether A4 (30 mol %), and with 4-(dimethylamino)benzoic acid (383) as an additive. Co-catalyst 383 favored the γ- vs α-attack and the formation of the desired product. After the VMcR, an intramolecular aldol condensation provided cycloadducts 384 in modest to fair yields, but with excellent diastereo- and enantioselectivities. Both electron-withdrawing and electron-donating groups on the aromatic substituent of enals 382 were well tolerated, while the use of aliphatic, linear enals resulted in sluggish conversion. A transition state involving E,Z-configured dienolate 381′ attacking the less hindered face of iminium ion 382′ (Scheme 104) was proposed by the authors to justify the stereoconfiguration of the products.
Scheme 104.

In 2014, the Chen group exploited the electronic transfer through the C=C bonds of a polyconjugated system for the HOMO activation of the C5-position (ε) of 2-furfuryl ketones of type 385 (Scheme 105).283 Through the formation of trienamine species with the chiral bifunctional primary amine-thiourea ent-A31, the π-system of 385 was activated for an enantioselective Friedel–Crafts (FC) alkylation. This was an alternative strategy with respect to the other catalytic asymmetric FC reactions already present in the literature, that normally proceeded by lowering the LUMO energy of electrophilic partners. The reaction was developed with alkylidenemalononitriles 386, as electron-deficient alkene, to generate products of type 387 in high yields and enantioselectivities. The alkylation was completely ε-regioselective (C5-position of the furan system). Various aryl and alkyl groups on the alkylidenemalononitrile acceptors were well tolerated, while the lowest enantiomeric excesses were registered with heteroaryl groups. Various α′-alkyl substituents on the 2-furfuryl ketones 385 were compatible, as well as groups at the C4- and C3-positions of the furan ring. Other activated alkenes were explored as acceptor components. The reaction with compounds 388 and 389 (Scheme 105) also proceeded toward the alkylation at the 5-position of the furan, even if with definitely lower enantioselectivity, and moreover other bifunctional catalysts had to be used. Interestingly, a similar reaction with nitrostyrene gave the α-attack product, while the maleimide behaved as a dienophile in Diels–Alder cycloaddition with the furan system. These data revealed that regioselectivity in these processes is strongly influenced by both electrophilicity and structural characteristics of the alkenes.
Scheme 105.

In 2016, Melchiorre et al. developed the first enantioselective catalytic variant of a Diels–Alder reaction between transient photoenol o-quinodimethanes 390′, obtained by light irradiation at 365 nm of 2-alkyl benzophenones 390 (for the mechanism, see Scheme 73), and maleimides 338 (Scheme 106).284 The reaction was catalyzed by the bifunctional thiourea-amine C13 in a mixture of cyclohexane and toluene at −5 °C. With these experimental conditions, excellent results were obtained, above all by considering the high challenge of performing an enantioselective catalytic version of a photoactivated reaction; tetrahydronaphthalenols 391 were formed in good yields, with high enantioselectivity and exquisite diastereoselectivity. The generality of the method was demonstrated since there was a wide tolerance for substituents on the benzophenone derivatives 390 as well as on the maleimide acceptors. Only the N-unprotected maleimide (R3 = H) afforded the product with low enantioselectivity (50% ee). The authors carried out experiments to elucidate the role of the catalyst, discovering that the quinuclidine core and thiourea moiety of the catalyst C13 exert two opposite yet cooperative roles. The quinuclidine core interferes with the photoenolization mechanism, acting as an inhibitor of the photoenol Diels–Alder, while the thiourea moiety increases the dienophilic character of 338 upon H-bonding activation and indeed acts as a chiral catalyst, channeling the reaction toward the enantioselective pathway.
Scheme 106.

The following year, the same photoenol o-quinodimethanes 390′ were used as the nucleophilic components in enantioselective organocatalytic VMcRs. The Melchiorre group, in particular, developed organocatalytic β-benzylation of enals 306 (Scheme 107, eq 1),285 presenting this reaction as the first effective enantioselective catalytic variant of the photoenolization/Diels–Alder sequence. The enals were activated as iminium ions by the diphenylprolinol tert-butyldimethylsilylether A13 in 1,2-dichlorobenzene, and the addition of diphenylphosphoric acid as acidic additive was useful to increase the reaction yield. The transformation furnished compounds of type 392 in fair yields and good enantioselectivities. Different aliphatic groups at the β-positions of the enals proved to be viable substituents, as well as different substituents on both aromatic rings of 390 were well tolerated. Interestingly, Diels–Alder adducts were never detected, so the authors carried out density functional theory (DFT) studies to explain why Michael adducts were preferred over cycloaddition products. The studies suggested the importance of a water molecule as a proton shuttle to transfer a proton by the photoenol to the iminium ion nitrogen, favoring the formation of an intermediate that selectively carries to the Michael product.
Scheme 107.

Another example of enantioselective Michael addition of photogenerated o-quinodimethanes 390′ (derived from benzophenones 390) to enones 393 was reported by Ye and collaborators (Scheme 107, eq 2).286 The reaction was catalyzed by aminoester A33 in toluene, using acetic acid as an additive (in the absence of acid no products were formed). With these optimized conditions, different cyclohexenones as well as linear α,β-unsaturated ketones 393 were intercepted by the photoenol species 390′, to give adducts 394 in good yields and excellent enantioselectivities. The reaction proved quite general and was successfully applied to 3-substituted 2-cyclohexenones, providing asymmetric access to products bearing all-carbon quaternary centers. Instead, for the reaction with cyclopentenone, it was necessary to change the catalyst by using A34, which consigned the adducts in low yields, but with excellent enantioselectivities.
The organocatalytic activation of cyclobutenones through the addition of an organocatalyst to form a vinylenolate intermediate by cleavage of C–C bond was described in 2015 at the same time and, independently, by Chi et al. (see Scheme 77)252 and Zhang and co-workers.287 In place of an NHC catalyst, exploited by the former group, Zhang explored the use of chiral bifunctional phosphine P1, which in the presence of cyclobutenone 395 readily undergoes C–C bond cleavage forming the 1,4-dipolar linear intermediate 395′, able to react as a vinylogous Michael donor on isatylidenemalononitriles 396 (Scheme 108). The reaction is a 1,4-dipolar spiroannulation that provided enantioenriched 3-spirocyclohexenone 2-oxindoles of type 397 in good yield and moderate to good enantiomeric excesses. The strong double H-bond donor moiety (3,5-di(trifluoromethyl)phenylthiourea) in the catalyst P1 was fundamental for the good enantioselectivity; in fact, phosphine-derived catalysts with single H-bond donors were not able to give good stereocontrol. Even the NaI additive was demonstrated to be important to increase enantiomeric excess values.
Scheme 108.

4.3.2. Indirect Procedures
4.3.2.1. Acyclic Nucleophiles
The sole example of preformed linear silyl dienol ethers derived from ketones as nucleophiles toward activated C=C bonds was reported by Schneider et al. in 2012.288 The reaction is a catalytic, enantioselective vinylogous Mukaiyama–Michael addition of silyl dienol ethers 398 to α,β-unsaturated aldehydes 399, that provided chiral 1,7-dioxo compounds 400 (Scheme 109). The catalytic strategy exploited the conversion of enals into more reactive chiral α,β-unsaturated iminium ions, using prolinol-derived catalyst A4, together with p-nitrobenzoic acid 192 as cocatalyst. Compounds 400 were synthesized in good yields, and with excellent enantioselectivities, while the level of diastereoselection, measured when R1 = Me, was not particularly gratifying. The sterically demanding mesityl group on the carbonyl in 398 was fundamental to shield the α-position and obtain high levels of regiocontrol (γ- vs α-attack). In fact, when the phenyl was present in place of the mesityl group, high percentages of α-1,4-regioisomers were isolated.
Scheme 109.

4.3.2.2. Cyclic Nucleophiles
In 2011, Lupton and colleagues reported the first all-carbon NHC-catalyzed [4 + 2] cycloaddition between silyl dienol ethers of type 401 and α,β-unsaturated acid fluorides 402 to produce racemic 1,3-cyclodienes 403 (Scheme 110, eq 1), in moderate to excellent yields and with complete diastereocontrol.289 The NHC catalyst B32 furnished acyl azolium intermediates 402′, by nucleophilic substitution of the acyl fluorides, that reacted as Michael acceptors290 with the dienolates 401′. The Michael addition followed by an intramolecular aldol reaction furnishes the [4 + 2] cycloadducts 401′′, that release the NHC catalyst following lactonization reaction. Finally, the decarboxylation of intermediates 401′′′ provides the final cyclohexenes products 403. This strategy was not applicable to aldehyde-derived TMS dienol ethers or to those derived from acyclic ketones, because in both cases O-acylation was favored. The studies to elucidate the mechanism reaction291 confirmed that the intermediate 401′′ is formed by a stepwise mechanism and not via a concerted cycloaddition. Moreover, the β-lactone intermediate 401′′′ was revealed to be stable until −20 °C and could be reductively intercepted with organolithium reagents to furnish racemic compounds 404 and 405, respectively, bearing four contiguous stereocenters (Scheme 110, eq 2).
Scheme 110.

4.4. Other Reactions
4.4.1. Direct Procedures
4.4.1.1. Cyclic Pronucleophiles
The vinylogous reactivity of photoenol (E)-406′a, generated from o-alkylphenyl ketones 406 via UV irradiation at 365 nm, was exploited by Murakami et al. for a vinylogous carboxylation reaction with CO2 (1 atm) to form o-acylphenylacetic acids 407 (Scheme 111).292 The reaction was clean since it did not need any catalyst or additional reagents but the simple irradiation of the starting materials in DMSO. The light energy that brings the formation of the highly reactive intermediate (E)-406′a is the driving force of the process. The authors proposed a mechanism in which the (E)-406′a undergoes a [4 + 2] cycloaddition reaction with the C–O double bond of CO2 to afford a six-membered cycloadduct, that collapses to the final carboxylic acid by a ring opening reaction; indeed the reactivity of CO2 as a dienophile is unprecedented.
Scheme 111.

γ-C–N bond formation via dienol or dienolate is a quite uncommon strategy, even because the α-amination of dienolate intermediates is often privileged.293 In fact, the first example of a direct γ-regioselective amination protocol of enones was reported by Mohr and colleagues only in 2015.294 The reaction of conjugated ketones 408, treated in THF at −78 °C with lithium hexamethyldisilazide (LiHMDS) to generate the dienolate, with dialkylazodicarboxylates 409 as acceptor counterparts, furnished the γ-aminated racemic products 410 (Scheme 112). A polar additive, such as hexamethylphopshoramide (HMPA), was fundamental to obtain high levels of regioselectivity, even if the reason was not fully clarified. Enones lacking the alkoxy group at the β-position (411) were compatible with the protocol and provided the corresponding aminated products (±)-412 in good yields. The reaction was also successfully tested on linear enones, conjugated esters, and lactones.
Scheme 112.

5. Vinylogous Esters and Lactones
Chiral, enantiopure α,β-unsaturated esters, be they linear or cyclic, are one of the most widespread classes of chiral frameworks, constituting the structural core of a vast set of natural products which display an impressive range of biological activities important for the development of novel physiological and therapeutic agents.35 Furthermore, the possibility to easily access remotely enolizable esters and higher-order homologues by synthesis renders this compound class a preeminent source of vinylogous and hypervinylogous carbon nucleophiles to be exploited in fruitful additions to differently adorned electrophiles thus providing access to a vast plethora of chiral products. In this context, due to the high versatility of this class of compounds and the outburst of research fields such as organocatalysis and green chemistry, the past decade has witnessed, along with the development of a vast array of new asymmetric, vinylogous transformations, the application of older methodologies to the total synthesis of increasingly complex, bioactive compounds.34
As for the previously described functionalities, a picture of the starting (pro)nucleophilic ester and lactone structures reported in this section is outlined in Figure 3, subdivided between acyclic (top) and cyclic (bottom) representatives and marked in their reactive (pro)nucleophilic remote sites.
Figure 3.
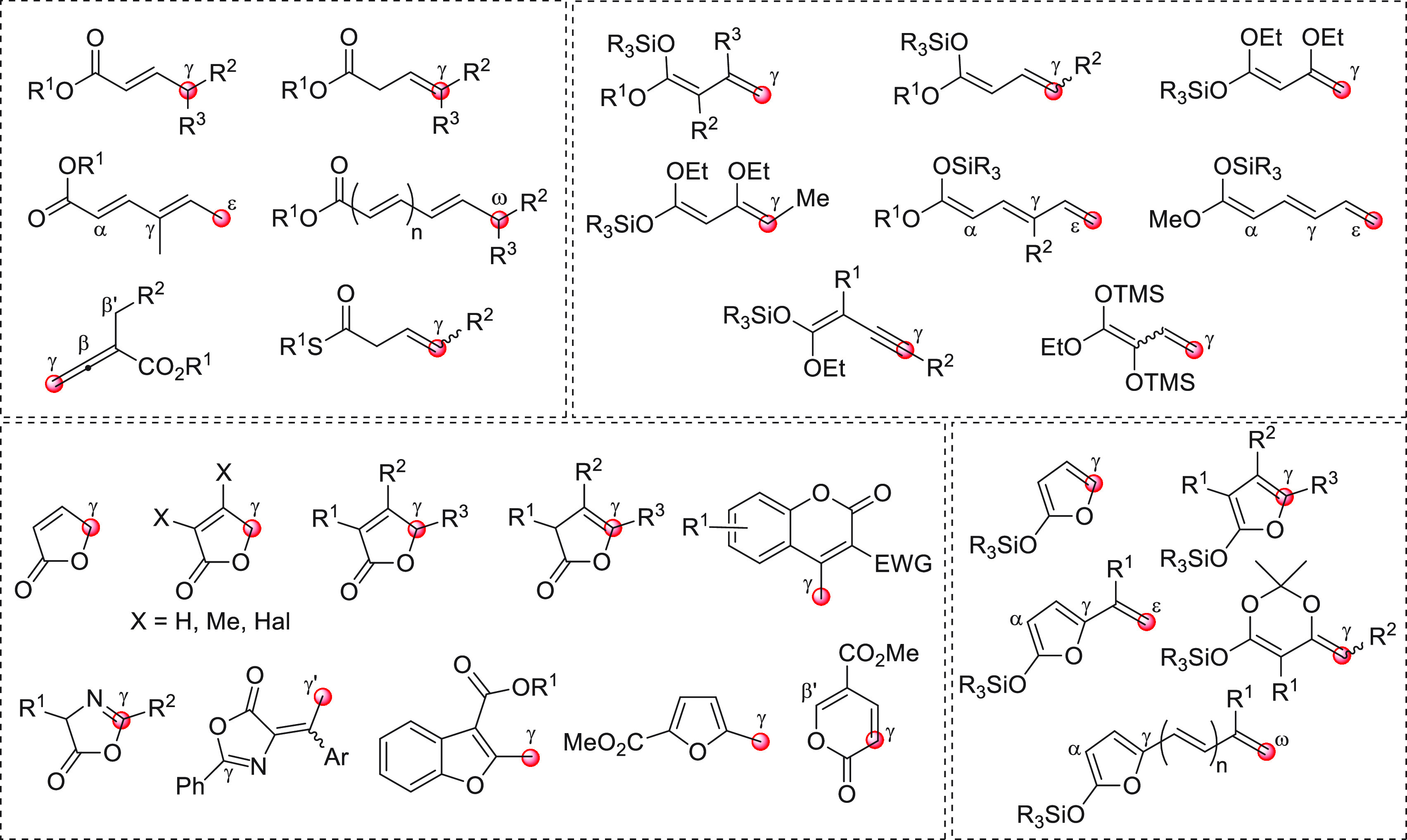
Collection of acyclic (above) and cyclic (below) (pro)nucleophilic esters at work in this section using direct and/or indirect procedures. Red circles denote the reactive (pro)nucleophilic carbon sites.
5.1. Additions to C=O Bonds
5.1.1. Direct Procedures
5.1.1.1. Acyclic Pronucleophiles
Starting from 2012, as part of their total synthesis program toward several members of the antimicrobial family called elansolids (Figure 4), the Kirschning’s group developed and exploited a direct, diastereoselective, and substrate-controlled Yamamoto bisvinylogous aldol reaction (BVAR) between (2E,4E)-ethyl-4-methylhexa-2,4-dienoate (413) and suitable chiral aldehydes of type 414 (Figure 4).295 The core structure of these natural polyketides features a bicyclo[4.3.0]nonane unit embedding a chiral polyene chain resulting in a macrolactone core in elansolids A1 and A2 or its linear seco-acid form in elansolids B1 and B2. Concerning the construction of the chiral polyene chain, Kirschning planned to use the (2S,3S) chirality within aldehydes 414 (representing the fragment C7–C11) to control the absolute R-configuration of the newly formed stereogenic center at C7, with the aid of the Yamamoto protocol.296−298
Figure 4.

Kirschning’s disconnection of the C6–C7 linkage of elansolids, envisaging a Yamamoto bisvinylogous aldol reaction (BVAR).
The Yamamoto bisvinylogous aldol reaction (BVAR) relies on the use of a bulky Lewis acid such as the C3-symmetric aluminum-tris-2,6-diphenylphenoxide M3 (ATPH) and lithium tetramethylpiperidine (LTMP) as the base (Scheme 113). Commonly, precomplexation of both the enolizable polyunsaturated ester compound and the aldehyde with 2.2 equiv of M3 at −78 °C is followed by the addition of an ethereal solution of stoichiometric LTMP that remotely deprotonates the ester to give an activated trienolate nucleophile that couples to the aluminum-precomplexed aldehyde via a bisvinylogous aldol reaction at the remote ε-terminus of the chain (Scheme 113, top). After aqueous work up, the expected homoallyl alcohol is provided. An extensive mechanistic study surveying differently substituted 2,3-syn and 2,3-anti-configured starting aldehydes 414 allowed Kirschning to elect silyloxy protected alkynyl (2S,3S)-414a as the best aldehyde candidate to directly access the 11-carbon long chiral polyene chain with the correct 7,8-anti relationship found in the elansolids.295
Scheme 113.

Indeed, the reaction between unsaturated ester 413 and (2S,3S)-414a under the standard Yamamoto BVAR conditions at −78 °C afforded the anti-Felkin adduct 415a, with a 74% yield and a dr (7,8-anti/7,8-syn) of 3.5:1 (Scheme 113, eq 1). More recently,297 in the effort to construct the eastern fragment of 20-deoxy-elansolid B1 (Figure 4), the same group obtained the desired 7,8-anti adduct indirectly, by exploiting a highly 7,8-syn diastereoselective Yamamoto BVAR between dienoate 413 and a iodovinyl aldehyde (2S,3S)-414b (Scheme 113, eq 2), accessing the 7,8-syn Felkin adduct 415b (71% yield, 20:1 dr). The C7 chiral inversion of 415b was then afforded by Dess–Martin oxidation-Luche reduction sequence, yielding a separable mixture of diasteoisomers in favor of 7-epi-415b (2:1 ratio). The procedure was then repeated with the undesired diastereoisomer, which finally allowed them to collect the 1,3-anti diastereoisomer 7-epi-415b in a combined 74% yield.
An intramolecular version of the Yamamoto VAR was exploited by Sammakia and co-workers in 2012 for the preparation of an advanced intermediate for the synthesis of (+)-peloruside A, a polyoxygenated 16-membered macrolide with potent antimicotic activity (Scheme 114).299 The reaction was applied to open intermediate 416 bearing pronucleophilic crotonate and nonenolizable furfural moieties that were cyclized using ATPH (M3, 2.2 equiv) and LTMP (2.0 equiv) in toluene/THF at −78 °C. The corresponding 16-membered macrolactone 417 was accessed in 86% yield as a 6:1 diastereomeric mixture.
Scheme 114.

The Sammakia’s group also developed a new aluminum-based Lewis acid, namely, aluminum tris(2,6-di-2-naphthylphenoxide) (ATNP, M4), capable of promoting the crossed Yamamoto VAR between methyl crotonate 418 and enolizable aldehydes of type 419 and 421 (Scheme 115, eqs 1 and 2).300 Indeed, the bulkier 2-naphthyl groups within M4, blocking the α-enolization of the aldehyde component, enabled the regioselective vinylogous enolization of the crotonate ester. As described in Scheme 115 (eq 1), using 3.3 equiv of M4, the direct, vinylogous aldol reaction between 418 and a series of achiral aliphatic and aromatic aldehydes of type 419 afforded the corresponding γ-adducts (±)-420 in good yields (up to 97%). Furthermore, under similar reaction conditions, addition of 418 to chiral α- and β-substituted aldehydes of type 421 (eq 2) proved also viable, providing the corresponding 5,6-anti-configured adducts 422 in good yields and stereoselectivity (up to 87% yield and up to 10:1 dr).
Scheme 115.

An attractive alternative to the common activation of linear unsaturated ester pronucleophiles via acid- or base-catalyzed conversion into their active (poly)enolate form resides in the generation of NHC-bonded dienolates (also called vinyl enolates) obtained by reacting the ester precursor with an active form of a suitable chiral N-heterocyclic carbene (NHC) precatalyst (vide previous sections). The annulation of these NHC-bonded vinyl enolates with substrates containing polar C–O and C–N double bonds paved new avenues to a variety of highly functionalized molecules.
In this context, a recent example for the asymmetric synthesis of spirocyclic heterocycles was developed by Yao and co-workers in 2015 (Scheme 116).301
Scheme 116.

Starting from readily available, linear α,β-unsaturated benzotriazole (Bt) esters of type 423 (Scheme 116), a formal NHC-catalyzed [4 + 2] cycloaddition reaction to N-alkyl isatins of type 424 was devised providing a series of chiral, enantioenriched spirocyclic oxindole dihydropyranones 425 featuring a tetrasubstituted carbon stereocenter. Indeed, a suitable optimization survey elected chiral triazolium B13 as the most effective precatalyst in promoting the reaction between 423 and 424 in toluene at −20 °C, in the presence of 1.2 equiv of Cs2CO3, providing the corresponding γ,i-locked [4 + 2] cycloadducts 425 in good yields (up to 87%) and enantioselectivities (up to 98% ee). The reaction proved to be quite general as it worked well on diverse benzotriazole 3-methylcinnamate derivatives and 5-methoxy or 4-Br-N-alkyl isatins.
5.1.1.2. Cyclic Pronucleophiles
The enantioselective VAR involving either α,β-unsaturated (e.g., furan-2(5H)-one) or β,γ-unsaturated furanone (e.g., α-angelicalactone) donors and chiral/achiral aldehyde acceptors has attracted particular attention in the chemical community being one of the most efficient tools to obtain chiral, γ-substituted, and γ,γ-disubstituted butenolides.
The first enantioselective, organocatalytic, and direct VAR reaction between dihalofuran-2(5H)-ones and aldehydes to give polyfunctionalized butenolides was reported by Terada’s group in 2010, using an axially chiral guanidine derivative as the catalyst (Scheme 117, eq 1).302 Catalyst C14, featuring a guanidine functionality (bearing a sterically demanding benzhydryl substituent Ar embedded into an axially chiral binaphthyl core), was found to be the most efficient tool to promote the enantioselective VAR between pronucleophilic dibromofuranone 426a and several differently substituted aromatic aldehydes of type 427. The reaction, performed in a 1:1 mixture of acetone/THF at −40 °C for at least 5 h, yielded the corresponding γ-homologated (5S,1′R)-configured products 428 with acceptable to good yields (up to 95%), and high diastereo- and enantiocontrol (up to 15.7:1 dr, up to 99% ee). Also dichlorinated furanone 426b proved to be a viable substrate in the VAR addition to benzaldehyde, yielding the corresponding, enantiopure adduct 429 with a high 90% yield even though the diastereocontrol was somehow hampered (3.3:1 syn/anti) by the less steric bias of the chlorine atoms with respect to bromine. To account for the observed stereoselectivity, a transition state was proposed, in which the guanidinium ion orchestrated the Si–Si approach of the dienolate to the aldehyde by hydrogen-bonding interactions.
Scheme 117.

More recently, Dudding and co-workers303 reported the development of a new, chiral, computationally designed seven-membered ring guanidine C15 as an effective organocatalyst for the asymmetric VAR between dibromofuranone 426a and a small panel of aromatic and aliphatic aldehyde acceptors 430 (Scheme 117, eq 2). The reaction carried out in THF at −10 °C yielded optically active, dibrominated γ-butenolides 431 in low to moderate yields (up to 66%), with acceptable diastereo- and enantioselectivity in favor of the (5R,1′S)-configured isomers.
The fruitful versatility of 3,4-dihalofuranones of type 426a and 426b was also exploited in 2011 by Lu and co-workers,304 who reported a diastereo- and enantioselective, direct VAR toward α-ketoesters catalyzed by the tertiary amine-thiourea bifunctional organocatalyst C16, accessing enantioenriched γ-substituted butenolides with the creation of a quaternary stereocenter (Scheme 118). Indeed, after an extensive catalyst screening, the authors found that l-tryptophan-derived catalyst C16 efficiently catalyzed the reaction between 3,4-dichlorofuran-2(5H)-one (426b) and a set of differently substituted tert-butyl glyoxylates of type 432 (Scheme 118, eq 1).
Scheme 118.

Consistently high diastereo- and enantioselectivities were observed for a wide range of aromatic, aliphatic, vinylic, and allyl-substituted α-ketoesters, accessing the corresponding γ-butenolides 433 in high yields with up to 32:1 dr in favor of the (5S,1′S)-configured products (up to 95% ee). Interestingly, the nonhalogenated furanone could also be used, although a much longer reaction time and a slightly lower enantioselectivity was observed (144 h; 69% yield, 10.1:1 dr, 82% ee, not shown). To account for the stereochemical outcome of the developed VAR, a transition-state was proposed (Scheme 118, bottom), highlighting the key role of hydrogen-bond interactions between the ketoester and the thiourea moiety of the catalyst orchestrating the selective attack of the C-γ of the dienolate to the Si face of the ketone, leading to the formation of the major stereoisomer. If the chlorine atoms were replaced by bulkier bromines as in 426a (Scheme 118, eq 2), an overall increase in diastereoselectivity was observed, probably due to tighter ion pairing between the catalyst and the dienolate, as a result of higher electron density on the enolate oxygen atom.
The just described direct VAR approaches involved the formation of active chiral quaternary ammonium dienolates as the key nucleophilic partners, as a result of the C-γ deprotonation of the dihalogenated (5H)-furan-2-ones operated by the catalyst. In this context, another attractive approach was proposed by Oudeyer, Levacher, and co-workers in 2013,305 who reported an asymmetric, direct, and anti-selective VAR of substituted (5H)-furan-2-ones of type 436 using a chiral quaternary ammonium aryloxide/N,O-bis(trimethylsilyl)acetamide (BSA, 439) couple as promoters (Scheme 119). This strategy relies on the in situ formation of chiral salt 439′ obtained by the combination of a silylated base such as 439 with the quinine-derived chiral ammonium salt directly accessible via in solution ion methatesis between the corresponding ammonium bromide C17 with sodium 4-methoxyphenoxide. The desired chiral quaternary ammonium dienolate 436′ is then obtained by the regioselective C-γ deprotonation of butenolide 436 which then may add to aliphatic or (hetero)aromatic aldehyde acceptors 437 to give the corresponding chiral δ-hydroxy butenolides 438 in good diastereomeric ratios (up to 19:1) and excellent enantioselectivities (up to 94% ee). Of note, despite the fact that unsubstituted (5H)-furan-2-one proved to be a viable pronucleophile giving the corresponding VAR adduct to benzaldehyde with comparable efficiency and stereoselectivity, the 4-methyl congener (not shown) reacted with benzaldehyde much less efficiently, with a poor 28% yield though with a good level of enantioselectivity (83% ee).
Scheme 119.

The first direct, organocatalyzed enantioselective VAR of an unsubstituted (5H)-furan-2-one using a cinchona alkaloid-based thiourea was achieved by Feng and co-workers in 2010 (Scheme 120, eq 1).306 After an extensive screening of bifunctional tertiary amine-thioureas, 9-epi-quinine thiourea C2 was elected as the catalyst of choice to guide the reaction between (5H)-furan-2-one 440 and a panel of differently substituted aromatic aldehydes 441 (with the sole exception of aliphatic cyclohexyl carbaldehyde) in Et2O at 30 °C, to provide anti-configured butenolides (5R,1’S)-442 with good yields (up to 93%) and satisfactory levels of diastereo- (up to 5.7:1 anti/syn) and enantioselectivity (up to 83% ee).
Scheme 120.

Inspired by Feng’s work, slightly better results were achieved by Hirashima and Miura in 2017 with a novel polydentate cinchona-squaramide organocatalyst C18 bearing a chiral perfluorobutane sulfonamide group at one end (Scheme 120, bottom).307 Indeed, under the optimized reaction conditions (10 mol % catalyst C18 in Et2O at rt), excess of pronucleophilic furanone 440 selectively added to a set of almost exclusively aromatic aldehydes of type 441, affording the corresponding anti-configured-γ-butenolides 442 in high yields (up to 96%) and high to excellent diastereo- and enantioselectivities (up to 13.3:1 dr, up to 97% ee). Although the transition states of the above direct VARs are still to be completely elucidated, the rationalization of the observed high stereocontrol could be found in the H-bond networking these multidentate H-bond donor catalysts engage with both the dienolate and the aldehyde components in a stereodefined manner. In this context both groups proposed similar transition states, in which the aldehyde component is activated through a directed double hydrogen bonding operated by either the thiourea moiety in C2 or the mixed squaramide-sulfonamide nitrogen atoms in C18; the quinuclidine moiety in the catalyst deprotonates furanone 440 to generate the corresponding dienolate intermediate whose selective attack to the Re face of the aldehyde is well orchestrated by the chiral backbone of both catalysts. The enantioselective, direct, VAR between furanone 440 and various aromatic aldehydes was also studied by Pansare and co-workers, who screened several bifunctional amino-thiourea and amino-squaramide organocatalysts to provide diastereomerically and enantiomerically enriched δ-hydroxy-5-substituted furanones of type 442 (Scheme 121, eq 1).308 Pansare’s survey unveiled the superior performance of the Rawal cyclohexanediamine squaramide C19 and stilbenediamine squaramide C20 over conventional aminothiourea catalysts, in accessing enantiopure, anti-configured butenolides 442 in good yields (up to 73%) and good diastereoselectivities (up to 8:1 dr). Subsequently, Pansare exploited the developed direct VAR to construct chiral, enantiopure δ-hydroxy butenolide adducts which served as the key starting materials in the total synthesis of several pharmaceutically relevant targets (Scheme 121, eqs 2 and 3).
Scheme 121.

In particular, benzaldehyde-derived adduct ent-442a, obtained from the direct VAR between 440 and benzaldehyde (441a) promoted by 20 mol % of squaramide ent-C20 (Scheme 121, eq 2) was used in the preparation of neurokinin receptor antagonists (+)-L-733,0606, (+)-CP-99,994, as well as (2S,3R)-3-hydroxypipecolic acid.309 Dioxolane derivative ent-442b, instead, served as key intermediate for the synthesis of antimalarial agents (+)-febrifugine and (+)-halofuginone (Scheme 121, eq 3).310
Finally, the versatility of α,β-unsaturated γ-butyrolactones as powerful pronucleophiles in the direct and γ-selective aldol condensation to aldehydes was further demonstrated by several successful examples of organo- and metal-catalyzed procedures giving access to condensed γ-ylidene-butenolide frameworks: due to the nonasymmetric nature of such processes though, these examples will not be commented on in this review.311−313,11 Switching to deconjugated β,γ-unsaturated butenolides, only a few reports appeared with the direct, catalytic VAR to functionalized ketones, which can offer the opportunity to construct challenging tertiary alcohols with vicinal tetrasubstituted stereogenic centers.314
Despite the interest arisen about α-angelicalactones as vinylogous pronucleophiles,35 only recently Feng and co-workers reported the first asymmetric VAR of a series of β,γ-unsaturated butenolides of type 443 to polyfunctionalized isatins 444, exploiting a C2-symmetric l-proline derived N,N’-dioxide ligand L9 in combination of Sc(OTf)3 as the most suited catalytic system (Scheme 122).315 Indeed, the vinylogous addition between 443 and 444, carried out in THF at 30 °C (for at least 72 h) in the presence of 3 Å MS, was perfectly orchestrated by the L9·Sc(III) complex, providing a series of enantiopure tertiary alcohols 445 containing not only the γ,γ-disubstituted butenolide motif but also the challenging tetrasubstituted oxindole framework. Based on the experimental investigations, a possible transition state was proposed in which L9 binds to the central scandium ion in a tetradentate manner to form two stereodefined six-membered chelate rings (Scheme 122) that efficiently guide the Re-Si approach between the C-γ of the in situ formed dienolate 443′ to the C3 carbonyl of isatin 444, affording products 445 with very high stereocontrol (up to >19:1 dr, and up to 99% ee).
Scheme 122.

An interesting direct, bisvinylogous aldol reaction (BVAR) of methyl 5-methylfuroate (446) toward either aldehydes 447 or a small panel of methyl ketones 449 was developed by Sammakia et al. applying a modified version of the Yamamoto’s protocol (Scheme 123, eqs 1 and 2).316 Furoate 446 is a quite interesting pronucleophile, since deprotonation of its exocyclic methyl group to generate the trienolate intermediate 446′ may be hampered by the presence of the relatively stable furan ring between the methyl and the ester group. Furthermore, trienolate 446′ bearing a highly reactive 2,5-dimethylene-2,5-dihydrofuran moiety, might in principle add to the activated aldehyde or ketone at the competing α, γ, or ε position, posing the issue of the regiocontrol of the reaction. After a brief optimization survey, Sammakia found that ATNP (M4, 3.3 equiv) with lithium tetramethylpiperidine (LTMP, 3.5 equiv) at −78 °C in 10:1 toluene/THF, promoted the reaction between furoate ester 446 and aldehydes 447 or ketones 449, to provide the corresponding aldol adducts (±)-448 or (±)-450 with moderate to high yields as the sole ε-adducts. (Interestingly, with several aliphatic, nonenolizable aldehydes, also Yamamoto’s bulky Lewis acid ATPH M3 was an effective promoter providing the products in high yields.) Of note, the reaction proved to be viable to a wide range of functionalized carbonyl acceptors, so that aromatic and enolizable aliphatic aldehydes and methyl ketones reacted with furoate 446 under the optimized reaction conditions. β-Alkoxy chiral aldehyde acceptors were also tested (not shown), giving the corresponding aldol adducts in acceptable yields albeit with low dr.
Scheme 123.

Among the varied repertoire of readily available cyclic lactone-type derivatives to be used as pronucleophilic vinylogous scaffolds in stereoselective transformations, azlactones (and in particular 4-alkylidene-oxazolones)317 are among the most useful starting materials for the synthesis of α-amino acids and heterocyclic scaffolds. However, until recently, the use of olefinic azlactones as pronucleophilic synthons in vinylogous aldol-type transformations remained elusive (vide infra). In this context, one of the few examples is given by the organocatalyzed, asymmetric formal hetero-Diels–Alder (HDA) reaction of olefinic azlactones of type 451 to isatins 452 achieved by Hu and co-workers via hydrogen-bond directed γ-addition (Scheme 124).318 This methodology provided an efficient access to S-configured spirooxindole dihydropyranones of type 453 in moderate to good yields (up to 88% yield) and excellent enantioselectivities (up to >99% ee). The reaction, catalyzed by bifunctional β-isocupreidine C21 (β-ICD, 20 mol %) was carried out in 1,2-dichloroethane at 30 °C and was quite efficient irrespective of the nature and position of the substituents on both the azlactone and the isatin substrates. A mechanism was also postulated envisaging both reaction partners to be synergistically activated by the bifunctional catalyst C21 to reach a stereodefined transition state in which a hydrogen-bonding network selectively guides the attack of azlactone dienolate 451′ on the Si face of isatin carbonyl. The formed aldolate adds then back to the azlactone carbonyl forming a [4 + 2] cycloadduct intermediate 451′′ which finally collapses to give the desired product 453 regenerating the catalyst.
Scheme 124.

A similar protocol was reported some years later by the same group (Scheme 125),319 who developed an efficient asymmetric [4 + 2] annulation between alkylidene azlactones of type 451 and α-keto esters 454 using chiral cinchonine-thiourea C22 as the bifunctional organocatalyst. Using this protocol, a wide range of the desired S-configured dihydropyranones 455 with a quaternary stereocenter were produced in generally good yields (up tp 88%) and excellent enantioselectivities (up to >99% ee). Here, the bifunctional thiourea catalyst enabled the synergistic activation of both reaction partners by forming a hydrogen bond-stabilized transition state in which the in situ formed dienolate 451′ was supposed to react on the Re face of the α-ketoesters 454 in a concerted or stepwise VAR to reach the desired product 455.
Scheme 125.

5.1.2. Indirect Procedures
5.1.2.1. Acyclic Nucleophiles
In 2010, Qu and co-workers reported the use of a novel BINOL–titanium–LiCl heterobimetallic complex (BTHL, M5, Scheme 126) as a useful catalyst to promote the enantioselective vinylogous Mukaiyama aldol reaction (VMAR) between Brassard’s diene 456 and a set of aromatic aldehydes of type 457 (Scheme 126, eq 1).320 In the optimized procedure, the catalytic system M5 (5 mol %), generated in situ from (R)-BINOL, Ti(Oi-Pr)4, H2O, and lithium chloride, carefully added in a 1:1:1:1 ratio, efficiently catalyzed the enantioselective VMAR affording the sole Z-configured (5R)-5-hydroxy-α,β-unsaturated ester derivatives 458 in good yields (up to 77%) and excellent enantioselectivities (up to 99% ee). Furthermore, VMAR addition of 456 to a more challenging aliphatic isobutyraldehyde (not shown) was also investigated, affording the corresponding adduct with 96% ee albeit with scarce efficiency (16% yield). A couple of years later,321 Qu exploited the same heterobimetallic catalytic system M5 in the development of a highly enantio- and diastereoselective VMAR between prostereogenic propionyl acetate-derived diene 459 and a set of aromatic and aliphatic aldehydes of type 460 (Scheme 126, eq 2). The reaction, carried out in the presence of catalyst M5 (10 mol %), yielded chiral, enantiopure δ-hydroxy-γ-methyl-β-methoxy acrylates of type 461 (up to 99% ee), in high yields (up to 96%) and almost complete diastereocontrol in favor of the Z-syn isomer (up to 99:1 dr). Of note, despite a strong positive nonlinear effect (NLE) being unveiled, suggesting a more complex active oligomeric titanium species in solution, a “Lewis acid assisted Lewis acid” model was proposed in which the lithium ion enhanced the Lewis acidity of the titanium center through associative interactions, by coordinating the oxygen of a μ-oxo bis-titanium complex, and allowing the metal to interact with the aldehyde carbonyl, discriminating its prochiral faces. Meanwhile, the diene would coordinate the weakly acidic lithium center in a chelating manner moving close to the aldehyde along a Si–Re trajectory (Scheme 126, bottom). Finally, this methodology was successfully exploited in the formal synthesis of polypropionate cystothiazole A and melithiazole C, a group of fungicidal β-methoxy (E)-acrylate antibiotics featuring a polypropionate chain attached to a thiazole or bis-thiazole core (not shown).
Scheme 126.

The development of catalytic, enantioselective VMAR involving linear, simple ester-derived dienol ethers such as silylketene acetal 462 (Scheme 127) is an issue of high importance in organic chemistry as it provides an efficient assembly of δ-hydroxy-α,β-unsaturated carbonyl compounds of type 465 and related polyketide frameworks, which are attractive targets for biology-driven research. Furthermore, it generally proved to be challenging since the absence of substituents in the α or β position may result in a lower α vs γ regioselectivity, or more pronounced product isomerization.
Scheme 127.

In this context, Feng’s group reported a catalytic, enantioselective VMAR promoted by a chiral N,N’-dioxide–indium salt complex between linear methyl crotonate-derived silyl ketene acetals 462 and differently functionalized aldehydes 463 (Scheme 127, eq 1).322 In particular, after an intense preliminary work, L-pipecolic acid-derived ligand L10 complexed with In(OTf)3 in a 2:1 ratio was elected as the best catalyst system for guiding the reaction between 462 and 463 in ethyl caproate as solvent at −20 °C, in the presence of 5-methyl salicylic acid 464 (20 mol %), to yield the corresponding γ-adducts 465 in high yield (up to 99% yield) and enantioselectivity (up to 98% ee). Irrespective of the nature and position of the substituents, both aromatic and α,β-unsaturated aldehydes proved to be viable substrates, together with linear and branched aliphatic aldehydes (albeit with less efficiency and enantioselectivity).
More recently, Feng et al. reported the application of chiral N,N′-dioxide/Y(OTf)3 and Sc(OTf)3 complexes as efficient catalysts for the bisvinylogous Mukaiyama aldol reaction of linear silyl ketene acetals 466 and 469 with α-ketoesters 467 and aromatic or aliphatic aldehydes 463, respectively (Scheme 127, eqs 2 and 3).323 It was found that l-proline derived ligand L9 (Scheme 122) in combination with Y(OTf)3 (1:1 molar ratio), efficiently catalyzed the enantioselective and ε-selective bisvinylogous aldol addition of trienolsilyl ketene acetal 466 to a vast panel of aromatic and aliphatic α-ketoesters of type 467. The corresponding polyunsaturated esters 468 were obtained in up to 95% yield and up to 98% ee, with very high >19:1 ε/α regiocontrol in most cases. Similarly, 10 mol % of L-ramipril derived ligand L11 in combination with Sc(OTf)3 was the best choice for the aldol addition of silyl trienolate 469 to aldehydes 463 (Scheme 127, eq 3). The reaction carried out in pentyl hexanoate as the solvent at −30 °C with the addition of 20 mol % of salicylic acid 130 provided the corresponding S-configured aldol adducts 470 in good overall yields (up to 95%) and stereoselectivity (up to 96% ee). The reason for the observed different specificity of metals toward aldehydes and α-ketoesters was postulated to be related to the fact that the bigger Y(III) atomic radius would better coordinate the bidentate α-ketoesters, while monodentate aldehydes would better coordinate with smaller Sc(III). A transition state, resembling the one proposed for the VMAR of cyclic butenolides to isatins (Scheme 122) was also proposed accounting for the selective Si face attack to the aldehyde (not shown).
In 2011 List and co-workers demonstrated the ability of axially chiral disulfonimide DSI (D1) to efficiently catalyze the vinylogous and bisvinylogous Mukaiyama aldol addition of linear silyl ketene acetals 471 and extended silyl congeners 474 to a wide set of aldehydes, via an asymmetric counterion-directed catalysis mechanism (ACDC, Scheme 128).324 Irrespective of the dienolate geometry, Z/E mixtures of silyl ketene acetal 471 (eq 1) reacted smoothly with aromatic and cinnamaldehyde derivatives 472 in the presence of disulfonimide D1 (5 mol %) in Et2O al −78 °C (for at least 3 days) giving almost exclusively the silylated γ-adducts 473 in good yields (up to 96%) with acceptable to high levels of stereocontrol (up to 96% ee). It was also found that aliphatic aldehydes were suitable substrates with promising reactivity, although reduced stereoselectivities were observed.
Scheme 128.

Comparable results were observed by applying a similar methodology between extended silyl trienolate derivatives 474 and aldehydes 472 (Scheme 128, eq 2). In this case, a catalytic, enantioselective, bisvinylogous Mukaiyama aldol reaction provided chiral ε-adducts 475 in acceptable yields (up to 75%) and enantiocontrol (up to 92% ee), but with a sensible drop of regioselectivity (up to 9:1 ε/α). This drop in regioselectivity was also predicted by density functional theory calculations (DFT) as reported in Figure 5: here, the reported Fukui’s values for the ε and α positions within extended trienolate 474a vary less than for the γ and α values of the corresponding vinylogous nucleophile 471a, envisaging a less distinct nucleophilic selectivity between the α and ε position.
Figure 5.

Calculated electron density values (Fukui’s numbers) of vinylogous nucleophiles as reported by List et al.
A 4-step ACDC pathway was proposed, in which chiral disulfinimide D1 actually acted as a precatalyst. According to this catalytic cycle (Scheme 129), D1 is initially activated by protodesilylation with the silyl ketene acetal involved in the reaction to form N-silyl disulfonimide complex D1′, that activates aldehydes 472 by O-silylation forming a chiral oxonium ion species 472′ coupled with the aza-anion of the chiral disulfonimide. At this point, a second molecule of silyl ketene acetal adds to chiral complex 472′ stereoselectively, to give silylated adducts 472′′ that finally release the products 473 while regenerating the silylated catalyst D1′.
Scheme 129.

An interesting advancement of the ACDC vinylogous process highlighted above was recently reported by List’s group unveiling a novel, catalytic, enantioselective VMAR between silyl alkynyl ketene acetals of type 476 and aryl aldehydes 477 (Scheme 130).325 For the purpose, a newly designed chiral disulfonimide D2 was elected as the best catalyst to deliver chiral tetrasubstituted allenoates of type 478 in high yields (up to 92%), with excellent regio- (>20:1 γ/α), diastereo-, and enantioselectivities (up to 27:1 dr, and up to 97% ee). This conceptually new “alkynylogous” transformation was challenging as it implied the selective γ-addition of an aldehyde acceptor through a nucleophilic enolate functionality transfer along a conjugated double and triple bond, generating a δ-hydroxy allenoate-moiety in a stereoselective manner. The developed reaction proved to be feasible to a broad range of aldehydes in combination with diverse alkynyl-substituted ketene acetals, and the obtained allenoate products were suitable substrates for a variety of further derivatizations accessing highly substituted enantiomerically enriched building blocks (not shown).
Scheme 130.

As the above methodologies demonstrate, the asymmetric VMAR is a powerful carbon–carbon bond-forming transformation with broad applicability that allows the construction of polyfunctionalized carbon chains in a convergent and stereoselective modality. Consequently, several syntheses were put forward in which the vinylogous Mukaiyama aldol reaction was successfully applied.
Polyketides, for example, proved to be attractive targets for biology-driven research.34,326 The established strategies for the total synthesis of polyketides often mimic the biosynthetic pathway by adding one propionate or acetate building block at a time. In this context, to reduce the number of transformations during a given synthesis, much effort has focused on the exploitation of vinylogous aldol or aldol-like processes which enable the stereoselective access to δ-oxy γ-homologated α,β-unsaturated ester derivatives with reliable predictability. In this context, chiral oxazaborolidinones (OXB)-promoted VMAR of linear silyl dienolates represent a good example of how a well-established asymmetric methodology may be exploited to the total synthesis of polyketide frameworks. OXB were independently developed by Yamamoto and Helmchen in 1990,327,328 and since then they have been applied in various asymmetric transformations including the Mukaiyama aldol reaction. Their utility in promoting enantioselective VMARs was first reported by Kalesse only in 2007 (e.g., section 3.1.2.1).85 In the past decade though, the usefulness of this chiral OXB-catalyzed VMAR was further assessed and exploited in natural product synthesis as demonstrated by Smith’s group in their total synthesis of (+)-irciniastatin A) and (−)-irciniastatin B, two cytotoxic secondary metabolites (Scheme 131, eq 1).329
Scheme 131.

To access the key tetrahydropyran precursor, the enantioselective VMAR between aliphatic α,α-disubstituted aldehyde 479 and linear silyl ketene acetal 471a was performed using L-tryptophan-derived B-phenyl-oxazaborolidinone M6 as the chiral promoter. Using Kalesse’s procedure (1 equiv of M6 in butyronitrile at rt), the corresponding vinylogous adduct 480 was obtained in good 66% yield and absolute enantiocontrol (>99% ee). More recently, in 2016 Kalesse used a similar reaction in the total synthesis of the highly potent anti-HIV natural product aetheramide A (Scheme 131, eq 2).330 These results could be rationalized by the originally proposed transition state for OXB-catalyzed VMARs (Scheme 131, bottom),30 according to which the observed enantioselectivity is based on the selective attack of the silyl nucleophile to the Re face of the aldehyde due to the shielding of the Si face operated by the indole moiety of the promoter.
Among the plethora of stereoselective transformations involving linear silyl ketene acetals as nucleophilic components, substrate controlled, diastereoselective VMARs represent one on the most exploited transformations for the construction of chiral polyketide frameworks since simple diastereoselectivities can be easily obtained through either Felkin or anti-Felkin controlled additions to α-chiral aldehydes. In this context, boron-centered Lewis acids proved to be superior catalysts in promoting diastereoselective VMARs, as in the case of Florence’s synthesis of the polyketide-derived macrolide Palmerolide C (Scheme 132, eq 1).331 Here, the C15–C24 subunit of the target was accessed through a syn-selective VMAR between 3-methyl silylbutenoate 483 and chiral aldehyde 484. Using BF3·OEt2 as a Lewis acid, the corresponding Felkin adduct 485 was obtained in a good 70% yield and 6:1 dr.
Scheme 132.

An additional level of complexity is added when nucleophiles exhibiting a terminal methyl group are employed. Such examples have been described by Kalesse et al. during the synthesis of the marine natural product (+)-tedanolide, a cytotoxic 18-membered marine polyketide isolated in 1984 (Scheme 132, eq 2). In 2012, Kalesse proposed an improved synthesis of tedanolide using a key diastereoselective VMAR between silyl ketene acetal (Z)-486 and chiral aldehyde 487, promoted by tris(pentafuorophenyl)borane B(C6F5)3 as the Lewis acid to afford the sole Felkin adduct 488 featuring an all-syn stereotriad, in 91% yield (>19:1 dr).332
The inherent directing effects of α-chiral aldehydes in Lewis acid-promoted diastereoselective VMARs were later investigated by Kalesse using silyl nucleophile (Z)-486 and Roche aldehyde 489 as model compounds (Schema 132, eq 3).333 Depending on the nature of the Lewis acid employed, three different stereotriads could be obtained, namely, the chelation-controlled (anti,syn)-490a, the (anti,anti)-490b, or the (syn,syn)-490c (Scheme 132, bottom). The highest selectivities were observed for the Felkin all-syn product 490c when Cy2BCl was used as the Lewis acid, proving again the superior ability of boron-centered Lewis acids to orchestrate the steroselective approach of the reagents.
In the field of catalytic, enantioselective Mukaiyama-type transformations, the concept of Lewis base activation of Lewis acids developed by Denmark in 1990 has been fruitfully applied to vinylogous transformations, and it is now one of the most reliable systems to promote enantioselective VMARs.334,335 Denmark’s catalytic system relies on the addition of a chiral Lewis base such as bisphosphoramide L12 (Scheme 133) to SiCl4 thus extending the silicon coordination sphere and generating a chiral catalytic complex with increased Lewis acidity that leads to successful activation of aldehydes while promoting for example enantioselective VMAR processes. Not surprisingly, it has found important applications in total synthesis, and few interesting examples involving the use of extended silyl ketene acetals as nucleophilic donors appeared recently.
Scheme 133.

In 2011, Nicolaou and co-workers applied Denmark’s L12·SiCl4 catalyst system during a synthetic and biological study on natural product monorhizopodin and its16-epi-analogue (Scheme 133, eq 1),336 which showed actin-binding properties without exhibiting cytotoxicity. Indeed, α,β-unsaturated aldehyde 492 treated with silyl ketene acetal 491a in the presence of 15 mol % of bisphosphoramide L12 and SiCl4 (1.1 equiv) afforded the corresponding optically pure secondary alcohol 493 in a good 69% yield and >90% ee.
Furthermore, in 2012 Winssinger and co-workers investigated a similar VMAR between dienolsilane 491a and the challenging crotonaldehyde substrate 494 during the syntheses of HSP90 inhibitors pochonins E and F (Scheme 133, eq 2).337 Again, chiral bisphosphoramide L12·SiCl4 complex promoted the enantioselective formation of the advanced intermediate 495 in 60% yield and excellent enantioselectivity (>94% ee).
Several studies by the Denmark group accounted for a transition state model in which the aldehyde binds to the hypervalent silicon center trans to one of the phosphoramides (Figure 6). This conformation places the aldehyde against one of the binaphthyl units and consequently allows exposure of the aldehyde’s Re face toward nucleophilic attack. Additionally, potential edge-to-face interactions between the aromatic aldehydes and the aromatic rings of the ligands can be used to rationalize the higher selectivity observed for unsaturated aldehydes as compared to simple aliphatic aldehydes.
Figure 6.

Catalytic complex involving Denmark’s SiCl4-bisphosphoramide L12 and an enal.
5.1.2.2. Cyclic Nucleophiles
In 2010, the Bolm group reported the development of a previously introduced copper-catalyzed enantioselective vinylogous Mukaiyama-type aldol reaction between 2-(trimethylsilyloxy)furan (TMSOF, 496a) and bidentate α-keto esters 497 affording the corresponding δ-hydroxy butenolides 498, bearing a quaternary stereogenic center bridging the lactone core to the carboxylate unit (Scheme 134).338,339 After a fine-tuning of the reaction conditions and an optimization of the modularly assembled ligand structure, several C1-symmetric aminosulfoximine L13/Cu(OTf)2 couples were tested, providing the desired products with high stereoselectivities (up to >99:1 dr and up to 99% ee) and excellent yields (up to 99%). Furthermore, this catalytic, enantioselective VMAR tolerated various electrophile/nucleophile combinations, spanning from S-, N-, and C-analogues of TMSOF to various alkyl and benzyl substituted keto esters 497, affording the corresponding thiobutenolide, lactam, and functionalized carbocycles in comparable yields and stereoselectivities (not shown).
Scheme 134.

The absolute configuration of the products was assigned as (5R,1′R), and the observed enantioselectivity was ascribed to a preferential attack of the Si face of TMSOF 496a to the Re face of activated electrophile 497′ accounting for a 5-membered cyclic bidentate complex in which copper coordinates the two carbonyl oxygen atoms of the keto ester.
Similarly to the previously accessed chiral esters, chiral α-functionalized phosphonic acid derivatives have attracted particular attention in the past decade, due to their valuable pharmaceutical applications as anticancer and antivirus agents.340 The pharmaceutical potential of these compounds has stimulated the development of several methodologies for the preparation of δ-hydroxy alkylbutenolide phosphonate analogues, accessible via stereoselctive VMARs using mainly TMSOF 496a as the lactone nucleophilic source. In this context, in 2011, Miao, Chen, et al. were the first to report a diastereoselctive VMAR between bidentate α-keto phosphonate 499 and 2-(trimethylsilyloxy)furan 496a catalyzed by Cu(OTf)2 (5 mol %) in the presence of 2,2,2-trifluoroethanol (TFE, 1.2 equiv) as additive in CH2Cl2 at 0 °C (Scheme 135, eq 1).341 The reaction proceeded rapidly affording the corresponding (O,P)-anti-configured 5-(hydroxyarylmethyl) furan-2(5H)-one phosphonates (±)-500 in high yields (up to 89%), with good to excellent diastereoselectivities (up to >99:1 dr). Similarly to the previously described work, the reaction was proposed to proceed via a staggered acyclic transition state in which Cu(OTf)2 coordinates the two oxygen atoms of the α-keto phosphonate (e.g., complex 499′), creating suitable steric effects that dictate the diastereoselective approach of the reactants.
Scheme 135.

Later in 2013, the same group devised an asymmetric version of such transformation exploiting a C2-symmetric bis(oxazoline) L14·Cu(OTf)2 catalyst complex (Scheme 135, eq 2).342 Indeed, the asymmetric VMAR between α-keto phosphonates 499 and TMSOF 496a carried out in dichloromethane at −78 °C in the presence of TFE as additive afforded the corresponding tertiary α-hydroxy phosphonates 500 in moderate to high yields (up to 86%), as well as high enantio- (up to 98% ee) and diastereoselectivities (up to 99:1) in favor of the (5R,1′R) configured isomers as assessed by X-ray crystallographic determination.
Almost in the same period, a very similar catalytic and highly stereoselective copper-catalyzed VMAR between α-keto phosphonates 499 and functionalized heterocyclic dienol silyl ketene acetals 501 was developed by Bolm and co-workers, exploiting the just mentioned C1-symmetric L13a·Cu(OTf)2 chiral catalytic complex (Scheme 135, eq 3).343 Here, (5R,1′S)-configured phosphonic γ-(hydroxyalkyl)butenolides 502 (and one N-Me lactam congener, not shown) were accessed with high yields and stereoselectivities showing a broad functional group tolerance for both the electrophilic and nucleophilic reactants.
The ability of siloxyfuran nucleophiles to act as d4 donor reagents in vinylogous, enantioselective aldol, and related processes was demonstrated by Casiraghi’s group, who investigated the VMAR of pyrrole- and furan-based dienoxysilanes in a catalytic, asymmetric format. In this context, the previously disclosed Denmark bisphosphoramide L12/SiCl4 couple proved to be a privileged catalytic system in promoting the enantioselective VMAR between a suitable silyloxyfuran derivativative and a set of aromatic aldehydes (not shown).344,345 Several years later, Curti, Del Rio, et al. applied Casiraghi’s methodology to the total synthesis of hydroxyphenyl γ-valerolactones, an important class of flavan-3-ol colonic metabolites (Scheme 136).346,347 Starting from 2-triisopropylsilyloxyfuran 496b and diverse alkoxy-substituted benzaldehydes 503, Denmark’s catalytic system L12·SiCl4 enabled the formation of the corresponding δ-hydroxybutenolides 504 in high yields (up to 98%) and generally good to high enantioselectivities. Interestingly, the diastereoselectivity of the process was highly dependent upon the nature and the substitution pattern of benzaldehydes 503; in fact, while 3′- and 4′-monosubstituted aldehydes yielded the expected anti-adducts 504, 3′,4′-bisilyloxy and 3′,4′,5′-trisilyloxybenzaldehyde congeners mainly furnished the C5-epimeric syn-configured adducts. Unexpectedly, while the majority of butenolide products 504 possessed the expected (1′S) absolute configuration (resulting from an attack on the expected Re face of the aldehyde), a striking and unprecedented enantiofacial inversion was experienced with polymethoxy- and polybenzyloxy-substituted aldehydes, for which (1′R)-configured anti-adduct were obtained (Si face attack). Selected δ-hydroxybutenolides 504 were then chosen as key precursors for the chemodivergent synthesis of chiral, enantioenriched hydroxyphenyl γ-valerolactone metabolites, as well as their racemic δ-valerolactone congeners (not shown).
Scheme 136.

In their continuing efforts to study the reactivity of vinylogous nucleophiles such as silyloxyfuran derivatives, the Casiraghi’s group also explored more extended adaptations of the classical heterocyclic siloxydienes. Focusing on densely unsaturated butenolides, in 2011 they reported a reliable catalytic, asymmetric bisvinylogous and hypervinylogous Mukaiyama-type aldol methodology using easily available extended furan-based silyloxy polyenes of type 505 and 508 (Scheme 137).348 Initially, the asymmetric bisvinylogous VMAR using trienolsilane 505 and a set of aromatic aldehydes 506, carried out in the presence of the Denmark’s bisphosphoramide L12·SiCl4 catalyst system and DIPEA (10 mol %) in CH2Cl2 at −78 °C, gave bisvinylogous 3′-hydroxybutenolides 507 with extraordinary ε-selectivity, in high isolated yields (up to 96%), and generally good to very high enantioselectivites (up to 99% ee) in favor of the (E,3′R)-configured isomer. This procedure was further applied to the more demanding VMAR between silyl polyenolates 508 and 4-bromobenzaldehyde 506a (Scheme 137, eq 2), demonstrating a perfect relay of the enolate reactivity over up to five conjugated double bonds and named this phenomenon “hypervinylogy”. This novel hypervinylogous Mukaiyama aldol reactions (HVMARs) displayed complete regioselectivity at the most remote ω-nucleophilic site of the substrates (ω vs α/γ/ε), good to moderate control of the product geometries (E vs Z), and excellent enantiocontrol (>96% ee). These findings contrasted with the previously reported results by List et al.324 (Scheme 128), where a sensible drop of regioselectivity was observed for the DSI-catalyzed bisvinylogoua VMAR of linear silyltrienolates. To account for the observed regioselectivity, DFT and Fukui functions calculations were performed on silyl (poly)enolate nucleophiles 505 and 508 (Scheme 137, bottom). Atomic Fukui indices at the carbon atoms of the reacting polyenes were in line with the data obtained by List, foreseeing a preferential electrophilic attack of the aldehydes at the terminal carbon atom of the silyloxy nucleophiles.
Scheme 137.

Switching the attention to the role of the medium (or solvent) in promoting vinylogous transformations, the past decade has witnessed a renewed interest toward alternative and potentially “greener” organic chemistry methodologies based on the use of safer and environmentally friendly reagents, catalysts, and reaction media. Water represents an interesting alternative to the more classical organic solvents due to its unique structure-dependent behavior that enhances hydrophobic interactions within transition states, even when the solubility of the reactants in water is low and the reaction takes place at the solid/liquid or liquid/liquid interfaces (in a pioneering work Sharpless introduced the expression “on-water conditions” to denote the rate acceleration observed in organic reactions when water-insoluble reactants are vigorously stirred in water suspension).349
In this context, Curti et al. and, more recently, De Rosa, Palombi, and co-workers demonstrated the feasibility of a diastereoselective “on water” VMAR applied to furan-based siloxydienes 496c and 496a (Scheme 138). In particular, in 2010, Curti, Casiraghi, et al. reported the first, uncatalyzed VMAR carried out “on water” conditions, using a salty water/methanol mixture as reaction medium (Scheme 138, eq 1).350 Ultrasonic irradiation of an emulsion of TBSOF (496c) and aromatic and cinnamic aldehydes 510 in a 1:1 brine/methanol mixture at 38–40 °C allowed the formation of the corresponding δ-hydroxy butenolides (±)-511 in high yields and good to excellent diastereoselectivity in favor of the 5,1′-syn configured adducts. Interestingly, the reaction between pyrrole-based dienoxysilane congeners and aromatic aldehydes proved also to be viable under the optimized condition, giving the corresponding vinylogous adduct in high yield but with an inverted anti-selectivity (see section 6). To account for the observed syn-selectivity, the authors speculated that, at the boundary between water and the dispersed droplets of the lipophilic reactants, water molecules worked as H-bond donor species that activate the aldehyde acceptor while controlling the mutual position of the reactants in a stacking synclinal transition state, ultimately leading to the favored syn-isomers (±)-511.351
Scheme 138.

Later, as part of a long-lasting venture toward unusual, environmentally friendly organocatalytic systems to promote stereoselective vinylogous transformations under solvent-free or aqueous media,352−354 De Rosa, Neri, and co-workers designed calixarene-based catalysts C23a–c (Scheme 138, bottom) to promote diastereoselective VMARs between TMSOF 496a and α-keto esters 512 under “on water” conditions. In a first report in 2016,355 they found that thioureido-calix[4]arene C23a or its calix[6]arene congener C23b (5 mol %) enabled the VMAR between 496a and a series of alkylbenzoylformates to yield the corresponding γ-adducts (±)-513 in acceptable yields with a slight preference for the 5,1′-anti-configured isomers (Scheme 138, eq 2). Interestingly, the authors found that lower conversions and a switch in diastereoselectivity in favor of syn-adducts could be observed when the same reaction was carried out in organic solvents under homogeneous catalysis. These observations and further NMR and computational studies unveiled the basis of the supramolecular control exerted by this type of catalysts, according to which the rate acceleration of the VMAR is closely related to the hydrophobicity of the calixarene skeleton and its ability to recognize the α-keto ester via H-bonding interactions with the thiourea group. The hydrophobic amplification of weak interactions between catalyst and substrates under “on-water” conditions inspired the De Rosa group to search for new supramolecular entities able to promote a distereoselective on-water VMAR. In 2017 these efforts ended up with the design of the simple tetraminocalix[4]arene C23c (Scheme 138),356,357 bearing weak H-bond-donor NH2 functionalities, capable to promote the VMAR of TMSOF 496a with polyfunctionalized α-keto esters 512, to afford δ-hydroxybutenolides (±)-513 in good yields (up to 99%) and acceptable diastereoselectivities.
In the context of organocatalyzed asymmetric methodologies,358 in 2010 Ma and Wang developed the asymmetric, vinylogous addition of TMSOF 496a to diverse aromatic aldehydes 514, using the bifunctional 9-epi-quinine-thiourea C2 as the catalyst of choice (Scheme 139, eq 1).359 The reaction, performed with C2 (20 mol %) in CHCl3 at −20 to 0 °C afforded a panel of diverse chiral, 5,1′-anti-configured δ-hydroxybutenolides 515 in good yields (up to 90%), and stereoselectivity (up to 9:1 dr, and up to 91% ee).
Scheme 139.

An almost similar procedure was reported in the same year by the Deng group,360 who successfully exploited readily accessible organic catalysts based on a carboxylate ammonium salt such as C2·TFA (Scheme 139, eq 2) prepared by mixing C2 with trifluoroacetic acid (TFA) in a 1:1 ratio. This newly developed chiral organic salt was able to effectively promote an anti-selective asymmetric VMAR between TMSOF 496a and diverse aryl, alkenyl, and alkyl aldehydes of type 516, affording the corresponding butenolides 517 with improved yields (up to 98%) and stereoselectivities (up to >20:1 dr, and up to 95% ee) even with the more challenging aliphatic aldehydes. Furthermore, catalyst C2·TFA was able to promote the VMAR of structurally hindered 5-substituted-2-trimethylsilyloxy furans 518 and aldehydes 519 generating chiral adducts 520 bearing adjacent tertiary-quaternary centers with comparable selectivities (not shown). To account for the ability of C2·TFA salt catalyst in promoting the reported anti-selective VMAR, a transition state was proposed based on the peculiar structure of the catalyst featuring a protonated quinuclidine unit and a carboxylate moiety linked to the thiourea through hydrogen-bond interactions (Scheme 139, bottom). The author postulated that the carboxylate ion could react with silyloxy furan 496a to form the corresponding trimethylsilyl ester and the 2-furoxy anion 496a′ while releasing a thiourea-NH functionality that could serve as a hydrogen bond donor to activate the aldehyde acceptor. With the nucleophilic anion and aldehyde in place, the reaction between the two reactants might proceed generating the observed anti-adducts.
As for the previously described linear silyl ketene acetals, cyclic silyloxy dienes are invaluable tools with which a myriad of densely functionalized nonracemic molecules could be constructed. Furthermore, the vinylogous aldol-type addition of these nucleophiles to carbonyl compounds (VMAR) has become one of the main routes for the assembly of chiral, enantiopure carbon chains of polyketide and polyketide-like natural products. In this context, paralleling the above-described methodological efforts to develop new, metal- or organocatalyzed asymmetric VMARs, the past decade has witnessed the exploitation of such chemistry on chiral aldehyde substrates, for the synthesis of chiral, enantioenriched bioactive compounds.
A diastereoselective BF3·OEt2-catalyzed VMAR between the dioxinone-derived dienolsilane 521a and suitable polysusbstituted chiral aldehydes of type 522 was expoited by Hoye’s group in 2010 in the total synthesis of the cytotoxic macrolide (−)-callipeltoside A (Scheme 140, entry 1).361 Here, the corresponding vinylogous Felkin-adduct (5,6-syn)-523 was obtained in a very good 90% yield as a single isomer. Similarly, Yadav et al. (2012)362 and Fuwa et al. (2015, 2016)363,364 applied the methodology with comparable results in the synthesis of either the C1–C14 macrolactone core of (−)-callipeltoside A (Scheme 140, entry 2), or the cytotoxic14-membered macrolide (−)-lyngbyaloside B (entry 3). A favorable 1,3-induced diastereoselective VMAR was studied by Forsyth’s group in the effort to synthesize the natural product phorboxazole A (entry 4).365 Indeed, during the preparation of the C31–C43 domain of the target, a highly efficient VMAR between 521a and a 3,4-dioxygenated aldehyde acceptor was devised using a stoichiometric Ti(Oi-Pr)2Cl2 as a Lewis acid, affording the corresponding adduct 526 in a good 90% yield and 4.4:1 dr..
Scheme 140.

An alternative diastereoselective VMAR was devised by Britton and co-workers in 2013 and applied to the total synthesis of the natural product (+)-solistatin (Scheme 140, entry 5).366 This strategy envisaged the use of a chiral aldehyde bearing a single chlorine atom introduced via enantioselective organocatalytic α-chlorination that acted as an easily removable achiral auxiliary. The Felkin stereodirecting influence of the chlorine atom resulted in the formation of the corresponding vinylogous adduct 527 with a good 63% yield as a 2:1 mixture of diastereoisomers in favor of the anti-configured isomer. Conversely, an anti-Felkin diastereoselective VMAR was applied by Scheidt in 2011 for the total synthesis of okilactomycin, an antitumor antibiotic agent.367 Using Kruger and Carreira’s copper-catalyzed VMAR368 between 521a and a pseudoephedrine-derived chiral aldehyde acceptor, a key β-hydroxy dioxinone fragment 528 was assembled in 70% yield and 10:1 diastereomeric ratio (Scheme 140, entry 6).
Highly substituted dioxinone-derived silyl dienolates were also exploited in diastereoselective VMARs with chiral aldehydes demonstrating, once again, the high versatility exerted by this family of silyl nucleophiles frequently used for the stereoselective construction of complex chiral carbon chains. An important application of such versatility was assessed by Meyers in 2016 reporting a practical and convergent route to a very large set of macrolide antibiotics.369
Here, two key diastereoselective VMARs between Z-configured dioxinone 529a and α-quaternarized chiral aldehydes were reported, generating the 4,5-syn aldol adducts 533 and 534 under different reaction conditions (Scheme 141, entries 1 and 2). In fact, the use of chelating magnesium bromide etherate (entry 1) or ZnCl2 (entry 2) as Lewis acids both yielded the corresponding (4S,5R,6R)-configured stereotriad with optimal yields (91% for both) and comparable diastereoselectivities (16:1 vs >20:1). A highly syn-selective MgBr2-catalyzed diastereoselective VMAR of methyl-dioxinone 530a was also exploited by Mulzer et al. in the total synthesis of epothilone D (entry 3).370 The reaction of 530a with a suitable PMB-protected α-hydroxy aldehyde yielded the corresponding anti-Felkin adduct 535 as a single isomer in almost quantitative yield. Finally, a titanium-catalyzed VMAR of γ,γ-disubstituted silyl ketene acetal 531a to a suitable 3-substituted chiral aldehyde was reported by Paterson et al. in 2013371 in the total synthesis of the macrocyclic polyketide (−)-rhizopodin (entry 4); a selective 1,3-chelate mechanism was here observed, allowing access to the corresponding 5,7-anti-configured adduct 536 with very high diastereocontrol (20:1 dr).
Scheme 141.

Turning to furan-based silyloxydienes, in 2016 Chen, Yang, et al.372,373 envisaged that the diastereoselective VMAR between 2-methyl-TBSOF 537a and a 2-methyl chiral aldehyde could give access to the C22–C23 syn-δ-hydroxybutanolide framework of the bioactive nortriterpenoid propindilactone G (Scheme 142, entry 1). The reaction carried out in the presence of BF3·Et2O (1.2 equiv), afforded the corresponding γ-adduct 540 in 60% yield, as a 3:1 syn/anti diastereomeric mixture. Regrettably, the resulting Felkin-type stereochemistries of the newly generated C22 and C23 stereogenic centers were opposite to those found in the natural product, and thus an alternative strategy needs to be devised. In 2011 Romo374 and co-workers reported a convergent total synthesis of the marine toxin (−)-gymnodimine envisaging a late-stage VMAR between silyloxyfuran 537b and a challenging chiral cycloexanone precursor to stereoselectively link the butenolide framework to the macrocyclic core of the target molecule. Brief exposure (ca. 1 min) of a mixture of 537b and the ketone acceptor to TiCl4 led to a smooth addition reaction, to provide the corresponding δ-hydroxy lactone 541 as a 1.1:1 diastereomeric mixture in 61% yield (entry 2).375
Scheme 142.

A diastereoselective methodology for preparing the butenolide–butyrolactone and furan–butyrolactone units embedded in complex natural furanocembranoid diterpenes was developed by Reiser et al. in 2012 (Scheme 142, entry 3).376 After a brief optimization survey, a highly selective (>99:1 dr) boron-catalyzed VMAR of 538a with an enantiopure cyclopropylcarbaldehyde yielded the corresponding syn-configured butenolide 542 structure in 65% yield in accordance with a proposed Felkin–Anh type model. This target served as key precursor for the synthesis of the northeastern sector of furanocembranoid bielschowskysin.377
A remarkable application of a diastereoselective VMAR involving a siloxyfuran nucleophile was exploited by Han, Jiang, and co-workers in 2016 in their report on the asymmetric total synthesis of (+)-19-dehydroxyl arisandilactone A (Scheme 143).378 It was serendipitously found that the BF3·Et2O-catalyzed VMAR between 537a and the strained polycyclic chiral aldehyde 543 afforded the arisandilactone A-precursor 544, as a result of a one-pot tandem reaction in which a typical diastereoselective VMAR forming the Felkin intermediate 543′ was followed by an intramolecular oxa-Michael reaction that generated a new tetrahydrofuran ring by cleavage of the cyclopropane carbon–carbon bond. The newly generated stereocenters at C20, C22, and C23 within product 544 formed a syn/syn stereotriad that was later epimerized to the corresponding anti/syn platform found in the target, via a DBU-catalyzed isomerization of the butenolide moiety (not shown).
Scheme 143.

In 2011 Porco Jr. and co-workers reported the total synthesis of tetrahydroxanthone natural products blennolides B and blennolide C in racemic formats, centered on a VMAR-type addition of siloxyfurans to an in situ generated benzopyrylium ion (Scheme 144).379 After a concise optimization survey, treatment of hydroxy chromone 545 with diisopropylsilyl ditriflate in the presence of 2,6-lutidine led to the formation of a benzopyrylium intemediate 545’ that, treated with 2-trimethylsiloxyfurans 496a and 538b at −78 °C, cleanly led to formation of chromone butenolides of type (±)-546 in good yields (up to 93%) and diastereoselectivity (up to 15:1 dr) after base-promoted desilylation. The regio- and diastereoselectivity of the vinylogous additions were probed using computational studies, which suggested the involvement of (Re*-Si*) Diels–Alder-like transition states.380−383
Scheme 144.

Another smart application of a VMAR-type addition of a siloxyfuran nucleophile to an in situ-formed oxocarbenium ion was introduced by Anderson and co-workers in the final step of their enantioselective total syntheses of rubriflordilactone A,384,385 a nortriterpenoid natural product which has attracted recent attention due to its moderate anti-HIV activity coupled with low cytotoxicity (Scheme 145, entry 1). After surveying several model systems, it was found that unstable chloropyran intermediate 547, obtained by treating the corresponding lactol precursor with a mixture of thionyl chloride and ZnCl2 (not shown), smoothly reacted via formation of an oxocarbenium ion with siloxyfuran 537b in the presence of substoichiometric ZnCl2 to afford a 1:1 mixture of the butenolide–tetrahydropyran framework of rubriflordilactone A 548 along with its C23-epimer in a 71% combined yield.
Scheme 145.

A related butanolide–tetrahydrofuran framework is also present in the DEFG ring system of the parent rubriflordilactone B, whose synthesis has been challenged by Peng in 2015 (Scheme 145, entry 2).386 Here, upon treatment of the acetate precursor 549 with catalytic Bi(OTf)3, the in situ generated cyclic oxonium ion quickly reacted with nucleophilic silyloxyfuran 537a to give the addition product 550 in 35% yield with the expected (22S,23S)-syn configuration.
An interesting VMAR-type reaction involving the addition of TMSOF 496a to an in situ generated sulfenium ion appeared in the total synthesis of the anti-inflammatory agents (−)-cephalosporolide E and (+)-cephalosporolide F by Raghavan and co-workers in 2016 (Scheme 145, entry 3).387 Treatment of a mixture of α-chloro sulfides 551 (obtained by α-chlorination with N-chlorosuccinimide in benzene from the corresponding sulfide, not shown) with 496a in the presence of ZnBr2 afforded the corresponding γ-adduct 552 as a diastereomeric mixture of all possible diastereomers in a 10:2:1:1 ratio respectively in a combined 76% yield. Of all, the (5R,1′R)- and (5S,1′R)-configured epimeric couple was found to be the major product of the reaction in accordance to a Felkin–Ahn model in which the medium-sized alkyne substituent would eclipse the sulfenium ion, while the trimethylsiloxy furan would approach it from the Si face, opposite to the bulky OTBS group.
5.2. Additions to C=N Bonds
5.2.1. Direct Procedures
5.2.1.1. Acyclic Pronucleophiles
Concerning the vinylogous, nucleophilic addition to the C=N functionality, asymmetric and catalytic strategies that can directly functionalize linear, α,β-unsaturated esters at the γ-position by exploiting vinylogous Mannich-type transformations represent useful solutions in organic synthesis, since they may provide γ-homologated δ-amino-derivatives in a chiral, enantiopure format.
With solid background on organocatalytic ester activation, Chi, Hu, et al. in 2013 developed the first N-heterocyclic carbene (NHC)-catalyzed direct γ-functionalization of linear α,β-unsaturated esters of type 553, that undergo stereoselective addition to glyoxal-derived hydrazones 554 in a highly efficient manner (Scheme 146). Following a careful optimization survey, l-leucine-derived chiral triazolium-based NHC precatalyst B33 was elected as the most suitable tool to access a wide panel of δ-lactams 555 bearing up to two new stereocenters in good yields (up to 91%) and excellent enantioselectivities (up to >99% ee). The authors also proved the usefulness of such optically active lactams by transforming a lactam derivative (R1 = Ph and R2 = H) into the corresponding chiral pipecolic acid (not shown).
Scheme 146.

5.2.1.2. Cyclic Pronucleophiles
Cyclic ester pronucleophiles could be engaged in γ,ipso-selective [4 + 2] annulations similar to those witnessed for their acyclic counterparts (see Scheme 146), but this time when aromatic cyclic vinylogous esters were employed, dearomative formation of oQDM dienolate intermediates had to be faced (see section 3). In this context, in 2016 Hu et al. reported the construction of optically pure heteroarene-fused δ-lactams 558 bearing a quaternary stereogenic center (Scheme 147),388 facing the dearomatizative γ-enolization of 2-methyl-heteroarene-3-carboxylic esters of type 556 induced by NHC B13 catalyst and DBU base. Highly reactive heterocyclic oQDM intermediates 556′ underwent highly enantioselective stepwise [4 + 2] annulation reactions with isatin-derived ketimines 557 to afford the constrained heteroarene-fused δ-lactams 558 in good yields (up to 81%) and high enantiocontrol (up to >99% ee).
Scheme 147.

A diastereoselective, direct, two-component vinylogous Mannich reaction between chiral N-tert-butanesulfinyl imines 560(389) and pronucleophilic dioxinone 559 was developed in 2017 by Chen, Zhang, et al. for the synthesis of δ-amino acid derivatives (Scheme 148).390 After a systematic screening of reaction conditions, it was found that the combination of lithium bis(trimethylsilyl)amide (LiHMDS) to generate the dioxinone-derived lithium dienolate, combined with BF3·OEt2 to activate the preformed imine component, proved to be the most efficient choice. A variety of aromatic and aliphatic aldimines of type 560 (Scheme 148, eq 1), ketimines 562 (eq 2), and isatin-derived ketimines 564 (eq 3), mostly bearing a chiral tert-butanesulfinyl auxiliary group at the nitrogen atom, were suitable substrates for this process, providing the corresponding adducts 561, 563, and 565 with varied levels of efficiency and generally with a good level of diastereocontrol.
Scheme 148.

To rationalize this BF3·Et2O-mediated vinylogous Mannich reaction, the authors provided a transition state model (Scheme 148, bottom), where the imine component, activated by the boron-centered Lewis acid, is engaged in a six-membered chairlike network with lithium ion that guides the Si face addition of the dioxinone-derived dienolate 559′ to the imine, leading to the S-configured products.
Recently, the same group reported a couple of works in which the just described direct, vinylogous Mannich-type addition to N-tert-butanesulfinyl imines was applied to the enantioselective synthesis of terpene indole alkaloids (−)-vindorosine391 and (−)-vindoline,392 whose challenging core structure is embedded in clinically important anticancer drugs such as vinblastine and vincristine (Scheme 149).
Scheme 149.

An initial study on model substrates, such as pronucleophilic dioxinones 559 and 566 and preformed indolyl N-tert-butanesulfinyl imines 567 confirmed the utility of the previously disclosed reaction conditions based on BF3·OEt2 and LiHMDS couple (Scheme 149). Indeed, treatment of 559 or 566 with lithium bis(trimethylsilyl)amide in THF at −78 °C, and subsequent reaction with indolyl-N-tert-butanesulfinylimine 567 activated by BF3·OEt2, resulted in the formation of the corresponding N-tert-butanesulfinylamines 568 on a gram scale, in high yields (up to 97%) and good to excellent diastereomeric ratios (up to >40:1). In particular, prostereogenic ethyl dioxynone 566, coupled with unsubstituted N-Boc indolyl imine 567a (R2 = H), gave the corresponding (4R,5S,Ss)-anti-adduct 568a in 83% yield predominantly, accompanied by small amounts of its (4S,5R,Ss)-anti-diastereoisomer (6.5:1 dr). Substrate 568a was then further elaborated to complete the total synthesis of the vindorosine and vindoline core, as planned (not shown). To account for the observed (4R,5S,Ss)-anti-selectivities resulting from a Si–Si approach, the authors recalled the previously described transition state (Scheme 148) adding an alternative, putative model in which the boron atom BF3 would not be involved (Scheme 149, bottom).
As already commented for the direct, vinylogous aldol reaction, one of the most exploited class of vinylogous pronucleophiles consists of conjugated or unconjugated furanone derivatives to access chiral, enantiopure γ-homologated butenolides. In this context, their use as vinylogous pronucleophiles in asymmetric Mannich-type addition to imine substrates affords chiral δ-amino γ-butenolides which are common motifs in a variety of natural products and pharmaceutical compounds, besides being also useful synthetic intermediates.
The synthetic utility of γ-butenolides led Shibasaki and co-workers to develop the first, direct, and catalytic asymmetric vinylogous Mannich reaction of furanones of type 569 with nonactivated N-thiophosphinoyl ketimines 570 (Scheme 150, eq 1).393
Scheme 150.

Capitalizing on their own previous ventures on the activation of such ketimines by soft Lewis acids through soft–soft interactions, Shibasaki envisaged that a cooperative catalytic system containing a soft Lewis acid and a hard Brønsted base would be a viable strategy to promote the catalytic generation of dienolates from the pronucleophilic γ-butenolide while simultaneously activating the ketimine component. Indeed, after a deep screening of Lewis acids and amine bases, it was found that several γ-crotonolactones of type 569 and N-thiophosphinoyl ketimines 570 smoothly reacted with a binary catalyst (10 mol %) composed of [Cu(CH3CN4)]PF6/(R,RP)-taniaphos (L15) using Et3N (50 mol %) in THF at 0 °C, to give optically pure, vinylogous Mannich adducts 571 in moderate to high yields (up to 92%) as the sole anti-isomers (dr > 20:1). The reaction proved to be quite general, so that both aromatic (acetophenone-derived) and aliphatic ketimines reacted with γ-crotonolactone and its 3- or 4-methyl congeners with almost equal efficiency.
An interesting alternative was developed several years later by Nakamura and co-workers, who reported the direct, enantioselective vinylogous Mannich reaction of unsubstituted γ-crotonolactone 440 to a series of aromatic and aliphatic N-phosphinoyl ketimines 572 using Zn(OTf)2 (10 mol %) coupled with an original quinine-derived picolinamide C24 used in combination with Et3N (1.0 equiv) as the base (Scheme 150, eq 2).394 Indeed, the reaction of 440 with a set of cyclic and acyclic ketimines 572 gave access to the corresponding optically active δ-phosphinolylamino-butenolides 573 in high yields (up to 99%) and with excellent diastereo- and enantioselectivities (up to 99:1 dr, and up to 88% ee). Furthermore, using the quasi-enantiomeric quinidine-derived amide C25 afforded the corresponding butenolides ent-573 with comparable yields and stereoselectivities. To clarify the observed selectivity of the direct VMnR between 440 and 572, MO calculations of the transition state were carried out, unveiling a network in the putative transition state (Scheme 150, bottom) in which the zinc(II) cation would coordinate two nitrogen atoms from the picolinoyl moiety, two oxygen atoms from the dienolate 440′, and a phosphoryl group. Also, the proton attached to the quinuclidine moiety coordinates the oxygen atom of the dienolate. In this stacked bonding network, the Si face of the dienolate approaches the Re face of the ketimine to avoid steric repulsion of the diphenylphosphinyl group to give the observed products.
In 2011 Feng and co-workers were the first who demonstrated how deconjugated butenolides such as α-angelica lactone 574 were also viable vinylogous nucleophiles to be used in metal-catalyzed enantioselective VMnR to provide chiral δ-amino γ,γ-disubstituted butyrolactones. (Scheme 151)395,314 Interestingly, in contrast with previously reported methodologies in which 574 was activated to the corresponding dienolate by base-promoted α-deprotonation, Feng envisioned the possibility to activate such donors through an in situ-generated O-bond metal dienolate by the sole Lewis acid catalysis. After having scrutinized a series of metal–ligand couples, the N,N′-dioxide L10·Sc(OTf)3 complex was elected as the best catalytic system to promote the VMnR between deconjugated α-angelica lactone 574 and a set of various aromatic N-anisidine aldimines of type 575, providing the corresponding δ-amino butenolide scaffolds 576 bearing adjacent quaternary and tertiary stereocenters, in good yields (up to 90%) and excellent diastereo- and enantioselectivities (up to 99:1 dr and up to 98% ee). Interestingly, the α,β-unsaturated furanone congener failed to undergo the VMnR and no product was observed under L10/Sc(OTf)3 catalysis. The electronic effects of the anisidine group were also investigated, revealing that the electronic property rather than the steric hindrance of substituents on the phenyl ring of aromatic aldimines had an impact on the enantiocontrol of the reaction; accordingly, the substrates with electron-withdrawing substituents gave higher ee values than those with electron-donating ones. A mechanism of this highly enantioselective process was proposed, suggesting that the use of the L10/Sc(OTf)3 couple is likely to generate a hexacoordinate scandium-centered intermediate 575′ from aldimine 575 After α-angelica lactone 574 is added, a new chiral scandium intermediate is generated, in which the Si–Si attack of the dienolate to the aldimine provided access to the targeted (5R,1′R)-configured γ,γ-disubstituted butenolides 576.
Scheme 151.

The first direct, vinylogous Mannich reaction which utilizes easily cleavable N-Cbz imines and either conjugated and unconjugated α,β- or β,γ-butenolides bearing a variety of substitution patterns was recently developed by the Trost group,396 exploiting their previously developed Zn-ProPhenol complex L16·Et2Zn whose ability in catalyzing other asymmetric transformations, including aldol reactions, had widely been proved.397 As described in Scheme 152 (eq 1), ProPhenol ligand L16 (10 mol %) and Et2Zn (20 mol %) in THF at rt efficiently catalyzed the VMnR between α-angelica lactone 574 and a series of Cbz-protected aromatic aldimines 577, yielding optically pure δ-amino butenolides 578 in good yields (up to 99%) and almost complete stereocontrol (up to >99.5% ee). Of note, under similar reaction conditions, this catalyst system successfully worked also with conjugated furanones 579 and aromatic aldimines 580 (Scheme 152, eq 2) affording chiral, enantiopure butenolides 581 in comparable yields and stereoselectivities. Based on previous studies on other Zn-ProPhenol catalyzed Mannich reactions, the authors proposed a transition state based on a two-point binding model in which the nitrogen and the carbamate oxygen of the imine binds to a zinc atom, generating a complex which directs the addition of the butenolide to the Re face of the imine (Scheme 152, bottom).
Scheme 152.

Finally, an organocatalyzed version of the direct, asymmetric, syn-selective VMnR of 3,4-dihalofuran-2(5H)-ones 426a and 426b with a series of N-tosyl aromatic aldimines 582 was developed by Xu, Wang, et al. in 2012, using natural quinine C26 as the catalyst (Scheme 153).398 A series of different aldimines derived from aromatic aldehydes bearing both electron-withdrawing and electron-donating groups were suitable electrophiles for the direct VMnR with halofuranones, giving the corresponding syn-configured δ-amino γ-butenolides 583 in excellent yields (up to 98%) and enantioselectivities (up to 95% ee). To account for the obtained selectivity and based on previous achievements on similar organocatalyzed reactions, a transition state was proposed (Scheme 153, bottom). Accordingly, the quinuclidine moiety of quinine activates the 3,4-dihalofuran-2(5H)-one 426, generating the nucleophilic dienolate 426′, while the hydroxyl group within the catalyst would activate the imine through hydrogen bonding interaction. The chiral quinine scaffold nicely orchestrates the Si–Si approach of the dienolate and the imine through noncovalent interactions, promoting the formation of the targeted butenolides 583.
Scheme 153.

5.2.2. Indirect Procedures
5.2.2.1. Acyclic Pronucleophiles
A highly selective metal-catalyzed, three-component vinylogous Mukaiyama-Mannich reaction (VMMnR) between acyclic silyl dienol ether 491b and aromatic aldehydes 584 and 2-aminophenol 585 was accomplished by Liu, Feng, et al. in 2010, using chiral N,N′-dioxide L17·Sc(OTf)3 complex as the catalyst (Scheme 154).399 A variety of aromatic aldehydes were found to be suitable substrates for the reaction and the desired δ-amino-α,β-unsaturated esters 586 were obtained in 90–99% yields with good to excellent enantioselectivities (up to >99% ee).
Scheme 154.

In the same year Schneider and co-workers400 reported the chiral Brønsted acid-catalyzed, two- or three-component enantioselective VMMnR of acyclic silyl dienolate 491a and 590 with several aromatic and aliphatic imines, providing enantioenriched δ-amino-α,β-unsaturated esters 589 and 592 in excellent stereoselectivities (Scheme 155, eqs 1 and 2). Capitalizing on a previous report,401 they discovered a superior second-generation Brønsted acid catalyst such as BINOL-based phosphoric acid D3, in promoting a high enantioselective three-component VMMnR starting from the respective aldehydes 587, p-anisidine 588, and silyl dienolate 491a in a solvent mixture of equal amounts of t-BuOH, 2-methyl-2-butanol, and THF at −50 °C, in the presence of 1 equiv of water (Scheme 155, eq 1).
Scheme 155.

Under these optimized conditions, aromatic, heteroaromatic and aliphatic aldehyde acceptors 587 were suitable substrates, generating the corresponding γ-adducts in high yields (up to 97%) and enantioselectivity (up to 97% ee). Furthermore, under similar reaction conditions when prostereogenic γ-substituted silyl dienolate 590 coupled to preformed imine 591, a two-component version of the reaction (Scheme 155, eq 2) provided the corresponding optically enriched anti-adducts 592 in good yields (up to 97%) and stereoselectivities. Interestingly, the reaction of the corresponding 3Z-configured silyl dienolate congener (3Z)-590 was much less efficient giving predominantly the syn-configured adduct 592 (1.5:1 syn/anti) in 41% yield and 72% ee (not shown). Mechanistic investigations including NMR spectroscopy and mass spectrometry unveiled the role of the protic reaction medium, which traps the cationic silicon species as silanols to regenerate the chiral Brønsted acid catalyst (not shown). The substrate scope of the catalytic, enantioselective VMMnR involving acyclic silyl ketene acetals was later extended to more challenging aliphatic aldimine electrophiles. In this context, a modified and highly useful protocol was developed using BINOL-based phosphoric acid D4 (10 mol % in THF, at −40 °C) as the catalyst of choice (Scheme 155, eq 3).402 Either substituted or unsubstituted silyl dienolates 491a and 590 were used in combination with a set of linear and branched aliphatic imines 593 providing the corresponding vinylogous adducts 594 in good to excellent yields (up to 96%) and high stereocontrol (up to 29:1 dr from 590, and up to 99% ee). Ester moieties, halogen atoms, and alkynes were also well tolerated in this process.
The disclosed VMMnR protocol served as the key stereoselective step in the total synthesis of 16 diversely substituted and enantioenriched indolizidine-based alkaloids (Scheme 156).403,404 Here, using differently substituted catalysts D4, D5, or D6 to promote the addition between simple (R1 = H) and prostereogenic (R1 = Me) dienolates 595 to a suitable in situ-formed aliphatic aldimine 596, the corresponding anti-configured γ-adduct intermediates were formed on a large scale. Upon treatment of the crude with acetic acid at reflux a subsequent cyclization step took place, providing key butyrolactam intermediates 597 in high overall yields (up to 93%) and excellent diastereo- and enantioselectivities (up to 28:1 dr and >99% ee).
Scheme 156.

Another smart application of Schneider’s protocol was implemented by Eastman, Wu, et al. in 2016 for the enantioselective formal syntheses of diverse dimeric and monomeric sulfur-containing quinolizidine triterpenoids nuphar alkaloids (Scheme 157).405 The syntheses involved, as the key step, the development of highly enantioselective Brønsted acid-catalyzed three-component VMMnR between supersilyl dienol ether 598, aliphatic aldehydes 599 or 601, and p-anisidine 588 as the amine source, to provide useful γ-adduct intermediates that were easily converted into the corresponding piperidine derivatives 600 or 602 via acetic acid or Pd-catalyzed annulations (Scheme 157, eqs 1 and 2).
Scheme 157.

Paralleling this chemistry, Schneider and co-workers also focused on the reactivity of bissilyldienediolate 603 (Scheme 158): an α-keto ester homoenolate equivalent which may react as a 1,3-zwitterionic synthon in cascade processes providing new synthetic opportunities.
Scheme 158.

In this context, Schneider, Boomhoff et al. in 2012 reported a novel, metal-catalyzed, one-pot [3 + 2] cycloannulation process generating tetrahydropyrrolo[2,1-b]benzoxazoles of type (±)-606 with two new stereogenic centers in good yields (up to 87%) as single isomers (Scheme 158, eq 1).406−409 In particular, the developed stepwise process consisted of a first, Yb(OTf)3-catalyzed three-component VMMnR between bissilyldienediolate 603, 2-aminophenols 605, and a set of diverse aromatic and aliphatic aldehydes 604, yielding the corresponding γ-adduct intermediates, which underwent fast hydrolytic cleavage to give a highly reactive α-keto ester 603′. This intermediate could then engage the imine in a cyclocondensation reaction generating the N,O-acetal 606 spontaneously, via formation of the cyclic iminium ion intermediate 603′′. Furthermore, a catalytic, enantioselective variant was preliminarily attempted on benzaldehydes 607, using the chiral Sc(OTf)3·ent-L10 as catalyst system of choice (Scheme 158, eq 2). Under slightly different reaction conditions, the corresponding products 608 could be obtained with good yields (91–75% respectively) and acceptable enantioselectivities (81–83% ee).
The same group used bis-silyldienolether 603 in the sequential addition to two imine substrates in a tandem, divergent vinylogous Mannich–Mannich–Pictet–Spengler process to generate chiral, polyfunctionalized hexahydropyrrolo[3,2-c]quinolines (±)-611 and (±)-614 in a one-pot operation (Scheme 159, eqs 1 and 2).410 Under nonaqueous conditions (DME at rt), Yb(OTf)3 (10 mol %) promoted a first VMMnR of 603 to a first added imine 609, generating the corresponding silyl enol ether intermediate that, after the addition of a substoichiometric loading of a second Brønsted acid catalyst such as TFA, engaged a second imine 610 in a “normal” Mannich reaction to provide a diamino α-keto ester intermediate 610′ (Scheme 159, eq 1).
Scheme 159.

This highly reactive intermediate then cyclized to give the corresponding iminium ion 610′′, which is finally trapped by the electron-rich anisidine moiety in a Pictet–Spengler reaction, affording the targeted pyrroloquinoline (±)-611. Under this protocol, a broad range of aromatic and aliphatic imines were tolerated, providing the corresponding products in good yields (up to 85% yield) and high diastereoselectivities (up to 49:1 dr).
Interestingly, a divergent option was viable by careful adjustment of reaction conditions (MeCN as solvent, at rt, using (PhO)2PO2H instead of TFA), so that 603 and added imines 612 and 613 (Scheme 159, eq 2) afforded the corresponding products (±)-614 in high yields (up to 93%) and moderate diastereoselectivity (up to 6.7:1 dr).
More recently, a conceptually similar stereocontrolled three-component [3 + 2]-cycloheteroannulation involving bis-silyldienediolate 603 and imines derived from 2-aminobenzoic acid and 2-aminobenzamide derivatives 616 and 620, catalyzed by Sm(OTf)3 in the presence of 2,4-dinitro benzenesulfonic acid (618, DNBSA), furnished highly substituted pyrrolo[1,2-a]benzoxazinones (±)-617 or pyrrolo[1,2-a]quinazolinones (±)-620, respectively, in good overall yields (Scheme 160, eqs 1 and 2).411−413
Scheme 160.

5.2.2.2. Cyclic Nucleophiles
The vinylogous Mukaiyama–Mannich reaction (VMMnR) of lactone-derived silyl ketene acetals with imines or iminium ions leading to δ-amino α,β-unsaturated carbonyl compounds is a useful transformation which has become a powerful methodology for the efficient synthesis of highly functionalized heterocycles, bioactive natural products, and pharmaceuticals. In this context, the past decade has witnessed with the concomitant development of new VMMnR methodologies (particularly in the field of asymmetric metal- and organocatalyzed transformations), the exploitation of previously assessed methodologies to the total synthesis of bioactive natural products.
In this context, vinylogous additions of 2-silyloxyfurans to C=N double bonds constitute very attractive processes, allowing the incorporation of oxygen functionalities into nitrogen-containing carbon frameworks. Furthermore, both enantioselective versions using chiral catalysts and diastereoselective versions using chiraly auxyliaries have been documented and clearly reported.
In 2010, the group of Shi and co-workers414 developed a silver(I)-catalyzed catalytic, asymmetric VMMnR of trimethylsiloxyfuran 496a with a set of fluorinated aldimines 621 in the presence of a chiral phosphine–oxazoline ligand (Sa,S)-L18 derived from S-BINOL (Scheme 161, eq 1). The reaction, carried out in THF at −78 °C, in the presence of EtOH (1.8 equiv) as a protic, silicon scavenging additive, afforded the corresponding 5,1′-anti-configured γ-adducts 622 in good to excellent yields (up to 99%), along with moderate to good enantioselectivities (up to 81% ee).
Scheme 161.

An extension of such protocol was later devised by the same group, who developed an anti-selective VMMnR between 496a and readily available fluorinated aldimines 623 bearing a chiral auxiliary group [(S)-1-phenylethyl group] (Scheme 161, eq 2).415 Using AgOAc-(Sa,S)-L18 (10 mol %) catalytic complex, EtOH (1.8 equiv) in THF at −78 °C, a set of “matched” chiral, fluorinated γ-butenolides of type 624 were accessed in high yields (up to 99%), good to excellent enantiomeric excesses (up to 99% ee), and up to >20:1 dr. In addition, 4-methyl-substituted siloxyfuran 538b was also tested as nucleophile in the VMMnR to 623, affording the corresponding adducts in comparable yields and stereoselectivites.
A similar silver(I)-catalyzed protocol was also applied to the catalytic, asymmetric VMMnR to N-Boc aldimines 625 (Scheme 161, eq 3).416 Indeed, treatment of 496a and imine 625 in the presence of catalytic quantities of chiral phosphine-oxazoline ligand (Ra,S)-L18 and AgOAc in CH2Cl2, in the presence of stoichiometric EtOH at −78 °C, provided the corresponding 5,1′-anti-configured δ-aminobutenolides 626 in good to high yields (up to 97%) along with moderate to good diastereo- and enantioselectivities (up to 7:1 dr and 86% ee).417−419
A conceptually different approach was devised by Huang et al. in 2011, who developed a diastereoselective VMMnR of cyclic sililoxyfuran 496c with preformed, aromatic, or aliphatic chiral N-tert-butanesulfinimines (t-BS-imines or Ellman’s imines) of type 627 to provide chiral, enantiopure δ-aminobutenolides 628 as versatile chiral precursors to access diverse functionalized heterocycles (Scheme 162, eq 1).420 Under the optimized reaction conditions, TBSOF 496c reacted in CH2Cl2 at −78 °C with either 627 or ent-627 in the presence of TMSOTf to afford the corresponding 5,1′-anti-configured 5-aminoalkylbutenolides 628 or ent-628 in good yields with dr ranging from 3:1 to 32:1. To account for the observed selectivity, a plausible transition state was described in which a monocoordinated complex between the Lewis acid and the sulfoxide moiety induced a preferred conformation of the imine which determined the favorable approach of 496c to the less sterically hindered Si face of the chiral imine 627. A similar methodology was exploited by the same group for the synthesis of diverse hydroxylated piperidine alkaloids and azasugar lactams (not shown).421
Scheme 162.

A rare example of syn-selective VMMnR involving N-tert-butanesulfinimine 629 was devised by the Huang’s group using 2-methyl-TBSOF 537a instead of 496c (Scheme 162, eq 2).422 Quite surprisingly, under the previously disclosed optimized conditions, TMSOTf (1.0 equiv) in CH2Cl2 at −78 °C afforded the optically pure, syn-configured adduct 630 in a high 97% yield. Butenolide 630 was then used as key intermediate in a three-pot, protecting group-free total synthesis of bioactive natural product (−)-pandamarilactonine-A.
Another highly regio- and diastereoselective TMSOTf-promoted VMMnR involving isatin-derived chiral N-tert-butanesulfinyl ketimines 632 and silyloxyfurans 631 was introduced by Singh and co-workers in 2014 (Scheme 162, eq 3).423−425 The method provided a clean entry to a wide range of challenging quaternary 3-aminooxindole butenolides 633 in good yields (up to 83%) and high diastereoselectivities (up to 99:1 dr). The authors proposed a syn-periplanar transition state where the bulky tert-butyl group hindered the Si face of the ketimine favoring the attack of the silyloxy furans on the Re face of 632, producing the observed anti-(5R,1′R)-configured adducts 633.
In 2013, an enantioselective VMMnR of silyloxufuran nucleophiles of type 634 and N- diphenylphosphinoyl ketimines of type 635 was successfully achieved by Nakamura and co-workers using Cu(II)-cinchona alkaloid picolinamide complex as the catalyst (Scheme 163).426
Scheme 163.

After a first optimization survey, 9-amino-9-deoxy-epi-cinchonidine-derived picolinamide C27 was elected as the best chiral ligand for the CuOAc-promoted VMMnR of a set of between methylated and nonmethylated silyloxyfurans 634 and aromatic, heteroaromatic, and also aliphatic ketimines to provide the corresponding (O,N)-anti-configured δ-amino-δ,δ-disubstituted butenolides 636 in high yields (up to 99%) and excellent diastereo- and enantioselectivities (up to 99:1 dr and up to 98% ee). To account for the observed (5R,1′S) absolute configuration of 636, a transition state was proposed in which two nitrogen atoms from the picolinamide moiety of C27 and two oxygen atoms coordinate copper(II) ion in a square plane manner, allowing the silyloxyfuran to attack the ketimine in the coordination sphere of the copper cation. The Si face of the dienolate approaches preferentially the Re face of the ketimine, thus avoiding steric repulsion of the diphenylphosphinoyl group.
A highly versatile, diastereoselective, one-step, and three-component VMMnR involving TMSOF 496a and a variety of nitrogen-containing heterocycles mainly of type 637 was described by Dodd and co-workers in 2014 (Scheme 164).427,428 Based on previous reports,429,430 the authors devised a highly diastereoselective procedure involving the use of acyl or sulfonyl chlorides as suitable activating agents of a plethora of different aza-heterocycles that, once acylated, generated a highly electrophilic iminium ion species of type 637′ that finally coupled to 496a in a diastereoselective manner. The racemic reaction products were generally obtained in high yields (up to 99%) and mainly as single diastereoisomers. Several aza-heterocycles were successfully employed for this reaction, such as isoquinolines, quinoline, phenanthridine, quinazoline, phthalazine, and β-carboline, while electrophiles included acetyl chloride, methyl chloroformate, methyl chloromalonate, 2-bromobutanoyl chloride, and arylsulfonyl chlorides. Furthermore, a N-Boc silyloxypyrrole nucleophile was tested under the optimized reaction conditions, giving appreciable results (not shown).
Scheme 164.

Huang and co-workers in 2011 applied a diastereoselective VMMnR between 2-methylsilyloxyfuran (537a) and chiral, bicyclic N,O-acetal 639 to access enantiopure tricyclic intermediate 640, as a key precursor in the total synthesis of the stemona alkaloid 9-epi-sessilifoliamide J (Scheme 165, eq 1).431 Among the Lewis acids tested, TMSOTf (1 equiv) gave the best results, providing a 3.5:1 mixture of the syn-configured γ-adduct 640 as a major isomer in a good 70% yield. A plausible transition state was proposed, in which the in situ-formed iminium ion 639′ (Scheme 165, bottom) added to the nucleophile through a favorable Diels–Alder-like Re–Re approach (not shown).
Scheme 165.

Several years later, the same group used a VMMnR in the total synthesis of (−)-sessilifoliamide J (Scheme 165, eq 2).432 Here, starting from chiral Cbz-protected N,O-acetal 641, the authors devised a two-component VMMnR with TMSOF (496a) as the vinylogous nucleophile. Thus, treatment of 496a and 641 with stoichiometric TMSOTf, in CH2Cl2 at −78 °C afforded the desired γ-adduct 642 (via formation of the cyclic iminium ion 641′) in a high 97% yield and a 5.6:1 dr in favor of the syn-configured isomer.
An interesting diastereoselective VMMnR in which the chiral information resides in the nucleophile was used by Gademann and co-workers, in their total synthesis of the securinega alkaloids virosaine A and the putatively related alkaloid bubbialidine (Scheme 165, eq 3).433 Following Busqué’s procedure,434 a vinylogous Mannich reaction between chiral, bicyclic dienolsilane 643 and N,O-acetal 644 was achieved using triisopropylsilyl triflate as a Lewis acid, generating a separable 1:1 mixture of two diastereoisomers in a combined 90% yield, from which the desired (R,R)-configured diastereoisomer 645 could be isolated.
More recently, Delair et al. used a BF3·Et2O-promoted, diastereoselective VMMnR between TMSOF 496a and a readily accessed chiral pyrrolidine N,O-acetal 646 to access butanolide 647 en route to the uncommon [6.6.5]-tricyclic ring system of the 8b-azaacenaphthylenes alkaloids (Scheme 165, eq 4).435 In the event, a mixture of two lactone isomers was obtained in a high 98% combined yield and 2.7:1 dr in favor of the desired syn-isomer 647.
Finally, List and co-workers reported a rare, highly enantioselective approach to the synthesis of δ-amino-β-ketoesters by a disulfonimide-catalyzed VMMnR utilizing N-Boc aldimines 648 and dioxinone-derived silyloxydiene 521a as reacting partners (Scheme 166).436 Of note, only 1 mol % of chiral disulfonimide precatalyst D2 served as an efficient promoter for the VMMnR, carried out in toluene at −50 °C. Using readily available aromatic or heteroaromatic N-Boc-protected imines, a variety of polyfunctionalized γ-adducts 649 were accessed in high yields (up to 98%) and high enantioselectivities (up to 97%). Regrettably, aliphatic imines were quite inert, and only a cyclohexyl derivative could react under the optimized reaction conditions, yielding the corresponding adduct in 60% yield and 20% ee.
Scheme 166.

5.3. Conjugate Additions to Electron-Poor C=C Bonds
5.3.1. Direct Procedures
5.3.1.1. Acyclic Pronucleophiles
An intriguing, yet useful alternative to the classical base-catalyzed activation of α,β-unsaturated esters via dienolate-like intermediates is based on the nucleophilic addition of phosphines to the β sp-carbon of suitable allenoate species of type XXX (Scheme 167). Due to the unique reactivity of electron-deficient allenes, the incorporation of electrophiles such as electron-deficient C=C, C=N, and C=O bonds has been widely employed in phosphine catalysis to access polyfunctionalized carbo- and heterocycles via Diels–Alder-like reactions.437
Scheme 167.

Indeed, as described in Scheme 167, α-brached allenoates of type XXX can react with electron poor alkenes XXXI to form cyclohexenes XXXII (via formal [4 + 2] cycloaddition) under the guidance of an appropriate phosphine catalyst. Mechanistically, under suitable reaction conditions, the generation of a phosphonium dienolate XXX′ by conjugate addition of the phosphine to the β-carbon of XXX, is followed by Michael addition to an activated olefin XXXI providing the zwitterionic intermediate XXXIII. A subsequent proton-transfer step facilitates tautomerization of the vinylphosphonium zwitterion XXXIV to an allylphosphonium zwitterion XXXV, which furnishes cyclohexene XXXII via intramolecular 6-endo cyclization followed by β-elimination of the phosphine (Scheme 167, γ-pathway). Alternatively, phosphonium dienolate XXX′ can isomerize into the vinylogous ylide XXX′′ (Scheme 167, β′-pathway), which adds to the olefin XXXI on its β′ carbon to provide the corresponding cyclohexene XXXII′′ through a reaction sequence similar to the previously described catalytic cycle. Despite the profound interest raised by these and other phosphine-catalyzed transformations, as testified by the several reviews published on this issue,438,439 only the γ-pathway annulations (formal [4 + 2]), involving a dienolate-like intermediate, may be regarded as a vinylogous process, thus legitimately placing this type of transformations in this review. In this context, after the seminal works by Kwon’s group who disclosed achiral phosphine-catalyzed [4 + 2] annulation of allenoates with activated imines and alkenes for the expedient construction of polyfunctionalized cyclohexenes and other heterocycles,440−442 the asymmetric phosphine-catalyzed [4 + 2] annulations of activated alkenes with allenoates evolved more slowly until recently, when several important examples have been reported.
In 2012, Lu and co-workers were the first to develop a highly enantioselective [4 + 2] annulation between allenoate 650a and a series of activated alkenes catalyzed by chiral amino acid-derived phosphines (Scheme 168, eqs 1 and 2).443 Their survey started by examining the catalytic effects of various amino acid-derived phosphines in the [4 + 2] annulation between allenoate 650a and several arylidenemalononitriles 651 (eq 1). O-Tris(trimethylsilyl)silyl (TTMSS) l-threonine-based phosphine–amide P2 was elected as more suitable catalyst, yielding the corresponding cis-configured cyclohexenes 652 in good overall yields (85–96%), moderate to good diastereoselectivity, and excellent enantioselectivity (up to 6.7:1 dr and 91–98% ee). The reaction was applied to a wide range of activated phenyl-, aryl-, and heteroarylidene malononitriles, while challenging alkenes derived from aliphatic aldehydes were found to be unsuitable for the annulation.
Scheme 168.

To further expand the scope of this asymmetric [4 + 2] annulation reaction, the authors switched to the more challenging annulation between allenoate 650a and isatin-derived ylidene malononitriles 653 to access chiral, enantiopure 3-spirocyclohexene-2-oxindoles 654, that are striking molecular frames of pharmaceutical interest (Scheme 168, eq 2). To this end, it was found that O-protected d-Thr-L-tert-Leu-derived phosphine P3 effectively catalyzed the formation of a number of 3-spirocyclohexene-2-oxindoles 654 in very high yields (89–96%), and with excellent diastereo- and enantioselectivities (up to 99:1 dr and 89–93% ee). The methyl ester group at the β′ position of allenoate 650a could also be replaced by other ester moieties or electron-poor aromatic rings, allowing the annulation to proceed smoothly to afford cyclization products in moderate yields, good dr and excellent ee (not shown).
More recently, the same group devised a phosphine-catalyzed [4 + 2] annulation process employing allenones of type 655 and β,γ-unsaturated α-keto esters 656 (Scheme 168, eq 3).444 By utilizing 10 mol % of the dipeptide-based bifunctional phosphine P4, almost optically pure 3,4-dihydropyrans 657 were obtained in high yields (71–95%). The reaction was applied to a wide range of β,γ-unsaturated α-keto esters bearing different aromatic groups, regardless of the steric and electronic properties of the substituents on the aromatic ring. Of note, different vinyl- and linear/branched alkyl-substituted α-keto esters were also tested, providing the desired products in good yields and nearly perfect enantioselectivities.
In the same year of Lu’s first work (2012), Zhao and co-workers reported the organocatalytic, enantioselective formal [4 + 2] cycloaddition between α-substituted dienoate 650b and β-substituted (E)-2-cyano acrylates 658 using chiral bifunctional N-acyl aminophosphine catalyst P5 (Scheme 169).445 γ-Regioselective access to polyfunctionalized cyclohexenes 659 was secured, bearing three contiguous stereocenters in high yield and excellent enantioselectivity.
Scheme 169.

Olefins 658 with electron-rich or electron-poor aryl substituents were well tolerated, while ortho-substituted derivatives, probably due to steric reasons, needed more elevated temperature (room temperature) to give satisfactory yields, albeit with a slight drop in the enantioselectivity. Interestingly, the challenging aliphatic isobutyraldehyde-derivative acrylate congener (R1 = isopropyl) was successfully coupled with 650b affording the corresponding isopropyl cyclohexene product in 92% yield and 97% ee. A tentative mechanism was also proposed to explain the stereochemical output observed. A cyclic six-membered transition state was considered responsible for the enantioselectivity of the first, vinylogous addition step favoring the Re face attack on the activated olefins. The newly formed stereocenter would then induce the chirality of the second one formed in the final cyclization step (not shown).
More recently, Guo et al. reported a γ-regioselective phosphine-catalyzed formal [4 + 2] cycloaddition reaction of allenoate 650b with unsaturated pyrazolones 660 under mild reaction conditions to afford challenging polyfunctionalized spiropyrazolone derivatives 661 in both racemic and enantioselective formats (Scheme 170, eq 1).446 Initially, the authors examined the reaction conditions by surveying a series of achiral phosphines as catalysts (not shown) and found that moderately nucleophilic MePPh2 (20 mol %) efficiently catalyzed the reaction between diethyl allenoate and differently substituted pyrazolones 660 in toluene at room temperature, providing the corresponding products (±)-661 as single 1′,5′-syn diastereoisomers in moderate to very high yields (49–99%). Switching to the asymmetric version of the reaction (Scheme 170, eq 1), several chiral phosphine derivatives were initially screened to catalyze the [4 + 2] annulation between 650b and 660, appointing thiourea-based bifunctional phosphine P6 as the most suited catalyst for the reaction. In fact, under the optimized reaction conditions, with P6 (20 mol %), an asymmetric variant of this [4 + 2] cycloaddition reaction was achieved, giving a set of polyfunctionalized (1′S,5′S)-configured chiral spiropyrazolone derivatives 661 in moderate to excellent yields with moderate to excellent diastereoselectivities and excellent enantioselectivities (89–95% ee). Again, a wide substitution pattern within the arylidene substituent of 660 was tolerated, with the sole exception of 3-Br and 4-Cl derivatives, which were less reactive. Interestingly, the minor 1′,5′-syn-configured diastereomers were nearly racemic (not shown).
Scheme 170.

The same group developed an enantioselective [4 + 2] annulation of barbiturate-derived alkenes 663 with allenoates 662 to access pharmaceutically relevant spirobarbiturates 664 as single 1′,5′-syn configured isomers, using a spirocyclic chiral phosphine P7 as the catalyst (Scheme 170, eq 2).447 Of note, a wide range of α-substituted allenoates bearing electron-rich, -neutral, and -deficient aromatic moieties and a series of aromatic barbiturate- and thiobarbiturate-derived alkenes (irrespective of their electronic properties) were tolerated, yielding various spirobarbiturate-cyclohexenes in good to excellent yields (56–95%), with excellent diastereo- and enantioselectivities (>20:1 dr, 90–99% ee). Interestingly, both the α-methyl-substituted allenoate and the cyclohexylydene aliphatic barbiturate derivative proved to be compatible substrates for this transformation.
5.3.1.2. Cyclic Pronucleophiles
In 2014 Kumagai, Shibasaki, et al. applied the soft Lewis acid/Brønsted base cooperative catalysis to enable the direct, catalytic, asymmetric vinylogous conjugate addition of α,β- and β,γ-unsaturated butyrolactones 665 and 668 to α,β-unsaturated thioamides 666 (Scheme 171, eqs 1 and 2).448
Scheme 171.

The reaction afforded the corresponding γ-homologated butenolides 667 and 669 bearing two consecutive tri- and tetrasubstituted stereogenic centers in a highly diastereo- and enantioselective fashion. In particular, the cooperative action of a soft Lewis acid such as chiral copper complex [Cu(CH3CN)4]PF6/(R)-Segphos (L19) and a Brønsted base such as Cy2NMe was the most suited catalytic system to promote the vinylogous Michael reaction (VMcR) between pronucleophilic γ-crotonolactones 665 and unsaturated thioamides 666 (eq 1).
The reaction of β-aryl- or β-heteroaryl-substituted α,β-unsaturated thioamides proceeded smoothly with a 5 mol % catalyst loading, regardless of the electronic nature of the substituents, leading to the corresponding γ-adducts 667 with high yields (78–98%) and almost complete diastereo- and enantioselectivity. Of note, even β-alkyl-substituted α,β-unsaturated thioamides were effective substrates, although 10 mol % of catalyst was required to complete the reaction. Interestingly, the same reaction was tested on other electron-deficient olefins such as unsaturated esters, amides, ketones, nitriles, imides, tosylated amides, thioesters, α-vinylidene malonates, and N-acylpyrrole derivatives, but all failed to give the desired product in considerable yield and stereoselectivity. A very similar reaction pattern was observed when α-angelicalactone-type pronucleophile 668 was used instead of 665 (Scheme 171, eq 2). In this case, the more common Et3N was used to complete the reaction with a 0.5–2 mol % catalyst loading. To account for the observed selectivity, a plausible catalytic cycle was proposed, envisaging the in situ formation of a vinylogous copper dienolate by the action of the tertiary amine and the weak assistance of the Cu/(R)-Segphos complex. Subsequent coordination of the thioamide 666 to the tetracoordinate copper center would produce the active transition state (Scheme 171, bottom), in which a favorable Re–Re substrate interaction takes place to generate the observed (5R,1′S)-configured products.
The above-described work is an example of how cooperative catalysis is emerging as a valuable tool in asymmetric synthesis, fueled by the identification of more efficient catalytic systems with even increasing substrate scopes. Many endeavors have been devoted during the past few years toward developing the right catalyst pairs giving rise to powerful bifunctional systems, and many types of organic catalysts have been used in combination with appropriate metals to achieve unprecedented enantioselective processes.
In this context, a nice example was reported by Wang’s group who devised a couple of cooperative catalytic systems that enabled the efficient, direct, asymmetric vinylogous Michael reaction of γ-aryl- and γ-alkyl-substituted deconjugated butenolides 670 to a series of aromatic and aliphatic enones 671 giving access to the corresponding chiral γ,γ-disubstituted butenolides 672 in good yields with high diastereo- and enantioselectivities (Scheme 172, eq 1).449 Indeed, after an initial survey to assess the viability of the reaction and the best Brønsted base/Lewis acid catalytic system, it was found that ternary (R)-BINOL L20/Al(Oi-Pr)3/quinine C26 and L20/La(Oi-Pr)3/C26 systems were the best choice, giving γ-adducts 672 in high yields and enantioselectivities (75–99% ee) as anti-configured isomers.
Scheme 172.

During the same year, a highly efficient N,N′-dioxide L21·Sc(OTf)3 complex has been developed by Feng and co-workers for the asymmetric VMcR of pronucleophilic γ-substituted-β,γ-unsaturated butenolides 673 to α,β-unsaturated γ-ketoesters 674, affording the corresponding γ,γ-disubstituted butenolide products 675 in moderate to good yields (up to 93%) with high dr (up to >19:1) and ee values (up to 97%) under mild reaction conditions (Scheme 172, eq 2).450 Under the optimal reaction conditions (5 mol % catalyst complex, in MeOAc at 0 °C), the substrate scope of the reaction was investigated. The ester group within acceptor 674 exhibited an influence on both diastereo- and enantioselectivity, with the bulkier tert-butyl group (R2 = t-Bu) giving the best results. The stereocontrol of the reaction was sensitive to neither the electronic property nor the steric hindrance of aromatic and aliphatic substituents (R3) on 674; however, m-substituted congeners generally gave lower yields than the parent o- and p-substituted derivatives. For what concerns the substituent within lactone 673, switching from Me to Et and n-C10H21, the diastereo- and enantioselectivity of the reaction increased but with lower efficiency. Unexpectedly, the reaction failed with the phenyl-substituted β,γ-unsaturated butanolide.
Switching to completely metal-free organocatalytic processes, the first direct, catalytic, enantioselective VMcR involving pronuclephilic deconjugated butenolides 676 was reported by the Alexakis’s group in 2011 (Scheme 173, eq 1).451,452 Indeed, under the optimized reaction conditions, Aminal-PYrrolidine (APY) catalyst A35 (15 mol %) was able to promote the VMcR between γ-methyl and γ-ethyl furanones with varied enals 677 in toluene at room temperature, affording a diastereomeric mixture of the corresponding γ-homologated butenolides 678 in good to high yields (60–95%) and very good enantioselectivity (up to 97% ee). A variety of substitution patterns at the enal β-position could be tolerated, so that both alkyl and phenyl substituents worked well under the optimized reaction conditions.
Scheme 173.

A sensible drop in diastereo- and enantioselectivity was observed when α-branched derivatives such as methacrolein were used as electrophilic components. Interestingly, preliminary mechanistic investigations allowed the authors to propose a mechanism in which the catalyst has the sole role of activating the electrophile via formation of the expected iminium ion 677′, while a thermodynamic enol equilibrium involving deconjugated butenolide 676 provides small quantities of dienol 676′ that undergoes a fast addition on the favored prostereogenic face of the electrophile not hindered by the aminal group of the catalyst (Scheme 173). More recently, the same group reported the direct, VMcR of unactivated α-angelica lactone 574 to enones 679 under iminium ion activation using the combination of chiral 9-amino-9-deoxy-epi-quinine A23 along with 2-hydroxy-1-naphthoic acid (681) as catalytic system (Scheme 173, eq 2).453 The reaction led to the formation of optically pure γ,γ-disubstituted butenolides 680 in high yields (up to 87%) and moderate-to-good anti diastereoselectivities (up to 12:1 dr).
An alternative approach based on noncovalent organocatalytic activation was explored by Huang, Tan, Jiang, et al. in 2012, in the effort to synthesize chiral, enantiopure γ,γ-disubstituted butenolides from easily accessible deconjugated butenolides 682 (Scheme 174).454 Following previous achievements from the same group about the peculiar ability of the 2-oxazolidinone amide moiety to interact with suitable groups such as a thiourea via H-bond,455 the authors have successively developed the direct, asymmetric VMcR of γ-aryl- and alkyl-substituted butenolides with suitable electrophiles such as (E)-4-oxo-4-arylbutenamides, (E)-oxazolidinone enoates, and (E) or (Z)-β-trifluoromethyl oxazolidinone enoate of type 683. Using L-tert-leucine-derived chiral bifunctional pyrrolidine-thiourea C28 (1 mol %) as the catalyst, various γ,γ-substituted butenolides 684 were obtained in good to excellent yields, with excellent enantio- and diastereoselectivities (up to 93% yield, 99% ee, and dr >99:1). According to experimental observations and DFT calculations, a suitable transition state was proposed (Scheme 174) in which the catalyst interacts through H-bond interactions with the electrophilic enamide in a bidentate manner. DFT calculations unveiled a considerable interaction between the oxazolidinone carbonyl and the α-H of the pendent pyrrolium moiety of the catalyst through a nonclassical C–H···O hydrogen bonding.
Scheme 174.

An important contribution that merges the covalent and noncovalent activation modes for the stereoselective synthesis of chiral, enantiopure γ,γ-disubstituted butenolides from prochiral precursors was reported by Dixon and co-workers in 2014 (Scheme 175).456 In particular, a highly diastereodivergent, asymmetric, and direct VMcR between β,γ-unsaturated butenolides 685 and α,β-unsaturated ketones 686 was devised, by employing stereochemically complementary yet nonenantiomeric primary amine catalytic systems. After an extensive optimization survey, it was found that catalyst A36, featuring a primary amine group, a vicinal secondary amine, and a sulphonamide moiety as a terminal hydrogen bond donor, perfectly catalyzed the reaction between α-angelica lactone derivatives 685 and enones 686 in the presence of o-nitrobenzoic acid 237 (20 mol %), to give almost enantiopure adducts 687 in high yields and high diastereoselectivity in favor of the anti-configured isomers (Scheme 175, eq 1).
Scheme 175.

Conversely, using bifunctional primary amine-thiourea catalyst A37, in combination with catalytic quantities of o-fluorobezoic acid (196) allowed the access to the corresponding syn-configured derivatives 688 with comparable results (Scheme 175, eq 2). A broad range of γ-substituted butenolides and functionalized enones, bearing both alkyl substituents and electron-rich/deficient aryl substituents, worked well under the optimized reaction conditions. Furthermore, cyclic enones were also tested with 2-cyclopentenone giving a relatively poor diastereoselectivity, as compared with 2-cyclohexenone or 2-cycloheptenone.
The observed catalyst-dependent diastereodivergence was ascribed to the different nature of polyamine catalysts A36 and A37 used to promote the stereoselective VMcR. In this context, the anti selective VMcR was envisaged to be the result of a favorable transition state in which a tight H-bonding interaction would preferentially control the position and orientation of the dienolate, overcoming the inherent steric hindrance of the reactants. Conversely, a weak and loose H-bonding interaction involving quinine thiourea catalyst A37 would allow a transition state in which the steric bias dominates the stereoselectivity, thus favoring syn selectivity.
Among the large variety of asymmetric Michael reactions developed so far, nitroalkenes have been one of the most widely used Michael acceptors, since they can undergo facile β-alkylation reactions and the corresponding Michael products may be interconverted to important organic functional groups. In this context, the direct, enantioselective Michael-type addition of α-angelica lactone pronucleophiles to nitroalkenes promoted by bifunctional, noncovalent organocatalysts, which provided useful γ-homologated butenolides with at least two contiguous stereocenters, has been widely explored in the past decade by several groups as described in Table 7. The first example of a direct, enantioselective VMcR involving γ-substituted deconjugated butenolides and nitroolefins was reported by Mukherjee and co-workers in 2012 (Table 7, entry 1),457,458 which unveiled the ability of a new quinine-derived bifunctional triiso-butyl catalyst C29 to promote the enanatioselective synthesis of densely functionalized, syn-configured butenolides with contiguous quaternary and tertiary stereocenters in excellent yield and high enantioselectivity (up to 98% ee) with perfect diastereoselectivity (>20:1 dr).
Table 7. Direct, Enantioselective VMcR of α-Angelica Lactone Pronucleophiles to Nitroalkenes, Catalyzed by Bifunctional Organocatalysts.
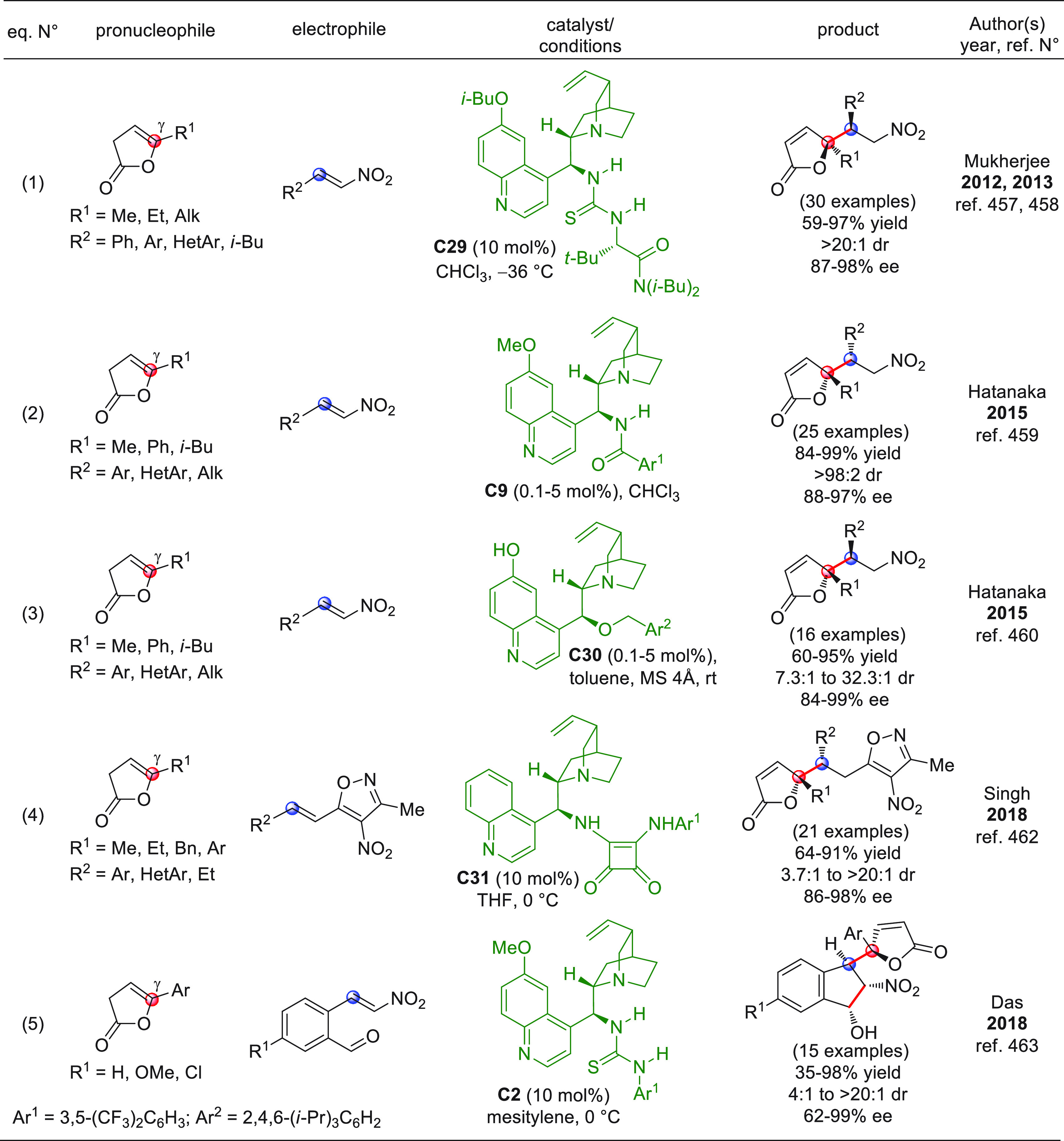
Some years later, Hatanaka’s group developed a novel bifunctional epi-quinine-derived 3,5-bis(CF3)benzamide catalyst C9 which showed a high catalytic activity (0.1–5 mol % loadings) in the asymmetric nitro-Michael addition reaction of a series of deconjugated lactones to a wide range of aromatic and aliphatic nitroalkenes (entry 2).459 The corresponding syn-configured adducts were obtained in high yields (up to 99%) with excellent diastereo- and enantioselectivities (>98:2 dr and up to 98% ee).
Capitalizing on these results, the same group turned attention toward the more challenging anti-selective nitro-Michael reaction (Table 7, entry 3).460,461 Remarkably, the requested catalyst-controlled switching of diastereoselectivity was achieved with the use of bulky 6′-OH-epi-quinine catalyst C30, that, mirroring the high catalytic activity of C9 in the previous reaction, promoted the addition between diverse sets of deconjugated lactones and nitroolefins, to access anti-configured Michael adducts in good yields (up to 95%) with excellent diastereo- and enantioselectivities (up to 32:1 dr and up to 99% ee).
More recently, highly unsaturated nitro-derivatives bearing a conjugated 4-nitro-5-vinyl isoxazole moiety were used by Singh and co-workers as electrophilic substrates for the enantioselective, vinylogous 1,6-conjugate addition between β,γ-unsaturated butanolide pronucleophiles giving access to a broad range of densely functionalized enantioenriched γ,γ-disubstituted butenolides (Table 7, entry 4).462 After a brief optimization survey, chiral bifunctional epi-quinine squaramide C31 was the best catalyst to provide syn-configured adducts in high yields (up to 91%), with excellent enantio- (up to 98% ee) and diastereoselectivities (up to >20:1).
An interesting cascade approach was recently developed by Das et al. to build polyfunctionalized, enantiopure (nitro)indanol scaffolds using a VMcR between γ-substituted deconjugated butenolides and o-formyl-β-nitro-styrenes using bifunctional epi-quinine-thiourea catalyst C2 (entry 5).463 The reaction, carried out in mesitylene at 0 °C, afforded the corresponding indanol products, which contain four contiguous stereocenters in good yields (up to 98%), in good diastereoselectivities (up to >20:1 dr), and with moderate to high enantioselectivities (up to 99% ee).
The high versatility of β,γ-unsaturated butenolides as γ-selective pronucleophilic species was further demonstrated by the development of a plethora of asymmetric vinylogous Michael-type addition reactions on diverse α,β-unsaturated carbonyl and sulfonyl acceptors other than nitroalkenes (Table 8). For example, in 2012 Mukherjee and Manna reported a highly diastereo- and enantioselective protocol for the direct VMcR of deconjugated alkyl-substituted butenolides to symmetrical N-arylmaleimides using the adamantane tertiary-amine/thiourea-based bifunctional catalyst C32 (5 mol %, Table 8, entry 1).464 The reaction afforded a varied set of enantioenriched succinimide derivatives in high yields (up to 99%) and diastereoselectivity (up to 18:1 dr). The scope of this reaction was further expanded on aryl-substituted butenolide congeners by Xu, Wang, et al. using only 1 mol % of a bifunctional squaramides catalyst (not shown), providing the corresponding succinimide derivatives in comparable yields and stereoselectivity, albeit with lower diastereocontrol (up to 4.5:1).465,466
Table 8. Direct, Enantioselective VMcR of α-Angelica Lactone Pronucleophiles to Differently Functionalized Michael Acceptors, Catalyzed by Bifunctional Organocatalysts.
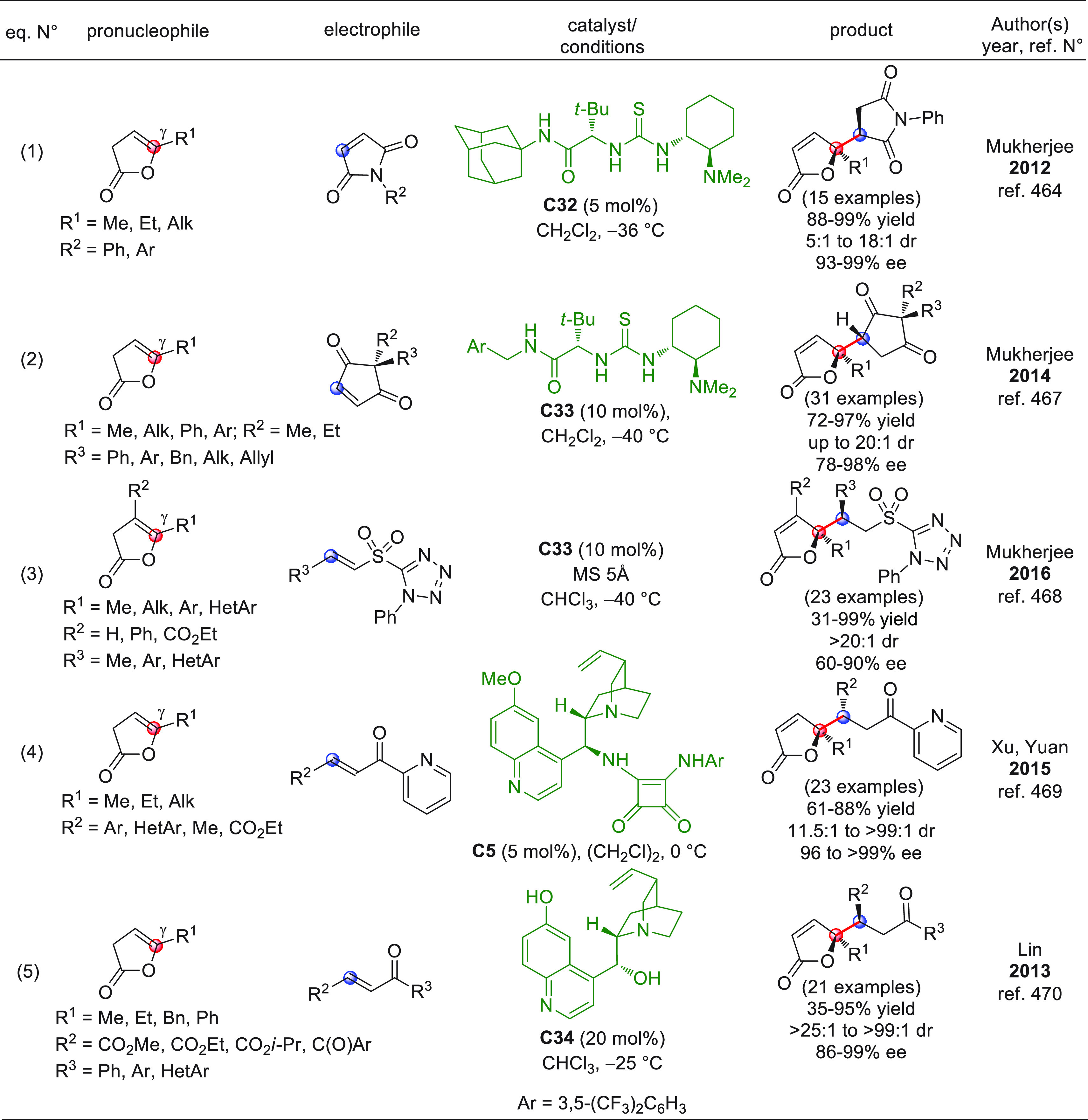
Also, Mukherjee and Manna presented an enantioselective, catalytic desymmetrization of 2,2-disubstituted cyclopentene-1,3-diones through the direct VMcR of deconjugated butenolides (Table 8, entry 2).467 Triggered by chiral, tertiary amino thiourea C33 (10 mol %), derived from the “matched” combination of (S)-tert-leucine and (1R,2R)-diaminocyclohexane, the reaction afforded ciclopentadione products featuring two quaternary and one tertiary stereocenter in high yields (up to 97%) and diastereoselectivity (>20:1).
More recently, Mukherjee et al. exploited the same catalyst C33 to promote a formal γ-allylation reaction of deconjugated butenolides based on a two-step sequence consisting of a catalytic diastereo- and enantioselective VMcR to vinyl sulfones followed by Julia–Kocienski olefination (Table 8, entry 3).468 This highly modular approach delivered densely functionalized butenolides containing a quaternary stereogenic center in excellent yields with good to high enantioselectivites.
Another interesting class of Michael acceptors used to functionalize α-angelica lactone derivatives through vinylogous transformations is represented by 2-enoylpyridines, bearing an α,β-unsaturated carbonyl moiety flanked by a pyridine core (Table 8, entry 4).469 In this context Xu, Yuan, et al. reported an asymmetric, anti-selective direct VMcR of alkyl-substituted deconjugated butenolides to 2-enoylpyridines catalyzed by the epi-quinine squaramide C5, to afford a number of γ,γ-disubstituted butenolide derivatives bearing two consecutive tri- and tetrasubstituted stereogenic centers, in acceptable yields (up to 88%) with excellent stereoselectivities (up to >99:1 dr and up to >99% ee).
As depicted in entry 5, 3-aroyl acrylates and 1,2-diaroylethylenes have been successfully used by Lin and co-workers,470 as Michael acceptors in a syn-selective organocatalytic, enantioselective, direct VMcR of deconjugated butenolides, to provide the corresponding γ,γ-dialkylated butenolides in high yields and almost complete distereo- and enantiocontrol. The reaction, promoted by bifunctional 6′-OH quinine derivative C34 (20 mol %), was quite general, since both alkyl- and aryl-substituted nucleophiles, as well as electron-rich or electron-deficient electrophiles, were compatible substrates under the optimized reaction conditions.
As Tables 7 and 8 demonstrate, β,γ-unsaturated furanones represent highly versatile pronucleophilic scaffolds that enable access to valuable γ,γ-disubstituted butenolides in a stereocontrolled manner.
Quite surprisingly, the use of conjugated α,β-unsaturated furan(5H)ones as pronucleophiles in direct, catalytic, vinylogous Michael-type transformations has been much less explored. One of such few examples was published by Ji, Wang, et al. in 2010, reporting an amine-thiourea promoted direct Michael addition of γ-butenolides to chalcones to access chiral, enantioenriched γ-alkylated butenolides (Scheme 176, eq 1).471 Indeed, under optimized reaction conditions, Takemoto’s amine-thiourea bifunctional catalyst C8 (20 mol %) was able to promote the VMcR between 440 and a series of aromatic and heteroaromatic chalcones 689 with the aid of LiOAc as a basic additive, to provide the vinylogous, syn configured adducts 690 in good to excellent diastereoselectivity and moderate to good enantioselectivities. Almost contemporarily, Liang, Ye, et al. reported a very similar transformation involving the direct, organocatalytic, asymmetric VMcR of 440 with 689 with a l-valine-derived multifunctional primary amine salt A38 as the catalyst (Scheme 176, eq 2).472 The reaction, in the presence of N-Boc-l-phenylalanine (10 mol %) as acidic additive, resulted in the formation of the corresponding ketobutenolides 690 with satisfactory yields and high diastereo- and enantioselectivities (up to 20:1 dr and 95–99% ee). Of note, differently from the previous methodology, several aliphatic ketones proved suitable substrates for this direct VMcR.
Scheme 176.

The allylic alkylation with Morita–Baylis–Hillman (MBH) adducts of type XXXVI (Scheme 177) through the catalysis of metal-free chiral Lewis bases has emerged as a powerful stereoselective strategy to deliver enantioenriched, multifunctional compounds. Capitalizing on the seminal works by the Krische473 and Shi’s groups,474 MBH carbonates or acetates proved to be easily activated by suitable chiral phosphines or tertiary amines via an SN2′-type mechanism affording a transient α,β-unsaturated carbonyl intermediate XXXVII (with the concomitant PGO– release) that may be engaged in 1,4-conjugate addition reactions with suitable nucleophiles.
Scheme 177.

Concerning the addition of vinylogous furan-derived dienolates, limitations on the use of unsubstituted preformed 2-silyloxyfurans boosted the development of new methodologies in which pronucleophilic α-angelica lactones of type XXXVIII were first in situ activated by a base to the corresponding dienolates, enabling the γ-selective 1,4-addition (VMcR) to XXXIX that, through a second SN2′-type pathway would provide the γ-alkylated butenolides XL featuring adjacent quaternary and tertiary stereocenters.
Along this line, one of the first examples of direct, enantioselective, vinylogous allylic alkylation of β,γ-unsaturated butenolides with ester- or ketone-derived MBH carbonates of type 692 to access γ,γ-disubstituted butenolides 693 was reported by Chen et al. in 2010 (Scheme 178, eq 1).475 After a first optimization survey, the C2-symmetric hydroquinidine catalyst C35 [(DHQD)2PYR] was elected as the best catalyst to promote the reaction between γ-substituted lactones 691 and aromatic MBH carbonates 692 in trifluomethylbenzene at 50 °C, giving access to the corresponding γ,γ-disubstituted butenolides 693 in moderate to good yields (up to 83%) and excellent stereoselectivities (dr >95:5 and up to 96% ee).
Scheme 178.

More recently, (2015) a very similar asymmetric, vinylogous allylic alkylation between 691 and a series of MBH carbonates of type 692 was reported by Sha, Wu et al. (not shown),476 using a chiral, bifunctional, squaramide-phosphine catalyst. Optically active γ,γ-disubstituted butenolides 693 were accessed in good-to-excellent yields (up to 98%) and excellent stereoselectivities (up to 99:1 dr, and up to 99% ee).
Chen et al. were also engaged in the development of an organocatalytic asymmetric assembly of isatin-derived Morita–Baylis–Hillman carbonates 694 and α-angelica lactones 691 to build the structurally challenging enantioenriched oxindole frameworks 695 (Scheme 178, eq 2).477 The reaction, carried out in mesitylene at −10 °C in the presence of 4 Å MS was efficiently catalyzed by β-isoquinidine C36 with the aid of slightly acidic (R)-BINOL additive (L20, 10 mol %), affording a set of multifunctional γ-adducts 695 in high yields (up to 91%) and stereoselectivities (up to 92% ee, dr >95:5).
Another useful family of pronucleophiles that showed a marked versatility in direct, vinylogous transformations are the 3-cyano-4-methylcoumarins 696, featuring an acidic γ-C(sp3)-H, which is enolizable under mild basic conditions thus enabling its γ-functionalization with suitable electrophilic partners. In this context, the first highly regio-, chemo-, and enantioselective direct VMcR of 3-cyano-4-methylcoumarin derivatives to a series of aromatic α,β-unsaturated ketones 697 was reported by Xie and co-workers in 2010, employing readily available 9-amino-9-deoxy-epiquinine A23 as the catalyst (Scheme 179).478 The reaction, carried out in CH2Cl2 at rt in the presence of A23 (20 mol %), afforded the corresponding S-configured, γ-homologated coumarins 698 in high yields (up to 91%) and enantioselectivity (up to 95% ee). A plausible mechanism was proposed (not shown), in which the chiral primary amine within the catalyst activated ketone 697 via formation of the corresponding iminium ion, while 3-cyano-4-methylcoumarin would be deprotonated by the quinuclidine moiety of the catalyst to generate the active dienolate species.
Scheme 179.

The just described application of 3-cyano-4-methylcoumarins as privileged vinylogous pronucleophiles in the enantioselective VMcR to unsaturated ketones paved the way to further applications and scope extensions. In this context, a successful organocatalyzed, enantioselective vinylogous allylic alkylation of coumarins was recently realized by Kayal and Mukherjee in 2017, using dimeric cinchona alkaloid C37 (QD)2PHAL, as the catalyst of choice (Scheme 180, eq 1).479 Under the optimized reaction conditions, C37 (10 mol %) promoted the reaction of differently substituted 4-methylcoumarins 699 with aromatic MBH carbonates 700 providing the corresponding γ-selective, functionalized coumarin 701 in good yields (up to 79%) and good to high enantioselectivities (up to 94% ee).
Scheme 180.

Similar results were later described by Kowalczyka and Albrecht in 2018 (Scheme 180, eq 2),480 who reported the allylic alkylation of ester- and nitrile-derived MBH carbonates 702 with 3-cyano-4-methylcoumarins 699 promoted by the dimeric C35 (DHQD)2PYR. Here, the reaction, carried out in CH2Cl2 at 40 °C afforded the corresponding γ-adducts 703 with improved yields (up to 98%) and enantioselectivities (up to 99% ee).
As witnessed for butenolide structures, oxazolones (azlactones) XLI (Scheme 181) are excellent templates for diversity-oriented syntheses of precious polyfunctionalized products, such as amino acids and heterocyclic structures.317 Therefore, the use of these building blocks in the field of organocatalysis constitutes an important tool in modern asymmetric synthesis. Azlactones of type XLI may be deprotonated at C4 by a suitable base, to provide the corresponding oxazole-dienolate XLI′ that reacts as a bidentate nucleophile with various electrophilic partners. Interestingly, depending on the substitution pattern of the azlactone, a γ-C2 vs α-C4 addition occurs, posing the key regioselectivity issue. When 2-aryl-substituted azlactones are used, mainly vinylogous γ-addition is observed affording azlactones XLII, whereas 2-tert-butylazlactones exclusively afford the nonvinylogous α-addition regioisomers of type XLIII (Scheme 181).
Scheme 181.

In this context, aryl-substituted oxazol-5-(4H)-ones (azlactones) of type 704 were exploited by Jørgensen and co-workers in 2010 as pronucleophilic scaffolds to demonstrate the excellent hydrogen-bond acceptor ability of unsaturated acyl phosphonates as electrophilic components in enantioselective, organocatalyzed Michael-type addition reactions (Scheme 182, eq 1).481 Indeed, it was found that bifunctional 9-epi-quinine-thiourea C2 selectively orchestrated the γ-addition of azlactones 704 to a series α,β-unsaturated acyl phosphonates 705 to afford the corresponding γ-alkylated products. Overall the reaction entailed a double nucleophilic reaction, and it was shown that the acyl phosphonates could serve as masked ester or amide equivalents, which upon quenching with a second nucleophile (MeOH, EtOH, morpholine, or 4-Br-benzylamine) provided a broad spectrum of optically active conjugate adducts 706 in good yields (up to 79% yields) and high enantioselectivities (up to 99% ee). Other different nonvinylogous carbon-based nucleophiles such as indoles and 1,3-dicarbonyl compounds were tested and the obtained results served as a base to postulate a general transition state involving azlactone pronucleophile addition, in which the oxazolone dienolate reacted as an electron-rich aromatic compound, which might form π–π interactions with the electron-poor aryl group of the adjacent thiourea motif (not shown).
Scheme 182.

In the same year, Moyano, Rios, et al. developed the first highly diastereo- and enantioselective organocatalytic entry of 2,2-disubstituted-2H-oxazol-5-ones 708 (Scheme 182, eq 2),482 via γ-selective vinylogous Michael addition of aryl-substituted oxazolones 704 to symmetric N-aryl maleimides 707. The C4-alkyl substitution in the pronucleophile, as well as several fluoroaryl substituents within the azlactone and maleimide scaffolds, worked well under the optimized reaction conditions, guided by Takemoto’s bifunctional thiourea catalysts C8, affording the corresponding adducts 708 with high yields (up to 99%) and very high regio- and stereocontrol (up to >25:1 dr, and up to 99% ee). Also, an (S)-isoleucine-derived chiral azlactone pronucleophile was tested, yielding the corresponding γ-adduct in 89% yield and a good 10:1 dr (not shown).
The reactivity scope of pronucleophilic azlactone and butyrolactone scaffolds in stereocontrolled vinylogous Michael-type additions was further expanded by Jørgensen and co-workers in 2013, developing a new, organocatalytic, enantioselective vinylogous 1,4- and 1,6-addition of methyl-substituted alkylidene azlactones and furanone congeners to unsaturated aldehydes (Scheme 183).483 Initially, the feasibility and scope of enolizable 2-phenyl-4-arylidene azlactones 709 for nucleophilic attack to both aryl- and alkyl-substituted enals 711 was tested. It was found that TBS-protected diphenylprolinol catalyst A13 (20 mol %) in the presence of Et3N (20 mol %) and brine (3 equiv) in CH2Cl2 at rt were the best reaction conditions, affording optically pure γ′-adducts 712 in good yields (up to 77% yield) and almost perfect stereocontrol (Scheme 183, eq 1).
Scheme 183.

Also, methyl- and iodo-substituted aromatic systems within the ylidene chain of 709 were tolerated giving comparable performance. Similar reaction conditions were then applied to the reaction between 5-phenyl-3-arylidene butyrolactone congeners 710 and enals 711. Here, catalyst A13 (20 mol %) with the aid of i-Pr2EtN (20 mol %) and brine (3 equiv) provided the corresponding S-configured γ′-homologated adducts 713 in high yields and stereoselectivity (eq 1). Finally, in order to extend the generality and the potential of the disclosed reaction, the authors applied the developed methodology to linear 2,4-dienals 714 (Scheme 183, eq 2). Remarkably, applying the latter conditions to the reaction between phenyl-substituted azlactones 709 or furanones 710 and alkyl substituted dienals 714, only the 1,6-addition took place, providing the corresponding enantioenriched γ′-adducts 715 and 716 with a full control of the double-bond geometry (>20:1 E/Z for both olefins). Interestingly, the 1,6-selectivity turned out to be dependent on the nature of the 2,4-dienal: in fact, addition of 709 or 710 to a phenyl-substitued dienal congener proceeded with lower conversions, yielding a mixture of 1,4- and 1,6-addition products (not shown). Suitable transition states were proposed by the authors to justify the observed regio- and enantioselectivities. Indeed, it was postulated that the negatively charged oxygen atom in the vinylogous dienolate species could be stabilized by the positively charged nitrogen atom of the iminium-ion intermediate, aligning the electrophilic β- or δ-carbon atom of the iminium-ion intermediate with the γ′-position of the dienolate promoting the high site-selective 1,4 vs 1,6 conjugate addition (Scheme 183). Furthermore, π–π stacking interaction between the β-aryl group of enal and the β′-aryl moiety of dienolate is proposed to be an important factor, stabilizing the transition state of the 1,4-addition, while the stereochemical outcome of the vinylogous additions toward enals and 2,4-dienals would be controlled by the shielding effect of the TBS-protected prolinol catalyst A13.
Finally, it is worth mentioning the direct, organocatalytic, enantioselective intermolecular cross- vinylogous Rauhut–Currier reaction of methyl coumalate 717 and α,β-unsaturated aldehydes 718 recently developed by Liu and Zu, in which the enals were activated via iminium ion catalysis to serve as the Michael acceptors and methyl coumalate was used as an activated diene to generate the latent enolate (Scheme 184).484 Indeed, prolinol silylether A13 in the presence of 2-trifluoromethylbenzoic acid (720) as cocatalyst promoted the conjugate addition of pronucleophilic ester 717 to a varied repertoire of aromatic and aliphatic enals 718 to provide enantioenriched products 719 in high yields (up to 99%) and enantioselectivity (up to 99%). Several alternative pathways were proposed with a common pattern: a first nucleophilic, conjugate addition of EtOH or the prolinol to the C6 of methyl coumalate 717 would generate a transient dienolate 717′, which could trigger the subsequent intermolecular 1,4-addition to the chiral, α,β-unsaturated iminium ion 718′. The resulting adduct 717′′ could then undergo rapid aromatization-driven elimination to deliver the final product 719.
Scheme 184.

5.3.2. Indirect Procedures
5.3.2.1. Acyclic Nucleophiles
One of the few VMMcR involving linear silyloxydienes was introduced in 2015 by Wu and Li, who developed an In(III)-catalyzed vinylogous addition of diverse γ-substituted (alkyl, aryl, benzyl) O- tris(trimethylsilyl)silyl vinylketene acetals 721 to 2,3-dihydro-4-pyridinones 722 (Scheme 184, eq 1).485
Interestingly, the supersilyl group (TTMSS) was found to be the best silyl group within the nucleophile to influence the γ vs α regiochemical control of the VMMcR promoted by In(OTf)3 (5–10 mol %) in toluene at −20 °C: in fact, the corresponding anti-configured γ-alkylated products of type (±)-723 could be obtained in good yields (up to 82%) and high diastereocontrol (up to 20.7:1). A plausible antiperiplanar transition state was proposed, in which a secondary orbital overlap (i.e., π–π stacking interactions) between the vinylketene moiety of 721 and the carbamate of the activated acceptor 722′ favored a Re–Re approach. Furthermore, γ-unsubstituted O-TBS vinylketene acetals 724 were also tested as nucleophiles in the VMMcR to various 2-substituted dihydropyridinones 725 (Scheme 185, eq 2). Under the same reaction conditions, a range of trans-disubstituted 4-piperidinones (±)-726 were accessed in comparable yields and diastereoselectivity, without the need of the more hindered supersilyl group. To account for the observed anti-facial selectivity, a combination of stereoelectronic and steric control was considered.
Scheme 185.

The author proposed a preferred half-boat conformation of activated 725′, with the C2 phenyl group adopting a pseudoaxial position to avoid diaxial 1,3-interactions with the N-Boc group. This would promote the vinylogous, bottom face addition of silyloxydiene 724, providing the observed anti-configured adducts.
In 2015, Schneider and Nareddy disclosed a highly enantioselective access to chiral 1-cyclopentenyl-α-keto esters 728 through the implementation of an organocatalytic, domino VMMcR/intramolecular Knoevenagel-type condensation of linear bis-silyl-1,3-dienediolate 603 to a varied set of aromatic and aliphatic α,β-unsaturated aldehydes 727 (Scheme 186).486
Scheme 186.

Using the Hayashi–Jørgensen prolinol TMS-ether A4 as the chiral organocatalyst, the reaction, carried out in Et2O under buffered conditions (pH 4), in the presence of an acidic additive such as 2,4-dinitrobenzoic acid 125 (1.0 equiv), afforded the 1-cyclopentenyl-α-keto esters 728 with high γ-regioselectivites (up to >20:1 γ:α), moderate to good yields (up to 65%), and excellent enantioselectivities (up to 98% ee). Mechanistically, this process involved a first, γ-selective intermolecular VMMcR between dienediolate 603 and the iminium ion derived by the covalent activation of the unsaturated aldehyde 727 by A4, generating a chiral, iminium ion intermediate 727′ that was then annulated by an intramolecular Knoevenagel-type condensation.
5.3.2.2. Cyclic Nucleophiles
An indirect vinylogous, allylic alkylation of Morita–Baylis–Hillman (MBH) acetates 729 with TMSOF 496a was developed by Shi and co-workers in 2011, introducing a series of multifunctional, chiral amide–phosphane scaffolds as useful organocatalysts (Scheme 187).487 Indeed, chiral amide-diphenylphosphane P8 promoted the reaction of 496a and 729 under mild reaction conditions in absolute MeOH or MeCN, giving access to the corresponding anti-configured γ-homologated butenolides 730 in good to excellent yields (42–98%) and high enantioselectivities (85–99%). The reaction was quite general, so that aromatic and heteroaromatic ketones and ester derived MBH acceptors 729 were good substrates for the reaction. As for the mechanism, it was proposed that a tandem SN2′/SN2′ substitution pathway could be operative (see Scheme 177), and NMR tracing experiments provided evidence for the existence of phosphonium ion intermediate 729′ being involved in the proposed Diels–Alder-like transition state (Scheme 187, bottom).
Scheme 187.

A highly efficient catalytic, asymmetric VMMcR of 2-silyloxyfuran with chalcone derivatives 731, catalyzed by a chiral N,N′-dioxide–scandium(III) complex, was reported by Feng et al. in 2011 (Scheme 188).488,489 In particular, 2-(tert-butyldimethylsilyloxy)furan (TBSOF, 496c) was coupled with a large set of differently substituted aromatic α,β-unsaturated ketones 731 in a t-BuOH/ethyl propionate mixture at 0 °C, under the guidance of Feng’s Sc(OTf)3-N,N′-dioxide ligand L22, to afford the sole 5,1′-anti-configured γ-adducts 732 in very high yields (up to 99%) and good to excellent enantioselectivities (up to 94% ee).
Scheme 188.

To account for the observed (5S,1′S)-stereoselectivity, a plausible transition state model was proposed in which the N-oxide and amide oxygen atoms of L22 coordinated to scandium ion in a tetradentate manner to form two six-membered chelate rings. The chalcone acceptors 731 bound to scandium in a favorable equatorial position, displaying the less hindered Si face to the attack by the Re face of the nucleophile.
Another asymmetric, metal-catalyzed VMMcR of silyloxyfurans 733 with α,β-unsaturated 2-acyl imidazoles 734 was recently reported by Singh et al. using either chiral Sc(III) or Er(III) complexes with a chiral, C2-symmetric ligand L23 (Scheme 189).490 In particular, the Sc(OTf)3·L23 catalyzed VMMcR of silyloxyfurans 733 and a vast set of aryl and Me-substituted acyl imidazoles 734, carried out in CHCl3 at rt in the presence of hexafluoroisopropanol (HFIP) as an additive, provided the expected γ-adducts 735 in good yields (up to 93%) with excellent diastereo- and enantioselectivity (up to >20:1 dr and 98% ee). Also, catalytic Er(OTf)3·L23 complex worked well under similar reaction conditions, affording several γ-adducts 735 in comparable yields and selectivities.
Scheme 189.

Switching to VMMcR involving cyclic electrophilic partners, the first enantioselective, Cu(II)-catalyzed asymmetric addition of 2-silyloxyfurans 736 to cyclic unsaturated oxo esters 737 was developed by Chabaud, Guillou, et al. in 2013 (Scheme 190, eq 1).491,492 Here, the C2-symmetric isopropyl-box (L14)·Cu(OTf)2 complex was elected as the best chiral catalytic system for the VMMcR involving 736 and a series of differently sized and substituted α,β-unsaturated oxo esters 737 providing the corresponding γ-adducts 738 in good yields (up to 81%) and excellent diastereocontrol (usually dr 99:1), albeit with modest to high enantioselectivities (59–96% ee), depending on the nature of the ester group and the substitution of the cyclic oxo ester.
Scheme 190.

A similar catalytic system was also exploited by Bolm and co-workers in 2015 for the synthesis of valuable enantioenriched, phosphorus-containing compounds through an asymmetric Cu(II)-catalyzed VMMcR between differently substituted cyclic dienol silanes 739 and α,β-unsaturated α-keto phosphonates 740 (Scheme 190, eq 2).493
Using the combination of the commercially available C2-symmetric bisoxazoline ligand L24 with copper(II) perchlorate hexahydrate salt [Cu(ClO4)2·6H2O], the VMMcR of 739 and 740 provided a variety of γ-homologated butenolides 741 in moderate to high yields (up to 82%) and high stereoselectivities (up to 99:1 dr and up to 98% ee). Of note, when the 4-methoxy-furan derivative was used as the nucleophilic partner, a different chemoselectivity was observed, providing the 1,2 aldol adduct instead of the 1,4, in a low 22% yield, 2.3:1 dr, and 97% ee (not shown).
An unprecedented vinylogous 1,6-Mukaiyama Michael/aza-Michael cascade of 2-silyloxyfurans 742 and in situ formed electrophilic azoalkenes of type 743′ was realized by Wang et al. in 2015 (Scheme 191, eq 1).494 After a deep optimization survey, Cu(II)·L24 complex was found to be the best catalytic system to enable the in situ formation of electrophilic azoalkenes 743′ from the corresponding alkyl chlorides 743 (with the aid of Na2CO3), triggering the γ-regioselective, 1,6-Mukaiyama Michael reaction of 742 to 743′. The aza-anion intermediate 743′′ thus formed cyclized via intramolecular conjugate addition, to afford the final butyrolactones 744 in good yields (up to 90%) with excellent stereoselectivity (up to 20:1 dr, and up to 99% ee).
Scheme 191.

Following this strategy, the same group developed a nickel-catalyzed 1,6-VMMcR/aza-Michael cascade of 2-silyloxyfurans 745 with N-sulfonyl-1-aza-1,3-dienes 746, affording the corresponding fused piperidine/lactone scaffolds (±)-747 with excellent diastereoselectivity (>20:1) and highly functional group tolerance (Scheme 191, eq 2).495
In addition to these metal-catalyzed asymmetric methodologies, a series of useful organocatalytic, enantioselective vinylogous Michael-type transformations were developed and applied to the total synthesis of valuable target molecules. Notably, enantioselective, iminium-catalyzed (LUMO-lowering) reactions of cyclic silyl ketene acetals to simple acrolein and methacrolein acceptors were introduced by Pihko and co-workers in 2012 and 2014, en route to the synthesis of the key C17–C28 segment of the cytotoxic marine natural products pectenotoxins (Scheme 192, eqs 1 and 2).496−498 Indeed, the authors envisaged the possibility to access key 5S-configured γ,γ-disubstituted butenolide intermediates 750 by implementing an asymmetric VMMcR between substituted furanone 748 and a suitable enal 749 like acrolein (R3 = H) or methacrolein (R3 = Me), promoted by a chiral secondary amine such as C2-symmetric 2,5-diphenylpyrrolidine ent-A19, which was the most suited for the purpose.
Scheme 192.

Although a wide range of iminium-catalyzed enantioselective reactions are known also in the field of vinylogous transformations, in the vast majority of cases the reactions have been restricted to β-substituted enals, so that enantioselective additions to α-substituted enals such as methacrolein (typically resulting an unreactive substrates) were quite challenging.499,500 In this context, Pihko disclosed that pyrrolidine ent-A19 (10 mol %) in the presence of 4-nitrobenzoic acid 192 (10 mol %) as cocatalyst and some amount of H2O (2.0 equiv) was an effective catalyst in promoting the VMMcR between silyloxyfurans 748 and β-substituted enals 749 (Scheme 192, eq 1), providing the corresponding γ-homologated products 750 in good yields (up to 90%) and stereoselectivities (up to 15.6:1 dr and up to 96% ee). Interestingly, the far from obvious rationalization of the observed enantioselectivity was supported by DFT computational studies, which unveiled the presence of attractive noncovalent interactions as key factors in controlling the enantiocontrol of the reaction. On these bases, a plausible transition state was proposed in which the Si face of silyloxyfuran 748 attacks the iminium intermediate via a Diels–Alder-like approach.
Another iminium-ion (LUMO-lowering activation), organocatalyzed VMMcR between γ-propargyl silyloxyfuran 751 and silyl enal 752 was exploited by Xie and co-workers in 2016 for the stereocontrolled construction of the C5-epi ABCDE-ring system of rubriflordilactone B (Scheme 192, eq 2).501 Irrespective of the bulkiness of the reagents, chiral prolinol silyl ethers A4 or A2/125 (20 mol %) catalyzed the VMMcR in the presence of H2O (2.0 equiv), allowing the construction of optically pure γ,γ-disubstituted butenolide 753 in good yield as a sole isomer (>20:1 dr).
Finally, a covalently activated, asymmetric, organocatalyzed VMMcR of 2-silyloxyfurans 754 to medium and large cyclic enones of type 755 was reported by Singh and co-workers in 2015 (Scheme 193).502 In this report, chiral, C2-symmetric primary diamine A24 (20 mol %) in the presence of trichloroacetic acid (TCA, 20 mol %) promoted the efficient syn-selective addition of 754 to 755, to provide the corresponding γ-homologated butenolide 756 in good yields (up to 92%) and high stereoselectivities (up to >99:1, and up to >99% ee).503−505
Scheme 193.

5.4. Other Reactions
5.4.1. Direct Procedures
5.4.1.1. Acyclic Pronucleophiles
In 2012, Tan and co-workers devised a guanidine-catalyzed, enantiodivergent, vinylogous allylic amination between linear, unconjugated β,γ-unsaturated thioesters 757 and dialkyl azodicarboxylates 758 (Scheme 194).506 Interestingly, the reaction catalyzed by chiral guanidine C37 and performed on (E)-757 afforded the corresponding 4S-configured γ-adducts 759 in good yields (up to 90%) and enantioselectivity (up to 96% ee). Coversely, starting from (Z)-757 and di-tert-butylazodicarboxylates 758a, under similar reaction conditions, the enantiodivergent access to 4R-configured adducts ent-759a could be assessed with comparable efficiency and selectivity. Computational studies were also performed envisaging a side-on mechanism with the formation of an s-trans dienolate (not shown). The theoretical study agreed well with experimental results providing an intuitive explanation for the inversion of the absolute configuration.
Scheme 194.

More recently, a Cu(II)-catalyzed, vinylogous aerobic oxidation of unsaturated esters with air was reported by Newhouse, Yin, et al. (Scheme 195).507 Under the optimized reaction conditions, a mild and operationally simple Cu(OTf)2-catalyzed vinylogous aerobic oxidation of β,γ-unsaturated ester 760 and the corresponding α,β-unsaturated congener 762 afforded γ-hydroxy ester derivatives (±)-761 in good yields (up to 86–92%) as single diastereoisomers (Scheme 195, eqs 1 and 2). Furthermore, under similar reaction conditions, bisvinylogous and hypervinylogous oxidations were performed, yielding the corresponding polyunsaturated products (±)-764 and (±)-766 with comparable yields and selectivities (Scheme 195, eq 3). Of note, tetramethylguanidine (TMG) was found to be crucial both as a base and as a key ligand to produce the active Cu(II) catalyst.
Scheme 195.

5.4.1.2. Cyclic Pronucleophiles
A rhodium-catalyzed, enantioselective, vinylogous epoxide-opening between methyl cyanocoumarin 767 and strained, olefinic epoxides 768 was recently devised by Lautens and co-workers, demonstrating, for the first time, how the principle of vinylogy can be applied also to the release of ring strain in polycyclic systems (Scheme 196, eq 1).508 Using a [Rh(cod)OH]2/L25 catalytic system, a series of chiral enantiopure (up to 99% ee) γ-adducts 769 were accessed in high yields (up to 98%). In general, the devised protocol tolerates a wide spectrum of substrates with varied electronic and steric properties.
Scheme 196.

Substituted methyl-cyanocoumarins 767 were also exploited by Antonchick, Waldmann, et al. as suitable pronucleophiles in a highly enantioselective copper-catalyzed vinylogous propargylic substitution, which afforded enantioenriched propargylic coumarins of type 771, which were also studied as autophagy inhibitors (Scheme 196, eq 2).509 Aromatic and aliphatic propargylic esters 770 reacted smoothly with substituted coumarins 767 under the guidance of the Cu(OTf)2/L26 catalytic system, to give the desired products with excellent yields (up to 96%) and enantioselectivities (up to 98% ee).
Transition metal-catalyzed, asymmetric allylic substitution reactions have emerged as an extremely powerful and versatile method for the synthesis of chiral, enantioenriched compounds from easily available starting materials through the enantioselective construction of carbon–carbon and carbon–heteroatom bonds. In this context, the first iridium-catalyzed enantioselective vinylogous allylic alkylation of coumarins has been recently reported almost contemporarily and independently by the groups of Mukherjee and Yin in 2018 (Scheme 197). In particular, using easily accessible linear allylic carbonates 773, as allylic electrophiles (eq 1), Mukherjee and co-workers510 found that [Ir(cod)Cl]2 complexed with chiral ligand L27 effectively promoted the vinylogous allylic alkylation of 4-methylcoumarins 772, in an exclusively branched-selective manner (>20:1 branched vs linear), affording the corresponding γ-adducts 774 in generally high yields (up to 98%) with an excellent level of enantioselectivity (up to 98%). Similarly, the catalytic asymmetric, vinylogous allylic alkylation of 4-methylcoumarins 772 and differently shaped, α,β-unsaturated lactones of type 776 was achieved by Yin et al.511 using allylic carbonates of type 775 and suitable ligands such as L27 or L28 to be complexed with [Ir(COD)Cl]2 (Scheme 197, eqs 2 and 3). Several γ-functionalized adducts of type 774 and 777 were obtained, in good yields, and with excellent regio- and enantioselectivities.
Scheme 197.

5.4.2. Indirect Procedures
5.4.2.1. Cyclic Nucleophiles
Cao, Wu, et al. introduced a novel decarboxylative formal [4 + 2] cycloaddition reaction between ethynyl benzoxazinanones 779 and 5-substituted 2-silyloxyfurans of type 778 (Scheme 198).512 The reaction, catalyzed by the chiral copper/L29 complex in the presence of ethyl morpholine (781) as a base, provided enantioenriched tetrahydroquinolines fused with a butyrolactone moiety 780, featuring three contiguous stereocenters, in high yields (up to 92%) and excellent diastereo- and enantioselectivities (up to 99:1 dr and up to 98% ee).
Scheme 198.

Concerning the mechanism, upon conversion of benzoxazinanone scaffolds 779 into the corresponding Cu–allenylidene species 779′, a first γ-selective addition of 778 to the C4 of 779′ took place affording the γ-homologated butenolide intermediate 779′′, that undergoes an intramolecular aza-Michael reaction providing the final tetrahydroquinoline targets 780. To account for the observed selectivity, a transition state was proposed in which the Si face of 778 approached the relatively less sterically hindered Re face of the Cu–allenylidene complex 779′.
A catalytic, enantioselective, Ir-catalyzed vinylogous allylic alkylation of trisubstituted allylic electrophiles such as allylic phosphates 783 with dioxinone-derived dienoxysilanes 782 was developed by Hartwig and Chen in 2016 (Scheme 199).513 By surveying different leaving groups within allylic electrophiles, the authors found that trisubstituted allylic phosphates 783 were suitable electrophiles for asymmetric allylation promoted by the Ir/phosphoramidite ligand L27 system, affording the corresponding branched allylated products 784 in good yields (up to 90%) and high stereoselectivity (12:1 to 20:1 branched vs linear; 5.1 to 8.1 γ vs α; up to 98% ee).
Scheme 199.

6. Vinylogous Amides and Lactams
Section 6 surveys original research appearing between 2010 and 2018 and dealing with the exploitation of α,β-unsaturated amides and lactams as vinylogous donors in asymmetric synthetic methodologies.
The ability to construct functionality-rich chiral amide- or lactam-based molecular entities in a chemo-, regio-, and stereocontrolled manner has long been the subject of extensive studies, given the widespread presence of these matrices in bioactive natural products as well as in nature-inspired synthetic intermediates and therapeutics. In this research domain, alongside the attention to the traditional and well-used aldol, Mannich, and Michael maneuvers, a growing interest is witnessed in those domino reactions where several reactive events are triggered, which comprise one or more vinylogous additions, eventually leading to cyclic or polycyclic products in an efficient and economic manner.
As already noted for other electron-rich donor classes (vinylogous aldehydes, ketones, and esters), the direct activation modalities of unsaturated amide and lactam precursors, characterized by in situ formation of di- or polyenolates, are highly represented during the period surveyed in this commentary, while the strategies based on indirect activation of these matrices (e.g., via silyl enol ether preformation) are limited in number but still used in target-oriented synthesis.
According to the main organization of this review article, acyclic and cyclic pronucleophiles displayed in Figure 7 will be sequentially discussed, where the electronic effects of the amide carbonyl system are transmitted to the remote position through conjugated double (or triple) bonds which either belong to acyclic chains or are inserted, at least partially, into a ring system.
Figure 7.

Panel of acyclic and cyclic amide pronucleophiles used in direct procedures (above) and preformed nucleophiles (silyl ketene N,O-acetals) exploited in indirect procedures (below). Red circles denote the reactive (pro)nucleophilic carbon site.
6.1. Additions to C=O Bonds
6.1.1. Direct Procedures
The direct catalytic asymmetric addition of vinylogous amide donors to carbonyl electrophiles (VAR) has emerged as a powerful and widely exploited strategy to assemble δ-hydroxy-α,β-unsaturated amides that represent advanced synthons in the preparation of natural product mimetics and therapeutics. The direct use of pronucleophilic molecular entities avoiding preactivation and protecting group installation processes is highly desirable for convenience and atom economy and has accordingly become the most applied strategy in catalytic asymmetric vinylogous reactions; for this reason, the studies focused on direct activation modalities of the amide precursors are much more frequent in the literature and, among these, those exploiting heterocyclic pronucleophiles have attracted the major interest.
6.1.1.1. Acyclic Pronucleophiles
The use of simple acyclic amide pronucleophiles in direct asymmetric aldol maneuvers is of great interest, even though poorly represented in 2010–2018, likely due to the intrinsic low reactivity of the generated dienolates. In most cases, these reactions constitute the first step of a reaction cascade leading to cycloaddition products. In 2014, based on previous investigations concerning the use of pyrazoleamides as donor species in Michael and Mannich addition reactions, Sha, Wu, and colleagues developed the first direct, enantioselective vinylogous aldol addition of allyl pyrazoleamides to isatins, as the initial step of a cyclization reaction cascade producing chiral spirocyclic dihydropyran-2-ones (Scheme 200, eq 1).514 β,γ-Unsaturated deconjugated amides 785 were chosen as precursors of the vinylogous enolates, since the tertiary amine catalysis was ineffective in promoting γ-enolization of the corresponding α,β-unsaturated counterparts. The pyrazoleamide function within pronucleophiles 785 was selected as an activating/directing group for enhancing stereocontrol, as well as a good leaving group enabling further transformations (in the event, the spirocyclization step). Aiming at the synthesis of alkaloidal bioactive compounds, isatins 786 were selected as electrophilic components to assemble chiral spiro-oxindole scaffolds. After a careful exploration of the tertiary amine-thiourea bifunctional organocatalysts and the reaction conditions, the authors succeeded in the construction of various chiral spirocyclic oxindole dihydropyranones 787 in excellent yields and good-to-excellent enantioselectivities, by using as low as 1 mol % of the Takemoto catalyst C8. The absolute S-configuration of the newly formed quaternary stereocenter was attributed on the basis of the literature optical rotation values, whereas the authors did not investigate the role of the organocatalyst in the formation of the sole quaternary stereocenter.
Scheme 200.

The same β,γ-unsaturated pyrazole amide donors 785 were exploited, two years later, by the same authors in vinylogous aldol-spirocyclization cascade reactions with o-quinone acceptors 788, in the presence of a chiral diamine-based thiourea organocatalyst, to produce spirocyclic pyranones 789 (Scheme 200, eq 2).515 The initial screening of the chiral promoter led to the selection of catalyst C38, whose H-bonding donor ability was judged crucial for the overall enantioselectivity of the process. The substrate scope was explored by reacting variously β-aryl (and β-alkyl) substituted allyl pyrazoleamides 785 with different 1,2-diketones 788 in the presence of C38 (5 mol %): spirocyclic dihydropyran-2-ones 789 were obtained in high yields (76–99%) and enantioselectivities ranging from 82 to 95%. Control experiments confirmed the role of the tertiary amine in the initial formation of the dienolate intermediate and the selective γ-reactivity of pyrazoleamides.
In their continuing effort toward the synthesis of chiral 5,6-dihydropyran-2-ones, Wu and colleagues envisaged that the pyrazoleamides of type 785 could act, once again, as γ-pronucleophiles in the asymmetric VAR-cyclization sequence with acyclic α-keto esters 790 to form chiral dihydropyranones 791 in a highly chemo- and enantioselective manner (Scheme 200, eq 3).516 The use of various allyl pyrazoleamides with a variety of γ-aryl α-keto esters in the presence of organocatalyst C22 (5 mol %) ensured the preparation of chiral cycloadducts 791 bearing quaternary stereocenters in high yields and enantioselectivities.
6.1.1.2. Cyclic Pronucleophiles
The use of α,β-unsaturated γ-butyrolactam pronucleophiles as donors in vinylogous aldol reactions, in principle, offers limitless possibilities to generate chiral highly functionalized nitrogen-containing heterocycles; N-Boc pyrrolinone, for example, has been widely employed as the immediate precursor of the corresponding pyrrole silyl dienolates, which served in a huge number of vinylogous indirect aldol, Mannich, and Michael transformations (vide infra). During the 2010–2018 period, however, the use of this simple nitrogen heterocycle in direct versions of such pivotal addition maneuvers has been marginally explored and just one contribution concerning the exploitation of pyrrolinone dienolates was found; on the other hand, contributions exploiting direct procedures based on heterocyclic scaffolds as 3-alkylidene oxindoles as starting materials are much more represented.
In 2014, Pettus and colleagues proposed a general diastereoselective metal-catalyzed VAR of tetramate pronucleophiles with various aldehydes (Scheme 201).517 Since the base-catalyzed aldol addition of tetramate-derived dienolates (obtained from 792) gave unsatisfactory results in terms of yield and stereoselectivity, the authors set up a one pot-two step Mukaiyama-type addition protocol by treating tetramates 792 with the Et3N/TMSOTf system, to in situ generate silicon dienolates, followed by addition of the aldehyde acceptors 793 and the catalytic Lewis acid (SnCl4). Syn-configured aldol adducts 794 were obtained with moderate-to-good efficiency and diastereoselectivity. An open-chain transition state was hypothesized by the authors, in which the carbonyl oxygen and the aryl-substituted tetramate nitrogen were both involved in the metal-chelation, accounting for the syn-selective outcome of the process. Moreover, the authors observed that the nature of the R1 substituent markedly affected both the efficiency and stereoselectivity of the process, with bromo-derivatives affording best results, and that the reaction was quite general with respect to the aldehyde structure.
Scheme 201.

Oxindole structures are common heterocyclic motives shared by many alkaloidal natural compounds and synthetic bioactive compounds. In particular, 3-alkylidene-2-oxindole frameworks (Scheme 202) have been the subject of great interest in the search of asymmetric synthetic methodologies to build oxindole and spirooxindole derivatives;518,519 however, if the electrophilic reactivity at the β-position has been widely explored in many examples of Michael addition processes, the vinylogous pronucleophilic character of these scaffolds, when a γ-enolizable site is present, has drawn attention only in recent years.
Scheme 202.

The first to realize the considerable potential of 3-alkylidene oxindoles as multifunctional γ-enolizable architectures was the group of Curti, Rassu, and Zanardi, who in 2012 exploited these scaffolds as vinylogous donors in direct organocatalyzed Michael reactions, as well as in indirect aldol and Mannich additions, where the corresponding silyl ketene N,O-acetals acted as vinylogous nucleophiles (vide infra).
A few years later, in 2015, Han and Chang reported on the use of 3-alkylidene-2-oxindoles of type 795 as pronucleophiles in direct addition to isatins 796 (Scheme 203, eq 1).520 In the event, the aldol-cyclization cascade was orchestrated by the bifunctional squaramide catalyst C5, producing the spirocyclic dihydropyran-2-ones 797 in moderate to excellent yields and fair enantioselectivities. It was hypothesized that, after the initial γ-deprotonation of the alkylidene oxindole moiety giving an s-cis dienolate, the addition of the nucleophile to the isatin carbonyl resulted in the formation of an aldolate, which in turn reacted with the oxindole carbonyl, leading to the unexpected opening of the lactam ring ultimately producing unsaturated lactones 797.
Scheme 203.

Expanding the scope of the vinylogous donor to prochiral or cycloalkylidene derivatives 798 (with R3, R4 corresponding to alkyl groups or belonging to a carbo- or a heterocycle), Chang and collaborators set up a diastereo- and enantioselective version of the aldol-cyclization reaction cascade previously described (Scheme 203, eq 2).521 Again, isatins of type 796 were selected as electrophilic counterpart and different cinchona-derived bifunctional organocatalysts were scrutinized. With 3-cycloalkylidene oxindoles 798 as the vinylogous donors, the best performing catalyst was the bifunctional squaramide C5, furnishing the bicyclic δ-lactones 799, bearing two vicinal new stereocenters. On the contrary, the use of 3-pentylidene indolinones 798 (R3 = Me, R4 = Et) required the deployment of the thiourea C2 and led to the enantiomeric spirooxindoles ent-799 (not shown). Scheme 203 (eq 2) shows the proposed transition state accounting for the observed configuration of compounds 799, where the ion pairing between the protonated amine catalyst and the oxindole dienolate would be operative, triggering the attack to the Re face of isatin; isatin, in turn, would be activated by the H-bonding network displayed by the squaramide moiety. The authors did not explain the reason for the enantioselectivity switch observed with thiourea C2 and concluded that other possible activation modes could not be excluded since no detailed mechanistic studies entailing DFT calculations were available.
In 2017, Singh and colleagues put in place an asymmetric aldol procedure exploiting 3-alkylidene oxindoles 800 as vinylogous pronucleophiles and α-keto esters 801 as electrophilic counterparts (Scheme 204).522 The authors hypothesized (and proved) that the cinchona-derived thiourea bifunctional organocatalyst C2 could be able, at the same time, to activate and orient the reactant partners leading to α,β-unsaturated aldol adducts 802 in a stereocontrolled manner. Among the tested organocatalysts, the thiourea C2 gave the best results, affording Z-configured δ-hydroxy esters 802 in yields ranging from 34 to 92% and enantiomeric excesses from 79 to 99%.
Scheme 204.

The utility of alkylidene oxindoles as vinylogous pronucleophiles in aldol addition-cyclization sequences is testified by another contribution proposed by Bencivenni and colleagues in 2018 (Scheme 205, eq 1).523 The authors reported on the vinylogous aldol-lactonization cascade involving alkylidene oxindoles 800 as donors and activated trifluoromethyl ketones 803 as acceptors in the presence of the bifunctional promoter C39. The organocatalytic aldol-lactonization proposed by Bencivenni represents a direct and atom economic strategy to access fluorinated α,β-unsaturated-δ-lactones as an alternative way to the cycloadditive methods based on NHC activation of enals reported by Chi and Song (vide infra). The authors selected the cinchona-derived organocatalyst C39 (Scheme 205, eq 1) to produce the concomitant γ-deprotonation of the alkylidene moiety to s-cis dienolate, the H-bond activation of the ketone, and the directioning of both the reagent partners during the aldol-cycloaddition sequence. The trifluoro-δ-lactones 804 were obtained in moderate to very good yields and excellent enantioselectivities, together with variable amounts of acyclic aldolate adducts (not shown). Assignment of the absolute configuration of the products relied upon TD-DFT calculations of electronic circular dichroism (ECD) spectra and corroborated the transition state proposed by the authors.
Scheme 205.

The efficacy of alkylidene oxindoles in additions to vinyl trifluoromethyl ketones was also demonstrated by Bencivenni et al. in a recent study that aimed at producing enantioenriched fluorinated allylic alcohols as precursors of molecular entities of biological interest (Scheme 205, eq 2).524 In this work, the authors had to face the challenging issues of chemo- and regioselectivity in addition to the stereoselectivity. In fact, when trifluoromethyl vinyl ketones of type 805 are used, both the 1,2- and 1,4-addition products can be in principle formed, and the pronucleophilic γ and γ′ positions within the oxindole 800 could compete with each other leading to a mixture of E/Z alkenes. The accurate scrutiny of various cinchona-derived catalysts revealed that both thioureas C39 and C40 (the quasi-enantiomeric 9-epi-quinidine-derived) were efficient in promoting the 1,2-addition of oxindoles 800 to ketones 805 and no traces of 1,4-adducts were found. When C39 was employed, R-configured allylic alcohols 806 were obtained, while the opposite enantiomers ent-806 were accessible by using C40 catalyst. To explain the remarkable stereocontrol of the process, the authors proposed a possible activation modality featuring the ion-pairing between the quinuclidine nitrogen of C39 and the oxindole s-cis dienolate and the concomitant thiourea H-bond activation with exposure of the ketone Si face to the nucleophilic attack (Scheme 205).
6.1.2. Indirect Procedures
The most common indirect procedures based on the use of amide (or lactam) donors entail the activation of the substrates as silicon-based di- or polyenolates before participating in the additive reaction. The past two decades have witnessed an extremely wide use of these Mukaiyama-type silyl derivatives as donors, especially in the vinylogous domain, to embody α,β-unsaturated carbonyl frameworks into linear, carbo- or heterocyclic scaffolds. The successful exploitation of extended silyl N,O-acetals is also due to their intrinsic propensity to privilege reactions at their remote sites, as demonstrated by many experimental pieces of evidence.6,34,35
6.1.2.1. Acyclic Nucleophiles
The use of chiral oxazolidinones derived from enantiopure amino acids as the auxiliaries to stereogovern the course of aldol additions (and related Mannich and Michael processes) has a long history starting in the early 1980s thanks to the pioneering works by Evans and co-workers.525 In 2004, Kobayashi, and later Hosokawa, successfully translated the concept of auxiliary-guided chirality transfer in the domain of vinylogous Mukaiyama-type aldol reactions, by investigating the preparation and use of vinyl ketene silyl N,O-acetals of type 807 to be used as vinylogous donors to access relevant, highly substituted polyketide units with a high level of stereocontrol (Scheme 206).526 In fact, the enolsilylation of α,β-unsaturated N-acyl oxazolidin-2-ones derived from l-valine afforded stable silyl dienolates of type 807, whose (E,E) double bond configuration and the conformational arrangement were ascertained by X-ray analysis and bidimensional NMR spectroscopy. The reaction between vinyl silyl ketene N,O-acetals 807 and a variety of aldehydes (aliphatic, aromatic, α,β-unsaturated) in the presence of TiCl4 efficiently produced anti-aldol adducts of type 809 (Scheme 206, eq 1).526 In the event, the authors proved that the presence of an α-methyl group in the acyl chain provided stability to the dienolate and was crucial to achieving a high level of stereoselectivity. On the basis of crystallographic and NMR data and energy-minimized conformation calculations, the authors proposed the transition state reported in Scheme 206, where the α-methyl group is disposed trans with respect to the oxazolidinone ring which, in turn, is almost perpendicular to the dienol ether plane with the l-valine isopropyl group shielding the upper face of the dienolate donor. This arrangement would likely guide the incoming aldehyde to approach the less hindered bottom face of the donor, leading to 6,7-anti-configured δ-hydroxy-α,β-unsaturated adducts of type 809.
Scheme 206.

The viability and predictability of the stereochemical outcome of this auxiliary-driven stereoselective VMAR strategy prompted the general exploitation of the Kobayashi reaction in asymmetric synthesis, as highlighted in the several publications appearing until 2010 and discussed in previous accounts.527,528
Over the 2010–2018 period, this fortunate trend carried on, and many other researchers besides the Kobayashi group exploited this strategy with remarkable achievements particularly in the field of natural product synthesis. In 2010, Hosokawa et al. exploited this approach to access the first total synthesis of epi-cochlioquinone A, a potent inhibitor of ACAT (acyl-CoA:cholesterol acyltransferase) (Scheme 206, eq 1).529 The vinyl silyl ketene N,O-acetal 807 was used as a donor in VMA addition to chiral (2S)-2-methyl butanal 808 (n = 0, R2 = Me, R3 = Et) in the presence of TiCl4 as Lewis acid, affording anti-aldol 809 in high yield and excellent control in the formation of the new stereocenters.
The viability of reactions exploiting remote asymmetric induction strategies and based on dienolate 807 was fully demonstrated in the convergent total synthesis of the antibiotic (+)-TMC-151C completed by Kobayashi and colleagues in 2011 (not shown).530 This compound showed a significant antitumor activity, along with an intriguing polyketide structure featuring three contiguous anti-homoallylic alcohol motifs that should be amenable to reiterative VMA additions of chiral oxazolidinones of type 807 to proper aldehydes. The initial attempts to reiterate the auxiliary-driven reactions of silyl dienolate 807 with the aldehyde counterparts in building up the new C–C bonds failed. Thus, the authors decided to adopt a convergent synthetic route entailing the independent synthesis of fragments C1–C6 and C7–C20 (target numbering) both featuring the intended anti-homoallylic alcohol motif, followed by their connection at a late stage by a silicon-tethered ring-closing metathesis reaction. In particular, l-valine-derived oxazolidinone 807 served as a donor in both the anti-selective VMARs giving the corresponding precursors of the C1–C6 and C7–C20 fragments of (+)-TMC-151C.
The vinyl silyl ketene N,O-acetal deriving from unnatural d-valine ent-807 was instead selected by Kobayashi et al. to accomplish the synthesis of the left-hand fragment (C9–C18) of the polyene antibiotic (−)-myxalamide A (Scheme 206, eq 2).531 The reaction of ent-807 with α,β-unsaturated aldehyde 808a promoted by titanium tetrachloride efficiently produced the anti-aldol 810 showing the expected anti-configuration at the newly formed stereocenters. Moreover, the authors observed that the reaction in which a catalytic amount of water (10 mol %) was added not only produced similar yield and stereoselectivity, but it also was markedly accelerated.
Going on in investigating the exploitation of silyl ketene acetals of type 807 in asymmetric VMAR, Kobayashi et al. explored the use of α-haloenals 808 (Scheme 206, n = 1, R1 = Hal, R2 = R3 = H) as acceptors to overcome the low reactivity of α,β-unsaturated aldehydes such as tiglic aldehyde (808, n = 1, R1 = Me, R2 = R3 = H).532 According to the established protocol, dienolate 807 reacted with two equivalents of haloenals 808 and TiCl4 affording anti-aldols of type 809 endowed with versatile δ-hydroxy-ε-halo functionalities along with two versatile double bonds. As previously demonstrated, even in this case, high yields could be obtained by prolonging reaction times or by adding a catalytic amount of water.533
The use of tiglic aldehyde as an acceptor in the stereoselective VMAR with silyl ketene acetal 807 was also exploited by Hosokawa and colleagues in 2013, during the synthesis of septoriamycin A, an antimalarial agent of microbial origin (not shown).534 As proof of the concept of the so-called wide range stereocontrol, the authors realized the divergent synthesis of four 2,4,6-trimethyloctanoate diastereoisomers by combining the initial Kobayashi reaction, that installed the chirality into the central core of the adducts with a stereoselective reduction, that transferred the chirality to the “surrounding” double bonds.
The chiral vinyl silyl ketene acetal derived from d-valine ent-807 was selected by Prusov and collaborators in 2012 as donor component in VMARs to accomplish the synthesis, and hence the structural elucidation, of eliamid, a secondary metabolite from myxobacteria with antifungal properties (not shown).535 Hoecker and Gademann in 2013 proposed the enantioselective synthesis of two natural products structurally related to piericidins, JBIR-02 and Mer-A2026B.536 Here the installation of the two and sole C9 and C10 stereocenters within the polyenic side-chain relied on the anti-selective VMAR of ent-807 with β-bromo-acrylaldehyde in the presence of TiCl4 (not shown). The crystallographic analysis of the intermediate adduct allowed the assignment of the absolute configurations of both the natural and the synthetic samples. More recently, Ohashi and Hosokawa carried out the Kobayashi reaction with compound 807 and acetaldehyde as the opening move in the synthesis of stoloniferol B and penicitol A (not shown).537 Also in this case, the VMA addition leading to anti-configured products ensured the efficient construction of the stereocenters within the targeted lactone and allowed the reassignment of the structure of the analogue fusaraisochromanone.
In the field of the total synthesis, structure elucidation, and biological evaluation of natural products, Kalesse and collaborators have long been active researchers, contributing to the success of the Kobayashi reactions based on the use of chiral oxazolidinones as vinylogous pronucleophiles. In 2012, they proposed the first total synthesis of pellasoren A with the aim of validating the stereochemical disposition of this natural product and establishing a model for its biosynthesis (Scheme 207, eq 1).538 The VMAR of (E,E) vinyl silyl ketene acetal 807 with chiral aldehyde acceptor 811 under optimized conditions was the key maneuver to install the anti-disposed C4–C5 stereocenters (target numbering) featuring the lactone moiety within the target compound.
Scheme 207.
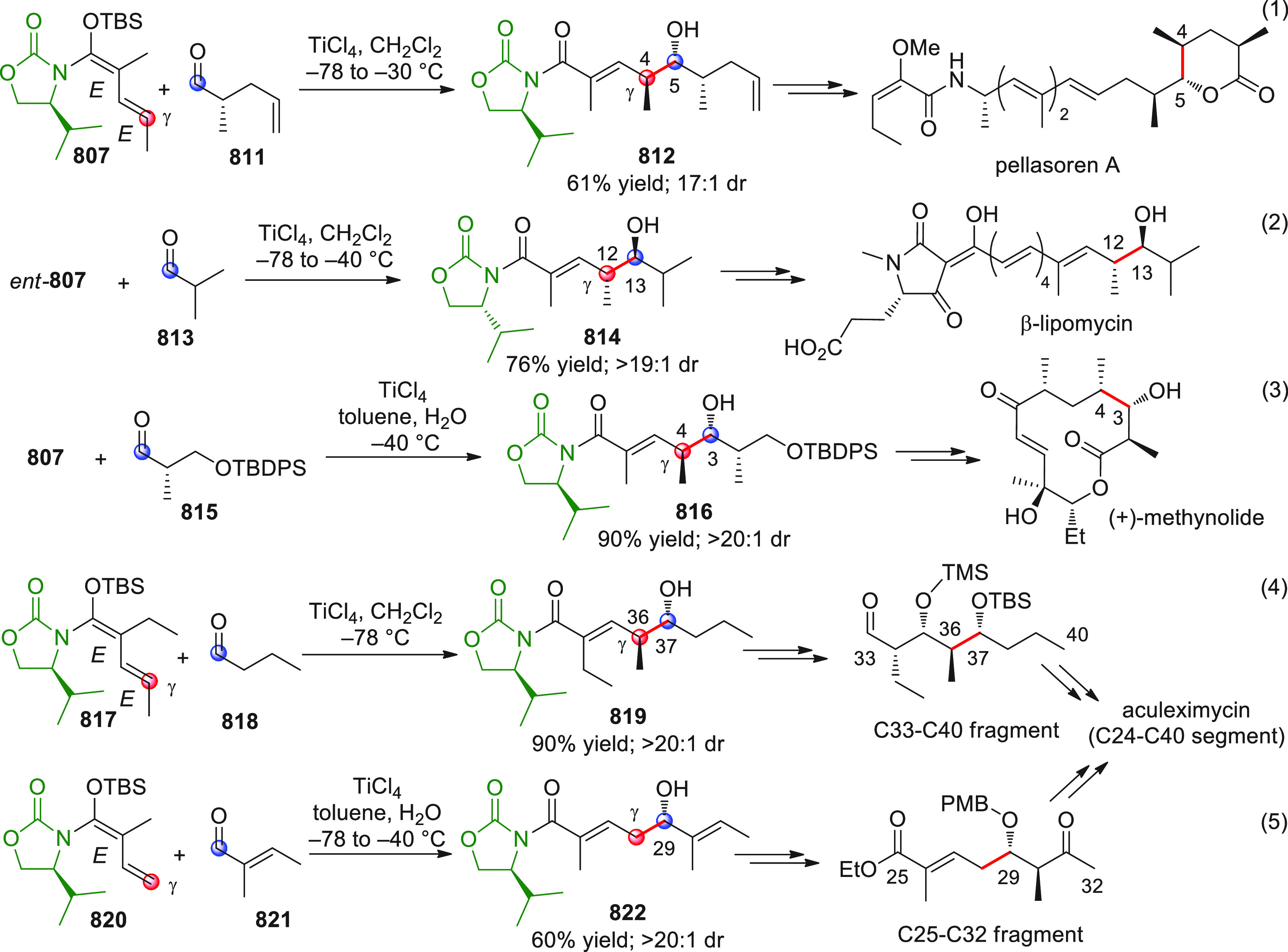
Along this line, Hartmann and Kalesse embarked on the synthesis of the antibiotic β-lipomycin with the aim of fully validating the C12 and C13 configurations predicted by statistical methods (Scheme 207, eq 2).539 In the event, a Kobayashi reaction between silyl ketene acetal ent-807 and isobutyraldehyde (813) was envisaged, providing the expected (12R,13S)-configured anti-aldol adduct 814 in good yield and stereoselectivity. As a whole, the proposed route not only provided access to the natural β-lipomycin product and its analogues, but it also assessed the proposed statistical method as a reliable tool in predicting the absolute configurations of methyl branches and secondary alcohols within modular polyketide carbon chains.
The total synthesis of (+)-methynolide recently proposed by the Kobayashi group, once again, was grounded on the exploitation of the well-known acetal 807 in stereoselective VMAR to β-silyloxy-substituted aldehyde 815 (Scheme 207, eq 3).540 In particular, the bulky silyl protecting group (preventing Ti-chelation), along with the use of toluene as a solvent, and the addition of catalytic water were required to obtain good yield (90%) of the desired (3S,4S)-anti-adduct 816 in >20:1 diastereomeric ratio, even though with very long reaction times (4.5 days).
Finally, the stereoselective synthesis of the C24–C40 segment of antibiotic aculeximycin, proposed by Hosokawa in 2015, was likewise set on the remote asymmetric induction strategy (Scheme 207, eqs 4 and 5).541 The authors chose to adopt a highly convergent approach (linking the two C33–C40 and C25–C32 fragments), in which the stereocontrolled formation of the C36, C37, and C29 asymmetric carbons (target numbering) relied on TiCl4-promoted VMARs of chiralized acetals 817 and 820 with proper aldehydes (818 and 821, respectively) under the usual Kobayashi reaction conditions; all the remaining stereocenters in the polyketide C24–C40 chain were generated exploiting substrate-controlled stereoselective reactions.
The utility of γ-methyl vinyl silyl ketene N,O-acetals of type 807 in anti-selective VMARs has been demonstrated in the many synthetic applications discussed so far. The development of complementary syn-selective VMARs employing analogous chiral oxazolidinone silyl dienolates greatly broadens the potential of this strategy in natural products synthesis. Syn-selective VMARs were first introduced by Kobayashi in 2009, who devised a strategy according to which a switch of facial selectivity was observed for α-heteroatom-substituted aldehydes in the TiCl4-promoted reactions of chiral oxazolidinone silyl dienolates.542
Further on in 2012, the Hosokawa group demonstrated that the stereochemical outcome of the Kobayashi reaction could depend on the amount of the added Lewis acid (Scheme 208, eq 1).543 In fact, the reactions employing (E,E)-vinyl silyl ketene N,O-acetal 807 (1.5 equiv) and TiCl4 (4 equiv) with a variety of both aromatic and aliphatic aldehydes 823 led to syn-aldol adducts 824 with an unexpected (and unexplained) switch of stereoselectivity. Although the reaction proceeded slowly with γ-oxy-aldehydes, syn-selectivity was observed in all cases, and aldols 824 were obtained in good to high yields and excellent levels of stereocontrol.
Scheme 208.

The utility of this methodology was demonstrated by Kanoh and colleagues during the synthesis of the C1–C18 macrolactone fragment of FD-891, a microbial macrolide that was reported to show remarkable antitumor activity.544 The authors exploited the VMA reaction between the enantiomer of 807 and a seven-carbon aldehyde (not shown) in the presence of an excess (5 equiv) of TiCl4 to form the C6–C7 bond (target numbering) and the corresponding stereocenters in high yield (95%). Unfortunately, the stereoselectivity of the aldol step was not reported, nor discussed by the authors.
The possibility to selectively access in a diastereodivergent manner to either anti- or syn-aldol products by simply varying the relative amounts of the same starting silyl ketene acetal and Lewis acid with respect to the aldehyde counterpart was fully demonstrated by Poulsen et al. during the studies on the synthesis of the enantiomer of cyclodepsipeptide BE-43547 A1.545 Starting from the same couple of substrates, a (Z,E)-configured vinyl silyl ketene N,O-acetal derived from l-valine and a suitable 13 carbon-long aldehyde, the reaction performed with TiCl4 (1 equiv) gave the corresponding anti-aldol in 87% yield and >20:1 diastereomeric ratio, while the reaction performed with an excess of the Lewis acid (4 equiv) gave the syn-aldol congener in 75% yield and >40:1 stereoselectivity, thus opening the way to the complete structural elucidation of the compounds belonging to the family of BE-43547 anticancer agents.
During their studies on remote asymmetric induction reactions, Hosokawa and colleagues discovered that the VMAR of the well-known 807 with acetals of type 825 in the presence of stoichiometric quantities of Lewis acid (BF3 etherate or TMSOTf) at different temperatures produced protected syn-aldol adducts 826 (Scheme 208, eq 2).546 The reactions involving acetals derived from aromatic and α,β-unsaturated aldehydes afforded the most efficient and stereoselective results, as compared to the reactions employing acetals from saturated aldehydes. This procedure turned out to be suitable for the one-pot conversion of aldehydes into the corresponding benzyl-protected syn-adducts (826, R2 = Bn), without erosion of yield and stereocontrol, and it was realized by adding dienolate 807 and TMSOTf (1 equiv) to a preformed mixture of the aldehyde of choice, BnOTMS and catalytic TMSOTf. According to the obtained results, the authors supposed that the reaction proceeded via an oxocarbenium ion through the preferred open-chain transition state reported in Scheme 208. Here, the largest R1 group of the acetal should be directed far away from the donor dienyl chain to minimize steric repulsion, while the oxonium hydrogen atom should point to the crowded area near the α-methyl group of the dienolate, thus exposing the oxonium Re face to the Si face of the donor. This transition state could also account for the variable selectivity observed depending on the bulkiness of the R2 protecting group.
Paralleling the chemistry already disclosed by Kobayashi concerning the syn-selective VMARs of vinyl silyl ketene acetals with α-heteroatom-substituted aldehydes,542 Chen and Yang established an efficient protocol based on the exploitation of vinyl silyl ketene acetals 827 derived from l-phenylalanine and ortho-substituted aromatic aldehydes 828 (Scheme 209, eq 1).547,548 The authors proved that syn-aldol adducts 829 could be stereoselectively obtained via a chelation-controlled VMAR and applied the protocol to the synthesis (and structural elucidation) of both the potent immunosuppressant NFAT-68 and the ansamacrolactam (+)-Q-1047H-A-A (Scheme 209).
Scheme 209.

Another example in which chelation-controlled syn-selective VMAR was successfully applied to the synthesis of nature-inspired bioactive compounds was described by Jürjens and Kirschning, who exploited the silyl dienol ether 827, a β-alkoxy-aldehyde, and Ti(Oi-Pr)Cl3 to realize the synthesis of a cytotoxic ansamycin hybrid (not shown).549
In the field of natural product synthesis, a relevant contribution is due to Symkenberg and Kalesse, who investigated the use of chiral (E,Z)-vinyl silyl ketene N,O-acetal 830 under the Kobayashi reaction conditions to obtain syn-adducts 831 (showing opposite stereochemistry at the new stereocenters with respect to compounds 829) in high yields and remarkable stereoselectivity (Scheme 209, eq 2).550 The authors successfully applied this protocol to the total synthesis of kulkenon (Scheme 209, bottom) and could revise and firmly assign the stereostructure of this polyketide macrolactone.551
The vinylogous aldol methodology leading to syn-configured polyketide synthons was also adopted by Dudley and collaborators to perform the enantioselective high-yielding construction of the C19–C20 bond within the side chain of palmerolide A (not shown).552
In 2010, Chen, Yang, et al. reported that the facial selectivity leading to syn-adducts in the Ti-mediated VMAR with dienolates of type 827 and chelating carbonyl acceptors 832 could be reversed by the addition of silver salts (Scheme 210).553 In fact, if the reaction was conducted with glyoxylate 832 (R1 = H, R2 = CO2Et) in the presence of stoichiometric quantities of TiCl4 and AgSbF6, (6S,7S)-configured anti-adduct 833 was produced in high yield and excellent stereocontrol. The postulated transition state is reported in Scheme 210 (left), where the presence of the silver cation likely allows the formation of a hexacoordinated complex between TiCl3+, the aldehyde, and the oxazolidinone carbonyl, favoring the Si-Si approach; on the other hand, in the absence of Ag+, the TiCl4–aldehyde complex could approach the diene according to the Re-Re trajectory, giving (6R,7R)-configured anti-adducts 834 (Scheme 210, right).
Scheme 210.

The remote asymmetric induction-based reactions employing phenylalanine-derived silyl dienolates 835 under Kobayashi conditions were adopted by Gademann and colleagues to advance the synthesis toward the macrolide antibiotic fidaxomicin, used in the treatment of resistant forms of tuberculosis, as well as an analogue, tiacumicin A (Scheme 211).554,555 In both cases, the convergent approach entailed the construction of the macrolactone core of the targets by joining the previously synthesized polyketide fragments. In the event, the issue of the C10–C11 bond construction (and implementation of the related stereocenters) was solved by employing the chiral vinyl silyl ketene acetal 835 that reacted with vinyl aldehyde 76a, affording anti-aldol 836 with excellent levels of stereocontrol. Then, the C10–C11 syn-configuration, required in the target, was obtained by a Mitsunobu reaction involving the reversal of configuration at C11.
Scheme 211.

In 2013, the Nagorny556 and Kuwahara557 research groups simultaneously and independently worked on the synthesis of lactimidomycin, a polyketide antibiotic showing antitumor and antifungal activities. In both cases, the C9–C10 bond of the side chain of lactimidomycin (not shown) was formed by a VMAR of a suitable vinyl silyl ketene acetal and acetaldehyde under Kobayashi conditions.
The total synthesis of nannocystin A, a 21-membered macrocyclic depsipeptide possessing a potent antitumor activity, was completed in parallel during 2016 by the groups of Ye558 and Chen.559 Both the research teams faced the construction of the seven carbon-long C5–C11 fragment within the final macrocycle by using VMARs of vinyl silyl ketene acetals deriving from d-amino acids (d-Phe or d-Val) and (E)-3-iodo-2-methylacrylaldehyde under usual Kobayashi conditions, to forge the C7–C8 bond and install the C7 stereocenter; moderate yields and stereoselectivities were obtained in all cases (not shown).
More recently, the Hosokawa group developed Z-configured crotonate-type silyl ketene N,O-acetals of type 837 equipped with a C5′-disubstituted oxazolidinone chiral auxiliary to address the remote asymmetric induction in aldol reactions (Scheme 212).560 As observed in previous studies,526 when α-methyl-lacking vinyl silyl ketene acetals were used, aldol adducts were obtained in moderate to low stereoselectivities, likely due to the preferred Z-configuration of the enolate double bond that keeps the reactive γ-site away from the auxiliary ring. The authors found that the TBDPS protecting group in 837 was crucial to improving the stability of the dienolate and that the oxazolidinone C5′ substituents were responsible for the modification of the overall dienolate conformation. Thus, the VMAR of Z-configured dienolate 837 with different aldehydes 823 turned out to be completely stereodivergent, affording either the 5R-configured O-silylated adducts 838 when SnCl4 was used (Scheme 212, eq 1) or the corresponding 5S-aldol products (not shown) when the reaction was performed in the presence of BF3·OEt2. The crystal structure analysis of 837 and NMR NOE experiments revealed that the substituents at C5′ in the oxazolidinone ring were actually efficient in determining the position of the valine isopropyl group and in “pushing down” the silyl group, thus directing one of the phenyl groups of TBDPS below the diene chain. Moreover, these studies revealed that, unlike BF3·OEt2, the Lewis acid SnCl4 promoted the isomerization of the Z-dienol ether to the more reactive E isomer, accounting for the observed stereodivergency. In order to prove the diene facial selectivity, the (Z,E)-configured γ-methyl dienolate 839 was exploited in VMARs with aldehydes 823 in the presence of tin tetrachloride and copper triflate (Scheme 212, eq 2). Also in this case, O-silylated anti-adducts 840 were obtained in good yields and variable stereoselectivity. Based on the overall results, the authors proposed a preferred transition state (Scheme 212, eq 1), where the Re face of the aldehyde approaches the upper face of the isomerized E diene. Another study by Hosokawa and colleagues analyzed the stereoselective VMA additions of TBS-protected dienol ether 839 to acetals 825 under the guidance of TMSOTf, that resulted in the formation of the δ-alkoxy-substituted syn-adducts 841 in good yields and stereoselectivities (Scheme 212, eq 3).561 The crystallographic and NMR studies corroborated the hypothesis that the chiral oxazolidinone ring was oriented 40 degrees away with respect to the diene plane and, as previously mentioned, the C5′ substituents could direct the silyl group toward the lower face of the diene (Scheme 212, eq 3); in this way the activated electrophile can approach the upper face of the donor, giving aldols 841 as the major products. The reaction could also be performed in a practical one-pot acetalization-addition procedure without decrease of both the efficiency and the stereocontrol.
Scheme 212.

The sole example of catalytic asymmetric VMAR involving silyl dienolates deriving from acyclic amides was proposed by Bolm in 2010.562 As previously described in the case of furan-based silyloxy dienes (vide supra), the copper aminosulfoximine complex (S)-L13a/Cu(OTf)2 (Scheme 135) was employed as the catalyst in the VMA addition of N,O-acetals obtained from unsaturated N-acyl morpholine to activated α-ketoesters. After optimization of the reaction conditions, the δ-hydroxy-α,β-unsaturated morpholino-amides were obtained, even if only moderate to good yields and no more than 91% enantiomeric excess could be achieved.
6.1.2.2. Cyclic Nucleophiles
As said before, chiral γ-butyrolactams are structural motives widely encountered in many important natural and non-natural bioactive compounds. These nitrogen-containing frameworks also represent important synthetic intermediates in assembling functionality-rich nitrogen heterocyclic structures.
Following their longstanding experience in the use of the popular heterocyclic silyloxy dienes, in 2010 Curti, Zanardi, et al. reported a study focused on the catalytic vinylogous aldol reaction of pyrrole- and furan-based silyl dienolates (the latter being already cited in section 5) with aromatic and heteroaromatic aldehydes.344 After a preliminary screening of various metal-based catalyst systems, it was found that the bisphosphoramide L12/SiCl4 couple was the optimal catalyst system to promote and govern the VMAR between silyloxy pyrroles 842 and aldehydes 843, producing the expected lactams 844 with high efficiency and stereocontrol (Scheme 213). It is noteworthy that the study revealed that the nature of the heteroatom substituents in the silyloxy diene scaffolds heavily affected the stereochemical reaction outcome; that is, the syn-configured adducts syn-844 were preferentially obtained with pyrroles bearing electron-withdrawing N-protecting groups (Boc, Ts, Cbz), while the use of dienes N-substituted with electron-donating groups (Bn, allyl, PMB) (as in the case of furans, vide supra) provided reversal of stereocontrol, giving rise to anti-844 adducts, preferentially. To account for the observed reaction diastereodivergence, dictated by the nature of the heteroatom substituent, a transition state model was proposed (Scheme 213, bottom right). With silicon-coordinating N-protecting groups (carbamates and sulfonylamides) able to bind to hypervalent silicon atom of the chiral catalyst, engagement of the Re face of the aldehyde carbonyl with the Re face of the pyrrole γ-site resulted in the preferential formation of syn-disposed (5S,1′S) adducts; on the contrary, lacking supplementary coordination at silicon, as in the case of Bn, allyl, or PMB N-substituents (or oxygen), steric effects prevailed, favoring the involvement of the pyrrole Si face with the preferential generation of (5R,1′S) anti-configured structures.
Scheme 213.

As previously described with furan-based silyl dienolates (see section 5), Curti, Casiraghi, et al. reported the stereoselective VMAR between the pyrrole counterparts and aromatic aldehydes on aqueous media giving equally successful results (Scheme 214).350 The uncatalyzed VMAR with silyloxy pyrrole 845 carried out in a mixture of brine and MeOH, under ultrasonic irradiation, was successfully applied to a number of aromatic aldehydes 843 leading to anti-configured δ-hydroxy-γ-lactams 846 in good yields, virtually complete γ-site selectivity, and moderate to good diastereoselectivity. However, a remarkable switch of diastereoselectivity was observed when passing from pyrrole (anti-selective) to furan silyl dienolates (syn-selective), highlighting, once more, how the nature of the heteroatom in the donor moiety critically affects the diastereocontrol of the reaction. On the basis of reports concerning similar on water processes, this reaction was postulated to occur at the boundary between water and the dispersed lipophilic phase, with critical H-bonding interactions between water molecules and the carbonyl acceptor, which likely governed the reciprocal position of the reactants in the transition state (Scheme 214); as opposite to furan nucleophiles, here the bulky, lipophilic N-tert-butoxycarbonyl group is shifted away from the water interface entering the inner space of the reactant droplets, thus reverting the diastereocontrol.
Scheme 214.

Contrary to the wide exploitation witnessed during the last 20 years of the past century dealing with the use of popular pyrrole silyloxydienes of type 842 or 845 (Schemes 213 and 214) in VMARs to chiral acceptors (according to the so-called chiron approach), in recent years the interest for these type of substrate-controlled stereoselective methodologies has gradually faded, likely due to the concomitant raising of asymmetric catalytic methodologies. Only two examples of target-oriented exploitation of the VMA reaction between silyloxy pyrroles and chiral aldehyde acceptors were found; in the first contribution by Zambrano, Battistini, et al.,563 the well-known vinylogous aldol addition of tert-butyldimethyl silyloxypyrrole (842a, R1 = Boc, TBSOP) to d-glyceraldehyde acetonide was chosen as the opening move in a short reaction sequence that led to the asymmetric synthesis of 1-deoxy-7,8-di-epi-castanospermine and, at the same time, served in the unambiguous reassignment of the stereostructure of this alkaloid (not shown).
The same VMAR between TBSOP (842a) and d-glyceraldehyde was the key step in the asymmetric total synthesis of (+)-N-acetyl norloline reported by Huang and colleagues in 2016.564 In this instance, the VMAR served to efficiently install three of the four stereogenic centers featuring the target pyrrolizidine alkaloid (not shown).
In 2012, Rassu, Curti, Casiraghi, and colleagues reported the very first example of the synthesis of 3-alkenyl-2-silyloxy indoles 847 and their use in asymmetric VMAR with aromatic aldehydes 843 (Scheme 215).565 A simple procedure to access N,O-silyl ketene acetals 847 was developed, using triethylamine and TBSOTf as enolizing-silylating reagents from the corresponding 3-alkylidene oxindoles; then, the addition of stable dienolates 847 to a variety of aromatic aldehydes was explored. Using silicon tetrachloride and diisopropyl ethylamine in CH2Cl2 in the presence of DMF, the corresponding racemic vinylogous aldol adducts 848 were obtained in moderate yields, with complete γ-site selectivity and excellent diastereoselectivity favoring the Z-configured alkenes (Scheme 215, eq 1). During an exploratory trial, the enantioselective version of the process was also discovered, using Denmark’s bisphosphoramide catalyst L12 in combination with SiCl4 (Scheme 215, eq 2). Chiral nonracemic products of type 848a eventually formed in acceptable yields, with appreciable Z-diastereoselectivity and promising enantioselectivity (up to 90% enantiomeric excess). Based on precedents on the use of the L12/SiCl4 catalyst system in VMARs of enoxysilanes to aromatic aldehydes, the authors hypothesized a catalytic pathway involving the attack of the indole nucleophiles to the Re face of the aldehyde carbonyl groups resulting in the preferential formation of 4′R-configured adducts.
Scheme 215.

Carrying on their studies on the exploration of vinylogous reactivity of oxindole matrices, Curti, Zanardi, and co-workers reported the first example of a catalytic, enantioselective hypervinylogous Mukaiyama aldol reaction (HVMAR) involving highly unsaturated 2-silyloxindoles of type 849 as donor substrates (Scheme 216).566 The silyl polyenolates 849 were reacted with various aromatic and aliphatic aldehydes 823 in the presence of the previously disclosed bisphosphoramide L12/SiCl4 catalytic system, to access enantioenriched 3-polyenylidene homoallylic carbinols 850 in high yields, with excellent levels of regioselectivity, enantioselectivity, and double bond geometrical selectivity. The study was centered on the exploration of the vinylogous transmittal of the enolate lactam reactivity along the exocyclic polyene chain. Thus, 3-butadienyl silyloxyindoles and higher homologous were selected as nucleophiles with the aim of providing complete regiocontrol at the very remote position according to the concept of hypervinylogy. After careful preliminary work on variously protected 3-butadienyl derivatives 849 (n = 1) and benzaldehyde 823 (R5 = Ph), intended to profile the best reaction conditions, it was found that the L12/SiCl4 couple efficiently catalyzed the bisvinylogous reaction at −45 °C in the dark (to avoid the double bond isomerization) and that the nitrogen protecting group greatly impacted on the outcome of the reaction, with benzyl group giving the best results in terms of regio-, diastereo-, and enantioselectivity as compared to the Moc group. The N-benzyl substitution was the best choice also in the case of the trisvinylogous version of the reaction (849, n = 2, R2 = Me, R3 = H), while in the case of the tetravinylogous reaction, the n-propyl N-protecting group ensured the best results (849, n = 3, R2 = Me, R3 = H). The 13C NMR analysis of the chemical shifts of the remote C-ω sites and the conformational analysis carried out on selected polyenolate donor species enabled the rationalization of the obtained results suggesting that the vinylogous reactivity propagation of polyene donors was dependent on the electron-donating properties of the indole N-substituents as well as the coplanarity of the polyene chain, which is strictly related to the vinylogous transmittal properties.
Scheme 216.

6.2. Additions to C=N Bonds
During the period analyzed in the present review, the growing interest toward the asymmetric synthesis of Mannich products, that are chiral δ-amino α,β-unsaturated carbonyl derivatives, prompted the implementation of new asymmetric methodologies falling in the field of vinylogous additions to C=N bonds that, starting from either cyclic or acyclic α,β-unsaturated amides, opened the access to chiral functionality-rich vinylogous δ-aminated architectures as skillfull precursors in the synthesis of alkaloidal/aminated bioactive substances.
6.2.1. Direct Procedures
6.2.1.1. Acyclic Pronucleophiles
Aiming at developing direct asymmetric reactions of α,β-unsaturated carbonyl compounds to be used as vinylogous donors in Mannich additions to imine electrophiles, Yin and colleagues proposed the β,γ-unsaturated N-acylpyrazoles 851 and their bisvinylogous counterparts 854 as pronucleophilic substrates in direct Mannich additions to N-Boc aldimines 852 under the guidance of the copper(I)-L30 (or L15) catalytic systems (Scheme 217).567
Scheme 217.

A careful exploration of the model reaction led to the finding that regioselectivity with this type of substrates could be improved by increasing steric hindrance at the pyrazole C3′ and C5′ positions and by using the bulky copper(I) bisphosphine complex based on L30, the so-called (R)-DTBM-SEGPHOS, thus favoring addition at the γ-site. The authors evaluated the substrate scope of the direct catalytic vinylogous Mannich addition by testing aromatic, heteroaromatic, and aliphatic aldimines, achieving (5S)-configured vinylogous δ-amino derivatives 853 (Scheme 217, eq 1) with complete γ-selectivity (γ:α > 20:1) in good to high yields and stereoselectivities, even when γ-substituted acylpyrazoles 851 (R2 = alkyl) were used (>20:1 diastereomeric ratio in favor of 4,5-anti-adducts). The authors succeeded in the development of the bisvinylogous version of the Mannich reaction by reacting acylpyrazoles 854 (Scheme 217, eq 2) and aldimines 852; in this case, the ferrocene-based bisphosphine L15 performed as the best ligand in the presence of Cy2NMe as the base, giving the ε-adducts 855 regioselectively (ε:α > 20:1) and in an enantiocontrolled manner.
The same research group, aiming at the construction of halogenated allylic carbon stereocenters, in 2018 described the application of the above disclosed asymmetric Mannich reaction to γ-halogenated α,β-unsaturated N-acyl pyrazoles 856 and aldimines 852 (Scheme 217, eq 3).568 As in the case of pyrazoles 851, the bulky bisphosphine ligand L30 was the key to perfectly control the regioselectivity, as well as the diastereo- and enantioselectivity. The optimized reaction conditions found in the model reaction for the fluoro-derivatives 856 (X = F), were applied to various aromatic, heteroaromatic, and aliphatic aldimines 852 giving syn-adducts 857 in good to excellent yields and stereoselectivities; when γ-chloro-substituted pyrazoles 856 (X = Cl) were used, copper(I)/L30 catalyst (3 mol %) and Et3N were enough to catalyze the Mannich addition to aldimines with generally excellent results. The mild reaction conditions, the broad substrate scope, the good tolerance of functional groups, and remarkable regio- and stereoselectivities, all these features definitely demonstrated the robustness and versatility of the proposed methodology in the vinylogous (and bis-vinylogous) Mannich reaction domain.
6.2.1.2. Cyclic Pronucleophiles
The use of cyclic amide pronucleophiles in direct vinylogous Mannich reactions has been scantly investigated; just a couple of examples are reported in the literature in which the dienolate donors belong to the alkylidene oxindole family.
The first example concerns the vinylogous Mannich addition of alkylidene oxindoles 858 to chiral fluoroalkyl aldimines 859 disclosed by Qing et al. in 2015 (Scheme 218, eq 1).569 The reaction was carried out in the presence of the strong base KHMDS to promote the γ-enolization of oxindoles 858 at −78 °C, while sulfinyl imine 859 was activated by means of Ti(Oi-Pr)4 as the Lewis acid. The addition resulted in the formation of α-alkylidene-δ-amino-δ-fluoromethyl oxindoles 860 in moderate to good yields and complete γ-regioselectivity (γ:α > 99:1). The degree of diastereselection was good in terms of Z/E double bond preference and substrate facial control, with (3Z,3′S)-configured isomers formed as the major products. Substitution at C5 of the oxindole ring with both electron-donating and electron-withdrawing groups was well tolerated, as well as the various N-protecting groups having different electronic properties and steric hindrance. The authors explained the substrate-controlled stereoselectivity of the addition through a nonchelated transition state model in which the sulfinyl oxygen coordinates the titanium isopropylate thus sterically shielding the Re face of the imine acceptor (not shown). In the same study, the authors also reported the reaction of an alkylidene benzofuranone, a scantly exploited vinylogous pronucleophile, with a chiral trifluoromethyl sulfinyl imine that produced a Z-adduct with comparable efficiency and stereoselectivity as the nitrogen counterpart.
Scheme 218.

In the second contribution authored by Kim and co-workers, the alkylidene oxindoles of type 858 were reacted with N,N-dimethylformamide dimethyl acetal 861 as the electrophilic partner, which at the same time generated the iminium ion acceptor and the methoxide ion acting as the base in deprotonating the oxindole γ-methyl group (Scheme 218, eq 2).570 The reaction was performed in DMF at 80 °C resulting in (E,E)-configured Mannich adducts 3-dimethylamino-2-propenylidene oxindoles 862 quite efficiently, after methanol elimination.
6.2.2. Indirect Procedures
6.2.2.1. Cyclic Nucleophiles
The vinylogous asymmetric Mukaiyama-type Mannich reactions of pyrrole-based silicon dienolates with imines provide an effective and straightforward way to construct rare α,β-unsaturated γ,δ-diaminocarbonyl frameworks, which are crucial motives in the synthesis of many natural and synthetic compound classes. In the past 2010–2018 period, the use of linear acyclic silyl ketene N,O-acetals as vinylogous donors in Mannich-type processes has been almost completely disregarded, while a limited number of studies involving silyloxy pyrroles (or oxindoles), classified as cyclic silyl ketene acetals, was found and commented on here.
In this research domain, Zanardi, Curti, and co-workers developed anti-selective, catalytic asymmetric Mannich addition protocols to access δ-aminated entities, by exploiting siloxy pyrroles as donors and preformed or in situ generated aldimine acceptors (Scheme 219).571,572 Excellent results in terms of regio-, diastereo-, and enantioselectivity were achieved utilizing, as the catalyst of choice, tert-leucine-derived ligand L31 in complex with silver(I) acetate, a system ideated and exploited by the Hoveyda and Snapper group a few years ago.573−575 As shown in Scheme 219, two different optimal protocols were elaborated, according to the nature of aldimine acceptors. When aromatic aldehyde substrates were involved, the vinylogous Mannich addition of N-Boc protected pyrrole 845 and preformed N-aryl imines 863 with ligand L31 and AgOAc (10 mol % each) performed well, while the reaction of alkyl (and hydroxyalkyl) aldehyde substrates 867 entailed a three-component sequential addition protocol, where the imine acceptors were formed in situ (from aldehydes 867 and o-thiomethyl-p-anisidine 866) prior to the addition of the catalyst and the N-Cbz candidate 865. Invariably, both protocols performed well, returning the expected anti-configured Mannich adducts 864 and 868 in high yields, excellent diastereomeric ratio, and moderate to good enantioselectivities. The synthetic versatility of the unsaturated lactam adducts was demonstrated by the transformation of a hydroxylated lactam, deriving from the reaction between pyrrole 865, d-glyceraldehyde and amine 866, into an unprecedented bicyclic furopyrrolone product, reminiscent of the structure of the naturally occurring (+)-goniofufurone (not shown). Based on the pioneering studies by Hoveyda and Snapper on silver-catalyzed asymmetric VMMnR of furan silyl dienolates, the authors proposed a transition state (Scheme 219, bottom left) accounting for the observed anti-configuration of the Mannich products. In the event, the o-thiomethyl group and the nitrogen atom within the imine acceptor participate in the tetracoordinated silver complex, thus exposing the imine Re face to the Si face of the incoming silyloxy pyrrole, with the catalyst amide carbonyl acting as a silicon scavenger; the tightly organized complex was judged responsible for the Lewis acid activation of the substrate and the Lewis base activation of the enolsilane, while determining the relative orientation of the reacting partners with the imine anti-disposed with respect to the bulky tert-butyl substituent within the catalyst ligand.
Scheme 219.

A few years later, following their longstanding interest in the exploitation of amino acid-based chiral phosphine catalysts, Hoveyda, Snapper, and co-workers applied the vinylogous Mukaiyama–Mannnich addition protocol to the same pyrrole 845 with preformed o-methylthio-p-anisidine imino-derivatives in the presence of silver(I) acetate and isoleucine-derived phosphine catalyst (not shown).419 The process turned out to be viable and enantioselective with aryl, alkyl, and alkynyl imines, even in the case of in situ formation of the Mannich acceptor.
The development of reliable and efficient chemical methodologies that fulfill such representative keywords as water, solvent-free, environment awareness, practicality, and atom economy is one of the most challenging issues of contemporary organic synthesis. Along this line, Zanardi et al. presented the first catalyst-free three-component vinylogous Mukaiyama–Mannich reaction (VMMnR) employing pyrrole silyl dienolates of type 842a, o-anisidine 869, and diverse aldehydes 823, which performed well in both aqueous and solvent-free environments (Scheme 220).576 Both lipophilic and hydrophilic, aromatic and aliphatic aldehydes proved to be competent substrates giving access to a wide collection of racemic α,β-unsaturated δ-aminolactams 870 in high isolated yields, with excellent levels of γ-site selectivity and chemoselectivity, and moderate to high diastereoselectivity in favor of anti-configured adducts. Of note, aqueous and solvent-free protocols were particularly suited for protected alkoxy aldehydes and highly hydrophilic substrates, leading to the corresponding adducts with promising yields and favorable anti-diastereoselectivity. As for the role exerted by water in this vinylogous Mannich reaction, it was recognized to be a critical ingredient acting as both the proton donor, to activate the in situ generated imine, and the silicon scavenger, to sequestrate the silicon ion into an inactive R3SiOH species, the sole byproduct in these environmentally benign transformations. Soon after, the same authors envisaged taking full advantage of this three-component vinylogous Mannich reaction in planning the stereodivergent synthesis of certain stereoisomers of the anti-influenza agent oseltamivir carboxylate.577 The initial VMMnR of siloxy pyrrole 842a, d-glyceraldehyde acetonide 871 and amine 869 (Scheme 220, eq 2), which installed the entire carbon skeleton and heteroatom substituents of the target, was carried out according to the previously disclosed one-pot three-component protocol in both aqueous and solvent-free conditions, and afforded the expected adducts in high global yield (on-water, 95% yield; neat, 96% yield) as a mixture of the three separable diastereoisomers, namely, the anti,anti-configured product 872 (Scheme 220, eq 2), along with minor amounts of syn,anti- and syn,syn-diastereoisomers (not shown). The most abundant isomer 872 was then converted into the target 4-epi-oseltamivir carboxylate (14 steps, 20.2% overall yield), an unprecedented stereoisomeric variant of the antiviral drug, whose inhibitory activity toward influenza A virus neuraminidase was assayed.
Scheme 220.

The utility of the vinylogous Mannich addition of pyrrole 842a to chiral N-tert-butanesulfinimines 873 according to a classical substrate-controlled approach was demonstrated by Ye, Huang, et al. in the total synthesis of the amino-pyrrolizidine alkaloid (+)-absouline and a precursor of (+)-loline, both characterized by vicinal anti-diamine motives (Scheme 221).578,579 During a preliminary work, the authors explored the efficiency of the TMSOTf-promoted Mannich additions to diverse N-tert-butanesulfinimines 873 prepared from (RS)-tert-butanesulfinamide and aliphatic or aromatic aldehydes, affording the corresponding anti-adducts 874 in good to excellent yields and remarkable stereoselectivity. In the event, the imine of TIPS-protected 3-hydroxypropanal was used as starting material to prepare the 1-aminopyrrolizidine alkaloid (+)-absouline. In the second contribution, the same authors exploited the previously disclosed VMMn addition of pyrrole 842a to the sulfinyl imine of l-glyceraldehyde aiming to directly access the vicinal diamino motif present in the (+)-loline.
Scheme 221.

Following their interest in the full exploitation of γ-enolizable alkylidene oxindoles as extended carbon nucleophiles, Sartori, Zanardi, et al. explored the use of the oxindole N,O-silyl ketene acetals 875 in VMMnRs involving imine precursors 876 (Scheme 222).580 The propenyl silyloxy indoles 875, already utilized in vinylogous aldol maneuvers (vide supra), were coupled to di-Boc aminals 876 with catalytic TMSOTf (20 mol %) which triggered the in situ formation of the imine electrophiles. The reaction proved to be quite efficient (47–78% yields), giving the corresponding chiral δ-amino-2-oxindoles 877 in a racemic format with complete γ-selectivity (γ:α > 49:1) and excellent diastereoselectivity in favor of Z-adducts. The protocol worked well with both aromatic and aliphatic aminals, and a representative δ-amino-2-oxindole deriving from benzaldehyde aminal (R2 = Ph) served as a precursor in the preparation of indole-based architectures including spirocyclopropaneoxindole 878 and homotryptamine analogue 879 (Scheme 222).
Scheme 222.

6.3. Conjugate Additions to Electron-Poor C=C Bonds
Asymmetric vinylogous reactions enabling the straightforward access to amide structures decorated with electron-deficient appendages (e.g., carbonyl, nitro, or cyano groups), double bonds, and stereocenters at their γ- (or more remote) positions are of wide interest in synthetic chemistry, as they can represent key intermediates in the diversity-oriented synthesis of natural and non-natural compounds of biological interest. In this context, many efforts have been devoted to the development of catalytic asymmetric methodologies based on vinylogous 1,4 additions to activated double bonds which provide chiral high-value α,β-unsaturated δ-substituted amide scaffolds bearing additional carbonyl, nitro, or cyano substituents at their ε-position.
6.3.1. Direct Procedures
6.3.1.1. Acyclic Pronucleophiles
The elaboration of asymmetric synthetic methodologies based on vinylogous addition–cyclization cascades to directly access spirocyclic oxindoles has recently attracted growing interest, as already mentioned for the aldol domain (vide supra). As a continuation of their efforts toward the synthesis of spirooxindoles as privileged structural motives in bioactive compounds, Sha, Wu, and colleagues developed a catalytic enantioselective vinylogous Michael addition–cyclization sequence involving acyclic β,γ-unsaturated amides of type 880 or 883 as vinylogous pronucleophiles and isatylidene malononitriles 881 or 884 as electrophilic partners (Scheme 223).581
Scheme 223.

Initially, the authors explored the reaction between pyrazole amide 880 and alkylidene malononitrile 881 (Scheme 223, eq 1) as a model to screen for the best organocatalyst and reaction conditions. The reaction worked well in the presence of cinchona-derived catalyst C35 [(DHQD)2PYR, 2 mol %], affording the spirocyclic adduct 882. According to the postulated reaction mechanism, the dienolate formed by deprotonation of the quinuclidine base reacted at its γ-position with the isatylidene malononitrile acceptor; the resulting carbanion intermediate attacked the amide carbonyl providing, after elimination of the pyrazole unit, the spiro-compound, thus completing the reaction cascade. During the evaluation of the substrate scope, the authors found that the use of 1H-benzotriazole allyl amides 883 as pronucleophiles gave the best results in terms of both yields and enantioselectivities; moreover, the N-protecting group and the substitution pattern at the aromatic ring of the oxindole acceptor 884 were examined giving generally good yields and good to excellent enantioselectivities (Scheme 223, eq 2).
In 2016, Boyce and Johnson reported an example of stereoselective auxiliary-driven vinylogous Michael cascade exploiting silylglyoximide 886 as latently chiralized pronucleophile and nitroalkenes 887 as the acceptor partners (Scheme 224).582 The in situ formation of the Z magnesium dienolate 886′ was initiated by vinylation of acyl silane 886, followed by [1,2]-Brook rearrangement (the carbon-to-oxygen silyl migration); then the dienolate 886′ coupled to nitroalkenes 887, thus terminating the vinylogous Michael cascade. The three-component reaction sequence yielded functionality-rich Z-silyl enol ethers 888 with complete chemo- (only traces of Grignard addition to nitroalkene were detected), regio- (only γ-adducts were observed), and facial selectivity (>20:1 diastereomeric ratio). Even though the efficiency of the process was poor, with yields ranging from 40% to 75%, the scope of the nitroalkene was broad, tolerating aryl, heteroaryl, alkyl, and alkenyl substituents. Of note, the nitrodiene 887 having R = styryl exhibited 1,4-selectivity instead of the potentially competitive 1,6-addition, providing evidence for the possible transition state accounting for such a high level of stereoinduction. In fact, supported by DFT calculations, a highly organized “trans-decalin” model was proposed, where the magnesium ion strongly coordinates both the auxiliary carbonyl oxygen and the nitro group, driving the approach of the dienolate to the Si face of the nitrolefin having the R substituent in a favorable pseudoequatorial position. The developed methodology provided direct and stereoselective access to densely functionalized α-heterosubstituted nitro-derivatives 888 whose Z-enol ether configuration prevented the Henry cyclization, commonly observed with similar intermediates.266
Scheme 224.

6.3.1.2. Cyclic Pronucleophiles
Among the cyclic amide pronucleophiles, the N-Boc pyrrolinone has been considered and still represents a valuable heterocyclic building block; it is prone to quite easy γ-enolization, and it may be used as a versatile d4 synthon in direct, catalytic vinylogous Michael addition reactions with electron-poor alkenes, giving access to highly functionalized γ-substituted pyrrolinone scaffolds in an efficient and atom economical manner.
In this research field, Chen and co-workers in 2010583 reported the first direct organocatalytic conjugate addition of N-Boc-pyrrolinone to α,β-unsaturated aldehydes under iminium ion activation and, in the same year, Shibasaki and colleagues described the direct catalytic asymmetric Michael (and Mannich) addition of unsaturated γ-butyrolactams to nitroolefins (and aromatic aldimines) in the presence of a dinuclear nickel complex, achieving outstanding results in both cases (not shown).584,585
The organometallic catalysis in asymmetric vinylogous Michael additions was applied shortly after by Wang et al. in a work appearing in 2011 (Scheme 225).586 The approach devised by these researchers to direct the addition of N-Boc pyrrolinone 889 with a series of chalcones and related derivatives 890 entailed the use of the magnesium/3,3′-Ph2-BINOL L32 complex as the optimum catalyst system, which was prepared in situ by reacting the ligand L32 with dibutyl magnesium. The addition to a variety of enones afforded syn-configured products 891 in high yields, high diastereoselectivities, and variable enantioselectivities (54–98% enantiomeric excess). The selectivity of the reaction was slightly affected by the R1 substituent and highly dependent on the nature of the R2 group. Bidentate chelation between the in situ-generated N-Boc pyrrole dienolate and the chiral BINOL/magnesium complex was supposed to activate the nucleophile, while providing the chemical environment for high stereocontrol (Scheme 225, bottom).
Scheme 225.

On the other hand, Wang and co-workers adopted a different approach to govern the vinylogous Michael addition between pyrrolinone 889 and diverse chalcones 892, based on the use of the popular bifunctional cinchona thiourea catalyst C22 (Scheme 226).587 As shown in Scheme 226, the respective syn-configured Michael adducts 893 were accessed in high yields, with very high margins of diastereo- and enantioselectivity. The generality of the reaction was quite large, tolerating either electron-donating or electron-withdrawing enone substituents; heterocyclic systems were also reliable substrates, while alkyl-substituted chalcones failed to give appreciable results.
Scheme 226.

Based on previous investigations on the thiourea-tertiary amine catalytic mechanism,588−590 in a subsequent paper the same researchers postulated different possible models for the interaction of the bifunctional catalyst C22 with the reacting donor and acceptor partners and performed in-depth studies on the same reaction displayed in Scheme 226 by means of a combination of both experimental (NMR) and theoretical (DFT) approaches.591 In the event, the authors demonstrated a new dual activation pathway. The key feature of the proposed transition state is that the carbonyl group of the chalcone is activated by the more acidic NH of the thiourea moiety, while the ammonium nitrogen of the catalyst and the second NH of the thiourea simultaneously activate the N-Boc lactam nucleophile via hydrogen bonding, thus favoring the Si-Si approach that accounted for the observed syn-configuration of the products.
Many other researchers explored the utility of pyrrolinone 889 as the pronucleophilic substrate in organocatalyzed asymmetric direct VMcR to various acceptors such as nitroolefins, alkylidene malonates, enones, dienones, enoylpyridines, to cite but a few. The results are condensed in Table 9.
Table 9. Organocatalyzed Direct Vinylogous Conjugate Additions of N-Boc-pyrrolinone to Various Michael Acceptors.
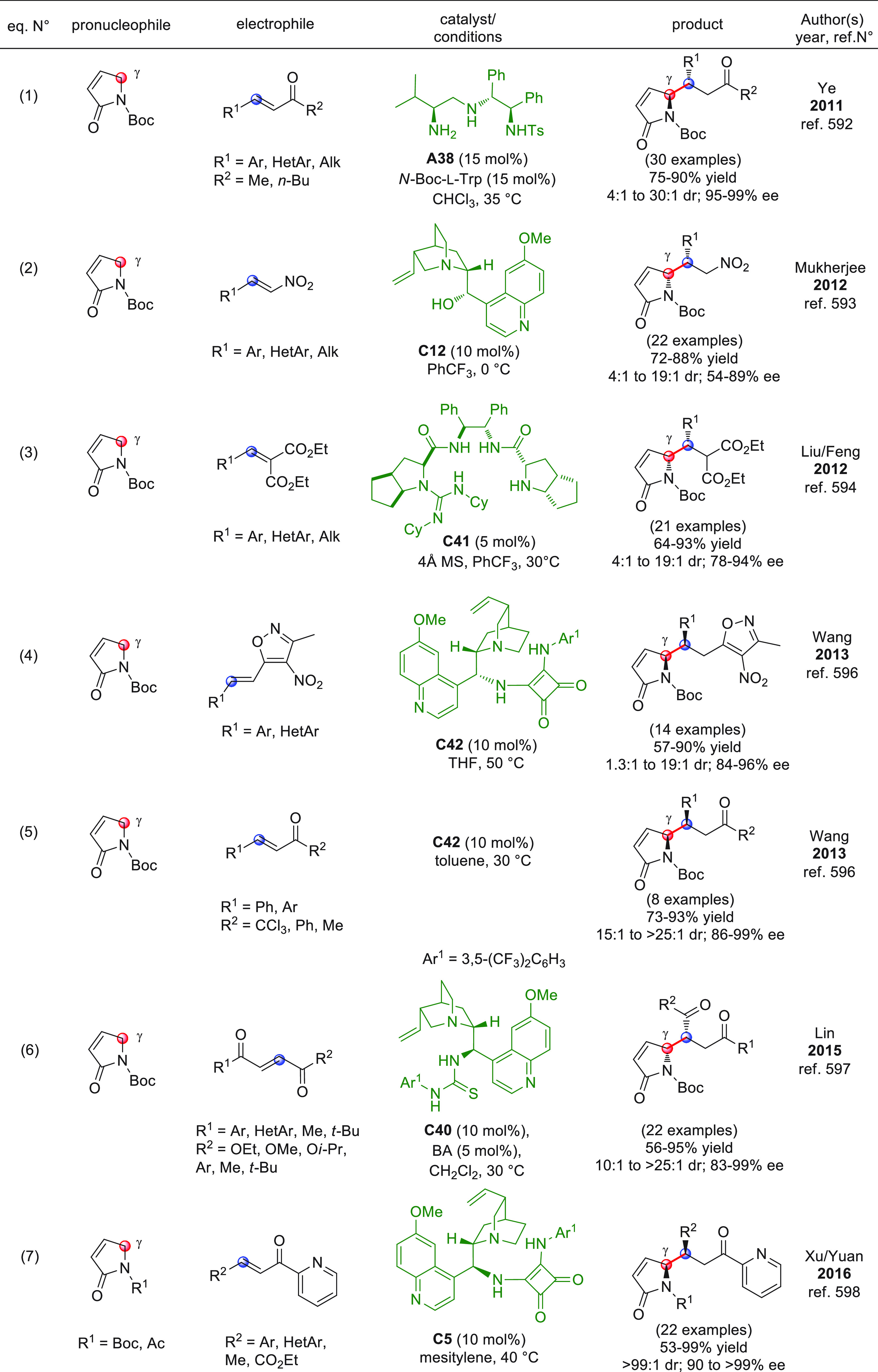
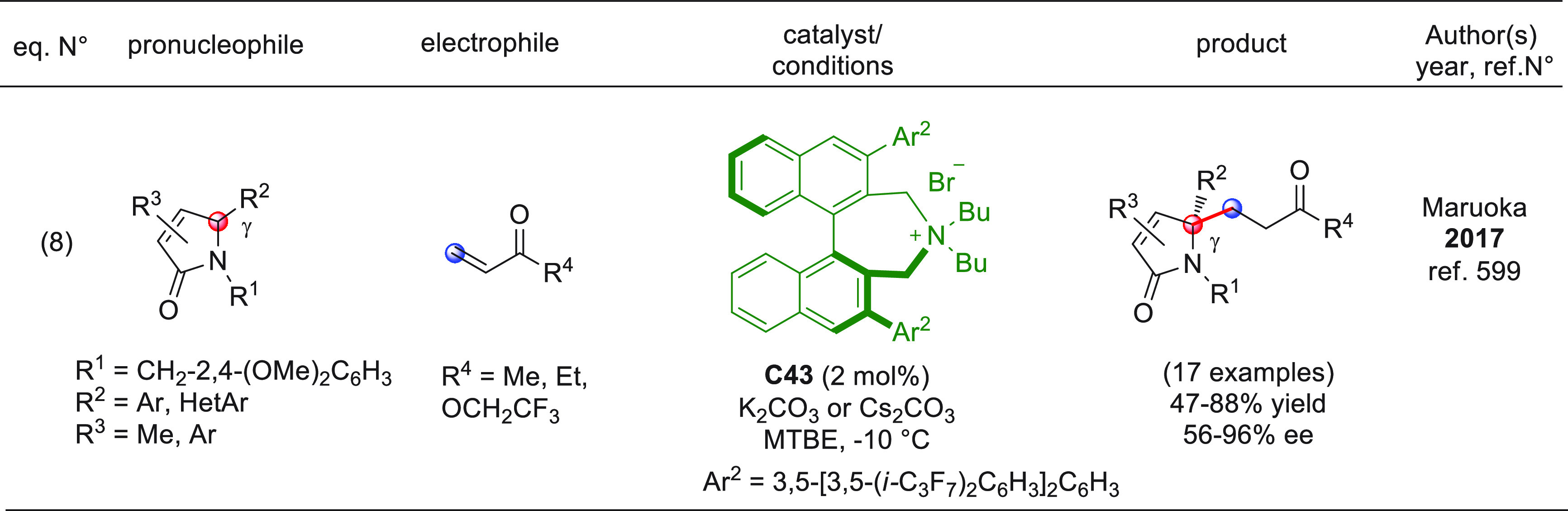
The trans-stilbene-derived chiral triamine catalyst A38 and N-Boc-L-tryptophan as an additive formed the catalyst system with which Ye et al. investigated the vinylogous Michael addition of N-Boc pyrrolinone to rather inert enone acceptors (Table 9, eq 1).592 The reaction scope proved to be large, encompassing aryl, heteroaryl, alkyl, and cycloalkyl enone substituents. In any case, uniformly high yields, diastereomeric ratios, and enantiomeric excesses were witnessed for the anti-configured pyrrolinone products. In the proposed transition state, LUMO-lowering iminium ion covalent activation of the acceptor by the primary amine of the catalyst, as well as HOMO-raising noncovalent activation of the dienolate donor via H-bonding network were simultaneously operative, along with the participation of the bulky amino acid L-tryptophan via ion pair interaction.
Mukherjee investigated the reaction of N-Boc pyrrolinone with a series of aryl, heteroaryl, and alkyl β-nitroolefins under the catalysis of quinidine derivative C12 (Table 9, eq 2).593 Syn-configured products were consistently obtained in good yields, excellent diastereoselectivity, and moderate enantiomeric excesses; on the other hand, the enantiomeric products could be accordingly prepared by using a dihydroquinine-derived organocatalyst. As for the mechanism, the authors postulated the activation of the nitroolefin by the quinidine hydroxyl group via H-bonding, whereas the tertiary amine moiety of the catalyst provided for the activation of the lactam pronucleophile.
Feng, Liu, et al. analyzed the behavior of the reaction involving N-Boc pyrrolinone and alkylidene malonate acceptors using the unsymmetrical guanidine-secondary amine multifunctional catalyst C41 (Table 9, eq 3).594 Optimal conditions were found using catalyst C41 (5 mol %) in combination with 4 Å molecular sieves in trifluoromethylbenzene. Reactions proved efficient and selective as far as diastereo- and enantioselectivity were concerned. A mechanistic rationale was proposed, based on control experiments and previous investigations.595 Thus, the basic guanidine in the catalyst provided γ-deprotonation of pyrrolinone to form the active dienolate; the N-Boc protecting group contributed in supplementary H-bonding to the amide on the same side of the guanidine moiety; meanwhile, the alkylidene malonate was activated through a network of hydrogen bonds with both the secondary amine and the second amide moiety of the catalyst.
The access to chiral substituted γ-butyrolactams by means of direct catalytic asymmetric Michael reactions was developed by Wang et al. by using α,β-unsaturated pyrrolinone as donor and 3-methyl-4-nitro-5-alkenyl-isoxazoles as bisvinylogous Michael acceptors (Table 9, eq 4).596 The quinine-derived squaramide C42(160) was identified as the best catalyst to govern the 1,6-Michael addition to various aryl nitro-isoxazoles, giving syn-adducts in high yields and remarkable diastereo- and enantioselectivity. The same bifunctional organocatalyst proved equally efficient in promoting the conjugate addition of N-Boc pyrrolinone to α,β-unsaturated trichloromethyl ketones (Table 9, eq 5).596 Excellent diastero- and enantioselectivities were achieved also in the case of chalcone having R1, R2 = Ph and with less reactive enone having R2 = Me.
α,β-Unsaturated γ-butyrolactam was also exploited as a pronucleophile in organocatalyzed vinylogous Michael additions to β-acyl acrylates and ene-diones by Lin and co-workers (Table 9, eq 6).597 The use of quinidine-derived bifunctional thiourea C40 in the presence of benzoic acid enabled the synthesis of the corresponding syn-configured butyrolactams in acceptable chemical yields; good-to-excellent diastereo- and enantioselectivities could be achieved with β-aroyl derivatives having R1 = aryl or heteroaryl, while with aliphatic keto esters (R1 = alkyl) the diastereoselectivity decreased.
Based on their previous studies on the organocatalyzed vinylogous conjugate additions of butenolides to enoylpyridines (vide supra, Table 8, eq 4), Xu, Yuan, et al. demonstrated the efficacy of the same quinine-derived squaramide C5 as the catalyst in Michael additions exploiting γ-butyrolactams as pronucleophilic reagents (Table 9, eq 7).598 The reaction was broad in scope concerning the nature of the aromatic ring substituents within the electron-poor acceptors, producing the corresponding adducts in high yields and excellent levels of stereocontrol; the N-acetyl protected lactam substrate gave efficiently the corresponding adduct, while with the N-methyl counterpart the reaction failed and no desired products were found. Based on experimental results and previous experience, the authors proposed a possible transition state model, emphasizing the role of the catalyst tertiary amine in activating the pyrrolinone dienolate and the role of squaramide NH groups in coordinating both the carbonyl and the pyridine nitrogen of the acceptor via H-bonding, thus precisely governing the stereoselective attack.
Aiming to the development of a catalytic enantioselective method to provide γ,γ-disubstituted γ-lactams, Maruoka and colleagues proposed the exploitation of chiral phase-transfer catalysis to promote the vinylogous Michael addition of γ-monosubstituted pyrrolinones to vinyl ketones (or esters) (Table 9, eq 8).599 During these studies, any attempt to selectively alkylate the substrates with benzyl bromide failed, while the conjugate addition to α,β-unsaturated carbonyl derivatives in the presence of chiral ammonium salt C43 worked well and stereoselectively led to unsaturated lactam products bearing a quaternary stereocenter at the γ-position. The introduction of aryl groups at the α-position did not compromise the reactivity and stereoselectivity, indicating the generality of the protocol. Moreover, the strategy was applied to trifluoroethyl acrylate acceptor (R4 = OCH2CF3) with just a modest erosion of the efficiency and selectivity.
The potential of unsaturated γ-butyrolactams as key precursors in asymmetric organocatalyzed synthetic strategies has been explored by Wang and collaborators, who proposed a direct approach to functionalize the pyrrolidinone scaffolds in β,γ-selective IEDDA [4 + 2] annulations (Scheme 227).600 The researchers set out to exploit unsaturated pyrazolones 894 or α,β-unsaturated imino derivatives 896 as electrophilic reagents and chiral bifunctional thioureas C44 or C45(160) as the organocatalysts. In light of their previous investigations,601 they envisaged that the dienolate deriving from lactam 889 might serve as electron-rich dienophile undergoing IEDDA annulations with proper electron-poor dienes of type 894 or 896; in fact, the reaction of 889 with phenyl-substituted pyrazolones 894 worked well in the presence of thiourea C44, while the alkyl-substituted counterparts required catalyst C45 in combination with benzoic acid, affording in one step tricyclic dihydropyranopyrrolidin-2-ones 895 in good to high yields and stereoselectivities (Scheme 227, eq 1). Wishing to explore the substrate scope, the protocol using catalyst C44 was extended to acyclic imino derivatives 896, providing a series of chiral aza-analogue architectures 897 in comparable efficient and stereoselective manner (Scheme 227, eq 2).
Scheme 227.

The authors hypothesized a transition state model (Scheme 227) accounting for the observed stereochemistry of the annulated products, in which a dual HOMOdienofile/LUMOdiene-activation was operative with the catalyst tertiary amine promoting the in situ dienolate formation and the thiourea moiety activating the pyrazolone acceptor. Although the experimental results were perfectly consistent with the hypothesis, a stepwise mechanism involving a sequential vinylogous Michael/oxa- or aza-Michael reaction cascade could not be ruled out.
A few years later, the issue of β,γ-functionalizing the pyrrolidinone scaffold was faced by Ye, Dixon, et al., who developed an unprecedented organocatalyzed reaction cascade initiated by a doubly vinylogous Michael addition (DVMcA) between γ-butyrolactams 898 and cyclic 2,4-dienones of type 899 or 901 (Scheme 228).602 The challenging simultaneous activation of the vinylogous donor 898 and the rather unresponsive dienones 899/901 acting as vinylogous Michael acceptors was obtained by the bifunctional diamine catalyst deriving from l-tert-leucine A34. When the challenging γ-to-δ 1,6-addition of protected γ-butyrolactams 898 involved the sterically congested β-substituted cyclohexenones 899, Michael adducts 900 were obtained with high regio- and stereoselectivity in the presence of A34 and p-anisic acid 235 (Scheme 228, eq 1). The remote transmission of the stereochemistry through the conjugate double bonds was ensured by the catalyst diamine functionalities responsible for the simultaneous covalent iminium ion activation of dienones and strong hydrogen-bonding interactions with the deprotonated butyrolactam. On the other hand, when 3-alkenyl 2-cyclopentenones 901 were used as substrates, the initial DVMcA was followed by a vinylogous Michael addition/isomerization cascade involving the γ′-position of cyclopentenone and the β-carbon of the lactam, thus affording tricyclic lactams 902 with four newly formed stereocenters (Scheme 228, eq 2).
Scheme 228.

The competence of β,γ-unsaturated γ-lactams in participating in [3 + 2] annulation reactions as multidentate nucleophiles was explored by Kalaitzakis, Vassilikogiannakis, et al. in 2018 during the asymmetric synthesis of a collection of pyrrolizidine bicyclic lactams (Scheme 229, eq 1).603 The authors set up a strategy to accomplish asymmetric and site-selective annulations with unconjugated γ-alkyl pyrrolinone 903 and α,β-unsaturated aldehydes 904 under silyl prolinol organocatalysis.
Scheme 229.

In light of the results, the authors hypothesized that the vinylogous Michael attack of the dienolates deriving from 903 to iminium-ion activated enals 904 was followed by the intramolecular N-hemiaminalization, thus providing bicyclic adducts 905 with remarkable levels of stereoselectivities, albeit in fair chemical yields. They postulated that the stereochemistry of 905 resulted from an ion-paired transition state (not shown) that forces the Si face of the lactam to attack the Si face of the activated enals in the first step of the reaction cascade; the aldehyde group is then trapped in the hemiaminal ring-closure.
The potential of covalent organocatalytic protocols in combination with tandem reactions was demonstrated some years before by Enders and co-workers in the rapid synthesis of a series of highly substituted 2-aza-bicyclooctadienones (Scheme 229, eq 2).604 These researchers developed a three-component quadruple reaction cascade of aryl α-ketoamides 906 and enals 907 in the presence of (S)-diphenylprolinol TMS ether A4. The cascade proceeded via an initial aza-Michael addition of the ketoamide nitrogen to iminium ion-activated enal, followed by the enamine-promoted aldol condensation forming the γ-butyrolactam intermediate 906′; this last compound participated as vinylogous donor in the Michael addition reaction with a second enal unit, forming the two stereocenters within the γ,γ-disubstituted pyrrolinone 906″ in a highly stereocontrolled manner; the final intramolecular aldol condensation via enamine catalysis completed the target bicyclic compounds 908. The authors noted that the high stereocontrol in the generation of the two stereocenters depended on the facial selectivity in the key vinylogous Michael addition step; here, the iminium ion-activated α,β-unsaturated aldehyde was attacked on its Re face by the dienolate thus generating both the new stereocenters and placing the two Ar3 substituents in a trans orientation. The whole process was orchestrated by the chiral amine organocatalyst A4 and produced in good yields complete diastereoselectivity and high enantioselectivity polysubstituted azabicyclooctadienones 908 (Scheme 229, eq 2).
As already noted, 3-alkylidene-2-oxindoles are among the most studied vinylogous heterocycles in Michael additions, which present their enolizable γ- (ε- or ω-) sites out (or far) from the cyclic indole core. Other useful heterocyclic scaffolds of this type are their aza-analogues (aza-alkylidene oxindoles and nitrone ylides deriving from isatin) along with alkylidene pyrazolinones.
It is worth pointing out here that most of the strategies taking advantage of the vinylogous nucleophilicity of alkylidene oxindoles entail direct executions in Michael-type addition reactions.
In the realm of racemic approaches based on alkylidene oxindoles, two contributions are due to the Shanmugam research group, the first of which appeared in the literature in 2010.605 Here, the authors reported an example of phosphine-catalyzed direct VMcA involving bromomethyl alkylidene oxindoles as vinylogous nucleophiles and maleimide or methyl acrylate as electrophilic partners (not shown). In the second example,606 the same researchers described the diastereoselective synthesis of a series of oxindole-appended vinyl cyclopropanes based on the use of sulfur ylides from bromoallylidene oxindoles and activated styrenes (not shown).
Going through the asymmetric versions of the conjugate additions involving alkylidene oxindoles, the first example of direct organocatalytic vinylogous Michael addition to nitroolefin acceptors was reported by Curti, Casiraghi, et al. in 2012 (Scheme 230, eq 1).607 The authors investigated the potential of 3-alkylidene-2-oxindoles 909 as viable γ-enolizable nucleophiles in conjugate additions to a variety of β-nitrostyrenes 910. The reaction was orchestrated by the bifunctional cinchona alkaloid thiourea C39 and delivered almost enantiopure γ-substituted Z-configured alkylidene oxindoles 911 with excellent levels of regio- (γ:α > 99:1), diastereo- (>20:1 diastereomeric ratio), and enantioselectivity (>99:1 enantiomeric excess). Provided that N-carbamoyl protecting groups within the oxindole substrates were used (Boc, Moc), the reaction scope and generality were remarkable, regardless of the presence of neutral, electron-withdrawing, or electron-donating substituents on the two reaction components. The authors demonstrated that the cooperativity between the basic and the acidic moieties in the bifunctional catalyst was a prerequisite for an efficient chirality transmittal.
Scheme 230.

One year later, in 2013, the same researchers reported another example of direct vinylogous Michael addition in which the acceptors were again nitroolefins 910, while the donors were prochiral alkylidene oxindoles of type 912, thus enabling the simultaneous installation of two stereocenters within the adducts 913 (Scheme 230, eq 2).608 Even in this case, the best results in terms of yield and diastereo- and enantioselectivity were obtained with thiourea C39, and the reaction furnished only Z-configured anti-configured products 913. The investigation of the substrate scope revealed that this vinylogous reaction was general, with the sole limitation that unprotected and N-alkyl substituted indoles did not give appreciable results. Based on the experimental evidence and several precedents with bifunctional organocatalysts,590,609 two possible models of the dual activation were proposed, in which both the nucleophilic and the electrophilic reaction components were activated by means of the bifunctional catalyst: the thiourea unit could activate the nitroalkene by double hydrogen bonding, while the quinuclidine base deprotonated the oxindole to afford the active dienolate species; an additional hydrogen bond between the carbonyl of the indole N-protecting group and the protonated quinuclidine base of the catalyst further contributed to the stabilization of the transition state (Scheme 230), thus ensuring stereocontrol and chirality transmittal from the catalyst to the product. An alternative transition state model involving electrophile activation by the protonated amine group of the catalyst and nucleophilic activation by the thiourea moiety could not be excluded.
Since these contributions, which launched the γ-enolizable alkylidene oxindoles as useful vinylogous donor matrices in asymmetric synthesis, a number of studies appeared dealing with the exploitation of these scaffolds in organocatalyzed vinylogous Michael additions to nitroalkene or α,β-unsaturated carbonyl acceptors which are listed in Tables 10 and 11, respectively.
Table 10. Asymmetric Vinylogous Conjugate Additions of γ-Enolizable 3-Alkylidene Oxindoles to Nitroalkene Acceptors.
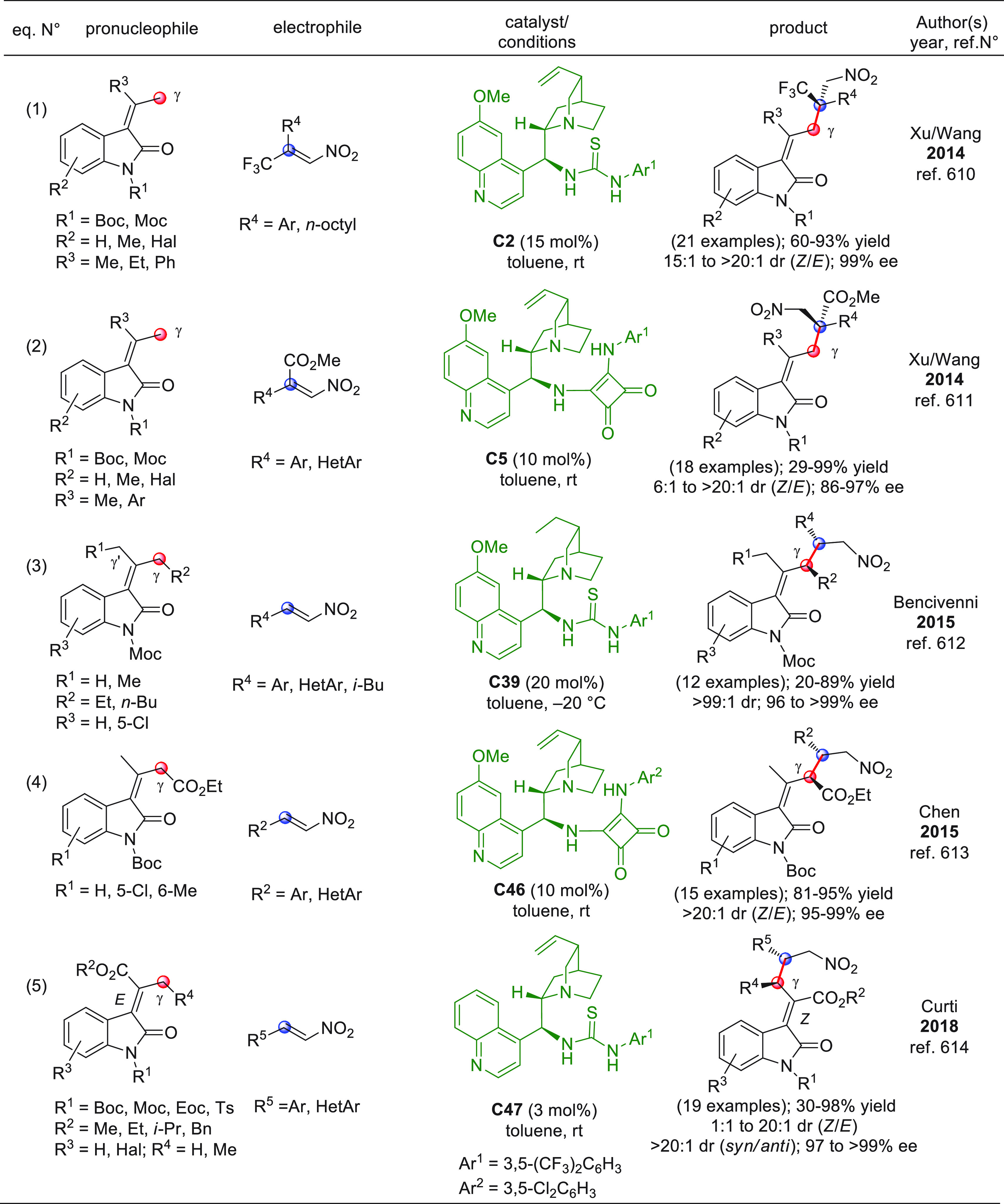
Table 11. Asymmetric Vinylogous Conjugate Additions of γ-Enolizable 3-Alkylidene Oxindoles to Unsaturated Carbonyl Acceptors.
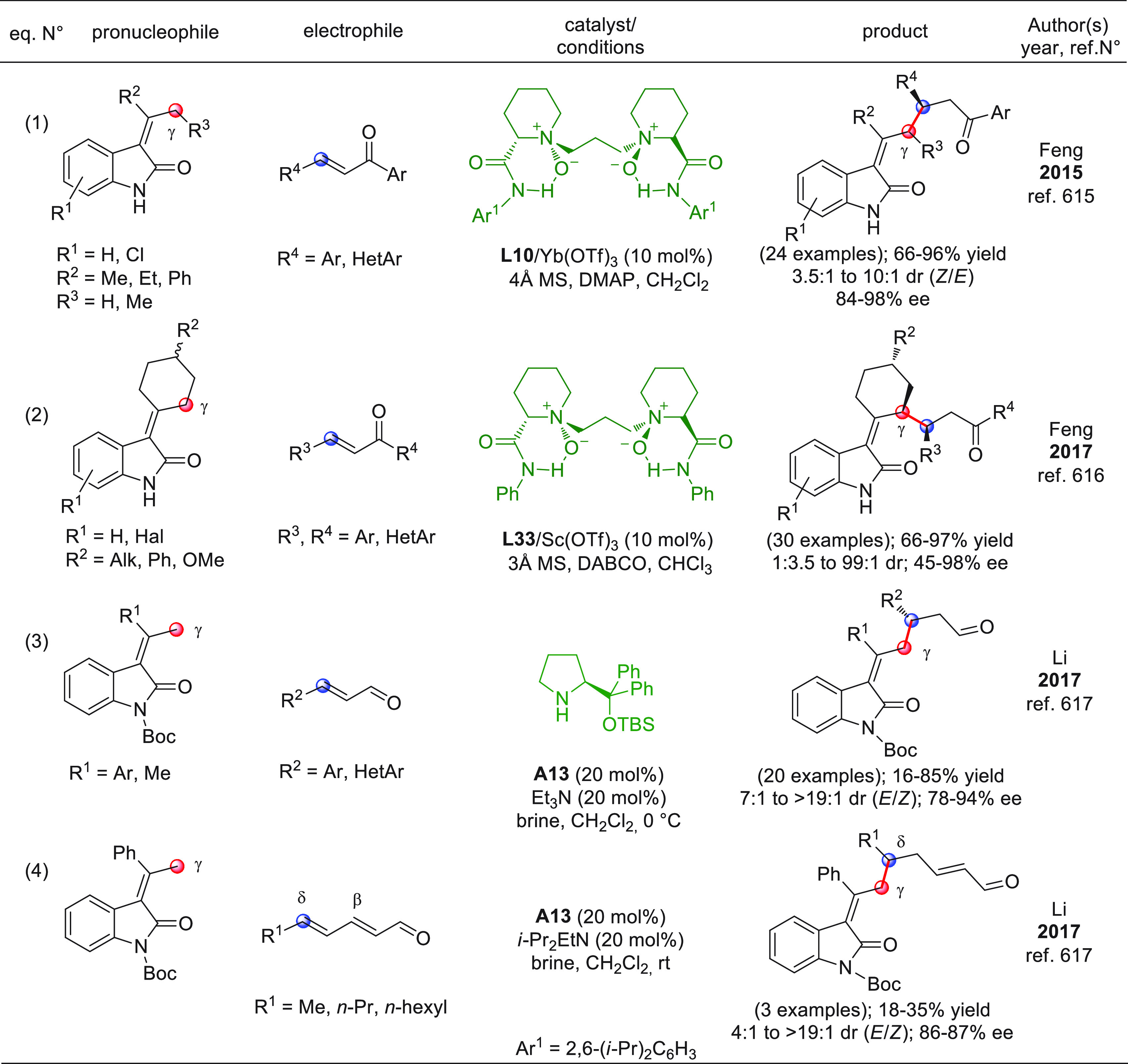
In 2014, Xu, Wang, et al. reported the vinylogous Michael addition reaction between alkylidene oxindoles and β,β-disubstituted nitroolefins catalyzed by bifunctional quinine thiourea C2 (Table 10, eq 1).610 A series of chiral oxindoles bearing trifluoromethylated all-carbon quaternary stereocenters was obtained in good yields, excellent Z/E ratio, and enantioselectivity.
The same authors published a further development of the protocol, in which the Michael addition of oxindoles to α-substituted β-nitroacrylates gave oxindole products with a quaternary stereocenter (Table 10, eq 2).611 The reaction required the use of quinine-derived bifunctional squaramide C5, which was postulated to simultaneously link via H-bonding the nitroalkene moiety and generate the oxindole dienolate by means of the tertiary amine unit.
Another example of direct, vinylogous Michael addition to β-nitroolefins was presented by Bencivenni and collaborators who studied the γ-functionalization of nonsymmetric alkylidene oxindoles having R1 ≠ R2 (Table 10, eq 3).612 The reactions were carried out at −20 °C to suppress the interconversion between E and Z isomers, and the organocatalyst of choice was the 9-epi-hydroquinine thiourea C39 being able to selectively deprotonate the oxindole γ-position (and not the γ′-site), as confirmed by isotope-effect experiments. The reactions involving Z-configured pronucleophiles proved to be completely regiocontrolled and extremely stereoselective, affording the corresponding products as single Z-diastereoisomers with excellent enantiocontrol. The chemical yields were generally good, with the exception of the reaction exploiting an aliphatic nitroalkene as acceptor. Similar results were achieved with E-configured substrates, providing Z-adducts, although in such cases regio- and stereoselectivity were decreased.
In 2015 Chen and co-workers proposed the dichloro-substituted squaramide catalyst C46 to promote the asymmetric Michael addition reaction of oxindolinylidene butanoates to nitroolefins (Table 10, eq 4).613 The use of catalyst C46 afforded the best results in terms of yield and diastereo- and enantioselectivity, and the reaction turned out to be efficient and selective with a variety of substituted nitrostyrenes.
Continuing on this theme, recently Curti and colleagues introduced (E)-3-(alkoxycarbonyl-2-alkyliden)-2-oxindoles as γ-enolizable pronucleophiles to be used in organocatalyzed Michael additions to nitroolefins (Table 10, eq 5).614 After extensive optimization studies, the thiourea C47 was selected to generate multidentate γ-dienolates that add to nitroalkenes and deliver vinylogous Z-configured products with excellent levels of site-selectivity and diastereo- and enantioselectivity. It is noteworthy that the reaction showed an unprecedented Z-selectivity, providing chiral alkylidene oxindoles with geometrically inverted C3–C2′ double bonds (the newly formed C–C bonds and the related stereocenters are oriented trans to the oxindole carbonyl), as opposed to most of the γ-homologated alkylidene oxindoles mentioned so far. The authors supposed that the ester handle within the oxindole pronucleophiles could play an active role in determining the geometry of the products and reported a mechanistic pathway supported by DFT calculations and accounting for the observed stereochemical outcome. According to their assumption, the alkoxycarbonyl group may function as an additional anchoring point with the catalyst quinuclidine portion stabilizing the s-trans conformation of the oxindole dienolate, while the thiourea would activate the electrophile by hydrogen bonding. Moreover, the synthetic utility of the Michael adducts was demonstrated by chemical transformations leading to a quaternary oxindolyl proline analogue and a chiral spyrocyclic furoindolone scaffold.
In 2015, Feng and co-workers reported the first example of direct, asymmetric VMcR of alkylidene oxindoles to α,β-unsaturated carbonyl acceptors (Table 11, eq 1).615 The reaction between unprotected oxindoles and chalcones was catalyzed by the ytterbium(III)-N,N′-dioxide chiral complex L10/Yb(OTf)3 in the presence of DMAP and molecular sieves. The yields of the reactions were generally high and the enantioselectivity was good, though the Z/E ratio for many adducts was not impressive. Two years later, the same authors envisaged exploiting a similar N,N′-dioxide chiral metal complex in vinylogous Michael additions to chalcones aiming at the deracemization of axially chiral cyclohexylidene oxindole compounds (Table 11, eq 2).616 In the event, the L33/Sc(OTf)3 complex served as the catalytic system in the one-step dynamic resolution of the starting unprotected racemic oxindoles, giving Michael products as the only Z-isomers in good yields and with high levels of diastereo- and enantiocontrol.
The asymmetric iminium ion activation of enals as electrophilic partners in vinylogous conjugate additions of alkylidene oxindoles was explored by Li and colleagues in 2017 (Table 11, eq 3).617 The TBS-protected diphenylprolinol A13 was chosen as the covalent organocatalyst to access the corresponding chiral Michael products in moderate to good yields and high enantioselectivities. Interestingly, the reaction of phenyl-substituted oxindole to 2,4-dienals resulted in 1,6-addition with complete δ-site preference and acceptable enantioselectivity, although yields turned out to be modest (Table 11, eq 4).
The sole example of an asymmetric annulation cascade involving 3-alkylidene oxindoles 914 and nitroolefin enoates 915 was reported by Feng and Li in 2017 (Scheme 231).618 The proposed reaction entailed a sequence of two Michael reactions, a first vinylogous conjugate addition to a nitroalkene moiety, followed by an intramolecular closure on the enoate portion. The procedure led enantioselectively to oxindoles 916 bearing three contiguous stereocenters on the newly formed chroman framework (X = O). The reaction cascade was orchestrated by the bifunctional squaramide C5, which was able to simultaneously generate the oxindole dienolate and activate the nitroolefin and the enoate moiety via H-bonding, according to the transition state proposed by the authors (Scheme 231, bottom).
Scheme 231.

Continuing on the theme of vinylogous indolinone chemistry, the ketimines and nitrones deriving from isatins can be regarded as aza-analogues of 3-alkylidene oxindoles (often these reagents are indicated as aza-alkylidene oxindoles) as they own a C=N bond which behaves as an inherently unsaturated π-system and participates in transferring the electronic properties of the indolinone core to the remote γ-site. Along this line, such isatin derivatives have found applications as precursors of aza-dienolates in vinylogous Michael additions to electron-poor olefins or in reaction cascades involving conjugate addition reactions. Apart from one example reported by Trivedi et al.619 in which the benzylimines deriving from isatin produced spirooxindole-pyrrolidines in racemic format (not shown), the remaining examples fall in the field of asymmetric organocatalyzed processes.
In 2015, Wang and colleagues disclosed that N-2,2,2-trifluoroethyl isatin ketimines 917 could be γ-deprotonated by means of bifunctional squaramide C31 to afford azomethine ylides (azadienolates) and take part to formal γ,α-regioselective [3 + 2] cycloadditions with nitroalkenes 910 (Scheme 232, eq 1).620 The scope of both substrates was explored and the reactions produced trifluoromethylated spiropyrrolidine oxindoles 918 in high yields and excellent enantio- and diastereoselectivities. The authors supposed the involvement of the tertiary amine catalyst in the formation and H-bonding of the azomethine ylide and the concomitant activation of the nitroalkene via the squaramide H-bonding, thus ensuring high stereocontrol.
Scheme 232.

On the other hand, Du, Chen et al. exploited stable and readily available N-benzyl nitrones of isatins 919 as precursors of the corresponding ylides in catalytic asymmetric γ,α-regioselective [3 + 2] cycloadditions to enals 920 (Scheme 232, eq 2).621 The reactions proceeded smoothly under the assistance of TMS-protected diphenyl prolinol A4 and a catalytic amount of trimethylamine, giving rise, after reduction with NaBH4, to N-hydroxy-pyrrolidinyl derivatives 921 in moderate to good yields and considerable enantioselectivity. The detection in the reaction of a Michael adduct intermediate from N-benzyl nitrone ylide (R1 = Me, R3 = Ph) and cinnamaldehyde confirmed the supposed stepwise mechanism for this cycloadditive process. Interestingly, a fine regioselectivity switch toward nonvinylogous α-conjugate addition could be obtained with crotonaldehyde by adding catalytic amounts of suitable metal salts.
Shortly after, the same authors set up an organocatalyzed protocol to perform aza-vinylogous-type Michael additions of nitrones 922 to nitroalkenes 910 (Scheme 232, eq 3).622 The reactions were run in the presence of bifunctional cinchonidine thiourea C47 and molecular sieves, and provided the vinylogous Michael products 923 under almost complete diastereo- and enantiocontrol. It is noteworthy that the Mannich-type cyclization step affording spiropyrrolidine scaffolds did not occur, contrary to the above procedures involving ketimines 917 and nitrones 919.
As previously mentioned, the Morita–Baylis–Hillman (MBH) carbonates belong to a group of valuable compounds that have been widely exploited as electrophiles in many examples of metal-free asymmetric allylic alkylations623 and other substitution reactions with various nucleophiles (vide supra). Although these procedures in most cases lead to overall alkylation products, they indeed proceed through Michael-type additions to electron-poor double bonds of MBH-intermediates, and therefore examples of this chemistry are judged to be nice examples of vinylogous processes and they are legitimately listed in and commented on in this section.
Li and colleagues reported the first example of an asymmetric allylic alkylation involving oxindoles 924 as the pronucleophilic reagents and MBH carbonates 925 as the electrophilic counterparts (Scheme 233).624 The reaction was promoted by the chiral bis-cinchona catalyst C48 activating the MBH carbonates, while the γ-deprotonation of oxindole donor relied on the in situ generated tert-butoxy anion released by compounds 925. The reaction scope was wide, and a series of γ-allyl-substituted alkylidene oxindoles 926 were prepared in moderate to good yields, with excellent enantioselectivity and Z/E selectivity.
Scheme 233.

The MBH carbonates 928 also served as competent substrates in the asymmetric allylic alkylation of trifluoroethylisatin ketimines 927, documented by Wang and collaborators in 2016 (Scheme 234).625 The best catalyst promoting the process was the β-isocupreidine C21 acting as a chiral Lewis base in the generation of the activated electrophiles from compounds 928; the products 929 were isolated in moderate yields and with remarkable diastereo- and enantioselectivities as a result of the nucleophilic attack of isatin ketimine dienolates to activated double bonds of MBH intermediates. The authors speculated on the possible reaction mechanism describing the process as a sequence of two SN2′-type reactions, that ultimately led to the chiral imines 929, useful precursors of biologically relevant fluorinated α-methylenelactams 930.
Scheme 234.

The umpolung of the MBH derivatives toward a nucleophilic reactivity can be obtained by using proper chiral tertiary phosphines or amines as Lewis bases able to form P or N ylide reagents. As demonstrated by Lu and co-workers, the in situ formed ylides from isatin MBH derivatives are capable to participate as 1,3-dipolar synthons in asymmetric [3 + 2] annulations and other cycloaddition maneuvers.626
In particular, the potential of MBH carbonates deriving from isatins (and acrylates or acrylonitriles) as vinylogous pronucleophiles in Michael-initiated cycloadditions has been explored by Ouyang, Chen, et al. in 2016 (Scheme 235).627 These researchers found that MBH carbonates 931 could alternatively undergo chemo- and stereocontrolled [2 + 1] (Scheme 235, eq 1) or [3 + 2] (Scheme 235, eq 2) annulations with activated alkenes 932, depending upon the nature of the Lewis base used. The reactions performed in the presence of α-isocupreine C49 as bifunctional catalyst efficiently provided cyclopropane derivatives 933 (or their enantiomers, if β-isocupreidine catalyst was employed) with high enantiomeric purity as a result of the [2 + 1] annulation (Scheme 235, eq 1). When bifunctional chiral phosphine P3 was used with the same couple of starting reagents 931 and 932, a switch in chemoselectivity was observed, and the [3 + 2] annulated spirooxindoles 934 were obtained in excellent yields and good to high enantioselectivity (Scheme 235, eq 2). The diastereoisomeric spirocycles 935 were prepared by using either bicyclic phosphine P9 or chiral base C50, with the latter showing better catalytic activity (Scheme 235, eq 3). DFT calculations were performed to rationalize the different annulation mechanisms and the related stereochemical outcomes; in every case, the first step was recognized as a vinylogous Michael addition of ylides 931′ to alkylidene diones 932, followed by an SN2 attack to the γ-position for the [2 + 1] cyclization (Scheme 235, eq 1); on the contrary, a second Michael reaction involving the oxindole α-position occurred to accomplish the [3 + 2] annulation process.
Scheme 235.

The same researchers also described an unexpected organocatalyzed domino process involving, once more, the isatin-derived MBH carbonates as pronucleophiles with unsaturated α-cyano ketones as activated alkene acceptors (Scheme 236).628
Scheme 236.

In the event, the highly electrophilic α-cyanoketones 936 were reacted with MBH carbonates 931 in the presence of β-isocupreidine C21, resulting in fused tetrahydrofuro-pyranoindoles 937 in enantiopure form (Scheme 236, eq 1). On the basis of previous experience with isatin ylides 931′, the authors supposed the initial generation of cyclopropane intermediates of type 931″ (not detected during the reaction) that subsequently underwent ring-opening, through O-attacking to the cyclopropane C3, followed by intramolecular oxa-Michael ring-closure, ultimately leading to the formation of the polyheterocyclic structures 937. During the exploration of the substrate scope, it was disclosed that the annulation pathway could switch to a conventional [3 + 2] version producing spirooxindoles 939 (Scheme 236, eq 2) when the MBH carbonates 938 were used under amine C51 catalysis. The DFT computational calculations suggested that a concerted mechanism for the ring-opening/ring-closure domino reactions was operative, accounting for the diastereocontrolled formation of polycyclic structures 937; regarding the sequential γ,α-Michael addition cascade, the authors concluded that the introduction of a stronger electron-withdrawing group at the C=C bond was the key to obtain the regioselective formation of spirooxindoles 939.
Prompted by the success of 3-alkylidene oxindoles as heterocyclic nucleophilic synthons in asymmetric vinylogous conjugate additions, in 2014 Zanardi, Rassu, and colleagues developed a new asymmetric strategy to functionalize α-alkylidenepyrazolinones by means of organocatalyzed 1,4-additions to nitroalkenes (Scheme 237).629 The reaction of variously substituted pyrazolinones 940 with aromatic nitroolefins 910 was orchestrated by quinine-based thiourea organocatalyst C39 and yielded to the corresponding γ-adducts 941 in high yields, with remarkable levels of enantio- and diastereoselectivities and geometrical selectivities. The pyrazolinones belonging to the enantiomeric series were efficiently accessed by using the quasi-enantiomeric quinidine-derived thiourea C40. Of note, prochiral pyrazolinone donors 940 bearing different enolizable γ and γ′ positions were competent substrates in the addition reaction giving the Z-configured regioisomers, irrespective of the geometry of starting ylidenes 940.
Scheme 237.

On the basis of the Michael adduct stereochemistry and chemical correlation experiments, and capitalizing on previous studies on related organocatalyzed vinylogous Michael reactions, the authors proposed a plausible mechanistic rationale entailing the γ-deprotonation of the pyrazolinone by the tertiary amine catalyst and the ion-pair interaction with the resulting dienolate; meanwhile, the thiourea moiety activated the electrophile by H-bonding. Thus, the observed Z versus E preference would be dictated by the s-cis conformation of the dienolate, while the anti/syn selectivity would be imparted by the Z-geometry of the donor double bond in the stereodetermining step.
The potential of alkylidene pyrazolinones 942 as vinylogous reagents in conjugate additions was also explored by Biju et al. during a study on the enantioselective synthesis of pyrazolone-fused spirocyclohexadienones 944 under oxidative NHC organocatalysis (Scheme 238).630 The authors demonstrated that a variety of pyrazolinones 942 reacted as dienolate donors 942′ with acyl azoliums 943′ generated from NHC salt ent-B18 and variously substituted cinnamaldehydes under oxidative conditions. The reactions were conducted in the presence of DBU as the base and quinone 45 as the oxidant and furnished the spirocyclic adducts 944 bearing an S-configured all-carbon quaternary stereocenter in moderate to good yields and good to excellent enantiomeric excess values. A plausible mechanism was postulated for this formal [3 + 3] spiroannulation, which involved a vinylogous Michael addition–spirocyclization–dehydrogenation reaction sequence. Accordingly, the γ-deprotonation of pyrazolone 942 by DBU was followed by the vinylogous intermolecular Michael addition of dienolate 942′ to the chiral acyl azolium ion 943′ leading to an intermediate (not shown) which, upon proton transfer, formed dienolate 942″; this last, in turn, reacted intramolecularly at its α-position by giving the C-acylation. The resulting spiro-compound intermediate formed the spirocyclohexadienone target 944 in the presence of the oxidant.
Scheme 238.

Inspired by the successful exploitation of arylidene pyrazolinones as bisnucleophile species in the synthesis of spirocyclic compounds, the same researchers ventured into the development of a one-pot organocatalyzed synthetic strategy to directly access pyrazolone spirocyclohexenols 946 (Scheme 239, eq 1).631 Biju and colleagues envisaged that the dienolate formed from pyrazolinones 945 would intercept the enals 920 activated as iminium ions by the chiral secondary amine A2; an intramolecular aldol maneuver would follow, giving rise to the spirocyclohexenols 946 (Scheme 239, eq 1). The authors succeeded in their plan and accomplished the synthesis of spirocyclic pyrazolinone targets in high yield and enantioselectivity, even though with moderate to good diastereoselectivities. A few months later, the Han and Li research group reported a very similar asymmetric γ,α-regioselective [3 + 3] cyclization involving the same pyrazolinones 945 and enals 920 via secondary amine organocatalysis (Scheme 239, eq 2).632 They adopted a protocol based on the use of prolinol A13 as the catalyst, in the presence of acetic acid or o-nitrobenzoic acid (237) achieving exactly the already reported cyclohexenols 946, even if with better yields and stereoselectivities.
Scheme 239.

The asymmetric synthesis of spirocyclohexene pyrazolones 948 bearing a nitro-substituent was the aim of the research performed by Xu et al., who exploited the bifunctional squaramide C5 to catalyze the γ,α-regioselective [3 + 3] cycloaddition reaction (Scheme 240).633 The well-known α-arylidene pyrazolinones 942 were selected as the γ,α-binucleophilic synthons to be used in the vinylogous Michael/Michael addition cascade with (E)-2-nitroallylic acetates 947. A series of spirocyclohexene pyrazolones 948 having a quaternary stereocenter were prepared in good yields and excellent stereoselectivities, thanks to the synergistic activation of the vinylogous pyrazolones and the α,β-unsaturated nitroacetates, as illustrated in Scheme 240. According to the proposed catalytic cycle, the Michael attack by the dienolate species forming the first stereogenic center was concurrent to the release of AcOH, and after tautomerization, a second intramolecular Michael reaction provided the final cyclohexene derivatives.
Scheme 240.

The research group of Guo also developed an asymmetric Lewis base-catalyzed γ,α-[3 + 3] cyclization strategy to access spiropyrazolone scaffolds 951, based on the exploitation of arylidene pyrazolones 949 as vinylogous donors and MBH carbonates 950 as the electrophilic precursors (Scheme 241).634 Here, the N-ylides formed from MBH carbonates 950 and the chiral Lewis base C52 could efficiently γ-deprotonate the pyrazolinones 949 and simultaneously generate the electrophilic Michael acceptors, thus avoiding the need of an additional base. Regarding the substrate scope, various aryl and heteroaryl substituted carbonates 950 proved competent in the reaction and high yields of the products along with good stereoselectivities were observed in most cases. The possible reaction mechanism consisted of the ylide formation, followed by the dienolate generation and the intermolecular vinylogous 1,4-attack affording intermediate 949′ (Scheme 241); after proton transfer, the resulting α-enolate 949″ completed the ring closure and regenerated the chiral catalyst.
Scheme 241.

6.3.2. Indirect Procedures
6.3.2.1. Acyclic Nucleophiles
In the period covered by this review the sole example demonstrating the utility of silicon dienolates from acyclic α,β-unsaturated amides in conjugate additions was proposed by the group of Schneider in 2014 (Scheme 242).635 These researchers profited from the Z-configured vinylketene silyl N,O-acetals 952 as vinylogous nucleophiles in the conjugate addition to enals 920. When pyrroles 952 were reacted with enals in the presence of diphenylprolinol silylether A4, variable mixtures of α- and γ-regioisomeric products were obtained; however, under optimized conditions, the best γ-regioselectivity was obtained when bulky diphenylmethylsilyl (DPMS)-substituted dienolates 952 were added to a range of enals 920; the vinylogous adducts 953 were obtained in generally high enantioselectivities when nonprochiral donors were used (952, R1 = H), while the presence of a γ-methyl appendage within the silyl dienol ethers resulted in moderate diastereopreference in favor of anti-stereoisomers.
Scheme 242.

6.3.2.2. Cyclic Nucleophiles
Among the many different transformations centered on the use of the popular silyloxypyrrole 842a as cyclic nucleophile in Michael reactions, it is worth mentioning the conjugate addition to 1,2-diaza-1,3-dienes of type 954, reported by Battistini, Zanardi, and colleagues during the synthesis of highly functionalized pyrrole-carboxylates 956 (Scheme 243).636
Scheme 243.

The one-pot three-step reaction cascade leading to the targeted pyrrole was performed in aqueous medium and consisted of an initial water-mediated diastereoselective vinylogous Mukaiyama-Michael coupling of pyrrole 842a to diazadiene 954, followed by a base-catalyzed intramolecular aza-Michael attack/ring-opening/aromatization sequence yielding the unsoluble pyrrole-carboxylate 956. The opening vinylogous Mukaiyama–Michael reaction was tested in either traditional organic solvents (in the presence of Lewis acid) or aqueous media; the crucial role exerted by water as both reaction environment and promoter was demonstrated by the remarkable results obtained in terms of reaction rate, stereoselectivity, efficiency, and general applicability of the protocol. Of note, this on-water procedure was conveniently extended to oxygen- and sulfur-containing nucleophiles with yields and diastereomeric ratios comparable to the nitrogen counterparts (not shown).
During an extensive study on the diastereodivergent synthesis of γ,γ-disubstituted butenolides by means of organocatalyzed vinylogous Michael reactions (vide supra), Dixon and co-workers examined the possibility to apply this catalyst-controlled stereodivergent strategy to the Mukaiyama–Michael reaction of pyrrole derivatives 957 (Scheme 244).456 The authors selected the Boc-protected γ-methyl (or γ-phenyl) silyloxypyrroles 957 as the vinylogous donors and various enones 958 as the acceptors; when the reactions were performed with chiral amine A38 as the catalyst, anti-configured lactams 959 were obtained in moderate yields and excellent enantiomeric excess, albeit with poor diastereoselectivities. The use of thiourea A37 as the catalytic system provided instead syn-configured lactams 959 as the major stereoisomers, with equally good results as for the efficiency and stereocontrol were concerned.
Scheme 244.

Finally, Li et al. reported the metal-catalyzed 1,6-conjugate addition of silyloxyindoles 960 to para-quinone methides 961 (Scheme 245).637 The reaction was performed in the presence of catalytic bismuth triflate, providing racemic α-alkylidene-δ-diaryl oxindoles 962 in excellent yields, with complete γ-site selectivity and preference for Z-configured isomers.
Scheme 245.

6.4. Other Reactions
In this section examples of vinylogous reactions are grouped which do not enter in the previously discussed additions to C=O, C=N, or activated C=C bonds and involving vinylogous, linear or cyclic, α,β-unsaturated amides (or lactams). Among these contributions, a couple of papers regarded asymmetric γ-selective aminations, alkylation, or acylation procedures, and only one example dealt with a stereoselective halogenation.
6.4.1. Direct Procedures
6.4.1.1. Acyclic Pronucleophiles
In a recent study, Huang, Zhang, et al. elaborated a metal-catalyzed protocol to regioselectively accomplish the γ-amination of N-acylpyrazoles 963 as acyclic pronucleophiles with azodicarboxylates 964 (Scheme 246).638 The authors noticed that the choice of the metal catalyst (silver versus zinc) was critical to control the regioselectivity; in fact, the use of a silver acetate/TMG system ensured the formation of racemic γ-aminated products 965 in moderate to good yields, while the alternative use of zinc acetate as catalyst allowed the access to the α-aminated analogues.
Scheme 246.

6.4.1.2. Cyclic Pronucleophiles
The asymmetric amination of oxindole-derivatives 966 with azodicarboxylates 964 was obtained by Chen and colleagues in 2015 by reacting butanoates 966 with aminating reagents 964 under the assistance of organocatalyst C53 in toluene (Scheme 247).639 The products 967 were prepared in good chemical yields, albeit with moderate enantioselectivities.
Scheme 247.

6.4.2. Indirect Procedures
6.4.2.1. Acyclic Nucleophiles
As described in section 6.1.2, the E,E-vinylketene silyl N,O-acetal 807 was used as the key reagent in syn- or anti-selective vinylogous Mukaiyama aldol reactions by a large number of researchers. The remote asymmetric induction strategy exploiting this type of reagents was also applied by the Hosokawa group to acylation,640 alkylation,641 and bromination642 reactions (Scheme 248), and the advanced intermediate adducts were successfully utilized in the total synthesis of various natural products.
Scheme 248.

6.4.2.2. Cyclic Nucleophiles
The synthesis of α-alkylidene-δ-diaryl-2-oxindoles 975 was accomplished by Singh et al. through metal-catalyzed vinylogous nucleophilic substitutions of diarylmethanols 974 with alkylidene silyloxyindoles 973 (Scheme 249).643
Scheme 249.

The reactions were catalyzed by indium triflate and proceeded efficiently giving the Z-configured γ-alkylated compounds 975, exclusively. The authors explained the observed Z-selectivity by the vinylogous attack of the s-cis conformer of silyloxyindoles on the diarylmethane cation intermediates, while steric concerns were invoked to account for the remarkable γ-selectivity.
7. Vinylogous Nitriles
As can be observed in Figure 8, most of the vinylogous pronucleophile nitriles are γ-enolizable-α,α′-dicyanoalkenes and they have long been exploited in catalytic asymmetric reactions.28,644 They are easily prepared by Knoevenagel-type condensation of carbonyl compounds and malononitrile, and the strong electron-withdrawing effect of the two nitrile moieties increases the acidity of the γ-protons, allowing a facile γ-enolization under very mild conditions. For this reason, they are generally more reactive than the corresponding carbonyl precursors and show a remarkable γ-selectivity in vinylogous addition reactions. Moreover, the γ vinylogous addition products may be prone to further manipulation, including cyclization, tandem processes, or even elimination, offering broad maneuvers for the synthesis of cyclic (or polycyclic) diversely functionalized structures via cascade reactions.
Figure 8.

Collection of linear and cyclic pronucleophilic nitriles at work in this chapter using the direct procedures. In the plain box the sole type of nucleophilic nitrile-derived silyl dienol ether used in indirect procedures. Red circles denote the reactive (pro)nucleophilic carbon site.
In this section, the separation between cyclic and acyclic (pro)nucleophiles is not always sharp, because in many cases methodological studies were carried out on both kinds of structures. The discussion of these works and the corresponding scheme has taken into account the prevailing type of structure in the paper.
7.1. Additions to C=O Bonds
7.1.1. Direct Procedures
7.1.1.1. Acyclic Pronucleophiles
Already in 2002, allylic cyanides were identified as viable pronucleophiles,645 bearing an α-proton with an acidity (pKa 21.1 in DMSO) suitable for achieving catalyst turnover via proton transfer. α-Selective additions to aldehydes,645 aldimines,646 and ketimines647 were reported, which exploited different activation strategies. However, only in 2009 Shibasaki et al. disclosed the vinylogous nucleophilicity of allylic cyanides 976, by developing the first direct catalytic and asymmetric addition to ketones, with a complete γ-regioselectivity.239 They developed a chiral soft Lewis acid/hard Brønsted base catalytic system consisting of [Cu(CH3CN)4]ClO4/L34/LiOAr, to synthesize alcohols with asymmetric tetrasubstituted carbon centers in good yields and enantioselectivities. The following year, the same authors developed a second generation catalytic system, with the addition of a hard Lewis base (L35) as a third catalytic component.240 The reaction between 976 and ketones 977 (Scheme 250) provided Z-configured alcohols 978 in good yields and with generally very high enantiomeric excesses. The bidentate bis(phosphine oxide) L35 substantially improved the catalytic performance, ensuring higher enantioselectivity in most cases with only 1 mol % catalyst loading. [Cu/L34]ClO4 serves as a soft Lewis acid and activates 976 through a soft–soft interaction, accelerating deprotonation. The deprotonation step is likely the rate-determining step and Li(OC6H4-p-OMe) is the active Brønsted base used to remove the α-hydrogen. The hard Lewis base L35 predominantly coordinates Li cations thanks to the hard–hard interaction (P=O···Li+) and accelerates the overall reaction rate by enhancing the Brønsted basicity of LiOAr. This cooperative catalysis could be applied only to allylic cyanides without substituents in β- or γ-positions; in fact, the steric factor is critical in preventing the addition to ketones, possibly because of increased steric demands in the transition state. When the ketone substrate possessed two alkyl substituents, the product was obtained in low yield and enantioselectivity (see product 978c), while when R1 was an aryl or a vinyl group, excellent enantiomeric excesses were observed.
Scheme 250.

Some years later, Shibasaki et al. applied this catalytic strategy to the addition of allylic cyanide to aldehydes.241 In this case, variable anti/syn mixtures of α- and γ-attack products were obtained and with lower enantiomeric excesses. The nature of the aldehyde, the hard Brønsted base, and the reaction time influenced the α/γ ratio. The conditions were optimized and the reaction was applied to the enantioselective synthesis of a key intermediate of fostriecin,648 a naturally occurring molecule with antitumor and antibiotic activity. As a corollary of these studies, in 2014 the same authors published a paper in which substrates of type 978 were directly converted to δ,δ-disubstituted unsaturated δ-valerolactones via a one-pot three-step sequence (not shown).649
7.1.1.2. Cyclic Pronucleophiles
While cyclic α,α-dicyanoalkene pronucleophiles have been largely used as vinylogous donors in Michael addition reactions (vide infra, section 7.3.1.2), their reactivity in aldol additions has been poorly investigated. Moreover, the two examples reported in these years are nonasymmetric procedures toward racemic compounds.
In 2010, Perumal et al. described the one-pot synthesis of functionalized spirooxindoles 981 via base-promoted vinylogous aldol addition/cyclization of cycloalkylidene malononitriles 979 with isatin derivatives 980 (Scheme 251).650 Products 981 were isolated in good yields as single diastereoisomers. Instead, when the seven-membered dicyanoolefin 982 was used, the reaction provided the aldol product 983 as a diastereoisomeric mixture, which did not undergo cyclization. This methodology was used by Shan et al. to prepare novel steroidal pyran-oxindole hybrids of type 985 (Scheme 252),651 with the aim of obtaining potent and selective cytotoxic agents.
Scheme 251.

Scheme 252.

7.1.2. Indirect Procedures
7.1.2.1. Acyclic Nucleophiles
Vinylogous additions of allylic nitrile to aldehydes were reported for the first time by Denmark and Wilson in 2012.652 Aliphatic aldehydes can readily undergo base-mediated self-condensation reactions, limiting the possibility to use basic reaction conditions, normally used in direct reactions to generate the enolate species. To overcome this problem, Denmark proposed the alternative strategy to preform the nucleophile species, the N-silyl vinylketene imines of type 986 (Scheme 253), by selective N-silylation of allylic nitrile anions. Compounds 986 were used as nucleophiles in enantioselective vinylogous aldol reactions with aldehydes 987, to generate δ-hydroxy α,β-unsaturated nitriles 988. The reaction was catalyzed by the Lewis base L12/SiCl4 complex and furnished E-configured products with high γ-regioselectivity (up to >97:3 γ:α), in moderate to good yields and with good to excellent enantioselectivities. With aromatic aldehydes, the catalyst loading could be lowered to 2.5 mol %. The γ-attack selectivity was mainly ascribed to the less steric encumbrance of the γ-site with respect to α-carbon atom of the ketene imine 986, while the absolute and relative configurations of the products were explained by the addition of the N-silyl vinylketene imines 986, in the s-trans conformation, to the Re face of the aldehyde (Scheme 253, left).
Scheme 253.

7.2. Additions to C=N Bonds
7.2.1. Direct Procedures
7.2.1.1. Acyclic and Cyclic Pronucleophiles
Even if an organocatalytic, asymmetric vinylogous Mannich reaction (VMnR) of α,α-dicyanoolefins to aldimines was successfully realized by Chen in 2007,653 the analogous reaction with ketimines was developed only in 2016 by Meng, Li, and co-workers (Scheme 254).654 Ketimines, in fact, are more stable than aldimines, and this makes their use as acceptors more challenging. Isatin N-Boc ketimines, however, are still good electrophiles because of the electron-withdrawing tert-butoxycarbonyl group, and they have been largely used in synthetic procedures, even because they furnish the oxindole backbone, an important structural motif in biologically and pharmacologically active compounds. The reaction between α,α-dicyanoolefins 989 and ketimines 990 was catalyzed by squaramide derived catalyst C54, to provide chiral 3,3′-substituted oxindoles 991 in good yields and good to excellent enantioselectivities. Different N-protecting groups (R4) were tolerated, even if the best results were obtained with benzyl groups. Finally, two cyclic α,α-dicyanoolefins were also investigated, and they afforded the desired products in good yields and enantioselectivities, but with almost no diastereoselection. A drawback of this strategy is the long reaction times, 3 to 8 days depending on the substrates. A potential transition-state structure was proposed by the authors (Scheme 254), in which the tertiary amine of the catalyst deprotonates the α,α-dicyanoolefin which holds, through coordination, the enolate-like structure in close proximity to the isatin N-Boc ketimine, in turn activated by N–H binding by the squaramide moiety.
Scheme 254.

Some months later, Chen et al. developed the same asymmetric VMnR between α,α-dicyanoolefins 992 and N-Boc isatin imines 993, with a different catalytic activation mode (Scheme 255, eq 1).655 The reaction was performed in toluene at −20 °C, with bifunctional amide phosphonium salt C55 (1 mol %) as a phase transfer catalyst and K2CO3 as a base. In these conditions, adducts 994 formed with very good yields and good to excellent enantiomeric excesses. Interestingly, the authors optimized the conditions for the removal of the malonic nitrile by treatment of the products 994 with KMnO4, obtaining the corresponding ketones; for this reason, they proposed these α,α-dicyanoolefins as surrogates of less reactive aryl methyl ketones. When α,α-dicyanoolefins derived from propiophenone (995), methyl tert-butylketone, or tetralone (not shown) were used, the reaction led to cyclized products of type 996 via a VMnR/intramolecular cyclization cascade sequence (Scheme 255, eq 2).
Scheme 255.

The last example of an asymmetric VMnR with α,α-dicyanoolefins as vinylogous nucleophiles was reported by Fustero, Pozo, et al. in 2018.656 Linear and cyclic pronucleophiles 997 reacted with chiral enantiopure fluorinated sulfinyl imines 998 under basic conditions to furnish compounds 999 as single diastereomers in low to good yields (Scheme 256). To account for the observed relative configuration of the products, the authors assumed that the reaction proceeds through a chelated transition state, where the potassium counterion in the enolate would form a chelate with the imine nitrogen atom in a chairlike modality, thus facilitating the attack of the nucleophile to the Re face of the imine.
Scheme 256.

7.3. Conjugate Additions to Electron-Poor C=C Bonds
7.3.1. Direct Procedures
7.3.1.1. Acyclic Pronucleophiles
In 2016, Tsogoeva et al. reported the unexpected discovery of an atom-economical metal-free domino transformation, which employed malononitrile and enolizable aldehydes 1000 in the presence of imidazole (15 mol %) and led in a single operation to complex compounds such as isoquinuclidine derivatives 1001 and their isomeric carbobicycles 1002, bearing an exocyclic imine group (Scheme 257).657 A first Knoevenagel reaction furnished α,α-dicyanoolefins 1000′, that underwent dimerization via VMcR giving intermediate 1000′′. The reaction of 1000′′ with the excess of malononitrile could follow two pathways, well elucidated by the authors, to provide either compounds 1001 or 1002, in low to moderate yields. When R1 in the aldehyde was an aromatic ring, the major products 1001 were isolated with excellent diastereoselectivities (>99:1), while when R1 was the methyl group, the lowest yield and diastereoselection of the panel compounds were registered.
Scheme 257.

Tsogoeva and colleagues exploited the same imidazole-catalyzed procedure for a three-component Knoevenagel/vinylogous Michael domino reaction of arylacetaldehydes, malononitrile, and trans-β-nitrostyrenes.658 Nitrostyrenes were able to intercept the in situ generated α,α-dicyanoolefins 1000′ to form the corresponding Michael adducts (such as 1003a–c in Figure 9) in good yield but with modest diastereoselection.
Figure 9.

Representative compounds prepared via three-component Knoevenagel/vinylogous Michael domino reaction between arylacetaldehydes, malononitrile, and trans-β-nitrostyrenes.658
In 2016, the Wang and Ye groups, independently, developed a mild and convenient strategy, an NHC-catalyzed formal [4 + 2] benzoannulation, for the synthesis of multisubstituted benzonitriles 1006 (Scheme 258 and Scheme 259 eq 1, respectively).659,660 The reaction mechanism is described in Scheme 258. The addition of the NHC catalyst to enal 1005 followed by deprotonation provides intermediate 1005′, which is oxidized by diphenoquinone 45 to intermediate XLIV. Vinylogous attack of γ-deprotonated α-cyano-β-methylenone 1004′ to XLIV furnishes intermediate XLV that, after an intramolecular aldol reaction and β-lactonization, yields the bicyclic compound XLVII and liberation of NHC catalyst. Decarboxylation of XLVII, followed by spontaneous oxidative aromatization affords benzonitriles 1006. The protocol is tolerant toward a wide range of substituents at the R2 position, while decreased, but still acceptable, yields were registered when R1 was an alkyl group.
Scheme 258.

Scheme 259.

A variant of this reaction was developed using bromoenals 1007 in place of enals (Scheme 259, eq 2).661 The reaction was performed without the presence of an oxidant such as 45. In fact, the formation of the intermediate of type XLIV (Scheme 258) was assured by the bromide elimination, while after the decarboxylation of intermediate of type XLVII, the DBU-promoted elimination of cyanide furnished 1,3,5-trisubstituted benzenes 1008 in good to excellent yields.
The methodology reported by Yang and co-workers for the construction of substituted carbazol-4-amine derivatives 1011 exploited the vinylogous addition of alkylidene malononitriles 1009 to 3-nitroindoles 1010, followed by cyclization/isomerization/elimination reactions (Scheme 260).662 This base-activated one-pot procedure provided carbazolamines 1011 in moderate to good yields. The reaction was performed in an achiral environment because the aromatization step leads to the loss of the stereogenic centers.
Scheme 260.

Another example of a base-mediated procedure in an achiral environment is the synthesis of densely functionalized pyrrolo-pyrazole systems 1014 via domino reaction of both linear and cyclic vinylmalononitriles 1012 with 1,2-diaza-1,3-dienes 1013 proposed by Favi and collaborators (Scheme 261).663 A plausible mechanism was provided by the authors. The overall transformation would involve at first a VMcR of 1012 to diazadienes 1013 to give intermediates 1012′. The aza-Michael cyclization of the azaallylic anion intermediate 1012′′, followed by an aza-cyclization and subsequent imine-enamine tautomerization would furnish adducts 1014 in moderate to good yields with high levels of chemo- and regioselectivity. An equal number of linear and cyclic alkylidene malononitriles were tested.
Scheme 261.

7.3.1.2. Cyclic Pronucleophiles
As stated before, the vinylogous adducts deriving from addition reactions of α,α-dicyanoalkene pronucleophiles to electrophiles are prone to further manipulation including cyclization, tandem processes, or even elimination. Most of these elaborations carry to aromatic products with the loss of the newly formed stereocenters. These works are less interesting from a methodological point of view but have their strength in the straitghforward formation of uncommon aromatic structures or substitution patterns. Some examples are reported in Figure 10, dealing with diversely functionalized products formed by domino reactions of cyclic α,α-dicyanoalkene pronucleophiles with Michael acceptors, through base activation in an achiral environment. As can be observed, the variety of the targeted chemotypes is wide and includes phenanthrene derivatives (1015 and 1016),664 5,6-dihydroquinoline motif (1017),665 pyrazolo[1,5-a]pyridines (1018),666 indenopyridine-fused spirocyclic systems (1019),667 spirocyclic oxindoles (1020, 1021, and 1022),668−670 and benzo[a]carbazole derivatives (1023 and 1024).671,672 Some of the compounds reported in Figure 10 still bear stereogenic centers, but they were isolated as racemic mixtures since the reactions were catalyzed by achiral bases (commonly Et3N, DIPEA, or DBU).
Figure 10.

Polycyclic structures obtained through base-activated domino reactions between cyclic α,α-dicyanoalkene pronucleophiles and Michael acceptors.
A similar base-promoted procedure was developed by Zhang and Zhang for the synthesis of angularly fused polycycles 1027 (Scheme 262).673 The reaction of cycloalkylidene malononitriles 1025 to enynes 1026 was catalyzed by DBU (20 mol %) in dichloroethane at room temperature and gave products 1027 in reasonable to very good yields and appreciable diastereocontrol. The first step of this one-pot tandem reaction is the vinylogous Michael addition of deprotonated 1025 to the electron-deficient enyne acceptors 1026. The allene intermediate of type 1025′ undergoes formal dehydro-Diels–Alder reaction to the target 1027, via stepwise anionic pathway (1025′′).
Scheme 262.

In a study of phosphine-catalyzed [4 + 2] annulation reactions between 1,4-dien-3-ones (1029) and α,α-dicyanoalkenes, He and co-workers reported also two examples of vinylogous addition reactions of cycloalkylidene malononitriles 1028 to dienones 1029, which provided anti-configured, racemic products 1030 (Scheme 263).674 The work represents a rare example of phosphine catalyzed VMcA, in which the enolate from 1028 reacts with the phosphine activated intermediate 1029′. The versatility of this procedure proved however limited since it could be applied just to dienones as acceptors, while a similar reaction between 1028 and chalcone (a monoenone) did not provide any addition product.
Scheme 263.

The organocatalyzed synthesis of polyfunctionalized spirooxindoles of type 1033 was reported by Perumal et al. featuring a domino reaction between vinylogous malononitriles 1031 and isatin-derived chalcone 1032 in the presence of l-proline (15 mol %) as the catalyst (Scheme 264, eq 1).675 The products were obtained in high yields as single trans diastereoisomers, but regrettably, no mention of their enantiomeric purity was made in the paper. In the same work, this procedure was also applied to the multicomponent reaction of 1031 with aromatic and heteroaromatic aldehydes 1034 and isoxazoles 1035 (Scheme 264, eq 2) to prepare spiroisoxazolones 1036, but again no information about the enantiomeric excesses of the products was reported.
Scheme 264.

The first organocatalyzed, enantioselective vinylogous Michael/cyclization reaction cascade of α,α-dicyanoalkenes with 3-alkylidene oxindole acceptors for the synthesis of enantiopure spirooxindoles was reported by Wang et al. in 2013 (Scheme 265).676 The authors presented the reaction using both acyclic and cyclic dicyanoalkenes. The rosin-derived bifunctional thiourea catalyst C44, bearing a pyrrolidine group, triggered the reaction between dicyanoolefins 1037 and electron-poor alkylidene oxindoles 1038, to provide spirooxindoles 1039 in high yields and excellent diastereo- and enantioselectivities (Scheme 265, eq 1). The same conditions applied to the more flexible monocyclic dicyanoolefins 1040 furnished spriooxindoles 1041 with equally optimal results (Scheme 265, eq 2), thanks to an unexpected final tautomerization process.
Scheme 265.

Structurally similar spirooxindoles of type 1043 were prepared in good yields, acceptable diastereoselectivities, and good to excellent enantioselectivities by Wang and co-workers, by exploiting the VMcR between dicyanoolefins 1031 and isatylidene malononitriles 1042 triggered by bifunctional thiourea catalyst C56 (Scheme 266, eq 1).677 The organocatalyst worked as both a Brønsted base to generate the γ-carbanion from 1031 and an activating unit of 1042 by hydrogen bonding between the thiourea moiety and the C=O group of the oxindole core.
Scheme 266.

One year later, in 2015, a similar vinylogous Michael addition–cyclization reaction between dicyanolefins 1040 or 1046 and N-methyl-protected isatylidene malononitrile 1044 (Scheme 266, eqs 2 and 3) was reported by Kesavan and collaborators for the access to spirooxindoles of type 1045 or 1047.678 The reaction was organocatalyzed by l-proline derived bifunctional thiourea C57 under mild reaction conditions (0 °C in toluene). Interestingly, a one-pot three component procedure, using dicyanoolefins 1040 as nucleophiles with N-unprotected isatin and malononitrile as Michael acceptors, was carried out, obtaining very good results in terms of yields and enantioselectivities.
The first direct asymmetric procedures of vinylogous Michael additions of cycloalkylidene malononitriles of type 1048 to nitroolefins 1049 were independently published by Deng et al. in 2005,679 Jørgensen et al. in 2006,680 and Chen et al. in 2007,681 with good results in terms of yields and enantioselectivities; in 2012, Chen and collaborators reported a new study of this reaction catalyzed by self-assembled organocatalytic systems (Scheme 267).682 As outlined in Scheme 267, formation of products 1050 was ensured by the combination of l-proline and thiourea-tertiary amine C2 (5 mol % each). However, this strategy did not give better results in terms of yields and enantioselectivities of the products with respect to previous publications.
Scheme 267.

In 2014, Zanardi and co-workers reported the first use of cycloalkylidene malononitriles of type 1051 as vinylogous pronucleophiles.683 These compounds can be in principle deprotonated at different positions (e.g., ε, ε′, and γ′-positions), offering multiple pronucleophilic sites and making the regioselectivity control of the reaction a challenging issue. The reaction of 1051 with α,β-unsaturated aldehydes 1052, triggered by prolinol-derived catalyst A13, led to the formation of isolable, ε-selective vinylogous Michael addition products 1053 (Scheme 268, eq 1). When the reaction solution was added with NHC-precatalyst B26 and KOAc base, a 1,6-Stetter closure consecutive to the first hypervinylogous Michael addition provided [3 + 2]-spiroannulated products 1054 with moderate to good yields, excellent diastereoselectivity, and very high levels of enantioselectivity. Interestingly, under similar reaction conditions (catalyst A4 instead of A13, CHCl3 instead of CH2Cl2) and using p-nitrophenol (1055) as cocatalyst, the reaction between 1051 and enals 1052 proceeded along a regiodivergent pathway, giving γ′,δ-selective [4 + 2] annulated products 1056 in high yields and excellent enantioselectivities (Scheme 268, eq 2). In this case, the vinylogous attack of the γ′ position of 1051 to iminium ion-activated 1052 was favored, followed by intramolecular attack of the enamine intermediate at the δ-site of 1051. Several control experiments indicated that the regiodivergence toward either ε,δ-[3 + 2] or γ′,δ-[4 + 2] products (eq 1 vs eq 2) was dictated by the presence or not of p-nitrophenol cocatalyst. When it is absent, a transition state of type XLVIII, carrying to compound 1053, is favored because of Coulombic interactions between the extended enolate 1051′ and the positively charged nitrogen atom of 1052′; on the other hand, when the phenol cocatalyst is present, a transition state of type XLIX, where the cross-conjugated enolate 1051″ approaches the iminium ion 1052′ along an endo Diels–Alder-like trajectory, is favored, thanks to the stabilization of 1051″ by hydrogen bonding with donor 1055.
Scheme 268.

An analogous formal [4 + 2]-cycloaddition between cyclohexenylidene malononitrile 1051 and enals 1057 under secondary amine organocatalysis for the access to bicyclooctanes 1058 was reported almost concurrently and independently by Chen and co-workers (Scheme 269).684 In this case, the authors optimized the reaction conditions not only for aromatic-group substituted enals (R3= Ar, HetAr) but also for alkyl-group substituted acroleins (R3 = Alk). In the first case, the reaction was catalyzed by TES-protected prolinol A12 and DIPEA, while in the second case the TMS-protected prolinol A4 in combination with benzoic acid was the best catalytic system to provide the products in good yields and with high enantiomeric excesses.
Scheme 269.

The following year, Zanardi and co-workers reported the use of allylidene malononitriles of type 1059 (Scheme 270, eq 1) as vinylogous donors, whose π-system conjugation propagates the nucleophilic character to the remote ε-position on the cycle.685 The reaction with both aliphatic and aromatic enals 1060, catalyzed by prolinol-derived catalyst A4, provided a series of fused polycycles 1061 in good yields, complete trans-diastereoselectivity, and very high enantioselectivities, via a formal [4 + 2]-eliminative cycloaddition reaction. The authors proposed a plausible mechanism, describing the reaction as a stepwise domino process involving a bis-vinylogous Michael/Michael/retro-Michael organocascade. After the first bisvinylogous Michael addition of 1059 to the iminium ion-activated acceptor 1060, an enamine intermediate is formed, which undergoes intramolecular Michael addition to the cycloadduct 1059a′. Finally, catalyst hydrolysis and malononitrile elimination (retro-Michael) deliver product 1061a. It is noteworthy that the authors performed also a one-pot procedure (Scheme 270, eq 2), using aldehyde 1060a in the presence of a stoichiometric amount of malononitrile for the in situ formation of allylidene malononitrile 1059a via Knoevenagel reaction, isolating the desired product 1061a in good yield and excellent enantiomeric excess, even if with lower dr with respect to the two-step procedure.
Scheme 270.

The first asymmetric and direct VMcR of α,α-dicyanoalkenes 1062 to 2-enoylpyridine N-oxides 1063 with a bifunctional thiourea/amine organocatalyst C47 was disclosed by Singh et al. in 2014 (Scheme 271).686 The products 1064 were obtained in good yields and selectivity. Two acyclic α,α-dicyanoalkenes proved equally successful.
Scheme 271.

In 2016, Yao et al. reported the direct vinylogous 1,6-conjugate addition of alkylidene malononitriles 1065 to p-quinone methides of type 1066 (Scheme 272).687 The reaction was catalyzed by chiral phosphine-thiourea organocatalyst P1 with triethylamine in diethyl ether or toluene and provided diarylmethine products 1067 in good yields, with high diastereocontrol and low to excellent enantioselectivities. Very low results in terms of enantioselectivity were obtained using cyclohexanone-derived dicyanoolefin (not shown). The role of the phosphorus atom in the catalytic cycle was unclear, but it was thought to play a role as a Lewis base on dicyanoolefin. The authors proposed the activation mode shown in Scheme 272, where the remote stereocontrol was achieved through intermolecular hydrogen-bond interaction between the thiourea catalyst and the p-quinone methides, while dicyanoolefin was more prone to attack the p-quinone methides from the Si face.
Scheme 272.

8. Other Vinylogous Pronucleophiles
8.1. Direct Procedures
8.1.1. Acyclic and Cyclic Pronucleophiles
This section groups in tabular format a selection of illustrative pieces of research in which quite unconventional pronucleophiles including α,β-unsaturated acyl chlorides,688,689 carboxylic acids,690−692 alkylnitroisoxazoles,693−698 vinylphenols,699−705 nitrotoluenes,706−708 and allylsulfones709 act as leading players in asymmetric vinylogous transformations. All the reported examples deal with the in situ generation of the reactive polyenolate-type donor species (direct activation modalities) which couple to suitable C=O, C=N, and activated C=C bonds at their vinylogous sites.
The first reported example deals with the use of γ-enolizable α,β-unsaturated acyl chlorides as vinylogous donors in tertiary amine-catalyzed [4 + 2] cycloaddition reactions with activated aldehydes as chloral (Table 12, eq 1).688 The authors demonstrated that the transformations proceeded through the formation of zwitterionic ammonium enolates of type L (eq 1), generated in situ from the starting acyl chlorides by use of the nucleophilic amine catalyst C59 and Sn(OTf)2 as Lewis acid cocatalyst. Enantioentriched δ-lactones were forged in variable yields, depending upon the nature of the R1 substituents within the donors. While this procedure proved quite limited as for the electrophilic scope, the authors identified in the same work an alternative catalyst combination (e.g., Er(OTf)3 in complex with chiral aliphatic β- or γ-amino alcohols), which was able to trigger the [4 + 2] cycloaddition of unsaturated acyl chlorides with unactivated aldehydes as benzaldehyde (not shown).
Table 12. Asymmetric Vinylogous Additions of Unconventional Pronucleophiles to C=O Bonds.
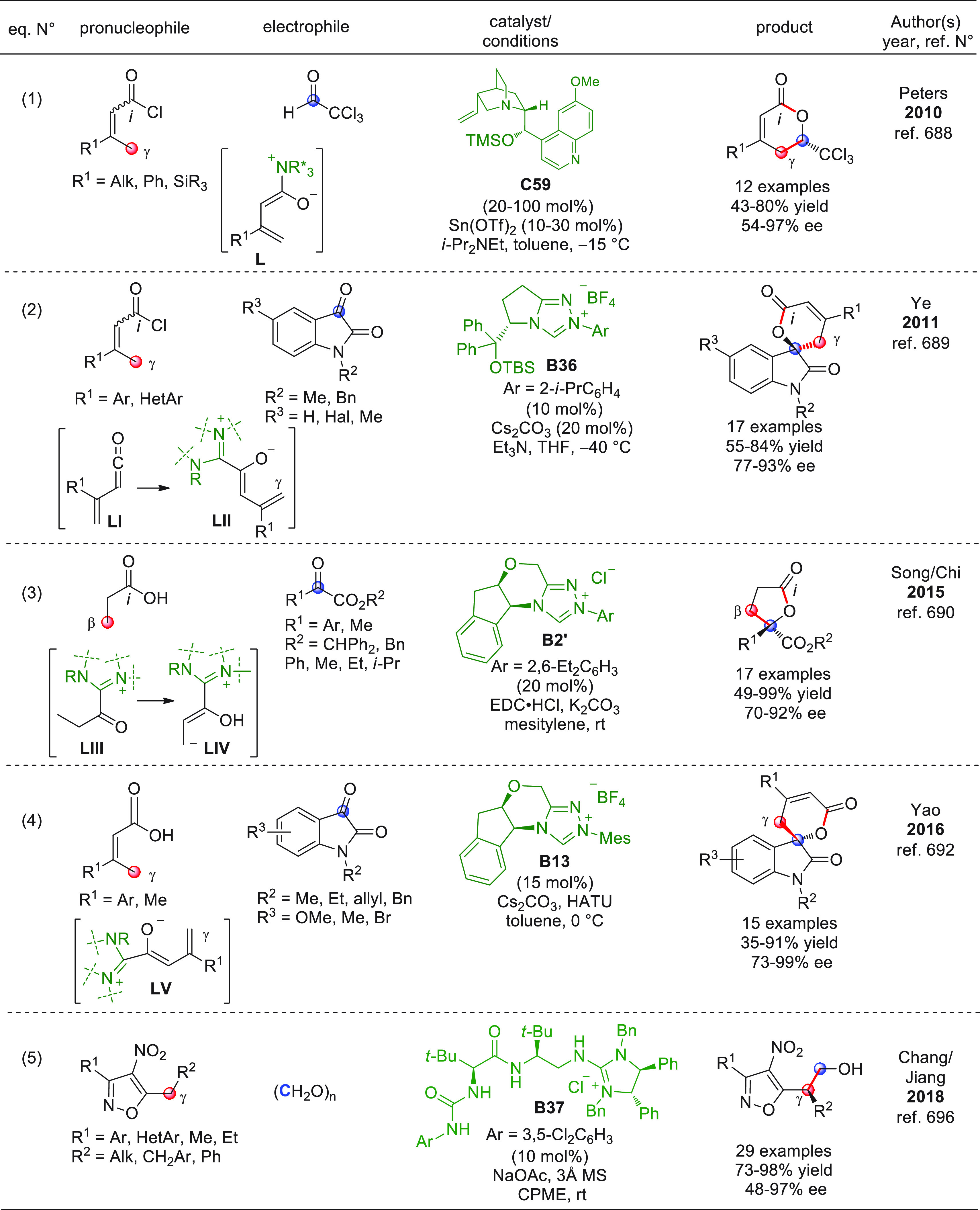
α,β-Unsaturated β-methylacyl chlorides were also employed in NHC-catalyzed [4 + 2] cyclization reactions with activated ketones such as isatins, which gave the corresponding spirocyclic unsaturated δ-lactones in good yields and enantiomeric excesses (Table 12, eq 2).689 It was proposed that NHC-bound vinyl enolates LII were operative, which were likely obtained in situ from vinyl ketenes LI, in turn generated by dehydrohalogenation of the starting acyl chloride with Et3N base. As in the previous case, the authors could not precisely define whether the δ-lactone products were the result of a concerted HDA cyclization or stepwise VAR followed by intramolecular closure.
Direct β-activation of propionic acid by NHC catalysis was developed by Song, Chi, et al. to be exploited in formal [3 + 2] cycloaddition with keto esters (Table 12, eq 3).690 Mechanistically, condensation of the three carbon-long acid with a coupling reagent (EDC) forms a carboxylic anhydride that reacts with the NHC catalyst B2′, to furnish the activated acyl intermediate LIII and hence the β-donor homoenolate LIV, ready for the coupling reaction with the carbonyl acceptor.
γ-Enolizable carboxylic acids (either α,β-unsaturated or saturated) were convenient γ-donor substrates to be exploited in NHC-catalyzed [4 + 2] cyclizations with isatins (Table 12, eq 4)692 or acylhydrazones (Table 13, eq 1).691 Treatment of the starting carboxylic acids with a condensing agent (HATU), NHC catalyst B13 or B38, and a base could release the corresponding activated intermediates, namely, dienolate LV (Table 12, eq 4) or dienolate LVII after oxidation of enolate LVI (Table 13, eq 1), which were ready for the asymmetric assemblage of the respective δ-lactone or δ-lactam products.
Table 13. Asymmetric Vinylogous Additions of Unconventional Pronucleophiles to Multiple C–N Bonds.
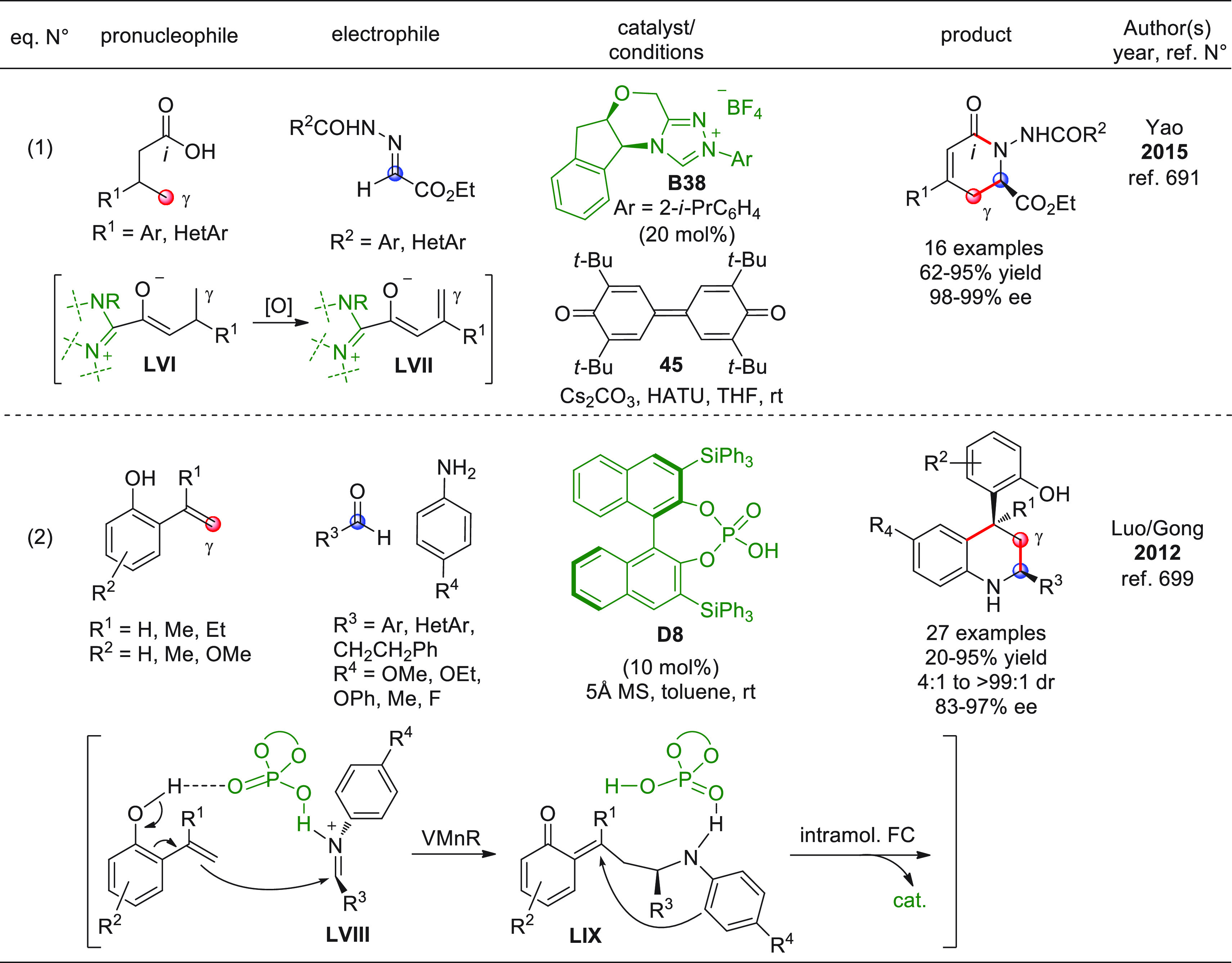
Direct asymmetric addition reactions of alkyl-substituted benzenes or heterocycles such as 5-alkyl-4-nitroisoxazoles could be effectively carried out by introducing nitro groups at ortho- and/or para-positions of the aromatic ring. In fact, the enhanced acidity of the C(sp3)-H benzylic protons induced by the presence of the nitro functionality could be exploited in either addition reactions to carbonyl compounds (Table 12, eq 5) or conjugate additions to electron-deficient alkenes (Table 14, eqs 1, 2, 3, and 6) under organocatalytic conditions. In other words, the leading functional group responsible for the pronucleophilic reactivity at the remote conjugated benzylic position is not a carbonyl function (as, for example, in Scheme 26, or Table 4, eqs 1 and 2) but rather the nitro function, according to vinylogous Henry-type nitroaldol or conjugate addition reactions involving the π-system of the (hetero)aromatic ring. Thus, for example, nitrotoluenes were employed in conjugate additions to iminium ion activated enals (Table 14, eqs 1 and 2)707,708 to afford the corresponding alkylated toluenes in high enantiomeric excesses. Using “elongated” nitrovinyl-nitrotoluenes such as those reported in Table 14, eq 3, efficient preparation of highly enantioenriched hexahydrophenanthrenes was at hand.706 Using, instead, 5-alkyl-4-nitroisoxazoles as heterocyclic vinylogous pronucleophiles, the VAR to paraformaldehyde (Table 12, eq 5) and VMcA to enals (Table 14, eq 6) were successfully realized.696,698
Table 14. Asymmetric Conjugate Additions of Unconventional Pronucleophiles to Activated C=C Bonds.
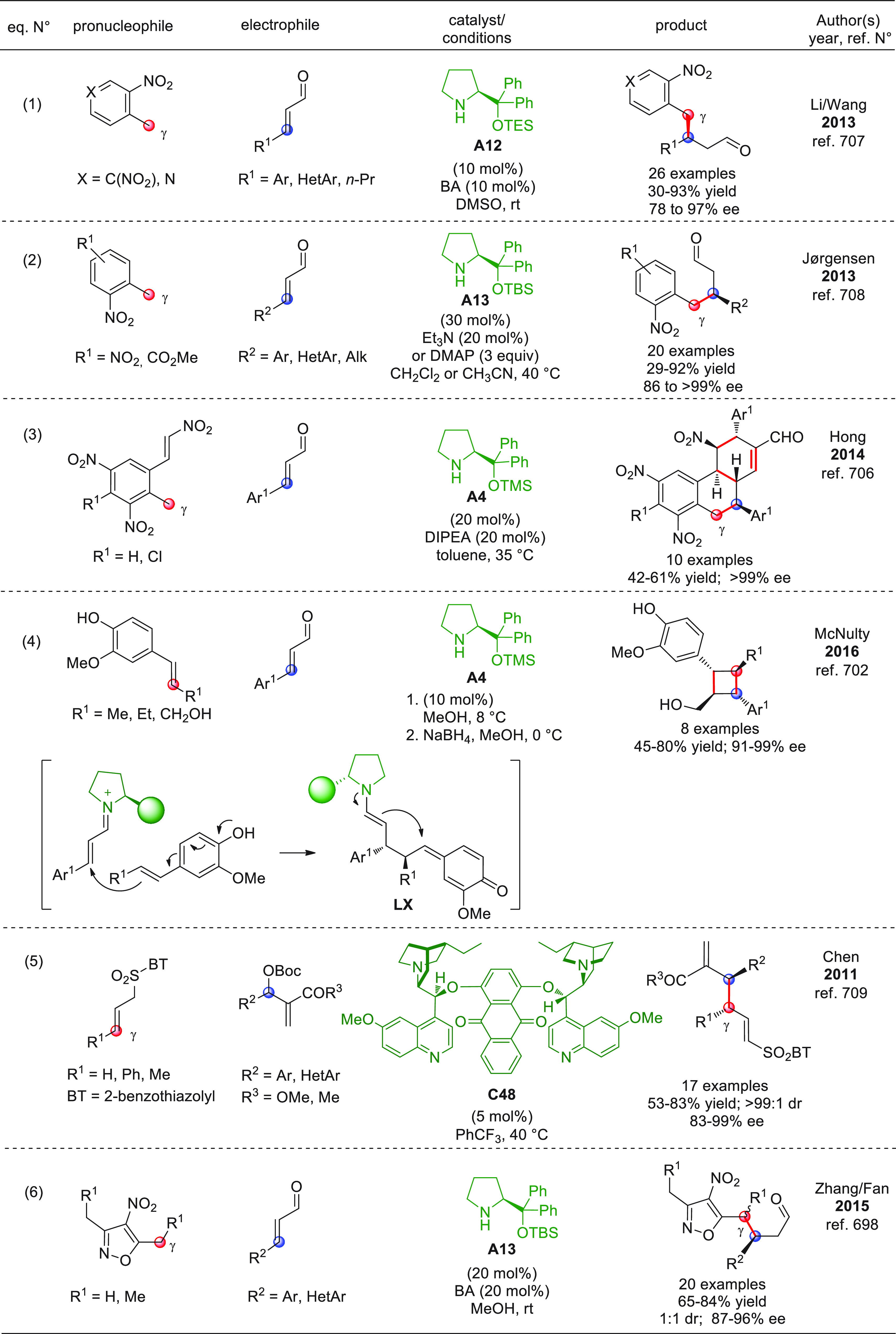
Another unconventional vinylogous pronucleophilic system is given by ortho- or para-substituted vinylphenol. Vinylphenols, together with vinylindoles,14 vinylpyrroles, and other π-extended electron-rich (hetero)aromatic substrates710,711 have been widely exploited in recent asymmetric synthesis programs as useful pronucleophilic synthons in remote Friedel–Crafts-type reactions. However, to stay focused on the subject of this review, we here restrict the field to o-vinyl- or p-vinylphenols, since their topology is strictly connected to dienol or polyenol species. Among these, we document just a few examples, where the presence of the OH group within the vinylphenol moiety proved to be essentially important to guide reactivity, exocyclic remote regioselectivity, and enantioselectivity.
In a first example, a Brønsted acid-catalyzed three-component Povarov reaction involving 2-hydroxystyrenes was developed, which provided an efficient method to access highly enantioenriched tetrahydroquinolines (Table 13, eq 2).699 The authors demonstrated that the phosphoric acid catalyst D8 acted as a bifunctional species activating both vinylphenol and the in situ-generated imine by hydrogen bonding interactions (species LVIII, eq 2). A two-step reaction pathway was operative, according to which a vinylogous Mannich reaction between vinylphenol and the imine acceptor occurred (LVIII to LIX), followed by intramolecular Friedel–Crafts reaction to the target compounds.
Secondary amine-catalyzed [2 + 2] cycloaddition between p-vinylphenols such as isoeugenol and cinnamaldehydes was developed by McNulty et al. in 2016, which afforded the corresponding tetrasubstituted cyclobutanes in good yields and enantiomeric excesses (Table 14, entry 4).702 Even in this case, it was found that the phenol group was crucially required to transmit the donor properties to the remote position with a postulated stepwise mechanism encountering a first attack of the vinylphenol to the aldehyde acceptor (activated as iminium ion by the amine catalyst A4), followed by intramolecular closure (via LX).
9. Concluding Remarks
In nearing to an end, this journey through the scientific publications dealing with the generation and use of π-extended enolate-type nucleophiles over the period of 2010–2018 calls for several final considerations.
The way in which vinylogous reactions were traditionally exploited has definitely been increased multifold or even surpassed by new concepts and new methods. Hence, the conventional generation of dienolates, via γ-deprotonation of unsaturated carbonyl compounds (with possible silyl enolate trapping), has been progressively supplemented with several ingenuous modalities where the starting pronucleophilic substrates are more complex in structure topology, chain length, substitution, and reactivity. A rapid glance to the general formulas of the (pro)nucleophilic species, reported at the beginning of each main chapter, confirms this. HOMO-raising activation modalities with chiral catalysts especially applied to π-extended carbonyl compounds (dienamine-, trienamine-, tetraenamine-activation, NHC-activation, noncovalent ion paired polyenolate activation, etc.) have resulted in hundreds of methodology-oriented papers revealing previously unexplored, remote C–C and C–X connections in an asymmetric context. Nonetheless, it is still hard for such innovative opportunities, involving aldehyde and ketone substrates, to find applications in target-oriented asymmetric syntheses, and we expect this issue to be addressed in the near future. On the other hand, the activation and use of π-extended ester/amide substrates are somehow more consolidated, and ample choice in indirect silyl ketene acetal-based procedures allowed many research groups to venture into successful target-oriented synthesis programs. A restricted area of vinylogy is occupied by the “other pronucleophiles”, including unconventional unsaturated acyl chlorides, carboxylic acids, alkylnitroisoxazoles, vinylphenols, nitrotoluenes, and allylsulfones, whose exploitation as vinylogous substrates in asymmetric synthesis is quite a novelty and is expected to fill the agenda of future research. As a further promising future direction, we expect that the photocatalytic generation of polyenolates will offer an interesting yet challenging opportunity to expand the arsenal of the ways multidentate carbonyl-related donor species are produced and exploited. Compared to our previous vinylogy-related review articles, we definitely noticed that researchers have been paying an increasing amount of attention, especially over the past 4–5 years, to the formulation of mechanistic hypotheses backed-up by solid calculations and experimental evidence; we expect this trend to become more established in the near future.
Finally, the extraordinarily wide spectrum of all the vinylogous products synthesized (which can be increased by postsynthesis transformations), vividly portrayed in the multitude of schemes and tables in this review article, bears witness to the fact that, in the context of contemporary organic synthesis, the application of vinylogy represents an excellent opportunity of selectively addressing molecular diversity beyond the boundaries of Nature.
Acknowledgments
We would like to dedicate this review article to the undying memory of Prof. Giovanni Casiraghi (1939–2016), for his invaluable contribution to the development of vinylogous aldol and related reactions. We are grateful to the University of Parma for the financial support of our projects based on the exploitation of the vinylogous principle in asymmetric synthesis (ZNRFNC_FIL, ZNRFNC_PRESTAZIONI_COMM, ZNRFNC_RICERCA_IST). Our sincere appreciation and gratitude are extended to our co-workers in the group, especially Gloria Rassu (CNR, Sassari, Italy) for her tireless work and lifelong passion for organic synthesis. Lucia Marzocchi is acknowledged for proofreading part of the manuscript.
Glossary
Abbreviations Used
- Ac
acetyl
- ACDC
asymmetric counterion-directed catalysis
- Alk
alkyl
- Ar
aryl
- ATNP
aluminum tris(2,6-di-2-naphthylphenoxide)
- ATPH
aluminum-tris(2,6-diphenylphenoxide)
- B
base
- BA
benzoic acid
- BB
Brønsted base
- BINOL
1,1′-bi-2-naphthol
- Bn
benzyl
- Boc
tert-butoxycarbonyl
- BSA
N,O-bis(trimethylsilyl)acetamide
- Bt
N-hydroxybenzotriazole
- BT
2-benzothiazolyl
- BTHL
BINOL-titanium-LiCl heterobimetallic complex
- t-Bu
tert-butyl
- BVAR
bisvinylogous aldol reaction
- Bz
benzoyl
- cat.
catalyst
- Cbz
benzyloxycarbonyl
- COD
1,5-cyclooctadiene
- CPME
cyclopentylmethyl ether
- CSA
camphorsulfonic acid
- Cy
cyclohexyl
- CyH
cyclohexane
- DA
Diels–Alder
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCE
1,2-dichloroethane
- DEA
N,N-diethylacetamide
- DEP
N,N-diethyl-3-pyridinecarboxamide
- DFT
density functional theory
- [DHQ]2PHAL
hydroquinine 1,4-phthalazinediyl diether
- [DHQD]2PYR
hydroquinidine-2,5-diphenyl-4,6-pyrimidinediyl diether
- DIPEA
diisopropylethylamine
- DMF
dimethylformamide
- DMP
Dess−Μartin periodinane
- DMAP
4-dimethylaminopyridine
- DMSO
dimethyl sulfoxide
- DNBA
2,4-dinitrobenzoic acid
- DNBSA
2,4-dinitrobenzenesulfonic acid
- DPMS
diphenylmethyl silyl
- DPTU
diphenyl thiourea
- DPP
diphenylphosphoric acid
- dr
diastereomeric ratio
- DSI
chiral disulfonimide
- DTBM-SEGPHOS
5,5′-bis[di(3,5-di-tert-butyl-4-methoxyphenyl)phosphino]-4,4′-bi-1,3-benzodioxole
- DVMcA
doubly vinylogous Michael addition
- ee
enantiomeric excess
- E1cB
elimination unimolecular conjugate base
- EDC
1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide
- Eoc
ethoxycarbonyl
- equiv
equivalent(s)
- EWG
electron-withdrawing group
- o-FBA
2-fluorobenzoic acid
- o-FBZA
2-fluorobenzaldehyde
- FC
Friedel–Crafts
- Hal
halogen
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate,
- HDA
hetero-Diels–Alder
- Hex
hexyl
- HetAr
heteroaryl
- HFIP
hexafluoroisopropanol
- HMDS
hexamethyldisilazide
- HOMO
highest occupied molecular orbital
- HVMAR
hypervinylogous Mukaiyama aldol reaction
- HWE
Horner–Wadsworth–Emmons
- ICD
isocupreidine
- IEDDA
inverse electron demand Diels–Alder
- IED-HDA
inverse electron demand hetero-Diels–Alder
- LA
Lewis acid
- LDA
lithium diisopropylamide
- LTMP
lithium tetramethylpiperidine
- LUMO
lowest unoccupied molecular orbital
- MBH
Morita–Baylis–Hillman
- Mes
2,4,6-trimethylphenyl (mesityl)
- MS
molecular sieves
- MTBD
7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene
- MTBE
methyl tert-butyl ether
- nd
not determined
- NHC
N-heterocyclic carbene
- NLE
nonlinear effect
- Ns
4-nitrobenzenesulfonyl
- Nu
nucleophile
- OXB
oxazaborolidinone
- PCC
pyridinum chlorocromate
- PG
protecting group
- Ph
phenyl
- Phe
phenylalanine
- PMB
4-methoxybenzyl
- PMP
4-methoxyphenyl
- i-Pr
isopropyl
- Pro
proline
- ProPhenol
2,6-bis[2-(hydroxydiphenyl)-1-pyrrolidinylmethyl]-4-methylphenol
- PTSA
p-toluenesulfonic acid
- oQDM
ortho-quinodimethane
- [QD]2PHAL
quinidine 1,4-phthalazinediyl diether
- rt
room temperature
- TANIAPHOS
(1-[(R)-(dimethylamino)[2-(diphenylphosphino)phenyl]methyl]-2-(diphenylphosphino)-(2R)-ferrocene)
- TBAF
tetra-n-butylammonium fluoride
- TBAT
tetrabutylammonium difluorotriphenylsilicate
- TBD
1,5,7-triazabicyclo[4.4.0]dec-5-ene
- TBDPS
tert-butyldiphenylsilyl
- TBS
tert-butyldimethylsilyl
- TBSOF
tert-butyldimethylsilyloxyfuran
- TCE
trichloroethanol
- TES
triethylsilyl
- TFA
trifluoroacetate, trifluoroacetic acid
- TFE
trifluoroethanol
- THF
tetrahydrofuran
- TIPS
triisopropylsilyl
- TIPSOF
triisopropylsilyloxyfuran
- TIPSOTf
triisopropylsilyl trifluoromethanesulfonate
- TMAA
tetrabutylammonium acetate
- TMAH
tetrabutylammonium hydroxide
- TMG
1,1,3,3-tetramethylguanidine
- TMS
trimethylsilyl
- TMSOF
trimethylsilyloxyfuran
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
- TPS
triphenylsilyl
- Tr
triphenylmethyl
- Trp
tryptophan
- Ts
4-methylphenylsulfonyl
- TTMSS
tris(trimethylsilyl)silyl
- VA
vinylogous aldol
- VAR
vinylogous aldol reaction
- VMAR
vinylogous Mukaiyama aldol reaction
- VMcR
vinylogous Michael reaction
- VMMcR
vinylogous Mukaiyama Michael reaction
- VMMnR
vinylogous Mukaiyama Mannich reaction
- VMnR
vinylogous Mannich reaction
Biographies
Claudio Curti is currently an Associate Professor of Organic Chemistry at the University of Parma, Italy (Food and Drug Department). He earned his Laurea degree in Pharmaceutical Chemistry and Technology in 2002 at the University of Parma. In 2005, he graduated from the postgraduate School of Chemical Synthesis at the University of Milan, Italy. In 2001, he joined the bioorganic synthesis group of the Department of Pharmacy (University of Parma) under the supervision of Prof. Giovanni Casiraghi, where he obtained his current position. His main research interests are in the field of asymmetric synthesis and organic chemistry methodology, focusing on the development of metal-based and organocatalytic, enantioselective vinylogous and hypervinylogous processes and their exploitation in the synthesis of multifunctional natural and natural-like compounds, including densely functionalized heterocycles and polyphenol metabolites.
Lucia Battistini is currently an Associate Professor of Organic Chemistry at the University of Parma, Italy (Food and Drug Department). She obtained her Laurea degree in Pharmaceutical Chemistry and Technology from the University of Parma in 1995. In 1999, she received her Ph.D. degree in Bioorganic Chemistry from the University of Torino, and in the same year she joined the group led by Prof. Giovanni Casiraghi at the Department of Pharmacy, University of Parma. Her current specific interests are focused on the development of new asymmetric methodologies for the synthesis of multifunctional natural and non-natural compounds of biological interest. Recently, her major scientific efforts have been devoted to the development of new classes of pseudopeptide ligands for molecular recognition and biomedical applications.
Andrea Sartori is currently an Associate Professor of Organic Chemistry at the University of Parma, Italy (Food and Drug Department). He graduated in Chemistry in 1997 at the University of Parma, and after one year of research fellowship at the Physics Department of the same Institution, he joined the group of Prof. D. N. Reinhoudt at the University of Twente (NL), where he received his Ph.D. in 2004. He returned to the University of Parma to work as a postdoctoral fellow at the Department of Pharmacy under the supervision of Prof. Giovanni Casiraghi. At this University in 2010 he obtained a position as Assistant Professor. His work focuses on the development of new asymmetric, vinylogous, and organocatalytic methodologies for the synthesis of chiral bioactive molecules and on the development of integrin pseudopeptide ligands for biomedical applications.
Franca Zanardi is Full Professor of Organic Chemistry in the Food and Drug Department of the University of Parma (Italy). She received her Laurea degree in Chemistry (1993) and her Ph.D. in bioorganic chemistry (1997) from the University of Parma under the supervision of Prof. Giovanni Casiraghi. She became an Assistant Professor in 1998 and Associate Professor in 2002. In 2017 she was the recipient of the Research Prize in Organic Chemistry-Life Sciences from the Organic Chemistry Division of the Italian Chemical Society. She currently leads the bioorganic synthesis group of the Food and Drug Department at the University of Parma. Her research interests concern the development of stereoselective, vinylogous methodologies addressed at the synthesis of biologically relevant chiral nonracemic molecules in the bio-organic and pharmaceutical domains. She is also involved in several research programs aimed at the synthesis and applications of biologically relevant pseudopeptides to be exploited as therapeutic/diagnostic tools in medicine.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrev.9b00481.
(PDF)
Author Contributions
† C.C., L.B., and A.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Vasseur A.; Bruffaerts J.; Marek I. Remote Functionalization through Alkene Isomerization. Nat. Chem. 2016, 8, 209–219. 10.1038/nchem.2445. [DOI] [PubMed] [Google Scholar]
- Tang R.-Y.; Li G.; Yu J.-Q. Conformation-Induced Remote meta-C-H Activation of Amines. Nature 2014, 507, 215–220. 10.1038/nature12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuson R. C. The Principle of Vinylogy. Chem. Rev. 1935, 16, 1–27. 10.1021/cr60053a001. [DOI] [Google Scholar]
- For a study on the γ/α site selectivity depending on the γ/α orbital coefficient of silyl/metal dienolates, see:; Denmark S. E.; Heemstra J. R. Jr.; Beutner G. L. Catalytic, Enantioselective, Vinylogous Aldol Reactions. Angew. Chem., Int. Ed. 2005, 44, 4682–4698. 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]
- Casiraghi G.; Zanardi F.; Appendino G.; Rassu G. Vinylogous Aldol Reaction: A Valuable, yet Understated Carbon-Carbon Bond-Forming Maneuver. Chem. Rev. 2000, 100, 1929–1972. 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]
- Casiraghi G.; Battistini L.; Curti C.; Rassu G.; Zanardi F. The Vinylogous Aldol and Related Addition Reactions: Ten Years of Progress. Chem. Rev. 2011, 111, 3076–3154. 10.1021/cr100304n. [DOI] [PubMed] [Google Scholar]
- Cordes M. H. C.; Kalesse M.. The Asymmetric Vinylogous Mukaiyama Aldol Reaction. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2019; pp 173–774. [Google Scholar]
- Boyko Y. D.; Dorokhov V. S.; Sukhorukov A. Y.; Ioffe S. L. Conjugated Nitrosoalkenes as Michael Acceptors in Carbon-Carbon Bond Forming Reactions: A Review and Perspective. Beilstein J. Org. Chem. 2017, 13, 2214–2234. 10.3762/bjoc.13.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millemaggi A.; Taylor R. J. K. 3-Alkenyl-Oxindoles: Natural Products, Pharmaceuticals, and Recent Synthetic Advances in Tandem/Telescoped Approaches. Eur. J. Org. Chem. 2010, 2010, 4527–4547. 10.1002/ejoc.201000643. [DOI] [Google Scholar]
- Silva E.; Silva A. 1,6-Conjugate Addition of Nucleophiles to α,β,γ,δ-Diunsaturated Systems. Synthesis 2012, 44, 3109–3128. 10.1055/s-0032-1316778. [DOI] [Google Scholar]
- Karak M.; Acosta J. A. M.; Barbosa L. C. A.; Boukouvalas J. Late-Stage Bromination Enables the Synthesis of Rubrolides B, I, K, and O. Eur. J. Org. Chem. 2016, 2016, 3780–3787. 10.1002/ejoc.201600473. [DOI] [Google Scholar]
- Ramachandran P. V.; Nicponski D.; Kim B. Total Regio- and Diastereocontrol in the Aldol Reactions of Dienolborinates. Org. Lett. 2013, 15, 1398–1401. 10.1021/ol400381q. [DOI] [PubMed] [Google Scholar]
- Urruzuno I.; Mugica O.; Zanella G.; Vera S.; Gómez-Bengoa E.; Oiarbide M.; Palomo C. α-Branched Ketone Dienolates: Base-Catalysed Generation and Regio- and Enantioselective Addition Reactions. Chem. - Eur. J. 2019, 25, 9701–9709. 10.1002/chem.201901694. [DOI] [PubMed] [Google Scholar]
- Mei G. J.; Shi F. Design and Application of 3-Alkyl-2-Vinylindoles in Bronsted Acid Catalyzed Reactions. Synlett 2016, 27, 2515–2524. 10.1055/s-0036-1588611. [DOI] [Google Scholar]
- Przydacz A.; Skrzyńska A.; Albrecht Ł. Breaking Aromaticity with Aminocatalysis: A Convenient Strategy for Asymmetric Synthesis. Angew. Chem., Int. Ed. 2019, 58, 63–73. 10.1002/anie.201808197. [DOI] [PubMed] [Google Scholar]
- Melchiorre P. Cinchona-Based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem., Int. Ed. 2012, 51, 9748–9770. 10.1002/anie.201109036. [DOI] [PubMed] [Google Scholar]
- Ramachary D. B.; Reddy Y. V. Dienamine Catalysis: An Emerging Technology in Organic Synthesis. Eur. J. Org. Chem. 2012, 2012, 865–887. 10.1002/ejoc.201101157. [DOI] [Google Scholar]
- Kumar I.; Ramaraju P.; Mir N. A. Asymmetric Trienamine Catalysis: New Opportunities in Amine Catalysis. Org. Biomol. Chem. 2013, 11, 709–716. 10.1039/C2OB26681D. [DOI] [PubMed] [Google Scholar]
- Vora H. U.; Wheeler P.; Rovis T. Exploiting Acyl and Enol Azolium Intermediates via N-Heterocyclic Carbene-Catalyzed Reactions of α-Reducible Aldehydes. Adv. Synth. Catal. 2012, 354, 1617–1639. 10.1002/adsc.201200031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.-Y.; Liu Q.; Chauhan P.; Enders D. N-Heterocyclic Carbene Catalysis via Azolium Dienolates: An Efficient Strategy for Remote Enantioselective Functionalizations. Angew. Chem., Int. Ed. 2018, 57, 3862–3873. 10.1002/anie.201709684. [DOI] [PubMed] [Google Scholar]
- Chauhan P.; Mahajan S.; Kaya U.; Hack D.; Enders D. Bifunctional Amine-Squaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions. Adv. Synth. Catal. 2015, 357, 253–281. 10.1002/adsc.201401003. [DOI] [Google Scholar]
- Uraguchi D.; Ooi T. Site-Selective Conjugate Addition Through Catalytic Generation of Ion-Pairing Intermediates. Top. Curr. Chem. 2015, 372, 55–83. 10.1007/128_2015_655. [DOI] [PubMed] [Google Scholar]
- Fang X.; Wang C.-J. Recent Advances in Asymmetric Organocatalysis Mediated by Bifunctional Amine–Thioureas Bearing Multiple Hydrogen-Bonding Donors. Chem. Commun. 2015, 51, 1185–1197. 10.1039/C4CC07909D. [DOI] [PubMed] [Google Scholar]
- Marcos V.; Alemán J. Old Tricks, New Dogs: Organocatalytic Dienamine Activation of α,β-Unsaturated Aldehydes. Chem. Soc. Rev. 2016, 45, 6812–6832. 10.1039/C6CS00438E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvi M.; Cassani C.; Moran A.; Melchiorre P. Secondary Amine-Catalyzed Asymmetric γ-Alkylation of α-Branched Enals via Dienamine Activation. Helv. Chim. Acta 2012, 95, 1985–2006. 10.1002/hlca.201200412. [DOI] [Google Scholar]
- Cui H. L.; Chen Y. C. α,α-Dicyanoalkenes: Versatile Vinylogous Nucleophiles for Organic Synthesis. Chem. Commun. 2009, 30, 4479–4486. 10.1039/b906201g. [DOI] [PubMed] [Google Scholar]
- Curti C.; Sartori A.; Battistini L.; Zanardi F. Exploring the Remote Reactivity of π-Extended Carbonyl Compounds: The Vinylogous Alkylidene Malononitrile Activation Strategy. Synlett 2018, 29, 266–281. 10.1055/s-0036-1589125. [DOI] [Google Scholar]
- Battistini L.; Curti C.; Rassu G.; Sartori A.; Zanardi F. Enolizable Alkylidene Heterocyclic and Carbocyclic Carbonyl Systems: Valuable Vinylogous Donor Substrates in Synthesis. Synthesis 2017, 49, 2297–2336. 10.1055/s-0036-1589487. [DOI] [Google Scholar]
- Dalpozzo R.; Mancuso R. Enantioselective Vinylogous Reactions of 3-Alkylidene Oxindoles. Synthesis 2018, 50, 2463–2472. 10.1055/s-0037-1609731. [DOI] [Google Scholar]
- Yin Y.; Jiang Z. Organocatalytic Asymmetric Vinylogous Michael Reactions. ChemCatChem 2017, 9, 4306–4318. 10.1002/cctc.201700941. [DOI] [Google Scholar]
- Schneider C.; Abels F. Catalytic, Enantioselective Vinylogous Michael Reactions. Org. Biomol. Chem. 2014, 12, 3531–3543. 10.1039/c4ob00332b. [DOI] [PubMed] [Google Scholar]
- Frías M.; Cieślik W.; Fraile A.; Rosado-Abón A.; Garrido-Castro A. F.; Yuste F.; Alemán J. Development and Application of Asymmetric Organocatalytic Mukaiyama and Vinylogous Mukaiyama-Type Reactions. Chem. - Eur. J. 2018, 24, 10906–10933. 10.1002/chem.201801866. [DOI] [PubMed] [Google Scholar]
- Hepburn H. B.; Dell’Amico L.; Melchiorre P. Enantioselective Vinylogous Organocascade Reactions. Chem. Rec. 2016, 16, 1787–1806. 10.1002/tcr.201600030. [DOI] [PubMed] [Google Scholar]
- Kalesse M.; Cordes M.; Symkenberg G.; Lu H.-H. The Vinylogous Mukaiyama Aldol Reaction (VMAR) in Natural Product Synthesis. Nat. Prod. Rep. 2014, 31, 563–594. 10.1039/C3NP70102F. [DOI] [PubMed] [Google Scholar]
- Mao B.; Fañanás-Mastral M.; Feringa B. L. Catalytic Asymmetric Synthesis of Butenolides and Butyrolactones. Chem. Rev. 2017, 117, 10502–10566. 10.1021/acs.chemrev.7b00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusseau X.; Chabaud L.; Guillou C. Synthesis of γ-Butenolides and α,β-Unsaturated γ-Butyrolactams by Addition of Vinylogous Nucleophiles to Michael Acceptors. Tetrahedron 2014, 70, 2595–2615. 10.1016/j.tet.2014.01.057. [DOI] [Google Scholar]
- Comprehensive Enantioselective Organocatalysis; Dalko P. I., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Lesch B.; Toräng J.; Vanderheiden S.; Bräse S. Base-Catalyzed Condensation of 2-Hydroxybenzaldehydes with α,β-Unsaturated Aldehydes - Scope and Limitations. Adv. Synth. Catal. 2005, 347, 555–562. 10.1002/adsc.200404239. [DOI] [Google Scholar]
- Liu K.; Chougnet A.; Woggon W.-D. A Short Route to α-Tocopherol. Angew. Chem., Int. Ed. 2008, 47, 5827–5829. 10.1002/anie.200801765. [DOI] [PubMed] [Google Scholar]
- Liu K.; Woggon W.-D. Enantioselective Synthesis of Daurichromenic Acid and Confluentin. Eur. J. Org. Chem. 2010, 2010, 1033–1036. 10.1002/ejoc.200901403. [DOI] [Google Scholar]
- Bröhmer M. C.; Bourcet E.; Nieger M.; Bräse S. A Unified Strategy for the Asymmetric Total Syntheses of Diversonol and Lachnone C. Chem. - Eur. J. 2011, 17, 13706–13711. 10.1002/chem.201102192. [DOI] [PubMed] [Google Scholar]
- Reyes E.; Uria U.; Carrillo L.; Vicario J. L. Enantioselective Cascade Reactions under N-Heterocyclic Carbene Catalysis. Synthesis 2017, 49, 451–471. 10.1055/s-0036-1589470. [DOI] [Google Scholar]
- Pellissier H. Recent Developments in Asymmetric Organocatalytic Domino Reactions. Adv. Synth. Catal. 2012, 354, 237–294. 10.1002/adsc.201100714. [DOI] [Google Scholar]
- For a review on the impact of organocascade catalysis in total synthesis, see:Grondal C.; Jeanty M.; Enders D. Organocatalytic Cascade Reactions as a New Tool in Total Synthesis. Nat. Chem. 2010, 2, 167–178. 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- For a related achiral secondary amine-triggered vinylogous aldol/aza-Michael domino reaction to racemic marmycin cores, see:Bourcet E.; Bröhmer M. C.; Nieger M.; Bräse S. Synthetic Studies towards Marmycins A and B: Development of the Vinylogous Aldol-Aza-Michael Domino Reaction. Org. Biomol. Chem. 2011, 9, 3136–3138. 10.1039/c1ob05124e. [DOI] [PubMed] [Google Scholar]
- Liu K.; Jiang X. Regioselective and Enantioselective Domino Aldol-Oxa-Michael Reactions to Construct Quaternary (Chroman) Stereocenters. Eur. J. Org. Chem. 2015, 2015, 6423–6428. 10.1002/ejoc.201501065. [DOI] [Google Scholar]
- Cassani C.; Melchiorre P. Direct Catalytic Enantioselective Vinylogous Aldol Reaction of α-Branched Enals with Isatins. Org. Lett. 2012, 14, 5590–5593. 10.1021/ol302711w. [DOI] [PubMed] [Google Scholar]
- Chatterjee I.; Bastida D.; Melchiorre P. Vinylogous Organocatalytic Triple Cascade Reaction: Forging Six Stereocenters in Complex Spiro-Oxindolic Cyclohexanes. Adv. Synth. Catal. 2013, 355, 3124–3130. 10.1002/adsc.201300667. [DOI] [Google Scholar]
- Frías M.; Padrón J. M.; Alemán J. Dienamine and Friedel-Crafts One-Pot Synthesis, and Antitumor Evaluation of Diheteroarylalkanals. Chem. - Eur. J. 2015, 21, 8237–8241. 10.1002/chem.201500660. [DOI] [PubMed] [Google Scholar]
- Orue A.; Uria U.; Reyes E.; Carrillo L.; Vicario J. L. Catalytic Enantioselective [5 + 2] Cycloaddition between Oxidopyrylium Ylides and Enals under Dienamine Activation. Angew. Chem., Int. Ed. 2015, 54, 3043–3046. 10.1002/anie.201410723. [DOI] [PubMed] [Google Scholar]
- Sohn S. S.; Rosen E. L.; Bode J. W. N-Heterocyclic Carbene-Catalyzed Generation of Homoenolates: γ-Butyrolactones by Direct Annulations of Enals and Aldehydes. J. Am. Chem. Soc. 2004, 126, 14370–14371. 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- Burstein C.; Glorius F. Organocatalyzed Conjugate Umpolung of α,β-Unsaturated Aldehydes for the Synthesis of γ-Butyrolactones. Angew. Chem., Int. Ed. 2004, 43, 6205–6208. 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]
- Sun L.-H.; Shen L.-T.; Ye S. Highly Diastereo- and Enantioselective NHC-Catalyzed [3 + 2] Annulation of Enals and Isatins. Chem. Commun. 2011, 47, 10136. 10.1039/c1cc13860j. [DOI] [PubMed] [Google Scholar]
- Dugal-Tessier J.; O’Bryan E. A.; Schroeder T. B. H.; Cohen D. T.; Scheidt K. A. An N-Heterocyclic Carbene/Lewis Acid Strategy for the Stereoselective Synthesis of Spirooxindole Lactones. Angew. Chem., Int. Ed. 2012, 51, 4963–4967. 10.1002/anie.201201643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawaz F.; Zaghouani M.; Bonne D.; Chuzel O.; Rodriguez J.; Coquerel Y. Design, Synthesis, and Organocatalytic Activity of N-Heterocyclic Carbenes Functionalized with Hydrogen-Bond Donors in Enantioselective Reactions of Homoenolates. Eur. J. Org. Chem. 2013, 2013, 8253–8264. 10.1002/ejoc.201301366. [DOI] [Google Scholar]
- Li J. L.; Sahoo B.; Daniliuc C. G.; Glorius F. Conjugate Umpolung of β,β-Disubstituted Enals by Dual Catalysis with an N-Heterocyclic Carbene and a Brønsted Acid: Facile Construction of Contiguous Quaternary Stereocenters. Angew. Chem., Int. Ed. 2014, 53, 10515–10519. 10.1002/anie.201405178. [DOI] [PubMed] [Google Scholar]
- Wang Z.-D.; Wang F.; Li X.; Cheng J.-P. N-Heterocyclic Carbene Catalyzed Annulation of Benzofuran-2,3-Diones and Enals: A Concise Synthesis of Spiro-Bis-Lactone. Org. Biomol. Chem. 2013, 11, 5634. 10.1039/c3ob41028e. [DOI] [PubMed] [Google Scholar]
- Seetha Lakshmi K.; Paul R.; Suresh E.; Nair V. N-Heterocyclic Carbene Mediated Homoenolate Annulation of Benzofuran-2,3-Diones: Stereoselective Synthesis of Bis-Spirofuranones. Synlett 2014, 25, 853–857. 10.1055/s-0033-1340715. [DOI] [Google Scholar]
- Jang K. P.; Hutson G. E.; Johnston R. C.; McCusker E. O.; Cheong P. H.-Y.; Scheidt K. A. Asymmetric Homoenolate Additions to Acyl Phosphonates through Rational Design of a Tailored N -Heterocyclic Carbene Catalyst. J. Am. Chem. Soc. 2014, 136, 76–79. 10.1021/ja410932t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman C. G.; Walker M. M.; Johnson J. S. Enantioconvergent Synthesis of Functionalized γ-Butyrolactones via (3 + 2)-Annulation. J. Am. Chem. Soc. 2015, 137, 122–125. 10.1021/ja511701j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an example of ynal-derived acyl azolium, see:Mahatthananchai J.; Kaeobamrung J.; Bode J. W. Chiral N-Heterocyclic Carbene-Catalyzed Annulations of Enals and Ynals with Stable Enols: A Highly Enantioselective Coates-Claisen Rearrangement. ACS Catal. 2012, 2, 494–503. 10.1021/cs300020t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J.; Xie X.; Han R.; Ma D.; Yang J.; She X. N-Heterocyclic Carbene (NHC)-Catalyzed/Lewis Acid Mediated Conjugate Umpolung of Alkynyl Aldehydes for the Synthesis of Butenolides: A Formal [3 + 2] Annulation. Chem. - Eur. J. 2013, 19, 4146–4150. 10.1002/chem.201204386. [DOI] [PubMed] [Google Scholar]
- ElSohly A. M.; Wespe D. A.; Poore T. J.; Snyder S. A. An Efficient Approach to the Securinega Alkaloids Empowered by Cooperative N-Heterocyclic Carbene/Lewis Acid Catalysis. Angew. Chem., Int. Ed. 2013, 52, 5789–5794. 10.1002/anie.201301849. [DOI] [PubMed] [Google Scholar]
- Lee A.; Scheidt K. A. A Cooperative N-Heterocyclic Carbene/Chiral Phosphate Catalysis System for Allenolate Annulations. Angew. Chem., Int. Ed. 2014, 53, 7594–7598. 10.1002/anie.201403446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For enolate formation from homoenolate sources, see:Kaeobamrung J.; Kozlowski M. C.; Bode J. W. Chiral N-Heterocyclic Carbene-Catalyzed Generation of Ester Enolate Equivalents from α,β-Unsaturated Aldehydes for Enantioselective Diels-Alder Reactions. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20661–20665. 10.1073/pnas.1007469107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo J.; Chen X.; Chi Y. R. Oxidative γ-Addition of Enals to Trifluoromethyl Ketones: Enantioselectivity Control via Lewis Acid/N-Heterocyclic Carbene Cooperative Catalysis. J. Am. Chem. Soc. 2012, 134, 8810–8888. 10.1021/ja303618z. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Yu C.; Li T.; Wang X. S.; Yao C. N-Heterocyclic Carbene/Lewis Acid Strategy for the Stereoselective Synthesis of Spirocyclic Oxindole-Dihydropyranones. Org. Lett. 2014, 16, 3632–3635. 10.1021/ol501224p. [DOI] [PubMed] [Google Scholar]
- Yao C.; Wang D.; Lu J.; Li T.; Jiao W.; Yu C. N-Heterocyclic Carbene Catalyzed Reactions of α-Bromo-α,β-Unsaturated Aldehydes/α,β-Dibromoaldehydes with 1,3-Dinucleophilic Reagents. Chem. - Eur. J. 2012, 18, 1914–1917. 10.1002/chem.201103358. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Yang L.; Deng Y.; Zhong G. Cooperative Catalysis of N-Heterocyclic Carbene and Brønsted Acid for a Highly Enantioselective Route to Unprotected Spiro-Indoline-Pyrans. Chem. Commun. 2015, 51, 8330–8333. 10.1039/C5CC02096D. [DOI] [PubMed] [Google Scholar]
- Rong X.; Yao H.; Xia W.; Du Y.; Zhou Y.; Liu H. Enantioselective Assembly of Spirolactones through NHC-Catalyzed Remote β-Carbon Addition of Enals with Isatins. ACS Comb. Sci. 2016, 18, 220–224. 10.1021/acscombsci.5b00197. [DOI] [PubMed] [Google Scholar]
- For a further example of NHC-catalyzed oxidative γ-addition of enals to isatins in a racemic context, see:Liu R.; Yu C.; Xiao Z.; Li T.; Wang X.; Xie Y.; Yao C. NHC-Catalyzed Oxidative γ-Addition of α,β-Unsaturated Aldehydes to Isatins: A High-Efficiency Synthesis of Spirocyclic Oxindole-Dihydropyranones. Org. Biomol. Chem. 2014, 12, 1885–1891. 10.1039/C3OB42008F. [DOI] [PubMed] [Google Scholar]
- Cheng J. T.; Chen X. Y.; Gao Z. H.; Ye S. N-Heterocyclic Carbene Catalyzed Generation and [4 + 2] Annulation of Unsubstituted Dienolate - Enantioselective Synthesis of Spirocyclic Oxindolodihydropyranones. Eur. J. Org. Chem. 2015, 2015, 1047–1053. 10.1002/ejoc.201403395. [DOI] [Google Scholar]
- Wu Z.; Li F.; Wang J. Intermolecular Dynamic Kinetic Resolution Cooperatively Catalyzed by an N-Heterocyclic Carbene and a Lewis Acid. Angew. Chem., Int. Ed. 2015, 54, 1629–1633. 10.1002/anie.201410030. [DOI] [PubMed] [Google Scholar]
- Sun J.; He F.; Wang Z.; Pan D.; Zheng P.; Mou C.; Jin Z.; Chi Y. R. Carbene-Catalyzed Enal γ-Carbon Addition to α-Ketophosphonates for Enantioselective Access to Bioactive 2-Pyranylphosphonates. Chem. Commun. 2018, 54, 6040–6043. 10.1039/C8CC03017K. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Gu L.; Gong Y. Direct Vinylogous Aldol Reaction Triggered by Tetrabutylammonium Fluoride: A Highly Regioselective and Diastereoselective Addition of Cyclic β-Haloenals to Aromatic Aldehydes. Synlett 2012, 23, 468–472. 10.1055/s-0031-1290313. [DOI] [Google Scholar]
- Zhang J. L.; Gong Y. F. Cross-Coupling Reactions of Two Different Activated Alkenes through Tetrabutylammonium Fluoride (TBAF) Promoted Deprotonation/Activation Strategy: A Regioselective Construction of Quaternary Carbon Centers. Org. Lett. 2011, 13, 176–179. 10.1021/ol1028332. [DOI] [PubMed] [Google Scholar]
- Chen X.; Yang S.; Song B. A.; Chi Y. R. Functionalization of Benzylic C(sp3)-H Bonds of Heteroaryl Aldehydes through N-Heterocyclic Carbene Organocatalysis. Angew. Chem., Int. Ed. 2013, 52, 11134–11137. 10.1002/anie.201305861. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Sun J.; Yan J.; Yang S.; Zheng P.; Jin Z.; Chi Y. R. Carbene-Catalyzed Indole 3-Methyl C(sp3)-H Bond Functionalization. J. Org. Chem. 2017, 82, 13342–13347. 10.1021/acs.joc.7b02436. [DOI] [PubMed] [Google Scholar]
- Janssen-Müller D.; Singha S.; Olyschläger T.; Daniliuc C. G.; Glorius F. Annulation of o-Quinodimethanes through N-Heterocyclic Carbene Catalysis for the Synthesis of 1-Isochromanones. Org. Lett. 2016, 18, 4444–4447. 10.1021/acs.orglett.6b02335. [DOI] [PubMed] [Google Scholar]
- Chen D. F.; Rovis T. N-Heterocyclic Carbene and Chiral Brønsted Acid Cooperative Catalysis for a Highly Enantioselective [4 + 2] Annulation. Synthesis 2016, 49, 293–298. 10.1055/s-0036-1588349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J.; Cheng Y.; Cheng J.; Li R.; Li P. Organocatalytic Asymmetric Benzylation and Aldol-Hemiacetalization of α,β-Unsaturated Trifluoromethyl Ketones: Efficient Enantioselective Construction of 3,4-Dihydroisocoumarins. Chem. - Eur. J. 2017, 23, 519–523. 10.1002/chem.201604920. [DOI] [PubMed] [Google Scholar]
- For an example in a racemic context featuring the addition reaction between 2-methyl-3,5-dinitrobenzaldehyde and isatin acceptors catalyzed by Et3N, see:Wang B.; Leng H. J.; Yang X. Y.; Han B.; Rao C. L.; Liu L.; Peng C.; Huang W. Efficient Synthesis of Tetrahydronaphthalene- or Isochroman-Fused Spirooxindoles Using Tandem Reactions. RSC Adv. 2015, 5, 88272–88276. 10.1039/C5RA15735H. [DOI] [Google Scholar]
- Mukaiyama T.; Ishida A. A Convenient Method for the Preparation of δ-Alkoxy-α,β-Unsaturated Aldehydes by Reaction of Acetals with 1-Trimethylsiloxy-1,3-Butadiene. Chem. Lett. 1975, 4, 319–322. 10.1246/cl.1975.319. [DOI] [Google Scholar]
- Gieseler M. T.; Kalesse M. Asymmetric Vinylogous Mukaiyama Aldol Reaction of Aldehyde-Derived Dienolates. Org. Lett. 2011, 13, 2430–2432. 10.1021/ol2006727. [DOI] [PubMed] [Google Scholar]
- Simsek S.; Horzella M.; Kalesse M. Oxazaborolidinone-Promoted Vinylogous Mukaiyama Aldol Reactions. Org. Lett. 2007, 9, 5637–5639. 10.1021/ol702640w. [DOI] [PubMed] [Google Scholar]
- Kena Diba A.; Noll C.; Richter M.; Gieseler M. T.; Kalesse M. Intramolecular Stereoselective Protonation of Aldehyde-Derived Enolates. Angew. Chem., Int. Ed. 2010, 49, 8367–8369. 10.1002/anie.201004619. [DOI] [PubMed] [Google Scholar]
- Gieseler M. T.; Kalesse M. Synthesis of Angiolam A. Org. Lett. 2014, 16, 548–551. 10.1021/ol403423r. [DOI] [PubMed] [Google Scholar]
- Laina-Martín V.; Humbrías-Martín J.; Fernández-Salas J. A.; Alemán J. Asymmetric Vinylogous Mukaiyama Aldol Reaction of Isatins under Bifunctional Organocatalysis: Enantioselective Synthesis of Substituted 3-Hydroxy-2-Oxindoles. Chem. Commun. 2018, 54, 2781–2784. 10.1039/C8CC00759D. [DOI] [PubMed] [Google Scholar]
- Frias M.; Mas-Ballesté R.; Arias S.; Alvarado C.; Alemán J. Asymmetric Synthesis of Rauhut-Currier Type Products by a Regioselective Mukaiyama Reaction under Bifunctional Catalysis. J. Am. Chem. Soc. 2017, 139, 672–679. 10.1021/jacs.6b07851. [DOI] [PubMed] [Google Scholar]
- He M.; Bode J. W. Catalytic Synthesis of γ-Lactams via Direct Annulations of Enals and N-Sulfonylimines. Org. Lett. 2005, 7, 3131–3134. 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]
- For a study on the NHC-catalyzed addition of homoenolates from α,β-unsaturated aldehydes to conjugated sulfonimines generated in situ, and subsequent methanolysis to give racemic GABA derivatives, see:Nair V.; Varghese V.; Babu B. P.; Sinu C. R.; Suresh E. A Novel Pseudo Four Component Reaction Involving Homoenolate for the Synthesis of γ-Aminobutyric Acid (GABA) Derivatives. Org. Biomol. Chem. 2010, 8, 761. 10.1039/b922981g. [DOI] [PubMed] [Google Scholar]
- Cardinal-David B.; Raup D. E. A.; Scheidt K. A. Cooperative N -Heterocyclic Carbene/Lewis Acid Catalysis for Highly Stereoselective Annulation Reactions with Homoenolates. J. Am. Chem. Soc. 2010, 132, 5345–5347. 10.1021/ja910666n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; DiRocco D. A.; Rovis T. N-Heterocyclic Carbene and Brønsted Acid Cooperative Catalysis: Asymmetric Synthesis of Trans-γ-Lactams. J. Am. Chem. Soc. 2011, 133, 12466–12469. 10.1021/ja205714g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H.; Tiwari B.; Mo J.; Xing C.; Chi Y. R. Highly Enantioselective Addition of Enals to Isatin-Derived Ketimines Catalyzed by N-Heterocyclic Carbenes: Synthesis of Spirocyclic γ-Lactams. Org. Lett. 2012, 14, 5412–5415. 10.1021/ol302475g. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Feng P.; Sun L.-H.; Cui Y.; Ye S.; Jiao N. N-Heterocyclic Carbene-Catalyzed Homoenolate Additions with N-Aryl Ketimines as Electrophiles: Efficient Synthesis of Spirocyclic γ-Lactam Oxindoles. Chem. - Eur. J. 2012, 18, 9198–9203. 10.1002/chem.201201375. [DOI] [PubMed] [Google Scholar]
- For a further example of γ-lactam-forming reactions between enals and N-sulfonyl ketimines catalyzed by ad hoc-prepared chiral azolium catalysts, see:Zheng P.; Gondo C. A.; Bode J. W. Late-Stage Diversification of Chiral N-Heterocyclic-Carbene Precatalysts for Enantioselective Homoenolate Additions. Chem. - Asian J. 2011, 6, 614–620. 10.1002/asia.201000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.; Yu X.; Wu J. Silver Triflate and N-Heterocyclic Carbene Co-Catalyzed Reaction of N′-(2-Alkynylbenzylidene)Hydrazide, Methanol with α,β- Unsaturated Aldehyde. Chem. Commun. 2010, 46, 6356–6358. 10.1039/c0cc01207f. [DOI] [PubMed] [Google Scholar]
- Siddiqui I.; Srivastava A.; Shamim S.; Srivastava A.; Waseem M.; Singh R. Highly Diastereoselective NHC-Catalyzed [4 + 3] Annulation of Enals, Aldehydes and N-Phenyl Urea/Thiourea for the Synthesis of Monocyclic Trans-1,3-Diazepanes. Synlett 2013, 24, 2586–2590. 10.1055/s-0033-1339894. [DOI] [Google Scholar]
- Xu W.-Y.; Iwaki R.; Jia Y.-M.; Zhang W.; Kato A.; Yu C.-Y. NHC-Mediated Cross-Coupling of Sugar-Derived Cyclic Nitrones with Enals: General and Efficient Synthesis of Polyhydroxylated Pyrrolizidines and Indolizidines. Org. Biomol. Chem. 2013, 11, 4622. 10.1039/c3ob40696b. [DOI] [PubMed] [Google Scholar]
- Wang M.; Huang Z.; Xu J.; Chi Y. R. N-Heterocyclic Carbene-Catalyzed [3 + 4] Cycloaddition and Kinetic Resolution of Azomethine Imines. J. Am. Chem. Soc. 2014, 136, 1214–1217. 10.1021/ja411110f. [DOI] [PubMed] [Google Scholar]
- Zheng P.-C.; Cheng J.; Su S.; Jin Z.; Wang Y.-H.; Yang S.; Jin L.-H.; Song B.-A.; Chi Y. R. Oxidative N-Heterocyclic Carbene-Catalyzed γ-Carbon Addition of Enals to Imines: Mechanistic Studies and Access to Antimicrobial Compounds. Chem. - Eur. J. 2015, 21, 9984–9987. 10.1002/chem.201501632. [DOI] [PubMed] [Google Scholar]
- For an example of NHC-catalyzed [3 + 3] annulation where cyclic methyl-substituted sulfonyl imines react as donor components with enal acceptors, see:Kravina A. G.; Mahatthananchai J.; Bode J. W. Enantioselective, NHC-Catalyzed Annulations of Trisubstituted Enals and Cyclic N-Sulfonylimines via α,β-Unsaturated Acyl Azoliums. Angew. Chem., Int. Ed. 2012, 51, 9433–9436. 10.1002/anie.201204145. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Wang J.; Xia W.; Shu S.; Jiao S.; Zhou Y.; Liu H. γ-Carbon Activation through N-Heterocyclic Carbene/Brønsted Acids Cooperative Catalysis: A Highly Enantioselective Route to δ-Lactams. Org. Lett. 2015, 17, 3850–3853. 10.1021/acs.orglett.5b01827. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Chen X.-Y.; Li S.; Jafari E.; Raabe G.; Enders D. N-Heterocyclic Carbene-Catalyzed [4 + 2] Annulation of β-Methyl Enals and Cyclic Trifluoromethyl Ketimines for the Asymmetric Synthesis of Dihydroquinazolinone Derivatives. Chem. Commun. 2017, 53, 11342–11344. 10.1039/C7CC06562K. [DOI] [PubMed] [Google Scholar]
- Ransborg L. K.; Overgaard M.; Hejmanowska J.; Barfüsser S.; Jørgensen K. A.; Albrecht Ł. Asymmetric Formation of Bridged Benzoxazocines through an Organocatalytic Multicomponent Dienamine-Mediated One-Pot Cascade. Org. Lett. 2014, 16, 4182–4185. 10.1021/ol501882x. [DOI] [PubMed] [Google Scholar]
- Li W.; Wei J.; Jia Q.; Du Z.; Zhang K.; Wang J. Asymmetric Synthesis of Tetrahydroquinolines through a [3 + 2] Cycloaddition Controlled by Dienamine Catalysis. Chem. - Eur. J. 2014, 20, 6592–6596. 10.1002/chem.201402089. [DOI] [PubMed] [Google Scholar]
- Izquierdo C.; Esteban F.; Parra A.; Alfaro R.; Alemán J.; Fraile A.; Ruano J. L. G. Control of the Dual Reactivity (Iminium-Dienamine) of β-Arylmethyl α,β-Unsaturated Aldehydes in Organocatalytic 1,3-Dipolar Cycloadditions with N-Benzoyl C,N-Cyclic Azomethine Imines. J. Org. Chem. 2014, 79, 10417–10433. 10.1021/jo5018519. [DOI] [PubMed] [Google Scholar]
- Liu J.-X.; Zhou Q.-Q.; Deng J.-G.; Chen Y.-C. An Asymmetric Normal-Electron-Demand Aza-Diels-Alder Reaction via Trienamine Catalysis. Org. Biomol. Chem. 2013, 11, 8175–8178. 10.1039/c3ob41698d. [DOI] [PubMed] [Google Scholar]
- For an example of a non-vinylogous α-selective aza-MBH reaction involving preformed silyl dienol ethers and imine acceptors under bifunctional organocatalysis, see:Frías M.; Carrasco A. C.; Fraile A.; Alemán J. A General Asymmetric Formal Synthesis of Aza-Baylis–Hillman Type Products under Bifunctional Catalysis. Chem. - Eur. J. 2018, 24, 3072. 10.1002/chem.201705892. [DOI] [PubMed] [Google Scholar]
- Nair V.; Vellalath S.; Poonoth M.; Suresh E. N-Heterocyclic Carbene-Catalyzed Reaction of Chalcones and Enals via Homoenolate: An Efficient Synthesis of 1,3,4-Trisubstituted Cyclopentenes. J. Am. Chem. Soc. 2006, 128, 8736–8737. 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- Chiang P. C.; Kaeobamrung J.; Bode J. W. Enantioselective, Cyclopentene-Forming Annulations via NHC-Catalyzed Benzoin-Oxy-Cope Reactions. J. Am. Chem. Soc. 2007, 129, 3520–3521. 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]
- For an example of intramolecular [3 + 2] annulation involving β-NHC-activated enals and chalcones giving cis-disposed cyclopentene-fused macrocycles, see:Seetha Lakshmi K. C.; Sinu C. R.; Padmaja D. V. M.; Gopinathan A.; Suresh E.; Nair V. A Novel Intramolecular Homoenolate Annulation Leading to the Formation of Cyclopentene-Fused Macrocycles. Org. Lett. 2014, 16, 5532–5535. 10.1021/ol5023239. [DOI] [PubMed] [Google Scholar]
- For a DFT study on the mechanism of NHC-catalyzed cycloannulation of homoenolate from butenal and 3-penten-2-one, see:Verma P.; Patni P. A.; Sunoj R. B. Mechanistic Insights on N-Heterocyclic Carbene-Catalyzed Annulations: The Role of Base-Assisted Proton Transfers. J. Org. Chem. 2011, 76, 5606–5613. 10.1021/jo200560t. [DOI] [PubMed] [Google Scholar]
- Cohen D. T.; Cardinal-David B.; Roberts J. M.; Sarjeant A. A.; Scheidt K. A. NHC-Catalyzed/Titanium(IV)-Mediated Highly Diastereo-and Enantioselective Dimerization of Enals. Org. Lett. 2011, 13, 1068–1071. 10.1021/ol103112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D. T.; Cardinal-David B.; Scheidt K. A. Lewis Acid Activated Synthesis of Highly Substituted Cyclopentanes by the N-Heterocyclic Carbene Catalyzed Addition of Homoenolate Equivalents to Unsaturated Ketoesters. Angew. Chem., Int. Ed. 2011, 50, 1678–1682. 10.1002/anie.201005908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X.; Jiang K.; Xing C.; Hao L.; Chi Y. R. A Highly Regio- and Stereoselective Cascade Annulation of Enals and Benzodi(Enone)s Catalyzed by N-Heterocyclic Carbenes. Angew. Chem., Int. Ed. 2011, 50, 1910–1913. 10.1002/anie.201007144. [DOI] [PubMed] [Google Scholar]
- Fan X.-W.; Cheng Y. N-Heterocyclic Carbene-Catalyzed Cascade Reaction of 2-Aroylvinylcinnamaldehydes with 2-Aroylvinylchalcones: Rapid Assembly of Six Contiguous Stereogenic Centers with High Diastereoselectivity. Org. Biomol. Chem. 2014, 12, 123–131. 10.1039/C3OB41656A. [DOI] [PubMed] [Google Scholar]
- Wang Z.-T.; Zhao Y.; Wang Z.-Y.; Cheng Y. N-Heterocyclic Carbene-Catalyzed Diastereoselective and Enantioselective Reaction of 2-Aroylvinylcinnamaldehydes with α,β-Unsaturated Imines: Complete Control and Switch of Diastereoselectivity by N-Substituents of Catalysts. J. Org. Chem. 2015, 80, 1727–1734. 10.1021/jo502668c. [DOI] [PubMed] [Google Scholar]
- Chen X.; Fang X.; Chi Y. R. Cis-Enals in N-Heterocyclic Carbene-Catalyzed Reactions: Distinct Stereoselectivity and Reactivity. Chem. Sci. 2013, 4, 2613–2618. 10.1039/c3sc50666e. [DOI] [Google Scholar]
- An alternative acyl anion-oxy-Cope rearrengement pathway was originally proposed for the formation of lactams of type 141; see:He M.; Bode J. W. Enantioselective, NHC-Catalyzed Bicyclo-β-Lactam Formation via Direct Annulations of Enals and Unsaturated N-Sulfonyl Ketimines. J. Am. Chem. Soc. 2008, 130, 418–419. 10.1021/ja0778592. [DOI] [PubMed] [Google Scholar]
- For an example of diastereoselective [3 + 2] annulation employing trans-enals and β,β-disubstituted α,β-unsaturated imines to give the “conventional” β-lactam products of type 141, see:Jiang K.; Tiwari B.; Chi Y. R. Access to Spirocyclic Oxindoles via N-Heterocyclic Carbene-Catalyzed Reactions of Enals and Oxindole-Derived α,β-Unsaturated Imines. Org. Lett. 2012, 14, 2382–2385. 10.1021/ol3008028. [DOI] [PubMed] [Google Scholar]
- McCusker E. O.; Scheidt K. A. Enantioselective N-Heterocyclic Carbene Catalyzed Annulation Reactions with Imidazolidinones. Angew. Chem., Int. Ed. 2013, 52, 13616–13620. 10.1002/anie.201307292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair V.; Sinu C. R.; Babu B. P.; Varghese V.; Jose A.; Suresh E. Novel Nucleophilic Heterocyclic Carbene Mediated Stereoselective Conjugate Addition of Enals to Nitrostyrenes via Homoenolate. Org. Lett. 2009, 11, 5570–5573. 10.1021/ol901918x. [DOI] [PubMed] [Google Scholar]
- Maji B.; Ji L.; Wang S.; Vedachalam S.; Ganguly R.; Liu X.-W. N-Heterocyclic Carbene Catalyzed Homoenolate-Addition Reaction of Enals and Nitroalkenes: Asymmetric Synthesis of 5-Carbon-Synthon δ-Nitroesters. Angew. Chem., Int. Ed. 2012, 51, 8276–8280. 10.1002/anie.201203449. [DOI] [PubMed] [Google Scholar]
- White N. A.; DiRocco D. A.; Rovis T. Asymmetric N-Heterocyclic Carbene Catalyzed Addition of Enals to Nitroalkenes: Controlling Stereochemistry via the Homoenolate Reactivity Pathway To Access δ-Lactams. J. Am. Chem. Soc. 2013, 135, 8504–8507. 10.1021/ja403847e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinu C. R.; Padmaja D. V. M.; Ranjini U. P.; Lakshmi K. C. S.; Suresh E.; Nair V. A Cascade Reaction Actuated by Nucleophilic Heterocyclic Carbene Catalyzed Intramolecular Addition of Enals via Homoenolate to α,β-Unsaturated Esters: Efficient Synthesis of Coumarin Derivatives. Org. Lett. 2013, 15, 68–71. 10.1021/ol303091m. [DOI] [PubMed] [Google Scholar]
- Izquierdo J.; Orue A.; Scheidt K. A. A Dual Lewis Base Activation Strategy for Enantioselective Carbene-Catalyzed Annulations. J. Am. Chem. Soc. 2013, 135, 10634–10637. 10.1021/ja405833m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H.; Jia W.-Q.; Sun L.-H.; Ye S. N-Heterocyclic Carbene Catalyzed [4 + 3] Annulation of Enals and o-Quinone Methides: Highly Enantioselective Synthesis of Benzo-ε-Lactones. Angew. Chem., Int. Ed. 2013, 52, 8607–8610. 10.1002/anie.201303903. [DOI] [PubMed] [Google Scholar]
- Wang M.; Rong Z.-Q.; Zhao Y. Stereoselective Synthesis of ε-Lactones or Spiro-Heterocycles through NHC-Catalyzed Annulation: Divergent Reactivity by Catalyst Control. Chem. Commun. 2014, 50, 15309–15312. 10.1039/C4CC07788A. [DOI] [PubMed] [Google Scholar]
- Guo C.; Schedler M.; Daniliuc C. G.; Glorius F. N-Heterocyclic Carbene Catalyzed Formal [3 + 2] Annulation Reaction of Enals: An Efficient Enantioselective Access to Spiro-Heterocycles. Angew. Chem., Int. Ed. 2014, 53, 10232–10236. 10.1002/anie.201405381. [DOI] [PubMed] [Google Scholar]
- Zhu T.; Zheng P.; Mou C.; Yang S.; Song B.-A.; Chi Y. R. Benzene Construction via Organocatalytic Formal [3 + 3] Cycloaddition Reaction. Nat. Commun. 2014, 5, 1–6. 10.1038/ncomms6027. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Que Y.; Li T.; Zhu L.; Yu C.; Yao C. N-Heterocyclic Carbene-Catalyzed [4 + 2] Cyclization of 2-Bromo-2-Enal with 3-Alkylenyloxindoles: Efficient Assembly of Spirocarbocyclic Oxindole. Org. Biomol. Chem. 2015, 13, 1829–1835. 10.1039/C4OB01706D. [DOI] [PubMed] [Google Scholar]
- Yao H.; Zhou Y.; Chen X.; Zhang P.; Xu J.; Liu H. N-Heterocyclic Carbene Catalytic [4 + 2] Cyclization of 3-Alkylenyloxindoles with Enals: γ-Carbon Activation for Enantioselective Assembly of Spirocarbocyclic Oxindoles. J. Org. Chem. 2016, 81, 8888–8899. 10.1021/acs.joc.6b01596. [DOI] [PubMed] [Google Scholar]
- Chen X.-Y.; Liu Q.; Chauhan P.; Li S.; Peuronen A.; Rissanen K.; Jafari E.; Enders D. N-Heterocyclic Carbene Catalyzed [4 + 2] Annulation of Enals via a Double Vinylogous Michael Addition: Asymmetric Synthesis of 3,5-Diaryl Cyclohexenones. Angew. Chem., Int. Ed. 2017, 56, 6241–6245. 10.1002/anie.201702881. [DOI] [PubMed] [Google Scholar]
- Han B.; He Z.-Q.; Li J.-L.; Li R.; Jiang K.; Liu T.-Y.; Chen Y.-C. Organocatalytic Regio- and Stereoselective Inverse-Electron-Demand Aza-Diels-Alder Reaction of α,β-Unsaturated Aldehydes and n-Tosyl-1-Aza-1,3-Butadienes. Angew. Chem., Int. Ed. 2009, 48, 5474–5477. 10.1002/anie.200902216. [DOI] [PubMed] [Google Scholar]
- Li J.-L.; Kang T.-R.; Zhou S.-L.; Li R.; Wu L.; Chen Y.-C. Organocatalytic Asymmetric Inverse-Electron-Demand Diels-Alder Reaction of Electron-Deficient Dienes and Crotonaldehyde. Angew. Chem., Int. Ed. 2010, 49, 6418–6420. 10.1002/anie.201002912. [DOI] [PubMed] [Google Scholar]
- Li J.-L.; Zhou S.-L.; Chen P.-Q.; Dong L.; Liu T.-Y.; Chen Y.-C. Asymmetric Diels-Alder Reaction of β,β-Disubstituted Enals and Chromone-Fused Dienes: Construction of Collections with High Molecular Complexity and Skeletal Diversity. Chem. Sci. 2012, 3, 1879–1882. 10.1039/c2sc20096a. [DOI] [Google Scholar]
- Albrecht Ł.; Dickmeiss G.; Weise C. F.; Rodríguez-Escrich C.; Jørgensen K. A. Dienamine-Mediated Inverse-Electron-Demand Hetero-Diels-Alder Reaction by Using an Enantioselective H-Bond-Directing Strategy. Angew. Chem., Int. Ed. 2012, 51, 13109–13113. 10.1002/anie.201207122. [DOI] [PubMed] [Google Scholar]
- For similar IED aza-DA cycloaddition reactions involving γ-enolizable enals as dienamine-activated γ,β-2π components and cyclic sulfonamide 4π dienes, see:Gu J.; Ma C.; Li Q. Z.; Du W.; Chen Y.-C. β,γ-Regioselective Inverse-Electron-Demand Aza-Diels-Alder Reactions with α,β-Unsaturated Aldehydes via Dienamine Catalysis. Org. Lett. 2014, 16, 3986–3989. 10.1021/ol501814p. [DOI] [PubMed] [Google Scholar]
- Weise C. F.; Lauridsen V. H.; Rambo R. S.; Iversen E. H.; Olsen M. L.; Jørgensen K. A. Organocatalytic Access to Enantioenriched Dihydropyran Phosphonates via an Inverse-Electron-Demand Hetero-Diels-Alder Reaction. J. Org. Chem. 2014, 79, 3537–3546. 10.1021/jo500347a. [DOI] [PubMed] [Google Scholar]
- Wang S.; Rodriguez-Escrich C.; Pericàs M. A. H-Bond-Directing Organocatalyst for Enantioselective [4 + 2] Cycloadditions via Dienamine Catalysis. Org. Lett. 2016, 18, 556–559. 10.1021/acs.orglett.5b03575. [DOI] [PubMed] [Google Scholar]
- Skrzyńska A.; Albrecht A.; Albrecht Ł. Aminocatalytic Strategy for the Synthesis of Optically Active Benzothiophene Derivatives. Adv. Synth. Catal. 2016, 358, 2838–2844. 10.1002/adsc.201600269. [DOI] [Google Scholar]
- Ramachary D. B.; Ramakumar K. Direct Organocatalytic Asymmetric Approach to Baylis-Hillman-Type Products through a Push-Pull Dienamine Platform. Eur. J. Org. Chem. 2011, 2011, 2599–2605. 10.1002/ejoc.201100075. [DOI] [Google Scholar]
- Albrecht Ł.; Dickmeiss G.; Acosta F. C.; Rodríguez-Escrich C.; Davis R. L.; Jørgensen K. A. Asymmetric Organocatalytic Formal [2 + 2]-Cycloadditions via Bifunctional H-Bond Directing Dienamine Catalysis. J. Am. Chem. Soc. 2012, 134, 2543–2546. 10.1021/ja211878x. [DOI] [PubMed] [Google Scholar]
- Talavera G.; Reyes E.; Vicario J. L.; Carrillo L. Cooperative Dienamine/Hydrogen-Bonding Catalysis: Enantioselective Formal [2 + 2] Cycloaddition of Enals with Nitroalkenes. Angew. Chem., Int. Ed. 2012, 51, 4104–4107. 10.1002/anie.201200269. [DOI] [PubMed] [Google Scholar]
- Qi L.-W.; Yang Y.; Gui Y.-Y.; Zhang Y.; Chen F.; Tian F.; Peng L.; Wang L.-X. Asymmetric Synthesis of 3,3-Spirooxindoles Fused with Cyclobutanes through Organocatalytic Formal [2 + 2] Cycloadditions under H-Bond-Directing Dienamine Activation. Org. Lett. 2014, 16, 6436–6439. 10.1021/ol503266q. [DOI] [PubMed] [Google Scholar]
- Halskov K. S.; Kniep F.; Lauridsen V. H.; Iversen E. H.; Donslund B. S.; Jørgensen K. A. Organocatalytic Enamine-Activation of Cyclopropanes for Highly Stereoselective Formation of Cyclobutanes. J. Am. Chem. Soc. 2015, 137, 1685–1691. 10.1021/ja512573q. [DOI] [PubMed] [Google Scholar]
- For examples of this chemistry before 2010, see:De Figueiredo R. M.; Fröhlich R.; Christmann M. Amine-Catalyzed Cyclizations of Tethered α,β-Unsaturated Carbonyl Compounds. Angew. Chem., Int. Ed. 2008, 47, 1450–1453. 10.1002/anie.200704688. [DOI] [PubMed] [Google Scholar]
- Hong B. C.; Wu M. F.; Tseng H. C.; Huang G. F.; Su C. F.; Liao J. H. Organocatalytic Asymmetric Robinson Annulation of α,β-Unsaturated Aldehydes: Applications to the Total Synthesis of (+)-Palitantin. J. Org. Chem. 2007, 72, 8459–8471. 10.1021/jo701477v. [DOI] [PubMed] [Google Scholar]
- Orue A.; Reyes E.; Vicario J. L.; Carrillo L.; Uria U. Enantio- and Diastereoselective Synthesis of Substituted Tetrahydro-1H-Isochromanes through a Dynamic Kinetic Resolution Proceeding under Dienamine Catalysis. Org. Lett. 2012, 14, 3740–3743. 10.1021/ol301602h. [DOI] [PubMed] [Google Scholar]
- Johansen T. K.; Gõmez C. V.; Bak J. R.; Davis R. L.; Jørgensen K. A. Organocatalytic Enantioselective Cycloaddition Reactions of Dienamines with Quinones. Chem. - Eur. J. 2013, 19, 16518–16522. 10.1002/chem.201303526. [DOI] [PubMed] [Google Scholar]
- Zhou X.-L.; Wang P.-S.; Zhang D.-W.; Liu P.; Wang C.-M.; Gong L.-Z. Enantioselective Functionalization of Inactive sp3 C–H Bonds Remote to Functional Group by Metal/Organo Cooperative Catalysis. Org. Lett. 2015, 17, 5120–5123. 10.1021/acs.orglett.5b02653. [DOI] [PubMed] [Google Scholar]
- Wang Z. Y.; Wong W.-T.; Yang D. Organocatalyzed Asymmetric Synthesis of Dihydrodibenzofurans Based on a Dienamine Process. Org. Lett. 2013, 15, 4980–4983. 10.1021/ol402288y. [DOI] [PubMed] [Google Scholar]
- Song A.; Zhang X.; Song X.; Chen X.; Yu C.; Huang H.; Li H.; Wang W. Construction of Chiral Bridged Tricyclic Benzopyrans: Enantioselective Catalytic Diels-Alder Reaction and a One-Pot Reduction/Acid-Catalyzed Stereoselective Cyclization. Angew. Chem., Int. Ed. 2014, 53, 4940–4944. 10.1002/anie.201402170. [DOI] [PubMed] [Google Scholar]
- Albrecht A.; Bojanowski J. Decarboxylative Aminocatalytic Cascade for the Synthesis of Dihydroxanthones. Adv. Synth. Catal. 2017, 359, 2907–2911. 10.1002/adsc.201700400. [DOI] [Google Scholar]
- Martín-Santos C.; Jarava-Barrera C.; Del Pozo S.; Parra A.; Díaz-Tendero S.; Mas-Ballesté R.; Cabrera S.; Alemán J. Highly Enantioselective Construction of Tricyclic Derivatives by the Desymmetrization of Cyclohexadienones. Angew. Chem., Int. Ed. 2014, 53, 8184–8189. 10.1002/anie.201402853. [DOI] [PubMed] [Google Scholar]
- Peña J.; Moro R. F.; Marcos I. S.; Sanz F.; Díez D. Highly Functionalised Cyclohexa-1,3-Dienes by Sulfonyl Nazarov Reagents. Tetrahedron 2014, 70, 4386–4394. 10.1016/j.tet.2014.04.076. [DOI] [Google Scholar]
- Xiao W.; Yin X.; Zhou Z.; Du W.; Chen Y.-C. Asymmetric α,γ-Regioselective [3 + 3] Formal Cycloadditions of α,β-Unsaturated Aldehydes via Cascade Dienamine–Dienamine Catalysis. Org. Lett. 2016, 18, 116–119. 10.1021/acs.orglett.5b03355. [DOI] [PubMed] [Google Scholar]
- Xie J.-K.; Wang Y.; Lin J.-B.; Ren X.-R.; Xu P.-F. Direct Noncovalent Activation of α,β-Unsaturated Aldehydes for the Stereodivergent Synthesis of Substituted Cyclohexenes. Chem. - Eur. J. 2017, 23, 6752–6756. 10.1002/chem.201701315. [DOI] [PubMed] [Google Scholar]
- The absolute configurations of stereocenters of this catalyst are those reported in the original paper, though some of them do not seem correct, given the impossibility to obtain this catalyst from the natural alkaloid sources.
- Jia Z.-J.; Jiang H.; Li J.-L.; Gschwend B.; Li Q.-Z.; Yin X.; Grouleff J.; Chen Y.-C.; Jørgensen K. A. Trienamines in Asymmetric Organocatalysis: Diels-Alder and Tandem Reactions. J. Am. Chem. Soc. 2011, 133, 5053–5061. 10.1021/ja1112194. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Gschwend B.; Albrecht Ł.; Hansen S. G.; Jørgensen K. A. Asymmetric Trienamine Catalysis for the Construction of Structurally Rigid Cyclic α,α-Disubstituted Amino Acid Derivatives. Chem. - Eur. J. 2011, 17, 9032–9036. 10.1002/chem.201101539. [DOI] [PubMed] [Google Scholar]
- Jia Z.-J.; Zhou Q.; Zhou Q.-Q.; Chen P.-Q.; Chen Y.-C. Exo-Selective Asymmetric Diels-Alder Reaction of 2,4-Dienals and Nitroalkenes by Trienamine Catalysis. Angew. Chem., Int. Ed. 2011, 50, 8638–8641. 10.1002/anie.201102013. [DOI] [PubMed] [Google Scholar]
- Prieto L.; Talavera G.; Uria U.; Reyes E.; Vicario J. L.; Carrillo L. Favoring Trienamine Activation through Unconjugated Dienals: Organocatalytic Enantioselective Remote Functionalization of Alkenes. Chem. - Eur. J. 2014, 20, 2145–2148. 10.1002/chem.201304666. [DOI] [PubMed] [Google Scholar]
- Zhu K.; Huang H.; Wu W.; Wei Y.; Ye J. Aminocatalyzed Asymmetric Diels–Alder Reaction of 2,4-Dienals and Rhodanine/Hydantoin Derivatives. Chem. Commun. 2013, 49, 2157–2159. 10.1039/c3cc00023k. [DOI] [PubMed] [Google Scholar]
- Zhang S.-J.; Zhang J.; Zhou Q.-Q.; Dong L.; Chen Y.-C. Aminocatalytic Asymmetric Exo-Diels-Alder Reaction with Methiodide Salts of Mannich Bases and 2,4-Dienals to Construct Chiral Spirocycles. Org. Lett. 2013, 15, 968–971. 10.1021/ol4002015. [DOI] [PubMed] [Google Scholar]
- Zhou Q.-Q.; Yuan X.; Xiao Y.-C.; Dong L.; Chen Y.-C. Aminocatalytic Asymmetric Diels-Alder Reaction of Phosphorus Dienophiles and 2,4-Dienals. Tetrahedron 2013, 69, 10369–10374. 10.1016/j.tet.2013.10.001. [DOI] [Google Scholar]
- Ma C.; Jia Z. J.; Liu J. X.; Zhou Q. Q.; Dong L.; Chen Y. C. A Concise Assembly of Electron-Deficient 2,4-Dienes and 2,4-Dienals: Regio- and Stereoselective Exo-Diels-Alder and Redox Reactions through Sequential Amine and Carbene Catalysis. Angew. Chem., Int. Ed. 2013, 52, 948–951. 10.1002/anie.201208349. [DOI] [PubMed] [Google Scholar]
- Ma C.; Gu J.; Teng B.; Zhou Q.-Q.; Li R.; Chen Y.-C. 1-Azadienes as Regio- and Chemoselective Dienophiles in Aminocatalytic Asymmetric Diels-Alder Reaction. Org. Lett. 2013, 15, 6206–6209. 10.1021/ol4030474. [DOI] [PubMed] [Google Scholar]
- Li X.; Lin M.-H.; Han Y.; Wang F.; Cheng J.-P. Asymmetric Diels-Alder Reaction of 3-Olefinic Benzofuran-2-Ones and Polyenals: Construction of Chiral Spirocyclic Benzofuran-2-Ones. Org. Lett. 2014, 16, 114–117. 10.1021/ol403094a. [DOI] [PubMed] [Google Scholar]
- Portalier F.; Bourdreux F.; Marrot J.; Moreau X.; Coeffard V.; Greck C. Merging Oxidative Dearomatization and Aminocatalysis: One-Pot Enantioselective Synthesis of Tricyclic Architectures. Org. Lett. 2013, 15, 5642–5645. 10.1021/ol402546h. [DOI] [PubMed] [Google Scholar]
- Li Y.; López-Delgado F. J.; Jørgensen D. K. B.; Nielsen R. P.; Jiang H.; Jørgensen K. A. Trienamine-Mediated Asymmetric [4 + 2]-Cycloaddition of α,β-Unsaturated Ester Surrogates Applying 4-Nitro-5-Styrylisoxazoles. Chem. Commun. 2014, 50, 15689–15691. 10.1039/C4CC08171D. [DOI] [PubMed] [Google Scholar]
- Hejmanowska J.; Dziȩgielewski M.; Kowalczyk D.; Albrecht Ł. Organocatalytic Enantioselective Approach to Spirocyclic Δβ,γ-Butenolides. Synlett 2014, 25, 2957–2961. 10.1055/s-0034-1378905. [DOI] [Google Scholar]
- Jia Z.-J.; Jiang K.; Zhou Q.-Q.; Dong L.; Chen Y.-C. Amine-N-Heterocyclic Carbene Cascade Catalysis for the Asymmetric Synthesis of Fused Indane Derivatives with Multiple Chiral Centres. Chem. Commun. 2013, 49, 5892–5894. 10.1039/c3cc42823k. [DOI] [PubMed] [Google Scholar]
- Cruz Cruz D.; Mose R.; Villegas Gómez C.; Torbensen S. V.; Larsen M. S.; Jørgensen K. A. Organocatalytic Cascade Reactions: Towards the Diversification of Hydroisochromenes and Chromenes through Two Different Activation Modes. Chem. - Eur. J. 2014, 20, 11331–11335. 10.1002/chem.201403505. [DOI] [PubMed] [Google Scholar]
- Yuan X.; Zhang S.-J.; Du W.; Chen Y.-C. Asymmetric Diels-Alder Cycloadditions of Trifluoromethylated Dienophiles Under Trienamine Catalysis. Chem. - Eur. J. 2016, 22, 11048–11052. 10.1002/chem.201600989. [DOI] [PubMed] [Google Scholar]
- Gómez C. V.; Cruz D. C.; Mose R.; Jørgensen K. A. Organocatalytic Cascade Reactions: Diversity-Oriented Synthesis for the Construction of Hydroisoquinoline Scaffolds. Chem. Commun. 2014, 50, 6035–6038. 10.1039/C4CC01231C. [DOI] [PubMed] [Google Scholar]
- Gu J.; Xiao B.-X.; Chen Y.-R.; Du W.; Chen Y.-C. Asymmetric Diels-Alder and Cascade Reaction of Quinone Imine Ketals and 2,4-Dienals: Construction of Chiral Benzo[de]Quinolone Derivatives. Adv. Synth. Catal. 2016, 358, 296–302. 10.1002/adsc.201500860. [DOI] [Google Scholar]
- Sellstedt M.; Schwalfenberg M.; Ziegler S.; Antonchick A. P.; Waldmann H. Trienamine Catalyzed Asymmetric Synthesis and Biological Investigation of a Cytochalasin B-Inspired Compound Collection. Org. Biomol. Chem. 2016, 14, 50–54. 10.1039/C5OB02272J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donslund B. S.; Nielsen R. P.; Mønsted S. M. N.; Jørgensen K. A. Benzofulvenes in Trienamine Catalysis: Stereoselective Spiroindene Synthesis. Angew. Chem., Int. Ed. 2016, 55, 11124–11128. 10.1002/anie.201605079. [DOI] [PubMed] [Google Scholar]
- Albrecht A.; Skrzyńska A.; Pietrzak A.; Bojanowski J.; Albrecht Ł. Asymmetric Aminocatalysis in the Synthesis of δ-Lactone Derivatives. Asian J. Org. Chem. 2016, 5, 1115–1119. 10.1002/ajoc.201600272. [DOI] [Google Scholar]
- Skrzyńska A.; Drelich P.; Frankowski S.; Albrecht Ł. Synthesis of γ,γ-Disubstituted Butenolides through a Doubly Vinylogous Organocatalytic Cycloaddition. Chem. - Eur. J. 2018, 24, 16543–16547. 10.1002/chem.201804650. [DOI] [PubMed] [Google Scholar]
- Albrecht Ł.; Cruz Acosta F.; Fraile A.; Albrecht A.; Christensen J.; Jørgensen K. A. Enantioselective H-Bond-Directing Approach for Trienamine-Mediated Reactions in Asymmetric Synthesis. Angew. Chem., Int. Ed. 2012, 51, 9088–9092. 10.1002/anie.201204790. [DOI] [PubMed] [Google Scholar]
- Monleõn A.; Glaus F.; Vergura S.; Jørgensen K. A. Organocatalytic Strategy for the Enantioselective Cycloaddition to Trisubstituted Nitroolefins to Create Spirocyclohexene-Oxetane Scaffolds. Angew. Chem., Int. Ed. 2016, 55, 2478–2482. 10.1002/anie.201510731. [DOI] [PubMed] [Google Scholar]
- Li Y.; Tur F.; Nielsen R. P.; Jiang H.; Jensen F.; Jørgensen K. A. Enantioselective Formal [4 + 2] Cycloadditions to 3-Nitroindoles by Trienamine Catalysis: Synthesis of Chiral Dihydrocarbazoles. Angew. Chem., Int. Ed. 2016, 55, 1020–1024. 10.1002/anie.201509693. [DOI] [PubMed] [Google Scholar]
- Zhou Q.-Q.; Xiao Y. C.; Yuan X.; Chen Y.-C. Asymmetric Diels-Alder Reactions of 2,4,6-Trienals via Tetraenamine Catalysis. Asian J. Org. Chem. 2014, 3, 545–549. 10.1002/ajoc.201400015. [DOI] [Google Scholar]
- Dieckmann A.; Breugst M.; Houk K. N. Zwitterions and Unobserved Intermediates in Organocatalytic Diels-Alder Reactions of Linear and Cross-Conjugated Trienamines. J. Am. Chem. Soc. 2013, 135, 3237–3242. 10.1021/ja312043g. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Nappi M.; Arceo E.; Vera S.; Melchiorre P. Asymmetric Catalysis of Diels–Alder Reactions with in Situ Generated Heterocyclic Ortho-Quinodimethanes. J. Am. Chem. Soc. 2011, 133, 15212–15218. 10.1021/ja206517s. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Nappi M.; Escudero-Adán E. C.; Melchiorre P. Multicatalytic Asymmetric Synthesis of Complex Tetrahydrocarbazoles via a Diels-Alder/Benzoin Reaction Sequence. Org. Lett. 2012, 14, 1310–1313. 10.1021/ol300192p. [DOI] [PubMed] [Google Scholar]
- Skrzyńska A.; Przydacz A.; Albrecht Ł. Organocatalytic Nonclassical Trienamine Activation in the Remote Alkylation of Furan Derivatives. Org. Lett. 2015, 17, 5682–5685. 10.1021/acs.orglett.5b02979. [DOI] [PubMed] [Google Scholar]
- Akutsu H.; Nakashima K.; Hirashima S.; Kitahara M.; Koseki Y.; Miura T. Asymmetric Conjugate Addition of 5-Benzylfurfurals to Nitroalkenes Using a Diaminomethylenemalononitrile Organocatalyst. Tetrahedron Lett. 2017, 58, 4759–4762. 10.1016/j.tetlet.2017.10.078. [DOI] [Google Scholar]
- He X.-L.; Zhao H.-R.; Duan C.-Q.; Du W.; Chen Y.-C. Remote Asymmetric Oxa-Diels–Alder Reaction of 5-Allylic Furfurals via Dearomatizative Tetraenamine Catalysis. Org. Lett. 2018, 20, 804–807. 10.1021/acs.orglett.7b03942. [DOI] [PubMed] [Google Scholar]
- Li X.; Wang S.; Li T.; Li J.; Li H.; Wang W. Formation of Dihydronaphthalenes via Organocatalytic Enatioselective Michael-Aldol Cascade Reactions with Arylalkanes. Org. Lett. 2013, 15, 5634–5637. 10.1021/ol402489e. [DOI] [PubMed] [Google Scholar]
- Enders D.; Hahn R.; Atodiresei I. Asymmetric Synthesis of Functionalized Tetrahydronaphthalenes via an Organocatalytic Nitroalkane-Michael/Henry Domino Reaction. Adv. Synth. Catal. 2013, 355, 1126–1136. 10.1002/adsc.201300039. [DOI] [Google Scholar]
- He X.-L.; Zhao H.-R.; Duan C.-Q.; Han X.; Du W.; Chen Y.-C. Asymmetric Benzylic Allylic Alkylation Reaction of 3-Furfural Derivatives by Dearomatizative Dienamine Activation. Chem. - Eur. J. 2018, 24, 6277–6281. 10.1002/chem.201800585. [DOI] [PubMed] [Google Scholar]
- Duan C.-Q.; He X.-L.; Du W.; Chen Y.-C. Asymmetric [4 + 2] Cycloadditions with 3-Furfural Derivatives and α-Cyano-α,β-Unsaturated Ketones. Org. Chem. Front. 2018, 5, 2057–2060. 10.1039/C8QO00351C. [DOI] [Google Scholar]
- Wang Y.; Lin J.-B.; Xie J.-K.; Lu H.; Hu X.-Q.; Xu P.-F. Dearomative Dienolate-Mediated Catalysis: A Remote Activation Strategy for Asymmetric Functionalization of Benzylic C–H Bonds of Heteroaryl Aldehydes. Org. Lett. 2018, 20, 5835–5839. 10.1021/acs.orglett.8b02523. [DOI] [PubMed] [Google Scholar]
- Hiltebrandt K.; Elies K.; D’hooge D. R.; Blinco J. P.; Barner-Kowollik C. A Light-Activated Reaction Manifold. J. Am. Chem. Soc. 2016, 138, 7048–7054. 10.1021/jacs.6b01805. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Rodríguez-Escrich C.; Johansen T. K.; Davis R. L.; Jørgensen K. A. Organocatalytic Activation of Polycyclic Aromatic Compounds for Asymmetric Diels-Alder Reactions. Angew. Chem., Int. Ed. 2012, 51, 10271–10274. 10.1002/anie.201205836. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Escrich C.; Davis R. L.; Jiang H.; Stiller J.; Johansen T. K.; Jørgensen K. A. Breaking Symmetry with Symmetry: Bifacial Selectivity in the Asymmetric Cycloaddition of Anthracene Derivatives. Chem. - Eur. J. 2013, 19, 2932–2936. 10.1002/chem.201300142. [DOI] [PubMed] [Google Scholar]
- Halskov K. S.; Johansen T. K.; Davis R. L.; Steurer M.; Jensen F.; Jørgensen K. A. Cross-Trienamines in Asymmetric Organocatalysis. J. Am. Chem. Soc. 2012, 134, 12943–12946. 10.1021/ja3068269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donslund B. S.; Halskov K. S.; Leth L. A.; Paz B. M.; Jørgensen K. A. Organocatalytic Asymmetric Strategies to Carbocyclic Structures by γ-Alkylation-Annulation Sequences. Chem. Commun. 2014, 50, 13676–13679. 10.1039/C4CC06556E. [DOI] [PubMed] [Google Scholar]
- Halskov K. S.; Donslund B. S.; Barfüsser S.; Jørgensen K. A. Organocatalytic Asymmetric Formation of Steroids. Angew. Chem., Int. Ed. 2014, 53, 4137–4141. 10.1002/anie.201400203. [DOI] [PubMed] [Google Scholar]
- Blom J.; Johansen T. K.; Jensen F.; Jørgensen K. A. Dynamic Resolution of 2-Cyclohexylidene Acetaldehydes through Organocatalytic Dienamine [4 + 2] Cycloaddition. Chem. Commun. 2016, 52, 7153–7156. 10.1039/C6CC02019D. [DOI] [PubMed] [Google Scholar]
- Stiller J.; Poulsen P. H.; Cruz D. C.; Dourado J.; Davis R. L.; Jørgensen K. A. Organocatalytic [4 + 2] Addition Reactions via Tetraenamine Intermediate. Chem. Sci. 2014, 5, 2052–2056. 10.1039/c4sc00081a. [DOI] [Google Scholar]
- Chintalapudi V.; Galvin E. A.; Greenaway R. L.; Anderson E. A Combining Cycloisomerization with Trienamine Catalysis: A Regiochemically Flexible Enantio- and Diastereoselective Synthesis of Hexahydroindoles. Chem. Commun. 2016, 52, 693–696. 10.1039/C5CC08886K. [DOI] [PubMed] [Google Scholar]
- For a brief, yet punctual overview on this subject, see:Palazzo T. A.; Mose R.; Jørgensen K. A. Cycloaddition Reactions: Why Is It So Challenging To Move from Six to Ten Electrons?. Angew. Chem., Int. Ed. 2017, 56, 10033–10038. 10.1002/anie.201701085. [DOI] [PubMed] [Google Scholar]
- Mose R.; Preegel G.; Larsen J.; Jakobsen S.; Iversen E. H.; Jørgensen K. A. Organocatalytic Stereoselective [8 + 2] and [6 + 4] Cycloadditions. Nat. Chem. 2017, 9, 487–492. 10.1038/nchem.2682. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Wang Z.-X.; Zhou Y.-C.; Xiao W.; Ouyang Q.; Du W.; Chen Y.-C. Switchable Regioselectivity in Amine-Catalysed Asymmetric Cycloadditions. Nat. Chem. 2017, 9, 590–594. 10.1038/nchem.2698. [DOI] [PubMed] [Google Scholar]
- Hayashi Y.; Gotoh H.; Honma M.; Sankar K.; Kumar I.; Ishikawa H.; Konno K.; Yui H.; Tsuzuki S.; Uchimaru T. Organocatalytic, Enantioselective Intramolecular [6 + 2] Cycloaddition Reaction for the Formation of Tricyclopentanoids and Insight on Its Mechanism from a Computational Study. J. Am. Chem. Soc. 2011, 133, 20175–20185. 10.1021/ja108516b. [DOI] [PubMed] [Google Scholar]
- Donslund B. S.; Monleón A.; Palazzo T. A.; Christensen M. L.; Dahlgaard A.; Erickson J. D.; Jørgensen K. A. Organocatalytic Enantioselective Higher-Order Cycloadditions of In Situ Generated Amino Isobenzofulvenes. Angew. Chem., Int. Ed. 2018, 57, 1246–1250. 10.1002/anie.201710694. [DOI] [PubMed] [Google Scholar]
- Donslund B. S.; Jessen N. I.; Bertuzzi G.; Giardinetti M.; Palazzo T. A.; Christensen M. L.; Jørgensen K. A. Catalytic Enantioselective [10 + 4] Cycloadditions. Angew. Chem., Int. Ed. 2018, 57, 13182–13186. 10.1002/anie.201807830. [DOI] [PubMed] [Google Scholar]
- Bertelsen S.; Marigo M.; Brandes S.; Dinér P.; Jørgensen K. A. Dienamine Catalysis: Organocatalytic Asymmetric γ-Amination of α,β-Unsaturated Aldehydes. J. Am. Chem. Soc. 2006, 128, 12973–12980. 10.1021/ja064637f. [DOI] [PubMed] [Google Scholar]
- Bergonzini G.; Vera S.; Melchiorre P. Cooperative Organocatalysis for the Asymmetric γ Alkylation of α-Branched Enals. Angew. Chem., Int. Ed. 2010, 49, 9685–9688. 10.1002/anie.201004761. [DOI] [PubMed] [Google Scholar]
- Stiller J.; Marqués-López E.; Herrera R. P.; Fröhlich R.; Strohmann C.; Christmann M. Enantioselective α- and γ-Alkylation of α,β-Unsaturated Aldehydes Using Dienamine Activation. Org. Lett. 2011, 13, 70–73. 10.1021/ol102559f. [DOI] [PubMed] [Google Scholar]
- Ikeda M.; Miyake Y.; Nishibayashi Y. Cooperative Catalytic Reactions Using Organocatalysts and Transition Metal Catalysts: Propargylic Allylation of Propargylic Alcohols with α,β-Unsaturated Aldehydes. Organometallics 2012, 31, 3810–3813. 10.1021/om300286b. [DOI] [Google Scholar]
- César da Silva R.; Chatterjee I.; Escudero-Adán E.; Weber Paixão M.; Melchiorre P. Synthesis of Cyclopropane Spirooxindoles by Means of a Vinylogous Organocatalytic Cascade. Asian J. Org. Chem. 2014, 3, 466–469. 10.1002/ajoc.201400014. [DOI] [Google Scholar]
- Yang W.; Ma D.; Zhou Y.; Dong X.; Lin Z.; Sun J. NHC-Catalyzed Electrophilic Trifluoromethylation: Efficient Synthesis of γ-Trifluoromethyl α,β-Unsaturated Esters. Angew. Chem., Int. Ed. 2018, 57, 12097–12101. 10.1002/anie.201806674. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Cruz D. C.; Li Y.; Lauridsen V. H.; Jørgensen K. A. Asymmetric Organocatalytic Thio-Diels-Alder Reactions via Trienamine Catalysis. J. Am. Chem. Soc. 2013, 135, 5200–5207. 10.1021/ja4007244. [DOI] [PubMed] [Google Scholar]
- Hejmanowska J.; Jasiński M.; Mlostoń G.; Albrecht Ł. Taming of Thioketones: The First Asymmetric Thia-Diels–Alder Reaction with Thioketones as Heterodienophiles. Eur. J. Org. Chem. 2017, 2017, 950–954. 10.1002/ejoc.201601307. [DOI] [Google Scholar]
- Hejmanowska J.; Jasiński M.; Wojciechowski J.; Mlostoń G.; Albrecht Ł. The First Organocatalytic, Ortho-Regioselective Inverse-Electron-Demand Hetero-Diels-Alder Reaction. Chem. Commun. 2017, 53, 11472–11475. 10.1039/C7CC06518C. [DOI] [PubMed] [Google Scholar]
- Chen X. Y.; Xia F.; Cheng J. T.; Ye S. Highly Enantioselective γ-Amination by N-Heterocyclic Carbene Catalyzed [4 + 2] Annulation of Oxidized Enals and Azodicarboxylates. Angew. Chem., Int. Ed. 2013, 52, 10644–10647. 10.1002/anie.201305571. [DOI] [PubMed] [Google Scholar]
- Ikota H.; Ishida T.; Tsukano C.; Takemoto Y. Synthesis of 3,3-Disubstituted Indoline-2-Thiones Catalysed by an N-Heterocyclic Carbene. Chem. Commun. 2014, 50, 8871–8874. 10.1039/C4CC04047C. [DOI] [PubMed] [Google Scholar]
- Han R.; Qi J.; Gu J.; Ma D.; Xie X.; She X. N-Heterocyclic Carbene-Catalyzed Formal [3 + 2] Annulation of Alkynyl Aldehydes with Nitrosobenzenes: A Highly Regioselective Umpolung Strategy. ACS Catal. 2013, 3, 2705–2709. 10.1021/cs400602v. [DOI] [Google Scholar]
- Chen J.; Yuan P.; Wang L.; Huang Y. Enantioselective β-Protonation of Enals via a Shuttling Strategy. J. Am. Chem. Soc. 2017, 139, 7045–7051. 10.1021/jacs.7b02889. [DOI] [PubMed] [Google Scholar]
- Wang M. H.; Cohen D. T.; Schwamb C. B.; Mishra R. K.; Scheidt K. A. Enantioselective β-Protonation by a Cooperative Catalysis Strategy. J. Am. Chem. Soc. 2015, 137, 5891–5894. 10.1021/jacs.5b02887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. H.; Barsoum D.; Schwamb C. B.; Cohen D. T.; Goess B. C.; Riedrich M.; Chan A.; Maki B. E.; Mishra R. K.; Scheidt K. A. Catalytic, Enantioselective β-Protonation through a Cooperative Activation Strategy. J. Org. Chem. 2017, 82, 4689–4702. 10.1021/acs.joc.7b00334. [DOI] [PubMed] [Google Scholar]
- Bencivenni G.; Galzerano P.; Mazzanti A.; Bartoli G.; Melchiorre P. Direct Asymmetric Vinylogous Michael Addition of Cyclic Enones to Nitroalkenes via Dienamine Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20642–20647. 10.1073/pnas.1001150107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Corrigendum:Bencivenni G.; Galzerano P.; Mazzanti A.; Bartoli G.; Melchiorre P. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4852. 10.1073/pnas.1302753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halskov K. S.; Naicker T.; Jensen M. E.; Jørgensen K. A. Organocatalytic Asymmetric Remote Aziridination of 2,4-Dienals. Chem. Commun. 2013, 49, 6382–6384. 10.1039/c3cc43506g. [DOI] [PubMed] [Google Scholar]
- Næsborg L.; Halskov K. S.; Tur F.; Mønsted S. M. N.; Jørgensen K. A. Asymmetric γ-Allylation of α,β-Unsaturated Aldehydes by Combined Organocatalysis and Transition-Metal Catalysis. Angew. Chem., Int. Ed. 2015, 54, 10193–10197. 10.1002/anie.201504749. [DOI] [PubMed] [Google Scholar]
- Afewerki S.; Córdova A. Combinations of Aminocatalysts and Metal Catalysts: A Powerful Cooperative Approach in Selective Organic Synthesis. Chem. Rev. 2016, 116, 13512–13570. 10.1021/acs.chemrev.6b00226. [DOI] [PubMed] [Google Scholar]
- Su Y. L.; Han Z. Y.; Li Y. H.; Gong L. Z. Asymmetric Allylation of Furfural Derivatives: Synergistic Effect of Chiral Ligand and Organocatalyst on Stereochemical Control. ACS Catal. 2017, 7, 7917–7922. 10.1021/acscatal.7b02667. [DOI] [Google Scholar]
- Fleming I.Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons, Ltd: Chichester, UK, 2010. [Google Scholar]
- Iriarte I.; Olaizola O.; Vera S.; Gamboa I.; Oiarbide M.; Palomo C. Controlling the α/γ-Reactivity of Vinylogous Ketone Enolates in Organocatalytic Enantioselective Michael Reactions. Angew. Chem., Int. Ed. 2017, 56, 8860–8864. 10.1002/anie.201703764. [DOI] [PubMed] [Google Scholar]
- Qiao B.; Huang Y. J.; Nie J.; Ma J. A. Highly Regio-, Diastereo-, and Enantioselective Mannich Reaction of Allylic Ketones and Cyclic Ketimines: Access to Chiral Benzosultam. Org. Lett. 2015, 17, 4608–4611. 10.1021/acs.orglett.5b02351. [DOI] [PubMed] [Google Scholar]
- Tong G.; Zhu B.; Lee R.; Yang W.; Tan D.; Yang C.; Han Z.; Yan L.; Huang K. W.; Jiang Z. Highly Enantio- and Diastereoselective Allylic Alkylation of Morita-Baylis-Hillman Carbonates with Allyl Ketones. J. Org. Chem. 2013, 78, 5067–5072. 10.1021/jo400496z. [DOI] [PubMed] [Google Scholar]
- Pierce M. D.; Johnston R. C.; Mahapatra S.; Yang H.; Carter R. G.; Cheong P. H. Y. Mechanism and Stereoselectivity of a Dual Amino-Catalyzed Robinson Annulation: Rare Duumvirate Stereocontrol. J. Am. Chem. Soc. 2012, 134, 13624–13631. 10.1021/ja3018219. [DOI] [PubMed] [Google Scholar]
- Bencivenni G.; Wu L. Y.; Mazzanti A.; Giannichi B.; Pesciaioli F.; Song M. P.; Bartoli G.; Melchiorre P. Targeting Structural and Stereochemical Complexity by Organocascade Catalysis: Construction of Spirocyclic Oxindoles Having Multiple Stereocenters. Angew. Chem., Int. Ed. 2009, 48, 7200–7203. 10.1002/anie.200903192. [DOI] [PubMed] [Google Scholar]
- Yazaki R.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Addition of Allyl Cyanide to Ketones. J. Am. Chem. Soc. 2009, 131, 3195–3197. 10.1021/ja900001u. [DOI] [PubMed] [Google Scholar]
- Yazaki R.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Addition of Allyl Cyanide to Ketones via Soft Lewis Acid/Hard Brønsted Base/Hard Lewis Base Catalysis. J. Am. Chem. Soc. 2010, 132, 5522–5531. 10.1021/ja101687p. [DOI] [PubMed] [Google Scholar]
- Otsuka Y.; Takada H.; Yasuda S.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Addition of Allylic Cyanides to Aldehydes for Expeditious Access to Enantioenriched Unsaturated δ-Valerolactones. Chem. - Asian J. 2013, 8, 354–358. 10.1002/asia.201201021. [DOI] [PubMed] [Google Scholar]
- Zhu B.; Zhang W.; Lee R.; Han Z.; Yang W.; Tan D.; Huang K. W.; Jiang Z. Direct Asymmetric Vinylogous Aldol Reaction of Allyl Ketones with Isatins: Divergent Synthesis of 3-Hydroxy-2-Oxindole Derivatives. Angew. Chem., Int. Ed. 2013, 52, 6666–6670. 10.1002/anie.201302274. [DOI] [PubMed] [Google Scholar]
- Jing Z.; Bai X.; Chen W.; Zhang G.; Zhu B.; Jiang Z. Organocatalytic Enantioselective Vinylogous Aldol Reaction of Allyl Aryl Ketones to Activated Acyclic Ketones. Org. Lett. 2016, 18, 260–263. 10.1021/acs.orglett.5b03412. [DOI] [PubMed] [Google Scholar]
- Han M. Y.; Luan W. Y.; Mai P. L.; Li P.; Wang L. Organocatalytic Asymmetric Vinylogous Aldol Reaction of Allyl Aryl Ketones to Silyl Glyoxylates. J. Org. Chem. 2018, 83, 1518–1524. 10.1021/acs.joc.7b02546. [DOI] [PubMed] [Google Scholar]
- Ray B.; Mukherjee S. Direct Catalytic Enantioselective Vinylogous Aldol Reaction of Allyl Ketones to Pyrazole-4,5-diones. J. Org. Chem. 2018, 83, 10871–10880. 10.1021/acs.joc.8b01566. [DOI] [PubMed] [Google Scholar]
- Bastida D.; Liu Y.; Tian X.; Escudero-Adán E.; Melchiorre P. Asymmetric Vinylogous Aldol Reaction via H-Bond-Directing Dienamine Catalysis. Org. Lett. 2013, 15, 220–223. 10.1021/ol303312p. [DOI] [PubMed] [Google Scholar]
- Tian X.; Melchiorre P. Control of Remote Stereochemistry in the Synthesis of Spirocyclic Oxindoles: Vinylogous Organocascade Catalysis. Angew. Chem., Int. Ed. 2013, 52, 5360–5363. 10.1002/anie.201301017. [DOI] [PubMed] [Google Scholar]
- Cuadros S.; Dell’Amico L.; Melchiorre P. Forging Fluorine-Containing Quaternary Stereocenters by a Light-Driven Organocatalytic Aldol Desymmetrization Process. Angew. Chem., Int. Ed. 2017, 56, 11875–11879. 10.1002/anie.201706763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaiano J. C. Laser Flash Photolysis Studies of the Reactions of Some 1,4-Biradicals. Acc. Chem. Res. 1982, 15, 252–258. 10.1021/ar00080a004. [DOI] [Google Scholar]
- Jung M. E.; Guzaev M. Studies toward the Enantiospecific Total Synthesis of Rhodexin A. J. Org. Chem. 2013, 78, 7518–7526. 10.1021/jo400909t. [DOI] [PubMed] [Google Scholar]
- Onyango E. O.; Jacobi P. A. Synthetic Studies on Furanosteroids: Construction of the Viridin Core Structure via Diels-Alder/retro-Diels-Alder and Vinylogous Mukaiyama Aldol-Type Reaction. J. Org. Chem. 2012, 77, 7411–7427. 10.1021/jo301232w. [DOI] [PubMed] [Google Scholar]
- Li B. S.; Wang Y.; Jin Z.; Zheng P.; Ganguly R.; Chi Y. R. Carbon-Carbon Bond Activation of Cyclobutenones Enabled by the Addition of Chiral Organocatalyst to Ketone. Nat. Commun. 2015, 6, 1–5. 10.1038/ncomms7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepburn H. B.; Magagnano G.; Melchiorre P. Light-Triggered Enantioselective Organocatalytic Mannich-Type Reaction. Synthesis 2016, 49, 76–86. 10.1055/s-0036-1588606. [DOI] [Google Scholar]
- Yang Y. Building Polyfunctional Piperidines: A Stereoselective Strategy of a Three-Component Mannich Reaction Inspired by Biosynthesis and Applications in the Synthesis of Natural Alkaloids (+)-241D; (−)-241D; Isosolenopsin A and (−)-Epimyrtine. RSC Adv. 2015, 5, 18894–18908. 10.1039/C4RA14418J. [DOI] [Google Scholar]
- Xiong X.-F.; Zhou Q.; Gu J.; Dong L.; Liu T.-Y.; Chen Y.-C. Trienamine Catalysis with 2,4-Dienones: Development and Application in Asymmetric Diels-Alder Reactions. Angew. Chem., Int. Ed. 2012, 51, 4401–4404. 10.1002/anie.201200248. [DOI] [PubMed] [Google Scholar]
- Chen P.-Q.; Xiao Y.-C.; Yue C.-Z.; Chen Y.-C. Trienamine Catalysis with Linear Deconjugated 3,5-Dienones. Org. Chem. Front. 2014, 1, 490–493. 10.1039/C4QO00079J. [DOI] [Google Scholar]
- Feng X.; Zhou Z.; Ma C.; Yin X.; Li R.; Dong L.; Chen Y. C. Trienamines Derived from Interrupted Cyclic 2,5-Dienones: Remote δ,ε-C = C Bond Activation for Asymmetric Inverse-Electron-Demand Aza-Diels-Alder Reaction. Angew. Chem., Int. Ed. 2013, 52, 14173–14176. 10.1002/anie.201307460. [DOI] [PubMed] [Google Scholar]
- Zhan G.; He Q.; Yuan X.; Chen Y. C. Asymmetric Direct Vinylogous Michael Additions of Allyl Alkyl Ketones to Maleimides through Dienamine Catalysis. Org. Lett. 2014, 16, 6000–6003. 10.1021/ol503017h. [DOI] [PubMed] [Google Scholar]
- Ramachary D. B.; Chowdari N. S.; Barbas C. F. Organocatalytic Asymmetric Domino Knoevenagel/Diels-Alder Reactions: A Bioorganic Approach to the Diastereospecific and Enantioselective Construction of Highly Substituted Spiro [5,5]Undecane-1,5,9-Triones. Angew. Chem., Int. Ed. 2003, 42, 4233–4237. 10.1002/anie.200351916. [DOI] [PubMed] [Google Scholar]
- Wu L. Y.; Bencivenni G.; Mancinelli M.; Mazzanti A.; Bartoli G.; Melchiorre P. Organocascade Reactions of Enones Catalyzed by a Chiral Primary Amine. Angew. Chem., Int. Ed. 2009, 48, 7196–7199. 10.1002/anie.200903280. [DOI] [PubMed] [Google Scholar]
- Feng X.; Zhou Z.; Zhou R.; Zhou Q. Q.; Dong L.; Chen Y. C. Stereodivergence in Amine-Catalyzed Regioselective [4 + 2] Cycloadditions of β-Substituted Cyclic Enones and Polyconjugated Malononitriles. J. Am. Chem. Soc. 2012, 134, 19942–19947. 10.1021/ja3106219. [DOI] [PubMed] [Google Scholar]
- Shi M. L.; Zhan G.; Zhou S. L.; Du W.; Chen Y. C. Asymmetric Inverse-Electron-Demand Oxa-Diels-Alder Reaction of Allylic Ketones through Dienamine Catalysis. Org. Lett. 2016, 18, 6480–6483. 10.1021/acs.orglett.6b03384. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Wang Y.; Yu T. Y.; Liang Y. M.; Xu P. F. Rationally Designed Multifunctional Supramolecular Iminium Catalysis: Direct Vinylogous Michael Addition of Unmodified Linear Dienol Substrates. Angew. Chem., Int. Ed. 2014, 53, 14128–14131. 10.1002/anie.201406786. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yu T. Y.; Zhang H. B.; Luo Y. C.; Xu P. F. Hydrogen-Bond-Mediated Supramolecular Iminium Ion Catalysis. Angew. Chem., Int. Ed. 2012, 51, 12339–12342. 10.1002/anie.201206881. [DOI] [PubMed] [Google Scholar]
- Guo Q.; Fraboni A. J.; Brenner-Moyer S. E. Direct Diastereo- and Enantioselective Vinylogous Michael Additions of Linear Enones. Org. Lett. 2016, 18, 2628–2631. 10.1021/acs.orglett.6b01050. [DOI] [PubMed] [Google Scholar]
- Yang D.; Wang L.; Han F.; Zhao D.; Zhang B.; Wang R. Direct Site-Specific and Highly Enantioselective γ-Functionalization of Linear α,β-Unsaturated Ketones: Bifunctional Catalytic Strategy. Angew. Chem., Int. Ed. 2013, 52, 6739–6742. 10.1002/anie.201301146. [DOI] [PubMed] [Google Scholar]
- Yang D.; Wang L.; Han F.; Zhao D.; Wang R. Design of a Combinational Magnesium Catalyst for the Stereocontrolled Cross Reaction of Enones. Chem. - Eur. J. 2014, 20, 8584–8588. 10.1002/chem.201403183. [DOI] [PubMed] [Google Scholar]
- Akula P. S.; Hong B.-C.; Lee G.-H. Catalyst- and Substituent-Controlled Switching of Chemoselectivity for the Enantioselective Synthesis of Fully Substituted Cyclobutane Derivatives via 2 + 2 Annulation of Vinylogous Ketone Enolates and Nitroalkene. Org. Lett. 2018, 20, 7835–7839. 10.1021/acs.orglett.8b03335. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Xu W.; Liu J.; Das D. K.; Yang S.; Perveen S.; Zhang H.; Li X.; Fang X. Enantioselective Intermolecular All-Carbon [4 + 2] Annulation via N-Heterocyclic Carbene Organocatalysis. Chem. Commun. 2017, 53, 13336–13339. 10.1039/C7CC08680F. [DOI] [PubMed] [Google Scholar]
- Sun F. G.; Sun L. H.; Ye S. N-Heterocyclic Carbene-Catalyzed Enantioselective Annulation of Bromoenal and 1,3-Dicarbonyl Compounds. Adv. Synth. Catal. 2011, 353, 3134–3138. 10.1002/adsc.201100622. [DOI] [Google Scholar]
- Yetra S. R.; Bhunia A.; Patra A.; Mane M. V.; Vanka K.; Biju A. T. Enantioselective N-Heterocyclic Carbene-Catalyzed Annulations of 2-Bromoenals with 1,3-Dicarbonyl Compounds and Enamines via Chiral α,β-Unsaturated Acylazoliums. Adv. Synth. Catal. 2013, 355, 1089–1097. 10.1002/adsc.201300219. [DOI] [Google Scholar]
- Ryan S.; Candish L.; Lupton D. W. Synlett 2011, 2011, 2275–2278. 10.1055/s-0030-1261226. [DOI] [Google Scholar]
- Di Iorio N.; Righi P.; Mazzanti A.; Mancinelli M.; Ciogli A.; Bencivenni G. Remote Control of Axial Chirality: Aminocatalytic Desymmetrization of N-Arylmaleimides via Vinylogous Michael Addition. J. Am. Chem. Soc. 2014, 136, 10250–10253. 10.1021/ja505610k. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Feng X.; Yin X.; Chen Y. C. Direct Remote Asymmetric Bisvinylogous 1,4-Additions of Cyclic 2,5-Dienones to Nitroalkenes. Org. Lett. 2014, 16, 2370–2373. 10.1021/ol500700d. [DOI] [PubMed] [Google Scholar]
- Feng X.; Zhou Z.; Yin X.; Li R.; Chen Y. C. Enantioselective Direct Bisvinylogous 1,6-Additions of β-Allyl-2-Cyclohexenone to α,α-Dicyanodienes through Trienamine Catalysis. Eur. J. Org. Chem. 2014, 2014, 5906–5909. 10.1002/ejoc.201402922. [DOI] [Google Scholar]
- Xiao Y. C.; Yue C. Z.; Chen P. Q.; Chen Y. C. Asymmetric Dearomatic Diels-Alder Reactions of Diverse Heteroarenes via π-System Activation. Org. Lett. 2014, 16, 3208–3211. 10.1021/ol501217u. [DOI] [PubMed] [Google Scholar]
- Xiao W.; Yang Q. Q.; Chen Z.; Ouyang Q.; Du W.; Chen Y. C. Regio- and Diastereodivergent [4 + 2] Cycloadditions with Cyclic 2,4-Dienones. Org. Lett. 2018, 20, 236–239. 10.1021/acs.orglett.7b03598. [DOI] [PubMed] [Google Scholar]
- Xiao W.; Zhou Z.; Yang Q. Q.; Du W.; Chen Y. C. Organocatalytic Asymmetric Four-Component [5 + 1+1 + 1] Cycloadditions via a Quintuple Cascade Process. Adv. Synth. Catal. 2018, 360, 3526–3533. 10.1002/adsc.201800636. [DOI] [Google Scholar]
- Jia Z. L.; Wang Y.; Zhao C. G.; Zhang X. H.; Xu P. F. Highly Enantioselective Construction of Hajos-Wiechert Ketone Skeletons via an Organocatalytic Vinylogous Michael/Stetter Relay Sequence. Org. Lett. 2017, 19, 2130–2133. 10.1021/acs.orglett.7b00767. [DOI] [PubMed] [Google Scholar]
- Jafari E.; Chauhan P.; Kumar M.; Chen X. Y.; Li S.; von Essen C.; Rissanen K.; Enders D. Organocatalytic Asymmetric Synthesis of Trifluoromethylated Tetrahydrocarbazoles by a Vinylogous Michael/Aldol Formal [4 + 2] Annulation. Eur. J. Org. Chem. 2018, 2018, 2462–2465. 10.1002/ejoc.201800034. [DOI] [Google Scholar]
- Möhlmann L.; Chang G. H.; Madhusudhan Reddy G.; Lee C. J.; Lin W. Organocatalytic Enantioselective Synthesis of Tetrahydrofluoren-9-ones via Vinylogous Michael Addition/Henry Reaction Cascade of 1,3-Indandione-Derived Pronucleophiles. Org. Lett. 2016, 18, 688–691. 10.1021/acs.orglett.5b03663. [DOI] [PubMed] [Google Scholar]
- Skrzyńska A.; Romaniszyn M.; Pomiklo D.; Albrecht L. The Application of 2-Benzyl-1,4-naphthoquinones as Pronucleophiles in Aminocatalytic Synthesis of Tricyclic Derivatives. J. Org. Chem. 2018, 83, 5019–5026. 10.1021/acs.joc.8b00170. [DOI] [PubMed] [Google Scholar]
- Li J.-L.; Yue C.-Z.; Chen P.-Q.; Xiao Y.-C.; Chen Y.-C. Remote Enantioselective Friedel-Crafts Alkylations of Furans through HOMO Activation. Angew. Chem., Int. Ed. 2014, 53, 5449–5452. 10.1002/anie.201403082. [DOI] [PubMed] [Google Scholar]
- Dell’Amico L.; Vega-Peñaloza A.; Cuadros S.; Melchiorre P. Enantioselective Organocatalytic Diels-Alder Trapping of Photochemically Generated Hydroxy-o-Quinodimethanes. Angew. Chem., Int. Ed. 2016, 55, 3313–3317. 10.1002/anie.201509472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Amico L.; Fernández-Alvarez V. M.; Maseras F.; Melchiorre P. Light-Driven Enantioselective Organocatalytic β-Benzylation of Enals. Angew. Chem., Int. Ed. 2017, 56, 3304–3308. 10.1002/anie.201612159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X.; Dong S.; Liu Z.; Wu G.; Zou C.; Ye J. Enantioselective Michael Addition of Photogenerated o-Quinodimethanes to Enones Catalyzed by Chiral Amino Acid Esters. Org. Lett. 2017, 19, 2322–2325. 10.1021/acs.orglett.7b00862. [DOI] [PubMed] [Google Scholar]
- Li Y.; Su X.; Zhou W.; Li W.; Zhang J. Amino Acid Derived Phosphine-Catalyzed Enantioselective 1,4-Dipolar Spiroannulation of Cyclobutenones and Isatylidenemalononitrile. Chem. - Eur. J. 2015, 21, 4224–4228. 10.1002/chem.201406475. [DOI] [PubMed] [Google Scholar]
- Gupta V.; Sudhir S. V.; Mandal T.; Schneider C. Organocatalytic, Highly Enantioselective Vinylogous Mukaiyama-Michael Reaction of Acyclic Dienol Silyl Ethers. Angew. Chem., Int. Ed. 2012, 51, 12609–12612. 10.1002/anie.201207058. [DOI] [PubMed] [Google Scholar]
- Ryan S. J.; Candish L.; Lupton D. W. N-Heterocyclic Carbene-Catalyzed (4 + 2) Cycloaddition/Decarboxylation of Silyl Dienol Ethers with α,β-Unsaturated Acid Fluorides. J. Am. Chem. Soc. 2011, 133, 4694–4697. 10.1021/ja111067j. [DOI] [PubMed] [Google Scholar]
- Samanta R. C.; Maji B.; De Sarkar S.; Bergander K.; Fröhlich R.; Mück-Lichtenfeld C.; Mayr H.; Studer A. Nucleophilic Addition of Enols and Enamines to α,β-Unsaturated Acyl Azoliums: Mechanistic Studies. Angew. Chem., Int. Ed. 2012, 51, 5234–5238. 10.1002/anie.201109042. [DOI] [PubMed] [Google Scholar]
- Ryan S. J.; Stasch A.; Paddon-Row M. N.; Lupton D. W. Synthetic and Quantum Mechanical Studies into the N-Heterocyclic Carbene Catalyzed (4 + 2) Cycloaddition. J. Org. Chem. 2012, 77, 1113–1124. 10.1021/jo202500k. [DOI] [PubMed] [Google Scholar]
- Masuda Y.; Ishida N.; Murakami M. Light-Driven Carboxylation of o-Alkylphenyl Ketones with CO2. J. Am. Chem. Soc. 2015, 137, 14063–14066. 10.1021/jacs.5b10032. [DOI] [PubMed] [Google Scholar]
- Yang X.; Toste F. D. Direct Asymmetric Amination of α-Branched Cyclic Ketones Catalyzed by a Chiral Phosphoric Acid. J. Am. Chem. Soc. 2015, 137, 3205–3208. 10.1021/jacs.5b00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Liu X.; Mohr J. T. Direct Regioselective γ-Amination of Enones. Org. Lett. 2016, 18, 716–719. 10.1021/acs.orglett.5b03689. [DOI] [PubMed] [Google Scholar]
- Schläger N.; Kirschning A. Substrate-Controlled Stereoselectivity in the Yamamoto Aldol Reaction. Org. Biomol. Chem. 2012, 10, 7721–7729. 10.1039/c2ob26185e. [DOI] [PubMed] [Google Scholar]
- Wang L.-L.; Kirschning A. Total Synthesis of Elansolids B1 and B2. Beilstein J. Org. Chem. 2017, 13, 1280–1287. 10.3762/bjoc.13.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. L.; Candito D.; Dräger G.; Herrmann J.; Müller R.; Kirschning A. Harnessing a p-Quinone Methide Intermediate in the Biomimetic Total Synthesis of the Highly Active Antibiotic 20-Deoxy-Elansolid B1. Chem. - Eur. J. 2017, 23, 5291–5298. 10.1002/chem.201605884. [DOI] [PubMed] [Google Scholar]
- Weber A.; Dehn R.; Schläger N.; Dieter B.; Kirschning A. Total Synthesis of the Antibiotic Elansolid B1. Org. Lett. 2014, 16, 568–571. 10.1021/ol403441c. [DOI] [PubMed] [Google Scholar]
- Gazaille J. A.; Abramite J. A.; Sammakia T. Toward the Synthesis of (+)-Peloruside A via an Intramolecular Vinylogous Aldol Reaction. Org. Lett. 2012, 14, 178–181. 10.1021/ol202966m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazaille J. A.; Sammakia T. The Vinylogous Aldol Reaction of Unsaturated Esters and Enolizable Aldehydes Using the Novel Lewis Acid Aluminum Tris(2,6-Di-2-Naphthylphenoxide). Org. Lett. 2012, 14, 2678–2681. 10.1021/ol300738f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que Y.; Li T.; Yu C.; Wang X.-S.; Yao C. Enantioselective Assembly of Spirocyclic Oxindole-Dihydropyranones through NHC-Catalyzed Cascade Reaction of Isatins with N -Hydroxybenzotriazole Esters of α,β-Unsaturated Carboxylic Acid. J. Org. Chem. 2015, 80, 3289–3294. 10.1021/jo502920w. [DOI] [PubMed] [Google Scholar]
- Ube H.; Shimada N.; Terada M. Asymmetric Direct Vinylogous Aldol Reaction of Furanone Derivatives Catalyzed by an Axially Chiral Guanidine Base. Angew. Chem., Int. Ed. 2010, 49, 1858–1861. 10.1002/anie.200906647. [DOI] [PubMed] [Google Scholar]
- Mirabdolbaghi R.; Hassan M.; Dudding T. Design and Synthesis of a Chiral Seven-Membered Ring Guanidine Organocatalyst Applied to Asymmetric Vinylogous Aldol Reactions. Tetrahedron: Asymmetry 2015, 26, 560–566. 10.1016/j.tetasy.2015.04.001. [DOI] [Google Scholar]
- Luo J.; Wang H.; Han X.; Xu L. W.; Kwiatkowski J.; Huang K. W.; Lu Y. The Direct Asymmetric Vinylogous Aldol Reaction of Furanones with α-Ketoesters: Access to Chiral γ-Butenolides and Glycerol Derivatives. Angew. Chem., Int. Ed. 2011, 50, 1861–1864. 10.1002/anie.201006316. [DOI] [PubMed] [Google Scholar]
- Claraz A.; Oudeyer S.; Levacher V. Chiral Quaternary Ammonium Aryloxide/N,O-Bis(Trimethyl-Silyl)Acetamide Combination as Efficient Organocatalytic System for the Direct Vinylogous Aldol Reaction of (5H)-Furan-2-One Derivatives. Adv. Synth. Catal. 2013, 355, 841–846. 10.1002/adsc.201201041. [DOI] [Google Scholar]
- Yang Y.; Zheng K.; Zhao J.; Shi J.; Lin L.; Liu X.; Feng X. Asymmetric Direct Vinylogous Aldol Reaction of Unactivated γ-Butenolide to Aldehydes. J. Org. Chem. 2010, 75, 5382–5384. 10.1021/jo100946d. [DOI] [PubMed] [Google Scholar]
- Sakai T.; Hirashima S.; Yamashita Y.; Arai R.; Nakashima K.; Yoshida A.; Koseki Y.; Miura T. Squaramide-Sulfonamide Organocatalyst for Asymmetric Direct Vinylogous Aldol Reactions. J. Org. Chem. 2017, 82, 4661–4667. 10.1021/acs.joc.7b00287. [DOI] [PubMed] [Google Scholar]
- Pansare S. V.; Paul E. K. Organocatalytic Asymmetric Direct Vinylogous Aldol Reactions of γ-Crotonolactone with Aromatic Aldehydes. Chem. Commun. 2011, 47, 1027–1029. 10.1039/C0CC04191B. [DOI] [PubMed] [Google Scholar]
- Pansare S. V.; Paul E. K. Synthesis of (+)-L-733,060, (+)-CP-99,994 and (2S,3R)-3-Hydroxypipecolic Acid: Application of an Organocatalytic Direct Vinylogous Aldol Reaction. Org. Biomol. Chem. 2012, 10, 2119–2125. 10.1039/c2ob06644k. [DOI] [PubMed] [Google Scholar]
- Pansare S.; Paul E. Synthesis of (+)-Febrifugine and a Formal Synthesis of (+)-Halofuginone Employing an Organocatalytic Direct Vinylogous Aldol Reaction. Synthesis 2013, 45, 1863–1869. 10.1055/s-0033-1338706. [DOI] [Google Scholar]
- Karak M.; Barbosa L. C. A.; Acosta J. A. M.; Sarotti A. M.; Boukouvalas J. Thermodynamically Driven, Syn-Selective Vinylogous Aldol Reaction of Tetronamides. Org. Biomol. Chem. 2016, 14, 4897–4907. 10.1039/C6OB00895J. [DOI] [PubMed] [Google Scholar]
- Karak M.; Barbosa L. C. A.; Maltha C. R. A.; Silva T. M.; Boukouvalas J. Palladium-Catalyzed Hydrodehalogenation of Butenolides: An Efficient and Sustainable Access to β-Arylbutenolides. Tetrahedron Lett. 2017, 58, 2830–2834. 10.1016/j.tetlet.2017.06.016. [DOI] [Google Scholar]
- Pereira U. A.; Moreira T. A.; Barbosa L. C. A.; Maltha C. R. A.; Bomfim I. S.; Maranhão S. S.; Moraes M. O.; Pessoa C.; Barros-Nepomuceno F. W. A. Rubrolide Analogues and Their Derived Lactams as Potential Anticancer Agents. MedChemComm 2016, 7, 345–352. 10.1039/C5MD00459D. [DOI] [Google Scholar]
- For a racemic DBU-catalyzed direct VAR between furan-2(3H)ones and isatins, see:Kumar A.; Kumar G.; Ramakrishna K.; Ramesh P.; Swetha A.; Meshram H. A Facile and Efficient Synthesis of Oxindole Motifs by Direct Vinylogous Aldol/Mannich Reaction of Isatins/Isatinimines with Furan-2(3H)-One. Synlett 2017, 28, 337–342. 10.1055/s-0036-1588341. [DOI] [Google Scholar]
- Tang Q.; Lin L.; Ji J.; Hu H.; Liu X.; Feng X. Catalytic Asymmetric Direct Vinylogous Aldol Reaction of Isatins with β,γ-Unsaturated Butenolides. Chem. - Eur. J. 2017, 23, 16447–16451. 10.1002/chem.201704100. [DOI] [PubMed] [Google Scholar]
- Hartwig W. T.; Sammakia T. Doubly Vinylogous Aldol Reaction of Furoate Esters with Aldehydes and Ketones. J. Org. Chem. 2017, 82, 759–764. 10.1021/acs.joc.6b02473. [DOI] [PubMed] [Google Scholar]
- de Castro P. P.; Carpanez A. G.; Amarante G. W. Azlactone Reaction Developments. Chem. - Eur. J. 2016, 22, 10294–10318. 10.1002/chem.201600071. [DOI] [PubMed] [Google Scholar]
- Gao T.-P.; Lin J.-B.; Hu X.-Q.; Xu P.-F. A Catalytic Asymmetric Hetero-Diels-Alder Reaction of Olefinic Azlactones and Isatins: Facile Access to Chiral Spirooxindole Dihydropyranones. Chem. Commun. 2014, 50, 8934–8936. 10.1039/C4CC03896G. [DOI] [PubMed] [Google Scholar]
- Gao T.-P.; Liu D.; Lin J.-B.; Hu X.-Q.; Wang Z.-Y.; Xu P.-F. Direct Construction of Chiral Quaternary Dihydropyranones through Highly Enantioselective Organocatalytic Hetero-Diels-Alder Reactions of Olefinic Azlactones. Org. Chem. Front. 2016, 3, 598–602. 10.1039/C6QO00018E. [DOI] [Google Scholar]
- Wang G.; Zhao J.; Zhou Y.; Wang B.; Qu J. Mild and Highly Enantioselective Vinylogous Aldol Reaction of Brassard’s Diene with Aromatic Aldehydes by Combined Lewis Acid Catalyst. J. Org. Chem. 2010, 75, 5326–5329. 10.1021/jo100674f. [DOI] [PubMed] [Google Scholar]
- Wang G.; Wang B.; Qi S.; Zhao J.; Zhou Y.; Qu J. Highly Enantio- and Diastereoselective Vinylogous Aldol Reaction by LiCl-Assisted BINOL-Titanium Species. Org. Lett. 2012, 14, 2734–2737. 10.1021/ol300946j. [DOI] [PubMed] [Google Scholar]
- Fu K.; Zheng J.; Lin L.; Liu X.; Feng X. Chiral N,N′-Dioxide–In(OTf)3-Catalyzed Asymmetric Vinylogous Mukaiyama Aldol Reactions. Chem. Commun. 2015, 51, 3106–3108. 10.1039/C4CC09820J. [DOI] [PubMed] [Google Scholar]
- Fu K.; Zhang J.; Lin L.; Li J.; Liu X.; Feng X. Chiral N,N′-Dioxide/Lanthanide(III) Complex Catalyzed Asymmetric Bisvinylogous Mukaiyama Aldol Reactions. Org. Lett. 2017, 19, 332–335. 10.1021/acs.orglett.6b03470. [DOI] [PubMed] [Google Scholar]
- Ratjen L.; García-García P.; Lay F.; Beck M. E.; List B. Disulfonimide-Catalyzed Asymmetric Vinylogous and Bisvinylogous Mukaiyama Aldol Reactions. Angew. Chem., Int. Ed. 2011, 50, 754–758. 10.1002/anie.201005954. [DOI] [PubMed] [Google Scholar]
- Tap A.; Blond A.; Wakchaure V. N.; List B. Chiral Allenes via Alkynylogous Mukaiyama Aldol Reaction. Angew. Chem., Int. Ed. 2016, 55, 8962–8965. 10.1002/anie.201603649. [DOI] [PubMed] [Google Scholar]
- Schetter B.; Mahrwald R. Modern Aldol Methods for the Total Synthesis of Polyketides. Angew. Chem., Int. Ed. 2006, 45, 7506–7525. 10.1002/anie.200602780. [DOI] [PubMed] [Google Scholar]
- Takasu M.; Yamamoto H. New Chiral Lewis Acid Catalysts Prepared from Simple Amino Acids and Their Use in Asymmetric Diels-Alder Reactions. Synlett 1990, 1990, 194–196. 10.1055/s-1990-21030. [DOI] [Google Scholar]
- Sartor D.; Saffrich J.; Helmchen G. Enantioselective Diels-Alder Additions with New Chiral Lewis Acids Derived from Amino Acids. Synlett 1990, 1990, 197–198. 10.1055/s-1990-21031. [DOI] [Google Scholar]
- An C.; Jurica J. A.; Walsh S. P.; Hoye A. T.; Smith A. B. Total Synthesis of (+)-Irciniastatin A (a.k.a. Psymberin) and (−)-Irciniastatin B. J. Org. Chem. 2013, 78, 4278–4296. 10.1021/jo400260m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstmann L.; Kalesse M. Total Synthesis of Aetheramide A. Chem. - Eur. J. 2016, 22, 11210–11212. 10.1002/chem.201602682. [DOI] [PubMed] [Google Scholar]
- Florence G. J.; Wlochal J. Synthesis of the Originally Proposed Structure of Palmerolide C. Chem. - Eur. J. 2012, 18, 14250–14254. 10.1002/chem.201203067. [DOI] [PubMed] [Google Scholar]
- Diaz N.; Zhu M.; Ehrlich G.; Eggert U.; Muthukumar Y.; Sasse F.; Kalesse M. An Improved Route to (+)-Tedanolide and Analysis of Its Subtle Effects Controlling Conformation and Biological Behaviour. Chem. - Eur. J. 2012, 18, 4946–4952. 10.1002/chem.201103038. [DOI] [PubMed] [Google Scholar]
- Landsberg D.; Hartmann O.; Eggert U.; Kalesse M. Diastereodivergent Vinylogous Mukaiyama Aldol Reaction. Synlett 2013, 24, 1105–1108. 10.1055/s-0033-1338933. [DOI] [Google Scholar]
- Denmark S. E.; Beutner G. L. Lewis Base Catalysis in Organic Synthesis. Angew. Chem., Int. Ed. 2008, 47, 1560–1638. 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Heemstra J. R. Lewis Base Activation of Lewis Acids: Catalytic, Enantioselective Vinylogous Aldol Addition Reactions. J. Org. Chem. 2007, 72, 5668–5688. 10.1021/jo070638u. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Jiang X.; Lindsay-Scott P. J.; Corbu A.; Yamashiro S.; Bacconi A.; Fowler V. M. Total Synthesis and Biological Evaluation of Monorhizopodin and 16-Epi-Monorhizopodin. Angew. Chem., Int. Ed. 2011, 50, 1139–1144. 10.1002/anie.201006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthikeyan G.; Zambaldo C.; Barluenga S.; Zoete V.; Karplus M.; Winssinger N. Asymmetric Synthesis of Pochonin E and F, Revision of Their Proposed Structure, and Their Conversion to Potent Hsp90 Inhibitors. Chem. - Eur. J. 2012, 18, 8978–8986. 10.1002/chem.201200546. [DOI] [PubMed] [Google Scholar]
- Frings M.; Atodiresei I.; Runsink J.; Raabe G.; Bolm C. Catalyzed Vinylogous Mukaiyama Aldol Reactions with Controlled Enantio- and Diastereoselectivities. Chem. - Eur. J. 2009, 15, 1566–1569. 10.1002/chem.200802359. [DOI] [PubMed] [Google Scholar]
- Frings M.; Atodiresei I.; Wang Y.; Runsink J.; Raabe G.; Bolm C. C1-Symmetric Aminosulfoximines in Copper-Catalyzed Asymmetric Vinylogous Mukaiyama Aldol Reactions. Chem. - Eur. J. 2010, 16, 4577–4587. 10.1002/chem.200903077. [DOI] [PubMed] [Google Scholar]
- Sevrain C. M.; Berchel M.; Couthon H.; Jaffrès P.-A. Phosphonic Acid: Preparation and Applications. Beilstein J. Org. Chem. 2017, 13, 2186–2213. 10.3762/bjoc.13.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. P.; Zhao X. N.; Miao Z. W.; Chen R. Y. Highly Diastereoselective Vinylogous Mukaiyama Aldol Reaction of α-Keto Phosphonates with 2-(Trimethylsilyloxy)Furan Catalyzed by Cu(OTf)2. Org. Biomol. Chem. 2011, 9, 6721–6726. 10.1039/c1ob05822c. [DOI] [PubMed] [Google Scholar]
- Hou G.; Yu J.; Yu C.; Wu G.; Miao Z. Enantio- and Diastereoselective Vinylogous Mukaiyama Aldol Reactions of α-Keto Phosphonates with 2-(Trimethylsilyloxy)-Furan Catalyzed by Bis(Oxazoline)-Copper Complexes. Adv. Synth. Catal. 2013, 355, 589–593. 10.1002/adsc.201200810. [DOI] [Google Scholar]
- Frings M.; Thomé I.; Schiffers I.; Pan F.; Bolm C. Catalytic, Asymmetric Synthesis of Phosphonic γ-(Hydroxyalkyl) Butenolides with Contiguous Quaternary and Tertiary Stereogenic Centers. Chem. - Eur. J. 2014, 20, 1691–1700. 10.1002/chem.201304331. [DOI] [PubMed] [Google Scholar]
- Curti C.; Ranieri B.; Battistini L.; Rassu G.; Zambrano V.; Pelosi G.; Casiraghi G.; Zanardi F. Catalytic, Asymmetric Vinylogous Mukaiyama Aldol Reactions of Pyrrole- and Furan-Based Dienoxy Silanes: How the Diene Heteroatom Impacts Stereocontrol. Adv. Synth. Catal. 2010, 352, 2011–2022. 10.1002/adsc.201000189. [DOI] [Google Scholar]
- Palombi L.; Acocella M. R.; Celenta N.; Massa A.; Villano R.; Scettri A. Highly Enantioselective Vinylogous Addition of 2-Trimethylsilyloxyfuran to Aldehydes Promoted by the SiCl4/Chiral Lewis Base System. Tetrahedron: Asymmetry 2006, 17, 3300–3303. 10.1016/j.tetasy.2006.11.041. [DOI] [Google Scholar]
- Curti C.; Brindani N.; Battistini L.; Sartori A.; Pelosi G.; Mena P.; Brighenti F.; Zanardi F.; Del Rio D. Catalytic, Enantioselective Vinylogous Mukaiyama Aldol Reaction of Furan-Based Dienoxy Silanes: A Chemodivergent Approach to γ-Valerolactone Flavan-3-ol Metabolites and δ-Lactone Analogues. Adv. Synth. Catal. 2015, 357, 4082–4092. 10.1002/adsc.201500705. [DOI] [Google Scholar]
- Mena P.; Bresciani L.; Brindani N.; Ludwig I. A.; Pereira-Caro G.; Angelino D.; Llorach R.; Calani L.; Brighenti F.; Clifford M. N.; et al. Phenyl-γ-Valerolactones and Phenylvaleric Acids, the Main Colonic Metabolites of Flavan-3-ols: Synthesis, Analysis, Bioavailability, and Bioactivity. Nat. Prod. Rep. 2019, 36, 714–752. 10.1039/C8NP00062J. [DOI] [PubMed] [Google Scholar]
- Curti C.; Battistini L.; Sartori A.; Lodola A.; Mor M.; Rassu G.; Pelosi G.; Zanardi F.; Casiraghi G. Catalytic, Asymmetric Hypervinylogous Mukaiyama Aldol Reactions of Extended Furan-Based Silyl Enolates. Org. Lett. 2011, 13, 4738–4741. 10.1021/ol2020626. [DOI] [PubMed] [Google Scholar]
- Narayan S.; Muldoon J.; Finn M. G.; Fokin V. V.; Kolb H. C.; Sharpless K. B. On Water”: Unique Reactivity of Organic Compounds in Aqueous Suspension. Angew. Chem., Int. Ed. 2005, 44, 3275–3279. 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]
- Curti C.; Battistini L.; Zanardi F.; Rassu G.; Zambrano V.; Pinna L.; Casiraghi G. Uncatalyzed, Diastereoselective Vinylogous Mukaiyama Aldol Reactions on Aqueous Media: Pyrrole vs Furan 2-Silyloxy Dienes. J. Org. Chem. 2010, 75, 8681–8684. 10.1021/jo101799e. [DOI] [PubMed] [Google Scholar]
- Woyciechowska M.; Forcher G.; Buda S.; Mlynarski J. General Switch in Regioselectivity in the Mukaiyama Aldol Reaction of Silyloxyfuran with Aldehydes in Aqueous Solvents. Chem. Commun. 2012, 48, 11029–11031. 10.1039/c2cc36656h. [DOI] [PubMed] [Google Scholar]
- Scettri A.; Sio V. D.; Villano R.; Manzo P.; Acocella M. R. Solvent-Free Mukaiyama and Mukaiyama-Michael Vinylogous Reactions of a Dioxinone-Derived Silyl Enol Ether Promoted by Lewis Bases. Tetrahedron Lett. 2010, 51, 3658–3661. 10.1016/j.tetlet.2010.05.016. [DOI] [Google Scholar]
- Acocella M. R.; Villano R.; Costabile C.; Scettri A. Vinylogous Aldol Reaction of a Silyloxydiene Promoted by Sulfoxides, as Lewis Bases: 1,4–Asymmetric Induction Mediated by a Remote Methylsulfinyl Group. Curr. Org. Chem. 2013, 17, 886–890. 10.2174/1385272811317080013. [DOI] [Google Scholar]
- Acocella M. R.; De Pascale M.; Maggio M.; Guerra G. Graphite Oxide as Catalyst for Diastereoselective Mukaiyama Aldol Reaction of 2-(Trimethylsilyloxy)Furan in Solvent Free Conditions. J. Mol. Catal. A: Chem. 2015, 408, 237–241. 10.1016/j.molcata.2015.07.033. [DOI] [Google Scholar]
- De Rosa M.; La Manna P.; Soriente A.; Gaeta C.; Talotta C.; Neri P. Exploiting the Hydrophobicity of Calixarene Macrocycles for Catalysis under “on-Water” Conditions. RSC Adv. 2016, 6, 91846–91851. 10.1039/C6RA19270J. [DOI] [Google Scholar]
- De Rosa M.; La Manna P.; Soriente A.; Gaeta C.; Talotta C.; Hickey N.; Geremia S.; Neri P. A Simple Tetraminocalix[4]Arene as a Highly Efficient Catalyst under “On-Water” Conditions through Hydrophobic Amplification of Weak Hydrogen Bonds. Chem. - Eur. J. 2017, 23, 7142–7151. 10.1002/chem.201701247. [DOI] [PubMed] [Google Scholar]
- De Rosa M.; La Manna P.; Soriente A.; Gaeta C.; Talotta C.; Hickey N.; Geremia S.; Neri P. Supramolecular Synthons in the γ-Hydroxybutenolides. CrystEngComm 2017, 19, 5079–5088. 10.1039/C7CE00953D. [DOI] [Google Scholar]
- Kong S.; Fan W.; Lyu H.; Zhan J.; Miao X.; Miao Z. Highly Diastereoselective Vinylogous Mukaiyama Aldol Reaction of Isatins with 2-(Trimethylsilyloxy)Furans Catalyzed by Quinine. Synth. Commun. 2014, 44, 936–942. 10.1080/00397911.2013.837926. [DOI] [Google Scholar]
- Zhu N.; Ma B.-C.; Zhang Y.; Wang W. Organocatalyzed Highly Enantioselective and Anti-Selective Construction of γ-Butenolides through Vinylogous Mukaiyama Aldol Reaction. Adv. Synth. Catal. 2010, 352, 1291–1295. 10.1002/adsc.201000099. [DOI] [Google Scholar]
- Singh R. P.; Foxman B. M.; Deng L. Asymmetric Vinylogous Aldol Reaction of Silyloxy Furans with a Chiral Organic Salt. J. Am. Chem. Soc. 2010, 132, 9558–9560. 10.1021/ja103331t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoye T. R.; Danielson M. E.; May A. E.; Zhao H. Total Synthesis of (−)-Callipeltoside A. J. Org. Chem. 2010, 75, 7052–7060. 10.1021/jo101598y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav J. S.; Haldar A.; Maity T. Towards the Synthesis of (−)-Callipeltoside A: Stereoselective Synthesis of the C1-C14 Macrolactone Core. Eur. J. Org. Chem. 2012, 2012, 2062–2071. 10.1002/ejoc.201101635. [DOI] [Google Scholar]
- Fuwa H.; Okuaki Y.; Yamagata N.; Sasaki M. Total Synthesis, Stereochemical Reassignment, and Biological Evaluation of (−)-Lyngbyaloside B. Angew. Chem., Int. Ed. 2015, 54, 868–873. 10.1002/anie.201409629. [DOI] [PubMed] [Google Scholar]
- Fuwa H.; Yamagata N.; Okuaki Y.; Ogata Y.; Saito A.; Sasaki M. Total Synthesis and Complete Stereostructure of a Marine Macrolide Glycoside, (−)-Lyngbyaloside B. Chem. - Eur. J. 2016, 22, 6815–6829. 10.1002/chem.201600341. [DOI] [PubMed] [Google Scholar]
- Wang B.; Hansen T. M.; Wang T.; Wu D.; Weyer L.; Ying L.; Engler M. M.; Sanville M.; Leitheiser C.; Christmann M.; et al. Total Synthesis of Phorboxazole A via de Novo Oxazole Formation: Strategy and Component Assembly. J. Am. Chem. Soc. 2011, 133, 1484–1505. 10.1021/ja108906e. [DOI] [PubMed] [Google Scholar]
- Halperin S. D.; Britton R. Chlorine, an Atom Economical Auxiliary for Asymmetric Aldol Reactions. Org. Biomol. Chem. 2013, 11, 1702–1705. 10.1039/c3ob27462d. [DOI] [PubMed] [Google Scholar]
- Tenenbaum J. M.; Morris W. J.; Custar D. W.; Scheidt K. A. Synthesis of (−)-Okilactomycin by a Prins-Type Fragment-Assembly Strategy. Angew. Chem., Int. Ed. 2011, 50, 5892–5895. 10.1002/anie.201102037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger J.; Carreira E. M. Apparent Catalytic Generation of Chiral Metal Enolates: Enantioselective Dienolate Additions to Aldehydes Mediated by Tol-BINAP Cu(II) Fluoride Complexes. J. Am. Chem. Soc. 1998, 120, 837–838. 10.1021/ja973331t. [DOI] [Google Scholar]
- Seiple I. B.; Zhang Z.; Jakubec P.; Langlois-Mercier A.; Wright P. M.; Hog D. T.; Yabu K.; Allu S. R.; Fukuzaki T.; Carlsen P. N.; et al. A Platform for the Discovery of New Macrolide Antibiotics. Nature 2016, 533, 338–345. 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prantz K.; Mulzer J. Synthesis of (Z)-Trisubstituted Olefins by Decarboxylative Grob-Type Fragmentations: Epothilone D, Discodermolide, and Peloruside A. Chem. - Eur. J. 2010, 16, 485–506. 10.1002/chem.200901567. [DOI] [PubMed] [Google Scholar]
- Dalby S. M.; Goodwin-Tindall J.; Paterson I. Total Synthesis of (−)-Rhizopodin. Angew. Chem., Int. Ed. 2013, 52, 6517–6521. 10.1002/anie.201301978. [DOI] [PubMed] [Google Scholar]
- Xu L. M.; You L.; Shan Z. H.; Yu R. C.; Zhang B.; Li Y. H.; Shi Y.; Chen J. H.; Yang Z. Asymmetric Total Synthesis of Propindilactone G, Part 1: Initial Attempts towards the Synthesis of Schiartanes. Chem. - Asian J. 2016, 11, 1406–1413. 10.1002/asia.201600129. [DOI] [PubMed] [Google Scholar]
- Zhang J. J.; You L.; Wang Y. F.; Li Y. H.; Liang X. T.; Zhang B.; Yang S. L.; Su Q.; Chen J. H.; Yang Z. Asymmetric Total Synthesis of Propindilactone G, Part 2: Enantioselective Construction of the Fully Functionalized BCDE Ring System. Chem. - Asian J. 2016, 11, 1414–1424. 10.1002/asia.201600130. [DOI] [PubMed] [Google Scholar]
- Kong K.; Moussa Z.; Lee C.; Romo D. Total Synthesis of the Spirocyclic Imine Marine Toxin (−)-Gymnodimine and an Unnatural C4-Epimer. J. Am. Chem. Soc. 2011, 133, 19844–19856. 10.1021/ja207385y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For another example of total synthesis involving a distereoselective VMAR between TMSOF and a cyclic aldehyde, see:Yamashita S.; Hayashi D.; Nakano A.; Hayashi Y.; Hirama M. Total Synthesis of Avermectin B1a Revisited.. J. Antibiot. 2016, 69, 31–50. 10.1038/ja.2015.47. [DOI] [PubMed] [Google Scholar]
- MacAbeo A. P. G.; Lehmann C. W.; Reiser O. Diastereoselective Synthesis of Enantiopure γ-Butenolide-Butyrolactones towards Pseudopterogorgia Lactone Furanocembranoid Substructures. Synlett 2012, 23, 2909–2912. 10.1055/s-0032-1317555. [DOI] [Google Scholar]
- For an example of VMAR between TMSOF and chiral aziridine acceptors, see:Lee H.; Kim J. H.; Lee W. K.; Cho J.; Nam W.; Lee J.; Ha H.-J. Highly Stereoselective Directed Reactions and an Efficient Synthesis of Azafuranoses from a Chiral Aziridine. Org. Biomol. Chem. 2013, 11, 3629–3634. 10.1039/c3ob27390c. [DOI] [PubMed] [Google Scholar]
- Han Y.-X.; Jiang Y.-L.; Li Y.; Yu H.-X.; Tong B.-Q.; Niu Z.; Zhou S.-J.; Liu S.; Lan Y.; Chen J.-H.; et al. Biomimetically Inspired Asymmetric Total Synthesis of (+)-19-Dehydroxyl Arisandilactone A. Nat. Commun. 2017, 8, 14233. 10.1038/ncomms14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T.; Johnson R. P.; Porco J. A. Jr. Vinylogous Addition of Siloxyfurans to Benzopyryliums: A Concise Approach to the Tetrahydroxanthone Natural Products. J. Am. Chem. Soc. 2011, 133, 1714–1717. 10.1021/ja110698n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For other synthetic applications of this methodology by the same group, see:Qin T.; Skraba-Joiner S. L.; Khalil Z. G.; Johnson R. P.; Capon R. J.; Porco J. A. Atroposelective Syntheses of (−) and (+) Rugulotrosin A Utilizing Point-to-Axial Chirality Transfer. Nat. Chem. 2015, 7, 234–240. 10.1038/nchem.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Iwata T.; Scharf A.; Qin T.; Reichl K. D.; Porco J. A. Asymmetric Synthesis of Gonytolide A: Strategic Use of an Aryl Halide Blocking Group for Oxidative Coupling. J. Am. Chem. Soc. 2018, 140, 5969–5975. 10.1021/jacs.8b02535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Also for similar LA mediated VMAR methodologies, see:Stubbing L. A.; Li F. F.; Furkert D. P.; Caprio V. E.; Brimble M. A. Access to 2-Alkyl Chromanones via a Conjugate Addition Approach. Tetrahedron 2012, 68, 6948–6956. 10.1016/j.tet.2012.05.115. [DOI] [Google Scholar]
- Liu J.; Li Z.; Tong P.; Xie Z.; Zhang Y.; Li Y. TMSI-Promoted Vinylogous Michael Addition of Siloxyfuran to 2-Substituted Chromones: A General Approach for the Total Synthesis of Chromanone Lactone Natural Products. J. Org. Chem. 2015, 80, 1632–1643. 10.1021/jo502571r. [DOI] [PubMed] [Google Scholar]
- Chaubet G.; Goh S. S.; Mohammad M.; Gockel B.; Cordonnier M.-C. A.; Baars H.; Phillips A. W.; Anderson E. A. Total Synthesis of the Schisandraceae Nortriterpenoid Rubriflordilactone A. Chem. - Eur. J. 2017, 23, 14080–14089. 10.1002/chem.201703229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh S. S.; Chaubet G.; Gockel B.; Cordonnier M.-C. A.; Baars H.; Phillips A. W.; Anderson E. A. Total Synthesis of (+)-Rubriflordilactone A. Angew. Chem., Int. Ed. 2015, 54, 12618–12621. 10.1002/anie.201506366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Duan S. M.; Wang Y.-W. Concise Synthesis of the DEFG Ring System in Rubriflordilactone B. Tetrahedron Lett. 2015, 56, 4509–4511. 10.1016/j.tetlet.2015.05.117. [DOI] [Google Scholar]
- Raghavan S.; Patel J. S.; Ramakrishna K. V. S. Synthesis of (−) Cephalosporolide E and (+) Cephalosporolide F Utilizing the Vinylogous Mukaiyama Type Reaction with an α-Chloro Sulfide. RSC Adv. 2016, 6, 72877–72884. 10.1039/C6RA13861F. [DOI] [Google Scholar]
- Xu J.; Yuan S.; Miao M. N-Heterocyclic Carbene Catalyzed [4 + 2] Annulation Reactions with in Situ Generated Heterocyclic Ortho-Quinodimethanes. Org. Lett. 2016, 18, 3822–3825. 10.1021/acs.orglett.6b01831. [DOI] [PubMed] [Google Scholar]
- Robak M. T.; Herbage M. A.; Ellman J. A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- Li G.; Xu X.; Tian H.; Liu X.; Chen W.; Yang X.; Zhang H. Asymmetric Synthesis of δ-Amino Acid Derivatives via Diastereoselective Vinylogous Mannich Reactions between N-tert-Butanesulfinyl Imines and Dioxinone-Derived Lithium Dienolate. RSC Adv. 2017, 7, 50822–50828. 10.1039/C7RA10529K. [DOI] [Google Scholar]
- Chen W.; Yang X.-D.; Tan W.-Y.; Zhang X.-Y.; Liao X.-L.; Zhang H. Total Synthesis of (−)-Vindorosine. Angew. Chem., Int. Ed. 2017, 56, 12327–12331. 10.1002/anie.201707249. [DOI] [PubMed] [Google Scholar]
- Chen W.; Tian H.; Tan W.; Liu X.; Yang X.; Zhang H. Total Synthesis of (−)-Vindoline. Tetrahedron 2019, 75, 1751–1759. 10.1016/j.tet.2018.11.046. [DOI] [Google Scholar]
- Yin L.; Takada H.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Vinylogous Mannich-Type Reaction of γ-Butenolides with Ketimines. Angew. Chem., Int. Ed. 2013, 52, 7310–7313. 10.1002/anie.201303119. [DOI] [PubMed] [Google Scholar]
- Nakamura S.; Yamaji R.; Hayashi M. Direct Enantioselective Vinylogous Mannich Reaction of Ketimines with γ-Butenolide by Using Cinchona Alkaloid Amide/Zinc(II) Catalysts. Chem. - Eur. J. 2015, 21, 9615–9618. 10.1002/chem.201500599. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Lin L.; Ji J.; Xie M.; Liu X.; Feng X. Catalytic Asymmetric Vinylogous Mannich-Type (AVM) Reaction of Nonactivated α-Angelica Lactone. Org. Lett. 2011, 13, 3056–3059. 10.1021/ol200939t. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Gnanamani E.; Tracy J. S.; Kalnmals C. A. Zn-ProPhenol Catalyzed Enantio- and Diastereoselective Direct Vinylogous Mannich Reactions between α,β- and β,y-Butenolides and Aldimines. J. Am. Chem. Soc. 2017, 139, 18198–18201. 10.1021/jacs.7b11361. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Bartlett M. J. ProPhenol-Catalyzed Asymmetric Additions by Spontaneously Assembled Dinuclear Main Group Metal Complexes. Acc. Chem. Res. 2015, 48, 688–701. 10.1021/ar500374r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.-L.; Bai J.-F.; Peng L.; Wang L.-L.; Jia L.-N.; Luo X.-Y.; Tian F.; Xu X.-Y.; Wang L.-X. Direct Asymmetric Vinylogous Mannich Reaction of 3,4-Dihalofuran-2(5H)-One with Aldimine Catalyzed by Quinine. J. Org. Chem. 2012, 77, 8338–8343. 10.1021/jo301115t. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Hui Y.; Zhou X.; Lin L.; Liu X.; Feng X. Highly Efficient Asymmetric Three-Component Vinylogous Mannich Reaction Catalyzed by a Chiral Scandium(III)-N,N’-Dioxide Complex. Adv. Synth. Catal. 2010, 352, 976–980. 10.1002/adsc.200900877. [DOI] [Google Scholar]
- Sickert M.; Abels F.; Lang M.; Sieler J.; Birkemeyer C.; Schneider C. The Brønsted Acid Catalyzed, Enantioselective Vinylogous Mannich Reaction. Chem. - Eur. J. 2010, 16, 2806–2818. 10.1002/chem.200902537. [DOI] [PubMed] [Google Scholar]
- Sickert M.; Schneider C. The Enantioselective, Brønsted Acid Catalyzed, Vinylogous Mannich Reaction. Angew. Chem., Int. Ed. 2008, 47, 3631–3634. 10.1002/anie.200800103. [DOI] [PubMed] [Google Scholar]
- Abels F.; Schneider C. A Modified and Highly Useful Protocol for the Brønsted Acid Catalyzed, Enantioselective, Vinylogous Mannich Reaction with Aliphatic Aldimines. Synthesis 2011, 2011, 4050–4058. 10.1055/s-0031-1289303. [DOI] [Google Scholar]
- Abels F.; Lindemann C.; Koch E.; Schneider C. A General Organocatalytic Approach toward the Enantioselective Total Synthesis of Indolizidine Based Alkaloids. Org. Lett. 2012, 14, 5972–5975. 10.1021/ol302871u. [DOI] [PubMed] [Google Scholar]
- Abels F.; Lindemann C.; Schneider C. A General Strategy for the Catalytic, Highly Enantio- and Diastereoselective Synthesis of Indolizidine-Based Alkaloids. Chem. - Eur. J. 2014, 20, 1964–1979. 10.1002/chem.201304086. [DOI] [PubMed] [Google Scholar]
- Li H.; Korotkov A.; Chapman C. W.; Eastman A.; Wu J. Enantioselective Formal Syntheses of 11 Nuphar Alkaloids and Discovery of Potent Apoptotic Monomeric Analogues. Angew. Chem., Int. Ed. 2016, 55, 3509–3513. 10.1002/anie.201600106. [DOI] [PubMed] [Google Scholar]
- Boomhoff M.; Schneider C. A Novel Three-Component [3 + 2] Cycloannulation Process for the Rapid and Highly Stereoselective Synthesis of Pyrrolobenzoxazoles. Chem. - Eur. J. 2012, 18, 4185–4189. 10.1002/chem.201200405. [DOI] [PubMed] [Google Scholar]
- More recently, this reaction has been studied “on flow” protocols and by mass spectrometry techniques and theoretical modeling calculations:Schulze S.; Pahl M.; Stolz F.; Appun J.; Abel B.; Schneider C.; Belder D. Liquid Beam Desorption Mass Spectrometry for the Investigation of Continuous Flow Reactions in Microfluidic Chips. Anal. Chem. 2017, 89, 6175–6181. 10.1021/acs.analchem.7b01026. [DOI] [PubMed] [Google Scholar]
- Stolz F.; Appun J.; Naumov S.; Schneider C.; Abel B. A Complex Catalytic Reaction Caught in the Act: Intermediates and Products Sampling Online by Liquid μ-Beam Mass Spectrometry and Theoretical Modeling. ChemPlusChem 2017, 82, 233–240. 10.1002/cplu.201600347. [DOI] [PubMed] [Google Scholar]
- Appun J.; Stolz F.; Naumov S.; Abel B.; Schneider C. Modular Synthesis of Dipyrroloquinolines: A Combined Synthetic and Mechanistic Study. J. Org. Chem. 2018, 83, 1737–1744. 10.1021/acs.joc.7b02466. [DOI] [PubMed] [Google Scholar]
- Boomhoff M.; Yadav A. K.; Appun J.; Schneider C. Modular, Flexible, and Stereoselective Synthesis of Pyrroloquinolines: Rapid Assembly of Complex Heterocyclic Scaffolds. Org. Lett. 2014, 16, 6236–6239. 10.1021/ol503167h. [DOI] [PubMed] [Google Scholar]
- Boomhoff M.; Ukis R.; Schneider C. A Highly Stereocontrolled, One-Pot Approach toward Pyrrolobenzoxazinones and Pyrroloquinazolinones through a Lewis Acid-Catalyzed [3 + 2]-Cycloannulation Process. J. Org. Chem. 2015, 80, 8236–8244. 10.1021/acs.joc.5b01293. [DOI] [PubMed] [Google Scholar]
- For other diastereoselective VMMnRs employing linear silyldienolates, see:Yang Y.; Phillips D. P.; Pan S. A Stereoselective Approach to 6-Alkylated Piperidinone & 2-Piperidine via Three-Component Vinylogous Mannich Reactions (VMR) and a Concise Synthesis of (S)-Anabasine. Tetrahedron Lett. 2011, 52, 1549–1552. 10.1016/j.tetlet.2011.01.090. [DOI] [Google Scholar]
- Liu Y.; Liu J.; Huang Y.; Qing F. L. Lewis Acid-Catalyzed Regioselective Synthesis of Chiral α-Fluoroalkyl Amines via Asymmetric Addition of Silyl Dienolates to Fluorinated Sulfinylimines. Chem. Commun. 2013, 49, 7492–7494. 10.1039/c3cc43741h. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Yuan Z.; Shi M. Axially Chiral Phosphine–Oxazoline Ligands in the Silver(I)-Catalyzed Asymmetric Mannich Reaction of Fluorinated Aldimines with Trimethylsiloxyfuran. Tetrahedron: Asymmetry 2010, 21, 943–951. 10.1016/j.tetasy.2010.05.025. [DOI] [Google Scholar]
- Zhao Q.-Y.; Yuan Z.-L.; Shi M. Highly Diastereo- and Enantioselective Vinylogous Mannich Reactions of Fluorinated Aldimines with Siloxyfurans. Adv. Synth. Catal. 2011, 353, 637–643. 10.1002/adsc.201000843. [DOI] [Google Scholar]
- Zhao Q. Y.; Shi M. Axially Chiral Phosphine-Oxazoline Ligands in Silver(I)-Catalyzed Asymmetric Mannich Reaction of N-Boc Aldimines with Trimethylsiloxyfuran. Tetrahedron 2011, 67, 3724–3732. 10.1016/j.tet.2011.03.046. [DOI] [Google Scholar]
- For other AgOAc-catalyzed VMMnRs yielding functionalized δ-amino-γ-butenolide adducts, see also:Zheng L. S.; Li L.; Yang K. F.; Zheng Z. J.; Xiao X. Q.; Xu L. W. New Silver(I)-Monophosphine Complex Derived from Chiral Ar-BINMOL: Synthesis and Catalytic Activity in Asymmetric Vinylogous Mannich Reaction. Tetrahedron 2013, 69, 8777–8784. 10.1016/j.tet.2013.07.105. [DOI] [Google Scholar]
- Shi Y. H.; Wang Z.; Shi Y.; Deng W. P. Facile and Highly Diastereoselective Synthesis of 3-Aminooxindoles via AgOAc-Catalyzed Vinylogous Mannich Reaction. Tetrahedron 2012, 68, 3649–3653. 10.1016/j.tet.2012.02.046. [DOI] [Google Scholar]
- Silverio D. L.; Fu P.; Carswell E. L.; Snapper M. L.; Hoveyda A. H. N-Substituted Tertiary and O-Substituted Quaternary Carbon Stereogenic Centers by Site-, Diastereo- and Enantioselective Vinylogous Mannich Reactions. Tetrahedron Lett. 2015, 56, 3489–3493. 10.1016/j.tetlet.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan S.-T.; Luo J.-M.; Du Y.; Huang P.-Q. Asymmetric Vinylogous Mannich Reactions: A Versatile Approach to Functionalized Heterocycles. Org. Lett. 2011, 13, 4938–4941. 10.1021/ol2020384. [DOI] [PubMed] [Google Scholar]
- Guo L.-D.; Liang P.; Zheng J.-F.; Huang P.-Q. A Concise and Divergent Approach to Hydroxylated Piperidine Alkaloids and Azasugar Lactams. Eur. J. Org. Chem. 2013, 2013, 2230–2236. 10.1002/ejoc.201201618. [DOI] [Google Scholar]
- Ye J. L.; Zhang Y. F.; Liu Y.; Zhang J. Y.; Ruan Y. P.; Huang P. Q. Studies on the Asymmetric Synthesis of Pandamarilactonines: An Unexpected Syn-Selective Vinylogous Mannich Reaction of N-tert-Butanesulfinimines. Org. Chem. Front. 2015, 2, 697–704. 10.1039/C5QO00098J. [DOI] [Google Scholar]
- Rao V. U. B.; Jadhav A. P.; Garad D.; Singh R. P. Asymmetric Vinylogous Mannich Reaction of Silyloxy Furans with N-tert-Butanesulfinyl Ketimines. Org. Lett. 2014, 16, 648–651. 10.1021/ol4037117. [DOI] [PubMed] [Google Scholar]
- For other peculiar, diastereoselective VMMnRs between silyloxyfurans and diverse imine- and nitrone-containing chiral auxiliaries, see:Tamura O.; Takeda K.; Mita N.; Sakamoto M.; Okamoto I.; Morita N.; Ishibashi H. Stereoselective Vinylogous Mannich Reaction of 2-Trimethylsilyloxyfuran with N-Gulosyl Nitrones. Org. Biomol. Chem. 2011, 9, 7411–7419. 10.1039/c1ob06067h. [DOI] [PubMed] [Google Scholar]
- Yu J.; Miao Z.; Chen R. Highly Efficient Asymmetric Vinylogous Mannich Reaction Induced by O-Pivaloylated D-Galactosylamine as the Chiral Auxiliary. Org. Biomol. Chem. 2011, 9, 1756–1762. 10.1039/c0ob01048k. [DOI] [PubMed] [Google Scholar]
- Hayashi M.; Sano M.; Funahashi Y.; Nakamura S. Cinchona Alkaloid Amide/Copper(II) Catalyzed Diastereo- and Enantioselective Vinylogous Mannich Reaction of Ketimines with Siloxyfurans. Angew. Chem., Int. Ed. 2013, 52, 5557–5560. 10.1002/anie.201301917. [DOI] [PubMed] [Google Scholar]
- Janody S.; Hermange P.; Retailleau P.; Thierry J.; Dodd R. H. Diastereoselective Three-Component Vinylogous Mannich Reaction of Nitrogen Heterocycles, Acyl/Sulfonyl Chlorides, and Silyloxyfurans/Pyrroles. J. Org. Chem. 2014, 79, 5673–5683. 10.1021/jo500827j. [DOI] [PubMed] [Google Scholar]
- For another application of such methodology to the synthesis of pharmaceutically relevant bicuculline derivatives, see:Ramerstorfer J.; Foppa V.; Thiery H.; Hermange P.; Janody S.; Berger M.; Dodd R.; Sieghart W. GABA A Receptor Subtype-Selectivity of Novel Bicuculline Derivatives. Curr. Med. Chem. 2015, 22, 771–780. 10.2174/0929867321666141106110356. [DOI] [PubMed] [Google Scholar]
- Razet R.; Thomet U.; Furtmüller R.; Chiaroni A.; Sigel E.; Sieghart W.; Dodd R. H. 5-[1’-(2’-N-Arylsulfonyl-1’,2’,3′,4’-Tetrahydroisoquinolyl)]-4,5-Dihydro-2(3H)-Furanones: Positive Allosteric Modulators of the GABA A Receptor with a New Mode of Action. J. Med. Chem. 2000, 43, 4363–4366. 10.1021/jm001049i. [DOI] [PubMed] [Google Scholar]
- Hermange P.; Dau M. E. T. H.; Retailleau P.; Dodd R. H. Highly Diastereoselective Three-Component Vinylogous Mannich Reaction between Isoquinolines, Acyl/Sulfonyl Chlorides, and Silyloxyfurans. Org. Lett. 2009, 11, 4044–4047. 10.1021/ol901452d. [DOI] [PubMed] [Google Scholar]
- Tuo S.-C.; Ye J.-L.; Wang A.-E.; Huang S.-Y.; Huang P.-Q. Concise Asymmetric Total Synthesis of 9-Epi-Sessilifoliamide J. Org. Lett. 2011, 13, 5270–5273. 10.1021/ol202140y. [DOI] [PubMed] [Google Scholar]
- Liu X.-K.; Ye J.-L.; Ruan Y.-P.; Li Y.-X.; Huang P.-Q. Total Synthesis of (−)-Sessilifoliamide J. J. Org. Chem. 2013, 78, 35–41. 10.1021/jo3014484. [DOI] [PubMed] [Google Scholar]
- Miyatake-Ondozabal H.; Bannwart L. M.; Gademann K. Enantioselective Total Synthesis of Virosaine A and Bubbialidine. Chem. Commun. 2013, 49, 1921–1923. 10.1039/c3cc38783f. [DOI] [PubMed] [Google Scholar]
- Bardají G. G.; Cantó M.; Alibés R.; Bayón P.; Busqué F.; de March P.; Figueredo M.; Font J. Diastereoselective Synthesis of Allosecurinine and Viroallosecurinine from Menisdaurilide. J. Org. Chem. 2008, 73, 7657–7662. 10.1021/jo801470u. [DOI] [PubMed] [Google Scholar]
- Kamath A.; Fabritius C.-H.; Philouze C.; Delair P. A New Approach to the 8b-Azaacenaphthylene Ring System. Org. Biomol. Chem. 2015, 13, 9834–9843. 10.1039/C5OB01013F. [DOI] [PubMed] [Google Scholar]
- Wang Q.; van Gemmeren M.; List B. Asymmetric Disulfonimide-Catalyzed Synthesis of δ-Amino-β-Ketoester Derivatives by Vinylogous Mukaiyama-Mannich Reactions. Angew. Chem., Int. Ed. 2014, 53, 13592–13595. 10.1002/anie.201407532. [DOI] [PubMed] [Google Scholar]
- For a seminal work, see:Lu X.; Zhang C.; Xu Z. Reactions of Electron-Deficient Alkynes and Allenes under Phosphine Catalysis. Acc. Chem. Res. 2001, 34, 535–544. 10.1021/ar000253x. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Xu X.; Kwon O. Phosphine Catalysis of Allenes with Electrophiles. Chem. Soc. Rev. 2014, 43, 2927–2940. 10.1039/C4CS00054D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y. C.; Kwon O. Advances in Nucleophilic Phosphine Catalysis of Alkenes, Allenes, Alkynes, and MBHADs. Chem. Commun. 2013, 49, 11588–11619. 10.1039/c3cc47368f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.-F.; Lan J.; Kwon O. An Expedient Phosphine-Catalyzed [4 + 2] Annulation: Synthesis of Highly Functionalized Tetrahydropyridines. J. Am. Chem. Soc. 2003, 125, 4716–4717. 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]
- Tran Y. S.; Kwon O. Phosphine-Catalyzed [4 + 2] Annulation: Synthesis of Cyclohexenes. J. Am. Chem. Soc. 2007, 129, 12632–12633. 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran Y. S.; Martin T. J.; Kwon O. Phosphine-Catalyzed [4 + 2] Annulations of 2-Alkylallenoates and Olefins: Synthesis of Multisubstituted Cyclohexenes. Chem. - Asian J. 2011, 6, 2101–2106. 10.1002/asia.201100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong F.; Han X.; Wang Y.; Lu Y. Highly Enantioselective [4 + 2] Annulations Catalyzed by Amino Acid-Based Phosphines: Synthesis of Functionalized Cyclohexenes and 3-Spirocyclohexene-2-Oxindoles. Chem. Sci. 2012, 3, 1231–1234. 10.1039/c2sc00963c. [DOI] [Google Scholar]
- Yao W.; Dou X.; Lu Y. Highly Enantioselective Synthesis of 3,4-Dihydropyrans through a Phosphine-Catalyzed [4 + 2] Annulation of Allenones and β,γ-Unsaturated α-Keto Esters. J. Am. Chem. Soc. 2015, 137, 54–57. 10.1021/ja5109358. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Chai Z.; Cao D.; Wang H.; Chen J.; Zhao G. Highly Enantioselective [4 + 2] Cycloadditions of Allenoates and Dual Activated Olefins Catalyzed by N-Acyl Aminophosphines. Org. Biomol. Chem. 2012, 10, 3195–3201. 10.1039/c2ob25295c. [DOI] [PubMed] [Google Scholar]
- Yang W.; Zhang Y.; Qiu S.; Zhao C.; Zhang L.; Liu H.; Zhou L.; Xiao Y.; Guo H. Phosphine-Catalyzed [4 + 2] Cycloaddition of Unsaturated Pyrazolones with Allenoates: A Concise Approach toward Spiropyrazolones. RSC Adv. 2015, 5, 62343–62347. 10.1039/C5RA11595G. [DOI] [Google Scholar]
- Liu H.; Liu Y.; Yuan C.; Wang G. P.; Zhu S.-F.; Wu Y.; Wang B.; Sun Z.; Xiao Y.; Zhou Q.-L.; et al. Enantioselective Synthesis of Spirobarbiturate-Cyclohexenes through Phosphine-Catalyzed Asymmetric [4 + 2] Annulation of Barbiturate-Derived Alkenes with Allenoates. Org. Lett. 2016, 18, 1302–1305. 10.1021/acs.orglett.6b00239. [DOI] [PubMed] [Google Scholar]
- Yin L.; Takada H.; Lin S.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Vinylogous Conjugate Addition of Unsaturated Butyrolactones to α,β-Unsaturated Thioamides. Angew. Chem., Int. Ed. 2014, 53, 5327–5331. 10.1002/anie.201402332. [DOI] [PubMed] [Google Scholar]
- Yang D.; Wang L.; Zhao D.; Han F.; Zhang B.; Wang R. Development of Metal/Organo Catalytic Systems for Direct Vinylogous Michael Reactions to Build Chiral γ,γ-Disubstituted Butenolides. Chem. - Eur. J. 2013, 19, 4691–4694. 10.1002/chem.201204466. [DOI] [PubMed] [Google Scholar]
- Ji J.; Lin L.; Zhou L.; Zhang Y.; Liu Y.; Liu X.; Feng X. N,N’-Dioxide-Scandium (III)-Catalyzed Asymmetric Michael Addition of β,γ-Unsaturated Butenolides to α,β-Unsaturated γ-Keto Esters. Adv. Synth. Catal. 2013, 355, 2764–2768. 10.1002/adsc.201300586. [DOI] [Google Scholar]
- Quintard A.; Lefranc A.; Alexakis A. Highly Enantioselective Direct Vinylogous Michael Addition of γ-Butenolide to Enals. Org. Lett. 2011, 13, 1540–1543. 10.1021/ol200235j. [DOI] [PubMed] [Google Scholar]
- For a similar VMcR between α,β-unsaturated furanones and enals promoted by α,α-diphenylprolinol silyl ether, see:Luo X.; Zhou Z.; Yu F.; Li X.; Liang X.; Ye J. Asymmetric Vinylogous Michael Reaction of α,β-Unsaturated Aldehyde with Buteno-4-Lactone. Chem. Lett. 2011, 40, 518–520. 10.1246/cl.2011.518. [DOI] [Google Scholar]
- Lagoutte R.; Besnard C.; Alexakis A. Direct Organocatalysed Double Michael Addition of α-Angelica Lactone to Enones. Eur. J. Org. Chem. 2016, 2016, 4372–4381. 10.1002/ejoc.201600707. [DOI] [Google Scholar]
- Zhang W.; Tan D.; Lee R.; Tong G.; Chen W.; Qi B.; Huang K.-W.; Tan C.-H.; Jiang Z. Highly Enantio- and Diastereoselective Reactions of γ-Substituted Butenolides Through Direct Vinylogous Conjugate Additions. Angew. Chem., Int. Ed. 2012, 51, 10069–10073. 10.1002/anie.201205872. [DOI] [PubMed] [Google Scholar]
- Zhao F.; Zhang W.; Yang Y.; Pan Y.; Chen W.; Liu H.; Yan L.; Tan C.-H.; Jiang Z. Synthesis of Sulfur-Substituted α-Stereogenic Amides and Ketones: Highly Enantioselective Sulfa-Michael Additions of 1,4-Dicarbonylbut-2-Enes. Adv. Synth. Catal. 2011, 353, 2624–2630. 10.1002/adsc.201100227. [DOI] [Google Scholar]
- Li X.; Lu M.; Dong Y.; Wu W.; Qian Q.; Ye J.; Dixon D. J. Diastereodivergent Organocatalytic Asymmetric Vinylogous Michael Reactions. Nat. Commun. 2014, 5, 4479. 10.1038/ncomms5479. [DOI] [PubMed] [Google Scholar]
- Manna M. S.; Kumar V.; Mukherjee S. Catalytic Enantioselective Construction of Quaternary Stereocenters by Direct Vinylogous Michael Addition of Deconjugated Butenolides to Nitroolefins. Chem. Commun. 2012, 48, 5193–5195. 10.1039/C2CC31700A. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Ray B.; Rathi P.; Mukherjee S. Catalytic Asymmetric Direct Vinylogous Michael Addition of γ-Aryl-Substituted Deconjugated Butenolides to Nitroolefins and N-Phenylmaleimide. Synthesis 2013, 45, 1641–1646. 10.1055/s-0033-1338705. [DOI] [Google Scholar]
- Sekikawa T.; Kitaguchi T.; Kitaura H.; Minami T.; Hatanaka Y. Catalytic Activity of Epi -Quinine-Derived 3,5-Bis(Trifluoromethyl)Benzamide in Asymmetric Nitro-Michael Reaction of Furanones. Org. Lett. 2015, 17, 3026–3029. 10.1021/acs.orglett.5b01224. [DOI] [PubMed] [Google Scholar]
- Sekikawa T.; Kitaguchi T.; Kitaura H.; Minami T.; Hatanaka Y. Anti-Selective Asymmetric Nitro-Michael Reaction of Furanones: Diastereocontrol by Catalyst. Org. Lett. 2016, 18, 646–649. 10.1021/acs.orglett.5b03539. [DOI] [PubMed] [Google Scholar]
- For another example of direct, asymmetric addition of phthalide derivatives to nitroolefins, see:Luo J.; Wang H.; Zhong F.; Kwiatkowski J.; Xu L. W.; Lu Y. Highly Diastereoselective and Enantioselective Direct Michael Addition of Phthalide Derivatives to Nitroolefins. Chem. Commun. 2013, 49, 5775–5777. 10.1039/c3cc42187b. [DOI] [PubMed] [Google Scholar]
- Rout S.; Joshi H.; Singh V. K. Asymmetric Construction of Remote Vicinal Quaternary and Tertiary Stereocenters via Direct Doubly Vinylogous Michael Addition. Org. Lett. 2018, 20, 2199–2203. 10.1021/acs.orglett.8b00493. [DOI] [PubMed] [Google Scholar]
- Deepake S. K.; Lanjewar A. B.; Thatikonda T.; Gonnade R. G.; Das U. Organocatalytic Asymmetric Cascade Reaction of γ-Substituted Deconjugated Butenolides with o-Formyl-β-Nitrostyrene. ChemistrySelect 2018, 3, 8189–8192. 10.1002/slct.201801467. [DOI] [Google Scholar]
- Manna M. S.; Mukherjee S. Catalytic Asymmetric Direct Vinylogous Michael Addition of Deconjugated Butenolides to Maleimides for the Construction of Quaternary Stereogenic Centers. Chem. - Eur. J. 2012, 18, 15277–15282. 10.1002/chem.201203180. [DOI] [PubMed] [Google Scholar]
- Guo Y.-L.; Jia L.-N.; Peng L.; Qi L.-W.; Zhou J.; Tian F.; Xu X.-Y.; Wang L.-X. Highly Enantioselective Direct Vinylogous Michael Addition of γ-Substituted Deconjugated Butenolides to Maleimides Catalyzed by Chiral Squaramides. RSC Adv. 2013, 3, 16973. 10.1039/c3ra43344g. [DOI] [Google Scholar]
- For another recent example of diastereoselective VMcR between deconjugated butenolides and maleimides, promoted by pepsin, see:Zhang M. J.; Li R.; He Y. H.; Guan Z. Pepsin-Catalyzed Vinylogous Michael Addition of Deconjugated Butenolides and Maleimides in Water. Catal. Commun. 2017, 98, 85–89. 10.1016/j.catcom.2017.03.021. [DOI] [Google Scholar]
- Manna M. S.; Mukherjee S. Remarkable Influence of Secondary Catalyst Site on Enantioselective Desymmetrization of Cyclopentenedione. Chem. Sci. 2014, 5, 1627–1633. 10.1039/C3SC53102C. [DOI] [Google Scholar]
- Simlandy A. K.; Mukherjee S. Catalytic Asymmetric Formal: γ-Allylation of Deconjugated Butenolides. Org. Biomol. Chem. 2016, 14, 5659–5664. 10.1039/C5OB02362A. [DOI] [PubMed] [Google Scholar]
- Wang Z.-H.; Wu Z.-J.; Huang X.-Q.; Yue D.-F.; You Y.; Xu X.-Y.; Zhang X.-M.; Yuan W.-C. Diastereo- and Enantioselective Direct Vinylogous Michael Addition of γ-Substituted Butenolides to 2-Enoylpyridines Catalyzed by Chiral Bifunctional Amine-Squaramides. Chem. Commun. 2015, 51, 15835–15838. 10.1039/C5CC06383C. [DOI] [PubMed] [Google Scholar]
- Das U.; Chen Y.-R.; Tsai Y. L.; Lin W. Organocatalytic Enantioselective Direct Vinylogous Michael Addition of γ-Substituted Butenolides to 3-Aroyl Acrylates and 1,2-Diaroylethylenes. Chem. - Eur. J. 2013, 19, 7713–7717. 10.1002/chem.201301332. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yu C.; Ji Y.; Wang W. Diastereo- and Enantioselective Organocatalytic Direct Conjugate Addition of γ-Butenolide to Chalcones. Chem. - Asian J. 2010, 5, 1303–1306. 10.1002/asia.201000014. [DOI] [PubMed] [Google Scholar]
- Huang H.; Yu F.; Jin Z.; Li W.; Wu W.; Liang X.; Ye J. Asymmetric Vinylogous Michael Reaction of α,β-Unsaturated Ketones with γ-Butenolide under Multifunctional Catalysis. Chem. Commun. 2010, 46, 5957–5959. 10.1039/c0cc01054e. [DOI] [PubMed] [Google Scholar]
- Cho C.-W.; Krische M. J. Regio- and Stereoselective Construction of γ-Butenolides through Phosphine-Catalyzed Substitution of Morita-Baylis-Hillman Acetates: An Organocatalytic Allylic Alkylation. Angew. Chem., Int. Ed. 2004, 43, 6689–6691. 10.1002/anie.200461381. [DOI] [PubMed] [Google Scholar]
- Jiang Y.-Q.; Shi Y.-L.; Shi M. Chiral Phosphine-Catalyzed Enantioselective Construction of γ-Butenolides through Substitution of Morita-Baylis-Hillman Acetates with 2-Trimethylsilyloxy Furan. J. Am. Chem. Soc. 2008, 130, 7202–7203. 10.1021/ja802422d. [DOI] [PubMed] [Google Scholar]
- Cui H.-L.; Huang J.-R.; Lei J.; Wang Z.-F.; Chen S.; Wu L. I.; Chen Y.-C. Direct Asymmetrie Allylic Alkylation of Butenolides with Morita-Baylis-Hillman Carbonates. Org. Lett. 2010, 12, 720–723. 10.1021/ol100014m. [DOI] [PubMed] [Google Scholar]
- Kang T.-C.; Zhao X.; Sha F.; Wu X.-Y. Highly Enantioselective Direct Allylic Alkylation of Butenolides with Morita-Baylis-Hillman Carbonates Catalyzed by Chiral Squaramide-Phosphine. RSC Adv. 2015, 5, 74170–74173. 10.1039/C5RA14667D. [DOI] [Google Scholar]
- Huang X.; Peng J.; Dong L.; Chen Y.-C. Asymmetric Assembly of 2-Oxindole and α-Angelica Lactone Units to Construct Vicinal Quaternary Chiral Centers. Chem. Commun. 2012, 48, 2439–2441. 10.1039/c2cc17777c. [DOI] [PubMed] [Google Scholar]
- Huang X.; Xu D.-C.; Chen C.; Wen Y.-H.; Xie J.-W.; Zhou F.-T. Organocatalytic and Direct Asymmetric Vinylogous Michael Addition of 3-Cyano-4-Methylcoumarins to α,β-Unsaturated Ketones. Tetrahedron Lett. 2010, 51, 6637–6640. 10.1016/j.tetlet.2010.10.055. [DOI] [Google Scholar]
- Kayal S.; Mukherjee S. Catalytic Enantioselective Vinylogous Allylic Alkylation of Coumarins. Org. Lett. 2017, 19, 4944–4947. 10.1021/acs.orglett.7b02421. [DOI] [PubMed] [Google Scholar]
- Kowalczyk D.; Albrecht Ł. Vinylogous Nucleophiles Bearing the Endocyclic Double Bond in the Allylic Alkylation with Morita-Baylis-Hillman Carbonates. Adv. Synth. Catal. 2018, 360, 406–410. 10.1002/adsc.201701185. [DOI] [Google Scholar]
- Jiang H.; Paixão M. W.; Monge D.; Jørgensen K. A. Acyl Phosphonates: Good Hydrogen Bond Acceptors and Ester/Amide Equivalents in Asymmetric Organocatalysis. J. Am. Chem. Soc. 2010, 132, 2775–2783. 10.1021/ja9097803. [DOI] [PubMed] [Google Scholar]
- Alba A.-N. R.; Valero G.; Calbet T.; Font-Bardía M.; Moyano A.; Rios R. Enantioselective Organocatalytic Addition of Azlactones to Maleimides: A Highly Stereocontrolled Entry to 2,2-Disubstituted-2H-Oxazol-5-Ones. Chem. - Eur. J. 2010, 16, 9884–9889. 10.1002/chem.201000239. [DOI] [PubMed] [Google Scholar]
- Dell’Amico L.; Albrecht Ł.; Naicker T.; Poulsen P. H.; Jørgensen K. A. Beyond Classical Reactivity Patterns: Shifting from 1,4- to 1,6-Additions in Regio- and Enantioselective Organocatalyzed Vinylogous Reactions of Olefinic Lactones with Enals and 2,4-Dienals. J. Am. Chem. Soc. 2013, 135, 8063–8070. 10.1021/ja4029928. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Zu L. Organocatalytic Enantioselective Cross-Vinylogous Rauhut–Currier Reaction of Methyl Coumalate with Enals. Angew. Chem., Int. Ed. 2018, 57, 9505–9509. 10.1002/anie.201805019. [DOI] [PubMed] [Google Scholar]
- Li H.; Wu J. Vinylogous Mukaiyama-Michael Reactions of Dihydropyridinones. Org. Lett. 2015, 17, 5424–5427. 10.1021/acs.orglett.5b02778. [DOI] [PubMed] [Google Scholar]
- Nareddy P. R.; Schneider C. A Highly Enantioselective, Organocatalytic [3 + 2]-Cycloannulation Reaction towards the de Novo-Synthesis of 1-Cyclopentenyl-α-Keto Esters. Chem. Commun. 2015, 51, 14797–14800. 10.1039/C5CC05967D. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Ma G. N.; Shi M. Diastereo- and Enantioselective Construction of γ-Butenolides through Chiral Phosphane-Catalyzed Allylic Alkylation of Morita-Baylis-Hillman Acetates. Eur. J. Org. Chem. 2011, 2011, 5146–5155. 10.1002/ejoc.201100704. [DOI] [Google Scholar]
- Zhang Q.; Xiao X.; Lin L.; Liu X.; Feng X. Highly Enantioselective Synthesis of γ-Substituted Butenolides via the Vinylogous Mukaiyama-Michael Reaction Catalyzed by a Chiral Scandium(III)-N,N′-Dioxide Complex. Org. Biomol. Chem. 2011, 9, 5748–5754. 10.1039/c1ob05558e. [DOI] [PubMed] [Google Scholar]
- For a diastereoselective NHC-catalyzed VMMcR between TMSOF and enones, see also:Wang Y.; Du G.-F.; Xing F.; Huang K.-W.; Dai B.; He L. N-Heterocyclic-Carbene-Catalysed Diastereoselective Vinylogous Mukaiyama/Michael Reaction of 2-(Trimethylsilyloxy)Furan and Enones. Asian J. Org. Chem. 2015, 4, 1362–1365. 10.1002/ajoc.201500348. [DOI] [Google Scholar]
- Rout S.; Das A.; Singh V. K. An Asymmetric Vinylogous Mukaiyama–Michael Reaction of α,β-Unsaturated 2-Acyl Imidazoles Catalyzed by Chiral Sc(III)– or Er(III)–Pybox Complexes. Chem. Commun. 2017, 53, 5143–5146. 10.1039/C7CC01763D. [DOI] [PubMed] [Google Scholar]
- Jusseau X.; Retailleau P.; Chabaud L.; Guillou C. Catalytic Enantioselective Vinylogous Mukaiyama-Michael Addition of 2-Silyloxyfurans to Cyclic Unsaturated Oxo Esters. J. Org. Chem. 2013, 78, 2289–2300. 10.1021/jo3020648. [DOI] [PubMed] [Google Scholar]
- The same group reported the diastereoselective VMMcR of silyloxyfurans to cyclic enones or unsaturated oxo esters:Chabaud L.; Jousseaume T.; Retailleau P.; Guillou C. Vinylogous Mukaiyama-Michael Reactions between 2-Silyloxyfurans and Cyclic Enones or Unsaturated Oxo Esters. Eur. J. Org. Chem. 2010, 2010, 5471–5481. 10.1002/ejoc.201000504. [DOI] [Google Scholar]
- Steinkamp A.-D.; Frings M.; Thomé I.; Schiffers I.; Bolm C. Asymmetric Copper-Catalyzed Vinylogous Mukaiyama Michael Addition of Cyclic Dienol Silanes to Unsaturated α-Keto Phosphonates. Chem. - Eur. J. 2015, 21, 7705–7708. 10.1002/chem.201500861. [DOI] [PubMed] [Google Scholar]
- Li J.; Huang R.; Xing Y. K.; Qiu G.; Tao H. Y.; Wang C. J. Catalytic Asymmetric Cascade Vinylogous Mukaiyama 1,6-Michael/Michael Addition of 2-Silyloxyfurans with Azoalkenes: Direct Approach to Fused Butyrolactones. J. Am. Chem. Soc. 2015, 137, 10124–10127. 10.1021/jacs.5b06509. [DOI] [PubMed] [Google Scholar]
- Liu K.; Chang X.; Wang C.-J. Nickel(II)-Catalyzed Cascade Vinylogous Mukaiyama 1,6-Michael/Michael Addition of 2-Silyloxyfuran with N -Sulfonyl-1-Aza-1,3-Dienes: Access to Fused Piperidine/Butyrolactone Skeletons. Org. Lett. 2016, 18, 6288–6291. 10.1021/acs.orglett.6b03150. [DOI] [PubMed] [Google Scholar]
- Kemppainen E. K.; Sahoo G.; Valkonen A.; Pihko P. M. Mukaiyama-Michael Reactions with Acrolein and Methacrolein: A Catalytic Enantioselective Synthesis of the C17-C28 Fragment of Pectenotoxins. Org. Lett. 2012, 14, 1086–1089. 10.1021/ol203486p. [DOI] [PubMed] [Google Scholar]
- Kemppainen E. K.; Sahoo G.; Piisola A.; Hamza A.; Kõtai B.; Pápai I.; Pihko P. M. Mukaiyama-Michael Reactions with Trans-2,5-Diarylpyrrolidine Catalysts: Enantioselectivity Arises from Attractive Noncovalent Interactions, Not from Steric Hindrance. Chem. - Eur. J. 2014, 20, 5983–5993. 10.1002/chem.201304240. [DOI] [PubMed] [Google Scholar]
- Siitonen J. H.; Pihko P. M. Total Synthesis of (+)-Greek Tobacco Lactone. Synlett 2014, 25, 1888–1890. 10.1055/s-0034-1378273. [DOI] [Google Scholar]
- For an achiral version of this pyrrolidine-catalyzed VMMcR applied to the synthesis of stemoamide alkaloids, see also:Zheng X.; Dai X.-J.; Yuan H.-Q.; Ye C.-X.; Ma J.; Huang P.-Q. Umpolung of Hemiaminals: Titanocene-Catalyzed Dehydroxylative Radical Coupling Reactions with Activated Alkenes. Angew. Chem., Int. Ed. 2013, 52, 3494–3498. 10.1002/anie.201210088. [DOI] [PubMed] [Google Scholar]
- Brito G. A.; Sarotti A. M.; Wipf P.; Pilli R. A. Cascade Cyclization Triggered by Imine Formation. Formal Synthesis of the Alkaloid (±)-Stemoamide and Its 9a-Epimer. Tetrahedron Lett. 2015, 56, 6664–6668. 10.1016/j.tetlet.2015.10.017. [DOI] [Google Scholar]
- Wang Y.; Li Z.; Lv L.; Xie Z. Synthetic Study of Rubriflordilactone B: Highly Stereoselective Construction of the C-5-Epi ABCDE Ring System. Org. Lett. 2016, 18, 792–795. 10.1021/acs.orglett.6b00057. [DOI] [PubMed] [Google Scholar]
- Jadhav A. P.; Bhaskara Rao V. U.; Singh P.; Gonnade R. G.; Singh R. P. Asymmetric Vinylogous Michael Reaction of Cyclic Enones with Silyloxy Furans. Chem. Commun. 2015, 51, 13941–13944. 10.1039/C5CC05617A. [DOI] [PubMed] [Google Scholar]
- For other diastereoselective VMMcR promoted by diverse non-metal catalysts or promoters, see:Scettri A.; Villano R.; Manzo P.; Acocella M. R. Mukaiyama-Michael Vinylogous Additions to Nitroalkenes under Solvent-Free Conditions. Cent. Eur. J. Chem. 2012, 10, 47–53. 10.2478/s11532-011-0122-7. [DOI] [Google Scholar]
- Acocella M. R.; Mauro M.; Falivene L.; Cavallo L.; Guerra G. Inverting the Diastereoselectivity of the Mukaiyama-Michael Addition with Graphite-Based Catalysts. ACS Catal. 2014, 4, 492–496. 10.1021/cs401053t. [DOI] [Google Scholar]
- Della Sala G.; Sicignano M.; Schettini R.; De Riccardis F.; Cavallo L.; Minenkov Y.; Batisse C.; Hanquet G.; Leroux F.; Izzo I. Switchable Diastereoselectivity in the Fluoride-Promoted Vinylogous Mukaiyama-Michael Reaction of 2-[(Trimethylsilyl)Oxy]Furan Catalyzed by Crown Ethers. J. Org. Chem. 2017, 82, 6629–6637. 10.1021/acs.joc.7b00743. [DOI] [PubMed] [Google Scholar]
- Wang J.; Chen J.; Kee C. W.; Tan C. H. Enantiodivergent and γ-Selective Asymmetric Allylic Amination. Angew. Chem., Int. Ed. 2012, 51, 2382–2386. 10.1002/anie.201107317. [DOI] [PubMed] [Google Scholar]
- Zhang H.-J.; Schuppe A. W.; Pan S.-T.; Chen J.-X.; Wang B.-R.; Newhouse T. R.; Yin L. Copper-Catalyzed Vinylogous Aerobic Oxidation of Unsaturated Compounds with Air. J. Am. Chem. Soc. 2018, 140, 5300–5310. 10.1021/jacs.8b01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh C. C. J.; Schmid M.; Peters B.; Fang X.; Lautens M. Exploiting Distal Reactivity of Coumarins: A Rhodium-Catalyzed Vinylogous Asymmetric Ring-Opening Reaction. Angew. Chem., Int. Ed. 2016, 55, 4600–4604. 10.1002/anie.201600654. [DOI] [PubMed] [Google Scholar]
- Xu H.; Laraia L.; Schneider L.; Louven K.; Strohmann C.; Antonchick A. P.; Waldmann H. Highly Enantioselective Catalytic Vinylogous Propargylation of Coumarins Yields a Class of Autophagy Inhibitors. Angew. Chem., Int. Ed. 2017, 56, 11232–11236. 10.1002/anie.201706005. [DOI] [PubMed] [Google Scholar]
- Sarkar R.; Mitra S.; Mukherjee S. Iridium-Catalyzed Enantioselective Direct Vinylogous Allylic Alkylation of Coumarins. Chem. Sci. 2018, 9, 5767–5772. 10.1039/C8SC02041H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.-Y.; Xiao J.-Z.; Yin L. Iridium-Catalyzed Direct Asymmetric Vinylogous Allylic Alkylation. Chem. Commun. 2018, 54, 11957–11960. 10.1039/C8CC07249C. [DOI] [PubMed] [Google Scholar]
- Chen H.; Lu X.; Xia X.; Zhu Q.; Song Y.; Chen J.; Cao W.; Wu X. Asymmetric Catalytic [4 + 2] Cycloaddition via Cu–Allenylidene Intermediate: Stereoselective Synthesis of Tetrahydroquinolines Fused with a γ-Lactone Moiety. Org. Lett. 2018, 20, 1760–1763. 10.1021/acs.orglett.8b00253. [DOI] [PubMed] [Google Scholar]
- Chen M.; Hartwig J. F. Iridium-Catalyzed Regio- and Enantioselective Allylic Substitution of Trisubstituted Allylic Electrophiles. Angew. Chem., Int. Ed. 2016, 55, 11651–11655. 10.1002/anie.201607053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.-Z.; Jiang Y.; Guan Y.-Q.; Sha F.; Wu X.-Y. Direct Enantioselective Vinylogous Aldol-Cyclization Cascade Reaction of Allyl Pyrazoleamides with Isatins: Asymmetric Construction of Spirocyclic Oxindole-Dihydropyranones. Chem. Commun. 2014, 50, 10790–10792. 10.1039/C4CC04235B. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Fu J.-H.; Li T.-Z.; Sha F.; Wu X.-Y. Enantioselective Direct Vinylogous Aldol-Cyclization Cascade Reaction between β,γ-Unsaturated Amides and o-Quinones. Org. Biomol. Chem. 2016, 14, 6435–6441. 10.1039/C6OB00893C. [DOI] [PubMed] [Google Scholar]
- Zhang Z.-G.; Fu J.-H.; Sha F.; Wu X.-Y. Highly Chemo- and Enantioselective Vinylogous Aldol/Cyclization Cascade Reaction to Construct Chiral 5,6-Dihydropyran-2-ones. Tetrahedron 2018, 74, 3557–3563. 10.1016/j.tet.2018.05.011. [DOI] [Google Scholar]
- David J. G.; Bai W.-J.; Weaver M. G.; Pettus T. R. R. A General Diastereoselective Catalytic Vinylogous Aldol Reaction among Tetramic Acid-Derived Pyrroles. Org. Lett. 2014, 16, 4384–4387. 10.1021/ol501715r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball-Jones N. R.; Badillo J. J.; Franz A. K. Strategies for the Enantioselective Synthesis of Spirooxindoles. Org. Biomol. Chem. 2012, 10, 5165–5181. 10.1039/c2ob25184a. [DOI] [PubMed] [Google Scholar]
- Hong L.; Wang R. Recent Advances in Asymmetric Organocatalytic Construction of 3,3′-Spirocyclic Oxindoles. Adv. Synth. Catal. 2013, 355, 1023–1052. 10.1002/adsc.201200808. [DOI] [Google Scholar]
- Han J.-L.; Chang C.-H. An Asymmetric Assembly of Spirooxindole Dihydropyranones through a Direct Enantioselective Organocatalytic Vinylogous Aldol-Cyclization Cascade Reaction of 3-Alkylidene Oxindoles with Isatins. Chem. Commun. 2016, 52, 2322–2325. 10.1039/C5CC08883F. [DOI] [PubMed] [Google Scholar]
- Han J.-L.; Tsai Y.-D.; Chang C.-H. Asymmetric Synthesis of Spirooxindole δ-Lactones with Vicinal Tertiary and Quaternary Stereocenters via Regio-, Diastereo-, and Enantioselective Organocatalytic Vinylogous Aldol-Cyclization Cascade Reaction. Adv. Synth. Catal. 2017, 359, 4043–4049. 10.1002/adsc.201701104. [DOI] [Google Scholar]
- Kumar K.; Jaiswal M. K.; Singh R. P. Asymmetric Vinylogous Aldol Reaction of α-Ketoesters with 3-Alkylidene Oxindoles. Adv. Synth. Catal. 2017, 359, 4136–4140. 10.1002/adsc.201700758. [DOI] [Google Scholar]
- Crotti S.; Di Iorio N.; Mazzanti A.; Righi P.; Bencivenni G. Enantioselective Synthesis of Trifluoromethyl α,β-Unsaturated δ-Lactones via Vinylogous Aldol-Lactonization Cascade. J. Org. Chem. 2018, 83, 12440–12448. 10.1021/acs.joc.8b01672. [DOI] [PubMed] [Google Scholar]
- Crotti S.; Belletti G.; Di Iorio N.; Marotta E.; Mazzanti A.; Righi P.; Bencivenni G. Asymmetric Vinylogous Aldol Addition of Alkylidene Oxindoles on Trifluoromethyl-α,β-Unsaturated Ketones. RSC Adv. 2018, 8, 33451–33458. 10.1039/C8RA06615A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D. A.; Stephen Clark J.; Metternich R.; Novack V. J.; Sheppard G. S. Diastereoselective Aldol Reactions Using β-Keto Imide Derived Enolates. A Versatile Approach to the Assemblage of Polypropionate Systems. J. Am. Chem. Soc. 1990, 112, 866–868. 10.1021/ja00158a056. [DOI] [Google Scholar]
- Shirokawa S.; Kamiyama M.; Nakamura T.; Okada M.; Nakazaki A.; Hosokawa S.; Kobayashi S. Remote Asymmetric Induction with Vinylketene Silyl N,O-Acetal. J. Am. Chem. Soc. 2004, 126, 13604–13605. 10.1021/ja0465855. [DOI] [PubMed] [Google Scholar]
- Hosokawa S. Recent Development of Vinylogous Mukaiyama Aldol Reactions. Tetrahedron Lett. 2018, 59, 77–88. 10.1016/j.tetlet.2017.11.056. [DOI] [Google Scholar]
- Hosokawa S. Remote Asymmetric Induction Reactions Using a E,E-Vinylketene Silyl N,O-Acetal and the Wide Range Stereocontrol Strategy for the Synthesis of Polypropionates. Acc. Chem. Res. 2018, 51, 1301–1314. 10.1021/acs.accounts.8b00125. [DOI] [PubMed] [Google Scholar]
- Hosokawa S.; Matsushita K.; Tokimatsu S.; Toriumi T.; Suzuki Y.; Tatsuta K. The First Total Synthesis and Structural Determination of Epi-Cochlioquinone A. Tetrahedron Lett. 2010, 51, 5532–5536. 10.1016/j.tetlet.2010.07.153. [DOI] [Google Scholar]
- Matsui R.; Seto K.; Sato Y.; Suzuki T.; Nakazaki A.; Kobayashi S. Convergent Total Synthesis of (+)-TMC-151C by a Vinylogous Mukaiyama Aldol Reaction and Ring-Closing Metathesis. Angew. Chem., Int. Ed. 2011, 50, 680–683. 10.1002/anie.201006230. [DOI] [PubMed] [Google Scholar]
- Fujita K.; Matsui R.; Suzuki T.; Kobayashi S. Concise Total Synthesis of (−)-Myxalamide A. Angew. Chem., Int. Ed. 2012, 51, 7271–7274. 10.1002/anie.201203093. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y.; Matsui R.; Suzuki T.; Nakazaki A.; Kobayashi S. Stereoselective Vinylogous Mukaiyama Aldol Reaction of α-Haloenals. Chem. Pharm. Bull. 2011, 59, 522–524. 10.1248/cpb.59.522. [DOI] [PubMed] [Google Scholar]
- Yamaoka M.; Nakazaki A.; Kobayashi S. Rate Enhancement by Water in a TiCl4-Mediated Stereoselective Vinylogous Mukaiyama Aldol Reaction. Tetrahedron Lett. 2010, 51, 287–289. 10.1016/j.tetlet.2009.10.140. [DOI] [Google Scholar]
- Nakamura T.; Harachi M.; Kano T.; Mukaeda Y.; Hosokawa S. Concise Synthesis of Reduced Propionates by Stereoselective Reductions Combined with the Kobayashi Reaction. Org. Lett. 2013, 15, 3170–3173. 10.1021/ol401406m. [DOI] [PubMed] [Google Scholar]
- Höfle G.; Gerth K.; Reichenbach H.; Kunze B.; Sasse F.; Forche E.; Prusov E. V. Isolation, Biological Activity Evaluation, Structure Elucidation, and Total Synthesis of Eliamid: A Novel Complex I Inhibitor. Chem. - Eur. J. 2012, 18, 11362–11370. 10.1002/chem.201201879. [DOI] [PubMed] [Google Scholar]
- Hoecker J.; Gademann K. Enantioselective Total Syntheses and Absolute Configuration of JBIR-02 and Mer-A2026B. Org. Lett. 2013, 15, 670–673. 10.1021/ol303502a. [DOI] [PubMed] [Google Scholar]
- Ohashi T.; Hosokawa S. Total Syntheses of Stoloniferol B and Penicitol A, and Structural Revision of Fusaraisochromanone. Org. Lett. 2018, 20, 3021–3024. 10.1021/acs.orglett.8b01048. [DOI] [PubMed] [Google Scholar]
- Jahns C.; Hoffmann T.; Müller S.; Gerth K.; Washausen P.; Höfle G.; Reichenbach H.; Kalesse M.; Müller R. Pellasoren: Structure Elucidation, Biosynthesis, and Total Synthesis of a Cytotoxic Secondary Metabolite from Sorangium Cellulosum. Angew. Chem., Int. Ed. 2012, 51, 5239–5243. 10.1002/anie.201200327. [DOI] [PubMed] [Google Scholar]
- Hartmann O.; Kalesse M. The Structure Elucidation and Total Synthesis of β-Lipomycin. Angew. Chem., Int. Ed. 2014, 53, 7335–7338. 10.1002/anie.201402259. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Fujimura M.; Fujita K.; Kobayashi S. Total Synthesis of (+)-Methynolide Using a Ti-Mediated Aldol Reaction of a Lactyl-Bearing Oxazolidin-2-one, and a Vinylogous Mukaiyama Aldol Reaction. Tetrahedron 2017, 73, 3652–3659. 10.1016/j.tet.2017.03.093. [DOI] [Google Scholar]
- Kato T.; Sato T.; Kashiwagi Y.; Hosokawa S. Synthetic Studies on Aculeximycin: Synthesis of C24–C40 Segment by Kobayashi Aldolization and Epoxide Rearrangements. Org. Lett. 2015, 17, 2274–2277. 10.1021/acs.orglett.5b00965. [DOI] [PubMed] [Google Scholar]
- Shinoyama M.; Shirokawa S.; Nakazaki A.; Kobayashi S. A Switch of Facial Selectivities Using α-Heteroatom-Substituted Aldehydes in the Vinylogous Mukaiyama Aldol Reaction. Org. Lett. 2009, 11, 1277–1280. 10.1021/ol9000312. [DOI] [PubMed] [Google Scholar]
- Mukaeda Y.; Kato T.; Hosokawa S. Syn-Selective Kobayashi Aldol Reaction Using the E,E-Vinylketene Silyl N,O-Acetal. Org. Lett. 2012, 14, 5298–5301. 10.1021/ol3024677. [DOI] [PubMed] [Google Scholar]
- Kanoh N.; Kawamata A.; Itagaki T.; Miyazaki Y.; Yahata K.; Kwon E.; Iwabuchi Y. A Concise and Unified Strategy for Synthesis of the C1–C18 Macrolactone Fragments of FD-891, FD-892 and Their Analogues: Formal Total Synthesis of FD-891. Org. Lett. 2014, 16, 5216–5219. 10.1021/ol502633j. [DOI] [PubMed] [Google Scholar]
- Villadsen N. L.; Jacobsen K. M.; Keiding U. B.; Weibel E. T.; Christiansen B.; Vosegaard T.; Bjerring M.; Jensen F.; Johannsen M.; Tørring T.; et al. Synthesis of ent-BE-43547A1 Reveals a Potent Hypoxia-Selective Anticancer Agent and Uncovers the Biosynthetic Origin of the APD-CLD Natural Products. Nat. Chem. 2017, 9, 264–272. 10.1038/nchem.2657. [DOI] [PubMed] [Google Scholar]
- Tsukada H.; Mukaeda Y.; Hosokawa S. Syn-Selective Kobayashi Aldol Reaction Using Acetals. Org. Lett. 2013, 15, 678–681. 10.1021/ol303519y. [DOI] [PubMed] [Google Scholar]
- Wang L.; Xi Y.; Yang S.; Zhu R.; Liang Y.; Chen J.; Yang Z. Asymmetric Total Synthesis and Structural Elucidation of NFAT-68. Org. Lett. 2011, 13, 74–77. 10.1021/ol102574d. [DOI] [PubMed] [Google Scholar]
- Yang S.; Xi Y.; Zhu R.; Wang L.; Chen J.; Yang Z. Asymmetric Total Syntheses of Ansamacrolactams (+)-Q-1047H-A-A and (+)-Q-1047H-R-A. Org. Lett. 2013, 15, 812–815. 10.1021/ol400038h. [DOI] [PubMed] [Google Scholar]
- Jürjens G.; Kirschning A. Synthesis of a Cytotoxic Ansamycin Hybrid. Org. Lett. 2014, 16, 3000–3003. 10.1021/ol5011278. [DOI] [PubMed] [Google Scholar]
- Symkenberg G.; Kalesse M. Syn-Selective Vinylogous Kobayashi Aldol Reaction. Org. Lett. 2012, 14, 1608–1611. 10.1021/ol300353w. [DOI] [PubMed] [Google Scholar]
- Symkenberg G.; Kalesse M. Structure Elucidation and Total Synthesis of Kulkenon. Angew. Chem., Int. Ed. 2014, 53, 1795–1798. 10.1002/anie.201309386. [DOI] [PubMed] [Google Scholar]
- Lisboa M. P.; Jones D. M.; Dudley G. B. Formal Synthesis of Palmerolide A, Featuring Alkynogenic Fragmentation and Syn-Selective Vinylogous Aldol Chemistry. Org. Lett. 2013, 15, 886–889. 10.1021/ol400014e. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Wang L.; Zhu R.; Deng L.; Yang Y.; Quan J.; Chen J.; Yang Z. An Unprecedented Silver Salt Effect Switches the Facial Selectivity in the Vinylogous Mukaiyama Aldol Reaction. Adv. Synth. Catal. 2010, 352, 2387–2393. 10.1002/adsc.201000420. [DOI] [Google Scholar]
- Miyatake-Ondozabal H.; Kaufmann E.; Gademann K. Total Synthesis of the Protected Aglycon of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). Angew. Chem., Int. Ed. 2015, 54, 1933–1936. 10.1002/anie.201409464. [DOI] [PubMed] [Google Scholar]
- Hattori H.; Kaufmann E.; Miyatake-Ondozabal H.; Berg R.; Gademann K. Total Synthesis of Tiacumicin A. Total Synthesis, Relay Synthesis, and Degradation Studies of Fidaxomicin (Tiacumicin B, Lipiarmycin A3). J. Org. Chem. 2018, 83, 7180–7205. 10.1021/acs.joc.8b00101. [DOI] [PubMed] [Google Scholar]
- Larsen B. J.; Sun Z.; Nagorny P. Synthesis of Eukaryotic Translation Elongation Inhibitor Lactimidomycin via Zn(II)-Mediated Horner–Wadsworth–Emmons Macrocyclization. Org. Lett. 2013, 15, 2998–3001. 10.1021/ol401186f. [DOI] [PubMed] [Google Scholar]
- Nagasawa T.; Kuwahara S. Formal Total Synthesis of Lactimidomycin. Org. Lett. 2013, 15, 3002–3005. 10.1021/ol401214f. [DOI] [PubMed] [Google Scholar]
- Liao L.; Zhou J.; Xu Z.; Ye T. Concise Total Synthesis of Nannocystin A. Angew. Chem., Int. Ed. 2016, 55, 13263–13266. 10.1002/anie.201606679. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Xu X.; Yang C.-H.; Tian Y.; Chen X.; Lian L.; Pan W.; Su X.; Zhang W.; Chen Y. Total Synthesis of Nannocystin A. Org. Lett. 2016, 18, 5768–5770. 10.1021/acs.orglett.6b02729. [DOI] [PubMed] [Google Scholar]
- Sagawa N.; Sato H.; Hosokawa S. Remote Asymmetric Induction Using Acetate-Type Vinylketene Silyl N,O-Acetals. Org. Lett. 2017, 19, 198–201. 10.1021/acs.orglett.6b03476. [DOI] [PubMed] [Google Scholar]
- Sagawa N.; Moriya H.; Hosokawa S. Syn Selective Vinylogous Mukaiyama Aldol Reaction Using Z,E-Vinylketene N,O-Acetal with Acetals. Org. Lett. 2017, 19, 250–253. 10.1021/acs.orglett.6b03549. [DOI] [PubMed] [Google Scholar]
- Frings M.; Goedert D.; Bolm C. Enantioselective Synthesis of Highly Functionalised Amides by Copper-Catalysed Vinylogous Mukaiyama Aldol Reaction. Chem. Commun. 2010, 46, 5497–5499. 10.1039/c0cc00996b. [DOI] [PubMed] [Google Scholar]
- Zambrano V.; Rassu G.; Roggio A.; Pinna L.; Zanardi F.; Curti C.; Casiraghi G.; Battistini L. Asymmetric Total Synthesis of 1-Deoxy-7,8-di-epi-Castanospermine. Org. Biomol. Chem. 2010, 8, 1725–1730. 10.1039/b924721a. [DOI] [PubMed] [Google Scholar]
- Ye J.-L.; Liu Y.; Yang Z.-P.; Huang P.-Q. The Asymmetric Total Synthesis of (+)-N-Acetyl Norloline. Chem. Commun. 2016, 52, 561–563. 10.1039/C5CC07480K. [DOI] [PubMed] [Google Scholar]
- Rassu G.; Zambrano V.; Tanca R.; Sartori A.; Battistini L.; Zanardi F.; Curti C.; Casiraghi G. 3-Alkenyl-2-Silyloxyindoles: An Enabling, Yet Understated Progeny of Vinylogous Carbon Nucleophiles. Eur. J. Org. Chem. 2012, 2012, 466–470. 10.1002/ejoc.201101446. [DOI] [Google Scholar]
- Curti C.; Sartori A.; Battistini L.; Brindani N.; Rassu G.; Pelosi G.; Lodola A.; Mor M.; Casiraghi G.; Zanardi F. Pushing the Boundaries of Vinylogous Reactivity: Catalytic Enantioselective Mukaiyama Aldol Reactions of Highly Unsaturated 2-Silyloxyindoles. Chem. - Eur. J. 2015, 21, 6433–6442. 10.1002/chem.201500083. [DOI] [PubMed] [Google Scholar]
- Zhang H.-J.; Shi C.-Y.; Zhong F.; Yin L. Direct Asymmetric Vinylogous and Bisvinylogous Mannich-Type Reaction Catalyzed by a Copper(I) Complex. J. Am. Chem. Soc. 2017, 139, 2196–2199. 10.1021/jacs.6b13042. [DOI] [PubMed] [Google Scholar]
- Zhong F.; Yue W.-J.; Zhang H.-J.; Zhang C.-Y.; Yin L. Catalytic Asymmetric Construction of Halogenated Stereogenic Carbon Centers by Direct Vinylogous Mannich-Type Reaction. J. Am. Chem. Soc. 2018, 140, 15170–15175. 10.1021/jacs.8b09484. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Yang Y.; Huang Y.; Xu X.-H.; Qing F.-L. Regio- and Diastereoselective Vinylogous Mannich Addition of 3-Alkenyl-2-Oxindoles to α-Fluoroalkyl Aldimines. Synlett 2014, 26, 67–72. 10.1055/s-0034-1379600. [DOI] [Google Scholar]
- Lee H. J.; Lee S.; Yu J.; Kim J. N. Vinylogous N,N-Dimethylaminomethylenation of 3-[(1-Substituted)Ethylidene]-Oxindoles with N,N-Dimethylformamide Dimethylacetal. Tetrahedron Lett. 2014, 55, 2450–2454. 10.1016/j.tetlet.2014.02.130. [DOI] [Google Scholar]
- Curti C.; Battistini L.; Ranieri B.; Pelosi G.; Rassu G.; Casiraghi G.; Zanardi F. Anti-Selective, Catalytic Asymmetric Vinylogous Mukaiyama Mannich Reactions of Pyrrole-Based Silyl Dienolates with N-Aryl Aldimines. J. Org. Chem. 2011, 76, 2248–2252. 10.1021/jo1021234. [DOI] [PubMed] [Google Scholar]
- Ranieri B.; Curti C.; Battistini L.; Sartori A.; Pinna L.; Casiraghi G.; Zanardi F. Diastereo- and Enantioselective Catalytic Vinylogous Mukaiyama-Mannich Reactions of Pyrrole-Based Silyl Dienolates with Alkyl-Substituted Aldehydes. J. Org. Chem. 2011, 76, 10291–10298. 10.1021/jo201875a. [DOI] [PubMed] [Google Scholar]
- Carswell E. L.; Snapper M. L.; Hoveyda A. H. A Highly Efficient and Practical Method for Catalytic Asymmetric Vinylogous Mannich (AVM) Reactions. Angew. Chem., Int. Ed. 2006, 45, 7230–7233. 10.1002/anie.200603496. [DOI] [PubMed] [Google Scholar]
- Mandai H.; Mandai K.; Snapper M. L.; Hoveyda A. H. Three-Component Ag-Catalyzed Enantioselective Vinylogous Mannich and Aza-Diels–Alder Reactions with Alkyl-Substituted Aldehydes. J. Am. Chem. Soc. 2008, 130, 17961–17969. 10.1021/ja807243t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland L. C.; Vieira E. M.; Snapper M. L.; Hoveyda A. H. Ag-Catalyzed Diastereo- and Enantioselective Vinylogous Mannich Reactions of α-Ketoimine Esters. Development of a Method and Investigation of Its Mechanism. J. Am. Chem. Soc. 2009, 131, 570–576. 10.1021/ja8062315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori A.; Dell’Amico L.; Curti C.; Battistini L.; Pelosi G.; Rassu G.; Casiraghi G.; Zanardi F. Aqueous and Solvent-Free Uncatalyzed Three-Component Vinylogous Mukaiyama-Mannich Reactions of Pyrrole-Based Silyl Dienolates. Adv. Synth. Catal. 2011, 353, 3278–3284. 10.1002/adsc.201100572. [DOI] [Google Scholar]
- Sartori A.; Dell’Amico L.; Battistini L.; Curti C.; Rivara S.; Pala D.; Kerry P. S.; Pelosi G.; Casiraghi G.; Rassu G.; et al. Synthesis, Structure and Inhibitory Activity of a Stereoisomer of Oseltamivir Carboxylate. Org. Biomol. Chem. 2014, 12, 1561–1569. 10.1039/c3ob42069h. [DOI] [PubMed] [Google Scholar]
- Ye J.-L.; Chen H.; Zhang Y.-F.; Huang P.-Q. A Versatile Access to Vicinal Diamine Motifs by Highly Anti-Selective Asymmetric Vinylogous Mannich Reactions: An Efficient Total Synthesis of (+)-Absouline. Org. Chem. Front. 2016, 3, 683–692. 10.1039/C6QO00022C. [DOI] [Google Scholar]
- Ye J.-L.; Liu Y.; Zhang Y.-F.; Yang Z.-P.; Huang P.-Q. Studies on the Second-Generation Approach to Loline Alkaloids: Synthesis of N-Bus-Norloline through N-tert-Butanesulfinyl Imine Based Asymmetric Vinylogous Mannich Reaction. Synthesis 2016, 48, 1684–1692. 10.1055/s-0035-1561432. [DOI] [Google Scholar]
- Ranieri B.; Sartori A.; Curti C.; Battistini L.; Rassu G.; Pelosi G.; Casiraghi G.; Zanardi F. 3-Alkenyl-2-Silyloxyindoles in Vinylogous Mannich Reactions: Synthesis of Aminated Indole-Based Scaffolds and Products. Org. Lett. 2014, 16, 932–935. 10.1021/ol4036598. [DOI] [PubMed] [Google Scholar]
- Li T.-Z.; Xie J.; Jiang Y.; Sha F.; Wu X.-Y. Enantioselective Vinylogous Michael/Cyclization Cascade Reaction of Acyclic β,γ-Unsaturated Amides with Isatylidene Malononitriles: Asymmetric Construction of Spirocyclic Oxindoles. Adv. Synth. Catal. 2015, 357, 3507–3511. 10.1002/adsc.201500469. [DOI] [Google Scholar]
- Boyce G. R.; Johnson J. S. An Asymmetric Vinylogous Michael Cascade of Silyl Glyoximide, Vinyl Grignard, and Nitroalkenes via Long Range Stereoinduction. J. Org. Chem. 2016, 81, 1712–1717. 10.1021/acs.joc.5b02693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.; Cui H.-L.; Xu S.; Wu L.; Chen Y.-C. Organocatalytic Direct Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactam to α,β-Unsaturated Aldehydes and an Illustration to Scaffold Diversity Synthesis. Chem. - Eur. J. 2010, 16, 10309–10312. 10.1002/chem.201001350. [DOI] [PubMed] [Google Scholar]
- Shepherd N. E.; Tanabe H.; Xu Y.; Matsunaga S.; Shibasaki M. Direct Catalytic Asymmetric Vinylogous Mannich-Type and Michael Reactions of an α,β-Unsaturated γ-Butyrolactam under Dinuclear Nickel Catalysis. J. Am. Chem. Soc. 2010, 132, 3666–3667. 10.1021/ja1002636. [DOI] [PubMed] [Google Scholar]
- Tanabe H.; Xu Y.; Sun B.; Matsunaga S.; Shibasaki M. Direct Catalytic Asymmetric Vinylogous Michael Reaction of α,β-Unsaturated γ-Butyrolactam under Dinuclear Nickel Schiff Base Catalysis. Heterocycles 2012, 86, 611–622. 10.3987/COM-12-S(N)58. [DOI] [Google Scholar]
- Lin L.; Zhang J.; Ma X.; Fu X.; Wang R. Bifunctional 3,3′-Ph2-BINOL-Mg Catalyzed Direct Asymmetric Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactam. Org. Lett. 2011, 13, 6410–6413. 10.1021/ol202713f. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Shao Y.-L.; Xu H.-S.; Wang W. Organocatalytic Direct Asymmetric Vinylogous Michael Reaction of an α,β-Unsaturated γ-Butyrolactam with Enones. J. Org. Chem. 2011, 76, 1472–1474. 10.1021/jo102223v. [DOI] [PubMed] [Google Scholar]
- Okino T.; Hoashi Y.; Takemoto Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. 10.1021/ja036972z. [DOI] [PubMed] [Google Scholar]
- Okino T.; Hoashi Y.; Furukawa T.; Xu X.; Takemoto Y. Enantio- and Diastereoselective Michael Reaction of 1,3-Dicarbonyl Compounds to Nitroolefins Catalyzed by a Bifunctional Thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. 10.1021/ja044370p. [DOI] [PubMed] [Google Scholar]
- Hamza A.; Schubert G.; Soós T.; Pápai I. Theoretical Studies on the Bifunctionality of Chiral Thiourea-Based Organocatalysts: Competing Routes to C–C Bond Formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. 10.1021/ja063201x. [DOI] [PubMed] [Google Scholar]
- Zhu J.-L.; Zhang Y.; Liu C.; Zheng A.-M.; Wang W. Insights into the Dual Activation Mechanism Involving Bifunctional Cinchona Alkaloid Thiourea Organocatalysts: An NMR and DFT Study. J. Org. Chem. 2012, 77, 9813–9825. 10.1021/jo302133n. [DOI] [PubMed] [Google Scholar]
- Huang H.; Jin Z.; Zhu K.; Liang X.; Ye J. Highly Diastereo- and Enantioselective Synthesis of 5-Substituted 3-Pyrrolidin-2-Ones: Vinylogous Michael Addition under Multifunctional Catalysis. Angew. Chem., Int. Ed. 2011, 50, 3232–3235. 10.1002/anie.201008255. [DOI] [PubMed] [Google Scholar]
- Choudhury A. R.; Mukherjee S. Organocatalytic Asymmetric Direct Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactam to Nitroolefins. Org. Biomol. Chem. 2012, 10, 7313–7320. 10.1039/c2ob25832c. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Dong S.; Liu X.; Lin L.; Feng X. Chiral Guanidine-Catalyzed Asymmetric Direct Vinylogous Michael Reaction of α,β-Unsaturated γ-Butyrolactams with Alkylidene Malonates. Chem. Commun. 2012, 48, 5040–5042. 10.1039/c2cc31470c. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Liu X.; Zhou L.; Lin L.; Feng X. Bifunctional Guanidine via an Amino Amide Skeleton for Asymmetric Michael Reactions of β-Ketoesters with Nitroolefins: A Concise Synthesis of Bicyclic β-Amino Acids. Angew. Chem., Int. Ed. 2009, 48, 5195–5198. 10.1002/anie.200901337. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Liu X.; Ma X.; Wang R. Organocatalyzed Asymmetric Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactam. Chem. Commun. 2013, 49, 9329–9331. 10.1039/c3cc44059a. [DOI] [PubMed] [Google Scholar]
- Chen Y.-R.; Das U.; Liu M.-H.; Lin W. Organocatalytic Enantioselective Direct Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactam to β-Acyl Acrylates and 1,2-Diacylethylenes. J. Org. Chem. 2015, 80, 1985–1992. 10.1021/jo5027047. [DOI] [PubMed] [Google Scholar]
- Wang Z.-H.; Wu Z.-J.; Yue D.-F.; You Y.; Xu X.-Y.; Zhang X.-M.; Yuan W.-C. Enantioselective Synthesis of Chiral α,β-Unsaturated γ-Substituted Butyrolactams by Organocatalyzed Direct Asymmetric Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactam to 2-Enoylpyridines. Org. Biomol. Chem. 2016, 14, 6568–6576. 10.1039/C6OB01191H. [DOI] [PubMed] [Google Scholar]
- Arlt A.; Toyama H.; Takada K.; Hashimoto T.; Maruoka K. Phase-Transfer Catalyzed Asymmetric Synthesis of α,β-Unsaturated γ,γ-Disubstituted γ-Lactams. Chem. Commun. 2017, 53, 4779–4782. 10.1039/C7CC01058C. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Liu L.; Zhang P.; Zhong Y.; Wang R. Catalytic Asymmetric β,γ Activation of α,β-Unsaturated γ-Butyrolactams: Direct Approach to β,γ-Functionalized Dihydropyranopyrrolidin-2-ones. Angew. Chem., Int. Ed. 2013, 52, 11329–11333. 10.1002/anie.201302622. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Wang L.; Kai M.; Zhu L.; Yao X.; Wang R. Asymmetric Inverse-Electron-Demand Hetero-Diels-Alder Reaction for the Construction of Bicyclic Skeletons with Multiple Stereocenters by Using a Bifunctional Organocatalytic Strategy: An Efficient Approach to Chiral Macrolides. Chem. - Eur. J. 2012, 18, 11465–11473. 10.1002/chem.201201102. [DOI] [PubMed] [Google Scholar]
- Gu X.; Guo T.; Dai Y.; Franchino A.; Fei J.; Zou C.; Dixon D. J.; Ye J. Direct Catalytic Asymmetric Doubly Vinylogous Michael Addition of α,β-Unsaturated γ-Butyrolactams to Dienones. Angew. Chem., Int. Ed. 2015, 54, 10249–10253. 10.1002/anie.201504276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzakis D.; Sofiadis M.; Triantafyllakis M.; Daskalakis K.; Vassilikogiannakis G. Asymmetric and Site-Selective [3 + 2]-Annulations for the Synthesis of High-Value Bicyclic Lactams. Org. Lett. 2018, 20, 1146–1149. 10.1021/acs.orglett.8b00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joie C.; Deckers K.; Raabe G.; Enders D. An Asymmetric Organocatalytic Quadruple Cascade to Tetraaryl-Substituted 2-Azabicyclo[3.3.0]octadienones. Synthesis 2014, 46, 1539–1546. 10.1055/s-0033-1340982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvakumar K.; Vaithiyanathan V.; Shanmugam P. An Efficient Stereoselective Synthesis of 3-Spirocyclopentene- and 3-Spiropyrazole-2-Oxindoles via 1,3-Dipolar Cycloaddition Reaction. Chem. Commun. 2010, 46, 2826–2828. 10.1039/b924066g. [DOI] [PubMed] [Google Scholar]
- Lingam K.; Shanmugam P.; Selvakumar K. Stereoselective Synthesis of Geometrically Strained, Oxindole-Appended Vinyl Cyclopropanes and Highly Substituted Cyclopentenes via Sulfur Ylide Cyclopropanation and Vinyl Cyclopropane Rearrangement. Synlett 2012, 2012, 278–284. 10.1055/s-0031-1290077. [DOI] [Google Scholar]
- Curti C.; Rassu G.; Zambrano V.; Pinna L.; Pelosi G.; Sartori A.; Battistini L.; Zanardi F.; Casiraghi G. Bifunctional Cinchona Alkaloid/Thiourea Catalyzes Direct and Enantioselective Vinylogous Michael Addition of 3-Alkylidene Oxindoles to Nitroolefins. Angew. Chem., Int. Ed. 2012, 51, 6200–6204. 10.1002/anie.201202027. [DOI] [PubMed] [Google Scholar]
- Rassu G.; Zambrano V.; Pinna L.; Curti C.; Battistini L.; Sartori A.; Pelosi G.; Zanardi F.; Casiraghi G. Direct Regio-, Diastereo-, and Enantioselective Vinylogous Michael Addition of Prochiral 3-Alkylideneoxindoles to Nitroolefins. Adv. Synth. Catal. 2013, 355, 1881–1886. 10.1002/adsc.201300168. [DOI] [Google Scholar]
- Tan B.; Lu Y.; Zeng X.; Chua P. J.; Zhong G. Facile Domino Access to Chiral Bicyclo[3.2.1]Octanes and Discovery of a New Catalytic Activation Mode. Org. Lett. 2010, 12, 2682–2685. 10.1021/ol1007795. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Wang G.; Jiang X.; Xu Z.; Lin L.; Wang R. Highly Enantioselective Organocatalyzed Vinylogous Michael-Type Reaction for the Construction of Trifluoromethylated All-Carbon Quaternary Stereocenters. Org. Lett. 2014, 16, 1394–1397. 10.1021/ol500157b. [DOI] [PubMed] [Google Scholar]
- Zhong Y.; Ma S.; Xu Z.; Chang M.; Wang R. Organocatalytic Asymmetric Vinylogous Michael Addition of 3-Alkylidene Oxindoles to α-Substituted β-Nitroacrylates: Facile Construction of a Chiral All-Carbon Quaternary Center. RSC Adv. 2014, 4, 49930–49933. 10.1039/C4RA09128K. [DOI] [Google Scholar]
- Di Iorio N.; Righi P.; Ranieri S.; Mazzanti A.; Margutta R. G.; Bencivenni G. Vinylogous Reactivity of Oxindoles Bearing Nonsymmetric 3-Alkylidene Groups. J. Org. Chem. 2015, 80, 7158–7171. 10.1021/acs.joc.5b01022. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Wang H.-F.; Chen W.-Q.; Chen W.-X.; Chen F.-E. Organocatalytic Asymmetric Vinylogous Michael Addition of 3-(2-Oxoindolin-3-Ylidene)Butanoates to Nitroalkenes Catalyzed by a Bifunctional Cinchona-Based Squaramide. Asian J. Org. Chem. 2015, 4, 619–621. 10.1002/ajoc.201500111. [DOI] [Google Scholar]
- Curti C.; Battistini L.; Sartori A.; Rassu G.; Pelosi G.; Lombardo M.; Zanardi F. (E)-3-(Alkoxycarbonyl-2-Alkyliden)-2-Oxindoles: Multidentate Pronucleophiles for the Organocatalytic, Vinylogous Michael Addition to Nitroolefins. Adv. Synth. Catal. 2018, 360, 711–721. 10.1002/adsc.201701164. [DOI] [Google Scholar]
- Xiao X.; Mei H.; Chen Q.; Zhao X.; Lin L.; Liu X.; Feng X. Direct Asymmetric Vinylogous Michael Addition of 3-Alkylidene Oxindoles to Chalcones Catalyzed by a Chiral N,N′-Dioxide Ytterbium(III) Complex. Chem. Commun. 2015, 51, 580–583. 10.1039/C4CC07204A. [DOI] [PubMed] [Google Scholar]
- Mei H.; Lin L.; Wang L.; Dai L.; Liu X.; Feng X. Highly Regio-, Diastereo- and Enantioselective Deracemization of Axially Chiral 3-Alkylideneoxindoles. Chem. Commun. 2017, 53, 8763–8766. 10.1039/C7CC05164F. [DOI] [PubMed] [Google Scholar]
- Feng J.; Li X.; Cheng J.-P. Enantioselective Organocatalyzed Vinylogous Michael Reactions of 3-Alkylidene Oxindoles with Enals. J. Org. Chem. 2017, 82, 1412–1419. 10.1021/acs.joc.6b02582. [DOI] [PubMed] [Google Scholar]
- Feng J.; Li X. Enantioselective Vinylogous Michael–Michael Cascade Reactions of 3-Alkylidene Oxindoles and Nitroolefin Enoates. J. Org. Chem. 2017, 82, 7317–7323. 10.1021/acs.joc.7b00938. [DOI] [PubMed] [Google Scholar]
- Ramesh P.; Rao K. S.; Trivedi R.; Kumar B. S.; Prakasham R. S.; Sridhar B. Highly Efficient Regio and Diastereoselective Synthesis of Functionalized Bis-Spirooxindoles and Their Antibacterial Properties. RSC Adv. 2016, 6, 26546–26552. 10.1039/C6RA00613B. [DOI] [Google Scholar]
- Sun Q.; Li X.; Su J.; Zhao L.; Ma M.; Zhu Y.; Zhao Y.; Zhu R.; Yan W.; Wang K.; et al. The Squaramide-Catalyzed 1,3-Dipolar Cycloaddition of Nitroalkenes with N-2,2,2-Trifluoroethylisatin Ketimines: An Approach for the Synthesis of 5′-Trifluoromethyl-Spiro[Pyrrolidin-3,2′-Oxindoles]. Adv. Synth. Catal. 2015, 357, 3187–3196. 10.1002/adsc.201500416. [DOI] [Google Scholar]
- Chen Y.-R.; Zhan G.; Du W.; Chen Y.-C. Regioselective Asymmetric Formal (3 + 2) Cycloadditions of Nitrone Ylides from Isatins and Enals. Adv. Synth. Catal. 2016, 358, 3759–3764. 10.1002/adsc.201600805. [DOI] [Google Scholar]
- Zhan G.; Shi M.-L.; Lin W.-J.; Ouyang Q.; Du W.; Chen Y.-C. Direct Asymmetric Aza-Vinylogous-Type Michael Additions of Nitrones from Isatins to Nitroalkenes. Chem. - Eur. J. 2017, 23, 6286–6289. 10.1002/chem.201701290. [DOI] [PubMed] [Google Scholar]
- Liu C.; Tan B.-X.; Jin J.-L.; Zhang Y.-Y.; Dong N.; Li X.; Cheng J.-P. Chiral Biscinchona Alkaloid Promoted Asymmetric Allylic Alkylation of 3-Substituted Benzofuran-2(3H)-Ones with Morita–Baylis–Hillman Carbonates. J. Org. Chem. 2011, 76, 5838–5845. 10.1021/jo200557b. [DOI] [PubMed] [Google Scholar]
- Feng J.; Li X.; Cheng J.-P. An Asymmetric Allylic Alkylation Reaction of 3-Alkylidene Oxindoles. Chem. Commun. 2015, 51, 14342–14345. 10.1039/C5CC06182B. [DOI] [PubMed] [Google Scholar]
- Li X.; Su J.; Liu Z.; Zhu Y.; Dong Z.; Qiu S.; Wang J.; Lin L.; Shen Z.; Yan W.; et al. Synthesis of Chiral α-Trifluoromethylamines with 2,2,2-Trifluoroethylamine as a “Building Block. Org. Lett. 2016, 18, 956–959. 10.1021/acs.orglett.5b03566. [DOI] [PubMed] [Google Scholar]
- Du Y.; Lu X.; Zhang C. A Catalytic Carbon–Phosphorus Ylide Reaction: Phosphane-Catalyzed Annulation of Allylic Compounds with Electron-Deficient Alkenes. Angew. Chem., Int. Ed. 2003, 42, 1035–1037. 10.1002/anie.200390266. [DOI] [PubMed] [Google Scholar]
- Zhan G.; Shi M.-L.; He Q.; Lin W.-J.; Ouyang Q.; Du W.; Chen Y.-C. Catalyst-Controlled Switch in Chemo- and Diastereoselectivities: Annulations of Morita-Baylis-Hillman Carbonates from Isatins. Angew. Chem., Int. Ed. 2016, 55, 2147–2151. 10.1002/anie.201510825. [DOI] [PubMed] [Google Scholar]
- Wang K.-K.; Wang P.; Ouyang Q.; Du W.; Chen Y.-C. Substrate-Controlled Switchable Asymmetric Annulations to Access Polyheterocyclic Skeletons. Chem. Commun. 2016, 52, 11104–11107. 10.1039/C6CC06148F. [DOI] [PubMed] [Google Scholar]
- Rassu G.; Zambrano V.; Pinna L.; Curti C.; Battistini L.; Sartori A.; Pelosi G.; Casiraghi G.; Zanardi F. Direct and Enantioselective Vinylogous Michael Addition of α-Alkylidenepyrazolinones to Nitroolefins Catalyzed by Dual Cinchona Alkaloid Thioureas. Adv. Synth. Catal. 2014, 356, 2330–2336. 10.1002/adsc.201300964. [DOI] [Google Scholar]
- Yetra S. R.; Mondal S.; Mukherjee S.; Gonnade R. G.; Biju A. T. Enantioselective Synthesis of Spirocyclohexadienones by NHC-Catalyzed Formal [3 + 3] Annulation Reaction of Enals. Angew. Chem., Int. Ed. 2016, 55, 268–272. 10.1002/anie.201507802. [DOI] [PubMed] [Google Scholar]
- Mondal S.; Mukherjee S.; Yetra S. R.; Gonnade R. G.; Biju A. T. Organocatalytic Enantioselective Vinylogous Michael-Aldol Cascade for the Synthesis of Spirocyclic Compounds. Org. Lett. 2017, 19, 4367–4370. 10.1021/acs.orglett.7b02085. [DOI] [PubMed] [Google Scholar]
- Leng H.-J.; Li Q.-Z.; Zeng R.; Dai Q.-S.; Zhu H.-P.; Liu Y.; Huang W.; Han B.; Li J.-L. Asymmetric Construction of Spiropyrazolone Skeletons via Amine-Catalyzed [3 + 3] Annulation. Adv. Synth. Catal. 2018, 360, 229–234. 10.1002/adsc.201701035. [DOI] [Google Scholar]
- Liu J.-Y.; Zhao J.; Zhang J.-L.; Xu P.-F. Quaternary Carbon Center Forming Formal [3 + 3] Cycloaddition Reaction via Bifunctional Catalysis: Asymmetric Synthesis of Spirocyclohexene Pyrazolones. Org. Lett. 2017, 19, 1846–1849. 10.1021/acs.orglett.7b00610. [DOI] [PubMed] [Google Scholar]
- Yang W.; Sun W.; Zhang C.; Wang Q.; Guo Z.; Mao B.; Liao J.; Guo H. Lewis-Base-Catalyzed Asymmetric [3 + 3] Annulation Reaction of Morita–Baylis–Hillman Carbonates: Enantioselective Synthesis of Spirocyclohexenes. ACS Catal. 2017, 7, 3142–3146. 10.1021/acscatal.7b00320. [DOI] [Google Scholar]
- Basu S.; Gupta V.; Nickel J.; Schneider C. Organocatalytic Enantioselective Vinylogous Michael Reaction of Vinylketene Silyl-N,O-Acetals. Org. Lett. 2014, 16, 274–277. 10.1021/ol403275k. [DOI] [PubMed] [Google Scholar]
- Battistini L.; Dell’Amico L.; Sartori A.; Curti C.; Pelosi G.; Casiraghi G.; Attanasi O. A.; Favi G.; Zanardi F. On-Water Vinylogous Mukaiyama-Michael Addition of Heterocyclic 2-Silyloxydienes to 1,2-Diaza-1,3-Dienes: One-Pot Three-Step Entry to Functionality-Rich Pyrroles. Adv. Synth. Catal. 2011, 353, 1966–1972. 10.1002/adsc.201100296. [DOI] [Google Scholar]
- Xie K.-X.; Zhang Z.-P.; Li X. Bismuth Triflate-Catalyzed Vinylogous Nucleophilic 1,6-Conjugate Addition of para-Quinone Methides with 3-Propenyl-2-Silyloxyindoles. Org. Lett. 2017, 19, 6708–6711. 10.1021/acs.orglett.7b03433. [DOI] [PubMed] [Google Scholar]
- Fu X.; Bai H.-Y.; Zhu G.-D.; Huang Y.; Zhang S.-Y. Metal-Controlled, Regioselective, Direct Intermolecular α- or γ-Amination with Azodicarboxylates. Org. Lett. 2018, 20, 3469–3472. 10.1021/acs.orglett.8b01183. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Chen W.-X.; Chen F.-E. Asymmetric Amination of 3-(2-Oxoindolin-3-Ylidene)Butanoates Catalyzed by a Cinchona-Derived Alkaloid. Asian J. Org. Chem. 2015, 4, 1044–1046. 10.1002/ajoc.201500299. [DOI] [Google Scholar]
- Takahashi Y.; Otsuka M.; Harachi M.; Mukaeda Y.; Hosokawa S. Stereoselective Acylation of the E,E -Vinylketene Silyl N,O-Acetal and Its Application to the Synthesis of Khafrefungin. Org. Lett. 2014, 16, 4106–4109. 10.1021/ol501805j. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Kubota K.; Ieki T.; Hosokawa S. Stereoselective Alkylation of the Vinylketene Silyl N,O-Acetal and Its Application to the Synthesis of Mycocerosic Acid. Org. Lett. 2016, 18, 132–135. 10.1021/acs.orglett.5b03422. [DOI] [PubMed] [Google Scholar]
- Sekiya S.; Okumura M.; Kubota K.; Nakamura T.; Sekine D.; Hosokawa S. Remote Asymmetric Bromination Reaction with Vinylketene Silyl N,O-Acetal and Its Application to Total Synthesis of Pellasoren A. Org. Lett. 2017, 19, 2394–2397. 10.1021/acs.orglett.7b00920. [DOI] [PubMed] [Google Scholar]
- Jadhav A. P.; Ali A.; Singh R. P. Vinylogous Nucleophilic Substitution of the Hydroxy Group in Diarylmethanols with 3-Propenyl-2-Silyloxyindoles: Towards the Synthesis of α-Alkylidene-δ-Diaryl-2-Oxindoles. Adv. Synth. Catal. 2017, 359, 1508–1514. 10.1002/adsc.201601265. [DOI] [Google Scholar]
- Cui H. L.; Chen Y. C. α,α-Dicyanoalkenes: Versatile Vinylogous Nucleophiles for Organic Synthesis. Chem. Commun. 2009, 4479–4486. 10.1039/b906201g. [DOI] [PubMed] [Google Scholar]
- Kisanga P. B.; Verkade J. G. P(RNCH2CH2)3N-Catalyzed 1,2-Addition Reactions of Activated Allylic Synthons. J. Org. Chem. 2002, 67, 426–430. 10.1021/jo0106492. [DOI] [PubMed] [Google Scholar]
- Aydin J.; Szabó K. J. Palladium-Pincer Complex Catalyzed C-C Coupling of Allyl Nitriles with Tosyl Imines via Regioselective Allylic C-H Bond Functionalization. Org. Lett. 2008, 10, 2881–2884. 10.1021/ol801070n. [DOI] [PubMed] [Google Scholar]
- Yazaki R.; Nitabaru T.; Kumagai N.; Shibasaki M. Direct Catalytic Asymmetric Addition of Allylic Cyanides to Ketoimines. J. Am. Chem. Soc. 2008, 130, 14477–14479. 10.1021/ja806572b. [DOI] [PubMed] [Google Scholar]
- Lewy D. S.; Gauss C.-M.; Soenen D. R.; Boger D. L. Fostriecin: Chemistry and Biology. Curr. Med. Chem. 2002, 9, 2005–2032. 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]
- Saito A.; Kumagai N.; Shibasaki M. Concise Enantioselective Synthesis of δ,δ-Disubstituted δ-Valerolactones. Tetrahedron Lett. 2014, 55, 3167–3171. 10.1016/j.tetlet.2014.04.006. [DOI] [Google Scholar]
- Babu T.; Karthik K.; Perumal P. A Facile, One-Pot Synthesis of Functionalized Spiro-Oxindoles via Vinylogous Aldol Reaction of Vinyl Malononitriles with Isatin Derivatives in Aqueous Media. Synlett 2010, 2010, 1128–1132. 10.1055/s-0029-1219588. [DOI] [Google Scholar]
- Yu B.; Qi P.-P.; Shi X.-J.; Shan L.-H.; Yu D.-Q.; Liu H.-M. Discovery of Novel Steroidal Pyran–Oxindole Hybrids as Cytotoxic Agents. Steroids 2014, 88, 44–52. 10.1016/j.steroids.2014.05.022. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Wilson T. W. Lewis Base Catalyzed Enantioselective Additions of an N-Silyl Vinylketene Imine. Angew. Chem., Int. Ed. 2012, 51, 3236–3239. 10.1002/anie.201108795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. Y.; Cui H. L.; Long J.; Li B. J.; Wu Y.; Ding L. S.; Chen Y. C. Organocatalytic and Highly Stereoselective Direct Vinylogous Mannich Reaction. J. Am. Chem. Soc. 2007, 129, 1878–1879. 10.1021/ja068703p. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Li Y.; Meng Q.; Li X. An Organocatalytic Enantioselective Vinylogous Mannich Reaction of α,α-Dicyanoolefins with Isatin: N-Boc Ketimines. Org. Chem. Front. 2016, 3, 709–713. 10.1039/C6QO00038J. [DOI] [Google Scholar]
- Cheng C.; Lu X.; Ge L.; Chen J.; Cao W.; Wu X.; Zhao G. Effective Asymmetric Vinylogous Mannich Reaction of Isatin Imines with α,α-Dicyanoolefins in the Presence of a Simple Chiral Amide Phosphonium Bifunctional Phase Transfer Catalyst. Org. Chem. Front. 2017, 4, 101–114. 10.1039/C6QO00546B. [DOI] [Google Scholar]
- Sanz-Vidal Á.; Torres J.; Soloshonok V. A.; Zhu Y.; Han J.; Fustero S.; del Pozo C. Asymmetric Vinylogous Mannich-Type Addition of α,α-Dicyanoalkenes to α-Fluoroalkyl Sulfinyl Imines. Adv. Synth. Catal. 2018, 360, 366–373. 10.1002/adsc.201701284. [DOI] [Google Scholar]
- Bock C. M.; Parameshwarappa G.; Bönisch S.; Neiss C.; Bauer W.; Hampel F.; Görling A.; Tsogoeva S. B. Generation of Complex Azabicycles and Carbobicycles from Two Simple Compounds in a Single Operation through a Metal-Free Six-Step Domino Reaction. Chem. - Eur. J. 2016, 22, 5189–5197. 10.1002/chem.201504798. [DOI] [PubMed] [Google Scholar]
- Grau D.; Grau B. W.; Hampel F.; Tsogoeva S. B. Three-Component Domino Knoevenagel/Vinylogous Michael Reaction: Entry to Challenging o-Terphenyls. Chem. - Eur. J. 2018, 24, 6551–6556. 10.1002/chem.201800048. [DOI] [PubMed] [Google Scholar]
- Jia Q.; Wang J. N-Heterocyclic Carbene-Catalyzed Convenient Benzonitrile Assembly. Org. Lett. 2016, 18, 2212–2215. 10.1021/acs.orglett.6b00844. [DOI] [PubMed] [Google Scholar]
- Zhang C. L.; Gao Z. H.; Liang Z. Q.; Ye S. N-Heterocyclic Carbene-Catalyzed Synthesis of Multi-Substituted Benzenes from Enals and α-Cyano-β-Methylenones. Adv. Synth. Catal. 2016, 358, 2862–2866. 10.1002/adsc.201600531. [DOI] [Google Scholar]
- Zhang C. L.; Ye S. N-Heterocyclic Carbene-Catalyzed Construction of 1,3,5-Trisubstituted Benzenes from Bromoenals and α-Cyano-β-Methylenones. Org. Lett. 2016, 18, 6408–6411. 10.1021/acs.orglett.6b03306. [DOI] [PubMed] [Google Scholar]
- Cao D.; Ying A.; Mo H.; Chen D.; Chen G.; Wang Z.; Yang J. [4 + 2] Annulation of 3-Nitroindoles with Alkylidene Malononitriles: Entry to Substituted Carbazol-4-amine Derivatives. J. Org. Chem. 2018, 83, 12568–12574. 10.1021/acs.joc.8b01876. [DOI] [PubMed] [Google Scholar]
- Attanasi O. A.; Favi G.; Geronikaki A.; Mantellini F.; Moscatelli G.; Paparisva A. Synthesis of Densely Functionalized 3a,4-Dihydro-1H-Pyrrolo[1,2-b]Pyrazoles via Base Mediated Domino Reaction of Vinyl Malononitriles with 1,2-Diaza-1,3-dienes. Org. Lett. 2013, 15, 2624–2627. 10.1021/ol401336s. [DOI] [PubMed] [Google Scholar]
- Alizadeh A.; Hosseini S. Y.; Sedighian H.; Bayat F. Synthesis of Phenanthrene Derivatives through the Reaction of an α,α-Dicyanoolefin with α,β-Unsaturated Carbonyl Compounds. Helv. Chim. Acta 2015, 98, 1426–1434. 10.1002/hlca.201500120. [DOI] [Google Scholar]
- Alizadeh A.; Yasub S.; Sedighian H.; Bayat F. Synthesis of 5,6-Dihydroquinolines and Succinates via the Reaction of α,α-Dicyanoolefins and Acetylenic Esters in a Ratio of 2:1. Tetrahedron 2015, 71, 7885–7891. 10.1016/j.tet.2015.08.033. [DOI] [Google Scholar]
- Alizadeh A.; Hosseini S. Y.; Vahabi A. H. Substituent Effects and Structural Features of α,α-Dicyanoolefins and Ketene Dithioacetals on Directing Polyfunctionalized 2,6-Dicyanoanilines or Pyrazolo[1,5-a]pyridines. Synlett 2014, 25, 2474–2479. 10.1055/s-0034-1379041. [DOI] [Google Scholar]
- Shakibaei G. I.; Bazgir A. A Highly Efficient One-pot Synthesis of Indenopyridine-fused Spirocyclic Systems. RSC Adv. 2016, 6, 22306–22311. 10.1039/C6RA00869K. [DOI] [Google Scholar]
- Hari Babu T.; Abragam Joseph A.; Muralidharan D.; Perumal P. T. A Novel Method for the Synthesis of Functionalized Spirocyclic Oxindoles by One-pot Tandem Reaction of Vinyl Malononitriles with Isatylidene Malononitriles. Tetrahedron Lett. 2010, 51, 994–996. 10.1016/j.tetlet.2009.12.082. [DOI] [Google Scholar]
- Lu Y.; Sun J.; Xie Y.; Yan C. Molecular Diversity of the Cyclization Reaction of 3-Methyleneoxindoles with 2-(3,4-Dihydronaphthalen-1(2H)-ylidene)malononitriles. RSC Adv. 2016, 6, 23390–23395. 10.1039/C6RA00476H. [DOI] [Google Scholar]
- Xie Y. J.; Sun J.; Yan C. G. Domino Reactions of Vinyl Malononitriles with 3-Phenacylideneoxindoles for Efficient Synthesis of Functionalized Spirocyclic Oxindoles. ACS Comb. Sci. 2014, 16, 271–280. 10.1021/co500006c. [DOI] [PubMed] [Google Scholar]
- Yamuna E.; Zeller M.; Jayarampillai K.; Prasad R. Regioselective Cycloaddition of Acetylenic Esters: a One-Pot Synthesis of Novel Dihydrobenzo[a]Carbazoles. Tetrahedron Lett. 2011, 52, 1649–1652. 10.1016/j.tetlet.2011.01.125. [DOI] [Google Scholar]
- Kripalaya Ratheesh A.; Sparkes H. A.; Prasad K. J. R. A New Strategy for the Synthesis of Diverse Benzo[a]carbazoles via a Divergent Catalytic Michael Reaction. Org. Biomol. Chem. 2018, 16, 2527–2540. 10.1039/C8OB00154E. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Zhang J. Base-Catalyzed Tandem Michael/Dehydro-Diels-Alder Reaction of α,α-Dicyanoolefins with Electron-Deficient 1,3-Conjugated Enynes: A Facile Entry to Angularly Fused Polycycles. Chem. - Eur. J. 2014, 20, 399–404. 10.1002/chem.201302960. [DOI] [PubMed] [Google Scholar]
- Zhou R.; Wang J.; Tian J.; He Z. Phosphine-Catalyzed [4 + 2] Annulation and Vinylogous Addition Reactions between 1,4-Dien-3-Ones and 1,1-Dicyanoalkenes. Org. Biomol. Chem. 2012, 10, 773–781. 10.1039/C1OB06187A. [DOI] [PubMed] [Google Scholar]
- Kiruthika S. E.; Perumal P. T.; Balachandran C.; Ignacimuthu S. An Easy Protocol for the Domino Synthesis of Diversely Functionalized Spirocarbocycles and Their Biological Evaluation. J. Chem. Sci. 2014, 126, 177–185. 10.1007/s12039-013-0560-1. [DOI] [Google Scholar]
- Shi X.-M.; Dong W.-P.; Zhu L.-P.; Jiang X.-X.; Wang R. Asymmetric Vinylogous Michael Addition/Cyclization Cascade Reaction for the Construction of Diversely Structured Spiro- Oxindole Skeletons. Adv. Synth. Catal. 2013, 355, 3119–3123. 10.1002/adsc.201300329. [DOI] [Google Scholar]
- Huang X.-F.; Zhang Y.-F.; Qi Z.-H.; Li N.-K.; Geng Z.-C.; Li K.; Wang X.-W. Organocatalytic Enantioselective Construction of Multi-Functionalized Spiro Oxindole Dienes. Org. Biomol. Chem. 2014, 12, 4372–4385. 10.1039/c4ob00545g. [DOI] [PubMed] [Google Scholar]
- Reddy Gajulapalli V. P.; Vinayagam P.; Kesavan V. Enantioselective Assembly of Functionalized Carbocyclic Spirooxindoles Using an L-Proline Derived Thiourea Organocatalyst. RSC Adv. 2015, 5, 7370–7379. 10.1039/C4RA13711F. [DOI] [Google Scholar]
- Xue D.; Chen Y.-C.; Wang Q.-W.; Cun L.-F.; Zhu J.; Deng J.-G. Asymmetric Direct Vinylogous Michael Reaction of Activated Alkenes to Nitroolefins Catalyzed by Modified Cinchona Alkaloids. Org. Lett. 2005, 7, 5293–5296. 10.1021/ol052283b. [DOI] [PubMed] [Google Scholar]
- Poulsen T. B.; Bell M.; Jørgensen K. A. Organocatalytic Asymmetric Allylic Carbon-Carbon Bond Formation. Org. Biomol. Chem. 2006, 4, 63–70. 10.1039/B514564C. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Zheng H.-T.; Liu T.-Y.; Yue L.; Chen Y.-C. Asymmetric Direct Vinylogous Carbon-Carbon Bond Formation Catalyzed by Bifunctional Organocatalysts. Tetrahedron 2007, 63, 5123–5128. 10.1016/j.tet.2007.04.011. [DOI] [Google Scholar]
- Chen W.-Y.; Ouyang L.; Chen R.; Li X. Asymmetric Direct Vinylogous Michael Reaction Catalyzed by Self-Assembled Organocatalyst. Synth. Commun. 2012, 42, 2585–2594. 10.1080/00397911.2011.563023. [DOI] [Google Scholar]
- Dell’Amico L.; Rassu G.; Zambrano V.; Sartori A.; Curti C.; Battistini L.; Pelosi G.; Casiraghi G.; Zanardi F. Exploring the Vinylogous Reactivity of Cyclohexenylidene Malononitriles: Switchable Regioselectivity in the Organocatalytic Asymmetric Addition to Enals Giving Highly Enantioenriched Carbabicyclic Structures. J. Am. Chem. Soc. 2014, 136, 11107–11114. 10.1021/ja5054576. [DOI] [PubMed] [Google Scholar]
- Li Q.-Z.; Gu J.; Chen Y.-C. Organocatalytic Asymmetric [4 + 2] Formal Cycloadditions of Cyclohexenylidenemalononitriles and Enals to Construct Chiral Bicyclo[2.2.2]Octanes. RSC Adv. 2014, 4, 37522–37525. 10.1039/C4RA07709A. [DOI] [Google Scholar]
- Brindani N.; Rassu G.; Dell’Amico L.; Zambrano V.; Pinna L.; Curti C.; Sartori A.; Battistini L.; Casiraghi G.; Pelosi G.; et al. Organocatalytic, Asymmetric Eliminative [4 + 2] Cycloaddition of Allylidene Malononitriles with Enals: Rapid Entry to Cyclohexadiene-Embedding Linear and Angular Polycycles. Angew. Chem., Int. Ed. 2015, 54, 7386–7390. 10.1002/anie.201501894. [DOI] [PubMed] [Google Scholar]
- Rout S.; Ray S. K.; Unhale R. A.; Singh V. K. Asymmetric Direct Vinylogous Michael Addition to 2-Enoylpyridine N-Oxides Catalyzed by Bifunctional Thio-Urea. Org. Lett. 2014, 16, 5568–5571. 10.1021/ol5025794. [DOI] [PubMed] [Google Scholar]
- Li X.; Xu X.; Wei W.; Lin A.; Yao H. Organocatalyzed Asymmetric 1,6-Conjugate Addition of para-Quinone Methides with Dicyanoolefins. Org. Lett. 2016, 18, 428–431. 10.1021/acs.orglett.5b03471. [DOI] [PubMed] [Google Scholar]
- Tiseni P. S.; Peters R. Catalytic Asymmetric Formation of δ-Lactones from Unsaturated Acyl Halides. Chem. - Eur. J. 2010, 16, 2503–2517. 10.1002/chem.200902896. [DOI] [PubMed] [Google Scholar]
- Shen L.; Shao P.; Ye S. N-Heterocyclic Carbene-Catalyzed Cyclization of Unsaturated Acyl Chlorides and Ketones. Adv. Synth. Catal. 2011, 353, 1943–1948. 10.1002/adsc.201100178. [DOI] [Google Scholar]
- Jin Z.; Jiang K.; Fu Z.; Torres J.; Zheng P.; Yang S.; Song B.-A.; Chi Y. R. Nucleophilic β-Carbon Activation of Propionic Acid as a 3-Carbon Synthon by Carbene Organocatalysis. Chem. - Eur. J. 2015, 21, 9360–9363. 10.1002/chem.201501481. [DOI] [PubMed] [Google Scholar]
- Que Y.; Xie Y.; Li T.; Yu C.; Tu S.; Yao C. An N-Heterocyclic Carbene-Catalyzed Oxidative γ-Aminoalkylation of Saturated Carboxylic Acids through in Situ Activation Strategy: Access to δ-Lactam. Org. Lett. 2015, 17, 6234–6237. 10.1021/acs.orglett.5b03223. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Yu C.; Li T.; Wang Y.; Lu Y.; Wang W.; Yao C. N-Heterocyclic Carbene-Catalyzed [4 + 2] Cyclization of α,β-Unsaturated Carboxylic Acids Bearing γ-H with Isatins: An Enantioselective Synthesis of Spirocyclic Oxindole-Dihydropyranones. Org. Biomol. Chem. 2016, 14, 1485–1491. 10.1039/C5OB02160J. [DOI] [PubMed] [Google Scholar]
- Moccia M.; Wells R. J.; Adamo M. F. A. An Improved Procedure to Prepare 3-Methyl-4-Nitroalkylenethylisoxazoles and Their Reaction under Catalytic Enantioselective Michael Addition with Nitromethane. Org. Biomol. Chem. 2015, 13, 2192–2195. 10.1039/C4OB02109F. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wei B.-W.; Lin H.; Zhang L.; Liu J.-X.; Luo H.-Q.; Fan X.-L. On Water” Direct Catalytic Vinylogous Henry (Nitroaldol) Reactions of Isatins for the Efficient Synthesis of Isoxazole Substituted 3-Hydroxyindolin-2-Ones. Green Chem. 2015, 17, 3266–3270. 10.1039/C5GC00503E. [DOI] [Google Scholar]
- Nagaraju S.; Satyanarayana N.; Paplal B.; Vasu A. K.; Kanvah S.; Kashinath D. Synthesis of Functionalized Isoxazole-Oxindole Hybrids via on Water, Catalyst Free Vinylogous Henry and 1,6-Michael Addition Reactions. RSC Adv. 2015, 5, 81768–81773. 10.1039/C5RA14039K. [DOI] [Google Scholar]
- Zhu B.; Li F.; Lu B.; Chang J.; Jiang Z. Organocatalytic Enantioselective Vinylogous Aldol Reaction of 5-Alkyl-4-Nitroisoxazoles to Paraformaldehyde. J. Org. Chem. 2018, 83, 11350–11358. 10.1021/acs.joc.8b01573. [DOI] [PubMed] [Google Scholar]
- Xia X.; Zhu Q.; Wang J.; Chen J.; Cao W.; Zhu B.; Wu X. Direct Asymmetric Vinylogous Mannich Addition of 3,5-Disubstituted-4-Nitroisoxazoles to Isatin-Derived Imines Catalyzed by a Bifunctional Phase-Transfer-Catalyst. J. Org. Chem. 2018, 83, 14617–14625. 10.1021/acs.joc.8b02439. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wei B.; Lin H.; Cui W.; Zeng X.; Fan X. Organocatalyzed Asymmetric Vinylogous Michael Reactions of 3,5-Dialkyl-Substituted 4-Nitroisoxazoles: A Direct Method for the Synthesis of Chiral Isoxazole Derivatives. Adv. Synth. Catal. 2015, 357, 1299–1304. 10.1002/adsc.201400833. [DOI] [Google Scholar]
- Shi F.; Xing G. J.; Tao Z. L.; Luo S. W.; Tu S. J.; Gong L. Z. An Asymmetric Organocatalytic Povarov Reaction with 2-Hydroxystyrenes. J. Org. Chem. 2012, 77, 6970–6979. 10.1021/jo301174g. [DOI] [PubMed] [Google Scholar]
- Shi F.; Xing G. J.; Zhu R. Y.; Tan W.; Tu S. A Catalytic Asymmetric Isatin-Involved Povarov Reaction: Diastereo- and Enantioselective Construction of Spiro[Indolin-3,2’-Quinoline] Scaffold. Org. Lett. 2013, 15, 128–131. 10.1021/ol303154k. [DOI] [PubMed] [Google Scholar]
- Zhang H. H.; Wang Y. M.; Xie Y. W.; Zhu Z. Q.; Shi F.; Tu S. J. Organocatalytic Chemo-, (E/Z)- and Enantioselective Formal Alkenylation of Indole-Derived Hydroxylactams Using o -Hydroxystyrenes as a Source of Alkenyl Group. J. Org. Chem. 2014, 79, 7141–7151. 10.1021/jo501293m. [DOI] [PubMed] [Google Scholar]
- Nielsen A. J.; Jenkins H. A.; McNulty J. Asymmetric Organocatalytic Stepwise [2 + 2] Entry to Tetra-Substituted Heterodimeric and Homochiral Cyclobutanes. Chem. - Eur. J. 2016, 22, 9111–9115. 10.1002/chem.201601842. [DOI] [PubMed] [Google Scholar]
- Suga H.; Hashimoto Y.; Toda Y.; Fukushima K.; Esaki H.; Kikuchi A. Amine-Urea-Mediated Asymmetric Cycloadditions between Nitrile Oxides and o-Hydroxystyrenes by Dual Activation. Angew. Chem., Int. Ed. 2017, 56, 11936–11939. 10.1002/anie.201705662. [DOI] [PubMed] [Google Scholar]
- Feng W.; Yang H.; Wang Z.; Gou B. B.; Chen J.; Zhou L. Enantioselective [3 + 2] Formal Cycloaddition of 1-Styrylnaphthols with Quinones Catalyzed by a Chiral Phosphoric Acid. Org. Lett. 2018, 20, 2929–2933. 10.1021/acs.orglett.8b00988. [DOI] [PubMed] [Google Scholar]
- Zhang Y. C.; Jiang F.; Wang S. L.; Shi F.; Tu S. J. Organocatalytic Chemo- and Regioselective Oxyarylation of Styrenes via a Cascade Reaction: Remote Activation of Hydroxyl Groups. J. Org. Chem. 2014, 79, 6143–6152. 10.1021/jo500859b. [DOI] [PubMed] [Google Scholar]
- Raja A.; Hong B.-C.; Lee G.-H. Organocatalytic Enantioselective Michael–Michael–Michael–Aldol Condensation Reactions: Control of Five Stereocenters in a Quadruple-Cascade Asymmetric Synthesis of Highly Functionalized Hexahydrophenanthrenes. Org. Lett. 2014, 16, 5756–5759. 10.1021/ol502821e. [DOI] [PubMed] [Google Scholar]
- Dell’Amico L.; Companyõ X.; Naicker T.; Bräuer T. M.; Jørgensen K. A. Asymmetric Organocatalytic Benzylation of α,β-Unsaturated Aldehydes with Toluenes. Eur. J. Org. Chem. 2013, 2013, 5262–5265. 10.1002/ejoc.201300899. [DOI] [Google Scholar]
- Li T.; Zhu J.; Wu D.; Li X.; Wang S.; Li H.; Li J.; Wang W. A Strategy Enabling Enantioselective Direct Conjugate Addition of Inert Aryl Methane Nucleophiles to Enals with a Chiral Amine Catalyst under Mild Conditions. Chem. - Eur. J. 2013, 19, 9147–9150. 10.1002/chem.201300304. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Lei Q.; Huang X.; Cui H.-L.; Zhou X.; Chen Y.-C. Lewis Base Assisted Brønsted Base Catalysis: Direct Regioselective Asymmetric Vinylogous Alkylation of Allylic Sulfones. Chem. - Eur. J. 2011, 17, 9489–9493. 10.1002/chem.201100534. [DOI] [PubMed] [Google Scholar]
- Duan G.-J.; Ling J.-B.; Wang W.-P.; Luo Y.-C.; Xu P.-F. Organocatalytic Formal [2 + 2] Cycloaddition Initiated by Vinylogous Friedel–Crafts Alkylation: Enantioselective Synthesis of Substituted Cyclobutane Derivatives. Chem. Commun. 2013, 49, 4625–4627. 10.1039/c3cc41785a. [DOI] [PubMed] [Google Scholar]
- Philipps A.; Fritze L.; Erdmann N.; Enders D. An Asymmetric Organocatalytic Quadruple Domino Reaction Employing a Vinylogous Friedel–Crafts/Michael/Michael/Aldol Condensation Sequence. Synthesis 2015, 47, 2377–2384. 10.1055/s-0034-1380197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.